
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu catalysts supported on CaO/MgO for glycerol conversion to lactic acid in alkaline medium employing a continuous flow reaction system
Arthur M. Bruno,
Thiago D. R. Simões ,
Mariana M. V. M. Souza and
Robinson L. Manfro*
,
Mariana M. V. M. Souza and
Robinson L. Manfro*
Escola de Química, Universidade Federal do Rio de Janeiro (UFRJ), Centro de Tecnologia, Bloco E, Sala 206, CEP 21941-909, Rio de Janeiro/RJ, Brazil. E-mail: robinson@eq.ufrj.br; Tel: +55-21-39387643
First published on 24th August 2020
Abstract
The production of lactic acid (LA) from glycerol in alkaline medium was investigated using Cu catalysts supported on CaO, MgO and xCaO/MgO (x = 5, 10, 15 wt%), employing a continuous flow reaction system over a period of 30 h. In addition to assessing the effect of the composition of the catalytic support, the influence of the temperature (200–260 °C), NaOH/glycerol molar ratio (0.5–1.5), hydroxide type (NaOH and KOH), as well as the influence of concentration (10 and 20 vol%) and purity of glycerol was investigated. The catalysts were prepared by a wet impregnation method and characterized by XRF, XRD, N2 adsorption–desorption, H2-TPR and CO2-TPD. The catalytic tests showed that the use of NaOH results in higher yields to LA. Cu catalysts supported on xCaO/MgO exhibited better catalytic performance than the CuCa and CuMg catalysts. The LA yield increases with the increase of the reaction temperature from 200 to 240 °C, and then decreases with a subsequent increase to 260 °C. NaOH/glycerol molar ratios greater than 1.25 are not necessary, since high yield to LA (96.9%) was obtained in the catalytic test performed using a molar ratio of 1.25. The catalysts showed excellent stability without evidence of deactivation over the evaluated period.
1. Introduction
2-Hydroxypropanoic acid, also named lactic acid (LA), is widely used in the cosmetic industry as a moisturizer and in the textile industry as a mordant (i.e. a chemical that enhances the color durability). Moreover, LA is used in the dairy industry as a pH regulator, preservative or even as an inhibitor of bacterial spoilage. It is currently being used as a precursor of “green” solvents such as ethyl lactate, and also employed in the synthesis of poly lactic acid (PLA), which is increasingly used in the production of biodegradable packaging.1 In addition, LA is considered a platform molecule from which valuable industrial oxygenated compounds are obtained such as: acrylic acid, 2,3-pentanedione, pyruvic acid, propanoic acid, 1,2-propanediol and acetaldehyde.2Market studies conducted in the early 2000s pointed to a growth of 19% per year in the consumption of LA for chemical applications.3 The worldwide consumption of LA in 2005 was estimated at 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–150000 tons;3 in the year 2010 the worldwide demand for LA was around of 300000–400000 tons.4 The global LA market is expected to grow from 714.2 kilotons in 2013 to 1960.1 kilotons in 2020.5 According to the report of Global View Research (California, USA), the production of PLA is expected to direct the increased demand for LA in the coming years. The global LA market size was valued at 1.29 billion USD in 2016 and is expected to reach 9.8 billion USD in 2025.5 European demand for PLA in 2010–2012 was around of 25000 tons per year, and could potentially reach 650000 tons per year in 2025.6
000–150000 tons;3 in the year 2010 the worldwide demand for LA was around of 300000–400000 tons.4 The global LA market is expected to grow from 714.2 kilotons in 2013 to 1960.1 kilotons in 2020.5 According to the report of Global View Research (California, USA), the production of PLA is expected to direct the increased demand for LA in the coming years. The global LA market size was valued at 1.29 billion USD in 2016 and is expected to reach 9.8 billion USD in 2025.5 European demand for PLA in 2010–2012 was around of 25000 tons per year, and could potentially reach 650000 tons per year in 2025.6
The physical properties of PLA depend on the isomeric purity of LA (L(+)- or D(−)). For example, L(+)-LA with high purity gives PLA of high melting point and high crystallinity.7 Furthermore, pure L(+)-LA can be polymerized to a high crystal polymer suitable for fiber and oriented film production and is expected to be useful in the production of crystal liquids.8
Currently, commercial production of LA is based on chemical synthesis or fermentation processes.4,9 Production by the fermentation processes has the advantage that a pure isomers (L(+)- or D(−)-LA) can be obtained by choosing an appropriate strain of LA bacteria,10 while chemical synthesis always results in a racemic mixture of LA.11 The fermentation has a high yield (90%) into lactic acid but several problems are associated with the process, such as the high cost of culture media, due to the specific requirements of LA producing bacteria,12 inhibition by the product,13 and high cost of purification process, besides being a nonecological process.14 In addition, the fermentation route may not be able to meet the growing demand for LA of the chemical industry.15
Research of new alternatives for chemical routes to produce LA has been of great interest in the last decades.16 One of the alternative production routes is the hydrothermal conversion of glycerol to LA in alkaline medium which was first described by Kishida et al.17 In this study Kishida used an autoclave reactor and NaOH or KOH as catalyst and after 90 min at 300 °C an aqueous solution of glycerol was converted into sodium lactate with yield higher than 90 mol% with 100% of glycerol conversion, employing NaOH or KOH/glycerol molar ratio equal to 3.78.
High temperatures are required when homogeneous catalytic process is used. The high temperature required for the dehydrogenation of glycerol to glyceraldehyde may promote a degradation of LA and pyruvaldehyde adversely affecting the selectivity to LA.18 To overcome this problem, Roy et al.19 investigated the use of a suitable catalyst for dehydrogenation of glycerol in alkaline medium at low temperature. In this study, they observed that use of copper catalysts together with NaOH provides a promising one-pot, low temperature route for glycerol conversion to LA. In this way, several investigations have been carried out focusing on the study of the combined use of heterogeneous catalysts in alkaline medium.9
Heterogeneous catalysts usually employed for hydrothermal conversion of glycerol into LA are based on noble metals such as Pt, Au and Pd. In addition to these metals, the use of copper has also been studied, since copper is conventionally used as a catalyst for the dehydrogenation of ethanol.20 The glycerol dehydrogenation to glyceraldehyde is the first step in the production of LA from glycerol, hence the study of copper as catalyst has been evaluated besides the use of non-noble metals is more economical.
Arcanjo et al.21 investigated the use of Pt and Pd catalysts supported on activated carbon. In this study it was obtained yield to LA of 68% and 74% using 10% Pd/C and 5% Pt/C as catalysts, respectively (molar ratio NaOH/glycerol = 1.1, 230 °C, 3 h). Ftouni et al.22 obtained after 24 h of reaction at 180 °C a yield of LA up to 84% employing 1.2% Pt/ZrO2 as catalyst and NaOH/glycerol molar ratio equal to 1.8. Other studies using copper-based catalysts demonstrate similar or even better performances than noble metal catalysts. Yin et al.20 employing hydroxyapatite (HAP), MgO and ZrO2 as catalytic support for Cu (16 wt% Cu loading) obtained yield to LA of 81.9%, 80.1% and 32.0%, respectively (molar ratio NaOH/glycerol = 1.1, 230 °C, 2 h). The authors observed that Cu/MgO and Cu/HAP catalysts exhibited higher LA yield because of higher basicity compared to Cu/ZrO2 catalyst. Shen et al.23 evaluated the effect of different-sized Cu2O nanoparticles in the hydrothermal conversion reaction of glycerol to LA and obtained a yield of 82.4% to LA after 2 h (molar ratio NaOH/glycerol = 1.2, 230 °C). On the other hand, Yin et al.24 investigated the effect of different-sized Cu0 nanoparticles and observed that the smaller metallic Cu0 nanoparticles have higher catalytic activity for the conversion of glycerol to lactic acid. This result was associated with the rapid reaction over small-sized metallic Cu0 nanoparticles which inhibits product degradation reactions and polymer-like chemicals through the interaction between glycerol and products.
Most of the works in the literature of glycerol conversion to LA was carried out using batch reactor. Some exceptions are the studies presented by Zhang et al.25 and Shimanouchi et al.26 which employed continuous flow reactor but with alkaline homogeneous catalysis. In previous studies of our group,27 using a continuous-flow reaction system, it was observed that the catalyst with the highest basicity presents highest yields to LA. A yield of LA around of 90% was obtained in the catalytic tests carried out with 20 wt% Cu/MgO catalyst (NaOH/glycerol molar ratio = 1, 240 °C/35 atm, WHSV = 2 h−1).
The production of LA from glycerol has been highlighted due to the growing production of biodiesel since glycerol generation represents 10% of the total volume of biodiesel produced.28 Therefore, there is a surplus of glycerol in the world market and the development of technologies that use glycerol besides giving it a noble destination can value the productive chain of biodiesel, because the cost of the biodiesel's production is still much higher than that of diesel fuel.29
According to Renewables 2017 Global Status Report,30 the world production of biodiesel in 2016 was 30.8 billion liters, and may have generated 3.0 billion liters of glycerol, approximately. Estimates for several countries show that the production of biodiesel and glycerol will continue to increase worldwide due to the mandatory use. For example, in Brazil the increase in the compulsory addition of biodiesel to diesel oil occurs since 2008, when it became mandatory to add biodiesel to diesel oil. Initially the mandatory blending of biodiesel to diesel oil was 2% (B2), and through successive increases over the years in 2017 reached 8% (B8) and from March of 2020 the Brazilian government increased the percentage of biodiesel in diesel oil to 12% (B12). According to National Agency of Petroleum, Natural Gas and Biofuels (ANP), the production of biodiesel in 2019 in Brazil was 5.9 billion liters,31 and there is an expectation of an even higher production in 2020 due to the increase in demand for biodiesel promoted by the Brazilian government, which aims to reach the B15 in 2023 according to the National Energy Policy Council of the Ministry of Mines and Energy (CNPE).
The objective of this study was to investigate the production of LA from glycerol in alkaline medium, using copper catalyst supported on CaO/MgO at different calcium oxide loadings, in continuous flow reaction system over a period of 30 h. According to the literature, there is evidence that the use of catalytic supports with basic characteristics presents better performance in glycerol transformation to LA compared to other catalytic supports of lower basicity. Thus, we intend to develop a catalytic system with high catalytic performance making possible the use of a lower NaOH/glycerol molar ratio. Additionally, copper has been shown effective as catalyst for this reaction.19 Furthermore, we evaluated the performance of the KOH and NaOH bases, the effects of reaction temperature (200, 220, 240 and 260 °C), NaOH/glycerol molar ratio (0.5, 0.75, 1.0, 1.25, 1.5), glycerol concentration (10 and 20 vol%) and of the use of crude glycerol.
2. Materials and methods
2.1. Characterization of catalysts
The supported copper catalysts (CuMg, CuCa, Cu5CaMg, Cu10CaMg and Cu15CaMg) were prepared by wet impregnation method with a loading of 20 wt% of CuO in calcined sample and the CaO content in the mixed support (CaO/MgO) was 0, 5, 10, 15 and 100 wt%.The MgO support was obtained from calcination of magnesium nitrate (Mg(NO3)2·6H2O) (Vetec), which was pre-calcined in muffle at 350 °C for 2 h with the aim of removing most of the hydration water, and then calcined at 500 °C for 3 h under flowing air (60 mL min−1) using a heating rate of 10 °C min−1 (rate used at all steps of the synthesis). Using the wet impregnation methodology CaO was added to MgO in appropriate amounts to produce catalysts with 5 wt%, 10 wt% and 15 wt% of CaO in the calcined catalyst. To synthesize the mixed oxide supports (CaO/MgO), appropriate quantities of calcium nitrate (Ca(NO3)2·4H2O) (Vetec) were solubilized in 100 mL of distilled and deionized water and added to the MgO. This mixture was placed in a flask, which was coupled in a rotary evaporator (IKA RV 10 Digital) and kept in rotation (120 rpm) for 1 h to homogenize the mixture, and then the mixture was heated to 80 °C under vacuum and maintained at this temperature until complete evaporation of the water. In sequence, the supports were dried at 110 °C overnight and subsequently calcined at 700 °C for 3 h under flowing air (60 mL min−1). Finally, after steps of preparation of the catalytic supports (Mg, 5CaMg, 10CaMg and 15CaMg), CuO was impregnated over these supports employing adequate amounts of copper nitrate (Cu(NO3)2·3H2O) (Vetec). The impregnation of the CuO was carried out following the same procedure adopted to prepare the mixed oxide supports, except the calcination temperature, which was 500 °C.
For the synthesis of the CuCa catalyst, calcium oxide (CaO) was obtained by calcining calcium nitrate (Ca(NO3)2·4H2O – Vetec), which was pre-calcined in a muffle at 700 °C for 2 h, in order to decompose the nitrates and remove the water present, and in the sequence it was calcined at 700 °C for 3 h under flowing air (60 mL min−1). Then, the CaO was impregnated with CuO following the methodology already described above.
