
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New marine-derived indolymethyl pyrazinoquinazoline alkaloids with promising antimicrobial profiles†
Solida Long a,
Diana I. S. P. Resendeab,
Andreia Palmeiraab,
Anake Kijjoabc,
Artur M. S. Silvad,
Maria Elizabeth Tiritanabe,
Patrícia Pereira-Terrabc,
Joana Freitas-Silvabc,
Sandra Barreirof,
Renata Silvaf,
Fernando Remiãof,
Eugénia Pintobg,
Paulo Martins da Costa*bc,
Emília Sousa*ab and
Madalena M. M. Pintoab
a,
Diana I. S. P. Resendeab,
Andreia Palmeiraab,
Anake Kijjoabc,
Artur M. S. Silvad,
Maria Elizabeth Tiritanabe,
Patrícia Pereira-Terrabc,
Joana Freitas-Silvabc,
Sandra Barreirof,
Renata Silvaf,
Fernando Remiãof,
Eugénia Pintobg,
Paulo Martins da Costa*bc,
Emília Sousa*ab and
Madalena M. M. Pintoab
aLQOF – Laboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal. E-mail: esousa@ff.up.pt
bCIIMAR – Centro Interdisciplinar de Investigação Marinha e Ambiental, Terminal de Cruzeiros do Porto de Leixões, Av. General Norton de Matos S/N, 4450-208 Matosinhos, Portugal
cICBAS – Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal. E-mail: pmcosta@icbas.up.pt
dQOPNA – Química Orgânica, Produtos Naturais e Agroalimentares, Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro, Portugal
eCESPU, Instituto de Investigação e Formação Avançada em Ciências e Tecnologias da Saúde (IINFACTS), Rua Central de Gandra, 1317, 4585-116 Gandra PRD, Portugal
fUCIBIO-REQUIMTE, Laboratório de Toxicologia, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
gLaboratório de Microbiologia, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
First published on 21st August 2020
Abstract
Due to the emergence of multidrug-resistant pathogenic microorganisms, the search for novel antimicrobials is urgent. Inspired by marine alkaloids, a series of indolomethyl pyrazino [1,2-b]quinazoline-3,6-diones was prepared using a one-pot microwave-assisted multicomponent polycondensation of amino acids. The compounds were evaluated for their antimicrobial activity against a panel of nine bacterial strains and five fungal strains. Compounds 26 and 27 were the most effective against Staphylococcus aureus ATCC 29213 reference strain with MIC values of 4 μg mL−1, and a methicillin-resistant Staphylococcus aureus (MRSA) isolate with MIC values of 8 μg mL−1. It was possible to infer that enantiomer (−)-26 was responsible for the antibacterial activity (MIC 4 μg mL−1) while (+)-26 had no activity. Furthermore, compound (−)-26 was able to impair S. aureus biofilm production and no significant cytotoxicity towards differentiated and non-differentiated SH-SY5Y cells was observed. Compounds 26, 28, and 29 showed a weak antifungal activity against Trichophyton rubrum clinical isolate with MIC 128 μg mL−1 and presented a synergistic effect with fluconazole.
Introduction
Infectious diseases caused by microorganisms stand as a major threat to public health.1 Since antibiotics were first introduced as medicines, these drugs have been used to prevent or treat infections in several applications.2,3 Nonetheless, antibacterial resistance has increased dramatically, becoming an emergency in healthcare during the last 40 years.4–6 Among 50 emerging infectious agents that have been identified, 10% have developed resistance to multiple drugs including antibiotics such as vancomycin,7,8 methicillin,9 carbapenems,10 and cephalosporins.11–13 Despite enormous efforts, the number of therapeutically useful compounds aiming to circumvent resistance is continuously decreasing and no truly novel class of compounds has been introduced into therapy, causing the World to face the “post-antibiotic era”.14,15 In order to restrain the clinical consequences of the development and spread of antimicrobial resistance both the preservation of current antimicrobials through their appropriate use and the discovery and development of new agents are mandatory.16Promising antimicrobial agents for overcoming multidrug resistance are emerging from a variety of sources and methodologies,17–23 with the very recent example of artificial intelligence aiding this discovery.24 Several reports emphasized the discovery of new sophisticated antimicrobials from marine sources as a promising strategy to overcome the ever-increasing drug-resistant infectious diseases.25–29 Particularly, fungal alkaloids containing an indolomethyl pyrazino[1,2-b]quinazoline-3,6-dione scaffold were isolated from marine organisms and presented very interesting antimicrobial activities.30 For instance, glyantrypine (1, Fig. 1), isolated from Cladosporium sp. PJX-41, exhibited moderate inhibitory activity against a bacterium Vibrio harveyi (MIC = 32 μg mL−1)31 and neofiscalin A (2), obtained fromNeosartorya siamensis KUFC 6349, exhibited a potent antibacterial activity against Staphylococcus aureus and Enterococcus faecalis (MIC = 8 μg mL−1).32 Regarding antifungal activity, fumiquinazoline F (3) for instance, obtained from a culture of Aspergillus fumigatus LN-4 showed good activity (MIC = 12.5–50 μg mL−1) against several plant pathogenic fungi,33 and cottoquinazoline D (4) obtained from Aspergillus versicolor LCJ-5-4, showed moderate antifungal activity against Candida albicans (MIC = 22.6 μM).34
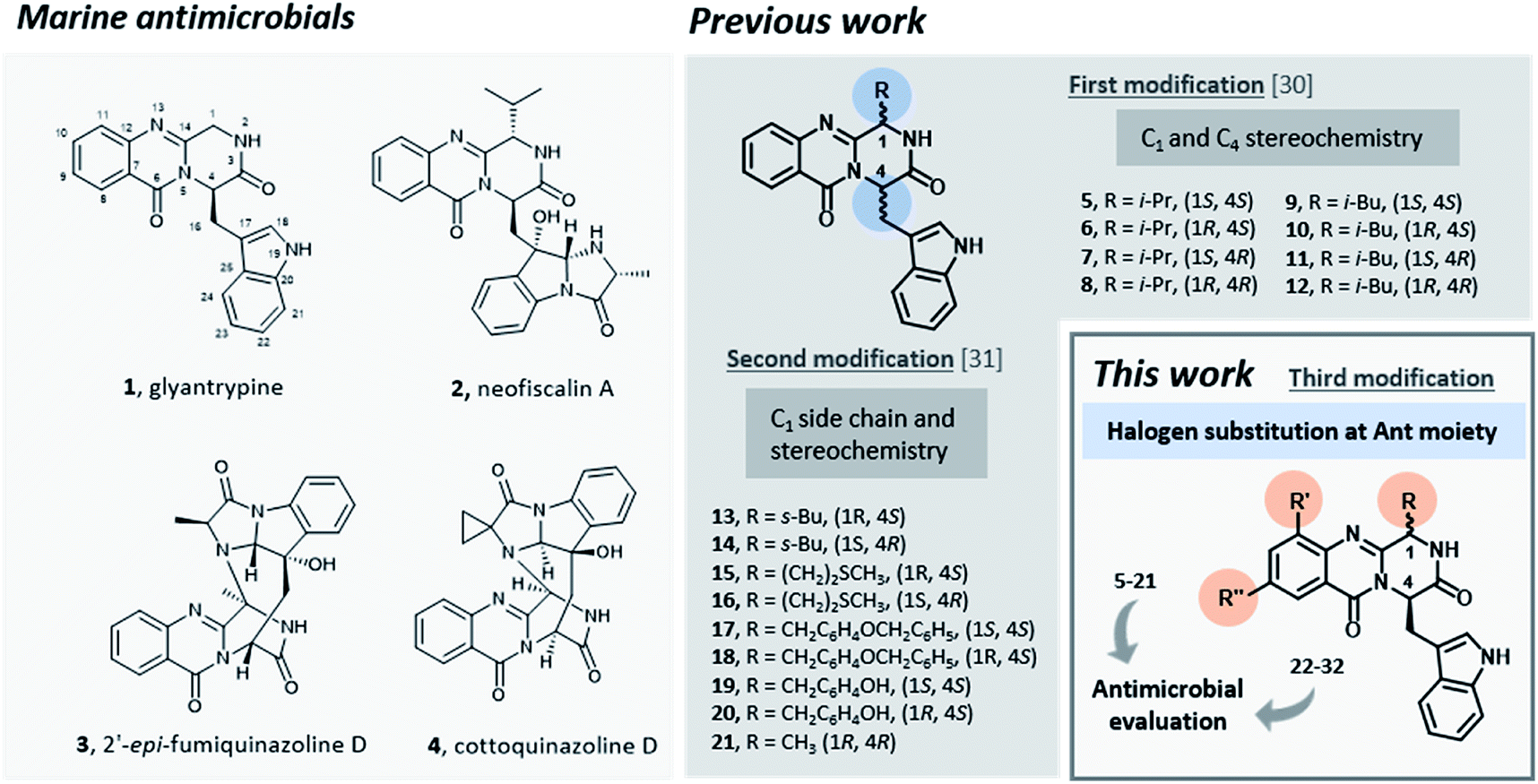 | ||
| Fig. 1 Marine antimicrobials 1–4 and rational of this work. | ||
Inspired by the relevant antibacterial activity of these quinazolinedione natural products30 and the fact that these pure alkaloids are obtained from the fugal culture in low yields,35 we have embarked on a synthesis of pyrazino[1,2-b]quinazoline-3,6-dione derivatives with a simpler indolomethyl moiety decreasing the number of stereogenic centers. Our first approach (5–12, Fig. 1) involved the synthesis of enantiomeric pairs of two members of this quinazolinone family (structural modifications at C-1 and C-4 stereochemistry), inspired by the marine-derived alkaloid fiscalin B (7).36 The second approach (13–21, Fig. 1) consisted in the synthesis of other derivatives of these natural alkaloids, but this time with modification of the C-1 side chain and the stereochemistry, by using different amino acids.37 Influenced by a large number of halogenated marine natural products with interesting antimicrobial activities isolated over the last few years,29,38 in the present work (22–32, Fig. 1) we present a third approach through the introduction of halogen atoms in the aromatic ring of anthranilic acid (Ant) which led to the discovery of promising antimicrobial agents within this series. The potential mechanism of action was studied using in silico docking on three potential antibacterial targets of antimicrobial alkaloids (S. aureus DNA gyrase B (GyrB), and S. aureus FtsZ), followed by molecular dynamics.
Results and discussion
Chemistry
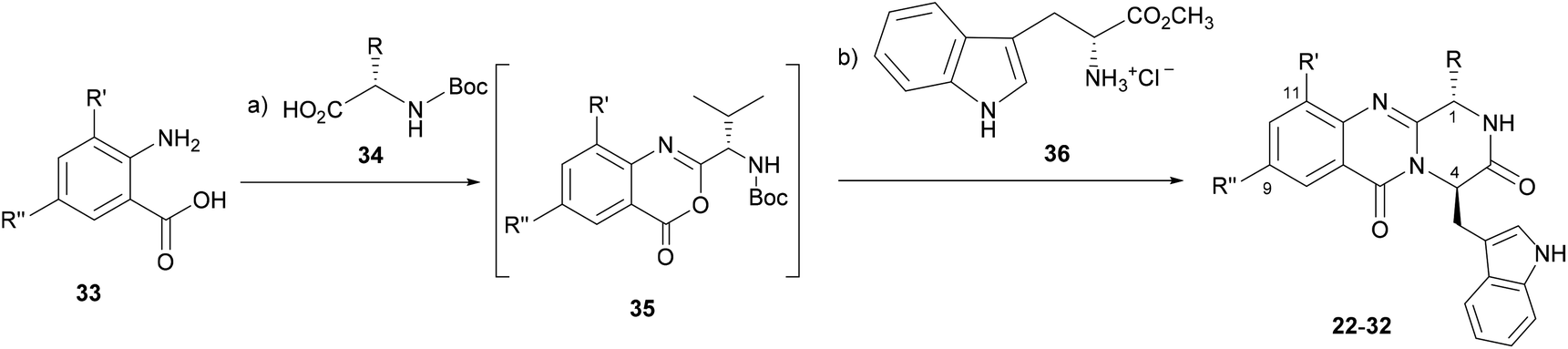
The eleven new indolomethyl pyrazino[1,2-b]quinazoline-3,6-dione derivatives 22–32 were synthesized by a previously described approach using a microwave-assisted multicomponent polycondensation of amino acids with modifications (Table 1).36,37,39 The coupling of halogenated commercial anthranilic acids (33) to N-protected L-α-amino acids (34), and further dehydrative cyclization using triphenyl phosphite [(PhO)3P], generated a kind of benzoxazin-4-ones intermediates 35 which, followed by the addition of D-tryptophan methyl ester (36) under microwave irradiation, furnished the desirable final products 22–32 (2–14% yield) with partial epimerization (Table 1). Starting from halogenated anthranilic acid 33, the reaction conditions had to be optimized to obtain the intermediates 35: the amount of (PhO)3P was increased from 1.2 eq. to 2.4 eq. and the reaction time was increased from 16 to 24 h. With these conditions we were able to increase the scope of this one-pot reaction to halogenated compounds. Interestingly, only anti isomers (1S,4R) were obtained by this methodology whereas the different side chains at C-1 were achieved by selecting diverse L-α amino acids–valine, leucine, and isoleucine. The purity of the compounds was determined, by a reversed-phase liquid chromatography (RP-LC, C18, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O; 50:50), to be on average higher than 95%, whereas that for 25 and 32 was 90%.
