
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot construction of carbohydrate scaffolds mediated by metal catalysts
Mana Mohan Mukherjee
 a,
Sajal Kumar Maity
b and
Rina Ghosh
*b
a,
Sajal Kumar Maity
b and
Rina Ghosh
*b
aLaboratory of Cell and Molecular Biology, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA
bDepartment of Chemistry, Jadavpur University, Kolkata 700032, India. E-mail: ghoshrina@yahoo.com; rina.ghosh@jadavpuruniversity.in
First published on 2nd September 2020
Abstract
Owing to the environmental concern worldwide and also due to cost, time and labour issues, use of one-pot reactions [domino/cascade/tandem/multi-component (MC) or sequential] has gained much attention among the scientific and industrial communities for the generation of compound libraries having different scaffolds. Inclusion of sugars in such compounds is expected to increase the pharmacological efficacy because of the possibility of better interactions with the receptors of such unnatural glycoconjugates. In many of the one-pot transformations, the presence of a metal salt/complex can improve the reaction/change the course of reaction with remarkable increase in chemo-/regio-/stereo-selectivity. On the other hand because of the importance of natural polymeric glycoconjugates in life processes, the development and efficient synthesis of related oligosaccharides, particularly utilising one-pot MC-glycosylation techniques are necessary. The present review is an endeavour to discuss one-pot transformations involving carbohydrates catalysed by a metal salt/complex.
 Mana Mohan Mukherjee | Dr Mana Mohan Mukherjee received his BSc degree from Krishnath College, India, and his MSc degree from University of Kalyani, India. He received his PhD degree on synthetic methodology development in carbohydrate chemistry and oligosaccharide synthesis from Jadavpur University, India under the supervision of Professor Rina Ghosh. He was then a National Post-doctoral Fellow at the Indian Association for the Cultivation of Science, Kolkata, India, associated with Professor Subrata Ghosh. After post doctoral research in Dr Paul Kovac's group at the Section on Carbohydrates, Laboratory of Bioorganic Chemistry, NIDDK, National Institutes of Health, Bethesda, where his work was on the development of synthetic glycoconjugate vaccine candidates against cholera, he is currently working in the same institute as a visiting fellow under Dr J. A. Hanover in the Laboratory of Cell and Molecular Biology. |
 Sajal Kumar Maity | Dr Sajal Kumar Maity was born in Howrah, India. He received his MSc from Calcutta University in 2004. He received his PhD on oligosaccharide synthesis from Jadavpur University, India under the supervision of Professor Rina Ghosh. He was a Dr D. S. Kothari postdoctoral fellow at Pune University, India. Then, he worked as a postdoctoral fellow in the Bio-organic unit at the University of Turku in the research group of Dr Tuomas Lönnberg, where his topic of research was covalently metallated oligonucleotide synthesis and studies of their photo-physical properties. Recently, he has returned to India and at present he is an associated honorary researcher with Professor Rina Ghosh at Jadavpur University pursuing research work. |
 Rina Ghosh | Dr Rina Ghosh is a Professor in the Department of Chemistry at Jadavpur University, Kolkata, India. She got her BSc and MSc degrees from Jadavpur University. After her PhD from the same University in the Carbohydrate field in 1986 under the late Professor Amalendu Das, followed by her two years post-doctoral research with Professor Nirmolendu Roy at the Indian Association for the Cultivation of Science, Kolkata, India on Synthetic carbohydrates, she continued her research career as a CSIR Pool officer at Jadavpur University. She joined as Lecturer in Chemistry at Bolpur College, West Bengal in 1992. She then moved to Tripura University as a faculty of Chemistry, in 1995, from where she came back to Jadavpur University, Kolkata in 1998, to join as a faculty of Chemistry. Her current research interest includes development of one-pot methods of protection–deprotection of carbohydrates and glycosylation reactions, synthesis of rare sugars, synthesis of bacteria related complex oligosaccharides utilising one-pot glycosylation reactions and studies of sugar based new surfactants. |
1. Introduction
At each and every moment, Nature is creating and/or regenerating chemical and biochemicals from its own feedstock with the help of metal catalysts and/or enzymes and coenzymes. In many of the corresponding biogenetic processes, cascade/domino one-pot reactions are involved to generate several new bonds in a process, and the reactions occur with exclusive specificity in terms of regio- and stereo-structures and sometimes even generating complex structures from simple starting renewable substrates. Nature takes care of its own necessary energy demanding cycles. The synthesis of organic compounds mostly uses multi-step reactions which involve extraction, purification (chromatographic or others) and characterisation of the product/s of each step. The demand for design and synthesis of such organic molecules in one-pot towards the generation of a library of compounds having different scaffolds, is gradually increasing in order to make the syntheses environmentally, economically and in terms of time, more viable. In several cases conversion of some of the multistep syntheses to the corresponding one-pot version is feasible, provided the reagents, solvents and conditions used in multistep processes are compatible with one-another and also with the products formed at subsequent steps. Learning from Nature's class-room, translating Nature's laboratory procedure into in vitro experiments and sometimes utilising Mother Nature's chiral feedstock, it has now been possible to create biologically potent chiral compound-libraries in one-pot, minimising cost, energy, labour and waste and thus addressing some of the green aspects relating to the environment.One-pot domino/cascade/tandem/multicomponent (MC) reactions are considered to be the most important synthetic tools for construction of structurally and regio- or stereo-chemically diverse molecules for generation of compound libraries. All these are greener approaches compared to their corresponding multistep versions, as all of these occur with high atom economy saving time, labour, money, energy and environment with minimisation of waste production. Several reviews have been published in the literature covering such one-pot reactions.1 In many of these one-pot reactions, metal salts or metal complexes play immense importance in formation of typical bond and help to carry out the transformations in highly chemo-, regio- and stereo-selective manner. Generally domino/cascade and tandem processes occur in one-pot maintaining same condition throughout the reaction. Such reactions involve usually mild reaction condition and proceed with formation of new other bonds between C–C or C–heteroatom with multiple consequential transformations on new functionalities, formed during the successive reactions, and sometimes is associated with rearrangement where the intermediates are not isolable. MC reactions are convergent chemical processes where more than two components react, the components along with the reagents are usually taken in the same pot at the beginning, but one/two component/s may also be added with some additional reagents with change in reaction condition sequentially during the reaction in the same pot. In MC reactions intermediates may be isolated.
Application of tandem/domino/cascade/sequential-/MC-one-pot reactions in carbohydrate field started its journey much later compared to those applied in other organic syntheses. Because of possibility of better interaction with the receptor biomolecules, tailor-made unnatural small glycoconjugates and sugar mimics are regarded as important targets with improved biological and pharmacological properties, and syntheses involving carbohydrates are gradually gaining the attention of researchers.2 Sugars like monosaccharides are important chiral inexpensive feedstock toward their utilisation in native form or in the form of their suitable derivative in the synthesis of medicinally and industrially potential chiral organic molecules.3 On the other hand, carbohydrates, particularly natural glycoconjugates involving biopolymers play immense importance in several life processes like cell–cell interaction, cell proliferation, transport mechanism, bacterial and viral infection and even in cancer metastasis.4 Instead of increasing the number of unplanned synthetic molecules toward drug development, nowadays there has now been creation of new combinatorial libraries which are designed based on natural product scaffolds.5
In the present comprehensive review we have concentrated our dissertation on one-pot reactions (tandem/domino/cascade/sequential/MC, even including those which involve 2/3/4 separate reactions occurring sequentially in one-pot) which are in some-way or other, metal catalysed and applied to carbohydrate field. Although L. F. Tietze suggested in a review on domino reactions1a not to use the term tandem, as according to his logic “Tandem means two at the same time and does not describe a time-resolved transformation”, but in this manuscript, in a few particular one-pot reactions we have used the terminology as were termed in the original papers. Our discussion will be focused mainly on the one-pot regio-/stereo-selective protection–deprotection of monosaccharides or on their suitable derivatives, on one-pot synthesis of sugar derived compounds and unnatural glycoconjugates, and also on the synthesis of oligosaccharides related to natural glycoconjugates utilising sequential one-pot glycosylations. Development of unnatural glycoconjugates has gained special interest of academic as well as industrial communities, as many of such compounds reveal themselves to be glycosidase or glycosyltransferase inhibitors. Use of inhibitors is now considered as one of the important avenues for disease management; several of such glycoconjugates already are or supposed to be considered as established drugs to treat diseases like bacterial and viral infections, metabolic diseases and even cancer.4b
A review on ‘One-pot construction of carbohydrate scaffolds mediated by metal catalysts’ is supposed to remain incomplete without inclusion of researches on metal catalysed one-pot conversion of carbohydrate biomass to other simple industrially important chemicals or bio-fuels. That the latter topic has gained tremendous attention during the past several years is reflected in the plethora of research publications in this direction. During the last few years a good number of reviews6 including several reviews in 2019,6h–s and one in 2020 (ref. 6t) have also been published on the above field. For this obvious reason we have excluded this topic from our discussion. We have also excluded the domino/tandem one-pot syntheses involving click reaction leading to macrocycles or oligosaccharide mimics,7 as those can be considered for a separate review. Although, for literature search we have depended on different online search engines with relevant keywords, but still in spite of our sincere effort a few publications may be missed particularly where this review-related one-pot reaction may have been employed as part of a total synthesis and published under different keywords.
2. One-pot protection–deprotection of sugars
Being polyhydroxylated compounds, synthetic advancement of carbohydrate molecules demands extensive functional group transformation/modification by means of suitable protection–deprotection of hydroxy groups. In a one-pot version, appropriate conditions are to be chosen and maintained throughout the reaction as much as possible. For management of protecting groups and anomeric leaving groups toward preparation of glycosyl donor and acceptor building blocks necessary for oligosaccharide synthesis, use of one-pot reactions are gradually increasing in place of some of their corresponding multistep approaches, and in the recent years with increase of sophistication also. One-pot synthesis of different glycosyl donor and acceptor building units in terms of regioselectivity and protecting groups, may however, need to be considered separately in some instances on case to case basis.2.1. Sequential protection of free sugar for synthesis of acetylated O-/S-glycosides
Per-O-acetylation of the free sugars is considered as the most commonly utilised protection profile adopted for further functional group manipulation, as these per-O-acetylated sugars can be easily transformed toward their corresponding S-/O-glycosides mediated by suitable Lewis acids. In the evolution of catalysts for per-O-acetylation reaction, there comes some Lewis acids that can in situ activate the anomeric acetate group toward further S-/O-glycosylation reaction, or that can be used in combination with traditionally employed acetate activation reagent BF3·Et2O for the same purpose.The first example of simple one-pot approaches in the two-step preparation of per-O-acetylated 1,2-trans thioglycosides (e.g., 3a/3b or 3c) from native monosaccharides (e.g., 1a/1b or 1c) via intermediate formation of the corresponding per O-acetylated sugars (2a/2b or 2c), was reported in 2003 ((A) in Scheme 1) by Hung's research group.8 In the seminal report, per-O-acetylation was carried out under neat condition using stoichiometric amount of Ac2O, and catalytic amount of Lewis acid, such as Cu(OTf)2, followed by treatment with BF3·Et2O, and the p-thiocresol as glycosyl acceptor, which resulted in the formation of the 1,2-trans thioglycosides in 66–75% overall yield. This was followed by a similar report by Dasgupta et al. in 2007 based on La(OTf)3 catalyst.9 The latter group achieved La(OTf)3 catalysed efficient one-pot conversion of free sugars under neat condition by sequential one-pot per-O-acetylation based on Ac2O followed by tandem O-/S-glycosylation in the presence of BF3·Et2O and 2-bromoethanol/p-methoxyphenol or p-cresol as the nucleophile.
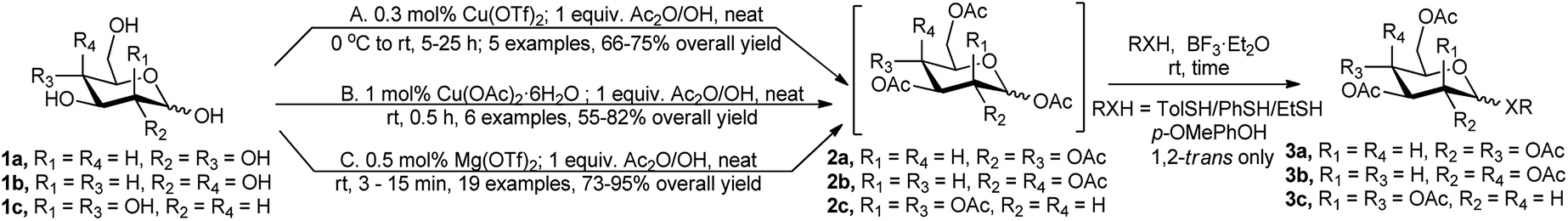 | ||
| Scheme 1 One-pot per-O-acetylation–thioglycosidation reaction. | ||
Another Cu(II) salt, viz Cu(OAc)2·6H2O was used by Chatterjee et al. for the same purpose in 2015.10 The principle for the reaction sequences is the same as the previous one; here they used 1 mol% Cu(OAc)2·6H2O as catalyst for per-O-acetylation step followed by thioglycosidation with p-thiocresol using 2 equivalents of BF3·Et2O ((B) in Scheme 1).
In a broader case study for this tandem sequence Mukherjee et al. used 0.5 mol% of Mg(OTf)2 as recyclable and robust catalyst for per-O-acetylation of unprotected mono and disaccharides, and subsequent one-pot transformation to the corresponding O-/S-glycosides, in reaction with various thiols and p-methoxyphenol, in the presence of 1.2 equivalents of BF3·Et2O ((C) in Scheme 1).11 The corresponding S-/O-glycosides were isolated in excellent yields and exclusive 1,2-trans selectivity without any sign of anomerisation. They also reported the recyclability of the catalyst used for further reactions. After work-up, the catalyst was recovered from the aqueous phase by evaporation, followed by drying overnight over P2O5. The recycle protocol was repeated five times, the percentage of the catalyst recovered was always more than 90% while the yields of phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside and phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-galactopyranoside were always more than 93% and 90%, respectively.