2.2. Catalyst characterization
The chemical composition of the catalysts was determined by X-ray fluorescence (XRF) using a Rigaku Primini spectrometer, equipped with X-ray generator tube of palladium.X-ray powder diffraction (XRD) patterns were recorded in a Rigaku Miniflex II X-ray diffractometer equipped with a graphite monochromator using CuKα radiation (λ = 1.54178 Å), X-ray tube operated at 30 kV with 15 mA and the diffractograms obtained were compared with the database of the Joint Committee on Powder Diffraction Standards (JCPDS). The measurements were carried out with steps of 0.05° using a counting time of 1 s per step and over the 2θ range of 5° to 90°. Reduced catalysts were analyzed after ex situ reduction under the same conditions used before the catalytic tests and spent catalysts were analyzed without any other treatment after reaction. The average crystallite sizes of the Cu0 were calculated by the Scherrer equation (eqn (1)), using the diffraction peak correspondent to (200) plane at 2θ = 50.4°:
 | (1) |
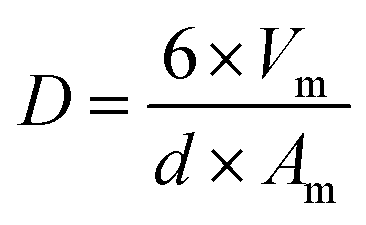 | (2) |
From the Cu dispersion data, it is possible to determine the Cu metal surface area per unit weight of catalyst (m−2 gcat−1) according to equation (eqn (3)):
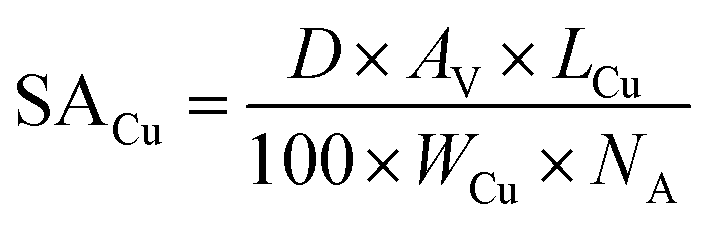 | (3) |
The textural characteristics were determined by N2 adsorption–desorption at −196 °C in a Micromeritics TriStar 3000. The specific area of the samples was obtained using the Brunauer–Emmett–Teller (BET) method and the pore volume obtained by Barrett–Joyner–Halenda (BJH) method. Prior to the analysis the samples were outgassed for 24 h at 250 °C in vacuum. The specific area of samples was calculated using the relative pressure range 0.06–0.20 and the pore volume was determined by BJH adsorption cumulative volume of pores between 2 nm and 60 nm of diameter.
The reducibility of the catalysts was analyzed by temperature-programmed reduction (TPR), carried out in a microflow reactor operating at atmospheric pressure. TPR analysis was carried out in a conventional apparatus equipped with a thermal conductivity detector (TCD). The samples (∼50 mg) were firstly dehydrated at 150 °C under argon (Ar) flow before the reduction. The samples were then cooled at room temperature. Afterwards, the heating was initiated up to 1000 °C with a rate of 10 °C min−1, using a mixture of 1.8% H2/Ar flowing at 30 mL min−1 through the sample. Decomposition of the peaks was performed using Gaussian shaped components. Agreement factors were in all cases higher than 0.98.
The catalyst basicity was investigated by temperature-programmed desorption of carbon dioxide (CO2-TPD), which was performed using a mass spectrometer QMG-220 (Pfeiffer). The samples of the catalytic supports were previously treated at 150 °C for 30 minutes under He flow (30 mL min−1) before adsorption of CO2. This prior treatment was not performed on the catalysts, since they were firstly reduced by a mixture of 1.8% H2/Ar up to 600 °C at a heating rate of 10 °C min−1 with an isothermal at 600 °C for 30 min. Carbon dioxide adsorption was carried out at room temperature with 10% CO2/He (30 mL min−1) for 30 min, followed by purging with He for 1 h. The desorption of chemisorbed CO2 was realized by heating up to 1000 °C at 20 °C min−1, under flowing pure He (30 mL min−1). The ratio m/z = 44 was used for quantification of carbon dioxide. Decomposition of the peaks was performed using Gaussian shaped components considering each shoulder as a peak. Agreement factors were in all cases higher than 0.98.
2.3. Catalytic tests
The glycerol conversion to lactic acid was carried out in a continuous system using a fixed-bed reactor of Inconel 625, with internal diameter of 0.5 cm and length of 15 cm. Aliquots were collected hourly for further HPLC analysis. All catalytic tests were performed during 30 h. However, before taking the 30 h of reaction, 2 h were taken for purging the reaction system. The period among 9 and 22 h of reaction was not analyzed (overnight). Although the first 2 h of reaction have been discarded due to the purging of the reaction system, in the first hours the results showed some fluctuation; however, after overnight period the results were much more stable, making it possible a consistent analysis of results.The catalysts were reduced in situ using a mixture of 30% H2/N2 with a total flow of 90 mL min−1 and a heating rate of 10 °C min−1 up to 600 °C for 30 min. The final reduction temperature was determined based on the results of the TPR analysis. It was evaluated reaction variables such as hydroxide type (KOH and NaOH), temperature (200 °C/20 atm, 220 °C/25 atm, 240 °C/35 atm and 260 °C/45 atm), NaOH/glycerol molar ratio (0.5, 0.75, 1.0, 1.25 and 1.5), glycerol concentration (10 and 20 vol%), as well as the use of crude glycerol with 85% of purity determined by HPLC analysis. The reagents were injected to the reactor by an HPLC pump (Eldex 1SAM). The pressure in the reactor was controlled using a diaphragm-type back pressure regulator (GO Regulator valve model BP-3). The mass of catalyst used was 1.25 g and the feed rate of 0.041 mL min−1, representing a weight hourly space velocity (WHSV) equal to 2 h−1 and as the catalyst bed is distributed in approximately 6 cm in the reactor, the residence time of glycerol is 29 min.
The liquid phase was analyzed in a Shimadzu Prominence HPLC with a Bio-Rad Aminex HPX-87 H column (300 × 7.8 mm) at 60 °C, using 0.01 M H2SO4 as eluent at 0.6 mL min−1, and refractive index (RID) and ultraviolet (UV) detectors. Samples for analysis were prepared by diluting aliquots of the reaction mixture in an aqueous solution of 0.01 M H2SO4, with dilution factor of the 1:100, thus, the final pH was very close to eluent of HPLC. The catalyst performance in terms of glycerol conversion, selectivity and yield to products i, was calculated according to eqn (4)–(6):
• Glycerol conversion:
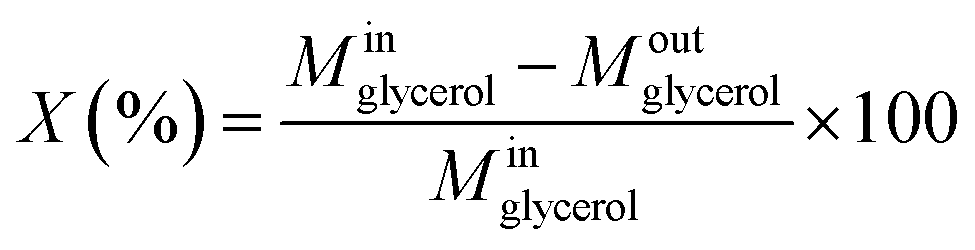 | (4) |
• Selectivity to product i:
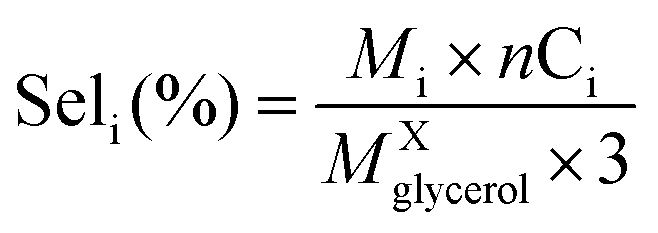 | (5) |
• Yield to product i:
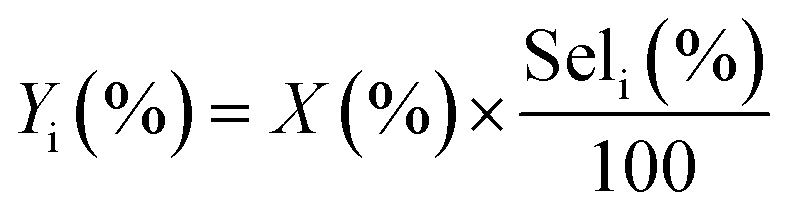 | (6) |
Turnover frequencies (TOF) values were calculated from the average formation rate (mol h−1) of LA obtained between 23 and 30 h of reaction divided by the number of metallic sites (mol), which was determined using the number of moles of Cu present in the mass of catalyst employed in the reaction multiplied by the dispersion, eqn (7).
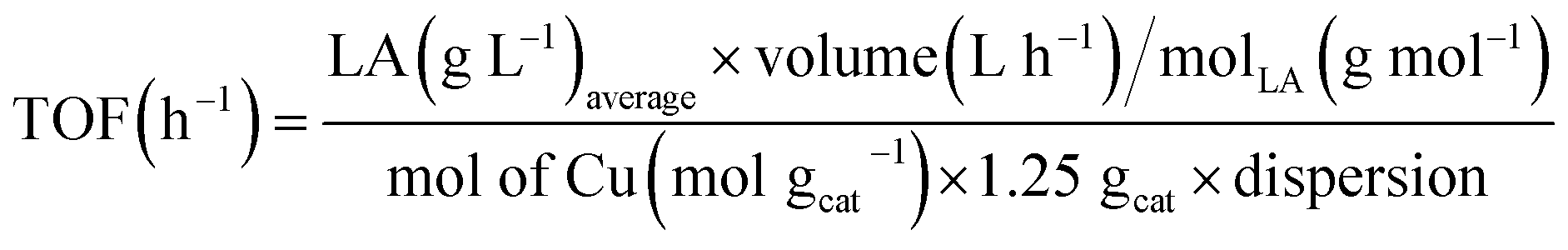 | (7) |
3. Results and discussion
3.1. Characterization of catalysts
The chemical composition of the catalysts calcined at 500 °C/3 h is presented in Table 1. The theoretical composition of CuO for all catalysts was 20 wt%. For the CuMg and CuCa catalysts, MgO and CaO were the only supports used, respectively. However, the catalysts Cu5CaMg, Cu10CaMg and Cu15CaMg were synthesized with different CaO loads, i.e. 5, 10 and 15 wt%, respectively. The real chemical composition of the catalyst was very similar to the nominal ones, with only minor differences that may be attributed to losses during synthesis and/or due to the imprecision of the equipment. Thus, it is observed that the synthesis method is effective in the preparation of catalysts with the desired chemical composition.| Catalyst | CuO (wt%) | CaO (wt%) | MgO (wt%) | SBET (m2 g−1) Cat/Supa | Vpore (cm3 g−1) Cat/Supa | Dpore (nm) Cat/Supa |
|---|---|---|---|---|---|---|
| a Cat = catalyst; Sup = support. | ||||||
| CuMg | 20.6 | — | 79.4 | 16/20 | 0.036/0.028 | 13.1/5.7 |
| CuCa | 21.0 | 79.0 | — | <10/<10 | 0.006/0.023 | 12.9/27.4 |
| Cu5CaMg | 21.8 | 5.7 | 72.5 | 12/13 | 0.023/0.036 | 11.8/18.1 |
| Cu10CaMg | 21.2 | 9.8 | 69.0 | 20/10 | 0.073/0.027 | 18.6/16.0 |
| Cu15CaMg | 19.4 | 15.2 | 65.4 | 10/15 | 0.031/0.028 | 19.5/7.1 |
Fig. 1 shows the XRD patterns of the catalytic supports, catalysts calcined at 500 °C/3 h, catalysts reduced at 600 °C/30 min, and spent catalysts after reactions (30 h). Except for CuCa catalyst, all the diffractograms of the catalytic supports, calcined and reduced catalysts exhibited the MgO phase with reflections at 2θ = 36.9°, 42.9°, 62.3°, 74.6° and 78.6° (JCPDS 45-0946). Fig. 1(C)–(E) show that after impregnation of MgO with CaO and subsequent calcination at 700 °C, the catalytic supports display a CaO crystalline phase with main diffraction peaks at 2θ = 32.2°, 37.3°, 53.8°, 64.1° and 67.3° (JCPDS 48-1467), and the intensity of the diffraction peaks increases with increasing CaO load. With these diffractograms, it is observed that the calcination temperature was effective in producing a crystalline CaO phase.