:H2O; 50:50), to be on average higher than 95%, whereas that for 25 and 32 was 90%.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R | R′ | R′′ | Yield (%) | [α]Db | erc | % purityd |
| a Reaction conditions: (a) dried-pyridine, (PhO)3P, 55 °C, 24 h; (b) dried-pyridine, (PhO)3P, 220 °C, 1.5 min.b Optical rotation.c er = enantiomeric ratio determined by enantioselective LC (column: amylose, Lux® 5 μm amylose-1, 250 × 4.6 mm, flow rate: 0.5 mL min−1, mobile phase: hexane:EtOH, 9:1).d % purity determined by RP-LC. |
|||||||
| 22 | i-Pr | H | Cl | 5 | −273 | 56:44 |
92 |
| 23 | i-Bu | H | Cl | 3 | +154 | 44:56 |
>99 |
| 24 | s-Bu | H | Cl | 2 | +130 | 46:54 |
93 |
| 25 | i-Pr | Cl | Cl | 5 | +140 | 43:57 |
90 |
| 26 | i-Bu | Cl | Cl | 4.5 | −169 | 60:40 |
>99 |
| 27 | s-Bu | Cl | Cl | 2.6 | −264 | 71:29 |
>99 |
| 28 | i-Pr | H | I | 4.1 | −175 | 51:49 |
95 |
| 29 | i-Pr | H | Br | 1.2 | −170 | 50:50 |
95 |
| 30 | i-Bu | H | I | 11.8 | −165 | 51:49 |
98 |
| 31 | i-Bu | H | Br | 13.8 | −243 | 51:49 |
98 |
| 32 | i-Bu | I | I | 3.5 | −229 | 54:46 |
90 |
Structure elucidation of compounds 22–32 was accomplished by 1D and 2D NMR (using CDCl3 or DMSO-d6 as solvents) spectral analysis and confirmed by HRMS. Particularly, the 1H NMR spectra of the monohalogenated derivatives, 22–24, and 28–31 whose halogen atoms (Cl, Br, or I) are at position 9, exhibited a signal corresponding to the resonance of H-8 in the form of a doublet at δH ca. 8.5 with a small coupling constant (4J = 2.4 Hz), evidencing a long range coupling with H-10; while the signal of H-10 appeared as double doublets at δ ca. 7.9 (3J = 8.7 Hz and 4J = 2.4 Hz). In addition, the signal corresponding to the resonance of H-11 appeared as doublet at δ ca. 7.5 (3J = 8.7 Hz), indicating a short-range coupling to H-10. Similarly, the 1H NMR spectra of the dihalogenated derivatives, i.e. 25–27 and 32, with two halogen atoms (Cl or I) at positions 9 and 11, also appeared as a doublet at δ ca. 7.76 (4J = 2.4 Hz) corroborating a long-range coupling between H-8 and H-10 (See in Material and methods).
It was observed also that the signals of the NH of the indole moiety (H-7′) and the amide group (H-2) appeared at higher frequencies when DMSO-d6 was used as a solvent. For example, for 25, 26, and 32, the signals H-2 and H-7′ appeared at δH 10.20 and 7.11 ppm, respectively, and this was due to the establishment of H-bonding (see the Experimental data).
Microbiology
| Compound | S. aureus ATCC 29213 | S. aureus 66/1 (MRSA) | E. faecalis ATCC 29212 | E. faecalis B3/101 (VRE) | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| Neofiscalin32 | 8 | 16 | 8 | 32 | 8 | 32 | 8 | 32 |
| 5–21 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 22 | 32 | >64 | >64 | >64 | 64 | >64 | >64 | >64 |
| (−)-22 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| (+)-22 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 23 | 32 | >64 | >64 | >64 | 32 | >64 | >64 | >64 |
| 23b | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 24 | 16 | >64 | >64 | >64 | 32 | >64 | >64 | >64 |
| 25 | 16 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 26 | 4 | >64 | 8 | >64 | >64 | >64 | >64 | >64 |
| (−)-26 | 4 | >64 | 4 | >64 | >64 | >64 | >64 | >64 |
| (+)-26 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 27 | 4 | >64 | 8 | >64 | >64 | >64 | >64 | >64 |
| 28 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 29 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 30 | 16 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 31 | 16 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 32 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
All the tested compounds exhibited a bacteriostatic activity, with minimal bactericidal concentrations (MBC) greater than 64 μg mL−1 (Table 2). Compound 24 was the most effective, with MIC values of 32 μg mL−1 and 16 μg mL−1 against E. faecalis ATCC 29212 and S. aureus ATCC 29213, respectively. In the range of concentrations tested, all these compounds were ineffective against E. faecalis B3/101, a VRE strain that was also resistant to ampicillin (Table 2). Regarding S. aureus, 26 and 27 inhibited the growth of methicillin-resistant S. aureus (MRSA) strain 66/1, with MIC values of 8 μg mL−1.
Synergistic effects with vancomycin and oxacillin were evaluated for MDR strains, but no effect was observed. These antibiotics are relevant in the treatment of infections caused by Enterococcus spp. and Staphylococcus aureus, respectively. The compounds showed activity only for Gram-positive strains and, overall, this activity was greater for reference strains than for clinically relevant strains, whether MDR or not. Regarding Gram-positive strains, the range was not equal for all compounds, with a greater number of compounds being active against S. aureus than E. faecalis. As for E. faecalis there appeared to exist an inverse relationship between the compound activity and a resistance against clinically important antibiotics; however, there was not a clear tendency for S. aureus. Noteworthy, the first series of compounds showed no relevant effect in the growth of non-malignant cells.
The ability to prevent biofilm formation was evaluated for compounds with antibacterial activity. Concentrations ranging from 2 × MIC to ¼ MIC were tested against S. aureus ATCC 29213, S. aureus 66/1 and E. faecalis ATCC 29212. The highest concentration tested was 64 μg mL−1, in order to keep a final in-test concentration of DMSO below 1%. The results were interpreted using a comparative classification that classifies adherence capabilities of tested strains into four categories: (i) non-adherent, (ii) weakly adherent, (iii) moderately adherent and, (iv) strongly adherent.42 Optical density cut-off values (ODc) for each microtiter plate were defined as three standard deviations above the mean OD of the negative control. The classification criteria are summarized in Table 3. This classification which uses the negative control as a starting point, instead of using the positive control as reference, reduces the risk of inconsistencies due to external factors that influence biofilm production.43 The compounds tested did not inhibit biofilm formation of E. faecalis ATCC 29212 (data not shown). S. aureus ATCC 29213 was classified as strong biofilm producer, and (−)-26 (2 × MIC, 8 μg mL−1) was able to impair this ability (Table 4), while 22–27, 30 and 31 were not able to prevent the formation of a strong biofilm (data not shown). The biofilm forming ability of S. aureus 66/1, which was also classified as a strong biofilm producer, was impaired by 26 (MIC, 8 μg mL−1 and 2 × MIC, 16 μg mL−1), (−)-26 (2 × MIC, 8 μg mL−1) and 27 (2 × MIC, 16 μg mL−1) (Table 5).
| Classification | Criteria |
|---|---|
| a OD, optical density; ODc, optical density cut-off value. | |
| Non-adherent | OD less than or equal to ODc |
| Weakly adherent | OD more than ODc and less than two-fold ODc |
| Moderately adherent | OD more than two-fold ODc and less than four-fold ODc |
| Strongly adherent | OD more than four-fold ODc |
| Comp. | Concentration (μg mL−1) | OD ± SD | Classification |
|---|---|---|---|
| a OD, optical density; SD, standard deviation; ODc, optical density cut-off value. The classification is based on the criteria from Table 3. Average OD value for negative control was 0.082 ± 0.006, therefore ODc equals 0.082 + (3 × 0.006) = 0.100; 2 × ODc = 0.201; 4 × ODc = 0.402. | |||
| None | 0 | 3.020 ± 0.046 | Strong |
| (−)-26 | 8 (2 × MIC) | 0.347 ± 0.177 | Moderate |
| (−)-26 | 4 (MIC) | 2.231 ± 0.952 | Strong |
| (−)-26 | 2 (½ MIC) | 3.132 ± 0.059 | Strong |
| (−)-26 | 1 (¼ MIC) | 3.119 ± 0.046 | Strong |
| Compound | Concentration (μg mL−1) | OD ± SD | Classification |
|---|---|---|---|
| a OD, optical density; SD, standard deviation; ODc, optical density cut-off value. The classification is based on the criteria from Table 3. Average OD value for negative control was 0.078 ± 0.007, therefore ODc equals 0.078 + (3 × 0.007) = 0.098; 2 × ODc = 0.196; 4 × ODc = 0.391. | |||
| None | 0 | 2.943 ± 0.098 | Strong |
| 26 | 16 (2 × MIC) | 0.112 ± 0.020 | Weak |
| 26 | 8 (MIC) | 0.131 ± 0.045 | Weak |
| 26 | 4 (½ MIC) | 0.453 ± 0.248 | Strong |
| 26 | 2 (¼ MIC) | 2.980 ± 0.549 | Strong |
| (−)-26 | 8 (2 × MIC) | 0.093 ± 0.013 | None |
| (−)-26 | 4 (MIC) | 0.884 ± 0.439 | Strong |
| (−)-26 | 2 (½ MIC) | 2.952 ± 0.437 | Strong |
| (−)-26 | 1 (¼ MIC) | 3.110 ± 0.210 | Strong |
| 27 | 16 (2 × MIC) | 0.285 ± 0.079 | Moderate |
| 27 | 8 (MIC) | 2.753 ± 0.676 | Strong |
| 27 | 4 (½ MIC) | 3.165 ± 0.192 | Strong |
| 27 | 2 (¼ MIC) | 2.755 ± 0.548 | Strong |
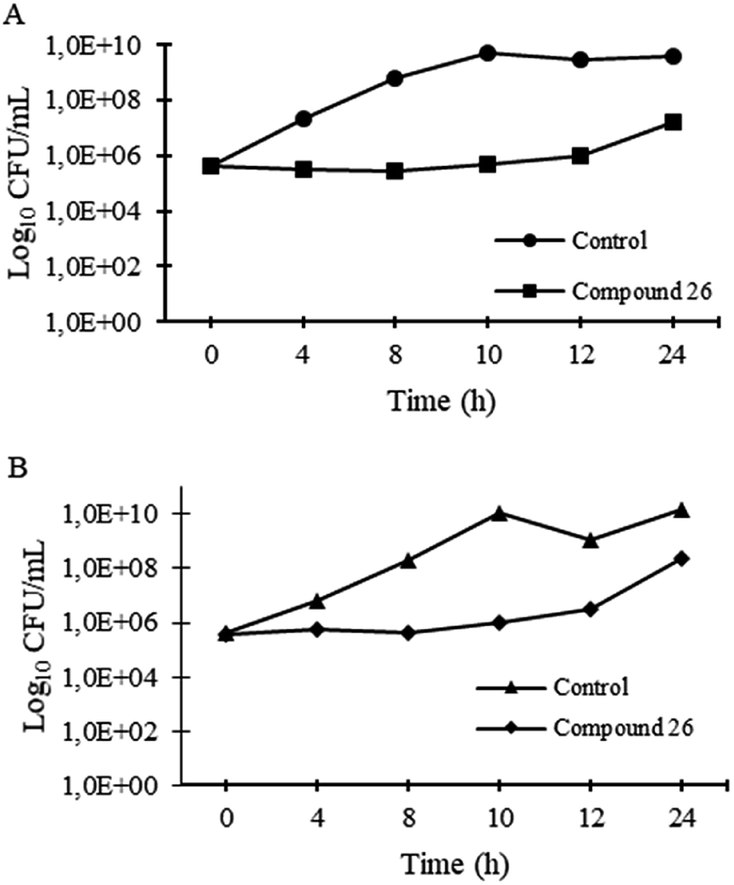 | ||
| Fig. 2 Time-kill kinetics of 26 at 64 μg mL−1 and untreated control for S. aureus ATCC 29213 (A) and S. aureus 66/1 (B) after 20 h incubation. | ||
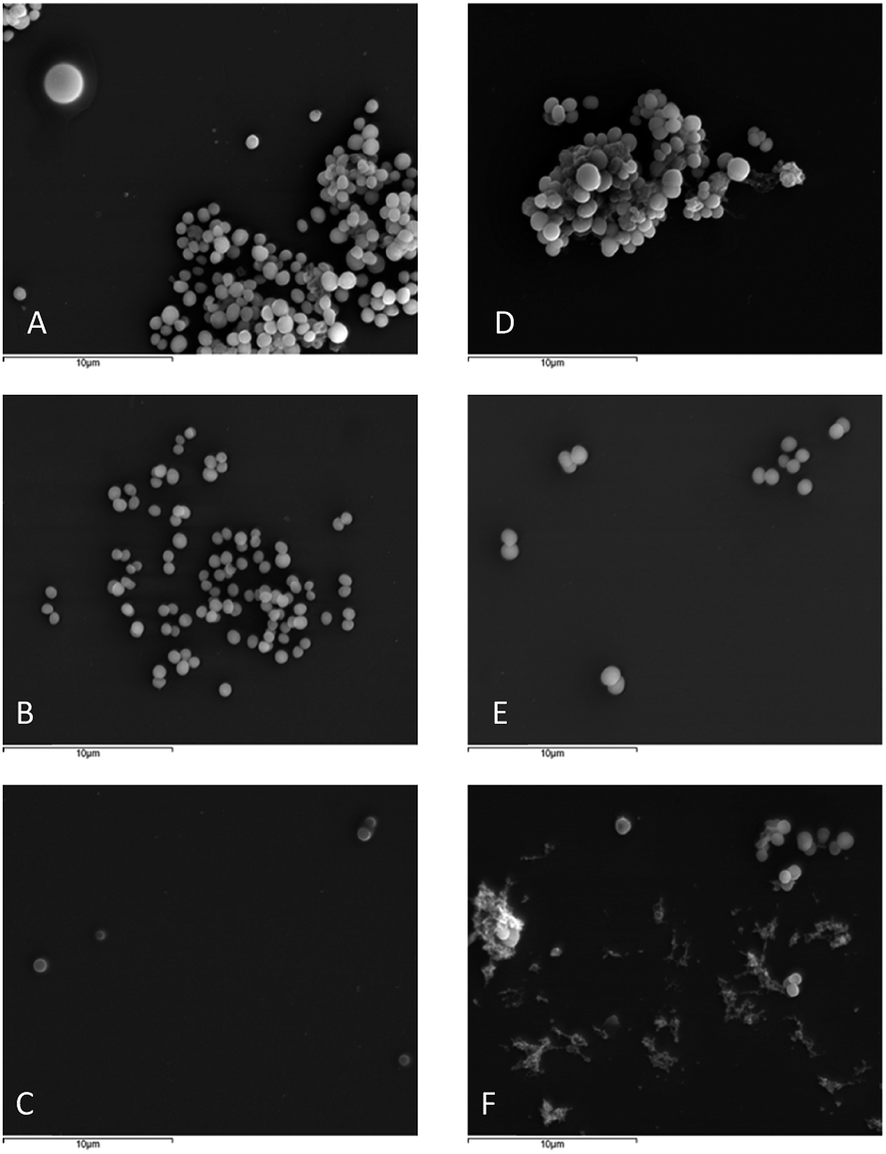 | ||
| Fig. 3 SEM of S. aureus ATCC 29213 untreated (A), treated with ½ MIC (2 μg mL−1) (B) and MIC (4 μg mL−1) (C) of 26; and S. aureus 66/1 untreated (D), treated with ½ MIC (4 μg mL−1) (E) and MIC (8 μg mL−1) (F) of 26 at 5000× amplification. | ||
In order to evaluate in vitro enantioselectivity activities, including antibacterial and antifungal activities, the most promising derivatives 22, 23, and 26 were obtained in milligram scale by a semi-preparative enantioselective liquid chromatography, employing a tris-3,5-dimethylphenylcarbamate amylose column with multiple injection in a 200 μL loop (see ESI† for details).
