
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective extraction and determination of aromatic amine metabolites in urine samples by using magnetic covalent framework nanocomposites and HPLC-MS/MS
Jingjing Yu *,
Bing Wang,
Junlan Cai,
Quanping Yan,
Sheng Wang,
Ge Zhao,
Junwei Zhao,
Lining Pan and
Shaofeng Liu*
*,
Bing Wang,
Junlan Cai,
Quanping Yan,
Sheng Wang,
Ge Zhao,
Junwei Zhao,
Lining Pan and
Shaofeng Liu*
Zhengzhou Tobacco Research Institute of CNTC, Zhengzhou 450001, China. E-mail: yujingjing307@163.com; sfliu@126.com; Fax: +86-371-67672575/2055; Tel: +86-371-67672575 ext. 2055
First published on 31st July 2020
Abstract
Purification and selective enrichment of ultra-low level metabolites in bio-samples is very important for HPLC-MS/MS analysis. A magnetic covalent organic framework (i.e. COFs) (TpPa-1) (i.e. 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde-p-phenylenediamine) material was synthesized and used for a magnetic solid phase extraction (MSPE) method in aromatic amine (AA) metabolites (i.e. 1-naphthylamine, 2-naphthylamine, 3-aminobiphenyl and 4-aminobiphenyl) in urine, and then HPLC-MS/MS was employed for analysis. The morphology, pore structure, surface area, chemical composition, magnetic properties, and thermal stability of the synthesized magnetic COFs were characterized with several analysis techniques, such as nitrogen adsorption–desorption isotherms, HRTEM, FTIR, XRD, and so on. MSPE conditions were optimized and analytical performance of the developed method was characterized. The limit of detection (LOD) and limit of quantification (LOQ) of four AAs ranged from 0.01 to 0.07 ng mL−1 and 0.04 to 0.22 ng mL−1, respectively. The recoveries of 1-NA, 2-NA, 3-ABP, 4-ABP were in the range of 81.9–105%, 87.8–102%, 101–120%, 88.3–117%, respectively. Good intra-day and inter-day precision were obtained with RSD of less than 5.8% and 9.9%, respectively. Furthermore, the synthesized magnetic COFs absorbent could be recycled in MSPE at least 5 times. Analytical results of AA metabolites in real urine samples with the developed method showed significant difference (p < 0.01) between smokers and nonsmokers. Thus, urinary AA metabolites could be exposure biomarkers for cigarette smoke.
1. Introduction
The FDA established a list of harmful and potentially harmful constituents in tobacco products and tobacco smoke in 2012.1 It contains a series of aromatic amine (AA) compounds, such as 1-naphthylamine (1-NA), 2-naphthylamine (2-NA) and 4-aminobiphenyl (4-ABP). What's more, 2-NA and 4-ABP belong to Group 1 (“carcinogenic to humans”) classified by the International Agency for Research on Cancer (IARC), while 1-NA is in Group 3 (“not classifiable as to its carcinogenicity to humans”).2 It has been reported that tobacco smoking is a major cause of bladder cancer3–5 in humans without occupational exposure, such as industries involving dyes, pharmaceuticals, pesticides and plastics, while AAs and their derivatives were important chemical intermediates.6–8 Urinary AAs could be used as exposure biomarkers of tobacco smoke. Some research has indicated that urinary AAs are higher in smokers than in nonsmokers.4,5,9,10AA metabolites can covalently bind to tissue macromolecules (proteins) and DNA to form adducts.4,5,11 And then the free, acetylated and glucuronidated AAs excreted in urine.11 Thus, as for the analysis of urinary AAs, acid, base, or enzyme hydrolysis process were used for deconjugating firstly.4,5,9,10 Owing to the ultra-low levels of AA metabolites in urine and the complexity of urine matrix, some approaches involving complex sample cleanup or derivatization4,5,11 were applied before GC-MS,4,5 GC-MS/MS10,11 or HPLC-MS/MS9 analysis. Derivatization reaction with AAs should be conducted for GC-MS or GC-MS/MS analysis due to the higher polarity of AAs. HPLC-MS/MS has been shown to be an efficient technique avoiding derivatization for the determination of polar compounds, such as AAs. Various sample clean-up procedures were applied for different sample matrix, such as conventional liquid–liquid extraction,12 solid-phase extraction (SPE) with SPE column9,13 and so on. For example, Saha's group12 have used HPLC-MS/MS technique to determine 6 AAs in cigarette smoke, and a liquid–liquid extraction and concentration process were applied, which was tedious and time-consuming. Our group9 has reported that a polycyclic aromatic hydrocarbons (PAHs)-molecular imprinting polymer (MIP) solid extraction column was used for the determination of AAs metabolite in urine samples of smokers and nonsmokers. The PAHs-MIP column could effectively purify the urine samples and decrease the matrix effect. However, the commercial SPE column was expensive and could not be recycled.
Recently, covalent organic frameworks (COFs) materials have been widely used as solid extraction adsorbent due to their fascinating properties including high specific surface area, excellent thermal stability, high porosity.14–20 Magnetic solid-phase extraction (MSPE) has attracted increasing attention due to its high extraction efficiency and recycled property. MSPE technique based on COFs materials is a perfect combine and has been successfully used in sample clean-up procedure.21–28 For example, Wang and coworkers21 reported a COF-based magnetic adsorbent for solid extraction of PAHs, and HPLC was employed for quantification. The COF-LZU1@PEI@Fe3O4 displayed high extraction efficiency for PAHs, which was mainly attributed to strong π–π stacking and hydrophobic interaction. Chen and coworkers22 used a core–shell structured magnetic COFs for MSPE for bisphenols from human serum samples. The developed method was fast, simple, highly efficient and sensitive, and the Fe3O4@COF nanocomposites exhibited excellent reusability.
In the present work, a magnetic COFs (TpPa-1) composite was prepared for MSPE technique, and then used for the extraction of AAs metabolite in urine. The as-prepared magnetic COFs material were characterized with different analysis techniques, such as nitrogen adsorption–desorption isotherm, high resolution transmission electron microscopy (HRTEM), X-ray diffraction analyses (XRD) and small angle X-ray scattering (SAXS), and so on. The conditions of MSPE were optimized and analytical performance was characterized. What's more, the reusability of as-prepared magnetic COFs material was studied in this work. Four AAs (i.e. 1-NA, 2-NA, 3-ABP and 4-ABP) in urine were determined and their urinary excretion levels for smokers and nonsmokers were investigated as well.
2. Materials and methods
2.1. Chemicals and reagents
1-NA, 2-NA, 3-ABP, 4-ABP, 1-NA-d7, 2-NA-d7, 3-ABP-d9 and 4-ABP-d9 were analytical standard and obtained from Dr. Ehrenstorfer (purity≥98%, Germany). Acetonitrile and methanol were HPLC grade and purchased from Dikama Corp. (USA). Formic acid was HPLC grade and obtained from TEDIA Company Inc. (USA). Hydrochloric acid and sodium hydroxide were of analytical reagent and obtained from ACROS ORGANICS (USA). Aqueous ammonia was of analytical reagent (CNW Technologies, Germany). All other reagents were analytical standard. Water was purified with a Milli-Q system (Millipore, Bedford, MA, USA).FeCl3·6H2O (97%) was ACS reagent from Sigma-Aldrich (USA), ethylene glycol (99.9%) was purchased from KEMIOU Chemical Reagents Ltd. (Tianjin, China). Anhydrous sodium acetate with purity of 99% was obtained from Alfa Aesar (USA). 1,6-Diaminohexane (98%) was obtained from Sigma-Aldrich (USA). 2,4,6-Trihydroxybenzene-1,3,5-tricarbaldehyde (Tp, 95%) was obtained from Ark Pharm, Inc. (United States). p-Phenylenediamine (Pa-1, 99%) was purchased from J&K Scientific Ltd. (China). Other reagents were analytical reagent, such as dimethylformamide and 95% ethyl alcohol.