Recently in a different approach Yan et al. have reported Dy(OTf)3 as recyclable catalyst for per-O-acetylation of free sugars followed by a semi-one-pot sequential conversion into their corresponding glycosyl hemiacetals (Scheme 2).12 They utilised 0.1 mol% of the catalyst under neat condition for per-O-acetylation step followed by additional use of 4.9 mol% of the same catalyst in methanol at 40 °C generating the glycosyl hemiacetals (4a/4b or 4c) within several hours in moderate to good yields.
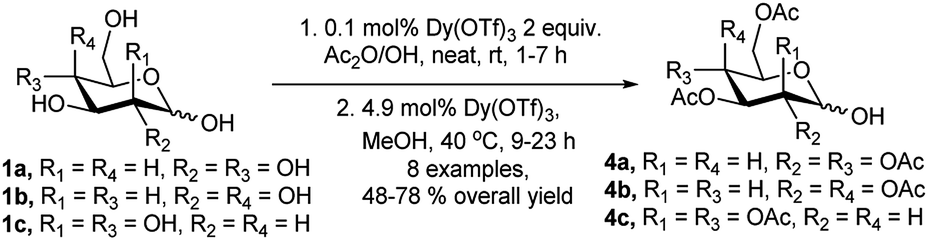 | ||
| Scheme 2 Dy(OTf)3-catalysed conversion of free sugars to per-O-acetylated hemiacetals. | ||
In 2006, Weng et al. published a very interesting report where MoO2Cl2 was employed as the sole catalyst for a one-pot four-step reaction. D-Glucose (1a) was acetylated using 3 mol% of the afore-mentioned catalyst, to form the per-O-acetylated glucose (2a) under neat condition, followed by one-pot thioglycosidation with p-thiocresol in dichloromethane (DCM) using 3 mol% of the foresaid catalyst generating the thioglycoside 5a, then subsequent deacetylation again after addition of 1 mol% of the catalyst in hot EtOH, and at last 4,6-O-benzylidenation reaction using 5 mol% of the catalyst afforded the final product 5 in 70–75% yield without isolation of any intermediate (Scheme 3).13
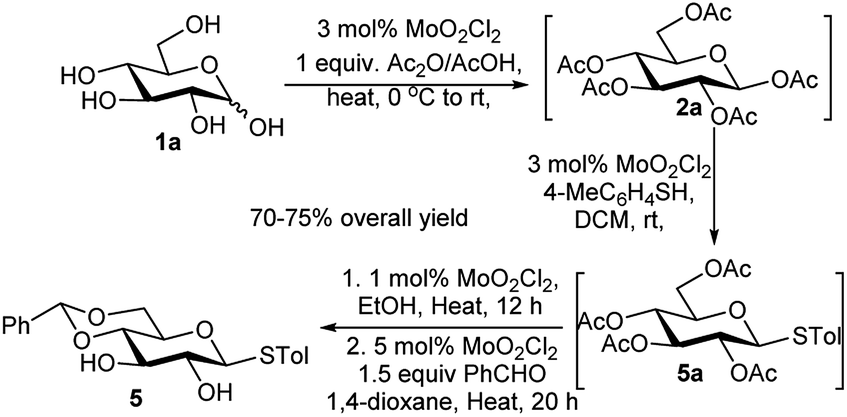 | ||
| Scheme 3 MoO2Cl2-catalysed one-pot multistep reaction. | ||
2.2. Sequential arylidenation–acetylation of S-/O-glycosides
Protection of 4,6-diols as 4,6-O-benzylidene is of immense importance in the synthesis of complex carbohydrates14a,b as these can be selectively transformed into the corresponding 6-deoxy sugars under oxidative condition.14c–f These benzylidene acetals can either be selectively transformed, under reductive conditions14g–i to their corresponding 4-O-benzylated or 6-O-benzylated analogue leaving the alternate hydroxy group free to be utilised further as glycosyl acceptor during oligosaccharide synthesis or to produce the corresponding 4,6-diol.14j–lGhosh et al. observed that although catalytic FeCl3 (20 mol%) could benzylidinate monosaccharide-based glycosides but 1.2 equivalents of FeCl3 was necessary along with 1.5 equivalents of benzaldehyde dimethylacetal and 4 Å MS in dry MeCN for 4′,6′-O-benzylidene formation of the corresponding disaccharide glycosides of lactose, maltose or cellobiose (e.g., 6); this followed by acetylation of the remaining hydroxy groups in the same pot using excess pyridine and Ac2O, furnished completely acetylated-4′,6′-O-acetalated glycosides (e.g., 7) in 61–84% overall yield ((A) in Scheme 4).15 Later the same group used 0.1 equivalent of Mg(OTf)2 as a single catalyst for 4,6- or 4′,6′-O-benzylidene formation of mono and disaccharide glycosides followed by acetylation of the remaining free hydroxy groups in the same pot ((B) in Scheme 4) in comparable overall yield, 73–84%.11
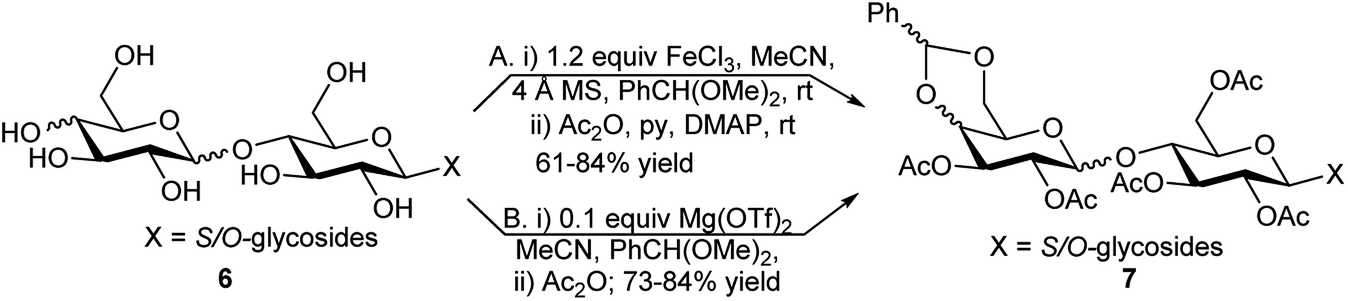 | ||
| Scheme 4 One-pot acetalation–acetylation reaction of disaccharide glycosides. | ||
Galan and co-workers employed Cu(OTf)2 as catalyst for one-pot synthesis of diverse sugar derivatives. The one-pot procedure involving 4,6-O-benzylidene ring formation was extended to traditional acetylation and finally in situ reductive benzylidene ring opening. When phenyl 1-thio glucoside (8a), or galactoside (8b) and NHTroc protected glucosamine substrate (8c) was subjected to one-pot 4,6-O-benzylidenation–acetylation reaction, the corresponding fully protected products 9a and 9b were obtained in 75%, and 9c in 76% yields (Scheme 5). Further regioselective reductive opening of the 4,6-O-benzylidene ring using Et3SiH as one-pot three-step reaction sequence resulted in their corresponding 4-hydroxy, 6-O-benzyl derivatives 10a in 60% yield and 10c and 10b in 50% yield (Scheme 5).16
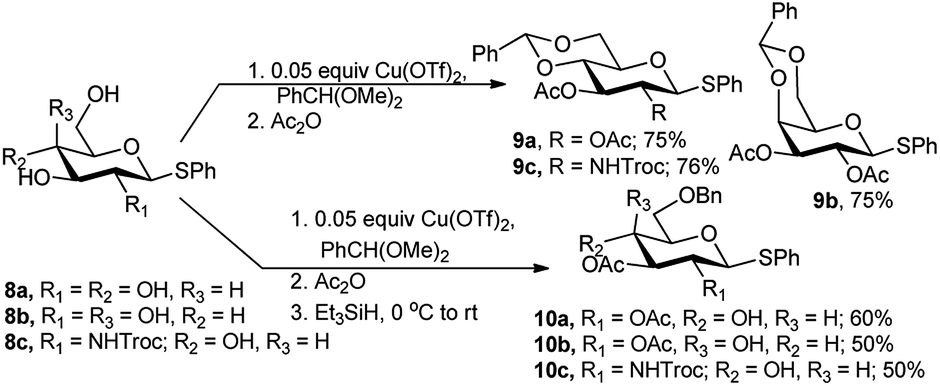 | ||
| Scheme 5 Cu(OTf)2-catalysed tandem acetalation/acetylation/reductive opening of acetal ring. | ||
2.3. Regioselective sequential protection of TMS-derived sugars involving acetalation
Regioselective sequential protection of TMS-derived sugars was first utilised by Hung and co-workers.17 TMS-protected sugars were chosen over normal free sugars to overcome the solubility barrier of native sugars. They used NEt3 and TMSCl for silylation of free sugar and then used catalytic amount of TMSOTf for large number of versatile one-pot transformation of carbohydrates involving acetalation reaction as the backbone reaction followed by several other one-pot reactions toward multiple directions. After this first report of such TMSOTf catalysed regioselective transformations, a plethora of reports were published thereafter.18 We are not going into the details of these type of reactions as “Si” is a metalloid. Inspired by these encouraging reports on TMSOTf-catalysed reactions, Beau and co-workers investigated metal catalysis for similar purposes. They reported a tandem process of benzylidenation – regioselective reductive etherification–acylation, catalysed initially by Cu(OTf)2,19a and in another case by FeCl3·6H2O,19b as a key step of orthogonally protected sugar derivatives. Scheme 6 illustrates the approach, with per-O-trimethylsilylated thioglucoside 11. In the presence of Cu(OTf)2 or FeCl3·6H2O, the per-O-silylated glucosides (11) underwent a sequential reaction with benzaldehyde and Et3SiH, providing 4,6-O-benzylidene-3-O-benzyl derivatives in situ, and subsequent reaction with Ac2O finally provided the 2-O-acetyl-4,6-O-benzylidene-3-O-benzyl glucopyranosides 12 in moderate to good yield. Moreover, after in situ generation of 4,6-O-benzylidene-3-O-benzyl free C-2-hydroxy intermediate 11a, followed by reductive opening of the corresponding 4,6-O-benzylidene acetal at O-4 or O-6 produced the respective 2,4-diol 13 or 2,6-diol 14 in good yields as well. Whereas, the O-6 ring opening using BH3·THF was not compatible with FeCl3·6H2O but well-suited with Cu(OTf)2.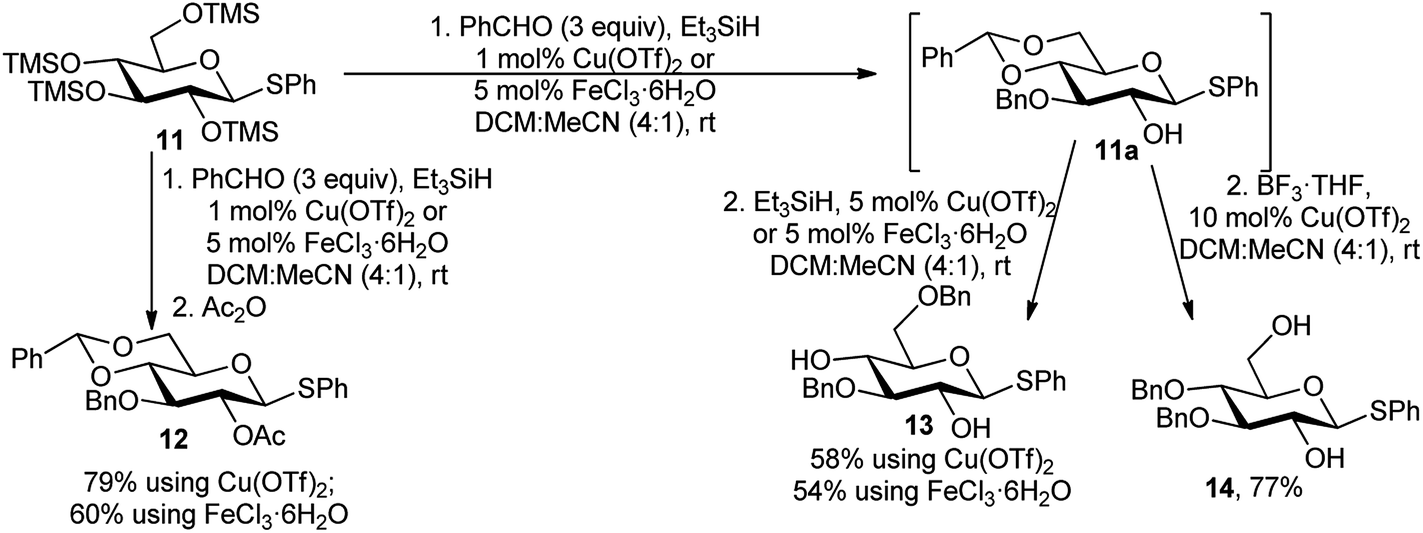 | ||
| Scheme 6 One-pot regioselective protection of TMS-derived glucose towards glycosyl donor (12) and acceptors (13 and 14). | ||
The proposed reaction mechanism for the regioselectivity of O-3 benzylation is that at room temperature, a 4,6-O- and 2,3-O-dibenzylidene intermediate could be generated, and then after regioselective reductive ring opening of the less stable 2,3-O-dibenzylidene acetal, the O-3 benzylated compound 11a was formed exclusively, non-upsetting the more stable 4,6-O-benzylidene ring (Scheme 7). While anhydrous FeCl3 is as effective as the aforesaid catalysts, and other iron salts such as Fe(acac)3, (FeCl3)2(TMEDA)3, Fe(NO3)3·9H2O, or FeCl2·4H2O were either inefficient or furnish lower yields.19b
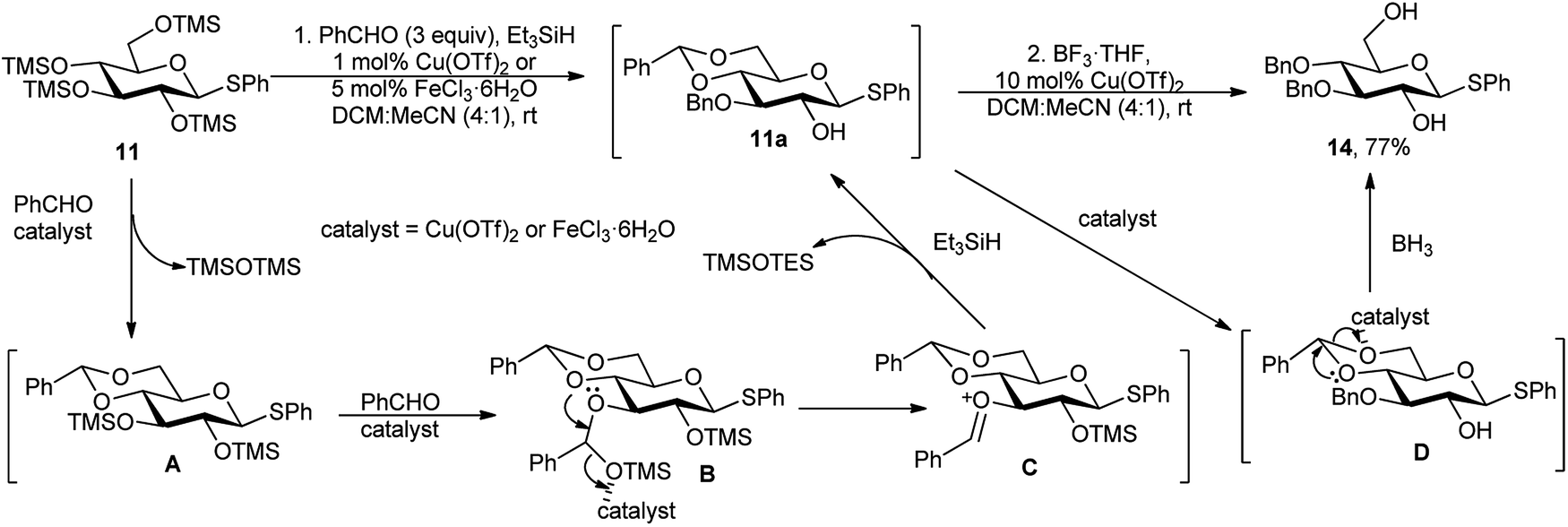 | ||
| Scheme 7 Probable mechanism for one-pot protection sequence of TMS derived sugar. | ||
Later, J.-M. Beau's group also demonstrated the catalytic efficacy of FeCl3·6H2O in one-pot synthesis of several disaccharide derivatives.19b,c Thus, one-pot reaction of C-2-symmetric per-O-silylated-α,α-D-trehalose 15 with 6 equivalents of benzaldehyde, 2.2 equivalents of Et3SiH, and 5 mol% of FeCl3·6H2O provided the symmetric benzylated compound 16, in 61% isolable yield (Scheme 8). This procedure could also be prolonged to a three-step process to obtain totally protected disaccharide 17 (Scheme 8). Thus, adding an excess of Ac2O (10 equivalents) and 5 mol% of FeCl3·6H2O furnished the expected 2,2′-di-O-acetylated-α,α-D-trehalose 17 in 41% yield. The shortcoming of this method was perceived with the one-pot bis-reductive benzylidene ring opening using 10 equivalents of Et3SiH and 15 mol% of the catalyst, which afforded the expected compound 18 in only a moderate overall yield of 28%.
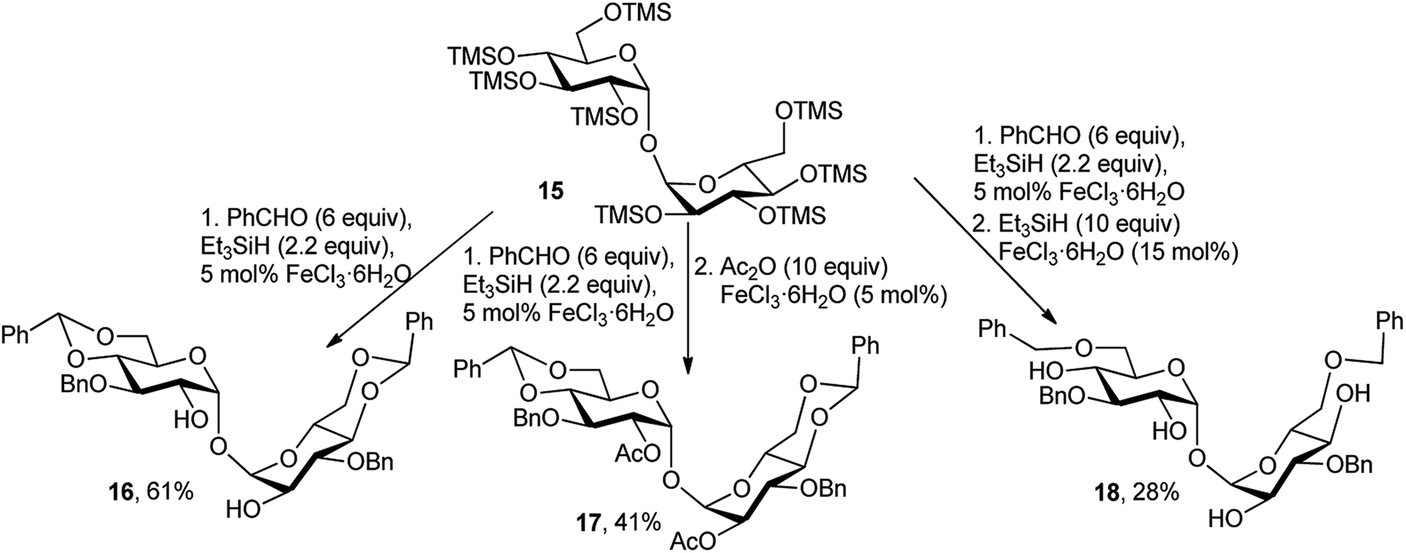 | ||
| Scheme 8 Tandem FeCl3·6H2O-catalysed reaction of TMS protected α,α-D-trehalose. | ||
The one-pot protocol with TMS derived sugar was also used for regioselective one-pot protection of 2-azido-2-deoxy-D-glucose derivative by Chang et al. in 2010.20 TMS protected 2-azido-2-deoxy-D-glucose substrate 19 was subjected to 4,6-O-benzylidene protection reaction followed by Cu(OTf)2 catalysed acetylation with excess Ac2O, and then reductive opening of the benzylidene ring with BH3·THF, afforded free 6-hydroxy-2-azido-2-deoxy-D-glucose derivative 20 in 59% overall yield. To alter the ring opening pattern, they replaced the reducing agent BH3·THF by Me2EtSiH and observed that the acetylation condition plays critical role in the outcome of the one-pot process. When 2.5 equivalents of Ac2O were used then 4-hydroxy group free product 21 was isolated in 45% yield, whereas use of 10 equivalents of Ac2O produced its corresponding acetylated analogue 22 in 62% overall yield (Scheme 9). They extended this one-pot reaction sequence toward generation of the hemiacetal 23, starting from the same tetra-O-TMS derivative 19.20 In this case, after formation of the 4,6-O-benzylidene ring, the 1,3-di-O-acetylation was accomplished under Cu(OTf)2-catalysis and further treatment with NH3 resulted in anomeric deacetylation with 59% overall yield (Scheme 9).
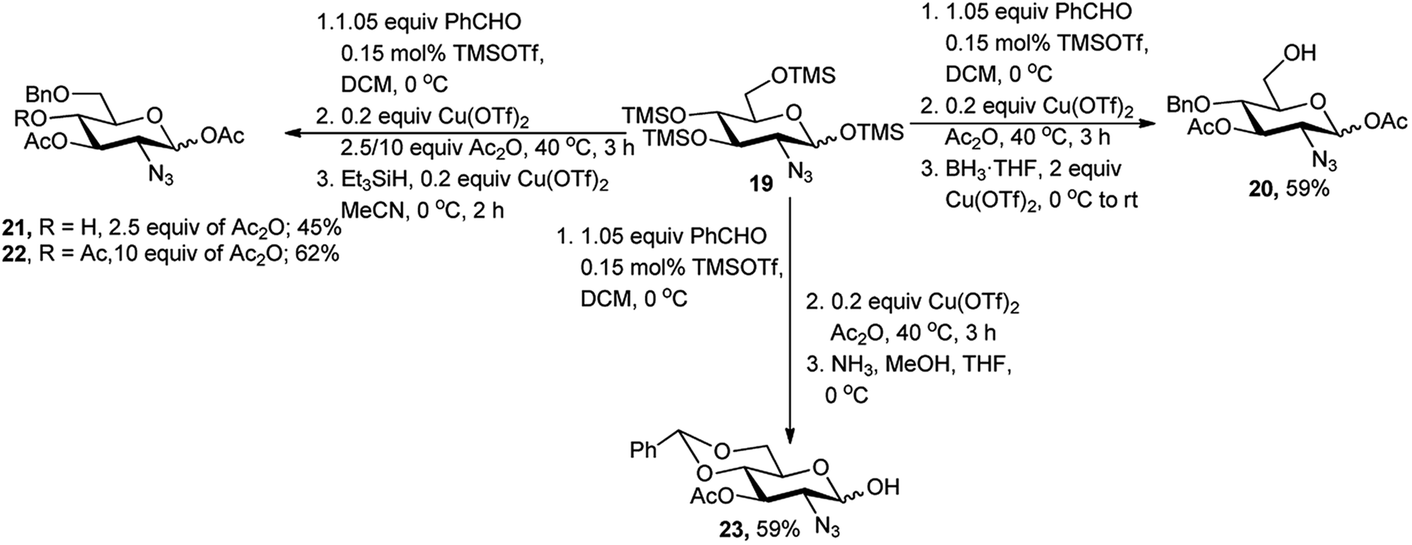 | ||
| Scheme 9 Regioselective sequential one-pot protection protocols of TMS derived 2-azido-2-deoxy-D-glucose. | ||
Instead of changing the promoter in the course of the one-pot protocol, a tandem catalysis with Cu(OTf)2 worked well during the entire process. Starting from the phenyl 1-thio-β-D-glucosamine derivatives 24 and 25, catalysis of the acetalation/acetylation steps proceeded properly with the azido substrate providing 81% of 26, using only 3 mol% of Cu(OTf)2 (Scheme 10).21 With benzaldehyde for the acetalation step, a stoichiometric amount of the copper salt was required for completion of the reaction with the acetamido substrate 27. Notwithstanding, use of benzaldehyde dimethyl acetal enabled a clean formation of 27 in 85% yield under catalytic conditions (Scheme 10). In the azido series, extension of the copper catalysis to a three-step sequence ending with a selective reductive opening of the benzylidene ring provided the 4-OH derivative 28 in 69% overall yield.
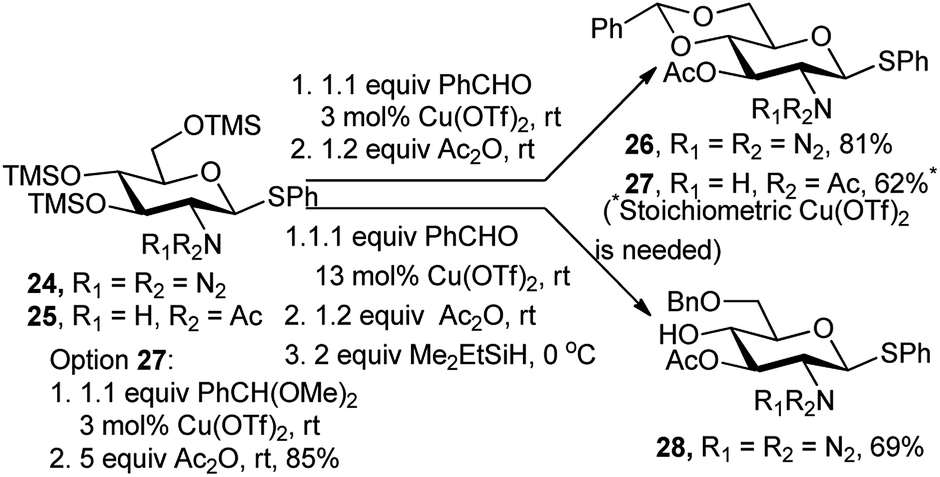 | ||
| Scheme 10 Cu(OTf)2-catalysed one-pot regioselective protection of D-glucosamine. | ||
2.4. Regioselective one-pot protection involving tin coordination
Use of stoichiometric amount of tin salt in regioselective protection of vicinal diols or spatially accessible diols (e.g. 4- and 6-hydroxy) has a long history in the carbohydrate field.22 Later, Demizu et al. reported catalytic use of tin salt for the first time, toward regioselective protection of polyols.23aThus, methyl glucoside (29) was selectively monobenzoylated using 0.05 equivalent of Me2SnCl2 as catalyst along with 1.2 equivalents of benzoyl chloride and 2 equivalents of N,N-diisopropylethylamine (DIPEA) in tetrahydrofuran (THF).
One-pot tosylation under similar condition using 1.1 equivalents of tosyl chloride produced the corresponding methyl 2-O-benzoyl-6-O-tosyl-α-D-glucopyranoside 30 in 63% overall yield. Interestingly, differentiation of the remaining 3,4-trans-diequatorial dihydroxy groups in 30 using Boc2O as the reagent under similar Me2SnCl2-catalysed conditions was successfully achieved to yield the 3-O-Boc protected product 31 in excellent yield (93% for this step and 32% for overall three steps) and regioselectivity (Scheme 11). Regioselective mono-benzoylation of 2-position followed by one-pot tosylation at 4-position of β-methyl xylopyranoside 32 resulted in compound 33 in 70% yield over two steps (Scheme 11). The excellent selectivity can be attributed to (a) the reversible interaction between Me2SnCl2 with the 1,2-cis-dioxygen atoms of the α-glucoside 29; (b) the increase of the proton acidity in the tin-coordinated intermediate which can be deprotonated easily by a weak base, such as DIPEA or 1,2,2,6,6-pentamethylpiperidine (PEMP);23b and (c) the nucleophilic addition on benzoyl chloride from the less hindered alkoxide to provide the corresponding 2-O-benzoyl derivatives for glucose and xylose in high yield. A similar phenomenon was observed in the case of the α-mannoside 34 (Scheme 11). The 3-O-functionalised product 35 was isolated via the proposed 2,3-cis-oriented five-membered ring intermediate 34a, presumably formed via elimination of 2 equivalents of HCl and further activation of the C-3 hydroxy group.23b
 | ||
| Scheme 11 Me2SnCl2-catalysed one-pot regioselective protection of polyols. | ||
The use of a catalytic amount of Bu2SnO under solvent-free conditions was first developed by Iadonisi and co-workers for organotin-catalysed benzylation and allylation of sugar polyols.24 Dong and Pei further demonstrated that regioselective benzylation could also be executed in toluene in the presence of organotin reagents, such as Bu2SnO, Bu2SnCl2, or Me2SnCl2, and also proposed a catalytic cycle as shown in Scheme 12.25 After the formation of dibutylstannylene acetal intermediate E between substrates and 0.1 equivalent of organotin, the intermediate E would further react with BnBr in the presence of 0.1 equivalent of tetrabutyl ammonium bromide (TBAB), leading to intermediate G where one position is benzylated and the other occupied by the tin species coordinated by a bromide. Deprotonated by a base, such as potassium carbonate, the un-benzylated substrate would also coordinate to the tetracoordinate tin atom to form pentacoordinate tin intermediate H. After tin species exchange through intermediates I and J, the final benzylated product would be generated from intermediate J, leading to the regeneration of dibutylstannylene acetal E from the un-benzylated substrate and further starting the next cycle.
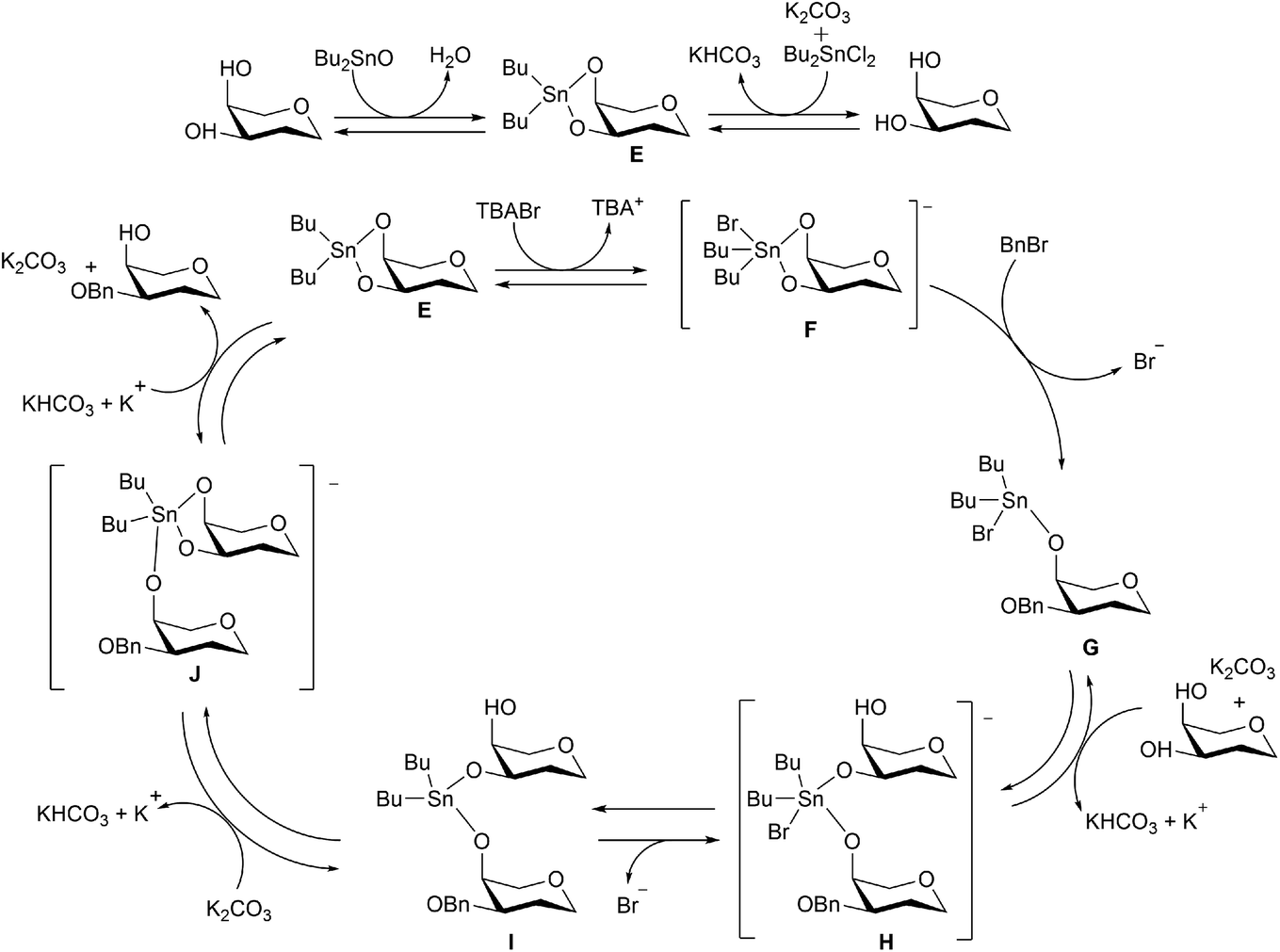 | ||
| Scheme 12 Catalytic cycle of organo-tin-catalysed reaction as proposed by Xu et al. (2014)25 (reproduced from ref. 25 with permission from John Wiley and sons, copyright 2020). | ||
Recently, orthogonal protection of saccharide polyols, under solvent-free condition and regioselective one-pot sequence employing TBDPS- or TBDMS-silylation at O-6 followed by Bu2SnO-catalysed benzylation or allylation, was reported by Iadonisi and co-workers (Scheme 13).26 This one-pot two-step protocol worked for the 2,3,4,6-tetraols 29 and 34, affording the 6-O-silylated and 3-O-benzylated/allylated diols 36, 38 and 39 and 6-O-silylated and 2-O-benzylated diol 37 in moderate to good yields (42–62%). The lower yields could be the results of the competitive reaction in the second silylation reaction due to residual etherification reagents from the first step for compounds 36 and 39 or the cleavage of the TBDPS group in the second benzylation step under high temperature for compound 37. They also showed that Bu2SnO-catalysed alkylation/silylation reaction combination is independent of their operational sequence.
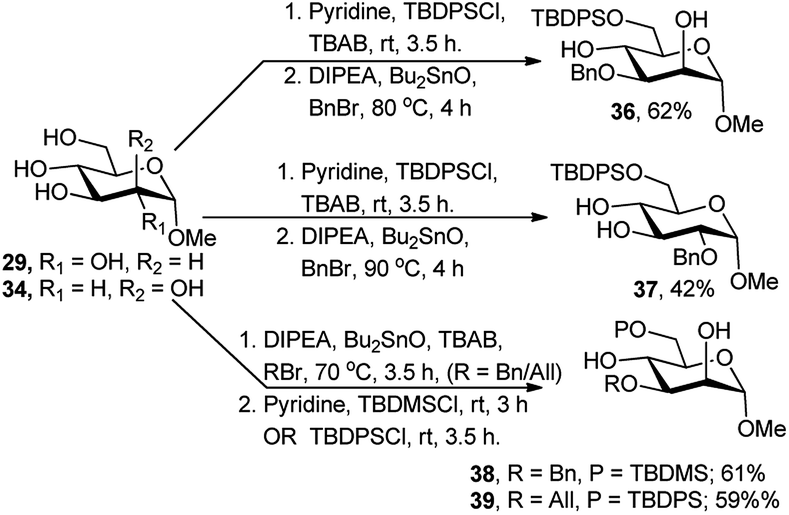 | ||
| Scheme 13 Bu2SnO-catalysed regioselective one-pot protection of 2,3,4,6-tetraols. | ||
3. Metal-catalysed tandem/domino/cascade/sequential one-pot reactions on glycal-based substrates
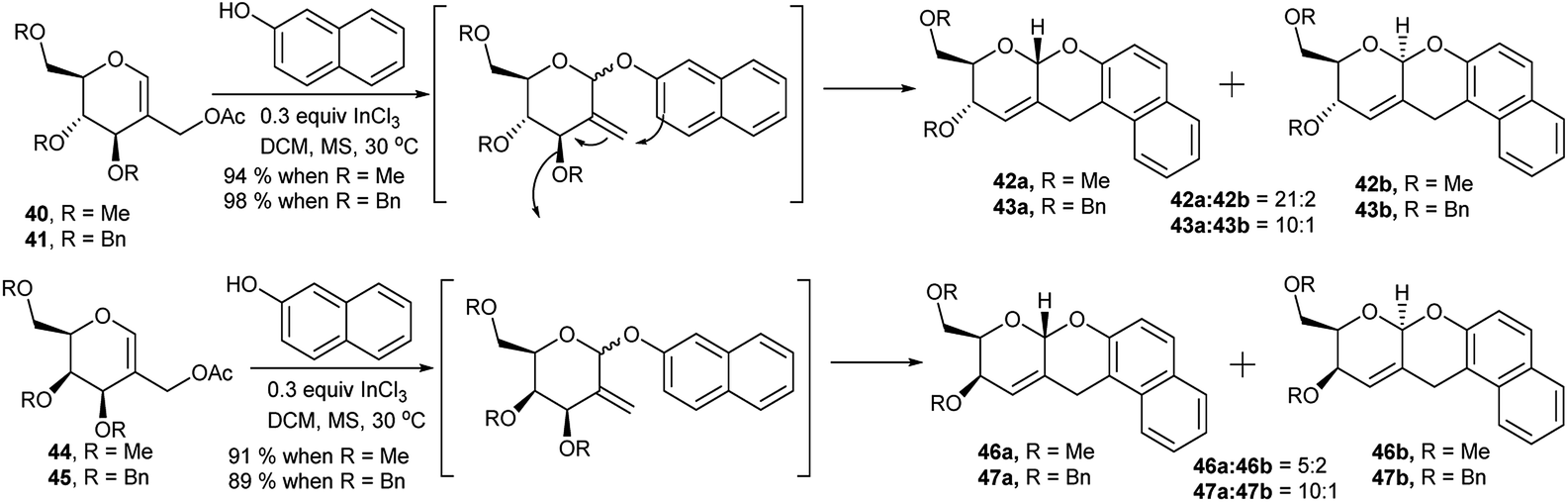
Glycals have been used as potential substrates for generation of sugar-based scaffolds.27 Lewis acid catalysed synthesis of sugar-fused chiral pyrano-pyran motifs was first reported by Balasubramanian et al. in 1993 and then after a decade in 2003.28 Role of InCl3 as metal catalyst toward such transformation of 2-C-acetoxymethyl glycals in more efficient fashion was observed by Ghosh et al. in 2005.29a Later on they prepared a number of such derivatives by coupling between 2-C-acetoxymethyl glycals and 2-napthol (Scheme 14) and studied the gelation property of one of these pyrano-pyran ring systems in detail.29b,c In the presence of catalytic amount of InCl3 methylated 2-C-acetoxy glucal (40) underwent Ferrier rearrangement with 2-napthol, followed by tandem cyclisation producing the corresponding products (42a and 42b) in excellent yield and stereoselectivity (94%, 42a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :42b = 21:2, Scheme 14) whereas the same for galactal analogue 44 resulted in good yield but poor stereoselectivity (91%, 46a:46b = 5:2, Scheme 14). For both benzylated glucal 41 and galactal (45) derivative, the reactions were almost equally good yielding and stereoselective (98% of 43 with 43a:43b = 10:1 and 89% of 47 with 47a:47b = 10:1, Scheme 14).
:42b = 21:2, Scheme 14) whereas the same for galactal analogue 44 resulted in good yield but poor stereoselectivity (91%, 46a:46b = 5:2, Scheme 14). For both benzylated glucal 41 and galactal (45) derivative, the reactions were almost equally good yielding and stereoselective (98% of 43 with 43a:43b = 10:1 and 89% of 47 with 47a:47b = 10:1, Scheme 14).
 | ||
| Scheme 14 InCl3-catalysed Ferrier rearrangement-tandem cyclisation of 2-C-acetoxy glycal. | ||
An easy efficient access to highly diastereoselective pyrano[3,2-b]-1-benzopyrans following a different one-pot route was developed by Yadav et al. (Scheme 15) where pyranopyrans (50) were generated from Sc(OTf)3 catalysed three-component one-pot reaction of glycals (49), trimethylorthoformate and substituted salicylaldehyde (48).30
 | ||
| Scheme 15 Sc(OTf)3-catalysed synthesis of pyrano[3,2-b]-1-benzopyrans. | ||
In continuation to the prior work on iodine catalysed Prins cyclisation on sugar based homoallylic alcohols in reaction with aldehydes toward generation of sugar annulated tetrahydropyrans,31a–d Yadav et al. further utilised Sc(OTf)3 for a similar application in other related sugar systems (51), which afforded high yields of highly stereoselective hexahydro-2H-furo[3,2-b]pyranopyrans (52),31e formed via tandem ene-Prins cyclisation routes (Scheme 16).
 | ||
| Scheme 16 Sc(OTf)3-catalysed ene-Prins reaction for generation of hexahydro-2H-furo[3,2-b]pyranopyrans. | ||
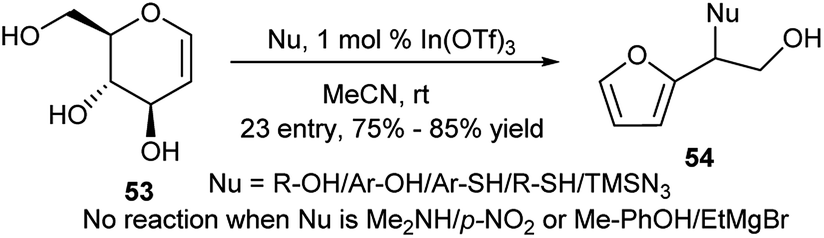
Another indium(III) salt, In(OTf)3 has been used by Mukherjee et al. for the one-pot transformation of the D-glucal toward racemic α-substituted α-hydroxymethylfurfuryl derivatives in the presence of nucleophiles.32 Thus, reactions of free D-glucal (53) with most of the O-, S-, C- and N-nucleophiles resulted in their corresponding furan-based products (54) in good yields (Scheme 17). However the reaction protocol is not valid for few nuleophiles like free amine, Grignard reagent, p-NO2/Me-phenols, but reason behind this is not known.
 | ||
| Scheme 17 In(OTf)3-catalysed synthesis of α-substituted furan derivatives. | ||
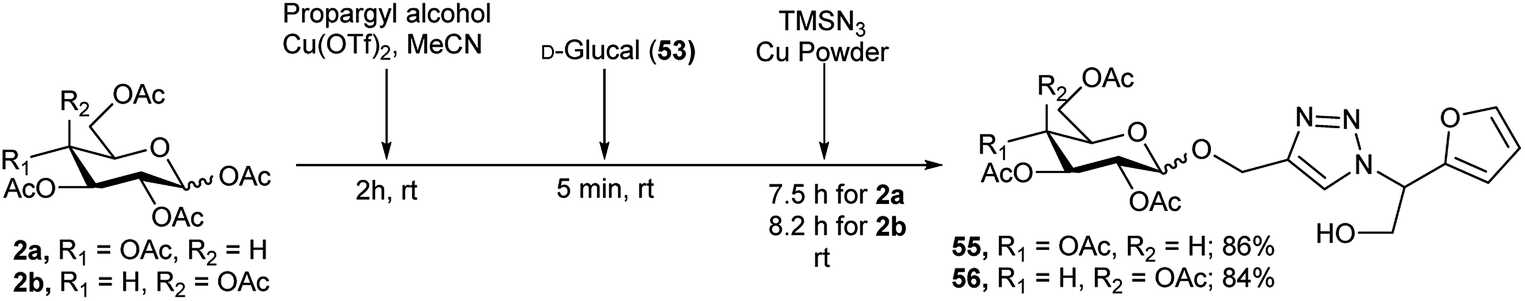
Extending this idea to another aspect where α-azido-α-hydroxymethyl furan was formed by the reaction of D-glucal with TMSN3 in combination with a click reaction of this azide with propargyl group, the same group then reported Cu-catalysed construction of various triazole fused furan-based glycoconjugates.33 Thus, they first converted per-O-acetylated glucose (2a)/galactose (2b) to its corresponding propargyl glycosides by reaction with propargyl alcohol in the presence of 10 mol% Cu(OTf)2 catalyst, then subsequent reaction with D-glucal (53) and TMSN3 resulted in their corresponding triazole-furan-based gluco (55)/galacto (56) derivatives in good yields (Scheme 18).
 | ||
| Scheme 18 Cu(OTf)2-catalysed one-pot MC-synthesis of triazole-furan-based glycoconjugates. | ||
In 2013, Mukherjee's group reported halogenated Lewis acid promoted tandem glycosylation–halogenation of aryl acetylenes with glycals to form stereo defined α,E-trisubstituted halo vinyl glycosides and their subsequent application in Pd-catalysed cross-coupling reactions.34
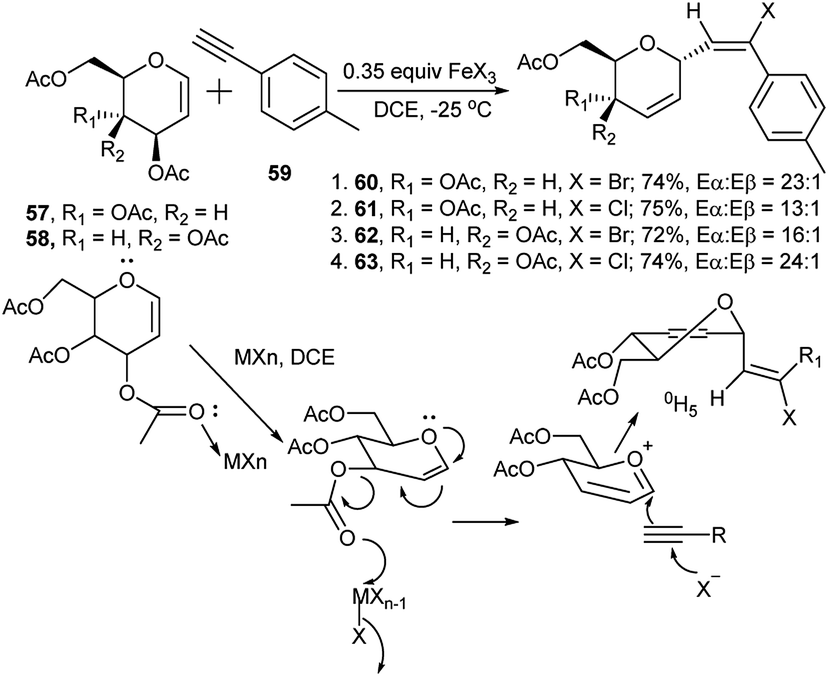
Reaction of tri-O-acetyl-D-glucal (57) with p-methyl phenyl acetylene (59) using 0.35 equivalent of FeBr3 at −25 °C produced the corresponding brominated C-vinyl glycoside 60 in 74% yield (Eα:Eβ = 23:1, entry 1, Scheme 19), whereas the same reaction with its galactal analogue (58) also resulted in the corresponding product 62 in almost similar yield albeit with relatively lower stereoselectivity (72%, Eα:Eβ = 16:1, entry 3, Scheme 19). Reaction of the same acetylene 59 with glucal 57 under FeCl3 catalysed reaction condition produced the corresponding chlorinated C-vinyl glycoside 61 in good yield and moderate stereoselectivity (75%, Eα:Eβ = 13:1, entry 2, Scheme 19) and that with galactal 58 resulted in the corresponding chlorinated product 63 in excellent yield and stereoselectivity (74%, Eα:Eβ = 24:1, entry 4, Scheme 19). As a plausible mechanism they presumed the possibility of the Lewis acid to play two major roles when phenyl acetylene and glucal triacetates were reacted. At first, the metal salt coordinates with the allylic acetoxy group of the glycal, making it more labile, and then the ring oxygen participates to generate a glycosyl oxocarbenium ion intermediate, that is subsequently attacked from the α-face by the alkyne which is concomitantly attacked by a halide ion, ultimately resulting in the formation of the corresponding product.
 | ||
| Scheme 19 FeCl3 or FeBr3-catalysed tandem halogenated C-vinyl glycoside synthesis using inactivated aryl acetylene. | ||
Palladium-catalysed, TEMPO mediated regio- and diastereo-selective domino Heck–Suzuki arylation of glycal (57/58) and pseudo glycals (71) was reported by Kusunuru et al.35 When glycals were allowed to react with a wide range of arylboronic acids using 10 mol% of Pd(OAc)2 as catalyst and 3 equivalents of TEMPO as radical initiator in acetic acid, the corresponding diaryl glycosides were obtained in excellent yield and diastereoselectivity. The resulting diaryls are C1–C2 (α,α) in the case of glycals and C1–C2 (β,β) for pseudoglycals and both were confirmed by NOSEY spectrum. Thus, reaction of D-glucal derivative 57 separately with arylboronic acids 64 and 65 at 30–40 °C produced the corresponding cis(α,α) 1,2-diarylated glucosides 67 and 68 in 76% and 60% respective yields (entries 1 and 2, Scheme 20). Similar reaction between D-galactal derivative 58 and arylboronic acids 64 and 66 separately resulted in the corresponding 1,2-diarylated galactosides 69 and 70 in 72% and 65% respective yields (entries 3 and 4, Scheme 20). Apart from the reaction of glycals, pseudoglycal 71, when subjected to react with p-methylphenylboronic acid 64 under the similar reaction conditions but at relatively lower temperature, produced its corresponding cis(β,β) 2,3-diarylated glycoside 72 in moderate yield (60%, Scheme 20). They also provided a possible reaction mechanism and a catalytic cycle of Pd(II)/Pd(0), showing involvement of TEMPO playing a key role in blocking syn-and anti-elimination of C2-PdOAc and C3-α/β-OAc from an anomeric aryl, 2-PdOAc intermediate (thus minimising the possibility of a side reaction generating C-aryl pseudoglycal); this was also corroborated by the corresponding quantum chemical analysis.
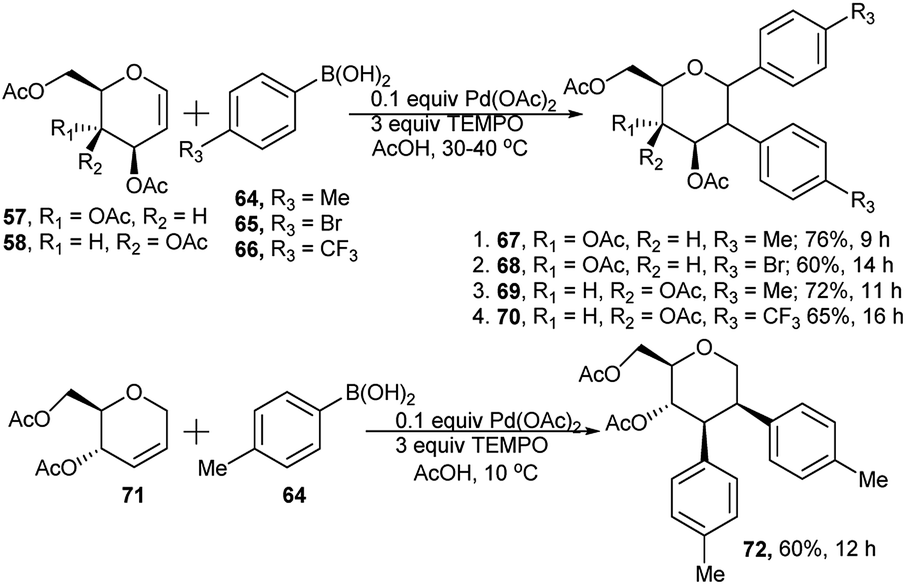 | ||
| Scheme 20 Pd(II)-catalysed domino Heck–Suzuki arylation of glycals. | ||
Yadav et al. has used glucal (57) as the sugar substrate for InCl3-catalysed three-component (3C) one-pot reactions with aryl amines and 1,3-dicarbonyl compounds (β-diketone/β-ketoester) in refluxing 1,2-dichloroethane (DCE) to generate the corresponding oxaaza bicyclononene scaffolds (73) in excellent yields and high diastereoselectivity. In situ generated β-enaminoketone or β-enaminoester is supposed to react further with the glycal to afford the bicycloheterocycles (Scheme 21).36a
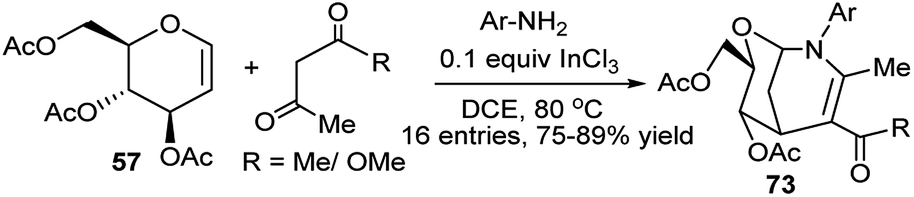 | ||
| Scheme 21 InCl3-catalysed three component preparation of oxaaza bicyclononene scaffolds. | ||
Yadav's group also reported a one-pot 3C condensation using 2-deoxyribose, acetyl acetone and aromatic amines to give similar 2-deoxyribose-based bicyclic aminols in good yields albeit moderate or no stereoselectivity.36b
A library of pentacyclic benzopyran fused pyranoquinolines (74) were prepared by another research group in low to high yields by Sc(OTf)3-catalysed one-pot 3C reactions of glycals (49a,b/57/58) with salicylaldehyde or its derivatives and aryl amines in acetonitrile at elevated temperature (Scheme 22).37a
 | ||
| Scheme 22 Sc(OTf)3-catalysed one-pot domino synthesis of pentacyclic benzopyran fused pyranoquinolines. | ||
This proceeded by domino Ferrier rearrangement and Povarov like reactions with polycyclisation, as also observed by this group earlier in another case.37b As proposed by the authors, the reaction proceeds initially by the formation of imino phenol K that remains in equilibrium with the corresponding enaminone K′, the latter then reacts with Sc(OTf)3 activated glycal generating intermediate L; L in its 5HO conformation undergoes intramolecular polycyclisation from the β-face of the pseudoglycal ring (Scheme 22). The reactions based on aniline or salicylaldehyde rings bearing electron withdrawing substituent proceeded smoothly giving the corresponding pentacyclic product in good yield, but aromatic rings bearing electron donor substituent generated the expected products in low yield. Further CAN mediated oxidative ring opening of one such pentacyclic product afforded an enantiopure chromenoquinoline, holding at C6, a trioxypropyl pendant.37a
Al(OTf)3 has been exploited as Lewis acid catalyst by Williams and co-workers for tandem reaction on D-galactal triacetate (58). This in reaction with phenol or its ring derivatives underwent tandem process as proposed by the authors: initial Ferrier rearrangement followed by tandem Friedel–Crafts C–C bond formation, assisted anchimerically by C4-OAc group, ultimately generating chiral bridged benzopyrans (75), as shown in Scheme 23.38a It is to be noted that D-glucal triacetate (57) when treated under the above condition underwent 1-C-arylation.38b Interestingly, further Al(OTf)3 catalysed reaction of 75 with Ac2O in the presence or absence of acetic acid afforded respectively sugar appended optically pure chromans (76) or chromenes (77).38a
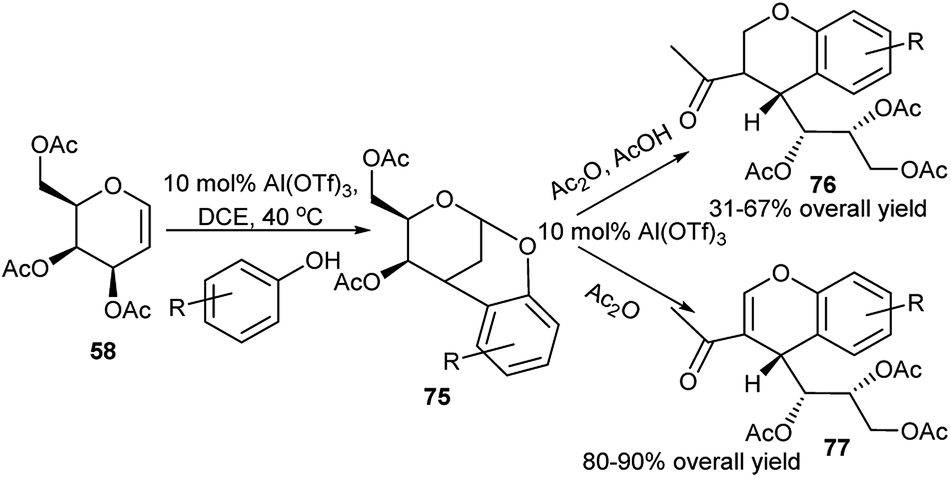 | ||
| Scheme 23 Al(OTf)3-catalysed tandem synthesis of chiral bridged benzopyrans, and sugar-appended chromans and chromenes. | ||
A synthetic protocol for highly substituted structurally diverse tetracyclic chromans 80 and isochromans 81 having sugar-aromatic hybrid structures, was developed by Werz's research group using diyne derived 2-bromoglycal system 78 or diyne-based 2-bromo-2,3-unsaturated sugar glycosides 79 by Pd(0) catalysed domino reaction involving oxidative addition, two successive carbopalladation and cyclisation steps (Scheme 24).39a,b
 | ||
| Scheme 24 Pd(0)-catalysed domino synthesis of chromans and isochromans. | ||
Inspired by the initial success of this domino protocol, this group further elaborated the method toward synthesis of chiral substituted biaryl systems from sugar-disubstituted tetra-alkynes39c and also synthesis of anthracycline aglycone mimics by domino carbopalladation of glycal-based bisalkynes.39d The mechanistic pathway as proposed39b by the authors is shown in detail in Scheme 25. Oxidative addition of Pd(0) into C–Br of the 2-bromoglycal substrate (78) generates an intermediate M, then after two successive steps involving cyclisation reactions via attack at alkyne carbon of carbapalladium intermediates (M and N) a new intermediate O is formed. Final Pd(II)-mediated intramolecular cyclisation of O followed by aromatisation of the resulting intermediate P affords the tetracyclic chroman 80.
 | ||
| Scheme 25 Proposed mechanistic pathway for one-pot conversation of 78 to 80. | ||
Yao et al. reported diversely-oriented synthesis of three different types of structurally drug like N-heterocyclic fused ring from one-pot cascade reaction between 2,3,6-tri-O-benzyl-D-glucal 49a and aromatic amines (Scheme 26).40 Reaction of D-glucal 49a with substituted aromatic amines produced their corresponding indolines in moderate yields. Reaction of 49a with N-benzyl-p-methylaniline in the presence of 30 mol% InBr3 and 1.2 equivalent water in nitromethane at 100 °C produced the corresponding indoline derivative 82 in 62% yield; whereas similar reaction between the same D-glucal 49a and aniline carrying electron withdrawing group at phenyl ring and allyl substitution at nitrogen produced their corresponding indoline derivative 83 in lower yield (40%, Scheme 26). On the other hand, similar reaction of D-glucal 49a with secondary aniline bearing an o-hydroxyl group produced the corresponding benzoxazine-fused heterocycles 84 or 85 in relatively higher yields (Scheme 26). The comparative higher yield for this second kind of products was attributed to a more energetically favoured formation of the six-membered fused ring system compared to the five-membered ring and the superior nucleophilicity of the hydroxy moiety present in the aniline substrate. Dy(OTf)3-catalysed reaction of the D-glucal 49a and N-benzyl-aniline produced in moderate yields tetrahydroquinoline-fused cyclo-pentanones in acetonitrile at 100 °C in the presence of 1.2 equivalent of water for 12 hours via Ferrier reaction followed by cascade interrupted Nazarov rearrangement. Under this condition N-benzyl-p-methoxy aniline and N-benzyl-p-hydroxyl aniline generated the corresponding fused rings 86 and 87 in 52% and 49% respective yields (Scheme 26).
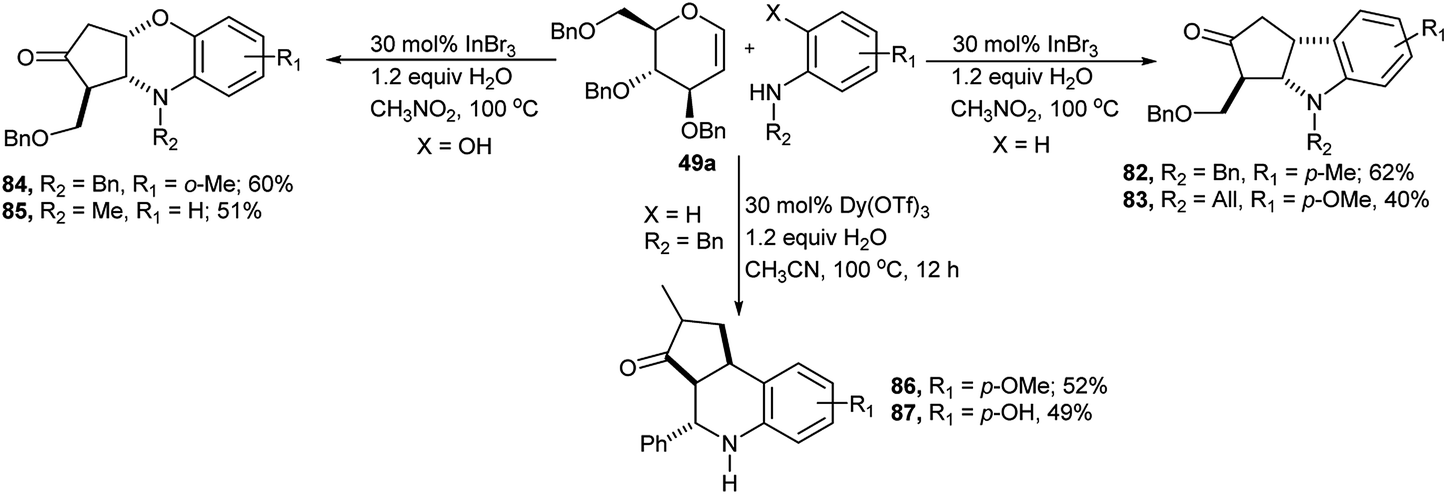 | ||
| Scheme 26 One-pot cascade transformation of D-glucal into drug like scaffolds. | ||
Holzapfel et al. employed pseudoglycal derived 1,6-enyne 88 for palladium-catalysed cascade carbocyclisation toward synthesis of enantiomerically pure tricyclic compounds (89–91). Under Wacker conditions in the presence of LiCl, the reactions proceeded with highly stereoselective ring expansion where two new C–Cl bonds were formed in the products (90a, 90b and 91). The products were thus dependent on the substrates and also on the corresponding reaction conditions (Scheme 27).41 This research group also reported interesting palladium-catalysed domino reactions in sugar 1,6-dienes and -enynes.42a,b
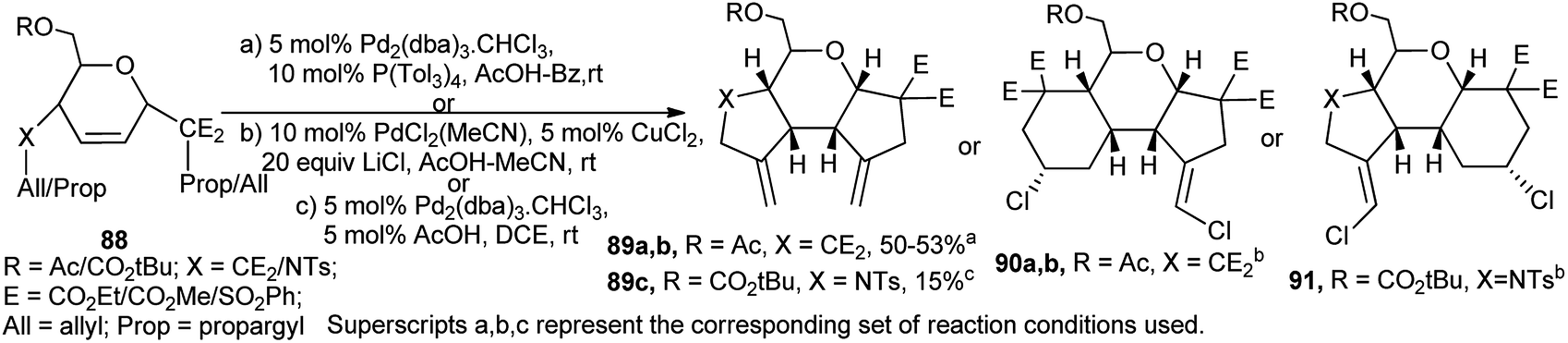 | ||
| Scheme 27 Pd2(dba)3-catalysed cascade reactions on sugar-based 1,6-enynes. | ||
Thus, the 1,6-diene, viz. 4-homoallyl substituted pseudoglycals 92 in the presence of PdCl2(MeCN)2 and CuCl2 in acetic acid-methanol at room temperature, underwent domino ring opening (to Q) – ring closure process forming an intermediate (R) which after elimination of HPdLnCl followed by double bond isomerisation finally afforded the corresponding functionalised C-glycals (dihydropyrans) 93a. In the presence of CO and the above set of reagents, alkoxycarbonylation of the terminal alkene and domino ring opening-ring closure finally produced functionalised chiral tetrahydropyrans 93b after methylation by diazomethane. But, under a Wacker condition in the presence of excess LiCl, 92 produced the chlorinated product 93c (Scheme 28).42a Under the Wacker condition, the reaction course was a bit different based on the sugar derived 1,6-enyne substrate, 94, where the substituted homopropargyl group was at C1 of the pseudoglycal, and a dimeric chlorovinyl appended product 95 was formed (Scheme 28).42b
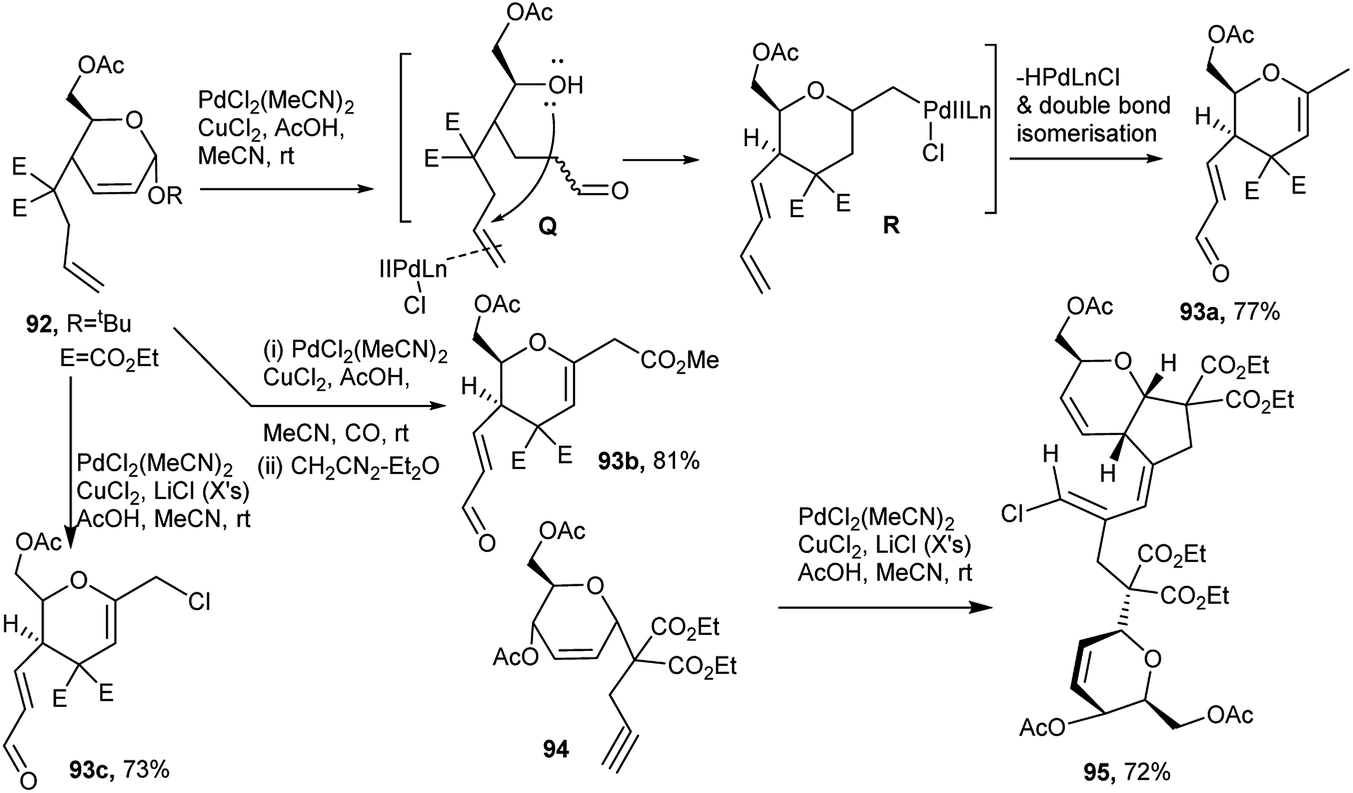 | ||
| Scheme 28 PdII–CuCl2-mediated domino reactions of sugar-based 1,6-diene and -enyne. | ||
Sugar derived exo-glycals having 2,3-oxirane were used by several researchers for one-pot generation of functionalised C-glycals under nucleophilic and electrophilic conditions. Thus, Gömez and co-workers utilised 1-exo-methylene-2,3-anhydrofuranoses 97, and obtained new functionalised C-glycals 98 in two-step one-pot reaction by Br2 and base from C-glycals 96, for Pd(0)-catalysed nucleophilic addition vis-à-vis epoxide opening (Scheme 29).43
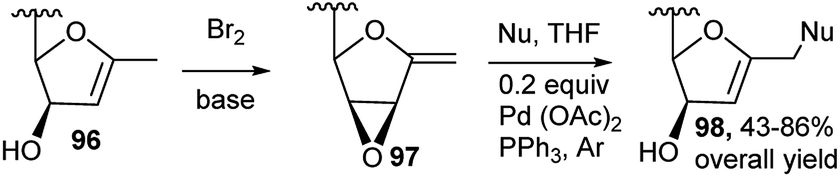 | ||
| Scheme 29 Pd-catalysed domino synthesis of functionalised C-glycals. | ||
They further employed vinyl oxiranes having exo-glycal skeleton 99 in the presence of Pd(0)-Et2Zn for reaction with carbonyl electrophiles. This proceeded by domino epoxide opening via formation of an allyl-Zn intermediate (100) from an initially formed π-allyl-Pd intermediate, followed by subsequent attack by the electrophile, and ultimately generated the corresponding new functionalised C-glycal derivatives 101 (Scheme 30).44 Although the protocol was compatible with highly oxygenated substrates and was highly regioselective in reaction with aldehyde electrophile generating always 1,5-diol, but the stereoselectivity varied from moderate to excellent depending upon the reaction partners.
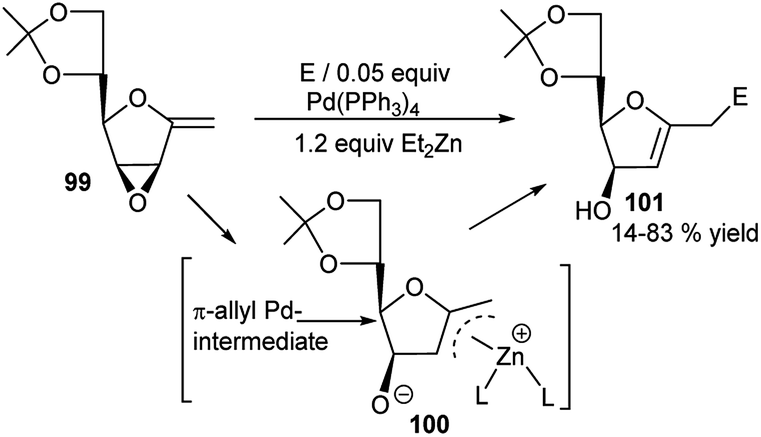 | ||
| Scheme 30 Pd(0)-catalysed electrophilic reactions of exo-glycal-based vinyl oxiranes. | ||
This group also developed a complete regio- and stereo-controlled one-pot 3C synthesis of 2-aminoglycosylidene type compounds 103 from Pd(0) catalysed reaction of mannose derived halo-alkenyl allylic-oxirane system 102, boronic acids or alkyl boronates and a variety of amines (Scheme 31).45
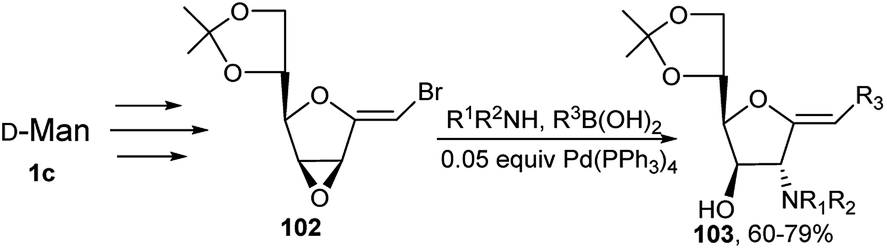 | ||
| Scheme 31 One-pot synthesis of furanose-based carbohydrate templates. | ||
Very recently Mishra et al. reported FeCl3-catalysed tandem synthesis of optically pure sugar-based benzimidazoles.46a Efficient reaction of benzylated or methylated glycal aldehydes (104) with varieties of substituted ortho-phenylenediamines in the presence of 20 mol% of FeCl3 catalyst in refluxing acetonitrile produced their corresponding sugar attached benzimidazoles (105) in moderate to excellent yield (Scheme 32). FeCl3 catalysed both cyclisation and aromatisation steps. It is to be noted that the efficacy of the tandem reaction is not hampered by carrying out the reaction under nitrogen. It is also to be mentioned that synthesis of similar sugar derived benzimidazoles was effected earlier based on VO(acac)2–CeCl3 combo catalyst in the presence of molecular oxygen.46b
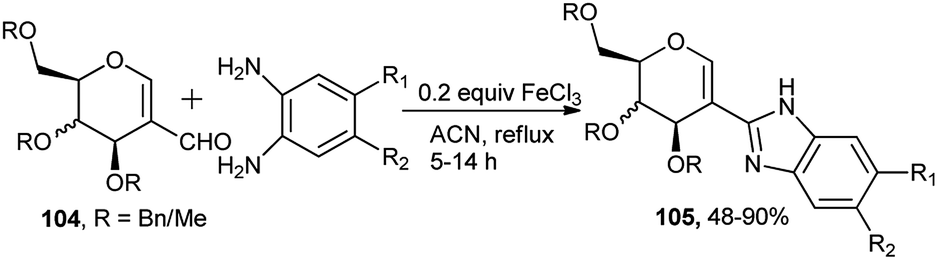 | ||
| Scheme 32 FeCl3-catalysed tandem synthesis of sugar-based benzimidazoles. | ||
4. Metal-catalysed domino/tandem/sequential one-pot (a) syntheses of unnatural nucleosides and (b) reactions on nucleosides
Chemical modification, either in the sugar part or in the base part gives rise to unnatural nucleosides or nucleoside analogues. Many of such compounds are already in use worldwide for treatment of virally infected or cancer patients; several other new nucleosides are under pre-clinical or clinical trial.47 More efforts are being made for development of new tailor-made nucleosides by synthesis or reactions on other nucleosides, utilising one-pot MC reactions.484.1. One-pot syntheses of novel nucleosides catalysed by metal salt/complex
In 2008, Siddique et al. reported clay K1049a,b-catalysed one-pot synthesis of 4-(3H)-quinazolinone N-nucleosides (107) from 3C reaction of unsubstituted anthranilic acid, ribosyl amine (106) and benzoic acid or its derivatives under solvent-free microwave conditions (Scheme 33).49c
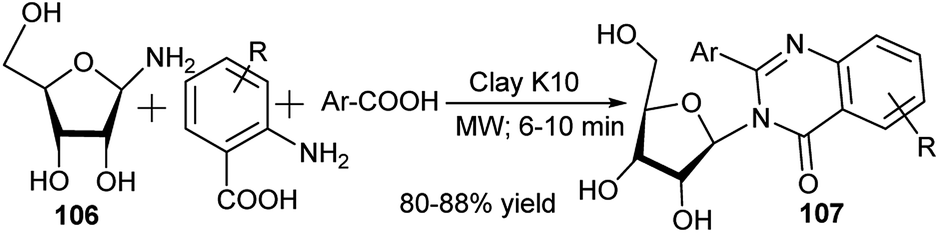 | ||
| Scheme 33 4-(3H)-Quinazolinone N-nucleoside synthesis. | ||
In addition to other established nucleoside analogous drugs, a new FDA approved drug called Efavirenz (Sustiva, 108) having benzoxazinone scaffold is also being used for treatment of AIDS. In order to have nucleoside and benoxazinone in one compound, Yadav and Rai in 2006 synthesised a library of new 4-aminobenzoxazinone N-nucleosides (110) in 68–80% yields by clay K10-catalysed microwave mediated 3C-coupling reactions under solvent free condition of ribosyl/deoxyribosyl urea (109), substituted salicylaldehydes and ammonium acetate (Scheme 34a).50a The authors proposed that the reaction proceeds through cycloisomerisation of an aldimine intermediate. In 2013 Rai and Singh reported the 3C one-pot synthesis of several 1,3-benzoxazine-2-thione N-nucleosides (112). They noted that D-ribose (111) in reaction with thiosemicarbazide and salicylaldehyde or its derivatives under microwave condition in the presence of clay K10 afforded the corresponding unnatural nucleosides in 61–69%; interestingly, the corresponding microwave mediated 2C-reactions of the pre-generated sugar anomeric thiosemicarbazides, obtained from combination of ribose and thiosemicarbazides catalysed by CeCl3·7H2O–NaI furnished the corresponding products in better (83–93%) yields (Scheme 34b).50b The resulting hydrazinated unnatural nucleosides were finally dehydrazinated.
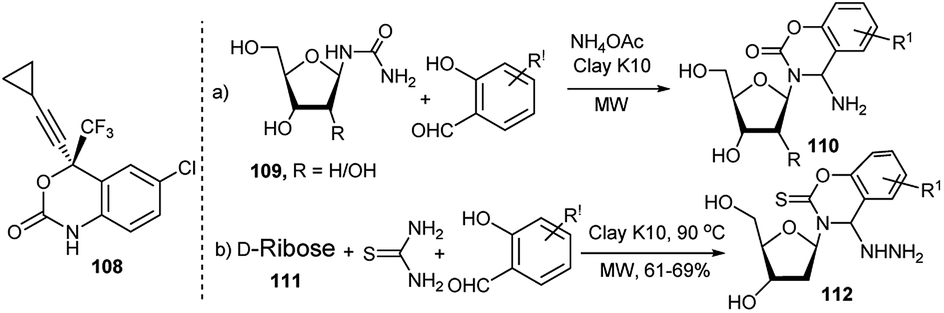 | ||
| Scheme 34 Clay K10-catalysed 3C-one-pot synthesis of 4-aminobenzoxazinone N-nucleosides (110) and 1,3-benzoxazine-2-thione N-nucleosides (112). | ||
For the synthesis of a group of unnatural fused triazolyl 2-quinolinone (FQuon) nucleosides 115, Bag et al. used N-benzyl-N(2-iodo-4-methylphenyl)propiolamide 113 for a CuI-catalysed one-pot click reaction with sugar anomeric azide 114 followed by tandem Ullmann type C–C coupling reaction for generation of 115 in its protected form (Scheme 35).51 Compound 115 is having both donor and acceptor H-bonding site and a conically projected benzene ring. The researchers further studied the photophysical properties of 115, which exhibited good interaction with BSA, good DNA duplex stabilisation property, as also supported by theoretical studies.
 | ||
| Scheme 35 One-pot tandem synthesis of triazolyl 2-quinolinone. | ||
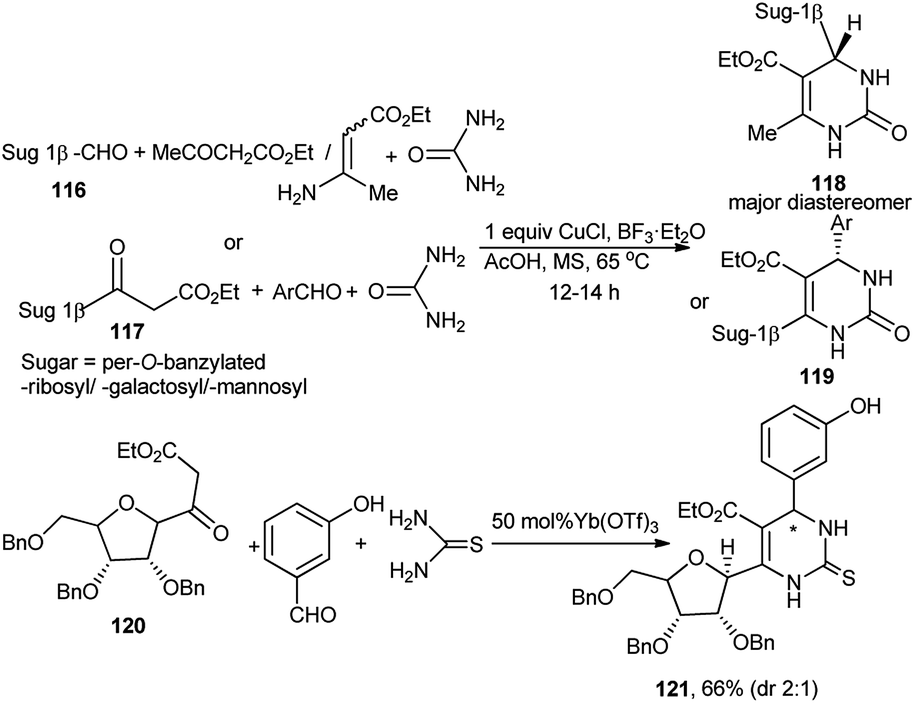
The Biginelli reaction has been widely used for preparation of dihydropyrimidinone scaffolds.52 A 3C promoter (CuCl/AcOH/BF3) system using stoichiometric Cu(I) was at first employed by Dondoni et al. for 3C one-pot diastereoselective synthesis of a series of N1/C4/C6-monoglycosylated and C4, C6 bisglycosylated dihydropyrimidinone (DHPone) glycoconjugates (118 and 119) from Biginelli reaction of sugar (pyranose/furanose) derived anomeric-carboxaldehyde 116, ethyl acetoacetate or sugar derived β-ketoester 117, benzaldehyde, respectively and urea53a–c in THF at 65 °C (Scheme 36). They also observed that when β-aminoacrylate was used instead of ethyl acetoacetate for the one-pot reaction based on sugar anomeric carboxadehyde; Yb(OTf)3 was established to be the best Lewis acid catalyst. Utilising the 3C promoter based protocol, this group also synthesised bis-C-glycosylated dihydropyrimidinones.53a They further noticed that whereas the above 3C promoter system was not compatible for the Biginelli reaction for preparation of the thiourea-based products, but use of 50 mol% Yb(OTf)3 could serve the purpose. Thus, Yb(OTf)3 catalysed the one-pot Biginelli reaction of ribofuranosyl-based ketoester 120 with 3-hydroxybenzaldehyde and thiourea generating the corresponding C6-glycosylated DHPone 121 in fair yield albeit lower diastereoselectivity (dr 2:1) (Scheme 36).53b,c
 | ||
| Scheme 36 One-pot synthesis of sugar derived DHP ones. | ||
4.2. Synthesis of novel nucleosides by reactions on other nucleoside derivatives
An ethynyl nucleoside (122) was utilised to prepare the corresponding C-3′-propargyl amine nucleoside (123a) by a CuBr-catalysed one-pot three-component reaction of 122 with diisopropyl amine and paraformaldehyde in refluxing THF (Scheme 37). Eventually further deprotected novel uridine analogue (123b) exhibited antitumor activity.54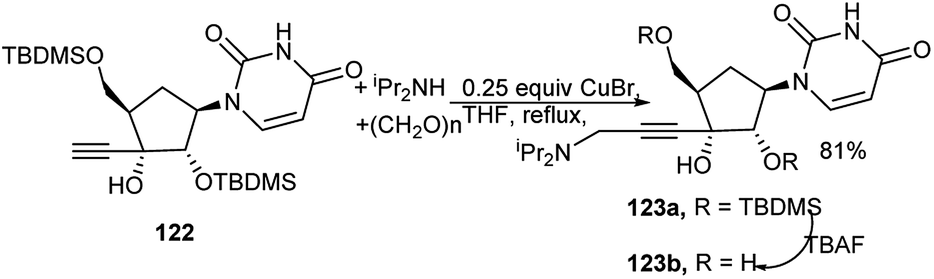 | ||
| Scheme 37 CuBr-catalysed 3C one-pot synthesis of uridine analogues. | ||
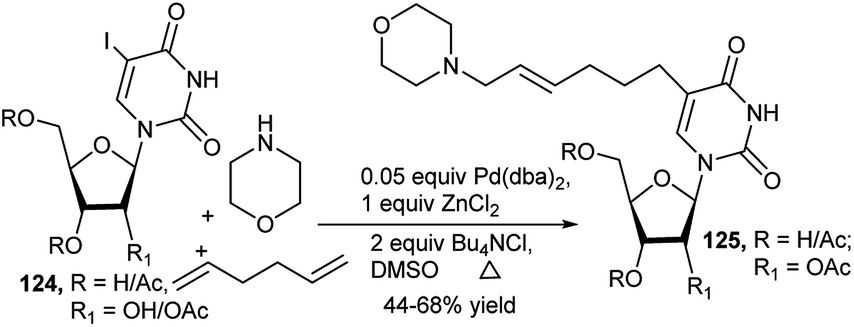
Larock et al. used commercially available 5-iodo-2′-deoxyuridine (124) for generation of tailor-made nucleosides (125) by one-pot MCR. Thus, Pd(0)-catalysed 3C-couplings of (124) with 1,2-, 1,3- or 1,ω-diene and different primary/secondary amines in the presence of ZnCl2 afforded a wide variety of C-5 aminoalkyl substituted nucleosides 125 (Scheme 38). It was observed that secondary amines are the most effective amines toward this end. Both acyclic and cyclic dienes can be used, and simple non-conjugated dienes provide good yields. But with increase in chain length/branching, yields get reduced. In the later case, pre-derivatisation of the sugar hydroxy groups was beneficial.55
 | ||
| Scheme 38 Palladium-catalysed one-pot synthesis of C-5 aminoalkyl substituted nucleosides. | ||
Synthesis of amino acid functionalised uracil nucleosides (127) was reported by Gheerardijn et al. by a Pd(0)-catalysed domino carboxamidation reaction of uracil nucleosides (126) using amino acid esters and carbon monoxide under pressure at 70 °C (Scheme 39).56 Pd2(dba)3 was utilised by Grigg et al. to catalyse 3C reactions of purine/thymidine/uridine-based allene, aryl/heteroaryl iodide and adamantyl amine or other amines for generation of trisubstituted Z-alkenes bearing the corresponding nucleoside skeleton. They further developed a Pd(0)-mediated MC-reaction protocol based on a 1,3,5,7-tetrakis(4-iodophenyl)adamantane, purine derived allene and different amines for generation of a tetrakis adamantane skeleton.57
 | ||
| Scheme 39 Pd(0)-catalysed domino synthesis of amino acid anchored uracil nucleosides. | ||
5. Metal-catalysed one-pot synthesis of sugar-based amino acids
Owing to be considered as building blocks for peptidomimetic research and also due to important biological properties of non-proteinogenic amino acids, their design and synthesis have gained much attention among the researchers worldwide toward their future potent application in the fields related to life science.58 4C Ugi reaction is the mostly applied isocyanide-based MC reaction for the synthesis of a wide variety of such compounds.59 Kunz and co-workers extensively studied 4C-Ugi reaction on sugar amines as chiral auxiliaries toward generation of chiral α-amino acid derivatives by asymmetric induction.60a–e For synthesis of chiral R-α-amino acid derivatives 129a, Kunz et al. first used 2,3,4,6-tetra-O-pivaloyl-β-D-galactopyranosyl amine 128a as chiral auxiliary for asymmetric synthesis of amino acids; later other sugars were also used. Thus, 128a60a,b or 2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyl amine 128b60c was initially utilised as the chiral amine for ZnCl2-mediated highly diastereoselective 4C-Ugi reaction employing a variety of aldehydes (aliphatic and aromatic), tert-butyl or phenyl isocyanide and different carboxylic acids (aliphatic and aromatic); in both of these reports the corresponding R-α-amino acid derivatives were obtained (129a or 129b) as shown in Scheme 40.60a–c Interestingly, when they made use of 2,3,4-tri-O-pivaloyl-β-L-arabinosyl amine 130 as the chiral amine, and reacted it in the presence of catalytic amount of ZnCl2 with a variety of aldehydes (aliphatic, aromatic and heteroaromatic), formic acid and tert-butylisocyanide in THF, in each case the corresponding S-α-amino acid derivative 131 was obtained in excellent yield (>95%) and high diastereoselectivity (Scheme 40).60c | ||
| Scheme 40 Diastereoselective synthesis of sugar appended R- or S-amino acid derivatives. | ||
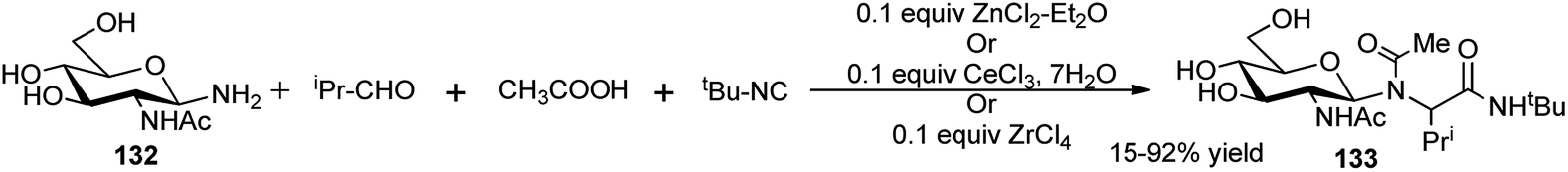
Thus, by proper choice of the sugar as chiral auxiliary, it is possible to generate ultimately both 2R- and 2S-α-amino acids after removal of the carbohydrate surrogate. Later on, Kunz and co-workers also undertook ZnCl2-mediated stereoselective combinatorial 4C-Ugi reactions on solid support based on polymer bound sugar amine derivatives for synthesis of α-amino acid derivatives;60d,e this time equivalent amount of the Lewis acid gave better results, and the performance in the solution phase reactions were better in terms of yields and stereoselectivities than those obtained in the solid phase reactions. Toward synthesis of optically pure α-amino acids, Ugi also studied in detail the effect of substituents and Lewis acids (type and load) on the yield and diastereoselectivity of 4C-sequential one-pot Ugi reactions on unprotected glycosyl amines (glucose, xylose and N-acetylglucosamine) as the chiral amine, aldehydes (aliphatic/aromatic), different acids including also N-protected amino acids and varied isocyanates. Best results in terms of yield (99%) and diastereoselectivity (de 95–99%) of the resulting α-amino acid derivative 133 were obtained based on GlcNAc-1-NH2 (132), iso-propylcarboxaldehyde, acetic acid and tert-butyl isocyanide in the presence of 10 mol% of ZnCl2·Et2O or CeCl3·7H2O in methanol using 4 Å molecular sieves (Scheme 41). A ZrCl4 catalysed similar reaction also could furnish almost quantitative yield of the corresponding α-amino acids albeit in lower de (80%).61
 | ||
| Scheme 41 Sugar-based α-amino acid derivative. | ||
Metal mediated one-pot reactions have also been used for generation of β-amino acids. Toward this end, an InCl3-catalysed asymmetric Mannich type 3C condensation reaction protocol in methanol was developed by Dondoni et al. based on formyl C-glycoside (134), benzyl amine and ketene silyl acetal (135) to afford a highly diastereoselective C-glucosyl/galactosyl/ribosyl/mannosyl-β-amino acid derivatives 136 (Scheme 42).62a,b For the mannose derived amino acid DCM was proved to be the best solvent (due to poor solubility of the corresponding imine intermediate).62b Dondoni's group further synthesised C-glycosylated β-amino acids (137) from one-pot Reformatsky reaction of sugar (glucosyl/mannosyl/ribosyl) aldehyde, p-methoxybenzyl amine and bromozinc enolate as the d2 synthon (Scheme 42).62b For the Reformatsky reaction, use of 5 mol% NiCl2(PPh3)2 in DCM was established as the best catalyst. Both of these reactions proceeded with complete asymmetric induction and with generation of the 3R-configured amino acid.
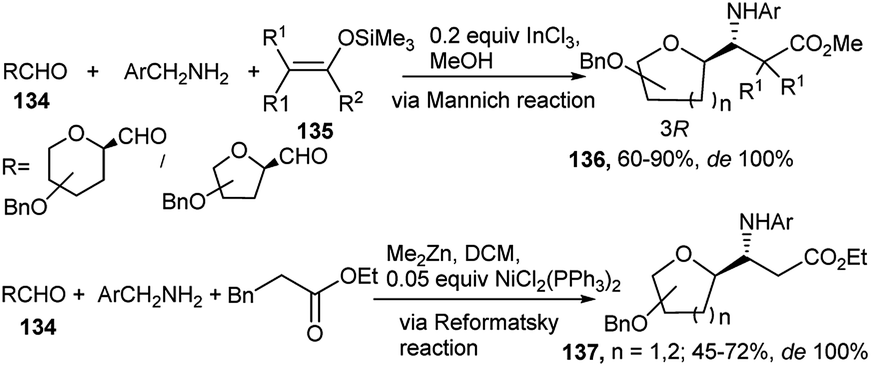 | ||
| Scheme 42 InCl3 and NiCl2(PPh3)2-catalysed diastereoselective synthesis of C-glycosyl β-amino acid derivatives. | ||
6. Metal-catalysed other miscellaneous domino/tandem/sequential/MC-one-pot reactions based on monomeric sugar substrates
Apart from the different types of product categories already discussed, a wide variety of scattered sugar scaffolds of different kinds have also been synthesised by metal-catalysed one-pot transformations, and those will be illustrated as miscellaneous examples in this section.A Bi(OTf)3-catalysed one-pot α-glycosylation–deprotection protocol was examined by Pastore et al. in dioxane–toluene–Et2O mixed solvent, based on mannosyl trichloroacetimidate glycosyl donor 138, bearing Fmoc protection on the C2–O (Scheme 43). After completion of the glycosylation reaction simple addition of triethylamine to the reaction mixture caused one-pot Fmoc deprotection; thus, this reaction sequence led to direct access to a glycosyl acceptor 139. This protocol has then been extended for iterative synthesis of linear and branched oligomannoses related to HIV gp120.63 Along with other transformations Hung's group has described a microwave assisted one-pot conversion of native sugars to 1,6-anhydrosugars 140 or thioglycosides 141 (Scheme 44). It proceeds with initial reaction of the free sugar with HMDS and TMSOTf generating the corresponding per-O-trimethylsilylated monosaccharide, which further reacts in situ with TMSOTf under microwave condition giving the corresponding anhydrosugar (140). Inspired by the success of this one-pot reaction they have further extended it for one-pot synthesis of fully protected thioglycosides from native sugars as shown in Scheme 44; here the initially formed per-O-silylated intermediate sequentially reacted with trimethyl(4-methylphenylthio)silane in the presence of ZnI2 under microwave condition affording the corresponding thioglycoside (141). They then applied the one-pot generated thioglycoside for in situ glycosylation with glycosyl acceptor in the presence of AgOTf, thus getting the corresponding disaccharide in one-pot from its glycosyl donor-based native sugar.64
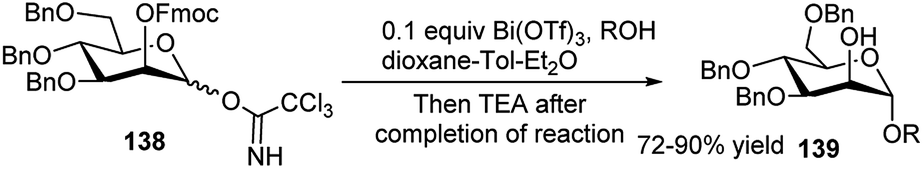 | ||
| Scheme 43 Bi(OTf)3-catalysed one-pot glycosylation–deprotection. | ||
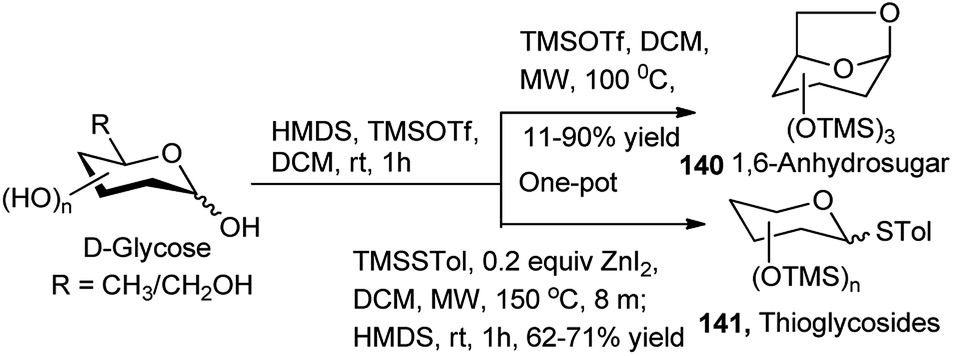 | ||
| Scheme 44 One-pot synthesis of 1,6-anhydrosugars and thioglycosides. | ||
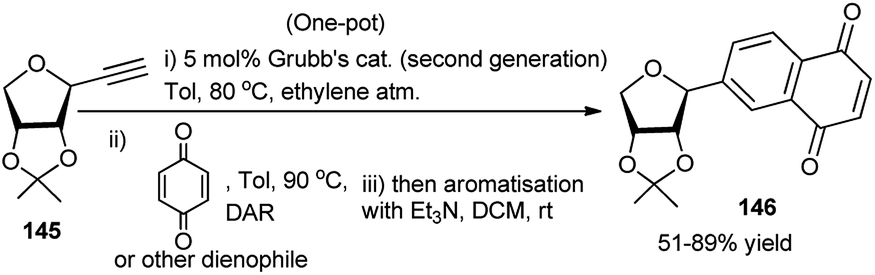
Recently, Messaoudi and co-workers have employed one Pd(II) complex for catalytic one-pot 3C reactions of thiosugars 142, bromo-iodo arenes/heteroarenes (143) and aryl boronic acid to prepare a variety of the corresponding unsymmetrical biaryl thioglycosides 144 as shown in Scheme 45.65 It is to be noted that a variety of biologically important natural products contain unique aryl S-glycosidic66 and aryl C-glycosidic scaffolds (Scheme 45).67 During their stepwise diversity-oriented synthesis of a variety of C-arylglycosides and spiro-C-aryl glycosides, Kaliappan's group prepared an intermediate compound (145) for its onward transformation to the corresponding C-aryl glycoside (146) by a sequential tandem Ru-catalysed enyne metathesis/Diels–Alder cycloaddition/aromatisation in one-pot (Scheme 46). The one-pot yields were however lower (30–35%) than the combined yields (>50%) of the respective stepwise conversion of 145 to the corresponding C-aryl glycosides.68
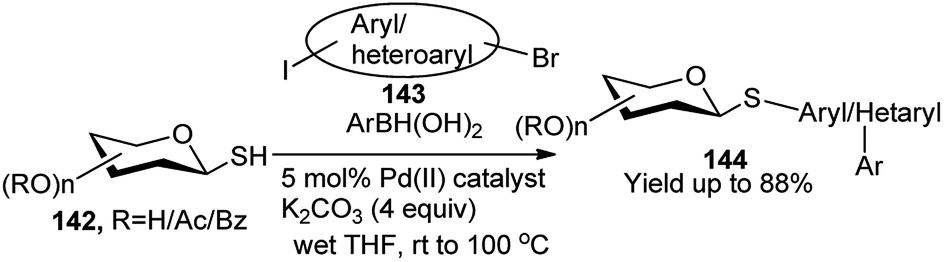 | ||
| Scheme 45 Palladium-catalysed tandem synthesis of unsymmetrical biaryl thioglycosides. | ||
 | ||
| Scheme 46 One-pot synthesis of C-aryl glycosides. | ||
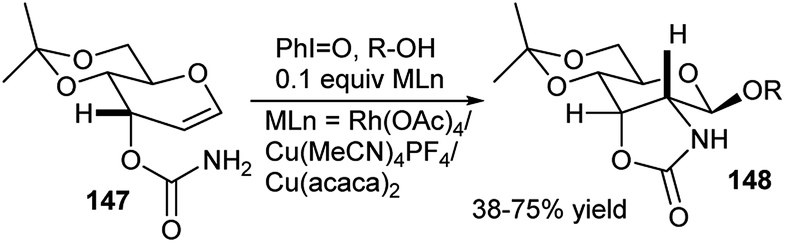
A highly diastereoselective tandem amidoglycosylation reaction was explored from reaction of allal 3-carbamates 147 with alcohol either under photochemical condition or using combined I(III)–MLn [M = Ru/Cu] reagent (Scheme 47). The researchers proposed that the reaction proceeds via formation of a Ru/Cu-acyl nitrenoid intermediate that undergoes tandem amidation–glycosylation with sequential C2–N bond formation-β-glycosylation furnishing the corresponding C2–N anchored glycosides 148.69
 | ||
| Scheme 47 Meta-catalysed one-pot amidoglycosylation. | ||
Mukhopadhyay's group used propargyl glycosides for 3C coupling reactions with aromatic aldehydes and aromatic amines in the presence of CuBr–RuCl3 combined catalyst under microwave condition for generation of the corresponding glycosylated propargyl amine derived glycoconjugates in high to excellent yields.70 A series of glycosylated N-sulfonylamidines 150 were prepared in high yields by this research group employing Cu(I) catalyst by 3C reactions of propargyl glycosides 149, different amines and sulfonyl azides in THF at room temperature (Scheme 48).71
 | ||
| Scheme 48 Cu(I)-catalysed one-pot synthesis of glycosylated N-sulfonylamidines. | ||
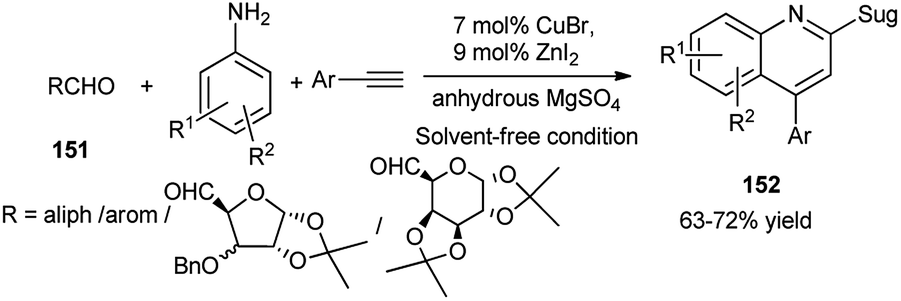
A variety of quinolines including sugar 152 were synthesised by Maity's research group by CuBr–ZnI2 dual catalysed one-pot oxidative 3C-coupling reactions of aldehydes including sugar derived aldehydes (151), aryl amines and aryl acetylenes in the presence of anhydrous MgSO4 under solvent free condition (Scheme 49). They further proved involvement of a Cu(I)–Cu(III) switching process, as evidenced from the peak at 529 nm in the UV-Vis spectrum of the corresponding reaction mixture. This was also corroborated by appearance of respective peaks of Cu(I) and Cu(III) from the XPS experiment. Based on these observations, they also proposed the mechanistic pathway including a catalytic cycle.72
 | ||
| Scheme 49 CuBr–ZnI2-catalysed one-pot synthesis of quinoline glycoconjugates. | ||
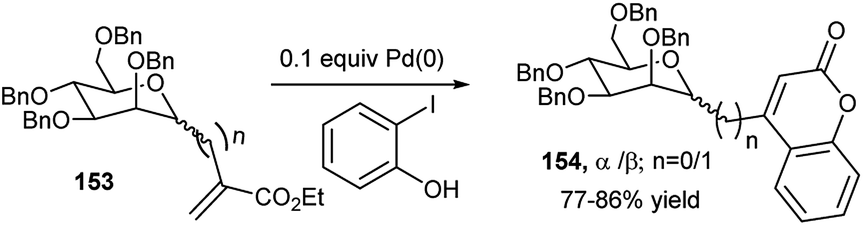
For the synthesis of C2 and C4-substituted quinoline-derived glycoconjugates, Mohan Das's group employed earlier peracetylated propargyl glycoside (glucose and galactose-based), aryl amine and substituted benzaldehyde in THF at 65 °C using CuBr–ZnI2 dual catalyst.73 A series of coumarin appended mannopyranosides (154) were prepared by Pd(0)-catalysed one-pot domino Heck reaction followed by lactonisation of methyl per-O-benzylated C-mannopyranosyl acrylates (153) and 2-iodophenols (Scheme 50).74
 | ||
| Scheme 50 Domino synthesis of C-mannopyranocoumarins. | ||
Previously, J. S. Yadav et al. developed an aqueous-mediated synthesis of sugar annulated pyrroles from 3C one-pot reactions of free aldoses, aryl amines and acetylacetone at 80 °C and isolated the acetylated products (155, Scheme 51) after in situ acetylation with Ac2O and dimethylaminopyridine.75
 | ||
| Scheme 51 Synthesis of sugar annulated pyrroles. | ||
Further reports were made by this group on the microwave-mediated clay K10-catalysed highly cis diastereoselective synthesis of thiosugar annulated bicyclic dihydropyrimidines (157, 158) (or thiones)76 or bicyclic tetrahydropyrimidinones,76 (159, 160) in high yields under solvent-free condition from 3C one-pot reactions of aldoses and 2-methyl-2-phenyl-1,3-oxathiolan-5-one (156) separately with urea/thiourea76 or amidines77 (Scheme 52). The mechanistic pathway of one reaction, as proposed by the researchers, is shown hereunder; the reaction proceeded via intramolecular domino cyclocondensation of the intermediate S, formed from reaction of xylose and 156 (Scheme 52).
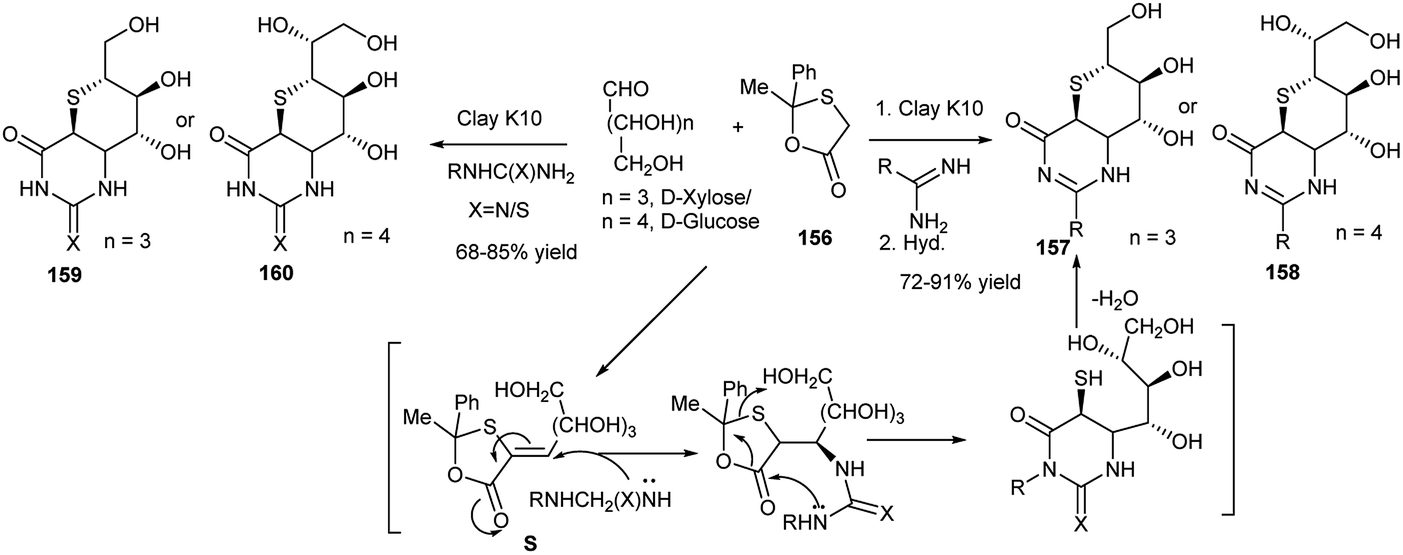 | ||
| Scheme 52 Microwave-mediated clay K10-catalysed one-pot synthesis of thiosugar annulated bicyclic dihydropyrimidines (157 and 158) and bicyclic tetrahydropyrimidinones (or thiones) (159 and 160). | ||
This group also explored the one-pot synthesis of a series of iminosugar-annulated perhydropyrimidines (161a and 161b, Scheme 53) using a Ce(III)-catalysed microwave mediated Biginelli reaction.78 They further synthesised 1,3-oxazin-2-ones 162 (or thiones, 163, Scheme 53) in high yields by clay K10-catalysed microwave enhanced reactions of D-glucose/D-xylose and semi(thiosemi)carbazide under solvent-free condition. They proposed that the reaction proceeds via one-pot domino cycloisomerisation, dehydrazination and dehydration of the initially formed sugar-semi(thiosemi)carbazones.79 They further utilised the tailor-made 1,3-oxazin-2-ones 162 (or thiones, 163) toward diversity-oriented synthesis of other 1,3-oxazin-2-one(thione) fused N-/O-heterocyclic compounds.
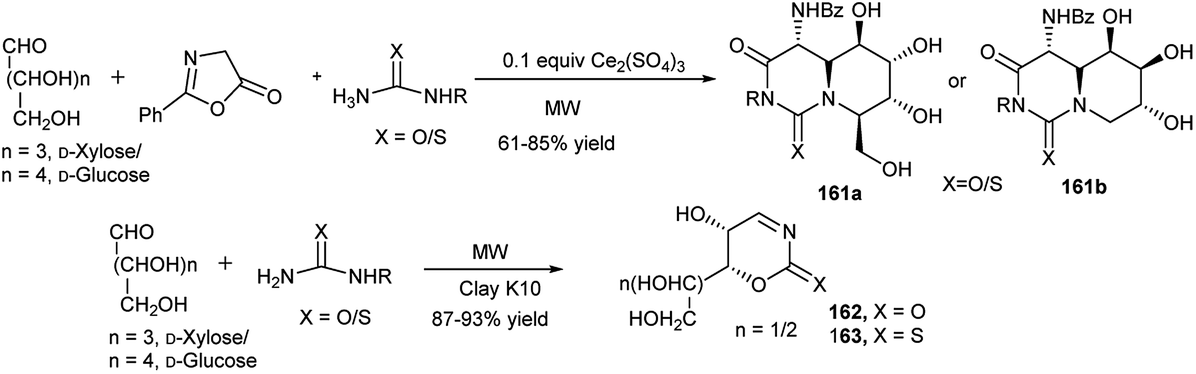 | ||
| Scheme 53 Iminosugar annulated perhydropyrimidines (161) and 1,3-oxazin-2-one/thione (162, 163) synthesis. | ||
Cao et al. described the synthesis of optically pure epimeric aminocyclopentitols (168 and 169) from the corresponding R-/S-tert-butyl sulphonamides (166 and 167), which were prepared by Fe(CO)5-catalysed intramolecular tandem isomerisation-Mannich reaction from sugar derived corresponding anomeric R-/S-tert-butyl sulphonamides (164 and 165) under photochemical condition in THF (Scheme 54).80
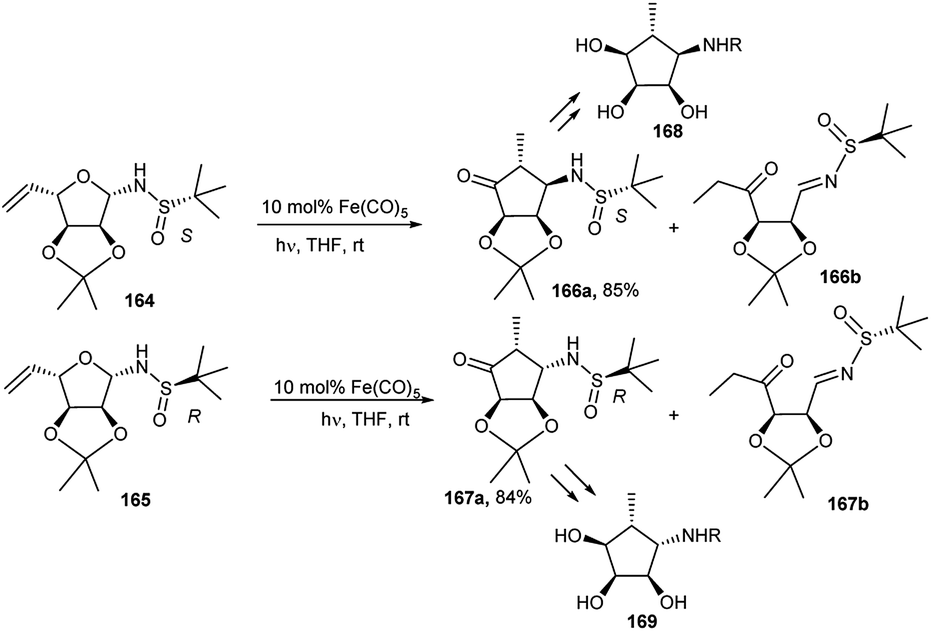 | ||
| Scheme 54 Fe(CO)5-catalysed intramolecular tandem isomerisation-Mannich reaction of sugar derived anomeric sulphonamides toward preparation of epimeric chiral aminocyclopentitols. | ||
An earlier report81 revealed that N-benzyl-β-aminocyclopentitol (168) has a much greater inhibitory activity of α-L-fucosidases from different sources than the corresponding α-amino epimer (169).
7. Metal-catalysed one-pot synthesis of oligosaccharides
During the past two decades, one-pot glycosylation strategies have been successfully utilised for the construction of various oligosaccharides. In general, one-pot glycosylation strategies mainly include orthogonal one-pot glycosylation, reactivity-based one-pot glycosylation, and preactivation-based iterative one-pot glycosylation, among which orthogonal one-pot is particularly more significant. Different combination of glycosyl donors and acceptors, for example, glycosyl trichloroacetimidate (TCAI) and thioglycoside,82 glycosyl N-phenyltrifluoroacetimidate (PTFAI) and thioglycoside,83 S-benzoxazolyl (SBox) glycoside and thioglycoside,84 glycosyl bromide and thioglycoside,85 glycosyl phosphites and thioglycoside, and glycosyl phosphates and thioglycoside86 have been exploited for the two-step, orthogonal one-pot synthesis of several oligosaccharides. Generally, common activator systems like TMSOTf and NIS/TMSOTf, or Cu(OTf)2 and NIS/TMSOTf, or Ag-salt and NIS/TMSOTf were used to accomplish the one-pot oligosaccharide construction. Different metal triflates and transition metals like Pd, Au, Ni, and Fe have been employed extensively as catalyst or promoter system for the activation of TCAI or PTFAI in glycosylation reaction. Interestingly, although these metal catalysts were used in chemoselective glycosylation, but hardly had been exploited to the one-pot synthesis of oligosaccharides. Only a few examples were found in literature. These metal catalysts were used exclusively or in combination with other promoter system in two-step, three-step, or four step orthogonal one-pot syntheses of oligosaccharides.In 2004, Fraser-Reid's research group87 developed a one-pot, double-differential glycosylation strategy based on an n-pentenyl ortho ester activation with NIS and catalytic amount of Yb(OTf)3. In this glycosidation protocol, an acceptor diol was chemo- and regioselectively glycosylated by using an n-pentenyl ortho ester which was activated by NIS/catalytic Yb(OTf)3 followed by subsequent addition of second glycosyl donor, which was either glycosyl trichloroacetimidate or thioglycoside and led to generate several branched oligosaccharides by one-pot under the action of the same activator system. For example, diol 171 or 172 was treated with n-pentenyl ortho ester 170 in the presence of NIS (2.5 equivalents) and Yb(OTf)3 (30 mol%), and after 10 minutes, when the initial glycosylation reaction was supposed to be completed by generation of the intermediate disaccharide 173 or 174, the second donor 175 or 176 was added to the reaction vessel to produce ultimately trisaccharide 177 or 178. Using the same protocol with same donor 170 and acceptor 179 resulted in the intermediate disaccharide 180, followed by addition of the second donor 175 or 176 finally generated the corresponding trisaccharide 181 in good yield (Scheme 55).
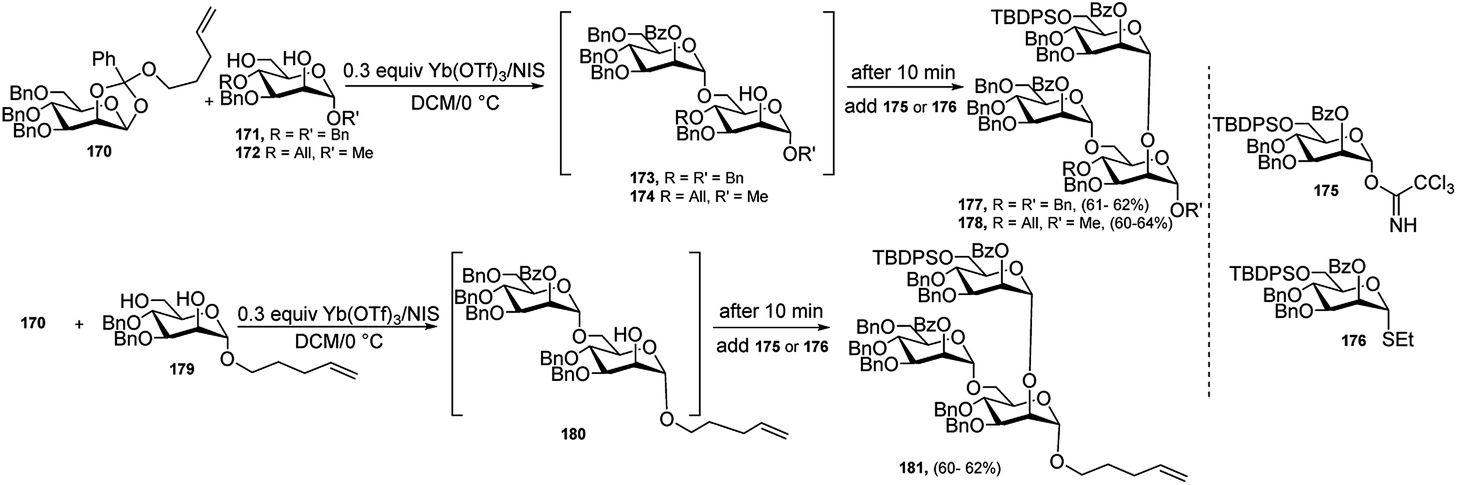 | ||
| Scheme 55 Yb(OTf)3-catalysed one-pot, double-differential glycosidation strategy. | ||
In 2006, M. Adinolfi et al.88 described the catalytic activity of Yb(OTf)3 in the selective activation of glycosyl trichloroacetimidate over glycosyl N-phenyltrifluoroacetimidate with similar protecting groups. In the same paper, they have extended the efficacy of this methodology for the multistep-glycosylation in one-pot. Two model oligosaccharides 186 and 191 were synthesised exploiting this method. Glycosyl donor 182 and acceptor 183 in acetonitrile at −30 °C in the presence of a catalytic amount of Yb(OTf)3 (0.03 equivalent) led to the consumption of the more reactive donor 182 and provided a disaccharide intermediate 184 with anomeric N-phenyltrifluoroacetimidate. On addition of acceptor 185 together with an additional amount of Yb(OTf)3 (0.07 equivalent) in the reaction mixture generated trisaccharide 186. Likewise, catalytic amount of Yb(OTf)3 (0.03 equivalent) was added to a mixture of mannosyl trichloroacetimidate 187 and mannosyl N-phenyltrifluoroacetimidate 188, at −30 °C offering in situ disaccharide intermediate 189, which was then coupled to mannose-based acceptor 190, in the presence of added catalytic amount of Yb(OTf)3 (0.07 equivalent), providing trisaccharide 191 in 40% overall yield (Scheme 56).
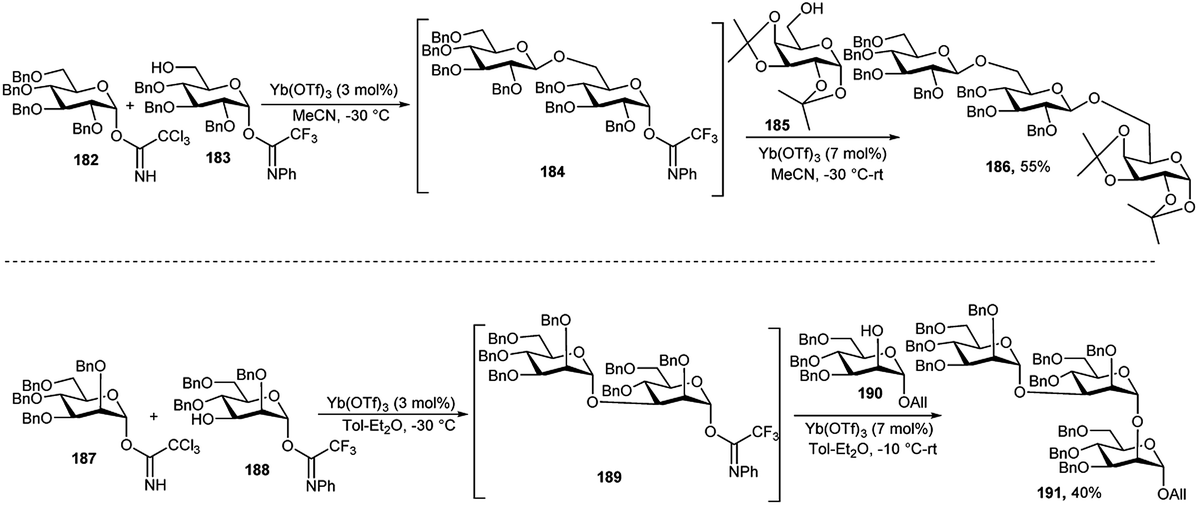 | ||
| Scheme 56 Yb(OTf)3-catalysed one-pot glycosylations toward trisaccharide derivatives. | ||
S. Valerio et al.89 demonstrated a one-pot synthesis of pentasaccharide intermediate 195 during the synthesis of an antitumor PI-88 pentasaccharide 196 using the above-mentioned glycosylation protocol in 2008, except instead of using Yb(OTf)3 only, Bi(OTf)3 was employed for the activation of PFTA. Thus, compound 192 and 188 were treated with a catalytic amount of Yb(OTf)3 (0.03 equivalent), and after the consumption of relatively more reactive mannosyl TCAI 193, a catalytic amount of Bi(OTf)3 (0.025 equivalent) and a trisaccharide acceptor 194 which was also prepared by one-pot manner following above-mentioned technique, were sequentially added, finally providing pentasaccharide 195 in excellent overall one-pot yield (Scheme 57).
 | ||
| Scheme 57 Synthesis of a pentasaccharide derivative by sequential metal-catalysed one-pot glycosylations. | ||
Yu and coworkers90 have recently demonstrated that 1,2-anhydrosugar could be activated by Ph3PAuOTf to undergo glycosylation in the presence of a suitable acceptor, and the resulting 2-OH, generated by in situ epoxide ring opening, can further be glycosylated in a one-pot fashion with a glycosyl ortho-hexynylbenzoate and additional portion of Ph3PAuOTf. During the assembly of cyclic triterpene saponin 202,91 Yu's group applied this technique to synthesise the intermediate 201. Compound 197 was allowed to react with 1,2-anhydroglucose 198 in the presence of PPh3AuOTf (0.2 equivalent) to provide intermediate 199 which then underwent a smooth reaction with ortho-hexynylbenzoate 200 in the presence of an additional portion of PPh3AuOTf (0.37 equivalent) leading finally to desired trisaccharide derivative 201 in a good 60% yield (Scheme 58).
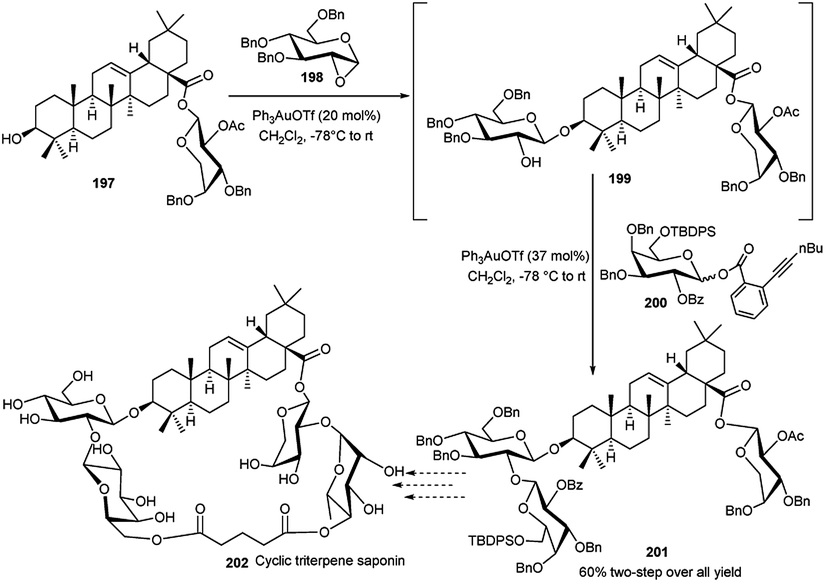 | ||
| Scheme 58 Synthesis toward cyclic triterpene saponin utilising Au(I)-catalysed sequential one-pot glycosylation reactions. | ||
Very recently, Sun's group92 has developed a chemoselective alkyne-activation-based glycosylation strategy employing o-alkynylbenzoate (ABz) and O-(p-methoxyphenylethynyl)phenyl (MPEP) glycosyl donor. Selective activation of ABz donor over MPEP with Ph3PAuNTf2 allowed them to construct complex oligosaccharides via one-pot orthogonal strategy, involving glycosyl trichloroacetimidate (TCAI), o-alkynylbenzoate (ABz) glycosyl donor and o-(p-methoxyphenylethynyl)phenyl (MPEP) glycoside. Two tetrasaccharide, for example, 208 and 209 were thus successfully synthesised. First, TCAI donor 203 and ABz glycosyl acceptor 204 were treated with TMSOTf, providing a disaccharide intermediate, which was then treated with Ph3PAuNTf2 and MPEP glycosyl acceptor 205, giving a trisaccharide intermediate having MPEP as a leaving group, which was further activated with NIS/TMSOTf in the presence of monosaccharide 206 that led to tetrasaccharide 208.