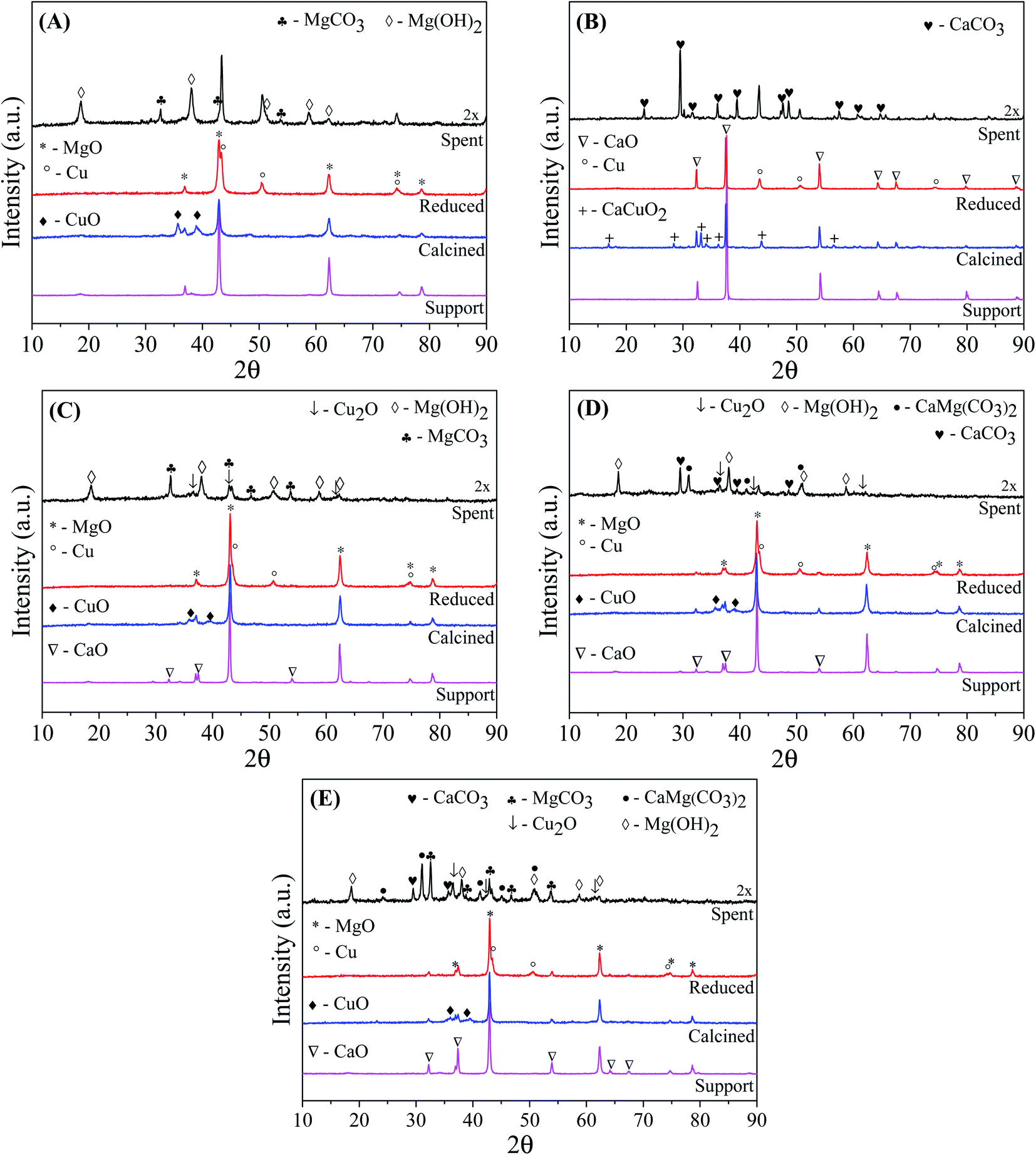 | ||
| Fig. 1 XRD patterns of the catalytic support, catalysts calcined, catalysts reduced and catalysts spent in the reactions performed at 240 °C/35 atm for 30 h with NaOH/glycerol molar ratio = 0.75. (A) CuMg, (B) CuCa, (C) Cu5CaMg, (D) Cu10CaMg and (E) Cu15CaMg. | ||
Mahdavi and Monajemi35 prepared CaO/MgO/Al2O3 catalysts by co-precipitation and obtained CaO and MgO crystalline phases by calcination temperature of 700 °C for 5 h. On the other hand, it is usual to use higher calcination temperatures (∼800 °C) due to the high stability of calcium carbonate (CaCO3).36 However, our synthesis process had the formation of CaCO3 minimized. It is observed in Fig. 1(A) and (C)–(E), that after impregnation of the catalytic supports with CuO and subsequent calcination at 500 °C/3 h the formation of a new phase of CuO takes place, with diffraction peaks at 2θ = 35.5° and 38.7° (JCPDS 48-1548). The low intensity of the peaks of CuO phase may be associated with a great dispersion of the CuO phase37 or it results from a phase with low crystallinity. In addition, it is noted that the addition of CuO promotes a reduction in the intensity of the diffraction peaks of the catalytic support, which may be related to a reduction in its crystallinity. Yin et al.20 synthesized copper catalysts supported on MgO and observed a loss of the crystallinity of the support with the increase of copper concentration in the catalysts, which was associated with high copper dispersion. After the addition of CuO over the Cu5CaMg catalyst, the CaO phase is no longer observed, probably due to the loss of crystallinity associated with the low CaO content.
The diffraction pattern of the calcined CuCa catalyst (Fig. 1(B)), in addition to presenting the characteristic peaks of the CaO phase, showed that the addition of CuO to the support generated a new crystalline phase of calcium copper oxide (CaCuO2), which has diffraction peaks at 2θ = 16.8°, 28.3°, 33.1°, 34°, 36.1°, 43.8° and 56.5° (JCPDS 48-0197). From the composition of this catalyst and the conditions used in calcination, the formation of this two phases (CaO and CaCuO2) is in accordance with the results predicted by Roth et al.,38 which studied the phase equilibrium diagram for the binary system CaO–CuO. Kaewpanha et al.39 prepared catalysts with different CuO loading supported in scallop shells (>99% CaO) by the wet impregnation method. After the calcination of the catalysts at 650 °C/3 h, it was obtained a mixture of phases, including CaO, CuO and CaCuO2. The formation of CaCuO2 was not observed in the Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts, which may be related to the low CaO concentration added to the catalyst and/or due to the strong interaction between CaO and MgO.
After catalyst reduction, it can be observed the absence of diffraction peaks associated with CuO and CaCuO2 phases, which demonstrates that the conditions used for activation of the catalysts are effective. The transformation of the CuO and CaCuO2 phases is accompanied by the formation of a new crystalline phase of metallic copper (Cu0), with main diffraction peaks at 2θ = 43.3°, 50.4° and 74.1° (JCPDS 04-0836).
Fig. 1 also shows the diffractograms of the catalysts used in the reactions after 30 h reaction at 240 °C/35 atm, using NaOH/glycerol molar ratio = 0.75 and 10 vol% glycerol solution. The diffractogram data for all the used catalysts were multiplied by 2, to make the visualization of the crystalline phases present clearer. The XRD of the spent CuMg catalyst (Fig. 1(A)) showed that the MgO phase was completely converted into two new crystalline phases: (1) magnesium hydroxide (Mg(OH)2), with main diffraction peaks at 2θ = 18.6°, 38.0°, 50.8°, 58.6° and 62.0° (JCPDS 07-0239) and (2) magnesium carbonate (MgCO3), with main diffraction peaks at 2θ = 32.6°, 42.9° and 53.8° (JCPDS 08-0479). In other studies of conversion of glycerol to LA in alkaline medium, a complete transformation of the MgO phase into Mg(OH)2 was also observed.20,27 The XRD of the spent CuCa catalyst showed that the CaO phase was completely transformed into calcium carbonate (CaCO3), which has main diffraction peaks at 2θ = 23.2°, 29.5°, 36.1°, 39.5°, 43.3°, 47.6° and 48.6° (JCPDS 72-1650). The catalysts composed by mixed support also showed a complete transformation of the crystalline structure of the supports. Thus, it can be seen that after the reactions, the Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts exhibited a mixture of several phases, such as Mg(OH)2, MgCO3, CaCO3, and in addition to these, a phase known as dolomite (CaMg(CO3)2) (JCPDS 75-1758) was also observed. Therefore, the catalytic support of all catalysts is completely transformed into different crystalline phases during the reaction.
It is observed that for both CuMg and CuCa catalysts, the metallic phase of copper (Cu0) is maintained even after the 30 h of reaction. Therefore, it is noted that these catalysts have excellent stability for the metallic phase. However, the diffractograms of the spent Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts exhibited a new phase of copper oxide (Cu2O) with low intensity which displays main diffraction peaks at 2θ = 36.4°, 42.3° and 61.3° (JCPDS 65-3288). Although there is an overlap of diffraction peaks in these spent catalysts, it is still possible to observe the peaks associated with metallic copper at positions 2θ = 43.3° and 50.4°. Therefore, despite the formation of Cu2O, the Cu0 phase is still observed after the reactions.
From the diffractograms of the reduced catalysts it was possible to determine the Cu average crystallite size and Cu dispersion, employing the Scherrer equation and Anderson correlation, respectively, besides the metallic surface area (Table 2). These results were determined using the diffraction peak at 2θ = 50.4°, which corresponds to (200) crystalline plane, due to the absence of interference with other crystalline phases. All catalysts exhibited similar crystallite size values, around 14 nm, demonstrating that both MgO and CaO supports have a good capacity of stabilizing the active phase, avoiding the sintering of copper particles during the CuO reduction. Another factor that may have contributed to this result may be associated with the dispersion of the CuO phase over the catalytic support, which is in agreement with the XRD results.
| Catalyst | Cu crystallite sizea (nm) | Db (%) | SACu (m2 gcat−1) | RDc (%) | No. of basic sitesd (μmol CO2 per gcat) | W:M:H strength basic sitese (%) |
Density of basic sites (μmol CO2 per m2) |
|---|---|---|---|---|---|---|---|
| a Calculated by the Scherrer equation using the (200) plane of Cu (2θ = 50.4°).b Estimated according to Anderson (eqn (2)).c Reduction degree (%) = H2 uptake (experimental)/H2 uptake (theoretical) × 100.d Amount of basic sites calculated from Gaussian deconvolution of CO2-TPD profiles.e Ratio of weak:medium:high strength basic sites (%), calculated from CO2-TPD profiles considering CO2 desorption peak (weak: peak below 400 °C; medium: peak between 400–600 °C; high: above 600 °C). |
|||||||
| CuMg | 14.6 ± 1.0 | 7.1 | 7.6 | 97 | 203 | 100:00:00 |
12.7 |
| CuCa | 14.1 ± 1.4 | 7.4 | 8.0 | 101 | 126 | 00:00:100 |
42.0 |
| Cu5CaMg | 14.4 ± 1.5 | 7.2 | 8.1 | 73 | 189 | 29:00:71 |
15.8 |
| Cu10CaMg | 14.8 ± 1.9 | 7.0 | 7.7 | 80 | 273 | 17:00:83 |
13.7 |
| Cu15CaMg | 12.9 ± 1.8 | 8.1 | 8.1 | 70 | 183 | 02:00:98 |
18.3 |
The catalysts also showed similar values of dispersion and metallic surface area, since these properties are intrinsically related to the crystallite size of Cu. The metallic dispersion values are in the range of 7.0 to 8.1%, with a metallic surface area around 8 m2 gcat−1. Zhang and He40 prepared Cu/CaO catalysts with low loading of Cu (about 2 wt%) by the wet impregnation method and obtained Cu particle size of 12.9 nm and dispersion of 9%. Using the same methodology, Moreira et al.27 obtained for the Cu/MgO (15 wt%) catalyst average Cu crystallite size of 12 nm with a dispersion of 6.3% and metallic surface area 7.5 m2 gcat−1.
The dispersion of the active phase (Cu) may be influenced by several factors, including the calcination temperature, copper loading and the synthesis methodology. Nagaraja et al.41 evaluated the influence of the synthesis methodology on Cu/MgO catalysts. Among the different methodologies used, coprecipitation, wet impregnation and solid–solid methods, coprecipitation method presented the highest copper dispersion (8.0%). Reddy et al.42 obtained for the Cu/MgO catalyst (20 wt%) prepared by coprecipitation method an average crystallite size of 64 nm. The conditions used in the preparation of the catalyst may have contributed to the big crystallite size. Although it used lower temperatures in calcination (450 °C) and reduction (280 °C) the high time employed in calcination (5 h) and reduction (3 h) may have contributed to the formation of larger particles.
Effects of sintering processes on the catalysts after reaction was possible to evaluate only for the CuCa catalyst since the other spent catalysts showed Cu diffraction peaks overlapping with other crystalline phases, which made the determination of the Cu crystallite size inadequate. Although a previous study carried out by the group has shown that the sintering process can be considered negligible,27 it was obtained an average Cu0 crystallite size of 51 nm for the CuCa spent catalyst, this is an indication that this catalyst has undergone sintering processes during the reaction.
Table 1 presents BET surface area, pore volume and pore diameter obtained from N2 adsorption isotherms of the supports and catalysts after calcination at 500 °C/3 h. It is observed that both the supports and the catalysts showed low values of specific area, in the range of 10 to 20 m2 g−1, except for CaO and CuCa, which presented specific areas below 10 m2 g−1, with values of 5 and 3 m2 g−1, respectively. The low specific areas may be associated with the methodology employed in the synthesis, as well as with the fact that the oxides of Cu, Ca and Mg are traditionally materials that have a low specific area. Low values of specific area have also been observed by other authors. Zhang et al.40 obtained for the Cu/MgO and Cu/CaO catalysts specific areas of 24.2 m2 g−1 and 0.6 m2 g−1, respectively. Manovic and Anthony43 obtained a specific area of 1.69 m2 g−1 for Cu/CaO (45 wt%) and Nagaraja et al.41 obtained 18 m2 g−1 for Cu/MgO prepared by wet impregnation method.
The N2 adsorption–desorption isotherms of the supports and catalysts exhibited a pattern type III, without hysteresis, with the exception of the supports of the CuMg and Cu15CaMg catalysts, which showed a pattern more similar to type IV with H3 hysteresis loop. In the type III isotherm, the adsorbed amount tends to infinity when P/P0 tends to 1, corresponding to the adsorption in multiple overlapping layers and occurs in non-porous or macroporous solids. The characteristics of the type IV isotherm are hysteresis loop, which is associated with capillary condensation in the mesopores and limiting uptake over a range of high P/P0.44 Although most synthesized materials did not exhibit isotherms characteristic of mesoporous materials, it is observed that the average pore diameter of the materials comprises the range of mesoporous materials (2–50 nm).44 Thus, we observed that the catalysts have a wide range of pore distribution.