2.2. Synthesis of amino-functionalized magnetic Fe3O4 nanoparticles
The amino-functionalized magnetic Fe3O4 nanoparticles were prepared by hydrothermal reaction.29 FeCl3·6H2O (4.0 g), anhydrous sodium acetate (8.0 g) and 1,6-diaminohexane (26.0 g) were added into ethylene glycol (120 mL) under mechanical agitation at 50 °C for 0.5 h until homogeneous solution was obtained. The mixture was added in Teflon-line stainless-steel autoclave, and sustained for 6 h at 200 °C. After the mixture was transferred into a flask, the synthesized amino-functionalized magnetic Fe3O4 nanoparticles was separated with the help of a magnet, and washed with water and 95% ethanol. The obtained magnetic nanoparticles dried in the air for 4 to 8 hours, and then dried in vacuum oven at 50 °C for 12 h.2.3. Synthesis of magnetic COFs material
The synthesis of magnetic COFs (TpPa-1) material was based on Schiff-base mechanism30,31 and the synthetic route was as follows. 0.0630 g Tp (0.30 mmol) was dissolved in a certain volume of ethanol (10–40 mL) by stirring. And then 0.1050 g the synthesized amino-functionalized magnetic Fe3O4 nanoparticles was added into the Tp solution, and then under mechanical agitation at 50 °C for 0.5 h. 0.0486 g Pa−1 (0.45 mmol) was dissolved in the same volume of ethanol as above (i.e. 10–40 mL). The obtained Pa-1 solution was added into the Tp mixture drop by drop for about 15 min under mechanical agitation in room temperature. The reaction was sustained for 12 h, and the brown colored magnetic COFs (TpPa-1) material was obtained.The obtained mixture was dried under vacuum at 50 °C, and then washed with dimethylformamide, 95% ethanol, and methanol successively. After each step, the synthesized magnetic nanoparticles could be collected from the mixture with the help of a magnet. The obtained magnetic COFs material was dried in vacuum oven at 50 °C for 12 h.
The synthesis process of COFs material (TpPa-1) was similar to that of magnetic COFs material, except for the addition of magnetic NH2–Fe3O4 nanoparticles.
2.4. Material characterization
Tristar II 3020 Surface Area Analyzer (Micromeritics Instrument Ltd. USA) was applied to obtain surface area and pore size distribution of the synthesized materials. The organo-functional group of the synthesized materials was characterized with Fourier transform infrared spectrometer (FTIR, BRUKER TENSOR 27, Germany). High resolution transmission electron microscopy (HRTEM, USA FEI TECNAI G2 F20) was used for the morphology characterization of the synthesized materials. X-ray diffraction analyses (XRD, Germany Bruker D8 venture) and small angle X-ray scattering (SAXS, Germany Bruker SAXS Nanostart) were used to characterize constitute and structure of the synthesized materials. The vibrating sample magnetometer (VSM) was used for magnetic sample characterization (PPMS-9(VSM), Quantum Design, USA). Thermogravimetric analysis (TGA) was measured with TGA IR thermogravimetric analyzer (NETZSCH STA 409 PC, Germany).2.5. Urine sampling
27 healthy volunteers containing 20 smokers and 7 nonsmokers were recruited into the study. All volunteers were informed about the aim of this study and freely gave written consent about the donation of urine. Smokers were between 18 and 65 years old, and smoked more than 5 Chinese cigarettes every day for more than 6 months. Urine samples of 24 hours for all persons were collected, and frozen at −18 °C immediately. Urinary creatinine was determined using an automatic biochemical analyzer (Beckman-coulter SYNCHRON CX9, USA).2.6. Sample ethical statement
All experiments were performed in accordance with the guidelines “Technical specification of management for field survey in large population-based cohort study (T/CPMA OO1-2019)” drafted by Chinese preventive medicine association, and approved by the ethics committee at Zhengzhou University. Informed consents were obtained from human participants of this study.2.7. Urine sample preparation and MSPE
Urine sample was thawed at room temperature. Before MSPE, sample was treated for acid hydrolysis as follow. 5 mL urine sample was put into 50 mL centrifuge tube, and 1 mL concentrated HCl was added and reacted in 80 °C water bath for 1 h. After cooling in an ice bath, 2.4 mL 5 mol L−1 NaOH solution were added to neutralize excess acid and the pH of final urine sample was adjusted to 12–13.As for MSPE, a certain amount of mixed internal standard solution (1-NA-d7, 2-NA-d7, 3-ABP-d9 and 4-ABP-d9) was added into the treated urine sample. Then, 20 mg magnetic COFs material was added, and the mixture was oscillated for 20 min with vortex apparatus (TALBOYS Advanced multi-tube vortexer, Germany) at room temperature. After extraction of the analytes, an external magnet was utilized to separate the magnetic materials. After urine was discarded, 5 mL water was used to wash the magnetic COFs material, and then 5 mL methanol each time was applied to elute analytes on the magnetic COFs material for three times with 3 min oscillation extraction each. Magnetic COFs material was separated with the help of a magnet. All the elution solution was mixed and dried to 1 mL with a nitrogen blowing concentrator (Caliper Life Sciences Turbovap II, USA) at 50 °C, and then 50 μL aqueous ammonia was added and mixed for HPLC-MS/MS analysis.
2.8. Instrument conditions
AA metabolites in urine were determined with an Agilent 1200 HPLC instrument (USA) coupled with an AB SCIEX Triple QUAD 5500 mass spectrometer (AB Sciex Instruments, Germany). A Waters Symmetry Shield™ RP18 column (150 mm × 2.1 mm i.d., 3.5 μm) was chosen for HPLC analysis with the flow rate of 300 μL min−1 and the injection volume was 10 μL. The mobile phase A was water with 0.1% formic acid, while B was acetonitrile with 0.1% formic acid. The gradient elution program was as follows: 0–3.0 min, 100% A; 3.1–17.0 min, 78% A and 22% B; 17.1–22.0 min, 100% B; precondition: 5 min.Mass detection conditions were as follows: ionization mode, positive ESI; ion spray voltage, 5000 V; ion source temperature, 550 °C; curtain gas, nitrogen; setting: 30 psi; ion source gas 1 (GS1), setting: 70 psi; ion source gas 2 (GS2), setting: 70 psi. Two ion pairs were chosen as qualitative ion pairs and a quantitative ion pair was used with multiple reaction monitoring (MRM) mode. The parameters of MRM containing precursor ion, production ion, dwell time, collision energy (CE), declustering potential (DP) for four AAs and the internal standards were shown in Table 1.
| Compounds | Precursor ion (m/z) | Production Ion (m/z) | Dwell time (ms) | CE (eV) | DP (V) |
|---|---|---|---|---|---|
| a The quantitative ion pair. | |||||
| 1-NA | 144.0 | 127.0a/77.0 | 50 | 30 | 100 |
| 2-NA | 144.0 | 127.0a/77.0 | 50 | 30 | 100 |
| 3-ABP | 170.0 | 153.0a/127.0 | 50 | 30 | 100 |
| 4-ABP | 170.0 | 153.0a/127.0 | 50 | 30 | 100 |
| 1-NA-d7 | 151.0 | 132.0a/81.0 | 50 | 30 | 100 |
| 2-NA-d7 | 151.0 | 132.0a/81.0 | 50 | 30 | 100 |
| 3-ABP-d9 | 179.0 | 160.0a/136.0 | 50 | 30 | 100 |
| 4-ABP-d9 | 179.0 | 160.0a/136.0 | 50 | 30 | 100 |
3. Results and discussion
3.1. Fabrication and characterization of magnetic COFs material
The synthesis of COFs (TpPa-1) material was based on the principle of Schiff-base reaction. Before the synthesis of COFs material, the amino group of magnetic Fe3O4 nanoparticles was reacted with aldehyde group of Tp, which could promote the generation of COFs on the surface of magnetic nanoparticles. The synthesis scheme of magnetic COFs material was shown in Fig. 1. Nitrogen adsorption–desorption isotherm was used to characterize surface area and pore volume of the synthesized magnetic COFs material, which was shown in Fig. 2. The magnetic COFs material exhibited a typical type-I reversible isotherm characteristic of microporous property. However, a sharp rise of adsorption volume at higher relative pressure (P/P0 = 1) for N2 adsorption–desorption isotherm indicated that large pore formed by particle packing. The BET surface area, pore volume and average pore diameter was shown in Table 2. The influence of temperature, stirring speed, reaction time, reaction solvents and solvent volume on the surface area of the synthesized magnetic COFs materials was investigated in this work. And only reaction solvent volume influenced surface area of the obtained materials obviously. With the decrease of reaction solvent volume, surface area of the obtained materials increased. The magnetic COFs material with the reaction solvent volume of 20 mL showed the highest surface area, which was applied for MSPE of urinary AAs. And the BET surface area was 192.1 m3 g−1, pore volume was 0.18 cm3 g−1, and average pore diameter was 3.8 nm. As for magnetic NH2–Fe3O4 nanoparticles, N2 adsorption–desorption isotherm indicated that large pore existence formed by particle packing. After the information of COFs material on the surface of magnetic NH2–Fe3O4 nanoparticles, the obtained magnetic COFs material showed the increased surface area. What's more, the magnetic COFs materials were synthesized with the reaction solvent volume of 20 mL for three times in this work. The BET surface area was 189.3, 192.1, 200.2 m3 g−1, respectively. And their extraction capacity for AAs metabolite in urine was investigated and the results were comparable. Thus, the preparation reproducibility of the magnetic COFs nanoparticles was acceptable. And the prepared magnetic COFs nanoparticles of three times were mixed for the MSPE experiment in the work. | ||
| Fig. 1 Synthesis scheme of magnetic COFs (TpPa-1) material. | ||
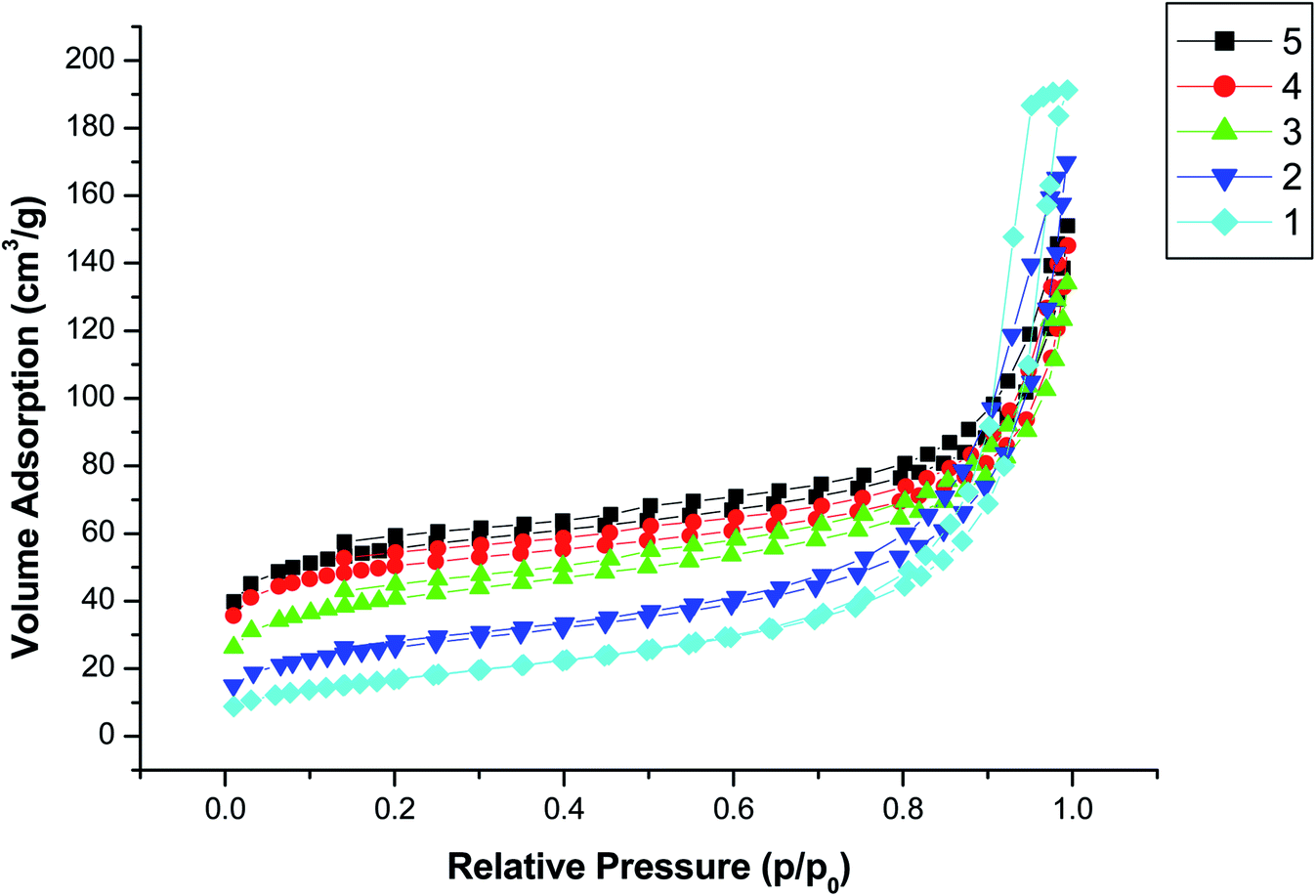 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of the synthesized materials. (1) Magnetic NH2–Fe3O4 material; (2–5) magnetic COFs materials (reaction solvent volume: 2–80 mL, 3–40 mL, 4–30 mL, 5–20 mL). | ||
| Magnetic materials | Solvent volume (mL) | BET surface area (m3 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|
| NH2–Fe3O4 | — | 62.3 | 0.29 | 15.6 |
| Magnetic COFs-2 | 80 | 94.3 | 0.26 | 8.3 |
| Magnetic COFs-3 | 40 | 143.0 | 0.18 | 4.4 |
| Magnetic COFs-4 | 30 | 174.1 | 0.18 | 3.9 |
| Magnetic COFs-5 | 20 | 192.1 | 0.18 | 3.8 |
Other structure analysis and characterization was performed for magnetic COFs material with the reaction solvent volume of 20 mL. HRTEM was applied to characterize morphologies of the synthesized magnetic materials as shown in Fig. 3. Magnetic NH2–Fe3O4 nanoparticles showed a uniform particle size of about 20 nm. The particle size of magnetic COFs materials (Fig. 3d) was larger than magnetic NH2–Fe3O4 nanoparticles (Fig. 3b) due to the formation of COFs on the surface of magnetic nanoparticles. And magnetic COFs material was a complex of COFs@ NH2–Fe3O4 nanoparticles and needle shaped COFs materials as shown in Fig. 3c.