Likewise, synthetically more challenging tetrasaccharide 209 having a 1,4-linked at the non-reducing end was synthesised following the same strategy of consecutive reactions among 203 with 204 followed by 205 and finally 207 (Scheme 59).
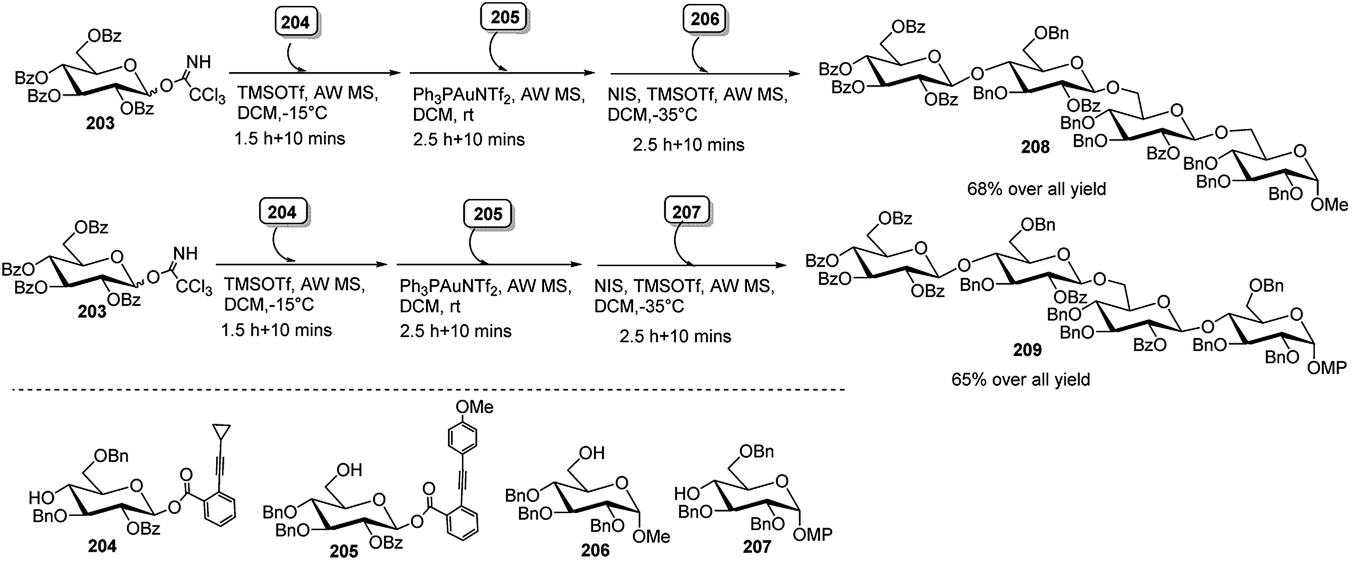 | ||
| Scheme 59 Synthesis of tetrasaccharide derivatives by Au(I)-catalysed sequential one-pot glycosylation reactions. | ||
Zu et al.93a used 5 mol% of Hg(NTf)2 as single catalyst for one-pot synthesis of gentiotetrasaccharide 215, found in lichens and can be employed as a bifidus factor to promote animal growth.93b Per-O-pivaloyl glycosyl trichloroacetimidate donor 210 and bifunctional glucosyl alkynylbenzoate acceptor 211 were treated with 5 mol% of Hg(NTf)2 for 5 minutes. Then, bifunctional enynyl glycosyl acceptor 212 was added to couple with the newly formed disaccharide intermediate, affording the trisaccharide in 5 minutes under the same Hg(NTf)2 catalyst. After that, pentenyl acceptor 213 was injected in the reaction mixture to react with the trisaccharide intermediate. After 30 minutes, the protected tetrasaccharide 214 was formed in 73% overall yield by use of a single catalyst in the reaction system (Scheme 60).
 | ||
| Scheme 60 Single catalyst-based one-pot synthesis of tetrasaccharide 214. | ||
Very recently Yang's group94 reported multistep orthogonal one-pot synthesis of the tetrasaccharide 218. Orthogonal glycosylation of trichloroacetimidate donor 203 with ynenoate acceptor 216 under catalysis of TMSOTf (0.2 equivalent) at −40 °C for 1.5 hours afforded the disaccharide intermediate donor, which was coupled with thioglycoside acceptor 217 under the catalytic activation by Ph3PAuOTf (0.2 equivalent) and TfOH (0.1 equivalent) at room temperature for another 1 hour to provide the intermediate trisaccharide. Coupling of the above intermediate trisaccharide with acceptor 206, promoted by NIS/TfOH at −20 °C for another 1 hour furnished the tetrasaccharide 218, in 66% overall yield (Scheme 61).
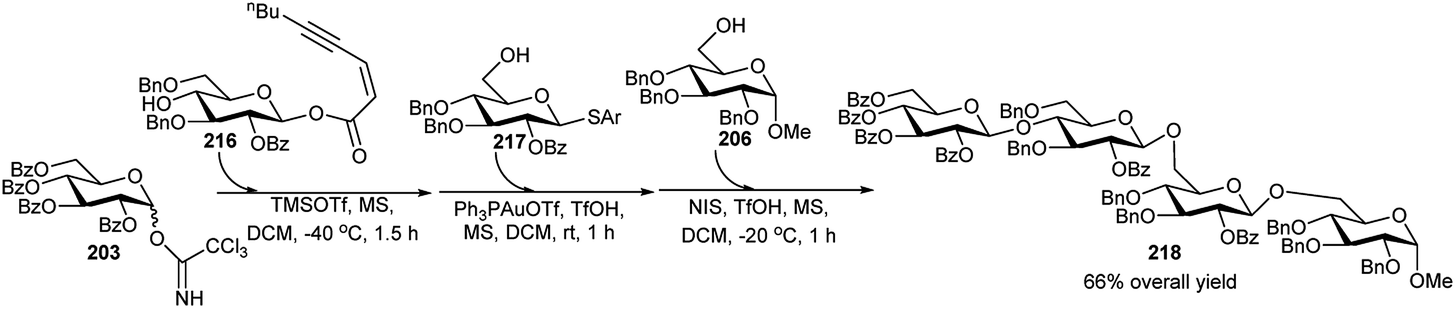 | ||
| Scheme 61 One-pot synthesis of tetrasaccharide-based on glycosyl ynenoate. | ||
Xiao's research group95 effectively employed glycosyl o-alkynylbenzoate (ABz), which can be activated under mild and neutral glycosylation condition, viz only needing Au(I) catalyst, for synthesis of oligosaccharides. They demonstrated a new and highly efficient orthogonal one-pot method for synthesis of oligosaccharides on the basis of glycosyl o-alkynylbenzoate (ABz). Different type of glycosyl donors including glycosyl TCAI,96 glycosyl PTFAI,97 p-toluene thioglycoside (STol),98 SBox glycoside,99 STaz glycoside,100 and glycosyl isoquinoline-1-carboxylate (IQC)101 were utilised in pairing with ABz to develop the corresponding orthogonal one-pot strategy (Scheme 62).
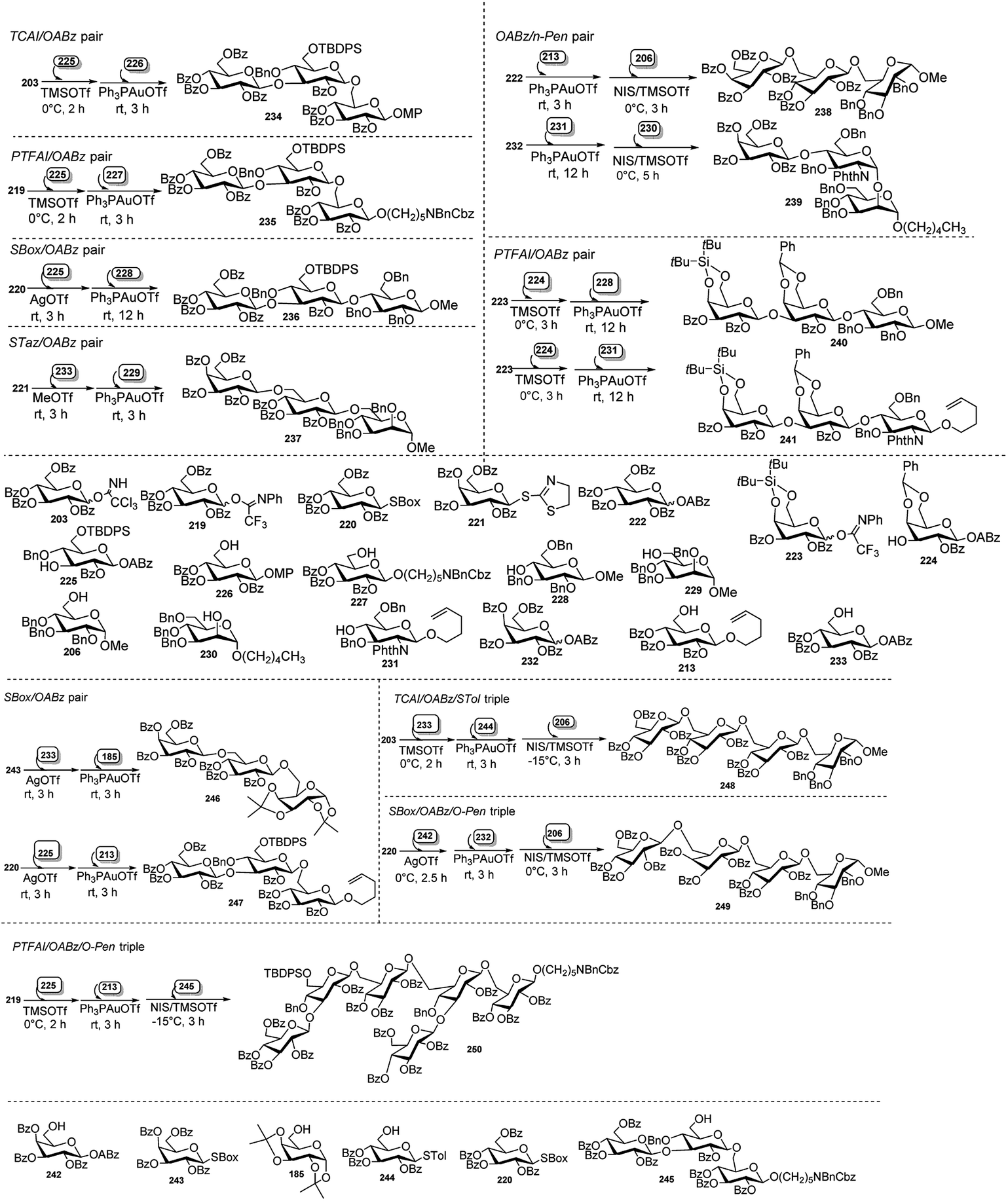 | ||
| Scheme 62 Different orthogonal one-pot strategies toward oligosaccharides. | ||
Recently, Ghosh et al.102 applied orthogonal one-pot glycosylation strategies for the synthesis of pentasaccharide 254, involving glycosyl trichloroacetimidate (TCAI) and thioglycoside. FeCl3 was successfully employed as a catalyst for the activation of glycosyl trichloroacetimidate in the sequential reaction. Pentasaccharide 254 was constructed by two different one-pot glycosylation approaches, one involving [1 + 2 + 2], and the other one [1 + 2 + 1 + 1]. In the [1 + 2 + 2] approach, 0.1 equivalent of FeCl3 was used as a catalyst for activation of rhamnopyranosyl trichloroacetimidate 251 in the presence of disaccharide acceptor 252 in the first step of the one-pot synthesis, providing a thioglycoside trisaccharide intermediate, which was further activated with NIS/FeCl3 with addition of the acceptor 253 leading to fully protected pentasaccharide 254 (Scheme 63). While in the second approach i.e., [1 + 2 + 1 + 1], rhamnopyranosyl trichloroacetimidate 251 was activated similarly with 0.1 equivalent FeCl3 in the presence of disaccharide 252, yielding a trisaccharide intermediate having a thio glycoside terminal. This was then activated in situ by Ph2SO/Tf2O at −60 °C followed by addition of monosaccharide 255 at −40 °C, generating a tetrasaccharide intermediate. This intermediate tetrasaccharide was finally subjected to another round of NIS/FeCl3-mediated glycosylation with acceptor 256 and ultimately providing pentasaccharide 254 in good yield (Scheme 63).
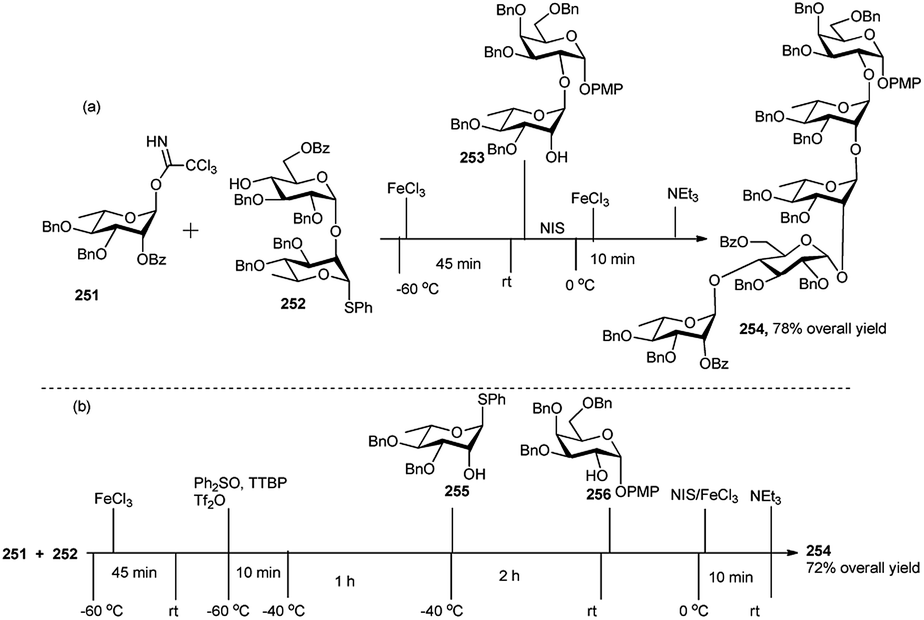 | ||
| Scheme 63 Synthesis of pentasaccharide derivative by (a) sequential 3C one-pot glycosylation reactions; (b) sequential 4C one-pot glycosylation reactions. | ||
8. Conclusion and perspectives
This review is an endeavour to illustrate the metal based one-pot (domino/cascade/tandem/sequential MC) reactions on native sugar feedstock or their suitable derivatives for generation of chiral intermediates for further synthetic application, or of medicinally potent glycoconjugates, or of oligosaccharides of biological relevance. Such metal catalysed single-pot reactions allow construction of library compounds having diverse structures with high regio-, chemo- and stereo-selectivity, sometimes generating high complexity in a simple manner. Because of operational simplicity, avoidance of isolation, purification and characterisation of intermediates, and involvement of much less solvents, time, labour and money unlike those in multistep syntheses, application of such one-pot reactions is increasing gradually.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Council of Scientific and Industrial Research (CSIR), New Delhi (Grant No. 02(0313)/17/EMR-II).Notes and references
- For reviews or books on Domino reactions: (a) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS; (b) H. Pellissier, Chem. Rev., 2013, 113, 442–524 CrossRef CAS; (c) Domino and intramolecular rearrangement reactions as advanced synthesis methods in glycosciences, ed. Z. J. Witczak and R. Bielski, John Wiley & Sons, 2016, p. 368 Search PubMed; For cascade reactions: (d) Metal catalysed cascade reactions, in Topics in organometallic chemistry, ed. T. J. J. Muller, Springer, Berlin, Germany, 2006, vol. 19, p. 339 Search PubMed; (e) K. C. Nicolaou, D. J. Edmonds and P. l. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS; (f) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC; (g) H. Ohno and S. Inuki, Synthesis, 2018, 50, 700–710 CrossRef CAS; For tandem reactions: (h) P. J. Parsons, C. S. Penkett and A. J. Shell, Chem. Rev., 1996, 96, 195–206 CrossRef CAS; (i) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS; (j) C. J. Chapman and C. G. Frost, Synthesis, 2007, 1–21 CAS; For one-pot multicomponent reactions: (k) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef; (l) J. D. Sunderhaus and S. F. Martin, Chemistry, 2009, 15, 1300–1308 CrossRef CAS; (m) S. Brauch, S. S. Van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC; (n) M. Koszytkowska-Stawińska and W. Buchowicz, Beilstein J. Org. Chem., 2014, 10, 1706–1732 CrossRef; (o) B. Rossi, N. Pastori, S. Prosperini and C. Punta, Beilstein J. Org. Chem., 2015, 11, 66–73 CrossRef; (p) H. C. Malinakova, Rep. Org. Chem., 2015, 5, 75–90 CrossRef CAS; (q) Md. M. Khan, R. Yousuf, S. Khan and Shafiullah, RSC Adv., 2015, 5, 57883–57905 RSC; (r) Md. M. Khan, S. Khan, Saigal and S. Iqbal, RSC Adv., 2016, 6, 42045–42061 RSC.
- (a) J. E. Jackman, C. A. Fierke, L. N. Tumey, M. Pirrung, T. Uchiyama, S. H. Tahir, O. Hindsgaul and C. R. Raetz, J. Biol. Chem., 2000, 275, 11002–11009 CrossRef CAS; (b) H. C. Hang and C. R. Bertozzi, J. Am. Chem. Soc., 2001, 123, 1242–1243 CrossRef CAS; (c) X. Li, T. Uchiyama, C. R. Raetz and O. Hindsgaul, Org. Lett., 2003, 5, 539–541 CrossRef CAS; (d) X. Liu, Y. Guo, Y. Li, Y. Jiang, S. Chubb, A. Azuma, P. Huang, A. Matsuda, W. Hittelman and W. Plunkett, Cancer Res., 2005, 65, 6874–6881 CrossRef CAS; (e) I. Velter, B. La Ferla and F. Nicotra, J. Carbohydr. Chem., 2006, 25, 97–138 CrossRef CAS; (f) A. Bernardi and P. Cheshev, Chem.–Eur. J., 2008, 14, 7434–7441 CrossRef CAS; (g) B. Lepenies, J. Yin and P. H. Seeberger, Curr. Opin. Chem. Biol., 2010, 14, 404–411 CrossRef CAS; (h) L. Bouché and H.-U. Reissig, Pure Appl. Chem., 2012, 84, 23–36 Search PubMed.
- E. Lenci, G. Menchi and A. Trabocchi, Org. Biomol. Chem., 2016, 14, 808–825 RSC.
- (a) Essentials of glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, C.S. Harbor Press, NY, 2009 Search PubMed; (b) P. H. Seeberger and R. D. Cummings, Glycans in Biotechnology and Pharmaceutical Industry, in Essentials of Glycobiology, Executive ed. A. Varki, C.S. Harbor Press, NY, 3rd edn, 2017, ch. 57, p. 823 Search PubMed; (c) P. M. Rudd and R. A. Dwek, Curr. Opin. Struct. Biol., 2006, 16, 559–560 CrossRef CAS.
- R. Breinbauer, I. R. Vetter and H. Waldmann, Angew. Chem., Int. Ed., 2002, 41, 2878–2890 CrossRef CAS.
- (a) L. Vilcocq, A. Cabiac, C. Especel, E. Guillon and D. Duprez, Oil Gas Sci. Technol. – Rev. IFP Energies nouvelles, 2013, 68, 841–860 CrossRef CAS; (b) A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC; (c) R. A. Sheldon, Curr. Opin. Green Sustain. Chem., 2018, 14, 89–95 CrossRef; (d) S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS; (e) L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS; (f) X. Liu and R. Wang, Int. J. Chem. Eng., 2018, 2018, 2316939 Search PubMed; (g) H. Zhang, Y. Hu, L. Qi, J. He, H. Li and S. Yang, Int. J. Chem. Eng., 2018, 2018, 7617685 Search PubMed; (h) X. Zou, C. Zhu, Q. Wang and G. Yang, Biofuels, Bioprod. Biorefin., 2019, 13, 153–173 CrossRef CAS; (i) J. Ma, S. Shi, X. Jia, F. Xia, H. Ma, J. Gao and J. Xu, J. Energy Chem., 2019, 36, 74–86 CrossRef; (j) H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS; (k) J. He, H. Li, S. Saravanamurugan and S. Yang, ChemSusChem, 2019, 12, 347–378 CrossRef CAS; (l) W. Khan, X. Jia, Z. Wu, J. Choi and A. C. K. Yip, Catalysts, 2019, 9, 127–150 CrossRef; (m) X. Jin, B. Yin, Q. Xia, T. Fang, J. Shen, L. Kuang and C. Yang, ChemSusChem, 2019, 12, 71–92 CrossRef CAS; (n) G. P. Perez, A. Mukherjee and M.-J. Dumont, J. Ind. Eng. Chem., 2019, 70, 1–34 CrossRef; (o) S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, J. Energy Chem., 2019, 32, 138–151 CrossRef; (p) Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499–3535 RSC; (q) W. Luo, W. Cao, P. C. A. Bruijnincx, L. Lin, A. Wang and T. Zhang, Green Chem., 2019, 21, 3744–3768 RSC; (r) B. C. E. Makhubela and J. Darkwa, Johnson Matthey Technol. Rev., 2018, 62, 4–31 CrossRef CAS; (s) K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 233, DOI:10.3390/en12020233; (t) N. A. S. Ramli and N. A. S. Amin, BioEnergy Res., 2020, 13, 693–736 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS.
- C.-A. Tai, S. S. Kulkarni and S.-C. Hung, J. Org. Chem., 2003, 68, 8719–8722 CrossRef CAS.
- S. Dasgupta, V. K. Rajput, B. Roy and B. Mukhopadhyay, J. Carbohydr. Chem., 2007, 26, 91–106 CrossRef CAS.
- D. Chatterjee, A. Paul, R. Rajkamal and S. Yadav, RSC Adv., 2015, 5, 29669–29674 RSC.
- M. M. Mukherjee, N. Basu, A. Chaudhury and R. Ghosh, RSC Adv., 2016, 6, 109301–109314 RSC.
- Y.-L. Yan, J.-R. Guo and C.-F. Liang, Chem.–Asian J., 2017, 12, 2471–2479 CrossRef CAS.
- S.-S. Weng, Y.-D. Lin and C.-T. Chen, Org. Lett., 2006, 8, 5633–5636 CrossRef CAS.
- (a) B. Ernst, G. W. Hart and P. Sinaÿ, Carbohydrates in Chemistry and Biology, Wiley-VCH, Weinheim, 2000, p. 1 Search PubMed; (b) P. J. Garegg, Preparative Carbohydrate Chemistry, ed. Hanessian, Marcel Decker, New York, 2nd edn, 1997, p. 3 Search PubMed; (c) J. Gelas, Adv. Carbohydr. Chem. Biochem., 1981, 39, 71–157 CrossRef CAS; (d) H.-S. Dang, B. P. Roberts, J. Sekhon and T. M. Smits, Org. Biomol. Chem., 2003, 1, 1330–1341 RSC; (e) D. Crich and Q. Yao, J. Am. Chem. Soc., 2004, 126, 8232–8236 CrossRef CAS; (f) A. Chaudhury, S. K. Maity and R. Ghosh, Beilstein J. Org. Chem., 2014, 10, 1488–1494 CrossRef; (g) P. J. Garegg, Pure Appl. Chem., 1984, 56, 845–858 CAS; (h) C.-R. Shie, Z.-H. Tzeng, S. S. Kulkarni, B.-J. Uang, C.-Y. Hsu and S.-C. Hung, Angew. Chem., Int. Ed., 2005, 44, 1665–1668 CrossRef CAS; (i) R. Panchadhayee and A. K. Misra, Synlett, 2010, 1193–1196 CAS; (j) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 3rd edn, 1999, p. 219 CrossRef; (k) P. J. Kocienski, Protecting Groups, Georg ThiemeVerlag, Stuttgart, Germany, 1st edn, 1994, p. 137 Search PubMed; (l) K. Jarowicki and P. J. J. Kocienski, J. Chem. Soc., Perkin Trans. 1, 2001, 2109–2135 RSC.
- N. Basu, S. K. Maity, S. Roy, S. Singha and R. Ghosh, Carbohydr. Res., 2011, 346, 534–539 CrossRef CAS.
- A.-T. Tran, R. A. Jones, J. Pastor, J. Boisson, N. Smith and M. C. Galan, Adv. Synth. Catal., 2011, 353, 2593–2598 CrossRef CAS.
- (a) C. –C. Wang, J. –C. Lee, S. –Y. Luo, S. S. Kulkarni, Y.-W. Huang, C.-C. Lee, K.-L. Chang and S.-C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS; (b) C.-C. Wang, S. S. Kulkarni, J.-C. Lee, S.-Y. Luo and S.-C. Hung, Nat. Protoc., 2008, 3, 97–113 CrossRef CAS.
- (a) A. A. Joseph, V. P. Verma, X. Y. Liu, C. H. Wu, V. M. Dhurandhare and C. C. Wang, Eur. J. Org. Chem., 2012, 744–753 CrossRef CAS; (b) M. A. Witschi and J. Gervay-Hague, Org. Lett., 2010, 12, 4312–4315 CrossRef CAS; (c) H.-W. Hsieh, M. W. Schombs, M. A. Witschi and J. Gervay-Hague, J. Org. Chem., 2013, 78, 9677–9688 CrossRef CAS; (d) S. S. Park and J. Gervay-Hague, Org. Lett., 2014, 16, 5044–5047 CrossRef CAS; (e) A. A. Joseph, C.-W. Chang and C.-C. Wang, Chem. Commun., 2013, 49, 11497–11499 RSC; (f) A. A. Joseph, V. M. Dhurandhare, C.-W. Chang, V. P. Verma, G. P. Mishra, C.-C. Ku, C.-C. Lin and C.-C. Wang, Chem. Commun., 2015, 51, 104–106 RSC; (g) C. C. Wang, J. C. Lee, S. Y. Luo, H. F. Fan, C. L. Pai, W. C. Yang, L. D. Lu and S. C. Hung, Angew. Chem., Int. Ed., 2002, 41, 2360–2362 CrossRef CAS; (h) R. Enugala, L. C. Carvalho and M. M. B. Marques, Synlett, 2010, 2711–2716 CAS; (i) Y.-C. Ko, C.-F. Tsai, C.-C. Wang, V. M. Dhurandhare, P.-L. Hu, T.-Y. Su, L. S. Lico, M. M. L. Zulueta and S.-C. Hung, J. Am. Chem. Soc., 2014, 136, 14425–14431 CrossRef CAS; (j) P. S. Patil, C.-C. Lee, Y.-W. Huang, M. M. L. Zulueta and S.-C. Hung, Org. Biomol. Chem., 2013, 11, 2605–2612 RSC.
- (a) A. Francais, D. Urban and J.-M. Beau, Angew. Chem., Int. Ed., 2007, 46, 8662–8665 CrossRef CAS; (b) Y. Bourdreux, A. Lemetais, D. Urban and J.-M. Beau, Chem. Commun., 2011, 47, 2146–2148 RSC; (c) A. Gouasmat, A. Lemétais, J. Solles, Y. Bourdreux and J.-M. Beau, Eur. J. Org. Chem., 2017, 3355–3361 CrossRef CAS.
- (a) K.-L. Chang, M. M. L. Zulueta, X.-A. Lu, Y.-Q. Zhong and S.-C. Hung, Org. Lett., 2010, 75, 7424–7427 CAS; (b) M. M. L. Zulueta, D. Janreddy and S.-C. Hung, Isr. J. Chem., 2015, 55, 347–359 CrossRef CAS.
- J.-M. Beau, Y. Bourdreux, G. Despras, A. Gouasmat, G. San Jose, D. Urban and B. Vauzeilles, Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, S. Vidal, Wiley-VCH Verlag, New York, 1st edn, 2019, vol. 7, pp. 201–226 Search PubMed.
- (a) S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS; (b) Y. Tsuda, M. E. Haque and K. Yoshimoto, Chem. Pharm. Bull., 1983, 31, 1612–1624 CrossRef CAS; (c) T. Ogawa and M. A. Matsui, Carbohydr. Res., 1977, 56, c1–c6 CrossRef CAS; (d) D. Wagner, J. P. Verheyden and J. G. Moffatt, J. Org. Chem., 1974, 39, 24–30 CrossRef CAS; (e) R. M. Munavu and H. H. Szmant, J. Org. Chem., 1976, 41, 1832–1836 CrossRef CAS; (f) M. A. Nashed and L. Anderson, Tetrahedron Lett., 1976, 17, 3503–3506 CrossRef.
- (a) Y. Demizu, Y. Kubo, H. Miyoshi, T. Maki, Y. Matsumura, N. Moriyama and O. Onomura, Org. Lett., 2008, 10, 5075–5077 CrossRef CAS; (b) W. Muramatsu, S. Tanigawa, Y. Takemoto, H. Yoshimatsu and O. Onomura, Chem.–Eur. J., 2012, 18, 4850–4853 CrossRef CAS.
- M. Giordano and A. Iadonisi, J. Org. Chem., 2014, 79, 213–222 CrossRef CAS.
- H. Xu, Y. Lu, Y. Zhou, B. Ren, Y. Pei, H. Dong and Z. Pei, Adv. Synth. Catal., 2014, 356, 1735–1740 CrossRef CAS.
- S. Traboni, E. Bedini and A. Iadonisi, Beilstein J. Org. Chem., 2016, 12, 2748–2756 CrossRef CAS.
- (a) R. Sagar and Y. D. Vankar, Tetrahedron Lett., 2008, 49, 4728–4730 CrossRef; (b) H. P. Kokatla, R. Lahiri, P. K. Kancharla, V. R. Doddi and Y. D. Vankar, J. Org. Chem., 2010, 75, 4608–4611 CrossRef CAS; (c) S. K. Yousuf, D. Mukherjee, L. Mallikharjunrao and S. C. Taneja, Org. Lett., 2011, 13, 576–579 CrossRef CAS; (d) L. V. R. Reddy, V. Kumar, R. Sagar and A. K. Shaw, Chem. Rev., 2013, 113, 3605–3631 CrossRef CAS; (e) Md. M. Khan, R. Yousuf, S. Khan and Shafiullah, RSC Adv., 2015, 5, 57883–57905 RSC; (f) H. H. Knife, Org. Biomol. Chem., 2019, 17, 4153–4182 RSC.
- (a) C. Booma and K. K. Balasubramanian, Tetrahedron Lett., 1993, 34, 6757–6760 CrossRef CAS; (b) N. G. Ramesh and K. K. Balasubramanian, Eur. J. Org. Chem., 2003, 4477–4487 CrossRef CAS.
- (a) R. Ghosh, A. Chackraborty, D. K. Maiti and K. Puranik, Tetrahedron Lett., 2005, 46, 8047–8051 CrossRef CAS; (b) R. Ghosh, A. Chackraborty, D. K. Maiti and V. G. Puranik, Org. Lett., 2006, 8, 1061–1064 CrossRef CAS; (c) S. Roy, A. Chackraborty, B. Chattopadhyay, A. Bhattacharya, A. K. Mukherjee, K. Puranik and R. Ghosh, Tetrahedron, 2010, 44, 8512–8521 CrossRef.
- J. S. Yadav, B. V. S. Reddy, L. Chandraiah, B. Jagannadh, S. K. Kumar and A. C. Kunwar, Tetrahedron Lett., 2002, 42, 4527–4530 CrossRef.
- (a) J. S. Jadav, B. V. S. Reddy, G. G. K. S. N. Kumar and T. Swamy, Tetrahedron Lett., 2007, 48, 2205–2208 CrossRef; (b) J. S. Jadav, B. V. S. Reddy, G. G. K. S. N. Kumar and G. M. Reddy, Tetrahedron Lett., 2007, 48, 4903–4906 CrossRef; (c) J. S. Jadav, B. V. S. Reddy, T. Maity and G. G. K. S. N. Kumar, Tetrahedron Lett., 2007, 48, 7155–7159 CrossRef; (d) J. S. Jadav, B. V. S. Reddy, T. Maity and G. G. K. S. N. Kumar, Tetrahedron Lett., 2007, 48, 8874–8877 CrossRef; (e) B. V. S. Reddy, A. V. Ganesh, A. S. Krishna, G. G. K. S. N. Kumar and J. S. Yadav, Tetrahedron Lett., 2011, 52, 3342–3344 CrossRef CAS.
- D. Mukherjee, S. K. Yousuf and S. C. Taneja, Org. Lett., 2008, 10, 4831–4834 CrossRef CAS.
- S. K. Yousuf, S. C. Taneja and D. Mukherjee, J. Org. Chem., 2009, 75, 3097–3100 CrossRef.
- M. Tatina, A. K. Kusunuru, S. K. Yousuf and D. Mukherejee, Chem. Commun., 2013, 49, 11409–11411 RSC.
- A. K. Kusunuru, C. K. Jaladanki, M. B. Tatina, P. V. Bharatam and D. Mukherjee, Org. Lett., 2015, 17, 3742–3745 CrossRef CAS.
- (a) J. S. Yadav, B. V. S. Reddy, M. Srinivas, C. Divyavani, A. C. Kunwar and C. Madavi, Tetrahedron Lett., 2007, 48, 8301–8305 CrossRef CAS; (b) J. S. Yadav, B. V. S. Reddy, G. Satheesh, G. Nara Simhulu, Y. Portier, C. Madhavi and A. C. Kunwar, Tetrahedron Lett., 2008, 49, 3341–3345 CrossRef CAS.
- (a) P. T. Moshapo, M. Sokamisa, E. M. Mmutlane, R. M. Mampab and H. H. Kinfe, Org. Biomol. Chem., 2016, 14, 5627–5638 RSC; (b) P. T. Moshapo and H. H. Kinfe, Synthesis, 2015, 47, 3673–3686 CrossRef CAS.
- (a) S. B. Simelane, H. H. Kinfe, A. Muller and D. B. G. Williams, Org. Lett., 2014, 16, 4543–4545 CrossRef CAS; (b) D. B. G. Williams, S. B. Simelane and H. H. Kinfe, Org. Biomol. Chem., 2012, 10, 5636–5642 RSC.
- (a) M. Leibeling, D. C. Koester, M. Pawliczek, S. C. Schild and D. B. Werz, Nat. Chem. Biol., 2010, 6, 199–201 CrossRef CAS; (b) D. C. Koester, M. Pawliczek, D. Kratzert, B. Dittrich and D. B. Werz, Bioorg. Med. Chem., 2010, 18, 3656–3667 CrossRef; (c) M. Leibeling and D. B. Werz, Chem.–Eur. J., 2012, 18, 6138–6141 CrossRef CAS; (d) M. Leibeling and D. B. Werz, Beilstein J. Org. Chem., 2013, 9, 2194–2201 CrossRef.
- H. Yao, R. William, S. Wang, J. He, T. Guo and X.-W. Liu, Chem.–Asian J., 2019, 14, 4024–4030 CrossRef CAS.
- C. W. Holzapfel and L. Marais, Tetrahedron Lett., 1997, 38, 8585–8586 CrossRef CAS.
- (a) C. W. Holzapfel and L. Marais, J. Chem. Res. (S), 1999, 190–191 RSC; (b) C. W. Holzapfel and L. Marais, S.-Afr. Tydskr. Chem., 1999, 52, 110–111 CAS.
- A. M. Gómez, A. Pedregosa, S. Valverde and J. C. López, Chem. Commun., 2002, 2022–2023 RSC.
- A. M. Gómez, A. Barrio, A. Pedregosa, S. Valverde and J. C. López, Tetrahedron Lett., 2003, 44, 8433–8435 CrossRef.
- A. M. Gómez, A. Barrio, A. Pedregosa, S. Valverde and J. C. López, J. Org. Chem., 2009, 74, 6323–6326 CrossRef.
- (a) S. Mishra, S. Roy, R. Ghosh, D. Dey and B. Hazra, J. Indian Chem. Soc., 2020, 97, 197–211 Search PubMed; (b) D. K. Maiti, S. Halder, P. Pandit, N. Chatterjee, D. De Joarder, N. Pramanik, Y. Saima, A. Patra and P. K. Maiti, J. Org. Chem., 2009, 74, 8086–8097 CrossRef CAS.
- (a) L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS; (b) Perspectives in Nucleosides and Nucleic Acid Chemistry, ed. M. V. Kisakürek and H. Rosemeyer, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- M. Koszytkowska-Stawińska and W. Buchowicz, Beilstein J. Org. Chem., 2014, 10, 1706–1732 CrossRef.
- (a) G. Nagendrappa, Appl. Clay Sci., 2011, 53, 106–138 CrossRef CAS; (b) B. S. Kumar, A. Dhakshinamoorthy and K. Pitchumani, Catal. Sci. Technol., 2014, 4, 2378–2396 RSC; (c) I. R. Siddiqui, S. A. Siddique, V. Srivastava, P. K. Singh and J. Singh, Arkivoc, 2008, xii, 277–285 Search PubMed.
- (a) L. D. S. Yadav and V. K. Rai, Tetrahedron Lett., 2006, 47, 395–397 CrossRef CAS; (b) V. K. Rai and N. Singh, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 247–255 CrossRef CAS.
- S. S. Bag, S. K. Das and H. Gogoi, Tetrahedron, 2018, 74, 2218–2229 CrossRef CAS.
- Suresh and J. S. Sandhu, Arkivoc, 2012, i, 66–133 Search PubMed.
- (a) A. Dondoni, A. Massi and S. Sabbatini, Tetrahedron Lett., 2001, 42, 4495–4497 CrossRef CAS; (b) A. Dondoni, A. Massi, S. Sabbatini and V. Bertolasi, J. Org. Chem., 2002, 67, 6979–6994 CrossRef CAS; (c) A. Dondoni and A. Massi, Mol. Diversity, 2003, 6, 261–270 CrossRef CAS.
- J. Dauvergne, A. Burger and J.-F. Biellmann, Nucleosides Nucleotides Nucleic Acids, 2001, 20, 1775–1781 CrossRef CAS.
- R. C. Larock, Y. Wang, X. Dong and T. Yao, Tetrahedron, 2005, 61, 11427–11439 CrossRef CAS.
- V. Gheerardijn, J. V. den Begin and A. Madder, Beilstein J. Org. Chem., 2014, 10, 2566–2572 CrossRef.
- R. Grigg, E. E. Elboray, M. F. Aly and H. H. Abbas-Temirek, Chem. Commun., 2012, 49, 11504–11506 RSC.
- K. Izawa, Farumashia, 2005, 41, 319–323 CAS.
- (a) I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1, 8–21 CrossRef; (b) I. Ugi and G. Kaufhold, Ann, 1967, 709, 11–28 CAS; (c) I. Ugi, S. Lohberger and R. Karl, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 2, p. 1083 Search PubMed; (d) A. Dömling and I. Ugi, Angew. Chem., Int. Ed. Engl., 2000, 39, 3168–3210 CrossRef; (e) I. Ugi, Pure Appl. Chem., 2001, 73, 187–191 CAS; (f) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef.
- (a) H. Kunz and W. Pfrengle, J. Am. Chem. Soc., 1988, 110, 651–652 CrossRef CAS; (b) H. Kunz and W. Pfrengle, Tetrahedron, 1988, 44, 5487–5494 CrossRef CAS; (c) H. Kunz, W. Pfrengle and W. Sager, Tetrahedron Lett., 1989, 30, 4109–4110 CrossRef CAS; (d) K. Oertel, G. Zech and H. Kunz, Angew. Chem., Int. Ed., 2000, 39, 1431–1433 CrossRef CAS; (e) G. Zech and H. Kunz, Chem.–Eur. J., 2004, 10, 4136–4149 CrossRef CAS.
- J. M. Drábik, J. Achatz and I. Ugi, Proc. Estonian Acad. Sci. Chem., 2002, 51, 156–168 Search PubMed.
- (a) A. Dondoni, A. Massi, S. Sabbatini and V. Bertolasi, Tetrahedron Lett., 2004, 45, 2381–2384 CrossRef CAS; (b) A. Dondoni, A. Massi and S. Sabbatini, Chem.–Eur. J., 2005, 11, 7110–7125 CrossRef CAS.
- (a) A. Pastore, M. Adinolfi, A. Iadonisi and S. Valerio, Carbohydr. Res., 2010, 345, 1316–1323 CrossRef CAS; (b) A. Pastore, M. Adinolfi, A. Iadonisi and S. Valerio, Eur. J. Org. Chem., 2010, 711–718 CrossRef CAS.
- Y.-C. Ko, C.-F. Tsai, C.-C. Wang, V. M. Dhurandhare, P.-L. Hu, T.-Y. Su, L. S. Lico, M. M. L. Zulueta and S.-C. Hung, J. Am. Chem. Soc., 2014, 136, 14425–14431 CrossRef CAS.
- S. Benmahdjoub, N. Ibrahim, B. Benmerad, M. Alami and S. Messaoudi, Org. Lett., 2018, 20, 4067–4071 CrossRef CAS.
- N. Ibrahim, M. Alami and S. Messaoudi, Asian J. Org. Chem., 2018, 7, 2026–2038 CrossRef CAS.
- T. Bililign, B. R. Griffith and J. S. Thorson, Nat. Prod. Rep., 2005, 22, 742–760 RSC.
- A. V. Subrahmanyam, K. Palanichamy and K. P. Kaliappan, Chem.–Eur. J., 2010, 16, 8545–8556 CrossRef CAS.
- E. Levites-Agababa, E. Menhaji, L. N. Perlson and C. M. Rojas, Org. Lett., 2002, 4, 863–865 CrossRef CAS.
- B. Roy, R. Raj and B. Mukhopadhyay, Tetrahedron Lett., 2009, 50, 5838–5841 CrossRef CAS.
- S. Mandal, H. M. Gauniyal, K. Pramanik and B. Mukhopadhyay, J. Org. Chem., 2007, 72, 9753–9756 CrossRef CAS.
- R. R. Mondal, S. Khamarui and D. K. Maiti, ACS Omega, 2016, 1, 251–263 CrossRef CAS.
- K. K. Kumar and T. Mohan Das, Carbohydr. Res., 2011, 346, 728–732 CrossRef.
- D. Giguere, P. Cloutier and R. Roy, J. Org. Chem., 2009, 74, 8480–8483 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, M. Srinivas, C. Divyavani, S. J. Basha and A. V. S. Sarma, J. Org. Chem., 2008, 73, 3252–3254 CrossRef CAS.
- L. D. S. Yadav, C. Awasthi, V. K. Rai and A. Rai, Tetrahedron Lett., 2007, 48, 4899–4902 CrossRef CAS.
- L. D. S. Yadav and A. Rai, Carbohydr. Res., 2009, 344, 2329–2335 CrossRef CAS.
- L. D. S. Yadav, A. Rai, V. K. Rai and C. Awasthi, Synlett, 2007, 1905–1908 CrossRef CAS.
- L. D. S. Yadav, V. P. Srivastava, V. K. Rai and R. Patel, Tetrahedron, 2008, 64, 4246–4253 CrossRef CAS.
- H. T. Cao, T. Roisnel and R. Grée, Eur. J. Org. Chem., 2011, 6405–6408 CrossRef CAS.
- A. Blaser and J.-L. Reymond, Org. Lett., 2000, 2, 1733–1736 CrossRef CAS.
- H. Yamada, T. Harada and T. Takahashi, J. Am. Chem. Soc., 1994, 116, 7919–7920 CrossRef CAS.
- Y. Hsu, H. H. Ma, L. S. Lico, J. T. Jan, K. Fukase, Y. Uchinashi, M. M. L. Zulueta and S. C. Hung, Angew. Chem., Int. Ed., 2014, 53, 2413–2416 CrossRef CAS.
- C.-C. Wang, J.-C. Luo, S.-Y. Lee, S. S. Kulkarni, Y.-W. Huang, C.-C. Lee, K.-L. Chang and S.-C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS.
- S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666–10667 CrossRef CAS.
- C.-H. Hsu, K.-C. Chu, Y.-S. Lin, J.-L. Han, Y.-S. Peng, C.-T. Ren, C.-Y. Wu and C.-H. Wong, Chem.–Eur. J., 2010, 16, 1754–1760 CrossRef CAS.
- K. N. Jayaprakash and B. Fraser-Reid, Org. Lett., 2004, 6, 4211–4214 CrossRef CAS.
- M. Adinolfi, A. Iadonisi and A. Ravidà, Synlett, 2006, 583–586 CAS.
- S. Valerio, A. Pastore, M. Adinolfi and A. Iadonisi, J. Org. Chem., 2008, 73, 4496–4503 CrossRef CAS.
- Y. Li, P. Tang, Y. Chen and B. Yu, J. Org. Chem., 2008, 73, 4323–4325 CrossRef CAS.
- Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem.–Eur. J., 2010, 16, 1871–1882 CrossRef CAS.
- Y. Hu, K. Yu, L.-L. Shi, L. Liu, J.-J. Sui, D.-Y. Liu, B. Xiong and J.-S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS.
- (a) Y. Zu, C. Cai, J. Sheng, L. Cheng, Y. Feng, S. Zhang, Q. Zhang and Y. Chai, Org. Lett., 2019, 21, 8270–8274 CrossRef CAS; (b) M. L. Corradi da Silva, M. Iacomini, E. Jablonski and P. A. J. Gorin, Phytochemistry, 1993, 33, 547–552 CrossRef CAS.
- X. Li, C. Li, R. Liu, J. Wang, Z. Wang, Y. Chen and Y. Yang, Org. Lett., 2019, 21, 9693–9698 CrossRef CAS.
- Y. Zhang, G. Xiang, S. He, Y. Hu, Y. Liu, L. Xu and G. Xiao, Org. Lett., 2019, 21, 2335–2339 CrossRef CAS.
- (a) R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CrossRef CAS; (b) R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef.
- (a) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC; (b) B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS.
- A. V. Demchenko, N. N. Malysheva and C. De Meo, Org. Lett., 2003, 5, 455–458 CrossRef CAS.
- A. V. Demchenko, P. Pornsuriyasak, C. De Meo and N. N. Malysheva, Angew. Chem., Int. Ed., 2004, 43, 3069–3072 CrossRef CAS.
- H.-Y. Wang, C. J. Simmons, S. A. Blaszczyk, P. G. Balzer, R. Luo, X. Duan and W. Tang, Angew. Chem., Int. Ed., 2017, 56, 15698–15702 CrossRef CAS.
- M. M. Mukherjee and R. Ghosh, J. Org. Chem., 2017, 82, 5751–5760 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |