
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDensity functional theory-based investigation of HCN and NH3 formation mechanisms during phenylalanine pyrolysis†
Baizhong Sun,
Chuanqun Liu,
Deyong Che,
Hongpeng Liu and
Shuai Guo *
*
School of Energy and Power Engineering, Northeast Electric Power University, Jilin 132000, China. E-mail: guoshuaidq@126.com
First published on 31st July 2020
Abstract
As sludge pyrolysis produces large amounts of toxic NH3 and HCN, many works have studied nitrogen transfer during this process, commonly employing amino acids as models of sludge protein. Herein, density functional theory is used to probe the production of HCN and NH3 during the pyrolysis of phenylalanine as a model, revealing the existence of two formation paths for each gas. In the first (lower-energy-barrier) NH3 formation path, the hydrogen bonding-assisted transfer of carboxyl group hydrogen to the amino group is followed by direct NH3 generation via decarboxylation, and the second (higher-energy-barrier) path features decarboxylation followed by the transfer of carboxyl group hydrogen to the adjacent carbon atom to form phenethylamine, the deamination of which affords NH3 and styrene. For HCN, the first (lower-energy-barrier) path features C2–C3 bond cleavage to afford dehydroglycine, which further decomposes to produce HCN, while in the second path, the decomposition of phenylalanine into phenethylamine, CO, and H2O is followed by internal hydrogen transfer in phenethylamine to generate HCN. The overall energy barrier of the two HCN formation paths exceeds that of NH3 formation paths, i.e., phenylalanine is more prone to afford NH3 than HCN upon pyrolysis.
1. Introduction
The growing demand for fossil fuels has drawn increased attention to alternative (e.g., renewable) energy sources such as sludge, which is a solid waste rich in volatile substances.1,2 Sludge pyrolysis is a promising sludge treatment method, allowing one to recover 80% of sludge energy, obtain valuable products such as bio-oil, biogas, and biochar, and reduce the amount of sludge by ∼93%.3 However, the nitrogen content of sludge (2.4–9.0 wt%) is much higher than that of traditionally used solid fuels (e.g., coal),4 resulting in the considerable release of nitrogen-containing gases (mainly NH3 and HCN) during pyrolysis. As NH3 and HCN are the main precursors of NOx, their direct emission causes acid rain and photochemical smog, thus adversely affecting human health5 and being a key issue limiting the development of the sludge pyrolysis technology.6 Therefore, the elucidation of NH3 and HCN formation mechanisms during sludge pyrolysis is a task of high practical importance.The transformation of nitrogen during sludge pyrolysis has been extensively studied,7–9 e.g., Cao et al.7 found that under rapid pyrolysis conditions, sludge nitrogen is mainly converted to nitrogen-containing gases such as NH3, HCN, and N2, the yields of which increase with increasing pyrolysis temperature. Tian et al.8 showed that the thermal cracking of sludge proteins affords three important intermediates, namely amine nitrogen, heterocyclic nitrogen, and nitrile nitrogen, the thermal cracking of which accounts for >80% of the HCN + NH3 production. Sludge nitrogen mainly exists in the forms of ammonium, nitrile, and protein nitrogen, with protein nitrogen having the largest share.9 As proteins have a very complex structure and a biomass type-dependent amino acid composition, their pyrolytic decomposition is difficult to model, and hence, simpler models (e.g., amino acids) are used.10,11 For example, Li et al.12 used thermogravimetric analyzer coupled with Fourier transform infrared spectrometry (TG-FTIR) to investigate the pyrolysis of phenylalanine (Phe) and tyrosine, revealing that their primary decomposition includes deamination and dehydration, while secondary decomposition mainly corresponds to the cleavage of cyclic dipeptides, with the gaseous products predominantly being NH3, H2O, CO2, CO, HNCO, HCN, and some organic compounds. Ramesh13 used Gas Chromatography-Mass Spectrometer (GC-MS) to study the pyrolysis of aspartic acid, asparagine, glutamic acid, glutamine, and pyroglutamic acid, demonstrating that these species first polymerize into peptides that subsequently decompose into lower-molecular-weight products. Choi et al.14 used GC-MS to study the dimerization of 20 amino acids, revealing that most amino acids that do not produce dimerization pyrolysis products have long side chains or ring structures.
Although the formation of HCN and NH3 during sludge and amino acid pyrolysis has been extensively investigated, the specific formation paths of these nitrogen-containing gases remain unclear and should therefore be further clarified, e.g., by quantum chemistry computations, which is commonly used to study the thermal decomposition mechanisms of various organic compounds. Liu et al.15 studied the pyrolytic decomposition of coal pyrrole to HCN, revealing that the corresponding energy barrier is substantially reduced in the presence of hydrogen radicals. Cristian et al.16 studied the formation of 2,5-diketopiperazine during the pyrolysis of proline, showing that the transition state corresponds to the (rate-determining) dehydration process. Saleh et al.17 employed density functional theory (DFT) calculations to study the decomposition of a single proline molecule, obtaining carboxylation and dehydration energy barriers of 297.1 and 304.6 kJ mol−1, respectively. Peng et al.18 studied the mechanism of aspartic acid pyrolysis and proposed a path for the formation of NH3 and HCN. At present, most research on amino acid pyrolysis has been performed at the experimental level, as exemplified by works probing the effects of pyrolysis temperature, heating rate, amino acid species, and catalyst on the distribution of pyrolysis products. However, the mechanism of amino acid pyrolysis remains underexplored, and hence, is herein probed by DFT calculations.
Phe is the main aromatic amino acid of sludge, undergoing insignificant dehydration during pyrolysis12 to afford small amounts of 3,6-dibenzyl-2,5-piperazinedione, which mainly undergoes unimolecular decomposition.19 As Phe does not form dimers even under the condition of high dilution.14 Phenylalanine is easily decomposed by several chemical pathways instead of condensation,20 the investigation of its unimolecular decomposition is expected to shed light on the mechanism of NH3 and HCN formation during sludge pyrolysis. Herein, the mechanism of Phe pyrolysis and the corresponding paths of NH3 and HCN formation are probed by DFT calculations, and the results provide a basis for the development of novel denitrification technologies.
2. Methods
The most stable configuration of Phe,21–24 which was also observed experimentally,23,24 was employed and optimized at the M06-2X/6-311G(d,p) level for further investigation, with the structures and atomic labels of Phe shown in the ESI.† The above conformation features intramolecular hydrogen bonds between the carboxyl group and the amino group as well as an additional stabilizing interaction between the p-electron system of the amino group and the aromatic ring. Moreover, another weaker interaction involving a carboxyl oxygen atom and an adjacent aromatic hydrogen was observed.23All calculations were performed using the Gaussian09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 software package. The Minnesota meta-exchange–correlation functional M06-2X26 and the 3-zeta basis set 6-311G(d,p) with polarization functions added to heavy and light atoms were selected as functional and basis sets, respectively, as the M06 suite of density functionals potentially contains important contributions from dispersion interactions.27 Unrestricted geometric optimizations were first performed on all initial guess structures to identify potential energy surface minima. Frequency calculations were performed on optimized geometries at the same level of theory, and all reactants and products were local minima on the potential energy surface without imaginary frequencies. Each transition state was a first-order saddle point on the potential energy surface with only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were carried out to check the correlation between the calculated initial states, transition states, and product geometries. After single-point energy calculations at the M06-2X/6-311G++(d,p) level, all values were corrected to account for zero-point vibrational energies.
25 software package. The Minnesota meta-exchange–correlation functional M06-2X26 and the 3-zeta basis set 6-311G(d,p) with polarization functions added to heavy and light atoms were selected as functional and basis sets, respectively, as the M06 suite of density functionals potentially contains important contributions from dispersion interactions.27 Unrestricted geometric optimizations were first performed on all initial guess structures to identify potential energy surface minima. Frequency calculations were performed on optimized geometries at the same level of theory, and all reactants and products were local minima on the potential energy surface without imaginary frequencies. Each transition state was a first-order saddle point on the potential energy surface with only one imaginary frequency. Intrinsic reaction coordinate (IRC) calculations were carried out to check the correlation between the calculated initial states, transition states, and product geometries. After single-point energy calculations at the M06-2X/6-311G++(d,p) level, all values were corrected to account for zero-point vibrational energies.
Rate constants, k(T), were evaluated using the conventional transition state theory.28 The transmission coefficient accounting for quantum tunneling corrections was calculated using the one-dimensional Eckart functional.29 Rate constants were fitted to modified Arrhenius parameters in the temperature region of 500–1200 K as
|
k(T) = ATnexp(−Ea/RT),
| (1) |
3. Results and discussion
During the decomposition of Phe, NH3 and HCN may be formed via numerous paths, including those involving dehydration, dehydrogenation, decarboxylation, deamination, isomerization, and tautomerization. Herein, the mechanisms of NH3 and HCN formation were divided into three groups according to the first step of Phe decomposition, namely 1,2-, 1,3-, and 1,4-hydrogen transfer (Fig. 1).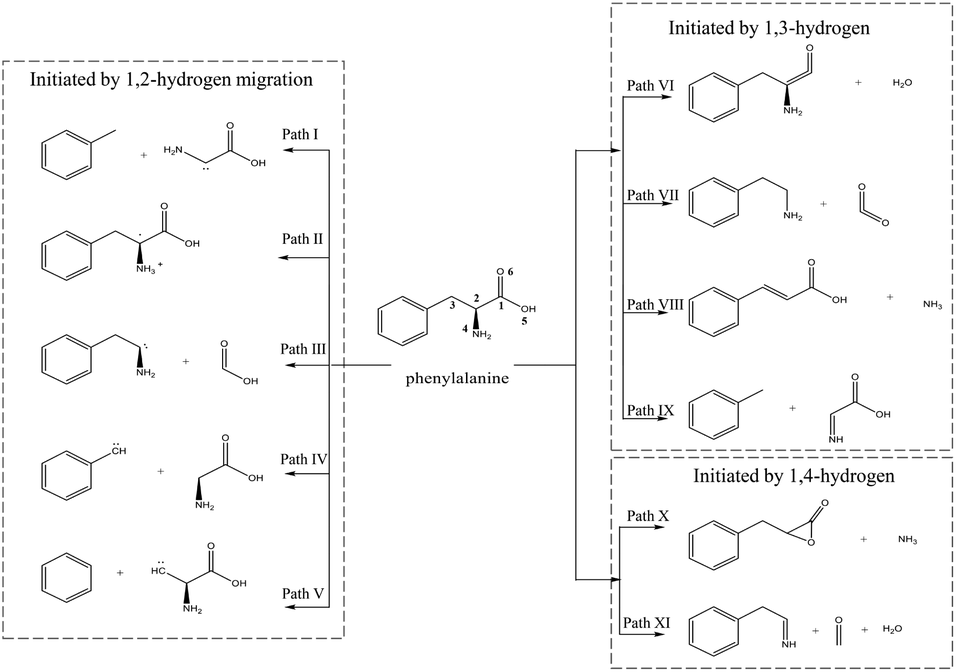 | ||
| Fig. 1 Initial interactions and pyrolysis mechanism of Phe. | ||
3.1 NH3 formation paths
According to Fig. 1, approximately 40 paths were analyzed and compared (see ESI†). Among them, the nine NH3 formation paths are presented in Fig. 2, with the four lowest total barriers observed for paths X-1, II-1, VII-1, and VIII-1 (203.74, 310.77, 304.85, and 325.87, respectively). Hence, only these four paths are discussed below, with their energy diagrams shown in Fig. 3.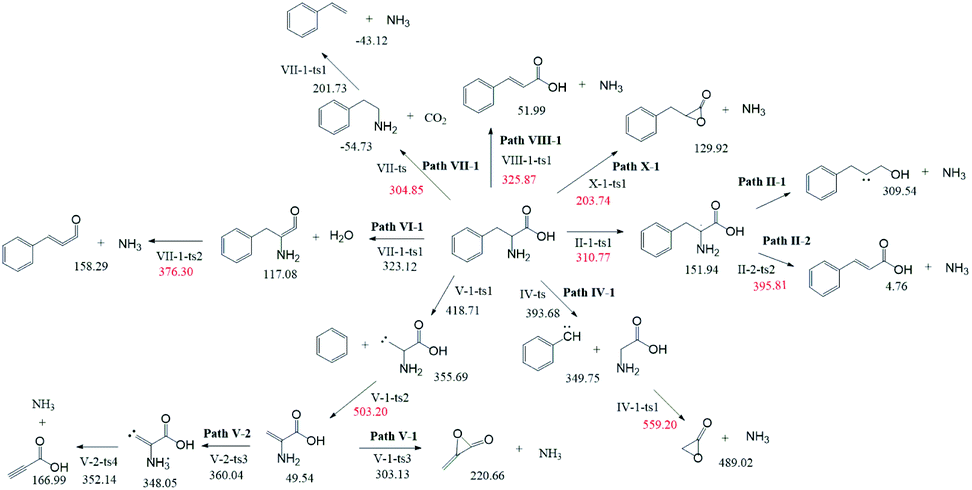 | ||
| Fig. 2 Possible NH3 formation paths. Numbers refer to energies in kJ mol−1. | ||
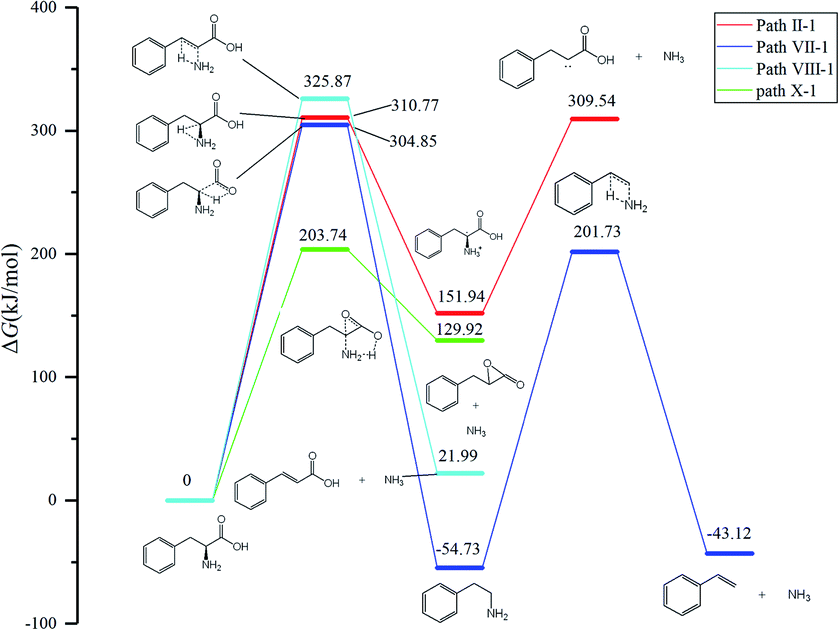 | ||
| Fig. 3 Energy diagrams of the four favorable NH3 formation paths. | ||
In path X-1, the carboxyl group hydrogen is transferred to the amino group, and subsequent Phe decomposition directly affords 3-benzyloxiran-2-one and NH3. This process passes through transition state X-1-ts1 (energy barrier = 203.74 kJ mol−1), and does not involve the formation of zwitterions, as they are not stable in the gas phase. Similarly, Rai et al.31 studied the zwitterion conformations of leucine and its hydrate, showing that zwitterions are not stable in the gas phase, as the dipole moment of leucine in the gas phase (5.73 D) is less than that in solution (7.37 D). Aldehydes with one carbon less than the parent amino acid were present in the pyrolysate of all members of this class,32 and benzyloxiran-2-one was expected to further decompose into phenylacetaldehyde, in agreement with the experimental detection of this species by Hidalgo et al.33 In turn, phenylacetaldehyde can decompose into benzaldehyde,34 which has been experimentally detected during the pyrolysis of Phe.35
In path II-1, the hydrogen on C1 is transferred to the amino group through 1,2-hydrogen migration to afford intermediate II-1-i1 via transition state II-1-ts1 (energy barrier = 310.77 kJ mol−1). Then, the C–N bond of II-1-i1 is cleaved to generate NH3, and the length of the C–N bond is relaxed scan. No transition states are observed for this process. The obtained product (II-1-i2, an unstable carbene) undergoes further reactions to generate carboxylic acids. According to Zang et al.,36 who observed this species for aspartic acid and Phe, the end product may be either a saturated or unsaturated carboxylic acid.
In path VII-1, Phe undergoes decarboxylation through a four-membered ring transition state (VII-ts), and the hydroxyl hydrogen at C1 is transferred to the amino group at C2 to form phenethylamine and CO2, in line with the findings of Li et al.,12 who observed the release of phenethylamine, CO2, and NH3 during the pyrolysis of Phe. Phenethylamine can undergo deamination via a four-membered ring transition state (VII-1-ts1) to form a double bond between C2 and C3, and the hydrogen at C3 can be combined with the amino group at C2 to form NH3 and styrene, in agreement with the experimental detection of these two species during Phe pyrolysis by Wang et al.35 Moreover, Patterson et al.19 found that styrene is the main product of phenethylamine pyrolysis. The potential barriers of this path (304.85 and 256.46 kJ mol−1) are similar to those observed for leucine.37 The initial reaction step is rate-determining, and the energy barrier is not high, which further confirms that path VII-1 plays an important role in Phe pyrolysis.
In path VIII-1, deamination occurs via a four-membered ring transition state (VIII-1-ts1), a double bond is formed between C2 and C3, and the hydrogen at C3 combines with the amino group at C2 to form intermediate 2-i1 and NH3 (barrier = 325.87 kJ mol−1). Cinnamic acid has good thermal stability, and a very small amount of cinnamic acid has been experimentally observed during Phe pyrolysis.19 Thus, the above route is not the main NH3 formation path, featuring the highest barrier among the four paths of NH3 generation.
Among the NH3 formation paths, path X-1 is the most favorable one in terms of the Gibbs free energy, featuring a barrier height of 203.74 kJ mol−1, but affords high-energy final products. The alternative path VII-1 has a larger energy barrier of 304.85 kJ mol−1 but affords lower-energy products and is exothermic. Thus, it can be concluded that path X-1 is the kinetically preferred path, while path VII-1 is the thermodynamically preferred path, i.e., that with the lowest free energy of products. Tian et al.38 claimed that the formation of NH3 at temperatures below 400–500 °C can be attributed to the pyrolysis of sludge amino structures to some extent.
3.2 HCN formation paths
Almost 30 possible HCN formation paths were identified (see ESI†), with the most favorable and feasible ones (Fig. 4) featuring the formation of dehydroglycine, methanimine, and isocyanide as intermediates. These intermediates subsequently decompose to afford HCN, with the lowest HCN generation barrier observed for isocyanide (110.2 kJ mol−1).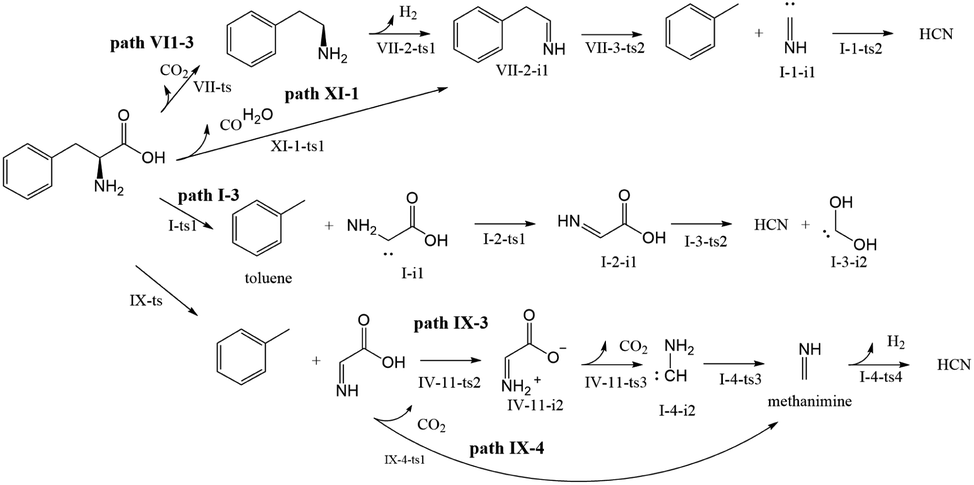 | ||
| Fig. 4 Probable HCN formation paths. | ||
As shown in Fig. 5, path I-3 features the breakage of the C2–C3 bond by 1,2-hydrogen transfer through a three-membered ring transition state (I-ts) to afford toluene and I-i1, a carbene with a non-bonding electron pair on C2. This carbene undergoes isomerization into I-2-i1 (dehydroglycine) via hydrogen transfer from N4 to C2. It is worth noting that in I-2-i1, the amino group and O6 are on the same side, and the O atom is more negatively charged than the C atom. Hence, the hydrogen atom on N4 is more easily transferred to O6 via the I-3-ts2 transition state than that to C1. Subsequently, the C1–C2 bond is broken to form HCN and a carbene, I-2-i2. As a result, the rate-determining step of path I-3 corresponds to hydrogen transfer from N4 to C2 (overall energy barrier = 369.28 kJ mol−1).
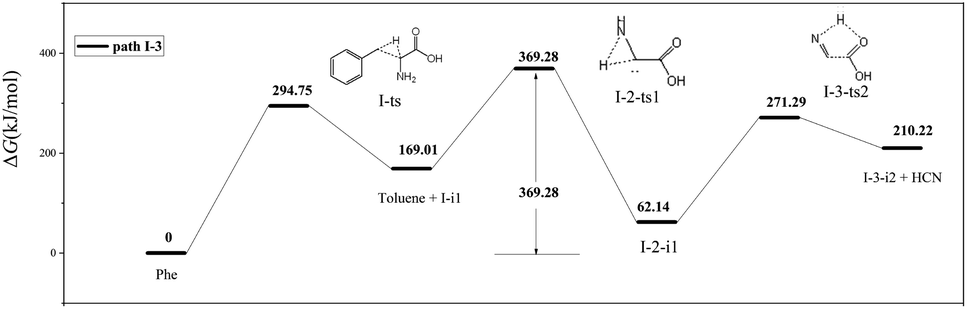 | ||
| Fig. 5 Pyrolytic reaction routes to HCN based on path I-3. | ||
In path VII-3 (Fig. 6), decarboxylation results in C1–C2 bond cleavage through a four-membered ring transition state (VII-ts). This reaction, producing phenethylamine and CO2, is considered to be important for the decomposition of amino acids.19,39 However, the amount of HCN produced during the pyrolysis of phenethylamine is very small.19 According to path VII-3, phenethylamine first undergoes rate-determining dehydrogenation to afford H2 and VII-2-i1 (energy barrier = 426.04 kJ mol−1), with subsequent decomposition of VII-2-i1 affording toluene and the HCN isomer, I-1-i1. The energy barrier for the isomerization of I-1-i1 into HCN through hydrogen transfer is 110.21 kJ mol−1, in agreement with the value of 129.20 kJ mol−1 suggested by Liu et al.15. The rate-determining step of path VII-3 is phenethylamine dehydrogenation, and the associated high total energy barrier of 426.04 kJ mol−1 suggests that the above path does not significantly contribute to HCN formation, in line with the results of Patterson et al.19
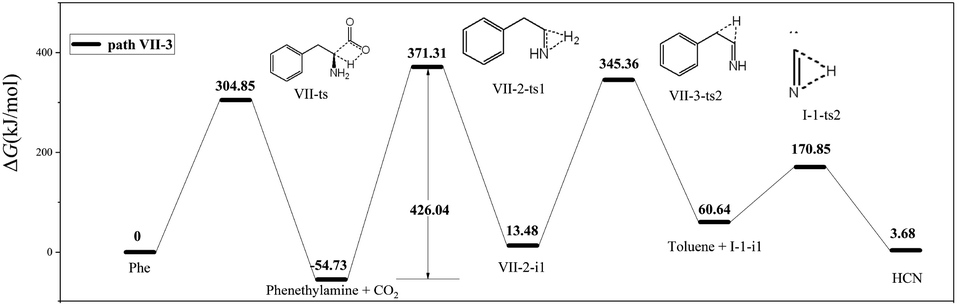 | ||
| Fig. 6 Pyrolytic reaction routes to HCN based on path VII-3. | ||
In path IX-3, the hydrogen on N4 is transferred to C3 to afford toluene and IV-9-i1 (dehydroglycine) (Fig. 7), a conformational isomer of I-2-i1 with the –OH group and N4 on the same side. The hydrogen atom on the carboxyl group is transferred to afford zwitterion IV-11-i2 through transition state IV-11-ts2. It is worth noting that although the Gibbs free energy of IV-11-i2 exceeds that of IV-11-ts2, IRC calculations reveal that the above generation of zwitterions from dehydroglycine is feasible. Subsequently, the C1–C2 bond is easily broken to form I-4-i2 and CO2, with the corresponding energy barrier equaling only 22.35 kJ mol−1. I-4-i2 undergoes hydrogen transfer via transition state I-4-ts3 to afford I-4-i3 aka methanimine (energy barrier = 182.41 kJ mol−1), which is viewed as the main HCN precursor, e.g., Johnson et al.40 reported that the pyrolysis of methanimine affords large amounts of HCN. Finally, methanimine is dehydrogenated to HCN via transition state I-4-ts4 (energy barrier = 404.64 kJ mol−1). Thus, the decomposition of Phe to generate HCN in path IX-3 requires two steps to overcome high energy barriers, and this path is therefore not favored.
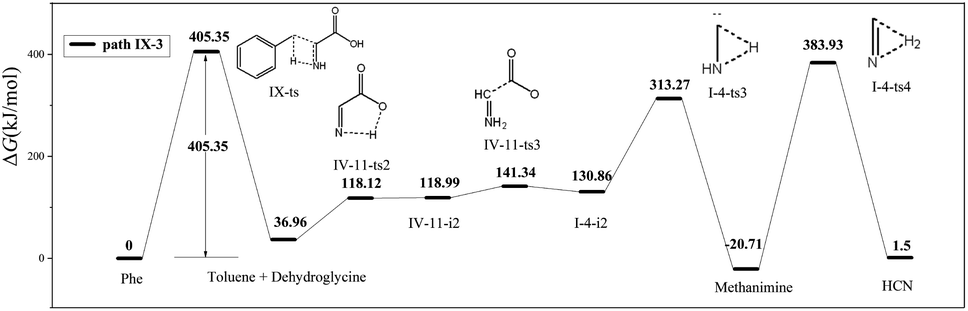 | ||
| Fig. 7 Pyrolytic reaction routes to HCN based on path IX-3. | ||
As shown in Fig. 8, the difference between paths IX-4 and IX-3 lies in the decarboxylation of IV-9-i1. In the former path, the carboxyl hydrogen is transferred to C2, and the C1–C2 bond is broken to generate CO2 and methanimine via transition state VI-10-ts2 (energy barrier = 309.12 kJ mol−1). This energy barrier is much higher than that of path IX-3, as the electronegativity of C is lower than that of N, i.e., hydrogen transfer to C is more difficult than to N. However, the intermediate (methanimine) of path IX-4 has better thermal stability than that (I-1-i1) of IX-3. Methanimine is then dehydrogenated to afford H2 and HCN, in agreement with path IX-3. Therefore, comparison of the energy barriers of the two decarboxylation routes of IV-9-i1 and the Gibbs free energy of products suggests that path IX-3 is kinetically preferred to path IX-4, while the latter path is preferred to the former from the viewpoint of thermodynamics.
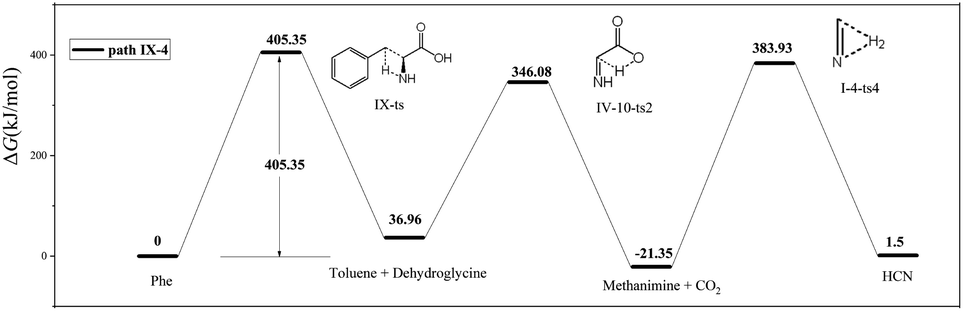 | ||
| Fig. 8 Pyrolytic routes to HCN based on path IX-4. | ||
In path XI-1 (Fig. 9), 1,4-hydrogen transfer from N4 to O5 is followed by the decomposition of the carboxyl group into CO and H2O. Among the initial steps of the other four paths, this step features the lowest energy barrier of 287.96 kJ mol−1 and is therefore most likely to occur. The subsequent decomposition of intermediate VII-2-i1 is consistent with path VII-3. The rate-determining step of path VII-3 is hydrogen atom transfer from C2 to C3 (overall energy barrier = 378.1 kJ mol−1).
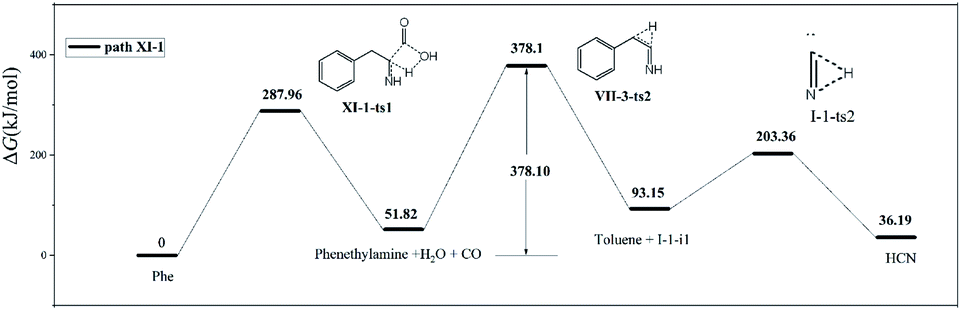 | ||
| Fig. 9 Pyrolytic routes to HCN based on path XI-1. | ||
Compared to path XI-1, path VII-3 has a higher energy barrier of the initial step and a higher total energy barrier. Therefore, the former HCN generation path is preferred to the latter. Experimentally, CO and H2O were detected alongside HCN during the pyrolysis of Phe.12
In summary, paths I-3 and XI-1 were found to have the lowest energy barriers (369.28 and 378.10 kJ mol−1, respectively) of all HCN formation routes, affording products with energies of 210.22 and 36.19 kJ mol−1, respectively. Thus, paths I-3 and XI-1 were concluded to be kinetically and thermodynamically favored, respectively.
3.3 Rate constant calculation
To simulate the conditions encountered in the actual pyrolysis environment as well as NH3 and HCN generation trends, we calculated the reaction rate constants for the most reasonable rate-determining steps of Phe decomposition. All rate constants were corrected according to Eckart's formula to find possible tunneling effects. As shown in Table 1, the initial reaction type of Phe pyrolysis had a significant effect on the formation of HCN or NH3.| No. | Path | Rate-determining step | A (s−1) | n | Ea/R (K) | Product |
|---|---|---|---|---|---|---|
| 1 | II-1 | II-1-ts1 | 1.213 × 107 | 2.15 | 304.29 | NH3 |
| 2 | VII-1 | VII-ts | 6.885 × 106 | 2.39 | 299.77 | NH3 |
| 3 | VIII-1 | VII-1-ts1 | 1.496 × 109 | 1.45 | 326.23 | NH3 |
| 4 | X-1 | X-1-ts1 | 8.827 × 1011 | 0.79 | 212.24 | NH3 |
| 5 | I-3 | I-2-ts1 | 1.302 × 104 | 2.77 | 180.22 | HCN |
| 6 | VII-3 | VII-2-ts1 | 5.903 × 10−13 | 7.46 | 366.52 | HCN |
| 7 | IX-3 | IX-ts | 16.13 | 3.80 | 383.53 | HCN |
| 8 | IX-4 | IX-ts | 16.13 | 3.80 | 383.53 | HCN |
| 9 | XI-1 | VII-3-ts2 | 1.860 × 107 | 2.01 | 320.10 | HCN |
Among all paths with internal hydrogen transfer as the starting reaction, path X-1 was the most favorable one, featuring an activation energy of 212.24 kJ mol−1 and affording NH3. Path VII-1 also generated NH3 with a higher activation energy of 299.77 kJ mol−1, and thus became more important at elevated temperature. Li et al.12 probed the pyrolysis of Phe and detected two infrared absorption peaks of NH3 in different temperature ranges, suggesting that the NH3 generation path is not unique and changes with temperature. Regarding HCN formation, the most favorable routes were identified as paths I-3 and XI-1, featuring activation energies of 180.22 and 320.10 kJ mol−1, respectively. Notably, the activation energies of the five HCN formation paths exceeded those of NH3 formation paths, which indicates that Phe is more prone to generate NH3 than HCN during pyrolysis. This conclusion is in line with the fact that the experimental yield of the former gas was shown to be much higher than that of the latter.41
Phe pyrolysis also produces nitrogen-containing heterocyclic compounds such as pyrrole, pyridine, and indole. Wornat et al.42 stated that N-heteroaromatic compounds have good thermal stability, and Liu et al.43 found that it is difficult to form HCN and NH3 during the pyrolysis of indole. The release of HCN during the pyrolysis of Phe is mainly due to the continued decomposition of small-molecular-weight intermediates (thermal cracking of volatiles7) and not to the secondary decomposition of nitrogen-containing heterocycles. Moreover, protein pyrolysis did not produce significant amounts of HCN.12
4. Conclusions
Herein, we theoretically studied the mechanism of Phe decomposition for 1,2-, 1,3-, and 1,4-hydrogen transfer as the initial step, revealing the availability of ∼40 paths to generate NH3 and HCN and arriving at the following conclusions.(1) Phe pyrolysis produces HCN and NH3 as two main nitrogen-containing products, with the production of the latter preferred to that of the former.
(2) NH3 is mainly produced via deamination, and the relative contributions of different paths strongly depend on temperature. Specifically, two major NH3 release paths are identified, with overall energy barriers differing by 87.53 kJ mol−1. In the preferred path (X-1), intramolecular hydrogen bonding between amino and carboxyl groups facilitates hydrogen transfer from the latter group to the former and thus decreases the related energy barrier, with subsequent deamination affording NH3 and 3-benzyloxiran-2-one. In the second path (VII-1), Phe is decarboxylated to form CO2 and phenethylamine, which is then deaminated to produce styrene and NH3.
(3) The release of HCN during Phe pyrolysis is mainly due to the continuous decomposition of small-molecular-weight intermediates, with the two major paths featuring overall energy barriers of 369.28 (I-3) and 378.10 kJ mol−1 (XI-I). Notably, the activation energy of the rate-determining step of path I-3 is much lower than for path XI-1, i.e., the former path is preferred to the latter.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Funds for Young Scholars of China (No. 51806033), National Key Technologies Research and Development Program (No. 2018YFB0905104) and Jilin Provincial Science and Technology Development Program (No. 20190201096JC).Notes and references
- Q. Zhang, H. Liu, G. Lu, L. Yi, H. Hu, H. Chi and H. Yao, Proc. Combust. Inst., 2017, 36, 4003–4010 CrossRef CAS.
- V. K. Tyagi and S. L. Lo, Renewable Sustainable Energy Rev., 2013, 25, 708–728 CrossRef CAS.
- J. A. Caballero, R. Front, A. Marcilla and J. A. Conesa, J. Anal. Appl. Pyrolysis, 1997, 40–41, 433–450 CrossRef.
- H. Liu, Q. Zhang, H. Hu, P. Liu, X. Hu, A. Li and H. Yao, Proc. Combust. Inst., 2015, 35, 2759–2766 CrossRef CAS.
- G. Bowatte, R. Tham, J. L. Perret, M. S. Bloom, G. Dong, N. Waidyatillake, D. Bui, G. G. Morgan, B. Jalaludin, C. J. Lodge and S. C. Dharmage, Int. J. Environ. Res. Public Health, 2018, 15, 257 CrossRef PubMed.
- H. Liu, L. Yi, H. Hu, K. Xu, Q. Zhang, G. Lu and H. Yao, Fuel, 2017, 195, 208–216 CrossRef CAS.
- J. P. Cao, L. Y. Li, K. Morishita, X. Bin Xiao, X. Y. Zhao, X. Y. Wei and T. Takarada, Fuel, 2013, 104, 1–6 CrossRef CAS.
- Y. Tian, J. Zhang, W. Zuo, L. Chen, Y. Cui and T. Tan, Environ. Sci. Technol., 2013, 47, 3498–3505 CrossRef CAS PubMed.
- M. C. Samolada and A. A. Zabaniotou, J. Waste Manag., 2014, 34, 411–420 CrossRef CAS PubMed.
- Q. Ren and C. Zhao, Environ. Sci. Technol., 2012, 46, 4236–4240 CrossRef CAS PubMed.
- Q. Ren, C. Zhao, X. Chen, L. Duan, Y. Li and C. Ma, Proc. Combust. Inst., 2011, 33, 1715–1722 CrossRef CAS.
- L. Jie, L. Yuwen, S. Jingyan, W. Zhiyong, H. Ling, Y. Xi and W. Cunxin, Thermochim. Acta, 2007, 467, 20–29 CrossRef.
- R. K. Sharma, W. G. Chan, J. Wang, B. E. Waymack, J. B. Wooten, J. I. Seeman and M. R. Hajaligol, J. Anal. Appl. Pyrolysis, 2004, 72, 153–163 CrossRef CAS.
- S. S. Choi and J. E. Ko, J. Anal. Appl. Pyrolysis, 2010, 89, 74–86 CrossRef CAS.
- J. Liu, X. Zhang, A. Shaw, Q. Lu, B. Hu, C. qing Dong and Y. ping Yang, J. Energy Inst., 2019, 92, 1468–1475 CrossRef CAS.
- C. Cervantes, J. R. Mora, L. Rincón and V. Rodríguez, Mol. Phys., 2020, 118 DOI:10.1080/00268976.2019.1594422.
- S. Rawadieh, I. Altarawneh, H. B. Alateyat and M. Altarawneh, Comput. Theor. Chem., 2013, 1018, 45–49 CrossRef CAS.
- P. Kang, W. Qin, Z. Q. Fu, T. P. Wang, L. W. Ju and Z. F. Tan, Int. J. Agric. Biol. Eng., 2016, 9, 166–176 Search PubMed.
- J. M. Patterson, E. P. Papadopoulos, N. F. Haidar and W. T. Smith, J. Org. Chem., 1973, 38, 663–666 CrossRef CAS PubMed.
- Y. Zubavichus, M. Zharnikov, A. Shaporenko, O. Fuchs, L. Weinhardt, C. Heske, E. Umbach, J. D. Denlinger and M. Grunze, J. Phys. Chem. A, 2004, 108, 4557–4565 CrossRef CAS.
- S. S. Bokatzian, M. L. Stover, C. E. Plummer, D. A. Dixon and C. J. Cassady, J. Phys. Chem. B, 2014, 118, 12630–12643 CrossRef CAS PubMed.
- G. Bouchoux, S. Bourcier, V. Blanc and S. Desaphy, J. Phys. Chem. B, 2009, 113, 5549–5562 CrossRef CAS PubMed.
- L. C. Snoek, E. G. Robertson, R. T. Kroemer and J. P. Simons, Chem. Phys. Lett., 2000, 321, 45–56 CrossRef.
- G. Von Helden, I. Compagnon, M. N. Blom, M. Frankowski, U. Erlekam, J. Oomens, B. Brauer, R. B. Gerber and G. Meijer, Phys. Chem. Chem. Phys., 2008, 10, 1248–1256 RSC.
- G. E. S. M. J. Frisch, G. W. Trucks and H. B. Schlegel, Gaussian, Revision D. 01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 215–241 Search PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- H. Eyring, J. Chem. Phys., 1935, 2, 107–115 CrossRef.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- A. K. Rai, X. Xu, Z. Lin and D. K. Rai, Vib. Spectrosc., 2011, 56, 74–81 CrossRef CAS.
- A. Ratcliff, E. E. Medley and P. G. Simmonds, J. Org. Chem., 1974, 39, 1481–1490 CrossRef PubMed.
- F. J. Hidalgo, M. M. León and R. Zamora, Food Chem., 2016, 209, 256–261 CrossRef CAS PubMed.
- L. C. Fong and V. A. Yaylayan, J. Agric. Food Chem., 2008, 56, 10697–10704 CrossRef PubMed.
- S. Wang, B. Liu and Q. Su, J. Anal. Appl. Pyrolysis, 2004, 71, 393–403 CrossRef CAS.
- X. Zang, J. C. Brown, J. D. H. Van Heemst, A. Palumbo and P. G. Hatcher, J. Anal. Appl. Pyrolysis, 2001, 61, 181–193 CrossRef CAS.
- S. Jabeen, Z. Zeng, M. Altarawneh, X. Gao, A. Saeed and B. Z. Dlugogorski, Int. J. Chem. Kinet., 2019, 51, 696–710 CrossRef CAS.
- F. J. Tian, B. Q. Li, Y. Chen and C. Z. Li, Fuel, 2002, 81, 2203–2208 CrossRef CAS.
- J. Hao, J. Guo, L. Ding, F. Xie, Q. Xia and J. Xie, J. Therm. Anal. Calorim., 2014, 115, 667–673 CrossRef CAS.
- W. R. Johnson and J. C. Kang, J. Org. Chem., 1971, 36, 189–192 CrossRef CAS PubMed.
- F. Wei, J. P. Cao, X. Y. Zhao, J. Ren, B. Gu and X. Y. Wei, Fuel, 2018, 218, 148 CrossRef CAS.
- M. J. Wornat, A. F. Sarofim, J. P. Longwell and A. L. Lafleur, Energy Fuels, 1988, 2, 775–782 CrossRef CAS.
- J. Liu, X. Zhang, B. Hu, Q. Lu, D. jia Liu, C. qing Dong and Y. ping Yang, J. Energy Inst., 2020, 93, 649–657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: This file contains further details of 9 NH3 and 30 HCN formation paths. It also includes cartesian coordinates (X, Y, Z) and vibrational frequencies (cm−1) for all the transition states. See DOI: 10.1039/d0ra05482h |
| This journal is © The Royal Society of Chemistry 2020 |