Fig. 2 shows the profiles of temperature-programmed reduction of the catalysts calcined at 500 °C for 3 h. It is observed that all catalysts showed peaks of reduction in the range of 365 °C to 685 °C. However, the peaks observed in the range of 570 °C to 685 °C were associated with the decomposition process CaCO3, which is formed due to CaO carbonation45 and/or due to the decomposition of residual carbonate, despite calcination at 500 °C, which is in accordance with previous studies.46,47 In addition, Kalinkin et al.48 observed that at room temperature, both CaO and Ca(OH)2 can react with humidity and CO2 of the air, forming amorphous CaCO3 on the surface of the particles, which is in accordance with the XRD results of the calcined CuCa catalyst (Fig. 1(B)) and with TPR results, since the decomposition of CaCO3 occurs in a range of 480 °C to 650 °C.49 The peaks obtained in the range of 365 °C to 470 °C were related to the reduction of CuO, which is above the range described in the literature (190 °C to 350 °C) for reducing of bulk CuO,47,50–52 and this may indicate the existence of a stronger interaction between the copper phases and the catalytic supports.
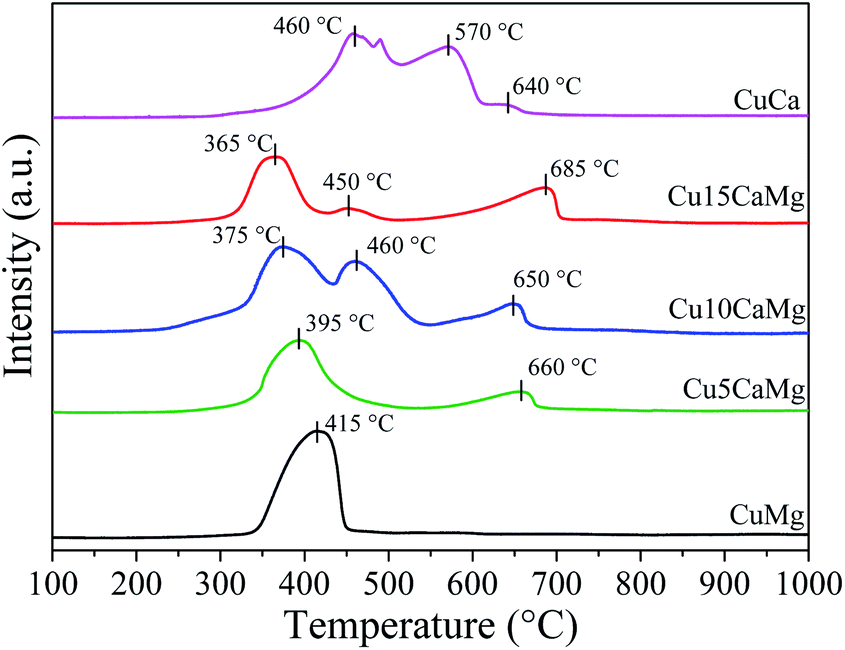 | ||
| Fig. 2 H2-TPR profiles of calcined catalysts. | ||
The CuMg catalyst exhibited a single reduction peak at 415 °C, indicating a homogeneous copper oxide phase and with strong interaction with the MgO support. Nagaraja et al.53 obtained for Cu/MgO (16 wt%) catalyst prepared by the wet impregnation a reduction profile similar to that found in this study, with a maximum reduction peak at 400 °C, approximately.
As it can be seen in Fig. 1(B), all Cu added to the pure CaO support led to the formation of the CaCuO2 phase, which showed a great stabilization for Cu, since the reduction profile of the CuCa catalyst had the highest temperatures. Halasz et al.54 investigated different types of cuprates through TPR analysis and observed that different cuprates, including Ca2CuO3, required higher reduction temperatures than CuO. Su et al.55 observed for Cu/CaO (10 wt%) catalyst a single reduction peak between 400–450 °C assigned to the reduction of Ca2CuO3 cuprate. The Cu5Ca, Cu10Ca and Cu15Ca catalysts showed main reduction peaks at lower temperatures, 395 °C, 375 °C and 365 °C, respectively. This may be indicative of a small increase in Cu dispersion because the higher dispersion offers larger reactive surface area and a higher concentration of defects where the reduction can start at a lower reduction temperature.51 However, it is observed that the Cu10Ca catalyst has a secondary reduction peak at 460 °C while the Cu15Ca catalyst has a small peak at 450 °C. A high CuO reduction temperature may also indicate less dispersion of CuO species,52 which is in agreement with the dispersion data presented in Table 2. Although the Cu10Ca catalyst has the main reduction peak at 375 °C, it has a second reduction peak at a higher temperature, thus, this result indicates that the catalyst has two distinct CuO phases.
The reduction degree (RD) of CuO shown in Table 2 was determined from the TPR analysis considering the consumption of H2 presented by the peaks in the range of 365–470 °C, and it was assumed that Cu2+ is reduced in one step (Cu2+ → Cu0) without formation of Cu2O. The CuMg and CuCa catalysts practically showed a complete reduction of CuO, while the Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts demonstrated degrees of reduction in the range of 70 to 80%. It is observed that low CaO load (5–15 wt%) promotes a decrease in the reduction degree. Similar results were observed by Xue et al.,56 where it was evaluated the influence of CaO addition on Cu/ZSM-5 catalysts. In this study it was observed that the reducibility of CuO decreases with the CaO addition, and this effect was associated with the coverage of the surface of CuO by CaO and the interactions between CaO and CuO, which are unfavorable for the reduction of CuO.
The basicity of the catalysts reduced at 600 °C/30 min was evaluated by CO2-TPD analysis and the desorption profiles are shown in Fig. 3. The peaks were deconvoluted and classified into three regions of temperature, according to the classification used by Aramendía et al.:57 up to 400 °C, 400–600 °C and above 600 °C, which were related to weak, medium and high strength basic sites, respectively. The CuMg catalyst displays only weak basic sites, with 3 peaks of CO2 desorption (135 °C, 190 °C and 255 °C), the main peak is located at 255 °C. These results are in accordance with the literature, since a wide range of CO2 desorption (50–400 °C) is commonly observed in catalysts supported on MgO.27,40,58,59 On the other hand, the CuCa catalyst mainly exhibits strong basic sites, with a narrower CO2 desorption band at higher temperature, in the range of 650 to 685 °C. It is widely accepted in the literature that CaO presents strong basic sites with CO2 desorption in the range of 500–750 °C.40,55,58,60
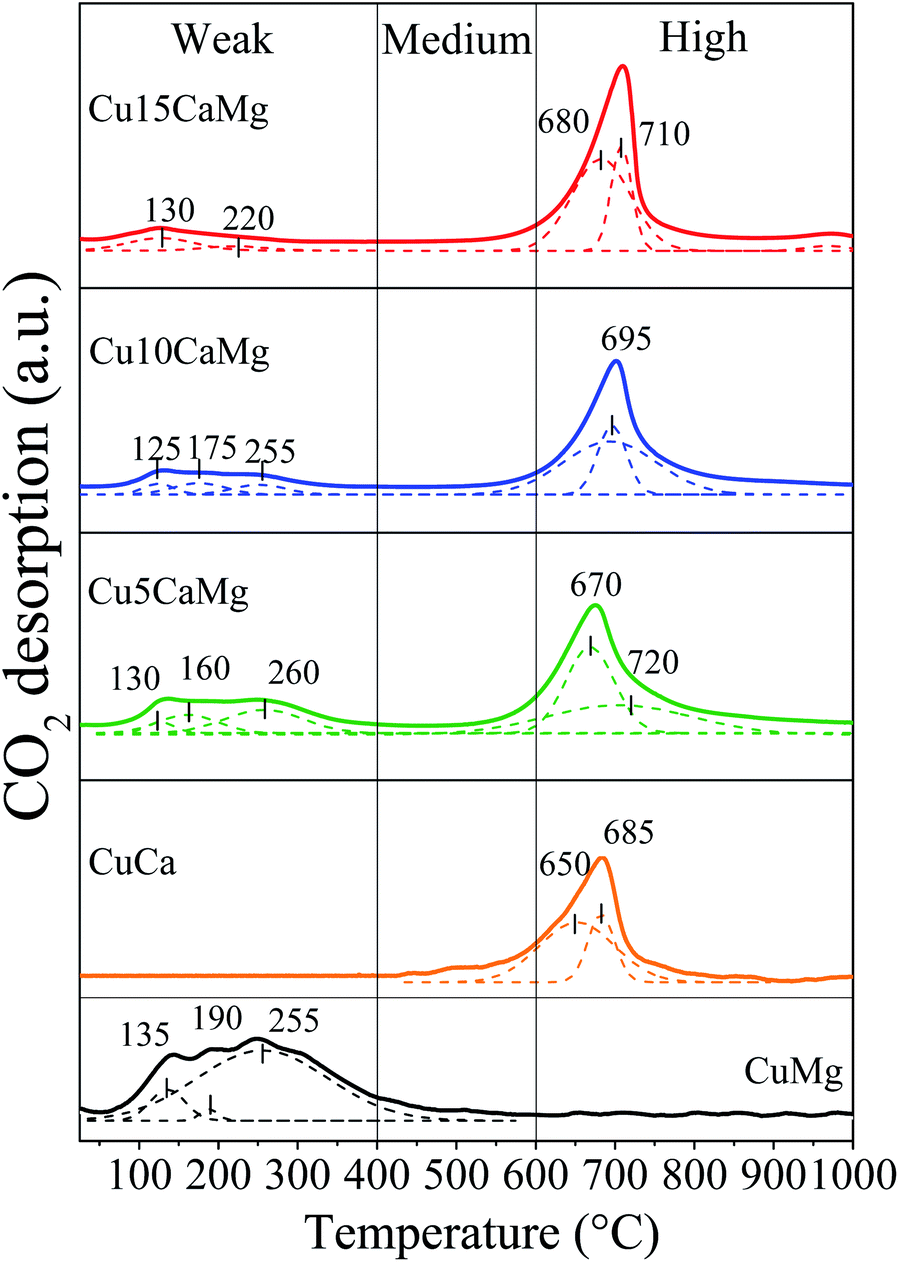 | ||
| Fig. 3 CO2-TPD profiles of the reduced catalysts. | ||
Catalysts composed of mixed oxides (Cu5CaMg, Cu10CaMg and Cu15CaMg) exhibited a wide range of CO2 desorption temperatures (75–850 °C), due to the presence of both types of sites: weak and strong basic, characteristic of the MgO and CaO phases, respectively. With successive increase in CaO load in the catalyst, it is observed that the strong basic sites become predominant, while the weak basic sites become less intense. Taufiq-Yap et al.58 observed this same behavior for CaO–MgO catalysts and this loss of weak basic sites was attributed to the dispersion of CaO on the surface of MgO, preventing the adsorption of CO2 on basic sites of MgO.
Table 2 shows the total amount of basic sites per gram of catalyst (μmol CO2 per gcat), the proportion of weak, medium and high strength basic sites and the densities of basic sites for each catalyst. Although the distribution of the strength of the basic sites was different among the catalysts, they presented a similar amount of basic sites, in the range of 130 to 270 μmol CO2 per gcat. The CuMg and CuCa catalysts showed values similar to those obtained by Zhang et al.,40 which obtained for the Cu/CaO and Cu/MgO catalysts prepared by wet impregnation 183 and 148 μmol CO2 per gcat, respectively. Although CaO promotes the formation of new strong basic sites in catalysts supported on CaO/MgO, there was no significant increase in the number of total basic sites, and this may be associated with a low proportion of CaO added and/or with the reduction of weak basic sites of the MgO. Furthermore, analyzing the strength distribution of the basic sites of catalysts, it is observed that the formation of strong basic sites is progressive with the increase in the CaO load, and concomitantly there is a reduction in the amount of weak basic sites. The catalysts showed a density of basic sites in the range of 12 to 18 μmol CO2 per m2, except for the CuCa catalyst, which showed a density of 42 μmol CO2 per m2. The density obtained for the CuCa catalyst may be overestimated because there is an inaccuracy in determining the specific area for materials with very low surface area. However, in general, it is observed that the addition of CaO to the catalysts promotes an increase in the density of basic sites.
3.2. Catalytic tests
Initially it was evaluated the influence of the NaOH and KOH bases and the choice of these bases was due to their strong basic character. Furthermore, among all types of base, the NaOH is the most widely used in the hydrothermal conversion reaction of glycerol to LA with or without catalysts and according to the study presented by Shen et al.63 KOH showed higher activity than NaOH in the hydrothermal conversion of glycerol to LA. The reaction conditions used to evaluate the effect of hydroxides (NaOH and KOH) on the reaction of LA production from glycerol in alkaline medium, employing continuous flow reaction system were: 10 vol% glycerol solution; base/glycerol molar ratio = 1; temperature/pressure = 240 °C/35 atm; CuMg catalyst (1.25 g) and feed flow = 0.041 mL min−1 (WSHV = 2 h−1).
Both hydroxides (NaOH or KOH) exhibited high glycerol conversions, with an average (23–30 h) glycerol conversion of 96.5% and 95.1%, respectively. Although the conversion of glycerol did not present significant differences, the average selectivity to LA was higher in the reaction performed with NaOH, which presented 90.4% of selectivity to LA, while the reaction carried out with KOH displayed an average selectivity to LA of 80.8%. Only 1,2-propanediol (1,2-PD) was obtained as byproduct in all catalytic tests and the average selectivity obtained was 12.4% (NaOH) and 7.4% (KOH). Since the conversion of glycerol obtained using the NaOH and KOH bases was practically the same and above 95%, the yield are close to the selectivity values. Thus, it was obtained the highest yield to LA (87.2%) in the catalytic test performed with NaOH, while the test performed with KOH showed a lower yield, 76.8%. The yield to 1,2-PD was of 11.9% (NaOH) and 7.0% (KOH). Our results indicate that NaOH has a better performance than KOH, although Shen et al.63 has observed the opposite. This difference may be associated with the reaction system used, once that Shen et al.63 used a batch system without heterogeneous catalysts.
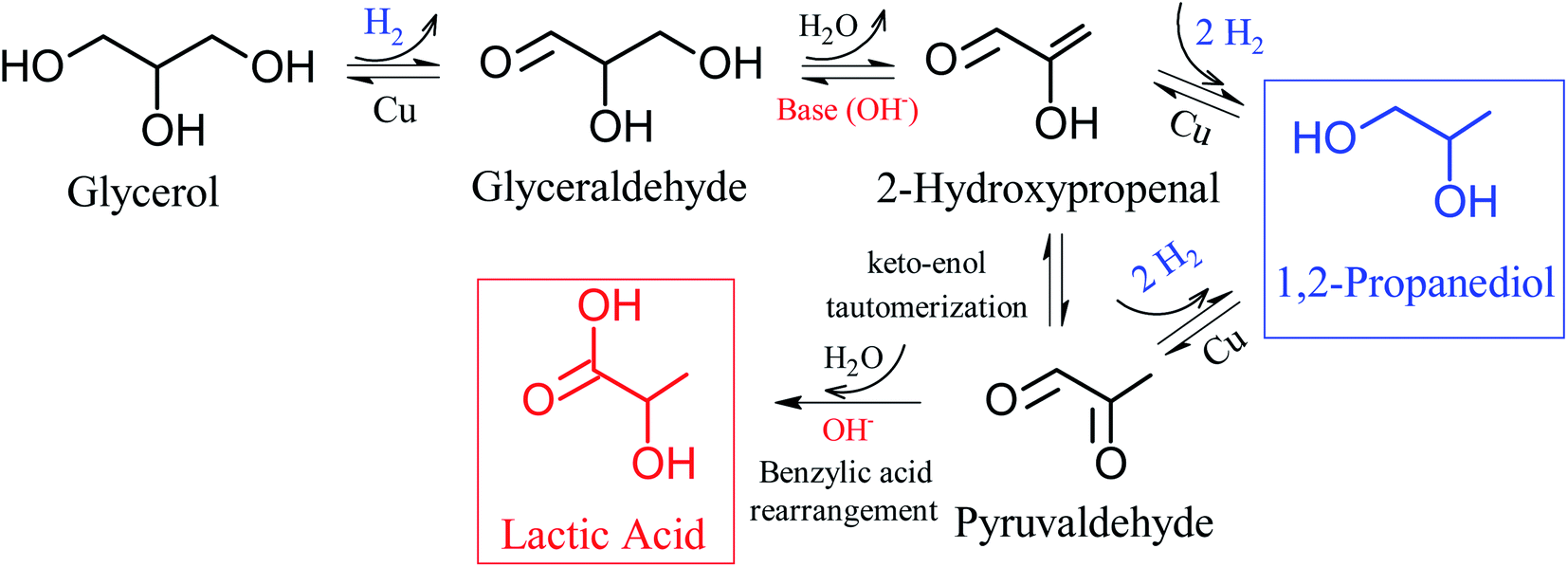
It is widely accepted in the literature that the hydrothermal conversion of glycerol to LA has as initial step the dehydrogenation of glycerol to glyceraldehyde. Subsequently glyceraldehyde undergoes dehydration to form 2-hydroxypropanal, which through a molecular rearrangement, known as keto–enol tautomerization, transforms the hydroxyl present in the 2-hydroxypropanal into a ketone forming pyruvaldehyde, and finally the pyruvaldehyde is converted to LA (lactate) by a mechanism similar to the rearrangement of benzylic acid (intra molecular Cannizzaro reaction);17,22,27,64–67 these steps are shown in Fig. 4. According to Ftouni et al.,22 the first step in the process that produces glyceraldehyde occurs on the metallic sites, while the other steps occur in solution in the presence of hydroxyl group (OH−). However, on the metallic sites the 2-hydroxypropenal and pyruvaldehyde may also be hydrogenated producing 1,2-PD.66,68 Furthermore, among transition metals, copper-based catalysts are efficient in glycerol hydrogenolysis under an atmosphere rich in H2 due to their high activity in the cleavage of C–O bonds associated with the low activity of cleavage of C–C bonds.69,70
| Catalyst | Xa (%) | Selectivitya (%) | Yielda (%) | TOFa,b (h−1) | ||
|---|---|---|---|---|---|---|
| LA | 1,2-PD | LA | 1,2-PD | |||
| a Reaction condition: 240 °C/35 atm; 10 vol% glycerol solution; NaOH/glycerol molar ratio = 0.75; 1.25 g catalyst; feed flow 0.041 mL min−1.b TOF values were based on the average rate of LA production (23–30 h). TOF = LA produced (mol h−1)/metallic Cu (mol). | ||||||
| CuMg | 81.9 | 63.9 | 36.5 | 52.3 | 29.9 | 8.0 |
| CuCa | 87.8 | 59.1 | 23.9 | 51.9 | 21.0 | 7.5 |
| Cu5CaMg | 96.4 | 78.0 | 20.7 | 75.2 | 19.9 | 10.7 |
| Cu10CaMg | 91.0 | 72.5 | 25.3 | 66.0 | 23.0 | 10.2 |
| Cu15CaMg | 96.4 | 68.1 | 27.7 | 65.7 | 26.7 | 10.7 |
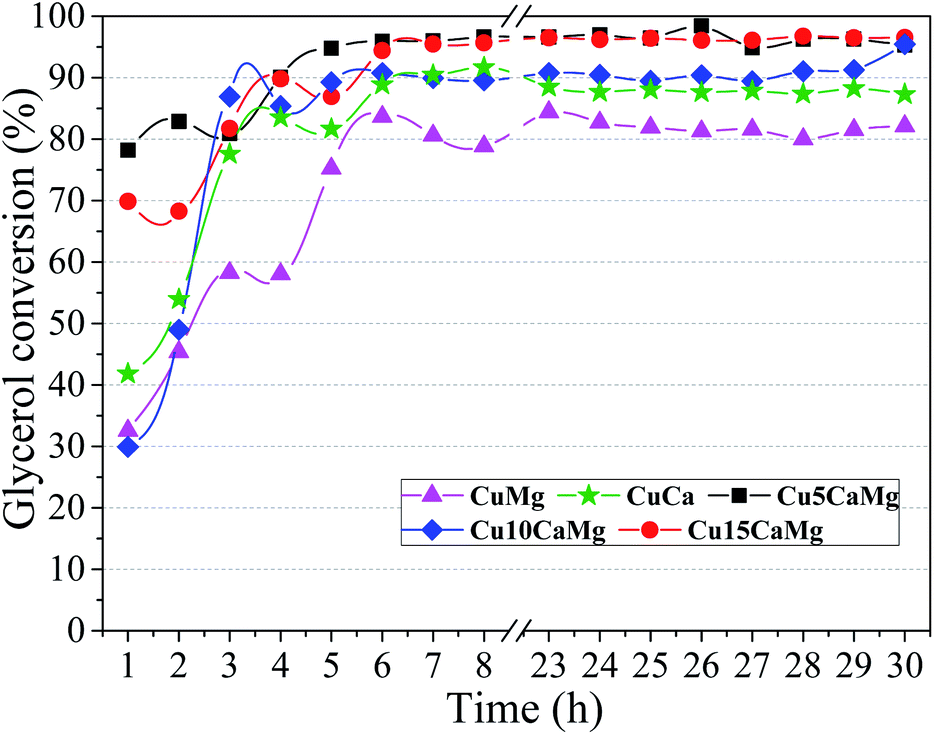 | ||
| Fig. 5 Glycerol conversion obtained from the different synthesized catalysts. Reaction condition: 1.25 g catalyst, 240 °C/35 atm, 10 vol% glycerol solution, NaOH/glycerol molar ratio = 0.75 and feed flow = 0.041 mL min−1 (WHSV = 2 h−1). | ||
Fig. 6(A) shows the selectivity to LA and 1,2-PD obtained during 30 h of reaction using different catalysts. It is observed that the catalysts composed of CaO/MgO have higher selectivities to LA than the CuMg and CuCa catalysts, the same behavior observed in the glycerol conversion. However, the highest selectivity is obtained with the addition of low CaO load (5 wt%), and subsequent additions seem to demonstrate a reduction in selectivity to LA, since the average selectivity decreased with successive increases in CaO (Table 3). Thus, the highest average selectivity to LA was obtained in the catalytic test performed with the Cu5CaMg catalyst (78.0%). This behavior may be associated with the basic sites of catalysts. With the addition of CaO to the catalyst, high strength basic sites are created; however, an increase in CaO loads drastically reduce proportion of weak strength basic sites, in this way, it is observed that a balance between weak and high strength basic sites should be maintained to obtain greater selectivities to LA, as well as superior glycerol conversions. Therefore, as shown in Table 2, a greater balance in the distribution of the basic sites is obtained in the Cu5CaMg catalyst.
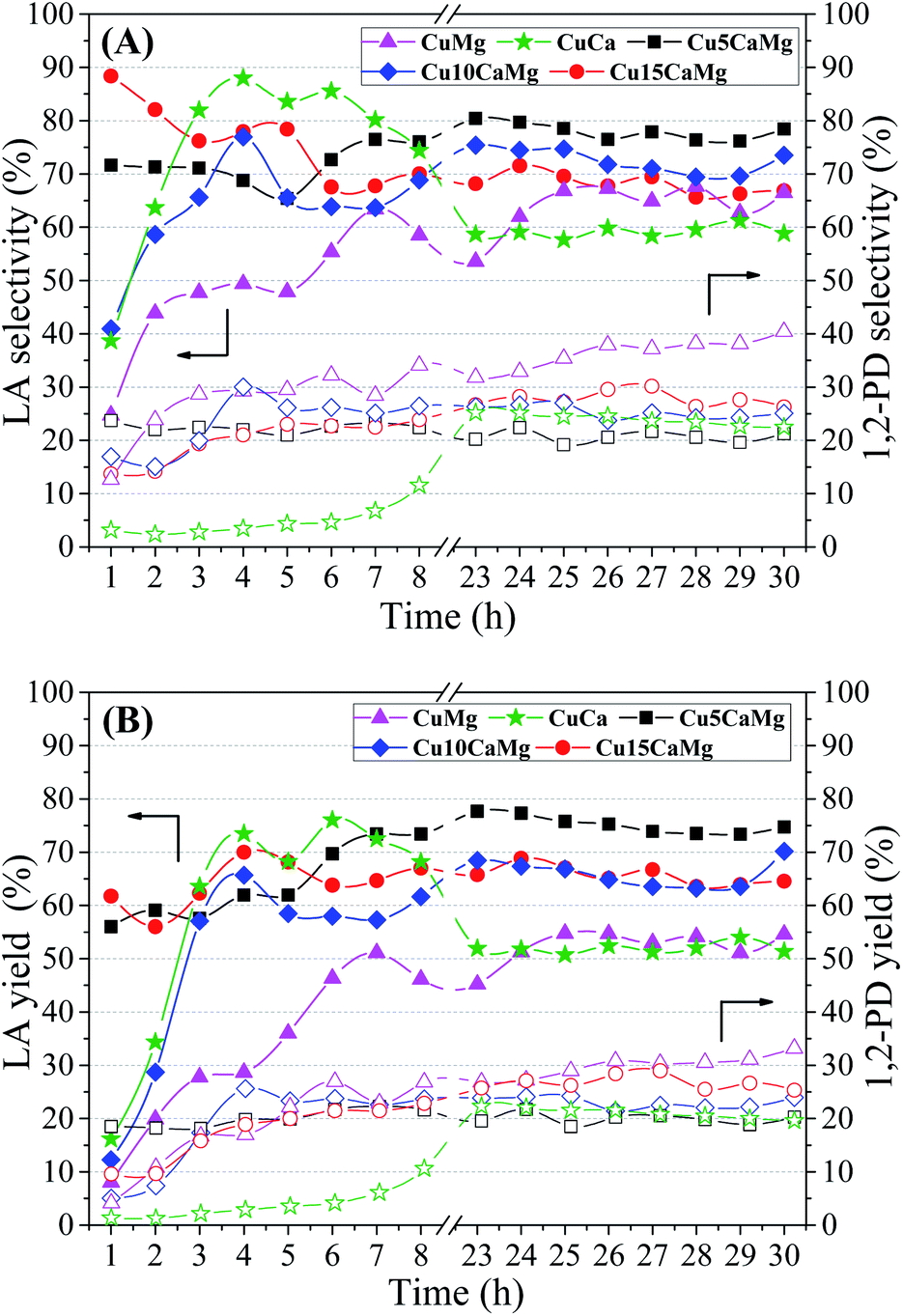 | ||
| Fig. 6 Lactic acid (filled symbols) and 1,2-propanediol (empty symbols) selectivity (A) and yields (B) obtained for the different catalysts evaluated. Reaction condition: 1.25 g catalyst, 240 °C/35 atm, 10 vol% glycerol solution, NaOH/glycerol molar ratio = 0.75 and feed flow = 0.041 mL min−1 (WHSV = 2 h−1). | ||
Regarding the selectivity to 1,2-PD, it is observed that the distribution of the basic sites has a behavior opposite to LA selectivity. The highest selectivity to 1,2-PD was obtained in the catalytic test carried out with the CuMg catalyst (36.5%), which presents only weak strength basic sites. Therefore, the effect of basic properties in relation to selectivity to 1,2-PD is not clear. The selectivities obtained for 1,2-PD may be associated with the aviability of metallic Cu0 (Fig. 4). Thus, the results presented by the CuCa and CuMg catalysts may be related to the greater reduction degree obtained (100%). In addition, CuMg and CuCa spent catalysts have not shown the formation of the Cu2O phase (Fig. 1). All catalysts, except CuCa, showed sum of selectivities (LA and 1,2-PD) close to 100%, this is an indication that gaseous products are not formed. The CuCa catalyst showed a selectivity sum of 83%, indicating that there may have been a greater conversion of glycerol to gaseous products and/or the generation of some unidentified liquid byproduct.
Fig. 6(B) shows the yield to LA and 1,2-PD obtained in the catalytic tests performed with the different catalytic supports. The Cu5CaMg catalyst showed the highest average yield to LA (∼75%), since it obtained high glycerol conversion and the highest average selectivity to LA among the catalysts. The Cu10CaMg and Cu15CaMg catalysts showed similar LA yields, around 65%, while CuMg and CuCa catalysts showed LA yields in the range of 52%. As shown in Fig. 1, the diffractograms of the Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts after reaction (spent) showed that a partial oxidation of Cu occurs, forming Cu2O. However, this did not result in a catalytic deactivation, because relatively stable values of LA yield were obtained (after overnight period).
This may indicate that the Cu oxide phase also has some catalytic activity in the reaction, which is in agreement with the literature.66,71 Moreover, the catalysts supported on mixed oxide (CaO–MgO) have reduction degree in the range of 70–80% and this incomplete reduction did not negatively impact the results of the catalytic tests, in accordance with the findings of Roy et al.19 and Yang et al.72 Li and Li73 evaluated different unsupported copper salts and bulk CuO in the reaction of glycerol conversion to LA in alkaline medium employing batch reactor. Although the CuBr2 salt was reduced during the reaction, the results obtained were slightly better than CuO at reaction temperature above 155 °C.
The turnover frequencies (TOF) values were calculated according to eqn (7) and the results are shown in Table 3. Catalytic tests performed with CuMg and CuCa catalysts exhibited the lowest TOF values. On the other hand, the catalytic tests carried out using the Cu catalysts supported on CaO/MgO exhibited an increase in the TOF value, but without a significant distinction among the Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts. This shows that the association of weak and high strength basic sites has a higher catalytic performance, than only weak (MgO) or high (CaO) strength basic sites. Thus, it is demonstrated that an addition of CaO greater than 5 wt% is not necessary, since the maximum catalytic performance is achieved in the smallest proportion. In addition, the highest LA yield was also obtained with the Cu5CaMg catalyst.
Similar results were observed by Yin et al.20 in the hydrothermal conversion of glycerol to LA. Although the Cu(16)/MgO catalyst exhibits almost double of basic sites, the better catalytic performance was obtained with Cu(16)/HAP catalyst, which may be associated with a wide distribution of basic sites (100–661 °C) presented by the Cu(16)/HAP catalyst, while the catalyst Cu(16)/MgO showed a more concentrated (202–396 °C) distribution of basic sites. Liu and Ye66 evaluated the influence of basic oxides (MgO, CaO and SrO) employing a batch reactor. It was observed that the catalytic activity of these oxides followed the same order of the basic strength (SrO > CaO > MgO); however, the performance of SrO is slightly better than CaO, not justifying its use because SrO is much more expensive than CaO.
| Temperature (°C)/pressure (atm) | Xa (%) | Selectivitya (%) | Yielda (%) | ||
|---|---|---|---|---|---|
| LA | 1,2-PD | LA | 1,2-PD | ||
| a Reaction conditions: 1.25 g Cu5CaMg catalyst; 10 vol% glycerol solution; NaOH/glycerol molar ratio = 0.75; feed flow 0.041 mL min−1. | |||||
| 200/20 | 72.0 | 61.4 | 16.3 | 44.2 | 11.7 |
| 220/25 | 97.4 | 64.9 | 15.7 | 63.2 | 15.3 |
| 240/35 | 96.4 | 78.0 | 20.7 | 75.2 | 19.9 |
| 260/46 | 90.8 | 61.3 | 22.3 | 55.7 | 20.2 |
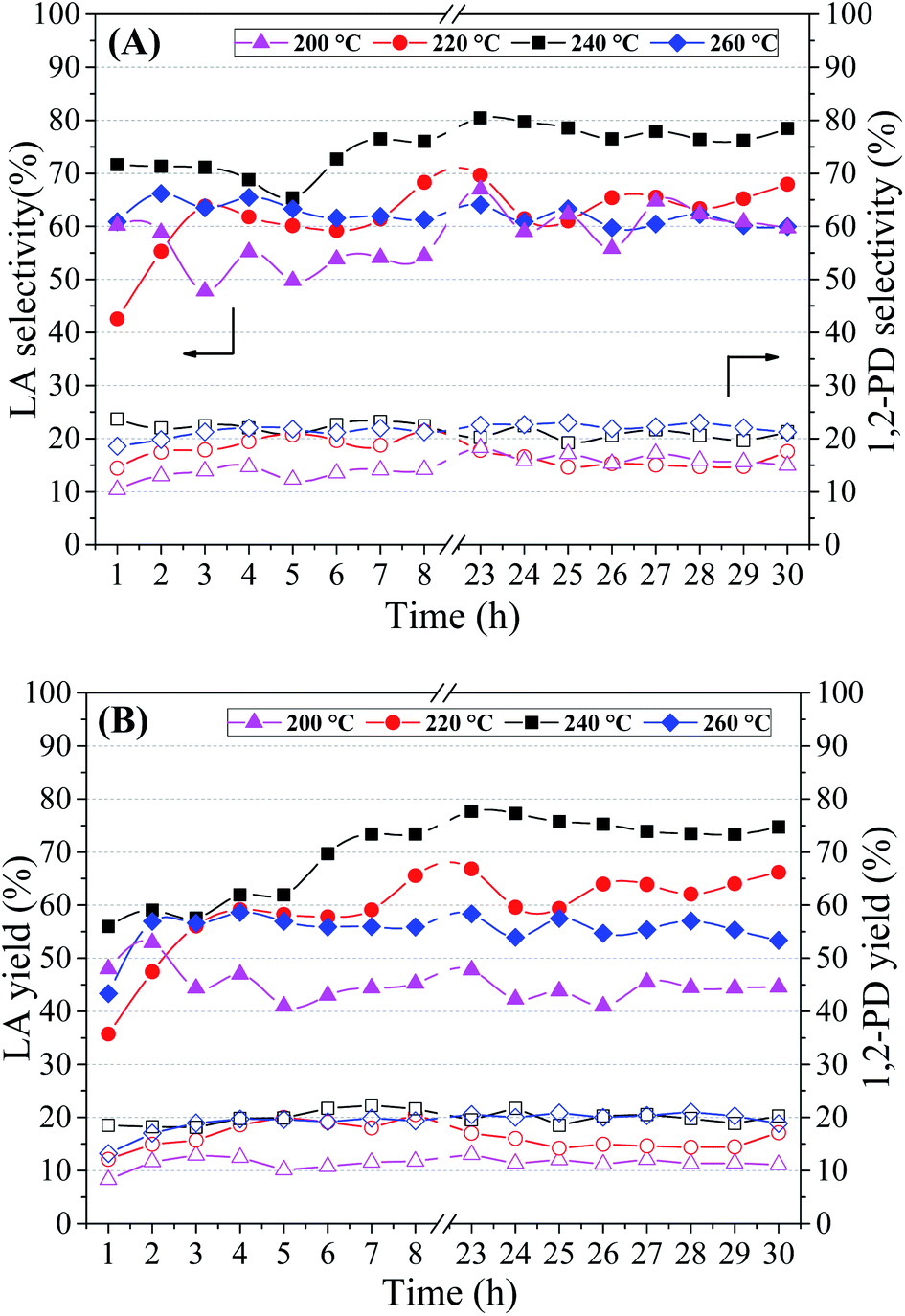 | ||
| Fig. 7 Lactic acid (filled symbols) and 1,2-propanediol (empty symbols) selectivity (A) and yields (B) at different temperatures/pressures (200 °C/20 atm; 220 °C/25 atm; 240 °C/35 atm; 260 °C/46 atm). Reaction conditions: 1.25 g Cu5CaMg catalyst, 10 vol% glycerol solution, NaOH/glycerol molar ratio = 0.75 and feed flow = 0.041 mL min−1 (WHSV = 2 h−1). | ||
The catalytic tests were performed employing the Cu5CaMg catalyst, which showed the best results in previous studies, 10 vol% glycerol solution, NaOH/glycerol molar ratio = 0.75, 1.25 g of catalyst and feed rate of 0.041 mL min−1 (WHSV = 2 h−1).
Table 4 shows the average conversion of glycerol (X) obtained in the period of 23–30 h of reaction. The lowest glycerol conversion (72%) was obtained at the lowest reaction temperature, and with subsequent increase in reaction temperature (220–260 °C) the glycerol conversion rises above 90%. Thus, to obtain high conversions of glycerol it is necessary to perform the catalytic tests at temperatures above 220 °C. The increase in pressure could negatively affect the conversion of glycerol, as was observed by Yang et al.72 In this study was evaluated the effect of pressure on the reaction of glycerol conversion to LA (NaOH/glycerol = 1; 180 °C; 8 h), and it was observed that the glycerol conversion reduced from 100% to 87.7% with an increase in pressure from 14 bar to 20 bar.
Fig. 7(A) shows that the highest selectivity to LA was obtained in the catalytic test carried out at 240 °C/35 atm, which presented values of LA selectivity close to 80%. Selectivity to LA in the range of 60 to 65% was obtained in the other conditions studied. Evaluating the Fig. 7(A), besides the reduction in selectivity to LA, it is not possible to observe the effect of temperature/pressure on LA selectivity. However, when evaluating the average selectivity to LA presented in Table 4, it is noted that the selectivity increases with the increase in temperature/pressure up to 240 °C. On the other hand, the selectivity to 1,2-PD shows an almost linear relationship with the increase in reaction temperature. This behavior may be associated with the hydrogenolysis of glycerol to 1,2-PD in an alkaline medium, a process widely studied in the literature.74,75 After reaching the maximum selectivity to LA at 240 °C, it is noted that the increase in temperature to 260 °C contributes negatively. This reduction in LA selectivity is in accordance with Ramírez-López et al.:18 they observed that the increase in temperature may promote the thermal decomposition of LA, as well as its intermediates. These effects are more evident when assessing yields of the products, Fig. 7(B). With successive increases in reaction temperature from 200 to 240 °C, the LA yield increases from 45 to 75%, respectively. However, in the catalytic test performed at 260 °C/46 atm there is an accentuated reduction in LA yield, with values in the range of 55%. The 1,2-PD yield also increases with rising temperatures, however, a maximum is reached (∼20%) at the highest temperatures (240–260 °C).
Roy et al.19 also observed an increase in the conversion of glycerol and in the yield to LA with the increase of the reaction temperature from 200 to 240 °C using Cu/SiO2 and batch reaction system. Li et al.76 evaluated the effect of temperature in a smaller range (100–175 °C), using commercial Cu–Zn–Al catalyst (63.5 wt% Cu) in batch reactor.
It was observed in the temperature range 100–145 °C that the glycerol conversion and LA yield increase rapidly with increasing reaction temperature, obtaining at 145 °C close to 100% of glycerol conversion with LA yield around of 90% (4 h of reaction; 1.2 g catalyst; NaOH/glycerol = 1.5). It is not possible to make a direct and precise comparison with our results, since the reaction conditions and the reaction system are different; however, we observed that it is possible to obtain high LA yields if the reaction conditions were adequate.
Yin et al.77 studied effect of reaction temperature on the conversion of glycerol into LA over Ni/graphite catalyst. With increasing the reaction temperatures from 180 to 230 °C, the conversions of glycerol increased from 34% to 95% and the yield of LA increased from 32% to 88%, approximately. However, in the catalytic test performed at 250 °C, although the conversion of glycerol was 100%, a reduction in the yield to LA (∼80%) was obtained. Thus, the appropriate reaction temperature should be less than 260 °C and among the temperatures evaluated, 240 °C proved to be the most suitable.
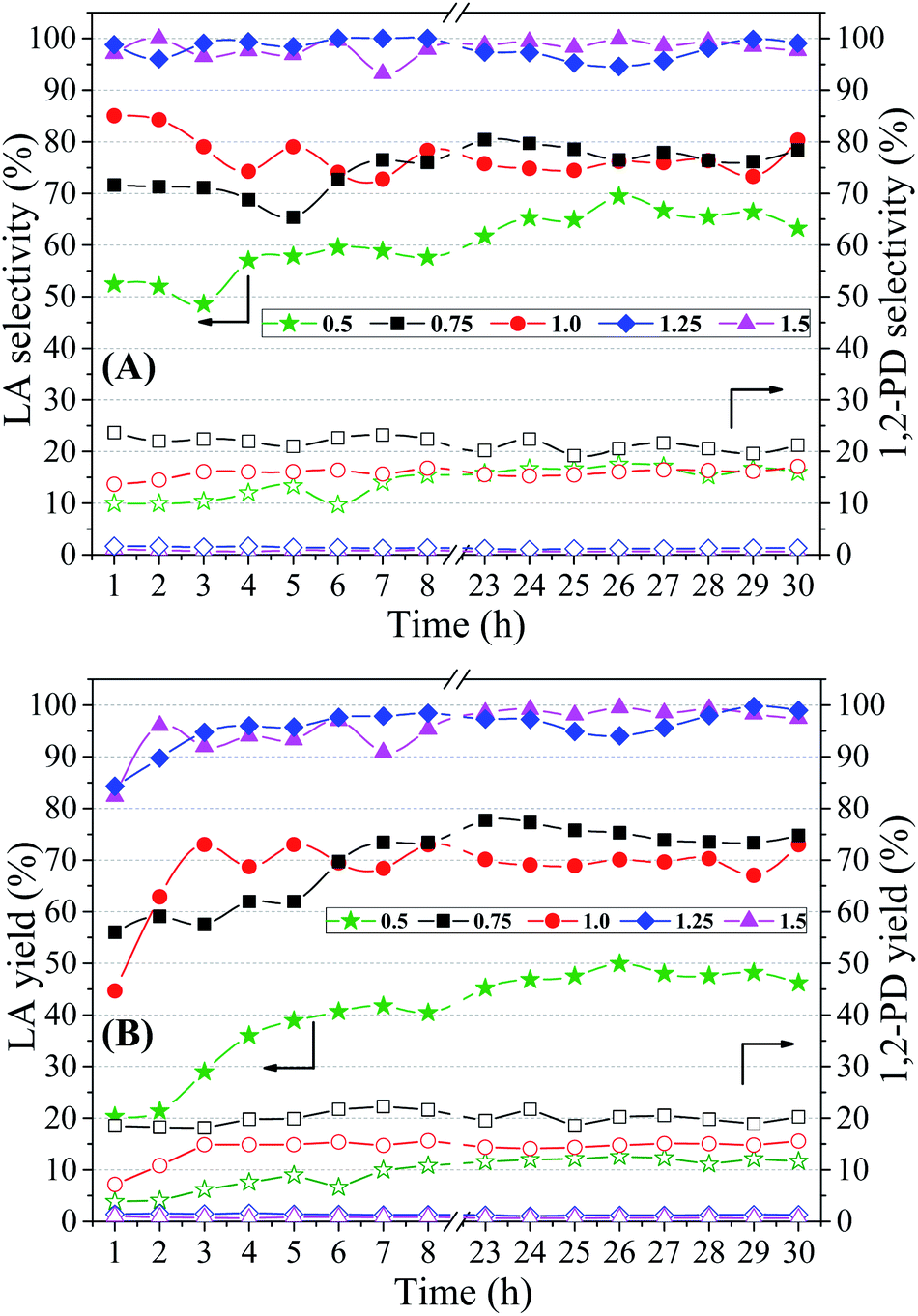 | ||
| Fig. 8 Lactic acid (filled symbols) and 1,2-propanediol (empty symbols) selectivity (A) and yields (B) at different NaOH/glycerol molar ratios (0.5, 0.75, 1.0, 1.25 and 1.5). Reaction conditions: 240 °C/35 atm, 1.25 g Cu5CaMg catalyst, 10 vol% glycerol solution and feed flow = 0.041 mL min−1 (WHSV = 2 h−1). | ||
| NaOH/glycerol molar ratio | Xa (%) | Selectivitya (%) | Yielda (%) | ||
|---|---|---|---|---|---|
| LA | 1,2-PD | LA | 1,2-PD | ||
| a Reaction conditions: 240 °C/35 atm; 10 vol% glycerol solution; Cu5CaMg catalyst (1.25 g); feed flow 0.041 mL min−1. | |||||
| 0.5 | 72.5 | 65.4 | 16.5 | 47.4 | 11.9 |
| 0.75 | 96.4 | 78.0 | 20.7 | 75.2 | 19.9 |
| 1.0 | 91.9 | 75.9 | 16.0 | 69.7 | 14.7 |
| 1.25 | 99.8 | 97.1 | 1.2 | 96.9 | 1.2 |
| 1.5 | 99.8 | 98.8 | 0.7 | 98.6 | 0.7 |
Analyzing Fig. 8(A), it is observed that the lowest selectivities to LA were obtained in the catalytic test performed with NaOH/glycerol molar ratio of 0.50. With the increase of the molar ratio to 0.75 and 1.00, it was observed an increase in LA selectivity (75–80%), but without much distinction between them. However, in the catalytic tests carried out with NaOH/glycerol molar ratios of 1.25 and 1.50, a significant increase in selectivity to LA was obtained, both reaching values close to 100%.
Regarding the selectivity to 1,2-PD, it is noted that with the increase in the molar ratio (0.75 to 1.5) the formation of 1,2-PD is continuously suppressed, reaching values below 1% (Table 5). Therefore, it is observed that the raise of the NaOH/glycerol molar ratio, besides promoting an increase in the conversion of glycerol and LA selectivity, minimizes the formation of 1,2-PD that is the main secondary reaction.
According to the literature, the addition of base is considered as a factor of great importance to initiate the reaction of transformation of glycerol to LA and to improve selectivity to LA. Some studies that evaluated the relationship between base and glycerol agree that the addition of bases, such as NaOH and KOH, potentiate the formation of LA.72,78,79
From Fig. 4, which shows reaction pathways for glycerol conversion into LA in alkaline medium, it is observed that the base (OH−) has a fundamental role in two steps, firstly in the dehydration of glyceraldehyde to 2-hydroxypropenal and posteriorly in the final step of the transformation of pyruvaldehyde in LA, through a molecular rearrangement also known as intra molecular Cannizzaro reaction. Thus, we observed that NaOH/glycerol molar ratios greater than 1.0, are necessary to obtain high selectivities to LA as well as to reduce secondary products.
Fig. 8(B) shows the yields obtained during 30 h of reaction under different NaOH/glycerol molar ratios and in Table 5 the average values (23–30 h) are presented. The lowest yield to LA was obtained in the catalytic test performed with the lowest NaOH/glycerol molar ratio (0.5), which is directly related to the poor results of glycerol conversion and selectivity to LA, obtained in this condition. Catalytic evaluations performed with NaOH/glycerol molar ratios of 0.75 and 1.0, a considerable gain in yield was obtained (∼20%). Greater gains in yield to LA were obtained when molar ratios greater than 1.0 were used, reaching yields close to 100% in the catalytic tests carried out with molar ratios of 1.25 and 1.5, Table 5. The yield to 1,2-PD shows the opposite behavior, since the increase in the concentration of NaOH in the reaction medium promotes a reduction in the yield to 1,2-PD; thus, it is observed that there is a shift of the reaction towards LA (Fig. 4), consequently minimizing the secondary reaction of 1,2-PD production.
The average conversion of glycerol obtained for the two concentrations was very close to each other, 96.4% and 94.7%, respectively to the concentrations of 10 and 20 vol%. However, the increase in glycerol concentration to 20 vol% promoted a loss of selectivity to LA, presenting an average selectivity of 59.3%, which represents a reduction of approximately 20 points in relation to the catalytic test performed with 10 vol% glycerol solution (78.0%), while the average selectivity to 1,2-PD showed a small increase to 24.4%. Therefore, the reduction of the selectivity to LA observed with the increase in the concentration of glycerol may be associated with the increase in the generation of 1,2-PD, as well as an increase in the glycerol conversion to gaseous products, since high values of glycerol conversion were obtained, and no new liquid byproducts were observed in the chromatograms. These results are in agreement with that observed by Yin et al.,77 where they obtained a reduction in the carbon balance from 96.8% to 83.8%, in the catalytic tests carried out with glycerol concentration of 3.6 vol% (0.5 M) and 21.9 vol% (3.0 M), respectively. This reduction in the carbon balance was related to the decomposition reactions of the products, with generation of CO2. Ramírez-López et al.18 also observed a reduction in selectivity and yield in LA with increased concentration of glycerol in the reaction medium (2.5 3.5 M). The authors related this result to an increase in the concentration of intermediate products and LA in the reaction medium and this has a negative effect, since it promotes an increase in decomposition rates.
The LA yield showed values around 56% for the catalytic test developed using a 20 vol% glycerol solution, representing a reduction of approximately 20 points, compared to the test performed with 10 vol% glycerol solution (75.2%). However, the most concentrated glycerol solution has been shown to positively affect the yield of 1,2-PD, which may be associated with a higher hydrogenation rate of 2-hydroxypropenal and/or pyruvaldehyde.
As previously reported in the synthesis of 1,2-PD, the presence of impurities such as salts, ash, methanol and water could affect the catalyst activity.80 Thus, in order to evaluate whether crude glycerol from biodiesel production could be used directly for synthesizing LA, a reaction using a crude glycerol obtained from a plant producing biodiesel from soybean oil was performed in the following conditions: 12 vol% crude glycerol solution (equivalent to 10 vol% pure glycerol solution), 240 °C/35 atm, NaOH/glycerol molar ratio of 1.25, feed flow of 0.041 mL min−1, 1.25 g of Cu5CaMg catalyst (WHSV = 2 h−1).
The conversion of crude glycerol showed high values with an average (23–30 h) conversion of 98.4%, very similar to that obtained with pure glycerol (99.8%). Therefore, the impurities present in the crude glycerol did not promote catalytic deactivation, since the conversion remained constant throughout the evaluated period (30 h). Usually such impurities (not analyzed) are salts, residual alcohol, soap, and fats. However, the selectivity to LA and 1,2-PD products was affected by the impurities present in the crude glycerol. The average (23–30 h) selectivity to LA was reduced from 97.1% (pure glycerol) to 90.2%, while the selectivity to 1.2-PD increased from 1.2% (pure glycerol) to 3.1%. Ftouni et al.22 evaluated the use of crude glycerol in a batch reactor at 180 °C using NaOH/glycerol molar ratio = 1.1 and Pt/ZrO2 catalyst. After 24 h of reaction it was observed that the selectivity to LA and 1,2-PD did not change, however the glycerol conversion was significantly impacted with the use of crude glycerol, which represented a reduction in the yield to LA of 72.2% (pure glycerol) to 52.5%, approximately. We also observed a reduction in yield to LA by using crude glycerol, however it was less pronounced, from 96.9% (pure glycerol) to 88.7%. This may suggest that the impact of impurities is less on the catalytic activity of copper than on platinum. Long et al.81 using 1.5 M sodium silicate as catalyst in a batch reactor at 300 °C, a slight reduction in LA yield was observed with the use of crude glycerol. Ramírez-López et al.18 evaluated the hydrothermal conversion of glycerol to LA at 280 °C with NaOH/glycerol molar ratio of 1.1 and did not observe differences between the tests performed with crude and pure glycerol.
4. Conclusions
Cu catalysts supported on CaO/MgO in different proportion was prepared by wet impregnation, containing 20 wt% of CuO. The XRD analysis showed that the calcination temperature of 700 °C was effective in the formation of the crystalline CaO phase, as well as demonstrated that the conditions employed in the reduction of CuO were adequate. Concerning the Cu average crystallite size and Cu dispersion, there were no significant differences among the catalysts, which showed values in the range of 14 nm and 7%, respectively. The XRD of the spent catalysts showed that new crystalline phases are formed after the catalytic test due to the transformation of the crystalline structure of the support, however, without promoting any loss of performance for the catalysts. In addition, it is noted that although the Cu2O formation occurs, the metallic copper phase remains after the reaction. The textural properties showed that the specific areas of all catalysts were similar and with low values (<20 m2 g−1).The H2-TPR analysis showed that the maximum reduction peaks of CuO of all catalysts were in the range 365 to 470 °C, values above the range considered in the literature for the CuO reduction. This fact was associated with a strong interaction between copper and supports. Through the CO2-TPD analysis it was observed that the combination of the supports (CaO and MgO) results in a catalyst with a wider distribution of basic sites, and the most balanced proportion between weak and high strength basic sites was obtained with the Cu5CaMg catalyst. In addition, it was observed that Cu catalysts with different CaO loads (5, 10 and 15 wt%) showed an increase in the density of basic sites in relation to the CuMg catalyst.
The catalytic tests showed that NaOH presents higher selectivities to LA than KOH, although both present high glycerol conversion. The Cu5CaMg, Cu10CaMg and Cu15CaMg catalysts showed superior catalytic performance in relation to the CuMg and CuCa catalysts, and this improved performance was associated with the wide distribution of basic sites. Although TOF analysis demonstrated that mixed support catalysts do not have much distinction among them, they had higher values (∼35%) than CuMg and CuCa catalysts. Therefore, it is observed that an addition of CaO greater than 5 wt% is not necessary to obtain an improvement in catalytic activity, because the highest LA yield was obtained in the catalytic test performed with the Cu5CaMg catalyst. Among the evaluated temperatures (200–260 °C), the condition of 240 °C/35 atm exhibited the best results in terms of glycerol conversion, selectivity and yield to LA. The increase in the NaOH/glycerol molar ratio demonstrated to be a relevant factor to achieve high values of glycerol conversion, selectivity and yield to LA, however, it was observed that in the molar ratio of 1.25 it was possible to obtain values close to the maximum with LA yield around 97%. In addition, it was noted that the increase in the molar ratio minimizes the formation of byproduct (1,2-PD). The increase in the concentration of glycerol in the feed from 10 vol% to 20 vol% showed to be disadvantageous to the reaction, due to the loss of selectivity to LA. In the catalytic test performed with crude glycerol, under the optimized conditions, it was observed that it is possible to replace the refined glycerol by crude glycerol without major losses in LA yield.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the GreenTec/EQ/UF for providing BET analysis, FAPERJ (#E-26/202.751/2019 and #E-26/010.100619/2018) and CNPq (#400706/2016-4) for the financial support granted to carry out this work.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- Y. Fan, C. Zhou and X. Zhu, Catal. Rev.: Sci. Eng., 2009, 51, 293–324 CrossRef CAS.
- Y. Wee, J. Kim and H. Ryu, Food Technol. Biotechnol., 2006, 44, 163–172 CAS.
- P. S. Panesar and S. Kaur, Int. J. Food Sci. Technol., 2015, 50, 2143–2151 CrossRef CAS.
- Grand View Research, Lactic Acid Market Analysis By Application (Industrial, F&B, Pharmaceuticals, Personal Care) & Polylactic Acid (PLA) Market Analysis By Application (Packaging, Agriculture, Transport, Electronics, Textiles), And Segment Forecasts, 2018–2025, 2017 Search PubMed.
- IEA Bioenergy Task 42 report, Bio-based Chemicals Value Added Products from Biorefineries, 2012 Search PubMed.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145–152 CrossRef CAS.
- H. Ohara and M. Yahata, J. Ferment. Bioeng., 1996, 81, 272–274 CrossRef CAS.
- N. Razali and A. Z. Abdullah, Appl. Catal., A, 2017, 543, 234–246 CrossRef CAS.
- Y. J. Wee, J. N. Kim and H. W. Ryu, Food Technol. Biotechnol., 2006, 44, 163–172 CAS.
- H. Oh, Y. J. Wee, J. S. Yun, H. H. Seung, S. Jung and H. W. Ryu, Bioresour. Technol., 2005, 96, 1492–1498 CrossRef CAS PubMed.
- H. Oh, Y.-J. Wee, J.-S. Yun and H.-W. Ryu, Appl. Biochem. Biotechnol., 2003, 105–108, 603–613 CrossRef CAS PubMed.
- S. K. Singh, S. U. Ahmed and A. Pandey, Process Biochem., 2006, 41, 991–1000 CrossRef CAS.
- D. Rathin and T. Shih-Perng, Fuels Chem. from Biomass, 1997, vol. 666, pp. 12–224 Search PubMed.
- L. Shen, Z. Yu, D. Zhang, H. Yin, C. Wang and A. Wang, J. Chem. Technol. Biotechnol., 2019, 94, 204–215 CrossRef CAS.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- H. Kishida, Z. Zhou, F. Jin, T. Moriya and H. Enomoto, Chem. Lett., 2005, 34, 1560–1561 CrossRef CAS.
- C. A. Ramírez-López, J. R. Ochoa-Gómez, M. Fernández-Santos, O. Gómez-Jiménez-Aberasturi, A. Alonso-Vicario and J. Torrecilla-Soria, Ind. Eng. Chem. Res., 2010, 49, 6270–6278 CrossRef.
- D. Roy, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2011, 1, 548–551 CrossRef CAS.
- H. Yin, C. Zhang, H. Yin, D. Gao, L. Shen and A. Wang, Chem. Eng. J., 2016, 288, 332–343 CrossRef CAS.
- M. R. A. Arcanjo, I. J. Silva, E. Rodríguez-Castellón, A. Infantes-Molina and R. S. Vieira, Catal. Today, 2017, 279, 317–326 CrossRef CAS.
- J. Ftouni, N. Villandier, F. Auneau, M. Besson, L. Djakovitch and C. Pinel, Catal. Today, 2015, 257, 267–273 CrossRef CAS.
- L. Shen, H. Yin, H. Yin, S. Liu and A. Wang, J. Nanosci. Nanotechnol., 2017, 17, 780–787 CrossRef CAS.
- H. Yin, H. Yin, A. Wang, L. Shen, Y. Liu and Y. Zheng, J. Nanosci. Nanotechnol., 2017, 17, 1255–1266 CrossRef CAS PubMed.
- G. Zhang, F. Jin, B. Wu, J. Cao, Y. S. Adam and Y. Wang, Int. J. Chem. React. Eng., 2012, 10, 1–22 Search PubMed.
- T. Shimanouchi, S. Ueno, K. Shidahara and Y. Kimura, Chem. Lett., 2014, 43, 535–537 CrossRef CAS.
- A. B. F. Moreira, A. M. Bruno, M. M. V. M. Souza and R. L. Manfro, Fuel Process. Technol., 2016, 144, 170–180 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Renewable Energy, 2008, 33, 1097–1100 CrossRef CAS.
- Y. Chisti, J. Biotechnol., 2013, 167, 201–214 CrossRef CAS PubMed.
- REN21, Renewables 2017: Global Status Report, 2017, vol. 72 Search PubMed.
- National Agency of Petroleum, Natural Gas and Biofuels (ANP), http://www.anp.gov.br/dados-estatisticos, accessed 5 April 2020 Search PubMed.
- J. R. Anderson, Structure of Metallic Catalysts, Academic Press, London, 1975 Search PubMed.
- O. Hinrichsen, T. Genger and M. Muhler, Chem. Eng. Technol., 2000, 23, 956–959 CrossRef CAS.
- S. Sato, J. Catal., 2000, 196, 195–199 CrossRef CAS.
- V. Mahdavi and A. Monajemi, J. Taiwan Inst. Chem. Eng., 2014, 45, 2286–2292 CrossRef CAS.
- M. C. G. Albuquerque, J. Santamaría-González, J. M. Mérida-Robles, R. Moreno-Tost, E. Rodríguez-Castellón, A. Jiménez-López, D. C. S. Azevedo, C. L. Cavalcante and P. Maireles-Torres, Appl. Catal., A, 2008, 347, 162–168 CrossRef CAS.
- L. Wang, J. R. Gaudet, W. Li and D. Weng, J. Catal., 2013, 306, 68–77 CrossRef CAS.
- R. S. Roth, C. J. Rawn, J. J. Ritter and B. P. Burton, J. Am. Ceram. Soc., 1989, 72, 1545–1549 CrossRef CAS.
- M. Kaewpanha, S. Karnjanakom, G. Guan, X. Hao, J. Yang and A. Abudula, J. Energy Chem., 2017, 26, 660–666 CrossRef.
- J. Zhang and D. He, J. Chem. Technol. Biotechnol., 2015, 90, 1077–1085 CrossRef CAS.
- B. M. Nagaraja, V. Siva Kumar, V. Shasikala, A. H. Padmasri, B. Sreedhar, B. David Raju and K. S. Rama Rao, Catal. Commun., 2003, 4, 287–293 CrossRef CAS.
- K. H. P. Reddy, R. Rahul, S. S. V. Reddy, B. D. Raju and K. S. R. Rao, Catal. Commun., 2009, 10, 879–883 CrossRef.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2011, 45, 10750–10756 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. a. W. Haul, L. Moscou, R. a. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- J. D. A. Bellido, J. E. De Souza, J. C. M'Peko and E. M. Assaf, Appl. Catal., A, 2009, 358, 215–223 CrossRef CAS.
- S. Casenave, H. Martinez, C. Guimon, A. Auroux, V. Hulea and E. Dumitriu, J. Therm. Anal. Calorim., 2003, 72, 191–198 CrossRef CAS.
- L. Dussault, J. C. Dupin, E. Dumitriu, a. Auroux and C. Guimon, Thermochim. Acta, 2005, 434, 93–99 CrossRef CAS.
- A. M. Kalinkin, E. V. Kalinkina, O. A. Zalkind and T. I. Makarova, Inorg. Mater., 2005, 41, 1073–1079 CrossRef CAS.
- Z. Mirghiasi, F. Bakhtiari, E. Darezereshki and E. Esmaeilzadeh, J. Ind. Eng. Chem., 2014, 20, 113–117 CrossRef CAS.
- G. C. Bond, S. N. Namijo and J. S. Wakeman, J. Mol. Catal., 1991, 64, 305–319 CrossRef CAS.
- C. J. G. Van Der Grift, A. F. H. Wielers, A. Mulder and J. W. Geus, Thermochim. Acta, 1990, 171, 95–113 CrossRef CAS.
- F. W. Chang, W. Y. Kuo and K. C. Lee, Appl. Catal., A, 2003, 246, 253–264 CrossRef CAS.
- B. M. Nagaraja, V. S. Kumar, V. Shashikala, A. H. Padmasri, S. S. Reddy, B. D. Raju and K. S. R. Rao, J. Mol. Catal. A: Chem., 2004, 223, 339–345 CrossRef CAS.
- I. Halasz, H. W. Jen, A. Brenner, M. Shelef, S. Kao and K. Y. Simon Ng, J. Solid State Chem., 1991, 92, 327–338 CrossRef CAS.
- M. Su, R. Yang and M. Li, Fuel, 2013, 103, 398–407 CrossRef CAS.
- H. Xue, X. Guo, S. Wang, C. Sun, J. Yu and D. Mao, Catal. Commun., 2018, 112, 53–57 CrossRef CAS.
- M. a. Aramendía, V. Borau, C. Jiménez, A. Marinas, J. M. Marinas, J. R. Ruiz and F. J. Urbano, J. Mol. Catal. A: Chem., 2004, 218, 81–90 CrossRef.
- Y. H. Taufiq-Yap, H. V. Lee, M. Z. Hussein and R. Yunus, Biomass Bioenergy, 2011, 35, 827–834 CrossRef CAS.
- A. O. Menezes, M. T. Rodrigues, A. Zimmaro, L. E. P. Borges and M. a. Fraga, Renewable Energy, 2011, 36, 595–599 CrossRef CAS.
- J. P. d. S. Q. Menezes, F. C. Jácome, R. L. Manfro and M. M. V. M. Souza, Catal. Lett., 2019, 149, 1991–2003 CrossRef CAS.
- L. S. Sharninghausen, J. Campos, M. G. Manas and R. H. Crabtree, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- S. A. Zavrazhnov, A. L. Esipovich, S. Y. Zlobin, A. S. Belousov and A. V. Vorotyntsev, Catalysts, 2019, 9, 1–21 CrossRef.
- Z. Shen, F. Jin, Y. Zhang, B. Wu, A. Kishita, K. Tohji and H. Kishida, Ind. Eng. Chem. Res., 2009, 48, 8920–8925 CrossRef CAS.
- C. Montassier, J. C. Ménézo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS.
- J. Ten Dam, F. Kapteijn, K. Djanashvili and U. Hanefeld, Catal. Commun., 2011, 13, 1–5 CrossRef CAS.
- L. Liu and X. P. Ye, Fuel Process. Technol., 2015, 137, 55–65 CrossRef CAS.
- A. M. Bruno, C. A. Chagas, M. M. V. M. Souza and R. L. Manfro, Renewable Energy, 2017, 118, 160–171 CrossRef.
- H. Mitta, P. K. Seelam, S. Ojala, R. L. Keiski and P. Balla, Appl. Catal., A, 2018, 550, 308–319 CrossRef CAS.
- Z. Huang, F. Cui, H. Kang, J. Chen and C. Xia, Appl. Catal., A, 2009, 366, 288–298 CrossRef CAS.
- A. Żelazny, K. Samson, R. Grabowski, M. Śliwa, M. Ruggiero-Mikołajczyk and A. Kornas, React. Kinet., Mech. Catal., 2017, 121, 329–343 CrossRef.
- R. Palacio, S. Torres, S. Royer, A. S. Mamede, D. López and D. Hernández, Dalton Trans., 2018, 47, 4572–4582 RSC.
- G.-Y. Yang, Y.-H. Ke, H.-F. Ren, C.-L. Liu, R.-Z. Yang and W.-S. Dong, Chem. Eng. J., 2016, 283, 759–767 CrossRef CAS.
- K.-T. Li and H.-H. Li, Appl. Biochem. Biotechnol., 2020, 191, 125–134 CrossRef PubMed.
- J. Feng, W. Xiong, B. Xu, W. Jiang, J. Wang and H. Chen, Catal. Commun., 2014, 46, 98–102 CrossRef CAS.
- S. Wang, Y. Zhang and H. Liu, Chem.–Asian J., 2010, 5, 1100–1111 CrossRef CAS PubMed.
- K. T. Li, J. Y. Li and H. H. Li, J. Taiwan Inst. Chem. Eng., 2017, 79, 74–79 CrossRef CAS.
- H. Yin, H. Yin, A. Wang and L. Shen, J. Ind. Eng. Chem., 2018, 57, 226–235 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
- H. S. A. De Sousa, F. D. A. A. Barros, S. J. S. Vasconcelos, J. M. Filho, C. L. Lima, A. C. Olivira, A. P. Ayala, M. C. Junior and A. C. Oliveira, Appl. Catal., A, 2011, 406, 63–72 CrossRef CAS.
- C. W. Chiu, M. A. Dasari, W. R. Sutterlin and G. J. Suppes, Ind. Eng. Chem. Res., 2006, 45, 791–795 CrossRef CAS.
- Y. D. Long, F. Guo, Z. Fang, X. F. Tian, L. Q. Jiang and F. Zhang, Bioresour. Technol., 2011, 102, 6884–6886 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |