
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed ortho-selective carbon–hydrogen amidation of benzamides with sulfonyl azides in ionic liquid†
Lin-Yu Jiao *ab,
Zi-Hui Ninga,
Qian Honga,
Xin-Hua Penga,
Xiao-Mei Yina,
Shanshan Liu*c,
Huiyong Chenab,
Zhuo Liab,
Ming Sunab and
Xiao-Xun Maab
*ab,
Zi-Hui Ninga,
Qian Honga,
Xin-Hua Penga,
Xiao-Mei Yina,
Shanshan Liu*c,
Huiyong Chenab,
Zhuo Liab,
Ming Sunab and
Xiao-Xun Maab
aSchool of Chemical Engineering, Northwest University, Xi'an, Shaanxi 710069, P. R. China. E-mail: lyjiao@nwu.edu.cn
bInternational Scientific and Technological Cooperation Base for Clean Utilization of Hydrocarbon Resources, Chemical Engineering Research Center of the Ministry of Education for Advance Use Technology of Shanbei Energy, Shaanxi Research Center of Engineering Technology for Clean Coal Conversion, Collaborative Innovation Center for Development of Energy and Chemical Industry in Northern Shaanxi, Northwest University, Xi'an, Shaanxi 710069, P. R. China
cShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an, Shaanxi 710021, P. R. China. E-mail: shanshanliu@sust.edu.cn
First published on 11th August 2020
Abstract
An efficient and convenient iridium(III) catalyzed ortho-C–H bond amidation of weakly coordinating benzamides treated with readily available sulfonyl azides as the amino source has been described. In this transformation, ionic liquids represents an ideal reaction medium, giving rise to a broad range of amidation products under mild conditions in the open air. This protocol offers moderate to excellent chemical yields, exclusive regioselectivities, and good functional group tolerance.
Introduction
Molecules containing nitrogen atoms are of great importance in the areas of natural products, materials science, biologically active compounds, and medicines.1 Among the current available strategies, Ullmann-type coupling reaction,2 Buchwald–Hartwig amination,3 and Chan–Lam coupling4 have been extensively investigated in the last decades towards the desired carbon–nitrogen bond formation and generally exhibited high transformation efficiency (Scheme 1a). However, these methodologies relied heavily on the prefunctionalized haloarenes, which would generate stoichiometric amounts of hydrogen halides and the corresponding salts as byproducts.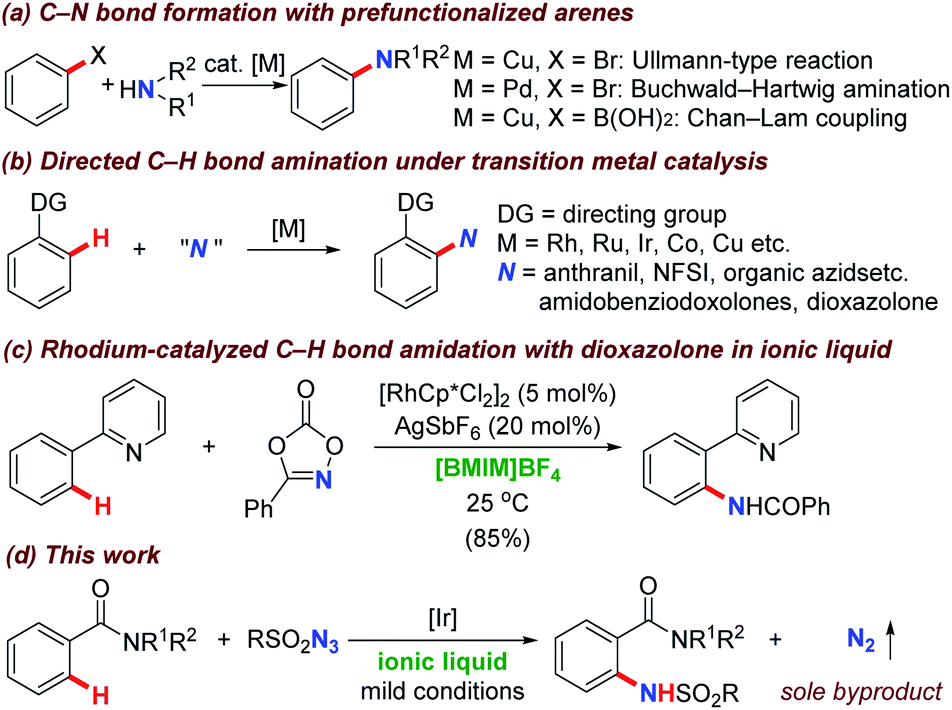 | ||
| Scheme 1 Strategies for the C–N bond formation under transition metal catalysis. | ||
Recently, the investigation of synthetic methodologies that accommodate selective functionalization of omnipresent carbon–hydrogen bonds to useful amine/amide functional groups under transition metal catalysis have been recognized as the most straightforward and powerful alternatives.5 Given the numerous reports concerning this topic, several different transition metals have been introduced to accomplish this target. Meanwhile, different nitrogen sources including anthranils, 1,4,2-dioxazol-5-ones, amidobenziodoxolones, N-fluorobenzenesulfonimide (NFSI), organic azides, and nitrosoarenes were successfully employed based on two distinct mechanisms (Scheme 1b).5–12 Particularly, the Chang group disclosed a significant breakthrough for the direct C(sp2)–H bond amidation via rhodium mediated nitrene insertion pathway deriving from sulfonyl azides in 2012,6 which represented a green process without the addition of external oxidants and released nitrogen gas as the sole byproduct. Consequently, many groups including Li,7 Sahoo,8 Jiao (N.),9 Ackermann,10 Glorius,11 and Kanai12 etc. continuously disclosed directed amidation methodologies and organic azides have been widely used as nitrogen source almost overnight.13 From then on, research efforts in this area have been devoted to the development of a number of amidation systems based on different combinations of transition metals (rhodium, ruthenium, iridium, cobalt, copper, as well as iron, et al.) and organic azides (sulfonyl-, aryl-, benzyl-, and alkyl-azides etc.). Notably, almost all of these transformations are conducted in organic solvents, in which the halogenated solvents such as 1,2-dichloroethane (DCE) and 1,2-dichlorobenzene (o-DCB) are most commonly choices.5–13 As far as we know, there are only limited reports focusing on C–N formation in environmental benign solvents.14
In the organic reactions, solvent consumption represented a major issue to lead to organic pollution. In order to encounter this problem, much efforts has been devoted to the environmental credentials of the reaction medium itself. As a consequence, a range of reaction mediums with greener character and more sustainable nature have been developed and evaluated over the past years.15 Ionic liquids, which are salts with room temperature or closed melting points and extremely high enthalpies of vaporization, have shown great potential with extraordinary properties through different cation/anion combinations compared with conventional organic solvents.15 The cations are normally dependent on the chain length while the counter anions are generally attributed to the enhancement of their water and air stabilities.16 Moreover, ionic liquids were introduced as reaction medium with great advantages including the lack of vapor pressure, non-volatility, non-flammability, and thermal stability over a wide temperature range. During the last years, ionic liquids have been successfully subjected into the C–H bond functionalization, which opened a new era to make this attractive strategy proceeding in an environmental benign fashion. Very recently, Wang and Wu developed a rhodium(III)-catalyzed C–H bond amidation with dioxazolones as amidating reagent (Scheme 1c).14b In this reaction, 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4) was employed as a green and recyclable reaction media, providing desired amidated product smoothly. This novel strategy, together with the pioneering works concerning sulfonyl azides as amidation precursors6–13 inspired us to examine the catalytic activity of transition metal for the directing group assisted C–H bond amidation process with organic azides in non-traditional solvent. Based on our previous experience on the transition metal catalyzed C–H bond functionalization,17 we were hopeful to take advantage of the aforementioned features in our protocol, thereby establishing an environmentally friendly approach that can be applied to a wide range of substrates in the absence of external oxidants to generate nitrogen gas as byproduct (Scheme 1d). We envisioned that ionic liquid could be an effective reaction medium to meet the requirements.
Results and discussion
To examine our hypothesis, we initiated the reaction between N-tertbutylbenzamide (1a) and tosyl azide (2a) as nitrogen source to identify of the reaction conditions. In this catalytic setup, [IrCp*Cl2]2, AgSbF6, and [BMIM]BF4 were chosen as catalyst, silver salt, and reaction medium rather than organic solvent, respectively. As shown in Table 1, the model substrates underwent C–H amidation as expected, although the corresponding products 3aa, namely N-(tert-butyl)-2-[(4-methylphenyl)sulfonamido]benzamide, was generated in low yield (18%) at room temperature (20 °C) (entry 1). However, it was noteworthy that the functionalization indeed occurred exclusively at the ortho-position of the starting material 1a, proving the amidated product 3aa in a single form with almost unreacted substrate recovery (78%) (isolated after purification by flash column chromatography on silica gel, data not shown in the table). The structure was confirmed unambiguously through NMR (nuclear magnetic resonance) and HR-MS (high resolution mass spectroscopic) analyses. Afterwards, other parameters such as reaction temperature, silver salts, additives, and ionic liquids were systematically investigated. The reaction temperature proven to be an important factor for the system. When the temperature was increased to 30 °C, the yield increased dramatically to 45% after shortened reaction time (entry 2). Silver salts screening indicated that the AgNTf2 was the best (entries 3–6), while AgBF4, which shared the identical anion with reaction medium, only generated a little amount of amidation product (entry 4). It is worth to mention that the amidation happened even when the catalyst loading reduced by half, albeit with slightly lower yield for longer reaction time (entry 7). Then, we attempted with the aid of doubled catalyst loading, but the yield was not substantially enhanced (entry 8). Importantly but not surprisingly, the starting material 1a remained untouched in the absence of the iridium catalyst (entry 9). In order to investigate the impact of additives on this reaction, we then examined different base and acid. However, the additive exhibited to be detrimental in this transformation, providing decreased or even traces amount of product (entries 10–12). This phenomenon indicated that the aforementioned combination of anion and cation of the ionic liquid (entry 3) was just enough to promote the amidation. Replace the anion of ionic liquid with [NTf2]− or [PF6]− resulted in an increasing of catalytic performance with [BMIM]PF6 representing the best result so far. Nevertheless, [BMIM]OTf displayed relative lower reactivity (entries 13–15). All the ionic liquids investigated were bearing neutral or inert anions such as [BF4]−, [PF6]−, [OTf]−, and [NTf2]−. However, the only role of these anions was to provide polarities to the reaction medium by enhancing the catalytic activities and they rarely involved into the catalysis. Additionally, they represented weakly coordinating properties to form hydrogen bonds to stabilize the catalytic system. It seems that the results showed almost an opposite trend with the basicity of them (basicity in decreasing order: [OTf]− > [BF4]− > [PF6]− > [NTf2]−) (entries 2 and 13–15).18 Remarkably, the result could be further improved when at 40 °C, with 1a consumed almost completely after 8 h to afford the tosyl amidation product in excellent yield (entry 16). However, the result at higher temperature (e.g. 50 °C) changed back into sluggish (entry 17). Therefore, it can be concluded that the optimal reaction conditions are as follows: the catalysis was performed with the aid of 5 mol% of [IrCp*Cl2]2 and AgNTf2 with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 at 40 °C in the open air, in which ionic liquid [BMIM]PF6 was the optimized reaction medium.
:4 at 40 °C in the open air, in which ionic liquid [BMIM]PF6 was the optimized reaction medium.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | x mol% | Silver salt | Additive | Solvent | Temperature (°C) | Time (h) | Yield (%)b |
| a Reaction conditions: N-tertbutylbenzamide 1a (0.40 mmol, 1.0 equiv.), tosyl azide 2a (0.60 mmol, 1.5 equiv.), indicated amount of catalyst [IrCp*Cl2]2, silver salts (4.0 equiv. of catalyst loading), indicated amount of additive in ionic liquid (0.60 mL, 0.67 M) as reaction medium at indicated temperature for indicated reaction time in the open air.b Isolated yield after purification by flash column chromatography on silica gel.c 10 mol% AgNTf2 was used.d 40 mol% AgNTf2 was used.e ND = not detected. | |||||||
| 1 | 5 | AgSbF6 | — | [BMIM]BF4 | 20 | 16 | 18 |
| 2 | 5 | AgSbF6 | — | [BMIM]BF4 | 30 | 8 | 45 |
| 3 | 5 | AgNTf2 | — | [BMIM]BF4 | 30 | 8 | 56 |
| 4 | 5 | AgBF4 | — | [BMIM]BF4 | 30 | 8 | 9 |
| 5 | 5 | AgOAc | — | [BMIM]BF4 | 30 | 8 | 38 |
| 6 | 5 | AgOTf | — | [BMIM]BF4 | 30 | 8 | 20 |
| 7 | 2.5 | AgNTf2c | — | [BMIM]BF4 | 30 | 24 | 40 |
| 8 | 10 | AgNTf2d | — | [BMIM]BF4 | 30 | 8 | 60 |
| 9 | 0 | AgNTf2 | — | [BMIM]BF4 | 30 | 24 | NDe |
| 10 | 5 | AgNTf2 | NaOAc (1.0 equiv.) | [BMIM]BF4 | 30 | 8 | 24 |
| 11 | 5 | AgNTf2 | Li2CO3 (1.0 equiv.) | [BMIM]BF4 | 30 | 8 | 36 |
| 12 | 5 | AgNTf2 | PivOH (1.0 equiv.) | [BMIM]BF4 | 30 | 8 | trace |
| 13 | 5 | AgNTf2 | — | [BMIM]NTf2 | 30 | 8 | 59 |
| 14 | 5 | AgNTf2 | — | [BMIM]PF6 | 30 | 8 | 72 |
| 15 | 5 | AgNTf2 | — | [BMIM]OTf | 30 | 8 | 34 |
| 16 | 5 | AgNTf2 | — | [BMIM]PF6 | 40 | 8 | 92 |
| 17 | 5 | AgNTf2 | — | [BMIM]PF6 | 50 | 8 | 80 |
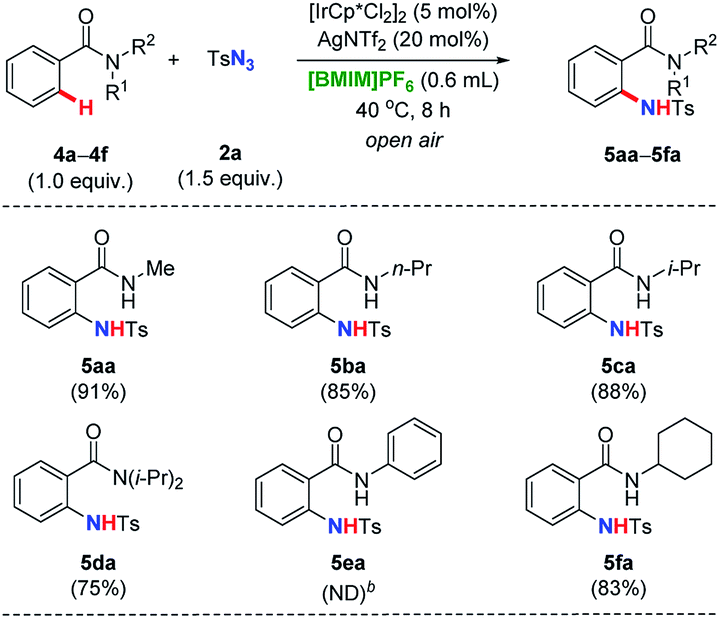
With the amidation conditions established, we were next encouraged to evaluate directing groups with different substituents on the nitrogen atom (Table 2). When the model t-butyl group of 1a was replaced by other alkyl moieties such as methyl (4a), n-propyl (4b), i-propyl (4c), as well as cyclohexyl (4f) groups, the reactivities baring a free N–H unit, although this effect was not especially decreased slightly, resulting in the corresponding products with very good yields (ranging from 83% to 91%). Furthermore, fully substituted benzamide 4d was less favored compared with 4c noticeable. Remarkably, substrate 4e with a phenyl instead of an alkyl group could no longer proceed under the identical reaction conditions, which provided even more available reactive sites in known cases.19
| a Reaction conditions: N-substituted benzamide 4a–4f (0.40 mmol, 1.0 equiv.), tosyl azide 2a (0.60 mmol, 1.5 equiv.), [IrCp*Cl2]2 (5.0 mol%), AgNTf2 (20 mol%), in [BMIM]PF6 (0.60 mL, 0.67 M) as reaction medium at 40 °C for 8 h in the open air, isolated yield reported after purification by flash column chromatography on silica gel.b ND = not detected. |
|---|
 |
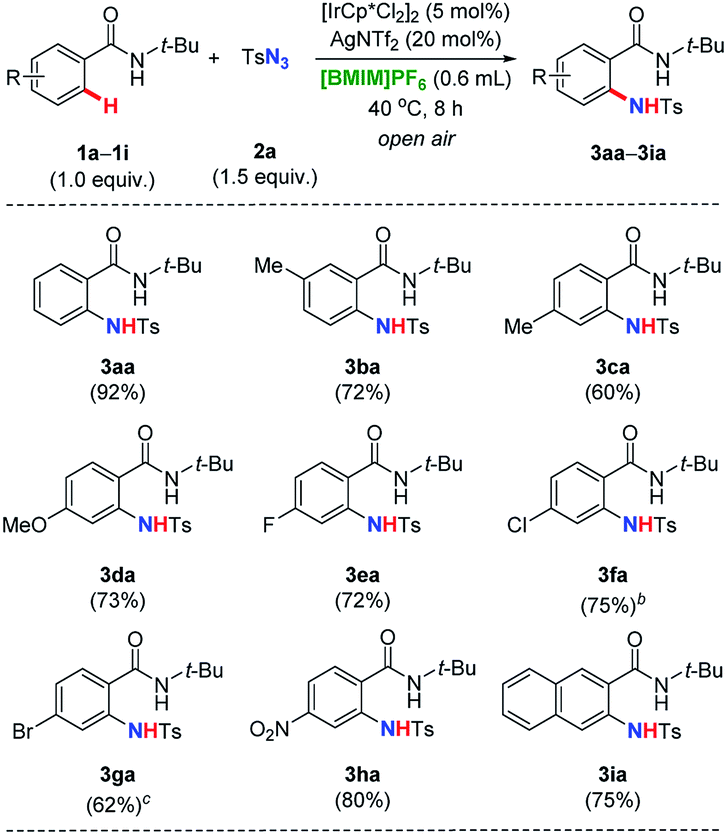
Afterwards, we turned our attention to explore the substrate scope and limitations of various benzamides 1a–1i with tosyl azide 2a (Table 3). Starting materials with substituent at the ortho-, meta-, and para-positions of the aromatic ring have been examined, respectively. The desired amidation was proceeded smoothly, leading to the corresponding product in good yields (1a–1i → 3aa–3ia). Importantly, it was observed that 1a still stood out to be the most effective substrate among its derivatives. The electronic properties of substitution (either electron-donating or -withdrawing group) on the aromatic ring generally played a small role to obtain good yields except for 1f and 1g, that the catalysis need to be conducted at elevated temperature (80 °C for the formation of 3fa and 60 °C for 3ga, respectively). Interestingly, when naphthalene baring the same directing group at the β-position introduced, a single amidating product was isolated in a 2,3-di-substituted fashion, further indicating the highly regioselectivity of our strategy (1i → 3ia).
| a Reaction conditions: N-tertbutylbenzamides 1a–1i (0.40 mmol, 1.0 equiv.), tosyl azide 2a (0.60 mmol, 1.5 equiv.), [IrCp*Cl2]2 (5.0 mol%), AgNTf2 (20 mol%), in [BMIM]PF6 (0.60 mL, 0.67 M) as reaction medium at 40 °C for 8 h in the open air, isolated yield reported after purification by flash column chromatography on silica gel.b Reaction proceeded at 80 °C.c Reaction proceeded at 60 °C. |
|---|
 |
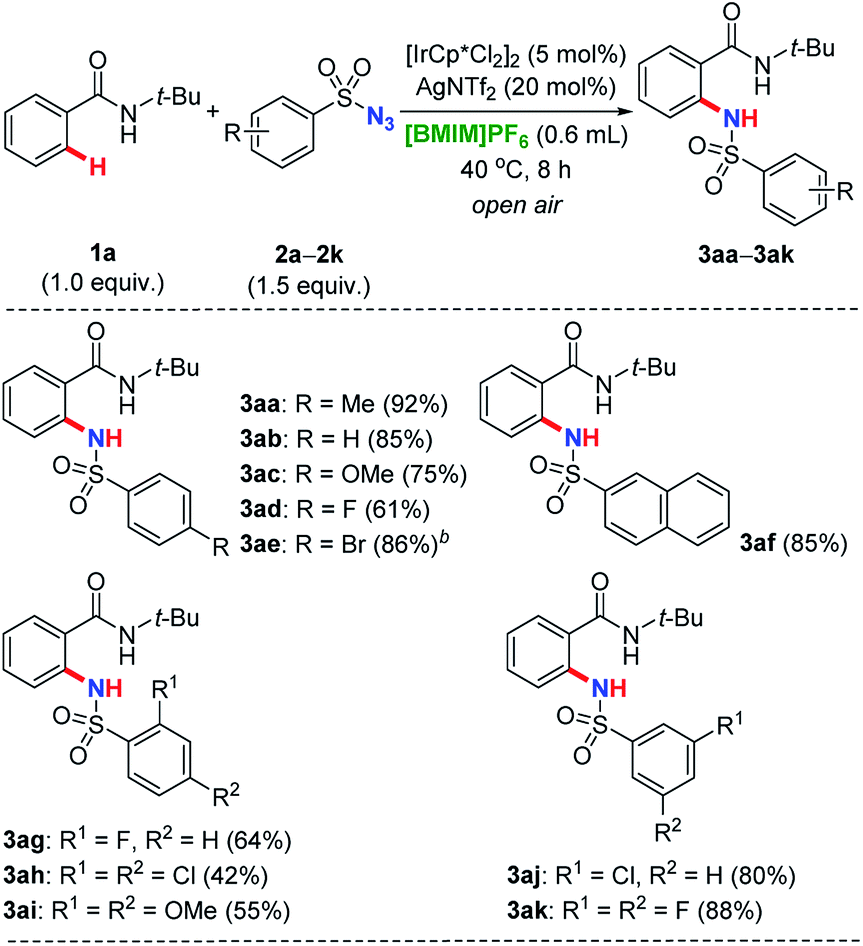
As shown in Table 4, besides 2a, a variety of other sulfonyl azides 2b–2k with varied substitution patterns as amidation reagents were investigated. These nitrogen sources demonstrated to be effective under iridium catalysis, providing ortho-selective substituted benzamide derivatives in moderate to excellent yields. Generally, functional groups commonly used in organic synthesis such as methyl (2a), hydrogen (2b), methoxyl (2c), fluoro (2d, 2g), bromo (2e) and chloro (2j), were well tolerated. This strategy has also been successfully applied for the naphthalene-2-sulfonyl azide (2f) to release desired product 3af. Compared with azides bearing mono-substituent at the aromatic core, disubstituted sulfonyl coupling partners both at 2- and 4-positions have performed reduced reactivities, regardless of the electronic properties (2h–2i → 3ah–3ai). It might be attributed to the effect of steric hindrance, since this phenomenon was not observed for the azide 2k containing two fluorine atoms both at the meta positions (2k → 3ak).
| a Reaction conditions: N-tertbutylbenzamide 1a (0.40 mmol, 1.0 equiv.), sulfonyl azide 2a–2k (0.60 mmol, 1.5 equiv.), [IrCp*Cl2]2 (5.0 mol%), AgNTf2 (20 mol%), in [BMIM]PF6 (0.60 mL, 0.67 M) as reaction medium at 40 °C for 8 h in the open air, isolated yield reported after purification by flash column chromatography on silica gel.b Reaction proceeded at 60 °C. |
|---|
 |
In order to make this methodology more general and applicable, we tried to employ benzyl azide 6a and alkyl azide 6b as well under the optimal reaction conditions. Gratifyingly, as illustrated in Scheme 2, the amidation underwent smoothly satisfyingly. These results provided forceful support of our achievements in this transformation.
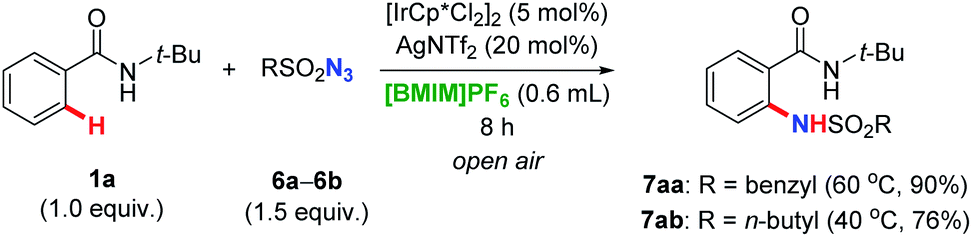 | ||
| Scheme 2 Benzyl and alkyl azides as nitrogen source for the amidation of benzamide 1a. | ||
A plausible mechanism of the amidation was described in Scheme 3 based on our experimental results and previous report.13c A cationic iridium(III) species A was generated in situ by the treatment of [IrCp*Cl2]2 with AgNTf2, and it could continuously participated in the C–H bond activation process of 1a to form a five-membered metallacycle intermediate B. Sequential cascade started with coordination of tosyl azide 2a and followed by the dissociation of nitrogen gas provided iridium ion–nitrene complex D. This resulting species was active enough to undergo migratory C(Ar)–Ir bond insertion to form E with a more stable six-membered ring. Finally, demetalation through the proton exchange with another molecule of starting material 1a made the successfully product 3aa delivery and regenerated the reactive A to involved in the next catalytic cycle.
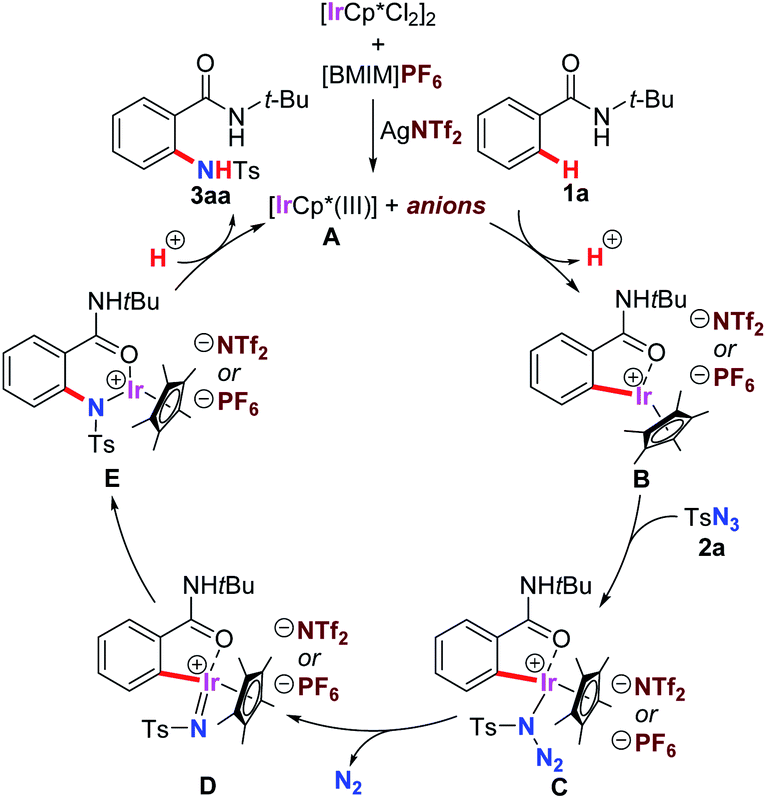 | ||
| Scheme 3 Proposed mechanism of the amidation. | ||
Conclusions
To sum up, we have described herein a catalytic transformation for the conversion of C(sp2)–H bond into C–N bond using organic azides as coupling partners under mild conditions. Benzamides and sulfonyl azides were undertaken for the first time in the greener ionic liquid under iridium catalysis, demonstrated great promise as an attractive alternative to the existing methodologies. The amidation was proceeded exclusively at the ortho-position relative to the directing group and generated nitrogen gas as the sole byproduct. This protocol allows the preparation of a range of sulfonyl amine substituted benzamides in moderate to excellent yields with a broad substrate scope. In addition, benzyl and alkyl sulfonyl azides were also tolerated well, demonstrating the reliable and practical features of this strategy. Furthermore, a mechanism was proposed accordingly. The exploration for further application to the synthesis of biologically active molecules, and detailed mechanism investigations are currently underway in our group.Experimental
General remarks
All reactions were performed in flame-dried glassware with magnetic stirring bar. Liquids and solutions were transferred with syringes. All the benzamide starting materials and sulfonyl azide coupling partners were obtained & used directly or prepared according to the known procedures.13c,20 Ionic liquids involved were purchased and used as received. Solvents were purified and dried following standard procedures. Technical grade solvents for extraction and chromatography (ethyl acetate, petroleum ether) were distilled prior to use. Analytical thin-layer chromatography (TLC) was performed on silica gel GF254 glass plates from Qingdao Haiyang Chemical Co,. Lt. Flash column chromatography was performed on silica gel (300–400 mesh) by Qingdao Haiyang Chemical Co,. Lt using the indicated solvents. Infrared (IR) spectra were recorded on a Bruker (VERTEX 70) FT-IR spectrophotometer by standard KBr-disk method and are reported (br = broad, vw = very weak, w = weak, m = medium, s = strong, vs = very strong) in wavenumbers (cm−1). High resolution mass spectrometry (HRMS) analyses were performed on an Agilent 6460 instrument. 1H, 13C, and 19F NMR spectra were recorded in CDCl3 on Bruker Avance III 400 MHz instrument. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) and are referenced to the residual solvent resonance as the internal standard (CHCl3: δ = 7.26 ppm for 1H and CDCl3: δ = 77.16 ppm for 13C). Data are reported as follows: chemical shift, multiplicity (br s = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz) and integration.General procedure for the iridium-catalyzed ortho-selective carbon–hydrogen bond amidation of benzamides with sulfonyl azides (GP 1)
A flame-dried Schlenk tube equipped with a magnetic stir bar is successively charged with the benzamides (0.40 mmol, 1.0 equiv.), sulfonyl azides (0.6 mmol, 1.5 equiv.), [IrCp*Cl2]2 (0.02 mmol, 5 mol%), and AgNTf2 (0.08 mmol, 20 mol%) in [BMIM]PF6 (0.6 mL, 0.67 mol L−1). The reaction mixture is stirred at the indicated temperature and time. The reaction is monitored by TLC. After completion of the reaction, the reaction mixture is allowed to cool to room temperature and diluted with dichloromethane (2.0 mL). Purification by flash column chromatography on silica gel using mixtures of petroleum ether and ethyl acetate as eluents affords the analytically pure product.N-(tert-Butyl)-2-(4-methylphenylsulfonamido)benzamide (3aa)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 20:1) afforded the analytically pure product (127 mg, 92%) as a colorless oil; Rf = 0.22 (petroleum ether:ethyl acetate = 20:1); IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 = 3396 (vs), 2964 (s), 1647 (vs), 1537 (vs), 1455 (m), 1389 (vs), 1232 (m), 1160 (vs), 952 (vs), 873 (s), 851 (w), 742 (s), 664 (vs), 563 (vs), 507 (s), 456 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.41 (s, 9H), 2.37 (s, 3H), 5.96 (br s, 1H), 7.04 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.19 (d, J = 7.3 Hz, 2H), 7.21 (dd, J = 8.4 Hz, J = 8.4 Hz, 2H), 7.64–7.68 (m, 3H), 10.85 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 28.6, 52.2, 121.5, 122.9, 123.5, 126.8, 127.1, 129.6, 132.1, 136.7, 138.5, 143.5, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H23N2O3S): calcd m/z 347.1424, found: 347.1421.
/cm−1 = 3396 (vs), 2964 (s), 1647 (vs), 1537 (vs), 1455 (m), 1389 (vs), 1232 (m), 1160 (vs), 952 (vs), 873 (s), 851 (w), 742 (s), 664 (vs), 563 (vs), 507 (s), 456 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.41 (s, 9H), 2.37 (s, 3H), 5.96 (br s, 1H), 7.04 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.19 (d, J = 7.3 Hz, 2H), 7.21 (dd, J = 8.4 Hz, J = 8.4 Hz, 2H), 7.64–7.68 (m, 3H), 10.85 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 28.6, 52.2, 121.5, 122.9, 123.5, 126.8, 127.1, 129.6, 132.1, 136.7, 138.5, 143.5, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H23N2O3S): calcd m/z 347.1424, found: 347.1421.
N-(tert-Butyl)-2-(phenylsulfonamido)benzamide (3ab)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and benzenesulfonyl azide (2b, 110 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (113 mg, 85%) as a colorless oil; Rf = 0.5 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3383 (vs), 3356 (vs), 2968 (s), 1633 (vs), 1535 (vs), 1450 (s), 1338 (vs), 1280 (s), 1163 (vs), 1091 (s), 937 (s), 871 (s), 758 (vs), 686 (s), 584 (vs), 464 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.41 (s, 9H), 6.00 (br s, 1H), 7.05 (dd, J = 7.4 Hz, J = 7.4 Hz, 1H), 7.35–7.42 (m, 4H), 7.51 (dd, J = 7.2 Hz, J = 7.2 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 7.4 Hz, 2H), 10.96 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.2, 121.1, 123.0, 123.8, 126.9, 127.0, 129.0, 132.2, 132.8, 138.4, 139.5, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H21N2O3S): calcd m/z 333.1267, found: 333.1270.
N-(tert-Butyl)-2-(4-methoxyphenylsulfonamido)benzamide (3ac)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 4-methoxybenzenesulfonyl azide (2c, 128 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (108 mg, 75%) as a colorless oil; Rf = 0.41 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3404 (vs), 2964 (s), 1637 (vs), 1527 (vs), 1460 (m), 1336 (vs), 1261 (vs), 1161 (vs), 1028 (s), 941 (m), 889 (m), 833 (s), 736 (m), 669 (w), 568 (vs), 459 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.43 (s, 9H), 3.83 (s, 3H), 5.99 (br s, 1H), 6.87 (d, J = 7.6 Hz, 2H), 7.05 (dd, J = 7.6 Hz, J = 7.4 Hz, 1H), 7.34–7.39 (m, 2H), 6.67 (d, J = 8.2 Hz, 1H), 7.71 (d, J = 7.6 Hz, 2H), 10.82 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.2, 55.6, 114.1, 121.5, 122.9, 123.5, 126.8, 129.2, 131.2, 132.2, 138.7, 162.9, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H23N2O4S): calcd m/z 363.1373, found: 363.1379.
N-(tert-Butyl)-2-(4-fluorophenylsulfonamido)benzamide (3ad)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 4-fluorobenzenesulfonyl azide (2d, 120 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (85 mg, 61%) as a colorless oil; Rf = 0.50 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3367 (vs), 3072 (s), 2972 (vs), 1635 (vs), 1546 (vs), 1494 (s), 1394 (w), 1332 (vs), 1296 (m), 1166 (vs), 1089 (s), 923 (w), 835 (vs), 765 (s), 661 (w), 563 (vs); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.44 (s, 9H), 5.96 (br s, 1H), 7.07–7.14 (m, 3H), 7.35–7.45 (m, 2H), 7.70–7.74 (m, 1H), 7.80–7.86 (m, 2H), 10.82 (d, J = 8.0 Hz, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.2, 116.1 (d, J = 23 Hz), 121.7, 122.8, 123.8, 126.7, 129.9 (d, J = 9.0 Hz), 132.3, 135.8, 138.5, 164.9 (d, J = 254 Hz), 167.9 ppm; 19F NMR (376 MHz, CDCl3, 298 K): δ = −105.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H20FN2O3S): calcd m/z 351.1173, found: 351.1176.
2-(4-Bromophenylsulfonamido)-N-tertbutylbenzamide (3ae)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 4-bromobenzenesulfonyl azide (2e, 157 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 60 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 10:1) afforded the analytically pure product (141 mg, 86%) as a colorless oil; Rf = 0.37 (petroleum ether:ethyl acetate = 8:1); IR (KBr): /cm−1 = 3381 (vs), 3087 (m), 2968 (vs), 1637 (vs), 1539 (vs), 1471 (s), 1390 (vs), 1269 (vs), 1166 (vs), 1089 (s), 1010 (s), 939 (m), 817 (s), 748 (vs), 559 (vs), 420 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.45 (s, 9H), 5.99 (br s, 1H), 7.07–7.15 (m, 1H), 7.36–7.43 (m, 2H), 7.53–7.57 (m, 2H), 7.63–7.76 (m, 3H), 11.01 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.3, 121.9, 122.9, 124.0, 126.8, 127.6, 128.6, 132.2, 132.3, 138.3, 138.7, 167.9 ppm; HRMS (APCI) exact mass for [M + Na]+ (C17H19BrN2NaO3S): calcd m/z 433.0192, found: 433.0187.
N-(tert-Butyl)-2-(naphthalene-2-sulfonamido)benzamide (3af)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and naphthalene-2-sulfonyl azide (2f, 140 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (130 mg, 85%) as a colorless oil; Rf = 0.53 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3398 (vs), 2976 (vs), 1930 (w), 1809 (w), 1639 (vs), 1531 (vs), 1369 (vs), 1269 (vs), 1128 (vs), 1070 (vs), 952 (vs), 806 (vs), 707 (vs), 653 (vs), 559 (vs), 472 (vs); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.34 (s, 9H), 5.93 (br s, 1H), 7.02 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.31–7.38 (m, 2H), 7.55–7.63 (m, 2H), 7.72–7.77 (m, 2H), 7.83–7.88 (m, 3H), 8.36 (s, 1H), 11.09 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.2, 121.7, 122.3, 123.1, 123.8, 127.0, 127.6, 127.9, 128.6, 128.9, 129.3, 129.4, 132.2, 132.3, 134.9, 136.7, 138.6, 168.2 ppm; HRMS (APCI) exact mass for [M + H]+ (C21H23N2O3S): calcd m/z 383.1424, found: 383.1420.
N-(tert-Butyl)-2-(2-fluorophenylsulfonamido)benzamide (3ag)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 2-fluorobenzenesulfonyl azide (2g, 121 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 3:1) afforded the analytically pure product (90 mg, 64%) as a colorless oil; Rf = 0.37 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3400 (s), 2966 (s), 2852 (m), 1647 (s), 1533 (s), 1473 (s), 1392 (s), 1265 (vs), 1220 (m), 1168 (vs), 1074 (vs), 948 (s), 777 (vs), 698 (m), 584 (vs), 457 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.46 (s, 9H), 5.94 (br s, 1H), 7.03 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.12 (dd, J = 8.8 Hz, J = 8.6 Hz, 1H), 7.23 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.32–7.38 (m, 2H), 7.50–7.55 (m, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.94 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 11.17 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.7, 52.3, 117.1 (d, J = 20 Hz), 120.1, 122.5, 123.2, 124.3 (d, J = 4 Hz), 126.7, 127.7 (d, J = 11 Hz), 130.8, 132.1, 135.1 (d, J = 9 Hz), 138.1, 159.0 (d, J = 262 Hz), 168.0 ppm; 19F NMR (376 MHz, CDCl3, 298 K): δ = −108.5 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H20FN2O3S): calcd m/z 351.1173, found: 351.1177.
N-(tert-Butyl)-2-(2,4-dichlorophenylsulfonamido)benzamide (3ah)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 2,4-dichlorobenzenesulfonyl azide (2h, 151 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 10:1) afforded the analytically pure product (67 mg, 42%) as a colorless oil; Rf = 0.62 (petroleum ether:ethyl acetate = 8:1); IR (KBr): /cm−1 = 3377 (vs), 3255 (m), 3093 (w), 2974 (m), 1639 (s), 1571 (m), 1454 (m), 1375 (s), 1269 (m), 1164 (vs), 948 (m), 823 (s), 705 (w), 622 (s), 568 (s), 493 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.50 (s, 9H), 6.04 (br s, 1H), 7.04 (dd, J = 7.4 Hz, J = 7.4 Hz, 1H), 7.31–7.41 (m, 3H), 7.42–7.48 (m, 1H), 7.54–7.59 (m, 1H), 8.06–8.15 (m, 1H), 11.54 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.7, 52.4, 118.6, 121.6, 122.9, 127.0, 127.2, 130.6, 131.6, 132.3, 132.9, 135.5, 137.9, 139.7, 168.0 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H19Cl2N2O3S): calcd m/z 401.0488, found: 401.0493.
N-(tert-Butyl)-2-(2,4-dimethoxyphenylsulfonamido)benzamide (3ai)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 2,4-dimethoxybenzenesulfonyl azide (2i, 146 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 10:1) afforded the analytically pure product (86 mg, 55%) as a colorless oil; Rf = 0.50 (petroleum ether:ethyl acetate = 8:1); IR (KBr): /cm−1 = 3325 (s), 3012 (w), 2974 (m), 1624 (vs), 1543 (s), 1490 (s), 1340 (m), 1265 (s), 1170 (vs), 1078 (s), 931 (m), 840 (m), 754 (m), 699 (m), 565 (vs), 462 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.53 (s, 9H), 3.88 (s, 3H), 3.92 (s, 3H), 6.03 (br s, 1H), 6.46 (d, J = 2.0 Hz, 1H), 6.55 (dd, J = 8.8 Hz, J = 2.1 Hz, 1H), 6.98 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.30–7.40 (m, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 8.8 Hz, 1H), 11.09 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.8, 52.0, 55.6, 56.1, 99.3, 104.1, 118.6, 119.6, 121.2, 121.9, 126.7, 132.0, 132.6, 139.3, 158.4, 164.9, 167.9 ppm; HRMS (APCI) exact mass for [M + H]+ (C19H25N2O5S): calcd m/z 393.1479, found: 393.1480.
N-(tert-Butyl)-2-(3-chlorophenylsulfonamido)benzamide (3aj)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 3-chlorobenzenesulfonyl azide (2j, 130 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (117 mg, 80%) as a colorless oil; Rf = 0.45 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3390 (vs), 2978 (s), 1637 (vs), 1531 (vs), 1492 (vs), 1460 (m), 1340 (vs), 1267 (vs), 1163 (vs), 1083 (s), 945 (s), 867 (s), 758 (s), 673 (s), 586 (vs), 499 (s); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.40 (s, 9H), 6.02 (br s, 1H), 7.07 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.31–7.41 (m, 3H), 7.46 (d, J = 7.6 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.75 (s, 1H), 11.11 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.3, 121.7, 122.8, 124.1, 125.2, 126.9, 127.1, 130.3, 132.4, 132.9, 135.1, 138.2, 141.3, 168.0 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H20ClN2O3S): calcd m/z 367.0878, found: 367.0881.
N-(tert-Butyl)-2-(3,5-difluorophenylsulfonamido)benzamide (3ak)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.4 mmol, 1.0 equiv.) and 3,5-difluorobenzenesulfonyl azide (2k, 132 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (130 mg, 88%) as a colorless oil; Rf = 0.65 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3408 (vs), 3064 (s), 2978 (s), 1641 (vs), 1541 (vs), 1440 (vs), 1348 (vs), 1294 (vs), 1161 (vs), 987 (vs), 886 (s), 750 (vs), 667 (s), 603 (vs), 540 (w), 497 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.45 (s, 9H), 6.03 (br s, 1H), 6.97 (dd, J = 8.4 Hz, J = 8.4 Hz, 1H), 7.13 (dd, J = 7.2 Hz, J = 7.2 Hz, 1H), 7.31–7.35 (m, 2H), 7.40 (d, J = 8.0 Hz, 1H), 7.49 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.71 (d, J = 7.4 Hz, 1H), 11.25 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.6, 52.4, 108.4 (t, J = 25 Hz), 110.8 (d, J = 12 Hz), 110.8 (d, J = 28 Hz), 121.9, 122.8, 124.3, 126.9, 132.5, 138.0, 142.8, 162.6 (dd, J = 253 Hz, J = 11 Hz), 167.9 ppm; 19F NMR (376 MHz, CDCl3, 298 K): δ = −105.6 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H19F2N2O3S): calcd m/z 369.1079, found: 369.1074.
N-(tert-Butyl)-5-methyl-2-(4-methylphenylsulfonamido)benzamide (3ba)
Prepared from N-(tert-butyl)-3-methylbenzamide (1b, 76.5 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (104 mg, 72%) as a colorless oil; Rf = 0.61 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3377 (vs), 2964 (s), 1637 (vs), 1543 (vs), 1454 (m), 1388 (vs), 1265 (s), 1164 (s), 1093 (vs), 952 (w), 831 (s), 715 (s), 678 (m), 588 (vs), 532 (s), 460 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.41 (s, 9H), 2.31 (s, 3H), 2.38 (s, 3H), 5.88 (br s, 1H), 7.11 (s, 1H), 7.20 (d, J = 7.8 Hz, 3H), 7.59 (d, J = 7.6 Hz, 1H), 7.64 (d, J = 7.8 Hz, 2H), 10.58 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 20.7, 21.5, 28.6, 52.1, 122.2, 123.4, 123.4, 127.1, 129.5, 132.8, 133.4, 135.9, 136.7, 143.3, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C19H25N2O3S): calcd m/z 361.1580, found: 361.1575.
N-(tert-Butyl)-4-methyl-2-(4-methylphenylsulfonamido)benzamide (3ca)
Prepared from N-(tert-butyl)-4-methylbenzamide (1c, 76.5 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 10:1) afforded the analytically pure product (87 mg, 60%) as a colorless oil; Rf = 0.31 (petroleum ether:ethyl acetate = 8:1); IR (KBr): /cm−1 = 3396 (vs), 3066 (w), 2964 (m), 1641 (vs), 1533 (vs), 1504 (w), 1386 (vs), 1269 (m), 1220 (m), 1164 (vs), 1089 (s), 962 (w), 813 (vs), 721 (vs), 538 (s), 453 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.42 (s, 9H), 2.35 (s, 3H), 2.39 (s, 3H), 5.86 (br s, 1H), 6.85 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 7.6 Hz, 3H), 7.52 (s, 1H), 7.69 (d, J = 7.6 Hz, 2H), 10.96 (br s, 0.01H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 21.6, 28.7, 52.0, 120.0, 122.0, 124.3, 126.5, 127.1, 129.5, 136.8, 138.7, 143.0, 143.3, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C19H25N2O3S): calcd m/z 361.1580, found: 361.1577.
N-(tert-Butyl)-4-methoxy-2-(4-methylphenylsulfonamido)benzamide (3da)
Prepared from N-(tert-butyl)-4-methoxybenzamide (1d, 82.9 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (110 mg, 73%) as a colorless oil; Rf = 0.58 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3398 (vs), 2962 (s), 2869 (w), 1633 (vs), 1535 (s), 1456 (w), 1336 (vs), 1160 (vs), 1269 (vs), 1163 (vs), 972 (s), 804 (s), 707 (vs), 644 (m), 592 (m), 541 (vs); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.41 (s, 9H), 2.37 (s, 3H), 3.80 (s, 3H), 5.79 (br s, 1H), 6.52 (d, J = 8.8 Hz, 1H), 7.19–7.24 (m, 4H), 7.71 (d, J = 7.6 Hz, 2H), 11.35 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 28.7, 52.0, 55.5, 105.4, 109.6, 114.5, 127.2, 128.0, 129.6, 136.8, 141.0, 143.5, 162.0, 168.0 ppm; HRMS (APCI) exact mass for [M + H]+ (C19H25N2O4S): calcd m/z 377.1530, found: 377.1532.
N-(tert-Butyl)-4-fluoro-2-(4-methylphenylsulfonamido)benzamide (3ea)
Prepared from N-(tert-butyl)-4-fluorobenzamide (1e, 78.1 mg, 0.4 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (105 mg, 72%) as a colorless oil; Rf = 0.20 (petroleum ether:ethyl acetate = 8:1); IR (KBr): /cm−1 = 3404 (vs), 3074 (w), 2968 (s), 1916 (w), 1641 (vs), 1497 (vs), 1361 (vs), 1269 (vs), 1155 (vs), 1018 (w), 950 (s), 825 (vs), 729 (vs), 636 (m), 543 (vs), 447 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.46 (s, 9H), 2.42 (s, 3H), 6.01 (br s, 1H), 6.69–6.74 (m, 1H), 7.26 (d, J = 7.6 Hz, 2H), 7.38–7.44 (m, 2H), 7.73 (d, J = 7.2 Hz, 2H), 11.29 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 28.6, 52.3, 107.7 (d, J = 26 Hz), 110.2 (d, J = 22 Hz), 118.1 (d, J = 3.0 Hz), 127.0, 128.9 (d, J = 11 Hz), 129.7, 136.4, 141.1 (d, J = 11 Hz), 143.9, 164.3 (d, J = 251 Hz), 167.5 ppm; 19F NMR (376 MHz, CDCl3, 298 K): δ = −104.9 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H22FN2O3S): calcd m/z 365.1330, found: 365.1333.
N-(tert-Butyl)-4-chloro-2-(4-methylphenylsulfonamido)benzamide (3fa)
Prepared from N-(tert-butyl)-4-chlorobenzamide (1f, 84.7 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 80 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (114 mg, 75%) as a colorless oil; Rf = 0.51 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3398 (vs), 3051 (m), 2970 (vs), 1928 (w), 1639 (vs), 1485 (vs), 1334 (vs), 1217 (vs), 1118 (vs), 1018 (w), 933 (vs), 813 (vs), 705 (vs), 626 (m), 538 (vs), 443 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.42 (s, 9H), 2.39 (s, 3H), 5.86 (br s, 1H), 6.98 (d, J = 8.4 Hz, 1H), 7.21–7.25 (m, 3H), 7.70–7.73 (m, 3H), 11.00 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.6, 28.6, 52.4, 120.5, 120.9, 123.3, 127.2, 127.8, 129.7, 136.5, 138.2, 140.0, 143.8, 167.4 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H22ClN2O3S): calcd m/z 381.1034, found: 381.1030.
4-Bromo-N-(tert-butyl)-2-(4-methylphenylsulfonamido)benzamide (3ga)
Prepared from 4-bromo-N-tertbutylbenzamide (1g, 102 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 60 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (105 mg, 62%) as a colorless oil; Rf = 0.64 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3394 (vs), 3078 (w), 2925 (vs), 1639 (vs), 1566 (m), 1481 (vs), 1336 (vs), 1217 (m), 1164 (vs), 1039 (w), 914 (vs), 738 (vs), 686 (s), 655 (m), 557 (vs), 424 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.44 (s, 9H), 2.42 (s, 3H), 5.90 (br s, 1H), 7.15–7.21 (m, 2H), 7.26 (d, J = 7.6 Hz, 2H), 7.73 (d, J = 7.6 Hz, 2H), 7.89 (s, 1H), 10.98 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.6, 28.6, 52.4, 121.0, 123.9, 126.3, 126.4, 127.2, 127.9, 129.7, 136.4, 140.0, 143.9, 167.4 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H22BrN2O3S): calcd m/z 425.0529, found: 425.0536.
N-(tert-Butyl)-2-(4-methylphenylsulfonamido)-4-nitrobenzamide (3ha)
Prepared from N-(tert-butyl)-4-nitrobenzamide (1h, 88.9 mg, 0.4 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 3:1) afforded the analytically pure product (125 mg, 80%) as a colorless oil; Rf = 0.27 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3413 (s), 3097 (m), 2976 (s), 2856 (w), 1647 (vs), 1533 (vs), 1458 (w), 1352 (vs), 1215 (m), 1161 (vs), 966 (s), 808 (s), 688 (s), 619 (w), 540 (vs), 433 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.48 (s, 9H), 2.41 (s, 3H), 6.21 (br s, 1H), 7.28 (d, J = 7.6 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.74–7.80 (m, 3H), 8.45 (s, 1H), 10.93 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.6, 28.5, 53.0, 114.7, 117.3, 126.8, 127.2, 128.2, 130.0, 136.0, 139.8, 144.5, 149.5, 166.4 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H22N3O5S): calcd m/z 392.1275, found: 392.1271.
N-(tert-Butyl)-3-(4-methylphenylsulfonamido)-2-naphthamide (3ia)
Prepared from N-(tert-butyl)-2-naphthamide (1i, 91 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (119 mg, 75%) as a colorless oil; Rf = 0.45 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3390 (vs), 3111 (w), 2962 (m), 1651 (vs), 1597 (m), 1531 (vs), 1446 (m), 1352 (vs), 1222 (w), 1163 (vs), 1089 (m), 923 (s), 709 (s), 653 (w), 572 (vs), 414 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.48 (s, 9H), 2.34 (s, 3H), 6.12 (br s, 1H), 7.15 (d, J = 7.6 Hz, 2H), 7.44 (dd, J = 7.2 Hz, J = 7.2 Hz, 1H), 7.55 (dd, J = 7.2 Hz, J = 7.2 Hz, 1H), 7.67 (d, J = 7.6 Hz, 2H), 7.78 (dd, J = 8.0 Hz, J = 7.6 Hz, 2H), 7.87 (s, 1H), 8.07 (s, 1H), 10.48 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 28.7, 52.4, 119.2, 124.1, 126.0, 127.1, 127.5, 127.6, 128.1, 128.4, 129.1, 129.5, 134.1, 134.7, 136.7, 143.4, 168.2 ppm; HRMS (APCI) exact mass for [M + H]+ (C22H25N2O3S): calcd m/z 397.1580, found: 397.1582.
N-Methyl-2-(4-methylphenylsulfonamido)benzamide (5aa)
Prepared from N-methylbenzamide (4a, 54.1 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (110 mg, 91%) as a colorless oil; Rf = 0.45 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3398 (vs), 2962 (vs), 2856 (m), 1732 (vs), 1649 (vs), 1539 (vs), 1456 (s), 1390 (vs), 1280 (vs), 1157 (vs), 1043 (w), 950 (s), 815 (s), 740 (vs), 565 (vs), 459 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 2.39 (s, 3H), 2.92 (s, 3H), 6.30 (br s, 1H), 7.03–7.10 (m, 1H), 7.23 (d, J = 7.9 Hz, 2H), 7.39 (d, J = 8.1 Hz, J = 8.1 Hz, 2H), 7.65–7.74 (m, 3H), 10.87 (br s, 1H) ppm; HRMS (APCI) exact mass for [M + H]+ (C15H17N2O3S): calcd m/z 305.0954, found: 305.0955.
2-(4-Methylphenylsulfonamido)-N-propylbenzamide (5ba)
Prepared from N-propylbenzamide (4b, 65.3 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (113 mg, 85%) as a colorless oil; Rf = 0.33 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3392 (vs), 3062 (w), 2966 (m), 1635 (vs), 1537 (s), 1452 (m), 1332 (s), 1211 (m), 1155 (vs), 1089 (vs), 916 (m), 808 (m), 761 (s), 699 (s), 563 (vs), 534 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 0.94 (t, J = 7.4 Hz, 3H), 1.52–1.61 (m, 2H), 3.35 (s, 3H), 3.28 (q, J = 6.8 Hz, 2H), 6.47 (br s, 1H), 7.03 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.18 (d, J = 8.0 Hz, 2H), 7.36 (dd, J = 7.8 Hz, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 8.0 Hz, 3H), 10.98 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 11.4, 21.5, 22.5, 41.7, 121.2, 121.7, 123.6, 126.9, 127.1, 129.5, 132.3, 136.5, 138.7, 143.6, 168.4 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H21N2O3S): calcd m/z 333.1267, found: 333.1265.
N-Isopropyl-2-(4-methylphenylsulfonamido)benzamide (5ca)
Prepared from N-isopropylbenzamide (4c, 65.3 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (117 mg, 88%) as a colorless oil; Rf = 0.69 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3384 (vs), 2979 (s), 1633 (vs), 1531 (vs), 1452 (m), 1332 (vs), 1259 (vs), 1213 (m), 1157 (vs), 1089 (vs), 929 (m), 829 (w), 763 (s), 624 (w), 563 (vs), 441 (vw); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.21 (d, J = 6.6 Hz, 6H), 2.35 (s, 3H), 4.07–4.19 (m, 1H), 6.21 (d, J = 7.0 Hz, 1H), 7.03 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.18 (d, J = 8.0 Hz, 2H), 7.36 (dd, J = 7.8 Hz, J = 7.8 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.62–7.67 (m, 3H), 10.97 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 22.4, 42.1, 121.4, 121.9, 123.6, 126.9, 127.1, 129.5, 132.3, 136.6, 138.7, 143.6, 167.6 ppm; HRMS (APCI) exact mass for [M + H]+ (C17H21N2O3S): calcd m/z 333.1267, found: 333.1262.
N,N-Diisopropyl-2-(4-methylphenylsulfonamido)benzamide (5da)
Prepared from N,N-diisopropylbenzamide (4d, 82.1 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (112 mg, 75%) as a colorless oil; Rf = 0.64 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3130 (vs), 2956 (s), 2821 (w), 1608 (vs), 1457 (vs), 1344 (vs), 1259 (w), 1211 (m), 1166 (vs), 1093 (s), 920 (m), 813 (s), 736 (vs), 621 (m), 567 (vs), 493 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.27 (br s, 12H), 2.37 (s, 3H), 3.66 (br s, 2H), 7.04–7.08 (m, 1H), 7.11 (ddd, J = 7.6 Hz, J = 7.6 Hz, J = 1.3 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 7.27–7.31 (m, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 8.34 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 20.6, 21.5, 121.9, 123.6, 126.3, 127.1, 127.9, 129.7, 130.1, 135.6, 137.4, 143.8, 169.0 ppm; HRMS (APCI) exact mass for [M + H]+ (C20H27N2O3S): calcd m/z 375.1737, found: 375.1734.
N-Cyclohexyl-2-(4-methylphenylsulfonamido)benzamide (5fa)
Prepared from N-cyclohexyl-2-(4-methylphenylsulfonamido)benzamide (4f, 81.3 mg, 0.40 mmol, 1.0 equiv.) and tosyl azide (2a, 131 μL, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (124 mg, 83%) as a colorless oil; Rf = 0.31 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3390 (vs), 2933 (vs), 2854 (s), 1631 (vs), 1529 (vs), 1448 (m), 1334 (vs), 1215 (m), 1149 (vs), 1089 (s), 937 (s), 813 (s), 754 (s), 671 (s), 563 (vs), 447 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.17–1.29 (m, 3H), 1.36–1.45 (m, 2H), 1.65–1.69 (m, 1H), 1.75–1.78 (m, 2H), 1.90–1.93 (m, 2H), 2.36 (s, 3H), 3.78–3.87 (m, 1H), 6.19 (d, J = 7.2 Hz, 1H), 7.03 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.18 (d, J = 8.0 Hz, 2H), 7.36 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.65–7.68 (m, 3H), 10.96 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 21.5, 24.9, 25.4, 32.7, 48.9, 121.3, 121.8, 123.5, 126.8, 127.1, 129.5, 132.3, 136.6, 138.8, 143.5, 167.5 ppm; HRMS (APCI) exact mass for [M + H]+ (C20H25N2O3S): calcd m/z 373.1580, found: 373.1584.
N-(tert-Butyl)-2-(phenylmethylsulfonamido)benzamide (7aa)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.40 mmol, 1.0 equiv.) and phenylmethanesulfonyl azide (6a, 118 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 60 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (125 mg, 90%) as a colorless oil; Rf = 0.45 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3371 (vs), 2970 (vs), 2920 (s), 1633 (vs), 1537 (vs), 1454 (m), 1334 (vs), 1394 (vs), 1280 (vs), 1159 (vs), 1029 (w), 945 (vs), 746 (vs), 630 (s), 546 (vs), 459 (m); 1H NMR (400 MHz, CDCl3, 298 K): δ = 1.45 (s, 9H), 4.39 (s, 2H), 6.18 (br s, 1H), 7.09 (ddd, J = 7.6 Hz, J = 7.6 Hz, J = 0.7 Hz, 1H), 7.24–7.32 (m, 5H), 7.38 (ddd, J = 7.2 Hz, J = 7.2 Hz, J = 1.2 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 10.93 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 28.7, 52.3, 58.4, 119.0, 120.8, 122.8, 127.1, 128.4, 128.8, 128.8, 130.8, 132.6, 139.6, 168.1 ppm; HRMS (APCI) exact mass for [M + H]+ (C18H23N2O3S): calcd m/z 347.1424, found: 347.1420.
N-(tert-Butyl)-2-(butylsulfonamido)benzamide (7ab)
Prepared from N-tertbutylbenzamide (1a, 70.9 mg, 0.40 mmol, 1.0 equiv.) and butane-1-sulfonyl azide (6b, 98 mg, 0.6 mmol, 1.5 equiv.) according to the GP 1 at 40 °C for 8 h. Purification by flash column chromatography on silica gel (petroleum ether:ethyl acetate = 8:1) afforded the analytically pure product (95 mg, 76%) as a colorless oil; Rf = 0.46 (petroleum ether:ethyl acetate = 3:1); IR (KBr): /cm−1 = 3375 (vs), 2964 (vs), 2873 (s), 1635 (vs), 1537 (vs), 1490 (s), 1328 (vs), 1265 (vs), 1145 (vs), 1097 (m), 943 (vs), 887 (s), 763 (vs), 661 (w), 520 (s), 460 (w); 1H NMR (400 MHz, CDCl3, 298 K): δ = 0.88 (t J = 7.4 Hz, 3H), 1.27–1.43 (m, 2H), 1.49 (s, 9H), 1.73–1.80 (m, 2H), 3.05–3.09 (m, 2H), 6.31 (br s, 1H), 7.10 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.42 (dd, J = 8.0 Hz, J = 8.0 Hz, 1H), 7.51 (d J = 7.6 Hz, 1H), 7.69 (d J = 8.0 Hz, 1H), 10.74 (br s, 1H) ppm; 13C NMR (101 MHz, CDCl3, 298 K): δ = 13.6, 21.4, 25.3, 28.7, 51.5, 52.3, 119.5, 121.5, 123.1, 127.4, 132.5, 139.1, 168.3 ppm; HRMS (APCI) exact mass for [M + H]+ (C15H25N2O3S): calcd m/z 313.1580, found: 313.1584.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the National Key R&D Program of China (2018YFB0604603), the Natural Science Basic Research Program of Shaanxi – Shaanxi Coal and Chemical Industry Joint Foundation (Program No. 2019JLM-20), and the National Natural Science Foundation of China (NSFC: 21606182, 21901150) for financial support.Notes and references
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed; (b) A. Ricci, Amino Group Chemistry, From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2007 Search PubMed.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- (a) A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS; (b) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS; (c) A. Ehrentraut, A. Zapf and M. Beller, J. Mol. Catal. A: Chem., 2002, 182–183, 515–523 CrossRef CAS; (d) R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- (a) D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863–3865 CrossRef CAS; (b) M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- For recent reviews and book chapter, see: (a) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (b) J. Jiao, K. Murakami and K. Itami, ACS Catal., 2016, 6, 610–633 CrossRef CAS; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (d) S. M. Ujwaldev, N. A. Harry, M. A. Divakar and G. Anilkumar, Catal. Sci. Technol., 2018, 8, 5983–6018 RSC; (e) Y. N. Timsina, B. F. Gupton and K. C. Ellis, ACS Catal., 2018, 8, 5732–5776 CrossRef CAS; (f) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (g) V. Arun, K. Mahanty and S. D. Sarkar, ChemCatChem, 2019, 11, 2243–2259 CrossRef CAS; (h) K. Tanaka, Rhodium Catalysis in Organic Synthesis: Methods and Reactions, Wiley-VCH Weinheim, 2019 CrossRef; (i) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (j) K. M. Korch and D. A. Watson, Chem. Rev., 2019, 119, 8192–8228 CrossRef CAS PubMed; (k) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS PubMed.
- J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953–8955 RSC.
- (a) M. Bhanuchandra, M. R. Yadav, R. K. Rit, M. R. Kuram and A. K. Sahoo, Chem. Commun., 2013, 49, 5225–5227 RSC; (b) M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638–1641 CrossRef CAS PubMed.
- (a) Q.-Z. Zheng and N. Jiao, Chem. Commun., 2013, 49, 5654–5656 RSC; (b) C. Tang, Y. Yuan, Y. Cui and N. Jiao, Eur. J. Org. Chem., 2013, 7480–7483 CrossRef CAS.
- V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef CAS PubMed.
- D.-G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed.
- B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491–1495 CrossRef CAS.
- For selected examples of the C–H bond amidation of benzamides, see: (a) J. Kim, J. Kim and S. Chang, Chem.–Eur. J., 2013, 19, 7328–7333 CrossRef CAS PubMed; (b) D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102–11109 CrossRef CAS PubMed; (c) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076–4085 CrossRef CAS; (d) G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2016, 55, 3781–3784 CrossRef CAS PubMed; for selected examples of C–H bond amidation directed with nitrogen atom, see: (e) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 CrossRef CAS PubMed; (f) M. Maraswami, G. Chen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 416–421 CrossRef CAS; for selected examples of C–H bond amidation directed by N-oxide, see: (g) H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed; (h) B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326–332 CrossRef CAS; for selected examples of C–H bond amidation directed by carbonyl group, see: (i) D. Mu, X. Wang, G. Chen and G. He, J. Org. Chem., 2017, 82, 4497–4503 CrossRef CAS PubMed; (j) X. Kong and B. Xu, Org. Lett., 2018, 20, 4495–4498 CrossRef CAS PubMed.
- For recent examples, see: (a) S. Lv, Y. Li, T. Yao, X. Yu, C. Zhang, L. Hai and Y. Wu, Org. Lett., 2018, 20, 4994–4997 CrossRef CAS PubMed; (b) Q. Ma, X. Yu, R. Lai, S. Lv, W. Dai, C. Zhang, X. Wang, Q. Wang and Y. Wu, ChemSusChem, 2018, 11, 3672–3678 CrossRef CAS PubMed; (c) L.-Y. Xie, S. Peng, L.-H. Lu, J. Hu, W.-H. Bao, F. Zeng, Z. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 7989–7994 CrossRef CAS; for recent examples related to C–C bond formation mediated in ionic liquid, see: (d) C. Zhang, X.-M. Chen, Y. Luo, J.-L. Li, M. Chen, L. Hai and Y. Wu, ACS Sustainable Chem. Eng., 2018, 6, 13473–13479 CrossRef CAS; (e) F. G. Cirujano, M. Stalpaert and D. E. De Vos, Green Chem., 2018, 20, 2481–2485 RSC; (f) A. F. Quivelli, P. Vitale, F. M. Perna and V. Capriati, Front. Chem., 2019, 7, 723 CrossRef CAS PubMed.
- (a) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed; (b) H. Olivier-Bourbigou and L. Magna, J. Mol. Catal. A: Chem., 2002, 182–183, 419–437 CrossRef CAS; (c) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed; (d) C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed; (e) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- (a) H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS PubMed; (b) D. Xiao, L. G. Hines, Jr., S. Li, R. A. Bartsch, E. L. Quitevis, O. Russina and A. Triolo, J. Phys. Chem. B, 2009, 113, 6426–6433 CrossRef CAS PubMed.
- (a) L.-Y. Jiao and M. Oestreich, Chem.–Eur. J., 2013, 19, 10845–10848 CrossRef CAS PubMed; (b) L.-Y. Jiao and M. Oestreich, Org. Lett., 2013, 15, 5374–5377 CrossRef CAS PubMed; (c) L.-Y. Jiao, P. Smirnov and M. Oestreich, Org. Lett., 2014, 16, 6020–6023 CrossRef CAS PubMed; (d) L.-Y. Jiao, A. V. Ferreira and M. Oestreich, Chem.–Asian J., 2016, 11, 367–370 CrossRef CAS PubMed; (e) L.-Y. Jiao, Z. Zhang, X.-M. Yin, Z. Li and X.-X. Ma, J. Catal., 2019, 379, 39–45 CrossRef CAS; (f) L.-Y. Jiao, X.-M. Yin, S. Liu, Z. Zhang, M. Sun and X.-X. Ma, Catal. Commun., 2020, 135, 105889 CrossRef; (g) L.-Y. Jiao, Z. Zhang, Q. Hong, Z.-H. Ning, S. Liu, M. Sun, Q. Hao, L. Xu, Z. Li and X.-X. Ma, Mol. Catal., 2020, 494, 111120 CrossRef CAS.
- D. R. MacFarlane, J. M. Pringle, K. M. Johansson, S. A. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905–1917 RSC.
- (a) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686–693 CrossRef CAS PubMed; (b) D. C. Simkó, P. Elekes, V. Pázmándi and Z. Novák, Org. Lett., 2018, 20, 676–679 CrossRef PubMed.
- J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2006, 128, 11693–11712 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Some experimental details and copies of NMR spectra for all of the amidation products. See DOI: 10.1039/d0ra05527a |
| This journal is © The Royal Society of Chemistry 2020 |