The pure enantiomers of 22, 23, and 26 were evaluated for antibacterial and antifungal activities. The enantiomer (−)-26 showed a MIC value of 4 μg mL−1 for reference strain S. aureus ATCC 29213, sensitive clinical isolate S. aureus 40/61/24, and methicillin-resistant strain S. aureus 66/1, while the enantiomer (+)-26 showed no effect (Table 2). Noteworthy, these derivatives showed higher potency than the natural product neofiscalin A (2), (tested by the same group with the same conditions).32,47,48 None of the pure enantiomers was active against the fungi tested.
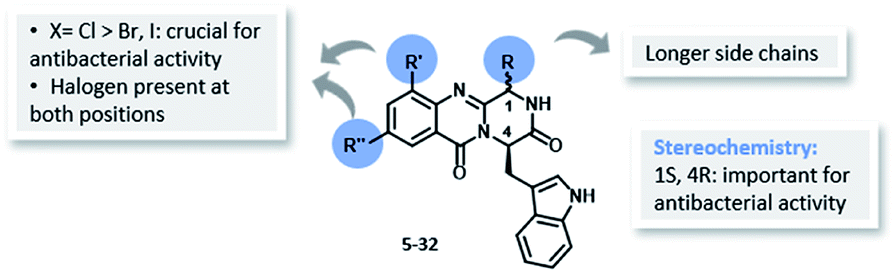 | ||
| Fig. 4 Structure–activity relationship for antibacterial activity of the library of quinazolinones 5–32. | ||
To understand the structural basis of the potential ligand binding to GyrB, and Fts, each prepared ligand was individually docked with the enzymes at their active site using AutodockVina. Test molecules present free binding energies on GyrB ranked from −9.3 to −9.6 kcal.mol−1 (Table 6), and all of the test compounds are predicted to establish more stable complexes with GyrB (lower docking score than test compound 08B; −7.8 kcal.mol−1). All the test compounds are predicted to have lower affinity to FtsZ than test compound 9PC (docking scores higher than −10.1 kcal.mol−1, respectively). Therefore, as GyrB has already been reported as a target for other quinazoline derivatives49,50 it is hypothesized as a possible target for the tested antimicrobial pyrazinoquinazolines. However, further target-specific testing will be performed in the near future to counterproof the in silico findings. s Spatial conformation of test compounds and key interactions of 26 in the active site of GyrB are presented in Fig. 5.
| Test molecules | Docking scores (kcal mol−1) | |
|---|---|---|
| 3u2d (GyrB) | 4dxd (FtsZ) | |
| a 08B (3u2d co-crystallized 4-bromo-5-methyl-N-[1-(3-nitropyridin-2-yl)piperidin-4-yl]-1H-pyrrole-2-carboxamide), and 9PC (4dxd co-crystallized 3-[(6-chloro[1,3]thiazolo[5,4-b]pyridin-2-yl)methoxy]-2,6-difluorobenzamide). | ||
| 22 | −9.5 | −9.5 |
| 23 | −9.6 | −9.5 |
| 24 | −9.3 | −8.5 |
| 25 | −9.6 | −7.8 |
| 26 | −9.6 | −9.7 |
| 27 | −9.5 | −6.9 |
| 28 | −9.3 | −8.4 |
| 29 | −9.5 | −9.1 |
| 30 | −9.5 | −7.7 |
| 31 | −9.4 | −9.1 |
| 32 | −9.4 | −8.1 |
| 08B | −7.8 | — |
| 9PC | — | −10.1 |
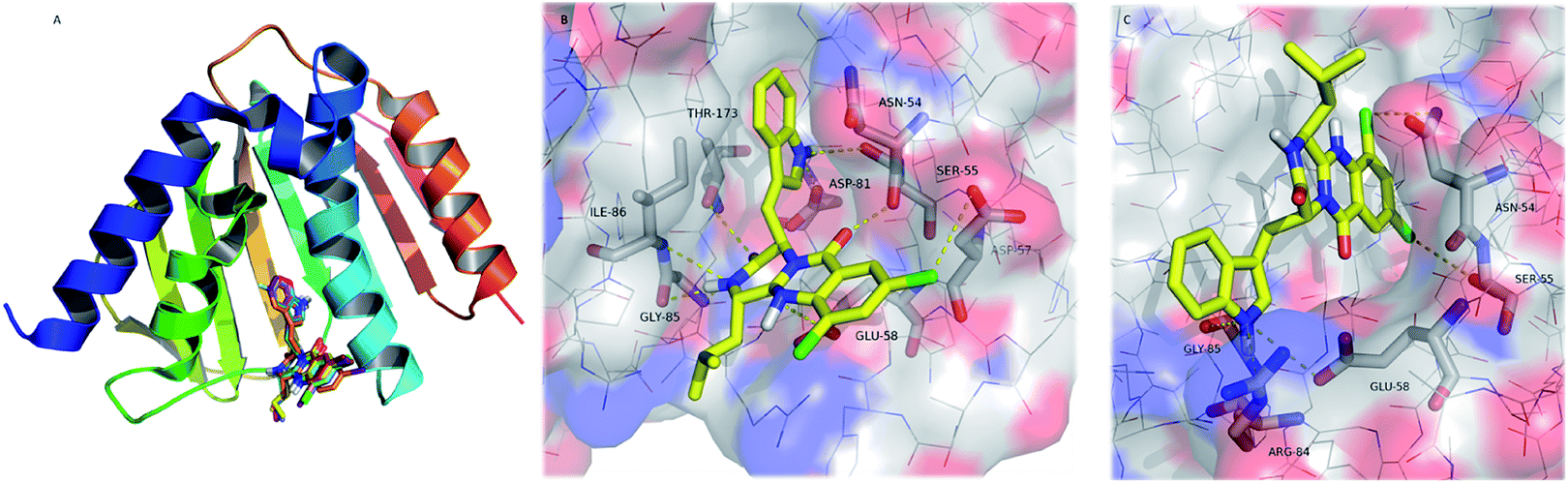 | ||
| Fig. 5 (A) Ribbon representation of GyrB (pdb code 3u2d), and docked 22–32 (sticks). (B and C) Detailed view of two representative top docked poses of 26 (yellow sticks) with polar interactions depicted as yellow broken lines (residues evolved are labelled); GyrB is represented as transparent surface, with carbons, oxygens, and nitrogens coloured grey, red, and blue, respectively. Hydrogens are omitted for simplification. | ||
The pyrazinoquinazoline moiety binds to the ATP binding site of GyrB, participating in hydrogen interactions with Ser-55, Glu-58, Gly-85, Ile-86 and Thr-173, and halogen interaction with Asp-57whereas the indole group is involved in hydrogen interactions with Asn-54 and Asp-81 (Fig. 5B). Alternatively, another docking pose suggests hydrogen interactions with Glu-58, Arg-84, and Gly-85, as well as halogen interactions with Asn-54 and Ser-55 (Fig. 5C). Some of these residues have already been described as being involved in the binding of known inhibitors to GyrB51–54 In both docking poses (Fig. 5B and C), the halogen atoms are predicted as being involved in the interaction with GyrB. In fact, currently, the halogenation of compounds has become an important strategy in drug design.55
Halogens are found to increase membrane permeability and the t1/2 by lowering metabolic degradation, and enhance binding affinity to targets.56,57 Halogens, especially chlorine and bromine, are present in a significant number of drugs, contributing favourably to ligand–protein interaction.58 Halogen bonding is a non-covalent interaction similar to the hydrogen bond (halogens act as hydrogen bond acceptors), and characterized by its directionality.59 There are several physico–chemical properties of chloride that may justify its more favourable contribution to activity when compared to bromide and iodide. In fact, chloride is more electronegative and it is a smaller atom, forming halogen bonds with specific lengths and angles, which contribute to a more appropriate fit to the target stereoelectronic profile. Furthermore, the presence of halogens has already been described as being responsible for the increase in the affinity of known inhibitors to the GyrB ATP binding site.60,61
However, molecular docking using a rigid target may be insufficient for the determination of the structure and the stability of the ligand:target complex. To gain a detailed insight in the energetic and geometric behaviour of the 26:GyrB complex in aqueous environment, a 2 ns MD simulation was performed based on two most stable complex structures obtained from the docking study considering the effects of the target flexibility and the explicit water solvation.
The final MD conformation of 26:GyrB complexes and potential energy plots of ligand conformations along the simulation time (Fig. 6) were obtained. When MD was performed on the 26's top docked conformation (Fig. 6A), the obtained result after 2200 ns simulation shows some differences from the original ligand conformation, namely the loss of the halogen interaction with Asp-57 (instead, the halogen binds to outside water molecules-not shown for simplification). Another high-ranked docking pose of 26 (with the chlorides inside the pocket) (Fig. 6B) was subjected to MD, showing that halogen interactions are established to Ile-51 and to Ser-129 via a water molecule; and hydrogen bonds are formed between 26 and Asn-54, Glu-58, and Arg-84.62 In conclusion, the halogenation of pyrazinoquinazolines was a successful approach in drug design of new antimicrobial compounds, potentially increasing the binding affinity and binding selectivity to GyrB.
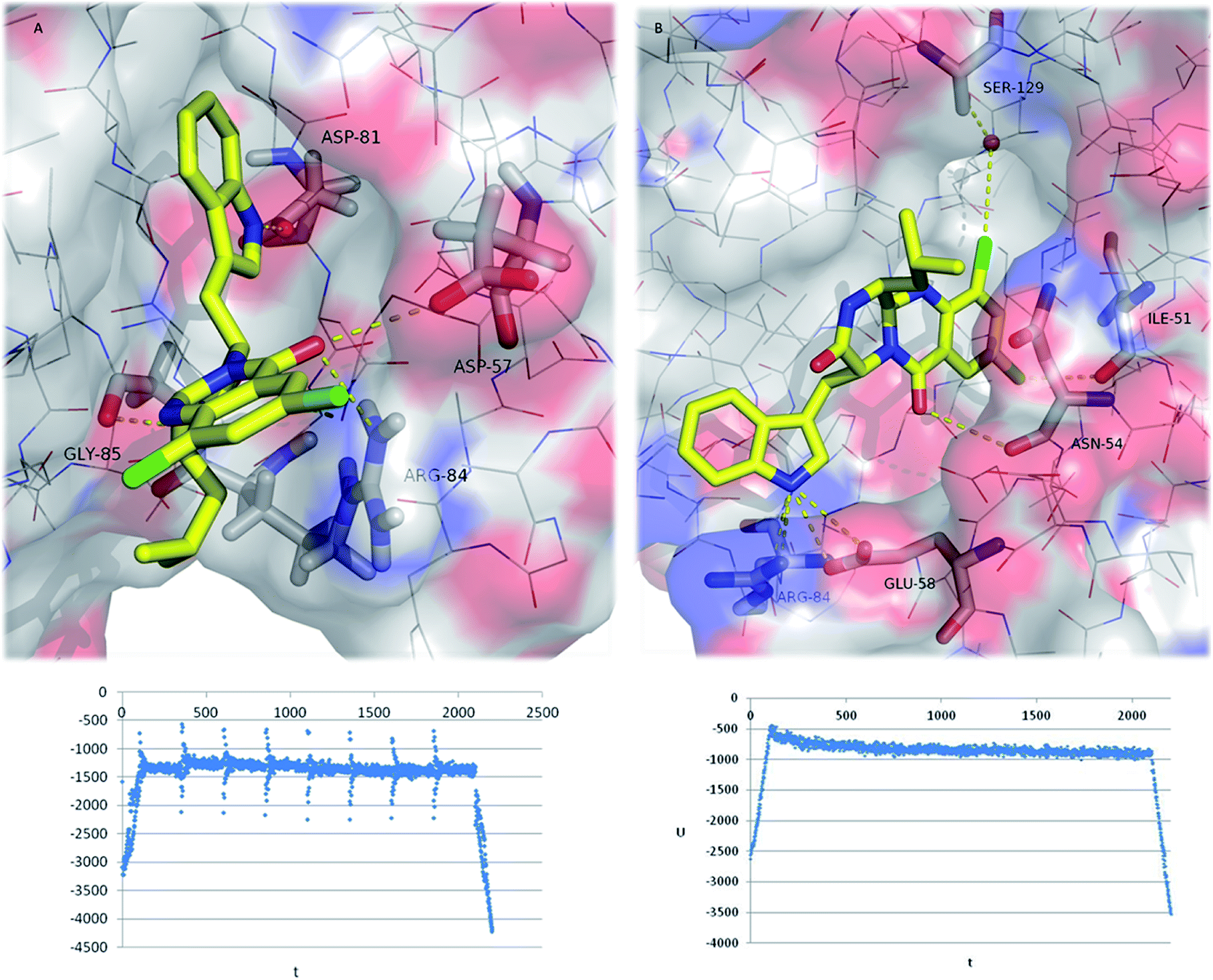 | ||
| Fig. 6 Final (2200 ps) MD conformations of 26:GyrB complexes (top ranked docking poses (A) and (B)). Polar interactions are depicted as yellow broken lines (residues evolved are labelled); GyrB is represented as transparent surface, with carbons, oxygens, and nitrogens coloured grey, red, and blue, respectively. Hydrogen atoms are omitted for simplification. Potential energy plot of the complexes during MD simulation is represented below each MD image (U = potential energy; t = time). | ||
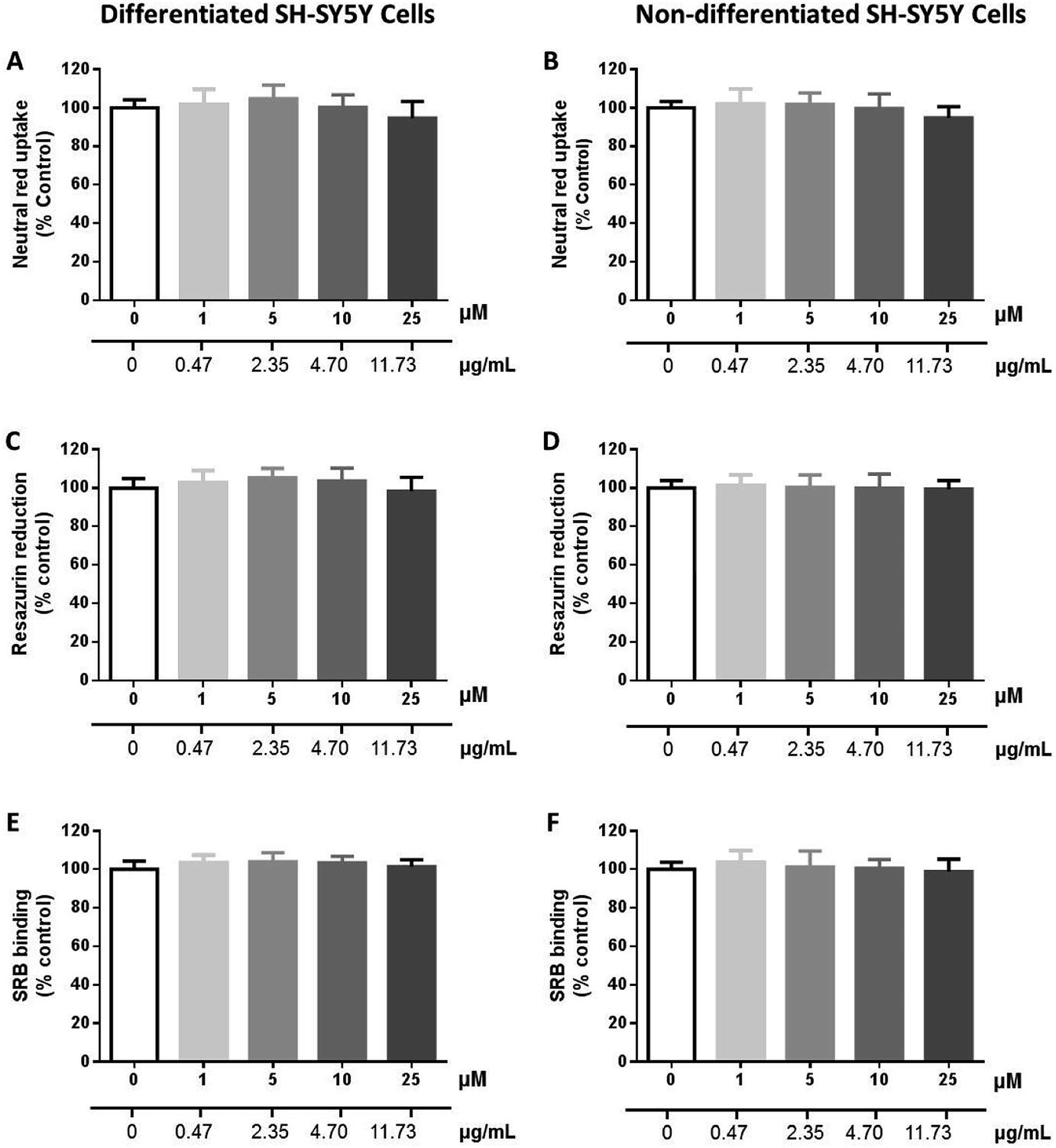 | ||
| Fig. 7 Cytotoxicity of 26 (0–25 μM) in differentiated (A, C and E) and non-differentiated (B, D and F) SH-SY5Y cells evaluated by the neutral red (NR) uptake (A and B), resazurin (REZ) reduction (C and D) and sulforhodamine B (SRB) binding (E and F) assays, 24 h after exposure. Results are expressed as mean ± SD from at least 3 independent experiments, performed in triplicate. Statistical comparisons were made using the parametric method of one-way ANOVA, followed by the Dunnett's multiple comparisons test. | ||
Conclusion
Among a new series of indolomethyl pyrazino[1,2-b]quinazoline-3,6-diones, 26 and 27 exhibited a potent antibacterial activity against S. aureus strains, with MIC values of 4 μg mL−1 for a reference strain and 8 μg mL−1 for a methicillin-resistant strain (S. aureus 66/1). Comparing with the marine natural product neofiscalin A (2), a two-fold reduction in the MIC values was observed. Regarding the structural complexity and synthetic pathways, the excellent and inspiring results obtained in the present study show that simpler molecules than neofiscalin A (2) are quite promising to find new agents to overcome MDR strains. In silico docking and molecular dynamics revealed that halogenation was a successful design strategy to discover new indolymethyl pyrazinoquinazoline with antimicrobial activity. Although the investigated biological activities of fiscalins are still in preliminary stages, there is no doubt that these are privileged structures with potential as drug candidates. Nevertheless, mainly due to the limited amounts isolated, most of the studies correspond to phenotypic screening assays and little is known about their molecular targets and pharmacokinetic properties. One limitation of the one-pot synthesis is related to the low yields obtained, which can be a drawback on the scale up of these compounds. The main challenge in synthesis is the intricate stereochemistry found in the more complex structures. Although the three-component one-pot assembly of this scaffold is a very efficient procedure to obtain some of these secondary metabolites, the high temperatures involved resulted in partial epimerization. Differentiation between stereoisomers was noted in their antimicrobial action but also occurs in drug disposition and is of particular significance for those processes that depend upon a direct interaction between the drug and a chiral biological macromolecule, e.g., active transport processes, binding to plasma and tissue proteins, and drug metabolism, deserving this issue special attention in future work.Experimental
Materials and methods
:H2O (50:50), and the flow rate was 1.0 mL min−1. Enantiomeric ratio was determined by enantioselective LC (LCMS-2010EV, Shimadzu, Lisbon, Portugal), employing a system equipped with a chiral column (Lux® 5 μm Amylose-1, 250 × 4.6 mm) and UV-detection at 254 nm, mobile phase was hexane:EtOH (90:10) and the flow rate was 0.5 mL min−1. For semi-preparative chromatography, a HLPC system consisted of a Shimadzu LC-6AD pump with a 200 μL loop was used with an amylose tris-3,5-dimethylphenylcarbamate coated with Nucleosil (500 A, 7 m, 20%, w/w) packed into a stainless steel (200 mm × 7 mm I.D. size) column, prepared in the UFSCar laboratory.63 Reagents used in cell culture, including Dulbecco's modified Eagle's medium (DMEM) high glucose, sodium bicarbonate, trypsin–ethylenediamine tetraacetic acid (EDTA) solution (0.25% trypsin/1 mM EDTA), retinoic acid (RA), 12-O-tetradecanoylphorbol-13-acetate (TPA), Trizma® base, neutral red (NR) solution, sulforhodamine B (SRB) and resazurin (REZ) were obtained from Sigma-Aldrich (Germany). Antibiotic mixture (10000 U mL−1 penicillin, 10000 μg mL−1 streptomycin) was obtained from Biochrom (Germany). Triton™ X-100 detergent solution was acquired from Thermo Fisher Scientific (Waltham, MA, USA). Heat inactivated fetal bovine serum (FBS), Hanks' balanced salt solution (HBSS) with or without calcium and magnesium [HBSS (+/+) or HBSS (−/−), respectively], and phosphate buffer solution with or without calcium and magnesium [PBS (+/+) or PBS (−/−), respectively] were obtained from Gibco (United Kingdom).:EtOAc (60:40) as a mobile phase. The preparative TLC was performed using DCM:Me2CO (95:5) as mobile phase. The major compound appeared as a black spot with no fluorescence under the UV light. The desired compounds were collected as yellow solids. Before analysis, compounds were recrystallized from MeOH.:44; mp: 200.3–202.4 °C; [α]D30 = −273 (c 0.05; CHCl3); νmax (KBr) 3277, 2924, 1682, 1592, 1470, 1323, 741 cm−1; 1H NMR (300 MHz, CDCl3): δH 8. 33 (d, 1H, J = 2.5 Hz, H-8), 8.33 (br, 1H, H-21), 7.70 (dd, 1H, J = 8.7, 2.5 Hz, H-10), 7.50 (d, J = 8.7 Hz, H-11), 7.39 (d, 1H, J = 8.0 Hz, H-26), 7.30 (d, J = 8.1 Hz, H-23), 7.12 (t, 1H, J = 8.0 Hz, H-24), 6.92 (t, 1H, J = 8.0 Hz, H-25), 6.63 (d, 1H, J = 2.3 Hz, H-20), 5.64 (dd, 1H, J = 5.4,2.7, H-4), 5.72 (s, 1H, H-2), 3.73 (dd, 1H, J = 15.0, 2.7 Hz, H-18a), 3.63 (dd, 1H, J = 15.0, 5.4 Hz, H-18b), 2.76 (d, J = 2.3 Hz, H-1), 2.60 (ddd, 1H, J = 13.9, 6.9, 2.3 Hz, H-15), 0.64 (d, 6H, J = 6.1 Hz, H-16 and H-17); 13C NMR (75 MHz, CDCl3): δC169.2 (C-3), 159.9 (C-6), 150.6 (C-14), 145.6 (C-12), 136.1 (C-22), 135.7 (C-10), 132.8 (C-9), 128.9 (C-8), 127.2 (C-27), 126.2 (C-11), 123.6 (C-20), 122.5 (C-24), 121.2 (C-7), 119.9 (C-25), 118.6 (C-26), 111.1 (C-23), 109.1 (C-19), 57.0 (C-4), 58.0 (C-1), 29.3 (C-16), 27.3 (C-18), 18.8 (C-16), 14.8 (C-17); (+)-HRMS-ESI m/z: 421.1442 (M + H)+, 443.1264 (M + Na)+ (calculated for C23H22N4O2Cl, 421.1432; C23H21N4O2ClNa, 443.1252).:56; mp: 208.8–210.1 °C; [α]D30 = +154 (c 0.15; CHCl3); νmax (KBr) 3277, 2924, 1682, 1592, 1470, 1323, 741 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 33 (d, 1H, J 2.4 Hz, H-8), 8.07 (br, 1H, H-21), 7.70 (dd, 1H, J = 8.7, 2.4 Hz, H-10), 7.54 (d, J = 8.7 Hz, H-11), 7.46 (d, 1H, J = 7.8 Hz, H-26), 7.29 (d, J = 7.8 Hz, H-23), 7.13 (t, 1H, J = 7.8 Hz, H-24), 6.98 (t, 1H, J = 7.8 Hz, H-25), 6.65 (d, 1H, J = 2.4 Hz, H-20), 5.65 (dd, 1H, J = 5.3, 2.7, H-4), 5.71 (s, 1H, H-2), 3.76 (dd, 1H, J = 15.1,2.7 Hz, H-18a), 3.63 (dd, 1H, J = 15.1, 5.3 Hz, H-18b), 2.70 (dd, J = 9.7, 2.3 Hz, H-1), 1.97 (ddd, 1H, J = 11.8, 7.7, 2.1 Hz, H-16), 1.39–1.30 (m, 2H, H-15), 0.77 (d, 3H, J = 6.4 Hz, H-17a), 0.28 (d, 3H, J = 6.5 Hz, H-17b); 13C NMR (75 MHz, CDCl3): δ 169.1 (C-3), 159.8 (C-6), 151.9 (C-14), 145.5 (C-12), 136.0 (C-22), 135.1 (C-10), 132.9 (C-9), 129.1 (C-8), 127.2 (C-27), 126.2 (C-11), 123.6 (C-20), 122.7 (C-24), 121.2 (C-7), 120.2 (C-25), 118.7 (C-26), 111.1 (C-23), 109.5 (C-19), 57.5 (C-4), 50.8 (C-1), 40.2 (C-15), 27.2 (C-18), 24.1 (C-16), 23.3 (C-17a), 19.7 (C-17b); (+)-HRMS-ESI m/z: 435.1579 (M + H)+, 457.1206 (M + Na)+ (calculated for C24H24N4O2Cl, 435.1588; C24H23N4O2ClNa, 457.1408).:54; mp: 209.1–211.2 °C; [α]D30 = +130 (c 0.03; CHCl3); νmax (KBr) 3277, 2924, 1682, 1592, 1470, 1323, 741 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 33 (d, 1H, J 2.4 Hz, H-8), 8.05 (br, 1H, H-21), 7.70 (dd, 1H, J = 8.7, 2.4 Hz, H-10), 7.49 (d, J = 8.7 Hz, H-26), 7.29 (d, J = 8.0 Hz, H-23), 7.13 (t, 1H, J = 8.0 Hz, H-24), 6.92 (t, 1H, J = 8.0 Hz, H-25), 6.63 (d, 1H, J = 2.4 Hz, H-20), 5.64 (dd, 1H, J = 5.3, 2.8, H-4), 5.80 (s, 1H, H-2), 3.72 (dd, 1H, J = 15.1, 2.8 Hz, H-18a), 3.62 (dd, 1H, J = 15.1, 5.3 Hz, H-18b), 2.69 (d, J = 2.2 Hz, H-1), 2.29 (ddd, 1H, J = 11.6, 7.7, 4.8 Hz, H-15), 0.99–0.79 (m, 2H, H-16), 0.70 (d, 3H, J = 7.7 Hz, H-17a), 0.63 (d, 3H, J = 7.2 Hz, H-17b); 13C NMR (75 MHz, CDCl3): δ 169.1 (C-3), 159.9 (C-6), 150.7 (C-14), 145.5 (C-12), 136.0 (C-22), 135.1 (C-10), 132.8 (C-9), 128.9 (C-8), 127.2 (C-27), 126.2 (C-11), 123.5 (C-20), 122.7 (C-24), 121.1 (C-7), 120.1 (C-25), 118.6 (C-26), 111.1 (C-23), 109.2 (C-19), 58.3 (C-1), 57.0 (C-4), 36.2 (C-15), 27.3 (C-18), 23.1 (C-16), 15.6 (C-17a), 12.0 (C-17b); (+)-HRMS-ESI m/z: 435.1580 (M + H)+, 457.1394 (M + Na)+ (calculated for C24H24N4O2Cl, 434.1588; C24H23N4O2ClNa, 457.1408).:57; mp: 232.9–235.1 °C; [α]D30 = +140 (c 0.038; CHCl3); νmax (KBr) 3293, 2954, 1671, 1611, 1511, 1465, 1240, 772, and 697 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 10.2 (br, 1H, H-21), 8. 20 (d, 1H, J 2.4 Hz, H-8), 7.83 (d, 1H, J = 2.4 Hz, H-10), 7.37 (d, 1H, J = 8.1 Hz, H-26), 7.33 (d, J = 8.1 Hz, H-23), 7.11 (s, 1H, H-2), 7.07 (t, 1H, J = 7.6 Hz, H-24), 6.87 (t, 1H, J = 7.6 Hz, H-25), 6.66 (d, 1H, J = 2.3 Hz, H-20), 5.50 (dd, 1H, J = 5.3, 2.9, H-4), 3.69 (dd, 1H, J = 14.9, 2.9 Hz, H-18a), 3.58 (dd, 1H, J = 14.9, 5.3 Hz, H-18b), 2.76 (d, J = 2.2 Hz, H-1), 2.60–254 (m, 1H, H-15), 0.71 (dd, 6H, J = 8.4, 7.2 Hz, H-16, H-17); 13C NMR (75 MHz, CDCl3): δ 169.2 (C-3), 159.9 (C-6), 150.6 (C-14), 145.7 (C-12), 136.0 (C-22), 135.1 (C-10), 132.8 (C-9), 128.9 (C-8), 127.2 (C-27), 126.2 (C-11), 123.6 (C-20), 122.7 (C-24), 121.2 (C-7), 120.1 (C-25), 118.6 (C-26), 111.1 (C-23), 109.2 (C-19), 58.1 (C-1), 57.0 (C-4), 29.3 (C-15), 27.3 (C-18), 18.8 (C-16), 14.8 (C-17); (+)-HRMS-ESI m/z: 455.1436 (M + H)+ (calculated for C23H21N4O2Cl2, 455.1041).:40; mp: 253.4–254.3 °C; [α]D30 = −169 (c 0.04; CHCl3); vmax (KBr) 3289, 2960, 1680, 1600, 1556, 1315, 757, 720 cm−1; 1H NMR (300 MHz, DMSO-d6): 10.22 (br, 1H, H-21), δ 8.13 (d, 1H, J 2.4 Hz, H-8), 7.75 (d, 1H, J = 2.4 Hz, H-10), 7.33 (d, 1H, J = 8.0 Hz, H-26), 7.25 (d, J = 8.0 Hz, H-23), 7.19 (br, H-2), 7.00 (t, 1H, J = 8.0 Hz, H-24), 6.82 (t, 1H, J = 8.0 Hz, H-25), 6.60 (d, 1H, J = 2.4 Hz, H-20), 5.42 (dd, 1H, J = 5.4, 2.9, H-4), 3.63 (dd, 1H, J = 15.0, 2.9 Hz, H-18a), 3.50 (dd, 1H, J = 15.0, 5.4 Hz, H-18b), 2.68 (dd, J = 7.3, 4.9 Hz, H-1), 1.94–1.86 (m, 1H H-16a), 1.50 (tt, 1H, J = 13.2, 6.5 Hz, H-15), 1.29–1.22 (m, 1H, H-16b), 0.56 (d, 3H, J = 6.6 Hz, H-17a), 0.35 (d, 3H, J = 6.6 Hz, H-17b); 13C NMR (75 MHz,DMSO-d6): δ 168.4 (C-3), 158.8 (C-6), 152.8 (C-14), 142.0 (C-12), 135.9 (C-22), 134.1 (C-10), 132.6 (C-9), 131.4 (C-8), 126.5 (C-27), 124.3 (C-11), 123.5 (C-20), 121.6 (C-24), 121.5 (C-7), 118.9 (C-25), 117.7 (C-26), 111.1 (C-23), 107.7 (C-19), 57.3 (C-4), 50.6 (C-1), 39.6 (C-15), 26.2 (C-18), 23.8 (C-16), 22.1 (C-17a), 20.5 (C-17b); (+)-HRMS-ESI m/z: 469.1186 (M + H)+, 491.1008 (M + Na)+ (calculated for C24H23N4O2Cl2, 469.1198; C24H22N4O2Cl2Na, 491.1018).:29; mp: 252.9–254.7 °C; [α]D30 = −264 (c 0.034; CHCl3); vmax (KBr) 3373, 3074, 2922, 1698, 1609, 1550, 1450, 1262, 794 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 33 (d, 1H, J 2.4 Hz, H-8), 8.05 (br, 1H, H-21), 7.70 (d, 1H, J = 2.4 Hz, H-10), 7.38 (d, 1H, J = 7.9 Hz, H-26), 7.29 (d, J = 7.9 Hz, H-23), 7.13 (t, 1H, J = 7.9 Hz, H-24), 6.92 (t, 1H, J = 7.9 Hz, H-25), 6.63 (d, 1H, J = 2.4 Hz, H-20), 5.64 (dd, 1H, J = 5.3, 2.8, H-4), 5.80 (s, 1H, H-2), 3.72 (dd, 1H, J = 15.0, 2.8 Hz, H-18a), 3.62 (dd, 1H, J = 15.0,5.3 Hz, H-18b), 2.69 (d, J = 2.2 Hz, H-1), 2.29 (ddd, 1H, J = 11.6, 7.9, 4.8 Hz, H-15), 0.99–0.79 (m, 2H, H-16), 0.70 (d, 3H, J = 7.3 Hz, H-17a), 0.63 (d, 3H, J = 7.3 Hz, H-17b); 13C NMR (75 MHz, CDCl3): δ 168.9 (C-3), 159.5 (C-6), 151.3 (C-14), 142.5 (C-12), 136.1 (C-22) 135.0 (C-10), 133.2 (C-9), 132.4 (C-8), 127.1 (C-27), 125.1 (C-11), 123.5 (C-20), 122.9 (C-24), 122.1 (C-7), 120.2 (C-25), 118.6 (C-26), 111.1 (C-23), 109.2 (C-19), 58.2 (C-1), 57.3 (C-4), 36.2 (C-15), 27.1 (C-18), 23.6 (C-16), 15.5 (C-17a), 12.1 (C-17b); (+)-HRMS-ESI m/z: 469.1186 (M + H)+, 491.1024 (M + Na)+ (calculated for C24H23N4O2Cl2, 469.1198; C24H22N4O2Cl2Na, 491.1018).:49; mp: 246.5–248.2 °C; [α]D30 = −175 (c 0.041; CHCl3); vmax (KBr) 3311, 3192, 2963, 1681, 1655, 1588, 1464, 1246, 828, and 741 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 71 (d, 1H, J 2.4 Hz, H-8), 8.04 (br, 1H, H-21), 8.02 (dd, 1H, J = 8.6, 2.1 Hz, C-10), 7.41 (d, 1H, J = 8.0 Hz, C-11), 7.30 (d, J = 8.4 Hz, H-26), 7.29 (d, J = 8.4 Hz, H-23), 7.13 (ddd, 1H, J = 8.0, 7.1, 0.9 Hz, H-24), 6.94 (ddd, 1H, J = 8.0, 7.1, 0.9 Hz, H-25), 6.61 (d, 1H, J = 2.4 Hz, H-20), 5.64 (dd, 1H, J = 5.4, 2.8, H-4), 5.67 (s, 1H, H-2), 3.73 (dd, 1H, J = 14.9, 2.7 Hz, H-18a), 3.61 (dd, 1H, J = 15.1, 5.4 Hz, H-18b), 2.64 (d, J = 2.4 Hz, H-1), 2.63–2.56 (m, 1H, H-15), 0.63 (d, 6H, J = 6.8 Hz, H-16 and H-17); 13C NMR (75 MHz, CDCl3): δ 169.1 (C-3), 159.5 (C-6), 151.0 (C-14), 146.3 (C-12), 143.5 (C-11), 136.0 (C-22), 135.7 (C-10), 129.0 (C-8), 127.2 (C-27), 123.5 (C-20), 122.7 (C-24), 121.7 (C-7), 120.2 (C-25), 118.7 (C-26), 111.1 (C-23), 109.3 (C-19), 91.4 (C-9), 58.1 (C-1), 57.0 (C-4) 29.7 (C-15), 27.3 (C-18), 18.8 (C-16), 14.8 (C-17); (+)-HRMS-ESI m/z: 513.0778 (M + H)+ (calculated for C23H22N4O2I, 513.0787).:50; mp: 236.5–238.0 °C; [α]D30 = −170 (c 0.03; CHCl3); vmax (KBr) 3292, 3193, 2958, 1681, 1666, 1592, 1466, 1237, 832, and 742 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 50 (d, 1H, J 2.2 Hz, H-8), 8.05 (br, 1H, H-21), 7.84 (dd, 1H, J = 8.7, 2.2 Hz, C-10), 7.41 (d, J = 8.7 Hz, H-11), 7.29 (dd, 2H, J = 8.0, 2.2 Hz, H-23 & H-26), 7.13 (ddd, 1H, J = 8.0, 7.1, 1.0 Hz, H-24), 6.93 (ddd, 1H, J = 8.0, 7.1, 1.1 Hz, H-25), 6.62 (d, 1H, J = 2.4 Hz, H-20), 5.64 (dd, 1H, J = 5.4, 2.8, H-4), 5.63 (s, 1H, H-2), 3.73 (dd, 1H, J = 14.9, 2.8 Hz, H-18a), 3.62 (dd, 1H, J = 15.0, 5.4 Hz, H-18b), 2.66 (d, J = 2.4 Hz, H-1), 2.60 (m, 1H, H-15), 0.65 (d, 3H, J = 6.5 Hz, H-16), 0.63 (d, 3H, J = 6.4 Hz, H-17); 13C NMR (75 MHz, CDCl3): δ 169.1 (C-3), 159.7 (C-6), 150.9 (C-14), 145.9 (C-12), 138.1 (C-10), 136.2 (C-22), 129.4 (C-8), 129.1 (C-11), 127.2 (C-27), 123.5 (C-20), 122.7 (C-7), 121.5 (C-24), 120.6 (C-25), 120.2 (C-9), 118.7 (C-26), 111.1 (C-23), 109.3 (C-19), 57.0 (C-4), 53.8 (C-1), 29.7 (C-15), 27.3 (C-18), 18.8 (C-16), 14.8 (C-17); (+)-HRMS-ESI m/z: 465.0987 (M + H)+, 487.0726 (M + Na)+ (calculated for C23H22N4O2Br: 465.0926; C23H21N4O2BrNa: 487.0746).:49; mp: 192.1–194.3 °C; [α]D30 = −165 (c 0.038; CHCl3); vmax (KBr) 3318, 2956, 1671, 1686, 1593, 1464, 1247, 790, and 740 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 70 (d, 1H, J 2.1 Hz, H-8), 8.03 (br, 1H, H-21), 8.03 (dd, 1H, J = 8.6, 2.1 Hz, C-10), 7.44 (d, J = 7.9 Hz, H-26), 7.33 (d, 1H, J = 8.6 Hz, H-11), 7.29 (d, J = 7.9 Hz, H-23), 7.13 (t, 1H, J = 7.9 Hz, H-24), 6.98 (t, 1H, J = 7.9 Hz, H-25), 6.68 (d, 1H, J = 2.4 Hz, H-20), 5.96 (s, 1H, H-2), 5.65 (dd, 1H, J = 5.2, 2.8, H–), 3.76 (dd, 1H, J = 15.0, 2.8 Hz, H-18a), 3.63 (dd, 1H, J = 15.0, 5.2 Hz, H-18b), 2.69 (dd, J = 9.6, 3.3 Hz, H-1), 2.02–1.92 (m, 1H, H-16), 1.40–1.30 (m, 2H, H-15), 0.79 (d, 3H, J = 6.5 Hz, H17a), 0.29 (d, 3H, J = 6.4 Hz, H-17b); 13C NMR (75 MHz, CDCl3): δ 169.5 (C-3), 159.4 (C-6), 152.1 (C-14), 146.3 (C-12), 143.4 (C-10), 136.1 (C-22), 135.7 (C-8), 129.2 (C-11), 127.1 (C-27), 123.5 (C-20), 122.9 (C-24), 121.8 (C-7), 120.4 (C-25), 118.7 (C-26), 111.2 (C-23), 109.5 (C-19), 91.5 (C-9), 57.4 (C-4), 51.0 (C-1), 40.1 (C-15), 27.1 (C-18), 24.1 (C-16), 23.3 (C17a), 19.7 (C-17b); (+)-HRMS-ESI m/z: 527.0936 (M + H)+, 549.0748 (M + Na)+ (calculated for C24H24N4O2I, 527.0944; C24H23N4O2INa, 549.0764).:49; mp: 227.0–228.2 °C; [α]D30 = −243 (c 0.037; CHCl3); vmax (KBr) 3284, 2959, 1686, 1658, 1599, 1433, 1245, 746, and 684 cm−1; 1H NMR (300 MHz, CDCl3): δ 8. 50 (d, 1H, J 2.3 Hz, H-8), 8.06 (br, 1H, H-21), 7.84 (dd, 1H, J = 8.5, 2.3 Hz, H-10), 7.47 (dd, 2H, J = 8.1, 1.9 Hz, H-26), 7.29 (d, J = 8.5 Hz, H-23), 7.14 (t, 1H, J = 8.0 Hz, H-24), 6.92 (t, 1H, J = 7.9 Hz, H-25), 6.65 (d, 1H, J = 2.4 Hz, H-20), 5.65 (dd, 1H, J = 5.2, 2.9, H-4), 5.71 (s, 1H, H-2), 3.76 (dd, 1H, J = 15.0, 2.9 Hz, H18a), 3.63 (dd, 1H, J = 15.0, 5.2 Hz, H-18b), 2.70 (dd, J = 9.7, 3.3 Hz, H-1), 2.07–1.89 (m, 1H, H-16), 1.38–1.21 (m, 2H, H-15), 0.77 (d, 3H, J = 6.3 Hz, H-17a), 0.28 (d, 3H, J = 6.5 Hz, H-17b); 13C NMR (75 MHz, CDCl3): δ 169.1 (C-3), 159.7 (C-6), 152.0 (C-14), 145.8 (C-12), 137.8 (C-10), 136.8 (C-22), 129.4 (C-8), 129.1 (C-11). 127.1 (C-27), 123.8 (C-20), 122.9 (C-7), 121.6 (C-24), 120.6 (C-9), 120.4 (C-25), 118.7 (C-26), 111.2 (C-23), 109.6 (C-19), 57.5 (C-4), 50.8 (C-1), 40.1 (C-15), 27.1 (C-18), 24.1 (C-16), 23.3 (C-17a), 19.7 (C-17b); (+)-HRMS-ESI m/z: 479.1086 (M + H)+, 501.0912 (M + Na)+ (calculated for C24H24N4O2Br, 479.1082; C24H23N4O2BrNa, 501.0900).:46; mp: 242.8–243.8 °C; [α]D30 = −229 (c 0.032; CHCl3); vmax (KBr) 3313, 2955, 1681, 1599, 1462, 1261, 772, and 669 cm−1; 1H NMR (300 MHz, DMSO-d6): 10.17 (br, 1H, H-21), δ 8. 62 (d, 1H, J 1.9 Hz, H-8), 8.55 (d, 1H, J = 1.9 Hz, H-10), 7.41 (d, 1H, J = 8.0 Hz, H-26), 7.33 (d, J = 8.0 Hz, H-23), 7.11 (br, H-2), 7.09 (t, 1H, J = 7.9 Hz, H-24), 6.91 (t, 1H, J = 7.9 Hz, H-25), 6.68 (d, 1H, J = 2.3 Hz, H-20), 5.50 (dd, 1H, J = 5.2, 2.9, H-4), 3.72 (dd, 1H, J = 14.9, 2.9 Hz, H-18a), 3.58 (dd, 1H, J = 15.0, 5.2 Hz, H-18b), 2.75 (dd, J = 6.6, 5.3 Hz, H-1), 2.11–1.95 (m, 1H, H15a), 1.68–1.53 (m, 1H H-15b), 1.38–1.23 (m, 1H, H-16), 0.62 (t, 3H, J = 6.5 Hz, H-17a), 0.47 (d, 3H, J = 6.6 Hz, H-17b); 13C NMR (75 MHz, DMSO-d6): δ 168.4 (C-3), 162.0 (C-6), 153.0 (C-12), 151.1 (C-10), 145.2 (C-12), 136.2 (C-22), 135.9 (C-8), 127.1 (C-27), 123.4 (C-20), 121.8 (C-7), 121.5 (C-24), 119.0 (C-25), 117.8 (C-26), 111.0 (C-23), 107.8 (C-19), 91.5 (C-9), 89.2 (C-11), 57.4 (C-4), 50.5 (C-1), 39.4 (C-15), 26.3 (C-18), 23.9 (C-16), 21.8 (C-17a), 20.6 (C-17b); (+)-HRMS-ESI m/z: 652.9915 (M + H)+, 674.9746 (M + Na)+ (calculated for C24H23N4O2Cl2, 652.9910; C24H22N4O2Cl2Na, 674.9730).Microbiology
Antimicrobial susceptibility testing
Biofilm formation inhibition assay
For compounds with antibacterial activity, their effect on biofilm formation was evaluated using the crystal violet method. Briefly, bacterial suspensions of 1 × 106 CFU mL−1 were prepared in Tryptone Soy broth (TSB-Biokar Diagnostics, Allone, Beauvais, France) supplemented with 1% (p/v) glucose (D(+)-glucose anhydrous for molecular biology, PanReac AppliChem, Barcelona, Spain); and four concentrations of compound were tested: 2 × MIC, MIC, ½ MIC and ¼ MIC, keeping final in-test concentration of DMSO below 1%. A control with inoculum and culture media, a control with appropriate concentration of DMSO, as well as a negative control (TSB alone) were included. Sterile 96-well flat-bottomed untreated polystyrene microtiter plates were used. After a 24 h incubation at 37 °C, the biofilms were heat-fixed for 1 h at 60 °C and stained with 0.5% (v/v) crystal violet (Química Clínica Aplicada, Amposta, Spain) for 5 min. The stain was resolubilized with 33% (v/v) acetic acid (acetic acid 100%, AppliChem, Darmstadt, Germany) and the biofilm biomass was quantified by measuring the absorbance of each sample at 570 nm in a microplate reader (Thermo Scientific Multiskan® EX, Thermo Fisher Scientific, Waltham, MA, USA).40,43 Three independent experiments were performed, in triplicate.Antimicrobial synergy testing
Time–kill kinetics assay
Time–kill kinetics of 26 was performed for S. aureus ATCC 29213 and MRSA S. aureus 66/1, according to CLSI guidelines.72 Briefly, a solution of 64 μg mL−1 of 26 was prepared in MHB2, as well as a control with appropriate concentration of DMSO; bacterial suspensions of 5 × 105 CFU mL−1 in MHB2 were added and incubated at 37 °C in an orbital shaker. Aliquots of 100 μL were taken at time intervals of 0, 2, 4, 8, 10, 12, and 24 h, and serially diluted (10-fold serial dilutions) in MHB2. Appropriate dilutions were inoculated into MH agar plates and incubated at 37 °C for 24 h. After incubation, colony counts were performed, and log10 CFU mL−1 was plotted against time.Scanning electron microscopy (SEM)
The effect of 26 on S. aureus ATCC 29213 and MRSA S. aureus 66/1 cellular surface was observed using scanning electron microscope.73 Briefly, solutions of 26 at MIC and ½ MIC were prepared in MHB2, as well as controls with appropriate concentration of DMSO; bacterial suspensions of 5 × 105 CFU mL−1 in MHB2 were added and incubated at 37 °C for 20 h. 500 μL of each sample was centrifuged at 4 °C for 6 min at 10000 rpm and washed 3 times with phosphate-buffered saline 0.01 M, pH 7.4 (Sigma, St. Louis, MO, USA), fixed in 2.5% (v/v) glutaraldehyde (glutaraldehyde solution 25%; Merck, Darmstadt, Germany) in 0.1 M sodium cacodylate buffer, pH 7.2 (Sodium cacodylate trihydrate ≥ 98%; Sigma, St. Louis, MO, USA) for 2h30 min at 4 °C, washed three times with sodium cacodylate buffer and post-fixed overnight in 1% (v/v) osmium tetroxide (OsO4 4% solution; Agar Scientific, Stansted, UK) in the same buffer. Subsequently, samples were washed two times with sodium cacodylate buffer and one time with ultrapure sterile water; following that, samples were resuspended in ultrapure sterile water, and transferred to 12 mm cover slips previously coated with 5% (v/v) APES solution [3-(triethoxysilyl)propylamine; Merck, Darmstadt, Germany] in acetone (Benzina Internacional, Maia, Portugal). The samples were then dehydrated in 30, 50, 70, 90, 95, 100% ethanol (Merck, Darmstadt, Germany) (3 times for 5 min for each concentration), and dried with a critical point dryer. Samples were coated with an Au/Pd thin film, by sputtering, using the SPI Module Sputter Coater equipment. The SEM/EDS examination was performed using a High resolution Scanning Electron Microscope with X-ray Microanalysis: JEOL JSM 6301F/Oxford INCA Energy 350.
Enantioselective liquid chromatography
:EtOH (50:50) at a final concentration 50 μg mL−1, and the injection volume was 10 μL. The HPLC system comprised a JASCO model 880-PU intelligent HPLC pump (JASCO corporation, Tokyo, Japan), equipped with a 7125 injector (Rheodyne LCC, Rohnert Park, CA, USA) fitted with a 20 μL LC loop, a JASCO model 880–30 solvent mixer involving a 875-UV intelligent UV/VIS detector, a system equipped with a chiral column (Lux® 5 μm amylose-1, 250 × 4.6 mm). The data acquisitions were performed using ChromNAC chromatography Data system (version 1.19.1) from JASCO Corporation (Tokyo, Japan). The mobile phase consisted of the mixture of n-hexane:EtOH (90:10, v/v), at a flow rate of 0.5 mL min−1. The mobile phase was prepared in a volume/volume ratio and degassed in an ultrasonic bath for at least 15 min before use. Chromatographic analyses were carried out in isocratic mode at 22 ± 2 °C, in duplicate. The UV detection was performed at a wavelength of 254 nm. The volume void time was considered to be equal to the peak of solvent front and was taken from each particular run. The enantiomeric ratios (e.r) were determined by the mean percentage of peak area of eluted peaks.:EtOH (50:50) at a concentration of 10 mg mL−1, and the injection volume was 100–200 μL. The HPLC system is similar to that described in quantitative analysis, equipped with an in-house column amylose tris-3,5-dimethylphenylcarbamate coated with Nucleosil (500 A, 7 mm, 20%, w/w) packed into a stainless steel (200 mm × 7 mm I.D. size) column, prepared in the UFSCar laboratory.63 Semi-preparative chromatographic separations were first achieved through multiple injection with 200 μL at a flow rate of 2 mL min−1. Chromatographic analyses were carried out in isocratic mode at 22 ± 2 °C. The UV detection was performed at a wavelength of 254 nm. The collected fractions were analyzed using the analytical column to determine their enantiomeric ratio/excess with the conditions described above.Cell-based studies
000 cells per cm2 and used 6 days after seeding. To obtain cells with a dopaminergic neuronal phenotype, SH-SY5Y cells were differentiated as previously described.74 Briefly, SH-SY5Y cells were seeded in complete DMEM medium containing 10 μM RA, and cultured for 3 days at 37 °C. After 3 days, 80 nM of TPA was added to the cultures, and cells were cultured for another 3 days, at 37 °C. The non-differentiated SH-SY5Y cells were maintained in complete DMEM medium for 6 days, mimicking the differentiation protocol but in the absence of both RA and TPA. The cells used in all experiments were taken between the 21st and 25th passages.000 cells per cm2, submitted or not to a RA and TPA differentiation protocol, and exposed, 6 days after seeding to 26 (0–25 μM), in fresh cell culture medium. Triton™ X-100 (0.1%) was used as positive control.:1) with 5% acetic acid], and the absorbance measured at 540 nm in a multiwell plate reader (PowerWaveX BioTek Instruments, Vermont, USA). The percentage of NR uptake relatively to that of the control cells (0 μM) was used as the cytotoxicity measure. Four independent experiments were performed, in triplicate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by national funds through FCT – Foundation for Science and Technology within the scope of UIDB/04423/2020, UIDP/04423/2020, and under the project PTDC/SAU-PUB/28736/2017 (reference POCI-01-0145-FEDER-028736), co-financed by COMPETE 2020, Portugal 2020 and the European Union through the ERDF and by FCT through national funds. S. L. thanks Erasmus Mundus Action 2 (LOTUS+, LP15DF0205) for full PhD scholarship. To Sara Cravo for technical support. To Centro de Apoio Cientifico e Tecnolόxico á Investigation (CACTI, University of Vigo, Pontevedra, Spain) for HRMS analysis.References
- D. E. Bloom and D. Cadarette, Infectious Disease Threats in the Twenty-First Century: Strengthening the Global Response, Front. Immunol., 2019, 10, 549 CrossRef PubMed.
- R. Laxminarayan, The State of the World's Antibiotics in 2018: A Keynote Lecture, Center of Disease Dynamics, Economics & Policy, Washington, D.C, 2018 Search PubMed.
- R. Gyawali and S. A. Ibrahim, Natural products as antimicrobial agents, Food Contr., 2014, 46, 412–429 CrossRef CAS.
- I. Sultan, S. Rahman, A. T. Jan, M. T. Siddiqui, A. H. Mondal and Q. M. R. Haq, Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective, Front. Microbiol., 2018, 9, 2066 CrossRef PubMed.
- S. H. Podolsky, The evolving response to antibiotic resistance (1945–2018), Palgrave Commun., 2018, 4, 124 CrossRef.
- C. L. Ventola, The antibiotic resistance crisis: part 1: causes and threats, P. T., 2015, vol. 40, pp. 277–283 Search PubMed.
- W. A. McGuinness, N. Malachowa and F. R. DeLeo, Vancomycin Resistance in Staphylococcus aureus Yale, J. Biol. Med., 2017, 90, 269–281 CAS.
- P. Courvalin, Vancomycin Resistance in Gram-Positive Cocci, Clin. Infect. Dis., 2006, 42, S25–S34 CrossRef CAS PubMed.
- P. D. Stapleton and P. W. Taylor, Methicillin resistance in Staphylococcus aureus: mechanisms and modulation, Sci. Prog., 2002, 85, 57–72 CrossRef CAS PubMed.
- F. S. Codjoe and E. S. Donkor, Carbapenem Resistance: A Review, Med. Sci., 2017, 6, 1 Search PubMed.
- S. H. Park, Third-generation cephalosporin resistance in gram-negative bacteria in the community: a growing public health concern, Kor. J. Intern. Med., 2014, 29, 27–30 CrossRef CAS PubMed.
- W. H. Organization, WHO publishes list of bacteria for which new antibiotics are urgently needed, Journal, 2017 Search PubMed.
- A. D. Russell, Antibiotic and biocide resistance in bacteria: Introduction, J. Appl. Microbiol., 2002, 92, 1S–3S CrossRef PubMed.
- C. A. Michael, D. Dominey-Howes and M. Labbate, The antimicrobial resistance crisis: causes, consequences, and management, Public Health Front, 2014, 2, 145 Search PubMed.
- WHO, Global antimicrobial resistance surveillance system (GLASS) report: early implementation 2016-2017, World Health Organization, Geneva, 2017 Search PubMed.
- J. H. Powers, Antimicrobial drug development – the past, the present, and the future, Clin. Microbiol. Infect., 2004, 10, 23–31 CrossRef PubMed.
- G.-F. Zha, S.-M. Wang, K. P. Rakesh, S. N. A. Bukhari, H. M. Manukumar, H. K. Vivek, N. Mallesha and H.-L. Qin, Discovery of novel arylethenesulfonyl fluorides as potential candidates against methicillin-resistant of Staphylococcus aureus (MRSA) for overcoming multidrug resistance of bacterial infections, Eur. J. Med. Chem., 2019, 162, 364–377 CrossRef CAS PubMed.
- H. Liu, S. Long, K. P. Rakesh and G.-F. Zha, Structure-activity relationships (SAR) of triazine derivatives: Promising antimicrobial agents, Eur. J. Med. Chem., 2020, 185, 111804 CrossRef CAS PubMed.
- K. P. Rakesh, M. H. Marichannegowda, S. Srivastava, X. Chen, S. Long, C. S. Karthik, P. Mallu and H.-L. Qin, Combating a Master Manipulator: Staphylococcus aureus Immunomodulatory Molecules as Targets for Combinatorial Drug Discovery, ACS Comb. Sci., 2018, 20, 681–693 CrossRef CAS PubMed.
- K. P. Rakesh, H. K. Kumara, B. J. Ullas, J. Shivakumara and D. Channe Gowda, Amino acids conjugated quinazolinone-Schiff’s bases as potential antimicrobial agents: Synthesis, SAR and molecular docking studies, Bioorg. Chem., 2019, 90, 103093 CrossRef CAS PubMed.
- K. P. Rakesh, H. K. Vivek, H. M. Manukumar, C. S. Shantharam, S. N. A. Bukhari, H.-L. Qin and M. B. Sridhara, Promising bactericidal approach of dihydrazone analogues against bio-film forming Gram-negative bacteria and molecular mechanistic studies, RSC Adv., 2018, 8, 5473–5483 RSC.
- L. Ravindar, S. N. A. Bukhari, K. P. Rakesh, H. M. Manukumar, H. K. Vivek, N. Mallesha, Z.-Z. Xie and H.-L. Qin, Aryl fluorosulfate analogues as potent antimicrobial agents: SAR, cytotoxicity and docking studies, Bioorg. Chem., 2018, 81, 107–118 CrossRef CAS PubMed.
- G.-F. Zha, J. Leng, N. Darshini, T. Shubhavathi, H. K. Vivek, A. M. Asiri, H. M. Marwani, K. P. Rakesh, N. Mallesha and H.-L. Qin, Synthesis, SAR and molecular docking studies of benzo[d]thiazole-hydrazones as potential antibacterial and antifungal agents, Bioorg. Med. Chem. Lett., 2017, 27, 3148–3155 CrossRef CAS PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackerman, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, A Deep Learning Approach to Antibiotic Discovery, Cell, 2020, 180, 688–702.e613 CrossRef CAS PubMed.
- C. C. Hughes and W. Fenical, Antibacterials from the sea, Chemistry, 2010, 16, 12512–12525 CrossRef CAS PubMed.
- N. Kasanah and M. T. Hamann, Development of antibiotics and the future of marine microorganisms to stem the tide of antibiotic resistance, Curr. Opin. Investig. Drugs, 2004, 5, 827–837 CAS.
- S.-H. Eom, Y.-M. Kim and S.-K. Kim, Marine bacteria: potential sources for compounds to overcome antibiotic resistance, Appl. Microbiol. Biotechnol., 2013, 97, 4763–4773 CrossRef CAS PubMed.
- E. Tortorella, P. Tedesco, F. Palma Esposito, G. G. January, R. Fani, M. Jaspars and D. de Pascale, Antibiotics from Deep-Sea Microorganisms: Current Discoveries and Perspectives, Mar. Drugs, 2018, 16, 355 CrossRef CAS PubMed.
- D. I. S. P. Resende, P. Pereira-Terra, A. S. Inácio, P. M. Da Costa, E. Pinto, E. Sousa and M. M. M. Pinto, Lichen xanthones as models for new antifungal agents, Molecules, 2018, 23, 2617 CrossRef PubMed.
- D. I. S. P. Resende, P. Boonpothong, E. Sousa, A. Kijjoa and M. M. M. Pinto, Chemistry of the fumiquinazolines and structurally related alkaloids, Nat. Prod. Rep., 2019, 36, 7–34 RSC.
- J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41, J. Nat. Prod., 2013, 76, 1133–1140 CrossRef CAS PubMed.
- L. J. Bessa, S. Buttachon, T. Dethoup, R. Martins, V. Vasconcelos, A. Kijjoa and P. M. da Costa, Neofiscalin A and fiscalin C are potential novel indole alkaloid alternatives for the treatment of multidrug-resistant Gram-positive bacterial infections, FEMS Microbiol. Lett., 2016, 363, 1–5 CrossRef PubMed.
- X.-J. Li, Q. Zhang, A.-L. Zhang and J.-M. Gao, Metabolites from Aspergillus fumigatus, an Endophytic Fungus Associated with Melia azedarach, and Their Antifungal, Antifeedant, and Toxic Activities, J. Agric. Food Chem., 2012, 60, 3424–3431 CrossRef CAS PubMed.
- Y. Zhuang, X. Teng, Y. Wang, P. Liu, G. Li and W. Zhu, New Quinazolinone Alkaloids within Rare Amino Acid Residue from Coral-Associated Fungus, Aspergillus versicolor LCJ-5-4, Org. Lett., 2011, 13, 1130–1133 CrossRef CAS PubMed.
- M. Prata-Sena, A. A. Ramos, S. Buttachon, B. Castro-Carvalho, P. Marques, T. Dethoup, A. Kijjoa and E. Rocha, Cytotoxic activity of Secondary Metabolites from Marine-derived Fungus Neosartorya siamensis in Human Cancer Cells, Phytotherapy Research, 2016, 30, 1862–1871 CrossRef CAS PubMed.
- S. Long, D. I. S. P. Resende, A. Kijjoa, A. M. S. Silva, A. Pina, T. Fernández-Marcelo, M. H. Vasconcelos, E. Sousa and M. M. M. Pinto, Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products, Mar. Drugs, 2018, 16, 261 CrossRef PubMed.
- S. Long, D. I. S. P. Resende, A. Kijjoa, A. M. S. Silva, R. Fernandes, C. P. R. Xavier, M. H. Vasconcelos, E. Sousa and M. M. M. Pinto, Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects, Molecules, 2019, 24, 534 CrossRef PubMed.
- W. G. Gribble, Biological Activity of Recently Discovered Halogenated Marine Natural Products, Mar. Drugs, 2015, 13, 4044–4136 CrossRef PubMed.
- J. F. Liu, P. Ye, B. Zhang, G. Bi, K. Sargent, L. Yu, D. Yohannes and C. M. Baldino, Three-component one-pot total syntheses of glyantrypine, fumiquinazoline F, and fiscalin B promoted by microwave irradiation, J. Org. Chem., 2005, 70, 6339–6345 CrossRef CAS PubMed.
- N. M. Gomes, L. J. Bessa, S. Buttachon, P. M. Costa, J. Buaruang, T. Dethoup, A. M. S. Silva and A. Kijjoa, Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis, Mar. Drugs, 2014, 12, 822–839 CrossRef PubMed.
- L. J. Bessa, A. Palmeira, A. S. Gomes, V. Vasconcelos, E. Sousa, M. Pinto and P. M. Da Costa, Synergistic effects between thioxanthones and oxacillin against methicillin-resistant staphylococcus aureus, Microb. Drug Resist., 2015, 21, 404–415 CrossRef CAS PubMed.
- S. Stepanović, D. Vuković, I. Dakić, B. Savić and M. Švabić-Vlahović, A modified microtiter-plate test for quantification of staphylococcal biofilm formation, J. Microbiol. Methods, 2000, 40, 175–179 CrossRef.
- S. Stepanović, D. Vuković, V. Hola, G. D. Bonaventura, S. Djukić, I. Cirković and F. Ruzicka, Quantification of biofilm in microtiter plates: overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci, APMIS, 2007, 115, 891–899 CrossRef PubMed.
- D. H. Bergey and J. G. Holt, Bergey's Manual of Determinative Bacteriology, Williams & Wilkins, Baltimore, Maryland, USA, 9th edn, 1994 Search PubMed.
- CLSI, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard-3rd edn, 2008 Search PubMed.
- CLSI, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi, Approved Standard-2nd edn, 2008 Search PubMed.
- S. Buttachon, A. Chandrapatya, L. Manoch, A. Silva, L. Gales, C. Bruyère, R. Kiss and A. Kijjoa, Sartorymensin, a new indole alkaloid, and new analogues of tryptoquivaline and fiscalins produced by Neosartorya siamensis (KUFC 6349), Tetrahedron, 2012, 68, 3253–3262 CrossRef CAS.
- W. War May Zin, C. Prompanya, S. Buttachon and A. Kijjoa, Bioactive Secondary Metabolites from a Thai Collection of Soil and Marine-Derived Fungi of the Genera Neosartorya and Aspergillus, Curr. Drug Deliv., 2016, 13, 378–388 CrossRef PubMed.
- S. Boyapati, U. Kulandaivelu, S. Sangu and M. R. Vanga, Synthesis, Antimicrobial Evaluation, and Docking Studies of Novel 4-Substituted Quinazoline Derivatives as DNA-Gyrase Inhibitors, Archiv der Pharmazie, 2010, 343, 570–576 CrossRef CAS PubMed.
- N. German, M. Malik, J. D. Rosen, K. Drlica and R. J. Kerns, Use of gyrase resistance mutants to guide selection of 8-methoxy-quinazoline-2,4-diones, Antimicrob. Agents Chemother., 2008, 52, 3915–3921 CrossRef CAS PubMed.
- J. Sun, P.-C. Lv, Y. Yin, R.-J. Yuan, J. Ma and H.-L. Zhu, Synthesis, structure and antibacterial activity of potent DNA gyrase inhibitors: N'-benzoyl-3-(4-bromophenyl)-1H-pyrazole-5-carbohydrazide derivatives, PloS one, 2013, 8, e69751 CrossRef CAS PubMed.
- M. Fujimoto-Nakamura, H. Ito, Y. Oyamada, T. Nishino and J. Yamagishi, Accumulation of mutations in both gyrB and parE genes is associated with high-level resistance to novobiocin in Staphylococcus aureus, Antimicrob. Agents Chemother., 2005, 49, 3810–3815 CrossRef CAS PubMed.
- A. E. Eakin, O. Green, N. Hales, G. K. Walkup, S. Bist, A. Singh, G. Mullen, J. Bryant, K. Embrey, N. Gao, A. Breeze, D. Timms, B. Andrews, M. Uria-Nickelsen, J. Demeritt, J. T. Loch III, K. Hull, A. Blodgett, R. N. Illingworth, B. Prince, P. A. Boriack-Sjodin, S. Hauck, L. J. MacPherson, H. Ni and B. Sherer, Pyrrolamide DNA gyrase inhibitors: fragment-based nuclear magnetic resonance screening to identify antibacterial agents, Antimicrob. Agents Chemother., 2012, 56, 1240–1246 CrossRef CAS PubMed.
- P. F. Chan, V. Srikannathasan, J. Huang, H. Cui, A. P. Fosberry, M. Gu, M. M. Hann, M. Hibbs, P. Homes, K. Ingraham, J. Pizzollo, C. Shen, A. J. Shillings, C. E. Spitzfaden, R. Tanner, A. J. Theobald, R. A. Stavenger, B. D. Bax and M. N. Gwynn, Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin, Nat. Commun., 2015, 6, 10048 CrossRef CAS PubMed.
- J. Fischer and C. R. Ganellin, Analogue-based Drug Discovery II, Wiley-VCH, Germany, 2010 Search PubMed.
- M. Z. Hernandes, S. M. Cavalcanti, D. R. Moreira, W. F. de Azevedo Junior and A. C. Leite, Halogen atoms in the modern medicinal chemistry: hints for the drug design, Curr. Drug Targets, 2010, 11, 303–314 CrossRef CAS PubMed.
- Y. Lu, Y. Liu, Z. Xu, H. Li, H. Liu and W. Zhu, Halogen bonding for rational drug design and new drug discovery, Expet Opin. Drug Discov., 2012, 7, 375–383 CrossRef CAS PubMed.
- P. M. J. Szell, S. Zablotny and D. L. Bryce, Halogen bonding as a supramolecular dynamics catalyst, Nat. Commun., 2019, 10, 916 CrossRef PubMed.
- F. Y. Lin and A. D. MacKerell Jr., Do Halogen-Hydrogen Bond Donor Interactions Dominate the Favorable Contribution of Halogens to Ligand-Protein Binding?, J. Phys. Chem. B, 2017, 121, 6813–6821 CrossRef CAS PubMed.
- M. Gjorgjieva, T. Tomasic, M. Barancokova, S. Katsamakas, J. Ilas, P. Tammela, L. Peterlin Masic and D. Kikelj, Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors, J. Med. Chem., 2016, 59, 8941–8954 CrossRef CAS PubMed.
- T. Tomasic, S. Katsamakas, Z. Hodnik, J. Ilas, M. Brvar, T. Solmajer, S. Montalvao, P. Tammela, M. Banjanac, G. Ergovic, M. Anderluh, L. Peterlin Masic and D. Kikelj, Discovery of 4,5,6,7-Tetrahydrobenzo[1,2-d]thiazoles as Novel DNA Gyrase Inhibitors Targeting the ATP-Binding Site, J. Med. Chem., 2015, 58, 5501–5521 CrossRef CAS PubMed.
- F. Collin, S. Karkare and A. Maxwell, Exploiting bacterial DNA gyrase as a drug target: current state and perspectives, Appl. Microbiol. Biotechnol., 2011, 92, 479–497 CrossRef CAS PubMed.
- S. A. Matlin, M. E. Tiritan, A. J. Crawford, Q. B. Cass and D. R. Boyd, HPLC with carbohydrate carbamate chiral phases: Influence of chiral phase structure on enantioselectivity, Chirality, 1994, 6, 135–140 CrossRef CAS.
- R. R. Simões, M. Aires-de-Sousa, T. Conceição, F. Antunes, P. M. da Costa and H. de Lencastre, High Prevalence of EMRSA-15 in Portuguese Public Buses: A Worrisome Finding, PloS One, 2011, 6, e17630 CrossRef PubMed.
- L. J. Bessa, A. Barbosa-Vasconcelos, Â. Mendes, P. Vaz-Pires and P. Martins da Costa, High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant, J. Water Health, 2014, 12, 426–435 CrossRef PubMed.
- CLSI, Performance standards for antimicrobial susceptibility testing, 27th edn, 2017 Search PubMed.
- CLSI, Performance Standards for Antimicrobial Disk Susceptibility Tests, 11th edn, 2012 Search PubMed.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard-10th edn, 2015 Search PubMed.
- M. Aureli, L. Mauri, M. G. Ciampa, A. Prinetti, G. Toffano, C. Secchieri and S. Sonnino, GM1 Ganglioside: Past Studies and Future Potential, Mol. Neurobiol., 2016, 53, 1824–1842 CrossRef CAS PubMed.
- D. Garzón, R. Cabezas, N. Vega, M. Ávila-Rodriguez, J. Gonzalez, R. M. Gómez, V. Echeverria, G. Aliev and G. E. Barreto, Novel Approaches in Astrocyte Protection: from Experimental Methods to Computational Approaches, J. Mol. Neurosci., 2016, 58, 483–492 CrossRef PubMed.
- F. C. Odds, Synergy, antagonism, and what the chequerboard puts between them, J. Antimicrob. Chemother., 2003, 52, 1 CrossRef CAS PubMed.
- A. Villanova, NCCLS. Methods for determining bactericidal activity of antibacterial agents; approved guideline. NCCLS document M26-A. , National Committee for Clinical Laboratory Standards, Wayne, PA, USA, 1999 Search PubMed.
- L. Albuquerque, A. R. M. Polónia, C. Barroso, H. J. C. Froufe, O. Lage, A. Lobo-da-Cunha, C. Egas and M. S. da Costa, Raineya orbicola gen. nov., sp. nov. a slightly thermophilic bacterium of the phylum Bacteroidetes and the description of Raineyaceae fam. nov, Int. J. Syst. Evol. Microbiol., 2018, 68, 982–989 CrossRef CAS PubMed.
- M. D. Arbo, R. Silva, D. J. Barbosa, D. D. da Silva, L. G. Rossato, M. d. L. Bastos and H. Carmo, Piperazine designer drugs induce toxicity in cardiomyoblast h9c2 cells through mitochondrial impairment, Toxicol. Lett., 2014, 229, 178–189 CrossRef CAS PubMed.
- R. Silva, E. Sousa, H. Carmo, A. Palmeira, D. J. Barbosa, M. Gameiro, M. Pinto, M. de Lourdes Bastos and F. Remião, Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat, Arch. Toxicol., 2014, 88, 937–951 CrossRef CAS PubMed.
- V. Vilas-Boas, R. Silva, C. Nunes, S. Reis, L. Ferreira, C. Vieira, F. Carvalho, M. d. L. Bastos and F. Remião, Mechanisms of P-gp inhibition and effects on membrane fluidity of a new rifampicin derivative, 1,8-dibenzoyl-rifampicin, Toxicol. Lett., 2013, 220, 259–266 CrossRef CAS PubMed.
- G. Repetto, A. del Peso and J. L. Zurita, Neutral red uptake assay for the estimation of cell viability/cytotoxicity, Nat. Protoc., 2008, 3, 1125–1131 CrossRef CAS PubMed.
- T. L. Riss, R. A. Moravec, A. L. Niles, S. Duellman, H. A. Benink, T. J. Worzella and L. Minor, in Assay Guidance Manual, ed. G. S. Sittampalam, A. Grossman, K. Brimacombe, M. Arkin, D. Auld, C. P. Austin, J. Baell, B. Bejcek, J. M. M. Caaveiro, T. D. Y. Chung, N. P. Coussens, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Haas, S. R. J. Hoare, J. Inglese, P. W. Iversen, S. D. Kahl, S. C. Kales, S. Kirshner, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, P. Saradjian, O. J. Trask Jr., J. R. Weidner, M. J. Wildey, M. Xia and X. Xu, Bethesda (MD), 2004 Search PubMed.
- E. A. Orellana and A. L. Kasinski, Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation, Bio-Protoc., 2016, 6, e1984 Search PubMed.
- C. M. Tan, A. G. Therien, J. Lu, S. H. Lee, A. Caron, C. J. Gill, C. Lebeau-Jacob, L. Benton-Perdomo, J. M. Monteiro, P. M. Pereira, N. L. Elsen, J. Wu, K. Deschamps, M. Petcu, S. Wong, E. Daigneault, S. Kramer, L. Liang, E. Maxwell, D. Claveau, J. Vaillancourt, K. Skorey, J. Tam, H. Wang, T. C. Meredith, S. Sillaots, L. Wang-Jarantow, Y. Ramtohul, E. Langlois, F. Landry, J. C. Reid, G. Parthasarathy, S. Sharma, A. Baryshnikova, K. J. Lumb, M. G. Pinho, S. M. Soisson and T. Roemer, Restoring methicillin-resistant Staphylococcus aureus susceptibility to beta-lactam antibiotics, Sci. Transl. Med., 2012, 4, 126ra135 Search PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- G. M. Morris, R. Huey and A. J. Olson, Using AutoDock for ligand-receptor docking, Curr. Protoc. Bioinf., 2008, 24, 8.14.1–8.14.40 Search PubMed.
- A. Cheng, S. A. Best, K. M. Merz and C. H. Reynolds, GB/SA water model for the Merck molecular force field (MMFF), J. Mol. Graph. Model., 2000, 18, 273–282 CrossRef CAS PubMed.
- S. Vilar, G. Cozza and S. Moro, Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- A. Hospital, J. R. Goñi, M. Orozco and J. L. Gelpí, Molecular dynamics simulations: advances and applications, Adv. Appl. Bioinf. Chem., 2015, 8, 37–47 Search PubMed.
- J. Mortier, C. Rakers, M. Bermudez, M. S. Murgueitio, S. Riniker and G. Wolber, The impact of molecular dynamics on drug design: applications for the characterization of ligand–macromolecule complexes, Drug Discov. Today, 2015, 20, 686–702 CrossRef CAS PubMed.
- J. B. Sturgeon and B. B. Laird, Symplectic algorithm for constant-pressure molecular dynamics using a Nosé–Poincaré thermostat, J. Chem. Phys., 2000, 112, 3474–3482 CrossRef CAS.
- M. Karplus and J. A. McCammon, Molecular dynamics simulations of biomolecules, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05319h |
| This journal is © The Royal Society of Chemistry 2020 |