 | ||
| Fig. 3 HRTEM image of synthesized magnetic materials ((a) and (b) magnetic NH2–Fe3O4 nanoparticles; (c) and (d) magnetic COFs material). | ||
FT-IR spectra showed chemical composition and structure information of magnetic nanomaterials. FT-IR spectra of COFs was shown as Fig. 4A. And the characteristic peak at 3500 cm−1 corresponding to –NH2 vibration, the adsorption bands at 1589 cm−1 was associated with carboxyl groups, and the peak at 1267 cm−1 was the characteristic adsorption related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, which indicated the CN vibration was formed for COFs materials. Compared with COFs material, the FI-IR spectra of magnetic COFs material (Fig. 4A) showed a new characteristic peak at 575 cm−1 which was corresponding to the Fe–O–Fe vibration, indicated that COFs material was warped on the magnetic NH2–Fe3O4 nanoparticles successfully. XRD spectra were shown in Fig. 4B, which gave the crystalline structure of the obtained magnetic COFs material. The diffraction peaks at 30.1°, 35.4°, 43.2°, 53.7°, 57.1° and 62.9° in the 2θ region of 30–40°, which can be assigned as (220), (311), (400), (422), (511) and (440) planes in the magnetic NH2–Fe3O4 nanoparticles.25 The diffraction peak at 0.6° in SXRD spectra might be related to the low crystallinity of magnetic COFs material. A magnetic hysteresis curve for magnetic NH2–Fe3O4 nanoparticles and magnetic COFs material were shown in Fig. 4C, and the saturation magnetic values were estimated to be 71.8 emu g−1 and 46.3 emu g−1, respectively. It was obviously, magnetic COFs material showed lower magnetic property than magnetic NH2–Fe3O4 nanoparticles owing to warping with COFs material. However, magnetic COFs material possessed great magnetic responsiveness and could be separated with external magnetic field. TGA curves were shown in Fig. 4D, which gave the mass ratios of COFs and Fe3O4 nanoparticles, and the thermal stability of magnetic COFs material. Magnetic NH2–Fe3O4 nanoparticles showed 4.7 wt% loss in the temperature range of 200–300 °C, which was attributed to the weight loss of the absorbed water, and the 5.2 wt% loss in the temperature range of 300–557 °C was due to the loss of amino functional groups on the surface. As for magnetic COFs material, the 6.1 wt% loss in the temperature between 200–430 °C was attributed to the weight of loss of the absorbed water, and 35 wt% loss in the range of 430–730 °C was due to the loss of COFs material. Obviously, the magnetic COFs material had excellent thermal stability under 430 °C.
N, which indicated the CN vibration was formed for COFs materials. Compared with COFs material, the FI-IR spectra of magnetic COFs material (Fig. 4A) showed a new characteristic peak at 575 cm−1 which was corresponding to the Fe–O–Fe vibration, indicated that COFs material was warped on the magnetic NH2–Fe3O4 nanoparticles successfully. XRD spectra were shown in Fig. 4B, which gave the crystalline structure of the obtained magnetic COFs material. The diffraction peaks at 30.1°, 35.4°, 43.2°, 53.7°, 57.1° and 62.9° in the 2θ region of 30–40°, which can be assigned as (220), (311), (400), (422), (511) and (440) planes in the magnetic NH2–Fe3O4 nanoparticles.25 The diffraction peak at 0.6° in SXRD spectra might be related to the low crystallinity of magnetic COFs material. A magnetic hysteresis curve for magnetic NH2–Fe3O4 nanoparticles and magnetic COFs material were shown in Fig. 4C, and the saturation magnetic values were estimated to be 71.8 emu g−1 and 46.3 emu g−1, respectively. It was obviously, magnetic COFs material showed lower magnetic property than magnetic NH2–Fe3O4 nanoparticles owing to warping with COFs material. However, magnetic COFs material possessed great magnetic responsiveness and could be separated with external magnetic field. TGA curves were shown in Fig. 4D, which gave the mass ratios of COFs and Fe3O4 nanoparticles, and the thermal stability of magnetic COFs material. Magnetic NH2–Fe3O4 nanoparticles showed 4.7 wt% loss in the temperature range of 200–300 °C, which was attributed to the weight loss of the absorbed water, and the 5.2 wt% loss in the temperature range of 300–557 °C was due to the loss of amino functional groups on the surface. As for magnetic COFs material, the 6.1 wt% loss in the temperature between 200–430 °C was attributed to the weight of loss of the absorbed water, and 35 wt% loss in the range of 430–730 °C was due to the loss of COFs material. Obviously, the magnetic COFs material had excellent thermal stability under 430 °C.
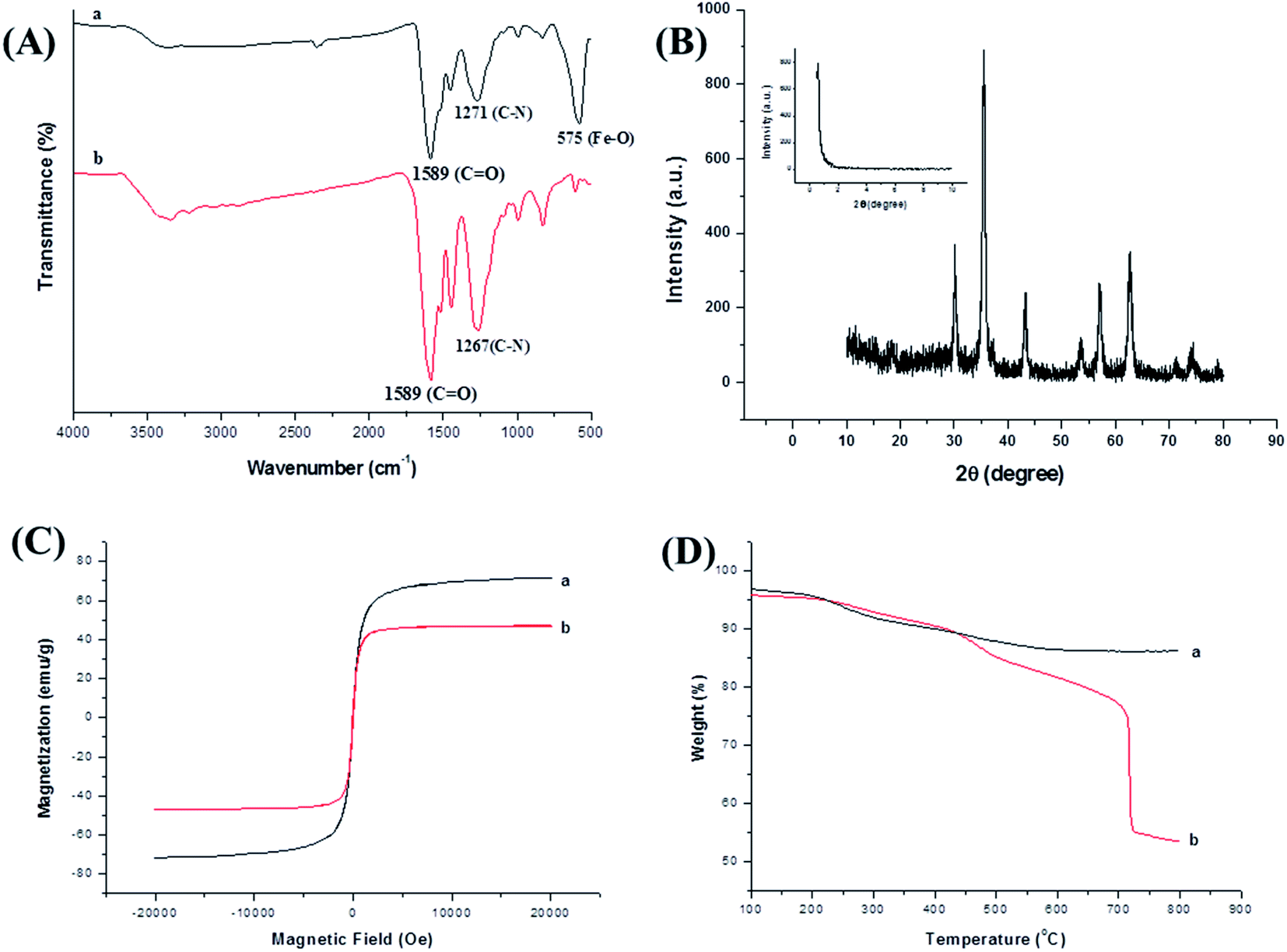 | ||
| Fig. 4 (A) FT-IR spectra of magnetic COFs (a) and COFs (b); (B) XRD spectra of magnetic COFs, and the inset was SXRD spectra; (C) hysteresis loops of magnetic COFs (a) and magnetic NH2–Fe3O4 nanoparticles (b); (D) TGA curves of magnetic NH2–Fe3O4 nanoparticles (a) and magnetic COFs (b). | ||
3.2. Optimization of MSPE conditions
Owing to the lower content of AA metabolites in urine sample, a mixed urine sample spiked with a certain amount of AAs standard solution was used to optimize MSPE conditions and characterize analytical performance of the developed method. To obtain the best extraction performance, some conditions of MSPE were optimized in this work, such as urine solution pH, NaCl amount, vortex extraction time, sorbent amount, different elution solvent and elution time.Firstly, sample solution pH was investigated. With the increase of sample solution pH, the extracted amount of urinary AAs increased, and the maximum was obtained at pH about 12.0 (Fig. 5A). The pKa of 4 AAs (1-NA, 2-NA, 3-ABP and 4-ABP) was 4.21 ± 0.10, 4.32 ± 0.10, 4.34 ± 0.10, 4.26 ± 0.10, respectively. Thus, they were almost completely molecular state when the solution pH of 12.0–13.0. What's more, the synthesized magnetic COFs (TpPa-1) material possessed imine groups in the framework, and showed molecular state in higher pH solution also. At higher pH solution (i.e. pH 12.0–13.0), AAs compounds could be easily adsorbed on the magnetic COFs materials due to π–π stacking. Thus, after acid hydrolysis, urine was adjusted to about pH 12.0–13.0 for MSPE in this work. To further improve extraction efficiency, a certain amount of NaCl was added into the treated urine for salting-out effect. As shown in Fig. 5B, with the increase of NaCl amount, the extracted amount of AAs enhanced until the addition amount of 2.0 g. When continuing to add NaCl, it could not be dissolved and a NaCl saturated solution was obtained. Thus, the amount of NaCl added was 2.0 g. Influence of vortex extraction time and magnetic absorbent amount used were studied also. As shown in Fig. 5C and D, the maximum extracted amount of AAs could be obtained after 20 min vortex extraction with 20 mg the synthesized magnetic COFs material.
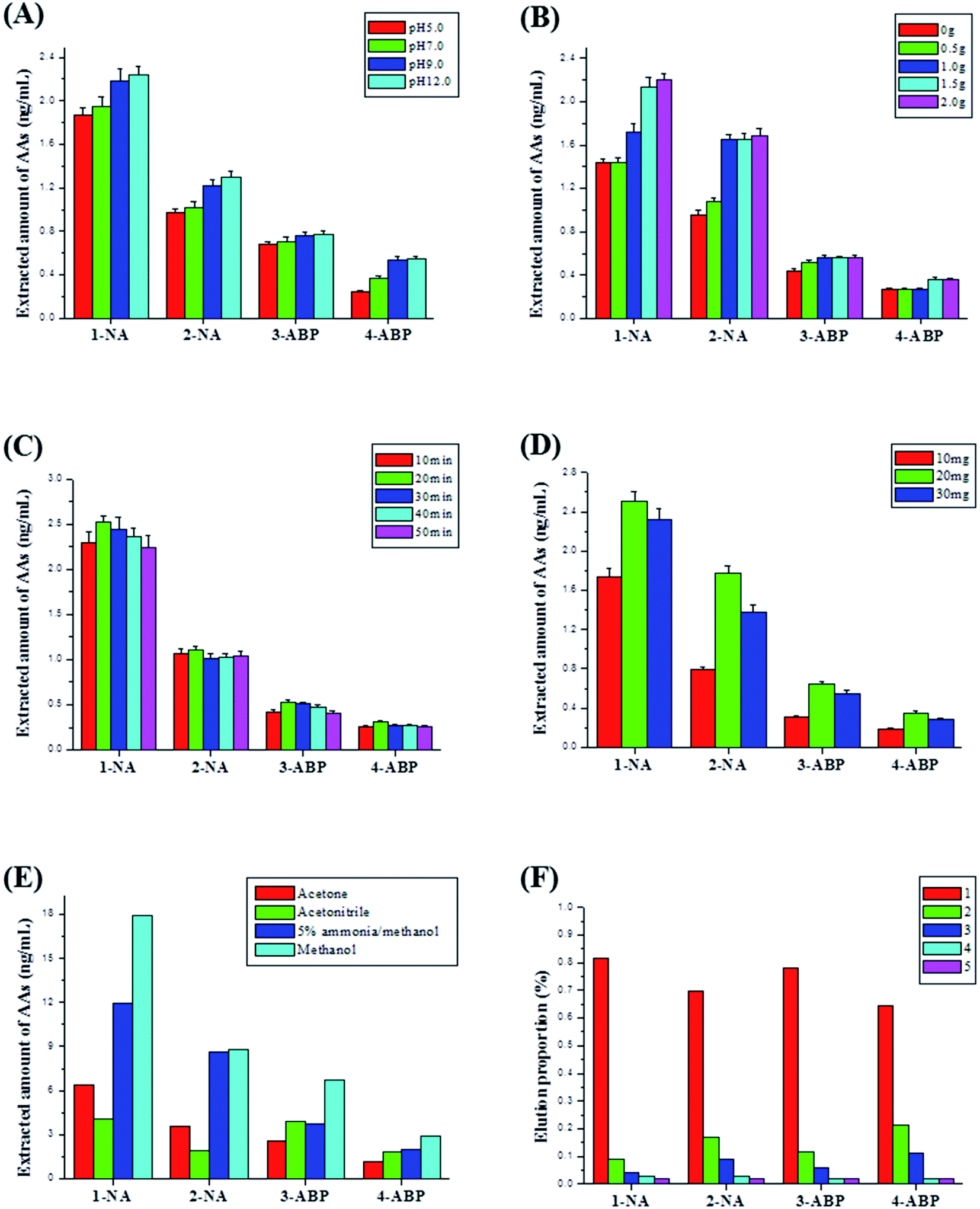 | ||
| Fig. 5 Effect of MSPE conditions on extraction performance of urinary 4 AAs: (A) sample solution pH, (B) NaCl amount, (C) vortex extraction time, (D) sorbent amount, (E) different elution solvent, (F) elution time. Data in (A)–(D) was average (±SD) after three repetition experiments. | ||
After vortex extraction, different solvent was used to elute, such as acetone, acetonitrile, 5% ammonia/methanol and methanol. As shown in Fig. 5E, the maximum extracted amount of AAs was obtained with methanol as elution solvent. The extracted amount of AAs after methanol elution was higher than that for 5% ammonia/methanol, which indicated there was mainly π–π stacking, not electrostatic interaction between AAs and magnetic COFs (TpPa-1) material. And as shown in Fig. 5F, after elution with 5 mL methanol, 80% 1-NA, 60% 2-NA, 70% 3-ABP and 60% 4-ABP was eluted from magnetic COFs. And after 3 times elution, over 95% of all AAs were eluted. Thus, 5 mL methanol each for three times was applied to elute AAs on magnetic COFs sorbent.
A spiked smoker urine sample was analyzed with HPLC-MS/MS technique after MSPE under the optimized conditions. As shown in Fig. 6, both 4 AAs and their corresponding isotope internal standard showed well-defined peaks, which could satisfy quantitative analysis.
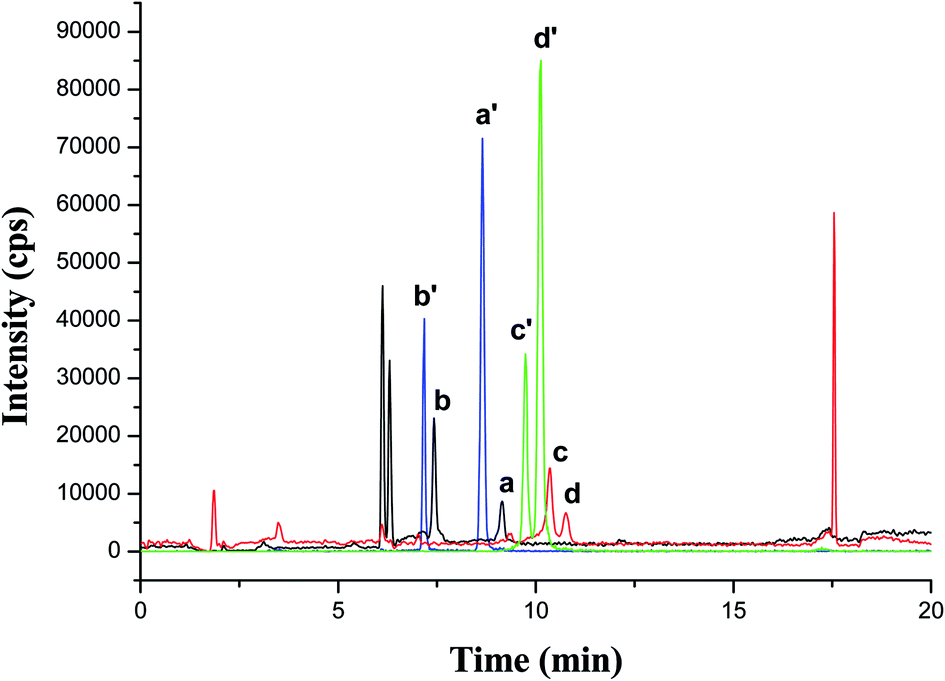 | ||
| Fig. 6 HPLC-MS/MS-MRM spectra of AAs and their corresponding internal standard in a spiked smoker urine sample. (a) 1-NA; (b) 2-NA; (c) 3-ABP; (d) 4-ABP; (a′) 1-NA-d7; (b′) 2-NA-d7; (c′) 3-ABP-d9; (d′) 4-ABP-d9. | ||
3.3. Analytical parameters
AAs standard working solution was prepared with 5% ammonia/methanol, which showed a well-defined chromatographic peak in HPLC-MS/MS, while standard working solution prepared with methanol gave a poor peak. A 5-point calibration curve was carried out by internal standard method. The concentration of 1-NA-d7, 2-NA-d7, 3-ABP-d9 and 4-ABP-d9 for each working solutions was 20 ng mL−1, 15 ng mL−1, 7 ng mL−1, 16 ng mL−1, respectively. Another calibration curve was prepared with urine matrix, which was calibrated by spiking a series of AAs standard solution in nonsmokers' urine extracts after MSPE based on magnetic COFs material. And the final concentration range was the same as that of calibration curve in solvent. The series of standard working solution prepared in solvent and urine matrix were analyzed using HPLC-MS/MS. And the linear concentration range of AAs and linear equation was shown in Table 3. According to the linear correlation coefficient (r), the linearity was good for calibration curves for both solvent and urine matrix (r > 0.99). What's more, as shown in Table 3, the slope ratios of two linear equations for four AAs are 1.00, 0.97, 1.01, 1.10, respectively. It indicated matrix effect was decreased significantly, which was attributed to that respective isotope internal standards of AAs were used, and urine sample was purified effectively with MSPE technique based on magnetic COFs material. Thus, the calibration curve in solvent could replace the calibration curve in urine matrix, and could be used for urine samples analysis.| Compounds | Linear concentration range (ng mL−1) | Calibration curve in solvent | Calibration curve in urine matrix | Slope ratio | LOD (ng mL−1) | LOQ (ng mL−1) |
|---|---|---|---|---|---|---|
| 1-NA | 0.5–25 | y = 0.102x − 0.014 (r = 0.9999) | y = 0.102x − 0.026 (r = 0.9996) | 1.00 | 0.07 | 0.22 |
| 2-NA | 0.25–12.5 | y = 0.259x − 0.0086 (r = 1) | y = 0.268x − 0.0080 (r = 0.9997) | 0.97 | 0.03 | 0.09 |
| 3-ABP | 0.10–5.0 | y = 0.276x + 0.00049 (r = 0.9998) | y = 0.274x + 0.051 (r = 0.9994) | 1.01 | 0.01 | 0.04 |
| 4-ABP | 0.05–2.5 | y = 0.0558x + 0.00036 (r = 0.9996) | y = 0.0507x + 0.035 (r = 0.9896) | 1.10 | 0.01 | 0.04 |
In addition, the lowest concentration standard working solution prepared in urine matrix was determined in HPLC-MS/MS for ten replicates. Limit of detection (LOD) and limit of quantification (LOQ) were obtained from three and ten times of the standard deviation of these determinations. As shown in Table 3, LODs and LOQs of four AAs ranged from 0.01 to 0.07 ng mL−1 and 0.04 to 0.22 ng mL−1, respectively. Compared with previous work, the LODs of this work were higher than those of four AAs metabolite in urine using a MIPs-SPE coupled with HPLC-MS/MS method in our previous work9 (i.e. 1.5–5 ng L−1). It was mainly because that the LODs in this work was conducted with the lowest concentration working solution prepared in urine matrix, but the LODs in ref. 9 was obtained with the lowest concentration working solution in solvent. However, the LOQs of AAs in this work were lower than the lowest concentration of calibration curve, which satisfied the requirement of quantitative analysis.
In order to assess the recovery, a smoker urine sample with spiking standard solution of AAs in three different concentration levels was analyzed. Each concentration level was performed with three replicate measurements. As is shown in Table 4, higher recoveries of 1-NA, 2-NA, 3-ABP, 4-ABP were obtained, which are in the range of 81.9–105%, 87.8–102%, 101–120%, 88.3–117%, respectively. Intra-day and inter-day precisions were investigated using a spiked urine sample (Table 4). The urine sample was analyzed 6 times on the same day to investigate the intra-day precision, and the RSDs were less than 5.8%. And the urine sample was analyzed on 5 different days to obtain the inter-day precision, and the RSDs were less than 9.9%.
| Compounds | Spiked level | Recovery (%) | Intra-day precision (n = 6, %) | Inter-day precision (n = 5, %) |
|---|---|---|---|---|
| 1-NA | Low | 81.9–95.2 | 4.5 | 9.8 |
| Mid | 100–105 | |||
| High | 95.7–101 | |||
| 2-NA | Low | 87.8–92.8 | 4.6 | 8.3 |
| Mid | 89.9–102 | |||
| High | 97.8–101 | |||
| 3-ABP | Low | 102–112 | 5.6 | 9.1 |
| Mid | 101–120 | |||
| High | 101–107 | |||
| 4-ABP | Low | 102–112 | 5.8 | 9.9 |
| Mid | 88.3–117 | |||
| High | 89.8–93.9 |
3.4. Recycle performance
The recycle performance of magnetic COFs material was investigated also. As shown in Fig. 7, the extracted amount of 4 AAs maintained over 90% after 5 times reuse. However, the extracted amount of AAs was less than 90% at 6 times reuse. Thus, the synthesized magnetic COFs material could be recycled as sorbent in MSPE process for at least 5 times.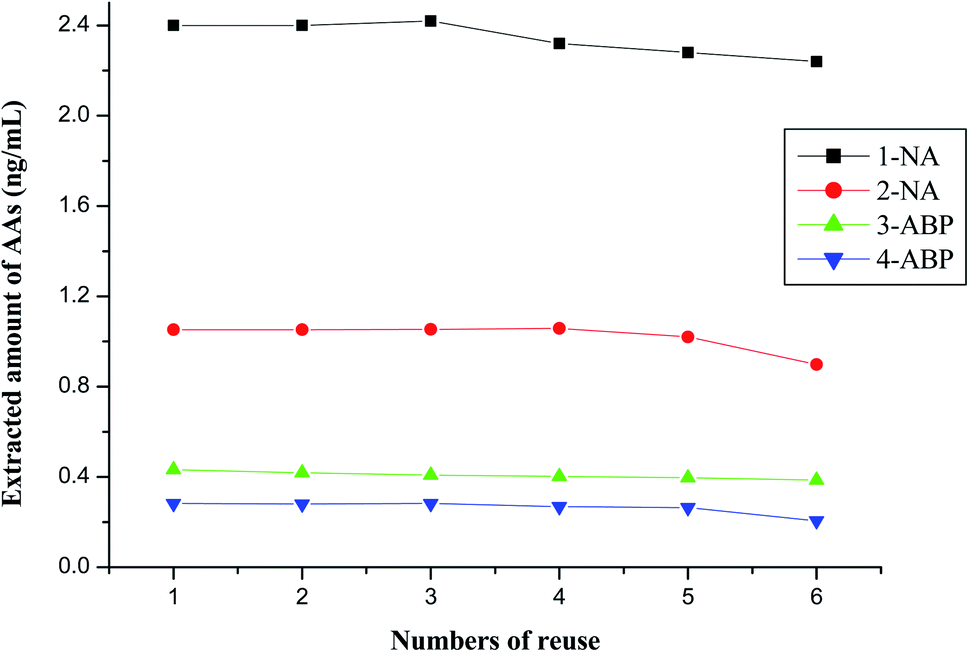 | ||
| Fig. 7 Extracted amount of AAs after several times reuse. | ||
Owing to the ultra-low content of AA metabolites in urine, a MIPs-SPE was chosen to purify urine for AA metabolites analysis,9 while a mixed type cation exchange SPE cartridge was suitable for purifying cigarette smoke sample13 nor urine sample9 for AAs analysis as shown in our previous work.9,13 Magnetic COFs material prepared in this work also could purify urine matrix and achieve the analysis of AA metabolites. However, compared with commercial SPE columns used in previous works which applied HPLC-MS/MS technique,9,13 the magnetic COFs material and MSPE showed some advantages. For example, the magnetic COFs material could recycle, which was more economical. And the sample pretreatment with MSPE technique was simple and practicable.
3.5. Human urine samples analysis
The developed method was applied to determine 4 AA metabolites in 27 urine samples collected from 20 smokers and 7 nonsmokers. And the result was calibrated with urinary creatinine and analysis of variance was applied for data processing. Table 5 and Fig. 8 showed the maximum, the minimum, the median, the average and variation range of 4 AA metabolites content in smoker and nonsmoker urine. It was obviously, all 4 AA metabolites content in smoker urine sample was higher than that for nonsmoker. And the P value between smoker and nonsmoker for 1-NA, 2-NA, 3-ABP and 4-ABP was 0.0009, 0.0006, 0.0008, and 0.0030 (i.e. p < 0.01), respectively, which indicated there was significant difference between the 4 AA metabolites content in smoker and nonsmoker urine. Thus, urinary AA metabolites could be as an exposure biomarker for cigarette smoke.| Compounds | AA metabolites content (pg mg−1 creatinine) | ||||
|---|---|---|---|---|---|
| Maximum | Minimum | Median | Average | ||
| Smoker | 1-NA | 492 | 9 | 169.73 | 189.60 |
| 2-NA | 1463 | 25 | 281.99 | 416.21 | |
| 3-ABP | 720 | 0 | 103.80 | 172.87 | |
| 4-ABP | 2184 | 14 | 264.82 | 419.27 | |
| Nonsmoker | 1-NA | 184 | 0 | 20.37 | 39.56 |
| 2-NA | 282 | 0 | 45.17 | 66.22 | |
| 3-ABP | 0 | 0 | 0 | 0 | |
| 4-ABP | 48 | 0 | 9.87 | 15.99 | |
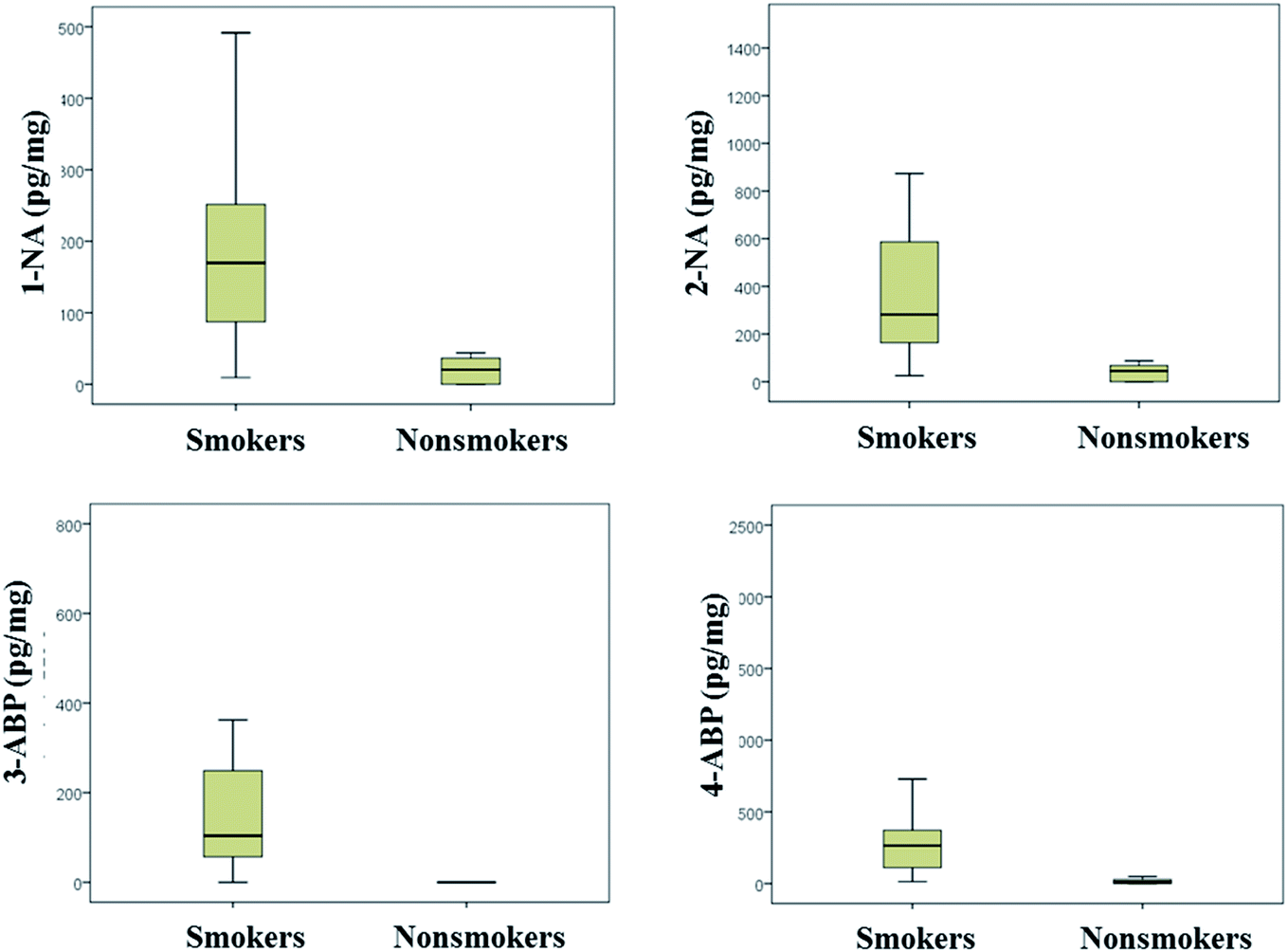 | ||
| Fig. 8 Average and variation range of AA metabolites in smoker and nonsmoker urine samples. | ||
4. Conclusion
A magnetic COFs (TpPa-1) material was synthesized and characterized in this work. The magnetic COFs material based MSPE technique was developed for the determination of AA metabolites (i.e. 1-NA, 2-NA, 3-ABP and 4-ABP) in urine with HPLC-MS/MS analysis. Under the optimum conditions of MSPE, the analytical performance of the developed method was characterized also. Calibration curves in solvent and urine matrix were conducted, and their slope was near. It indicated that matrix effect was decreased significantly attributing to the effective purification with MSPE based on magnetic COFs material. LODs and LOQs of four AAs ranged from 0.01 to 0.07 ng mL−1 and 0.04 to 0.22 ng mL−1, respectively. As for recovery, 1-NA, 2-NA, 3-ABP, 4-ABP were in the range of 81.9–105%, 87.8–102%, 101–120%, 88.3–117%, respectively. The intra-day and inter-day precisions were good also with RSDs of less than 5.8% and 9.9%, respectively. In addition, the synthesized magnetic COFs material could be recycled as sorbent in MSPE process for at least 5 times. The developed method was applied in the determination of four AAs in urine samples of smokers and nonsmokers. AA metabolites contents in smoker urine samples were higher than those for nonsmoker. And there were significant difference between smoker and nonsmoker urine (p < 0.01). Thus, urinary AA metabolites could be as an exposure biomarker for cigarette smoke.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from the National Natural Science Foundation of China (No. 31901768).References
- Food and Drug Administration, Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke; Established List, Docket No. FDA-2012-N-0143 Search PubMed.
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, IARC, 2013, http://www.monographs.iarc.fr/ENG/Classification/index.php, accessed 30 July 2013 Search PubMed.
- IARC Monographs on the evaluation of the carcinogenic risk of chemicals to humans. Chemicals industrial processes and industries associated with cancer in humans, Lyon, 1982 Search PubMed.
- G. Grimmer, G. Dettbarn, A. Seidel and J. Jacob, Sci. Total Environ., 2000, 247, 81–90 CrossRef CAS PubMed.
- K. Riedel, G. Scherer, J. Engl, H. W. Hagedorn and A. R. Tricker, J. Anal. Toxicol., 2006, 30, 187–195 CrossRef CAS PubMed.
- P. Kämpfer, S. Crettaz, S. Nussbaumer, M. Scherer, S. Krepich and O. Deflorin, J. Chromatogr. A, 2019, 1592, 71–81 CrossRef PubMed.
- B. J. Brüschweiler and C. Merlot, Regul. Toxicol. Pharmacol., 2017, 88, 214–226 CrossRef PubMed.
- T. Carreon, M. J. Hein, K. W. Hanley, S. M. Viet and A. M. Ruder, Occup. Environ. Med., 2014, 71, 175–182 CrossRef PubMed.
- J. J. Yu, S. Wang, G. Zhao, B. Wang, L. Ding, X. B. Zhang, J. P. Xie and F. W. Xie, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 958, 130–135 CrossRef CAS PubMed.
- J. J. Niu, X. L. Zhao, Y. Y. Jin, G. T. Yang, Z. H. Li, J. J. Wang, R. Q. Zhao and Z. Y. Li, J. Chromatogr. A, 2018, 1555, 37–44 CrossRef CAS PubMed.
- S. Mazumder, R. A. Ahamed, E. McGahee, L. Q. Wang and T. H. Seyler, J. Anal. Toxicol., 2019, 43, 25–35 CrossRef CAS PubMed.
- S. Saha, R. Mistri and B. C. Ray, J. Chromatogr. A, 2009, 1216, 3059–3063 CrossRef CAS PubMed.
- F. W. Xie, J. J. Yu, S. Wang, G. Zhao, Q. L. Xia, X. B. Zhang and S. S. Zhang, Talanta, 2013, 115, 435–441 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef CAS.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu and K. Suenaga, Science, 2016, 351, 365–369 CrossRef CAS PubMed.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 121, 5547–5550 CrossRef.
- R. Wang and Z. L. Chen, Microchim. Acta, 2017, 184, 3867–3874 CrossRef CAS.
- L. Chen, Y. T. He, Z. X. Lei, C. L. Gao, Q. Xie, P. Tong and Z. Lin, Talanta, 2018, 181, 296–304 CrossRef CAS PubMed.
- S. J. He, T. Zeng, S. H. Wang, H. Y. Niu and Y. Q. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 2959–2965 CrossRef CAS PubMed.
- Y. H. Yan, Y. J. Lu, B. C. Wang, Y. Q. Gao, L. L. Zhao, H. Z. Liang and D. P. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 26539–26545 CrossRef CAS PubMed.
- C. H. Gao, G. Lin, Z. X. Lei, Q. Zheng, J. S. Lin and Z. Lin, J. Mater. Chem. B, 2017, 5, 7496–7503 RSC.
- W. F. Zhang, Y. H. Zhang, G. R. Zhang, J. Y. Liu, W. D. Zhao, W. J. Zhang, K. Hu, F. W. Xie and S. S. Zhang, Analyst, 2019, 144, 5829–5841 RSC.
- W. F. Zhang, C. Lan, H. M. Zhang, Y. H. Zhang, W. J. Zhang, W. D. Zhao, C. Johnson, K. Hu, F. W. Xie and S. S. Zhang, J. Agric. Food Chem., 2019, 67, 3733–3743 CrossRef CAS PubMed.
- Y. H. Zhang, Y. Y. Song, J. Wu, R. J. Li, D. Hu, Z. A. Lin and Z. W. Cai, Chem. Commun., 2019, 55, 3745–3748 RSC.
- L. Y. Wang, J. Bao, L. Wang, F. Zhang and Y. D. Li, Chem.–Eur. J., 2006, 12, 6341–6347 CrossRef CAS PubMed.
- J. Tan, S. Namuangruk, W. F. Kong, N. Kungwan, J. Guo and C. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 1–7 CrossRef.
- S. J. He, T. Zeng, S. H. Wang, H. Y. Niu and Y. Q. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 2959–2965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |