
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the biosynthesis of Sch-642305 in the fungus Phomopsis sp. CMU-LMA†
Francesco Trenti‡
a,
Karen E. Lebea,
Emilie Adelinb,
Jamal Ouazzanib,
Carsten Schotte*a and
Russell J. Cox *a
*a
aInstitute for Organic Chemistry and BMWZ, Leibniz Universität Hannover, Schneiderberg 38, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bCentre National de la Recherche Scientifique, Institut de Chimie des Substances Naturelles ICSN, Gif-sur-Yvette, France
First published on 21st July 2020
Abstract
Sch-642305 is an unusual bicyclic 10-membered macrolide produced by the filamentous fungus Phomopsis sp. CMU-LMA for which no biosynthetic evidence exists. Here, we generate a draft genome sequence of the producing organism and discover the biosynthetic gene cluster responsible for formation of Sch-642305. Targeted gene disruptions together with reconstitution of the pathway in the heterologous host Aspergillus oryzae dissect key chemical steps and shed light on a series of oxidoreductions occuring in the pathway.
Introduction
Fungal polyketides are one of the most important classes of secondary metabolites, displaying both high structural diversity and a broad range of biological activities.1 Among these Sch-642305 (SCH 1, Scheme 1A) is an unusual bicyclic 10-membered macrolide produced by various filamentous fungi.2–4 It displays potent inhibitory activity against both bacterial DNA primase (EC50 = 70 μM) and HIV-1 Tat (IC50 = 1 μM), making it an attractive target in the ongoing search for novel treatments of bacterial and viral infections.2,4 Since its initial discovery from the filamentous fungus Penicillium verrucosum in 2003 (ref. 2) it has attracted the interest of synthetic chemists and several synthetic approaches towards SCH 1 have been reported.5–7 A ‘biomimetic’ synthesis of SCH 1 was reported by Snider and Zhou based on a key transannular Michael cyclization of the 14-membered lactone 2 to generate the bicyclic scaffold of SCH 1 (Scheme 1C and ESI Fig. S1†), although it is not known if the biosynthesis does, in fact, involve a transannular Michael reaction.8 Lactone 2 is structurally related to the fungal metabolite mutolide 3, a highly reduced polyketide 2,5,8-triene lactone possessing the same number of carbons as SCH 1 (Scheme 1C). Mutolide 3 was shown by labelling experiments to be derived from seven acetate/malonate units and both hydroxyl groups are derived from atmospheric oxygen, suggesting post-elongation oxidative tailoring (Scheme 1C).9 However, 3 has never been co-isolated from any SCH 1 producing strain.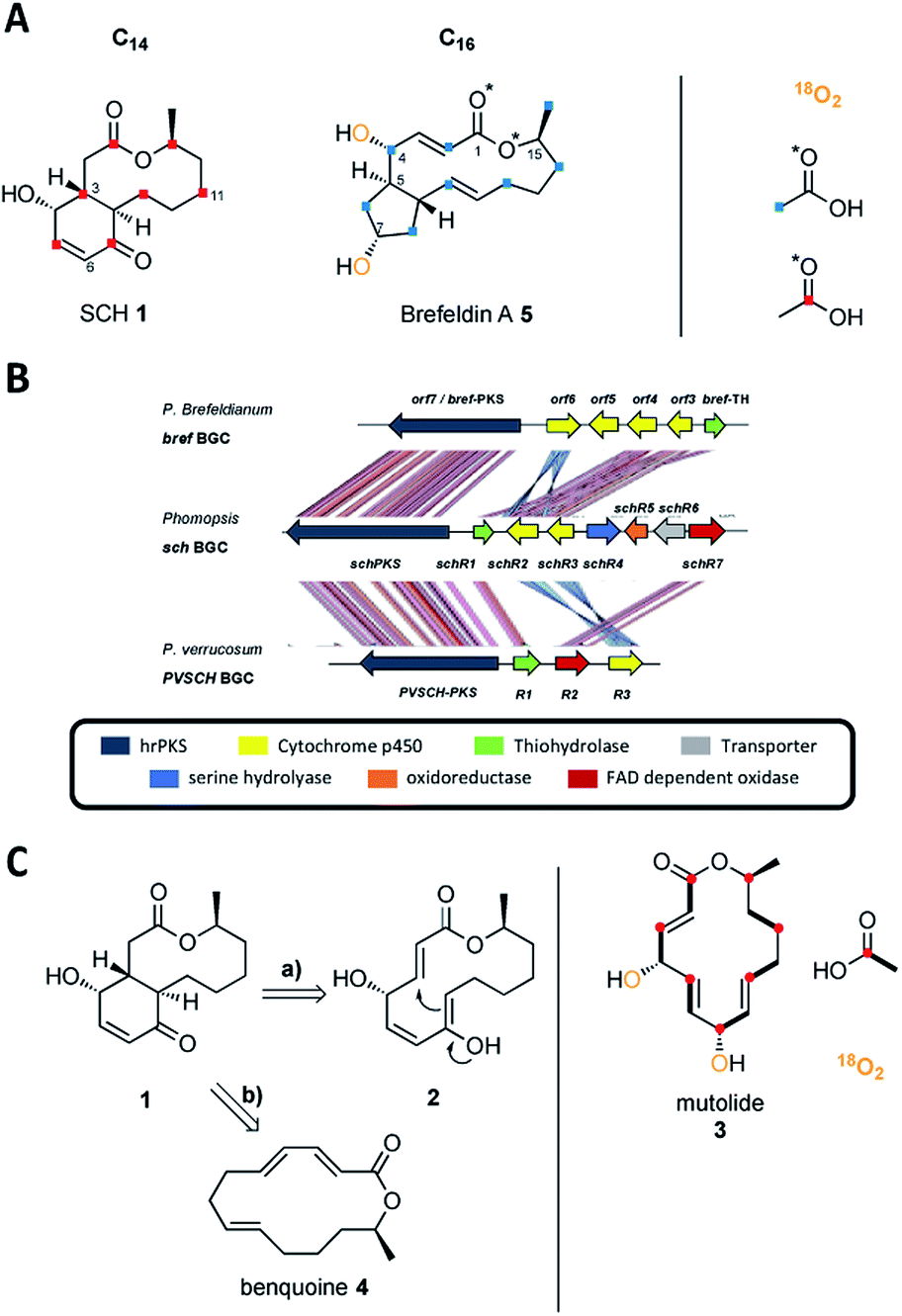 | ||
| Scheme 1 A) Incorporation of labelled acetates and 18O2 into SCH 1 and brefeldin A 5; (B) sch biosynthetic gene cluster and comparison to the bref BGC and PVSCH BGC; (C) hypothetical biosynthetic route to SCH 1 proposed by Snider and Zhou on the basis of the fungal metabolite mutolide 3 (a) and on the basis of benquoine 4 (b). Incorporation of labelled sodium [1-13C]-acetate and 18O2 is displayed for 3. | ||
Co-isolation of SCH 1 with the 14-membered 2,4,8-triene lactone benquoine 4 (Scheme 1C) from Phomopsis sp. CMU-LMA suggested 4 to be the true precursor of SCH 1 (ESI Fig. S2†) but to-date the biosynthetic pathway has remained elusive.3
SCH 1 is structurally related to the bicyclic polyketide brefeldin A 5 (Scheme 1A) that was isolated from various filamentous fungi such as Eupenicillium brefeldianum.10–12 Brefeldin A differs in that it is a 13-membered macrolide (not 10-membered) that is fused to a cyclopentane (not a 4-hydroxycyclohexenone) at the C-5 (not C-3) junction. Incorporation studies with labelled acetate and atmospheric oxygen established the polyketide origin of brefeldin A 5.13–15 As in mutolide 3, both hydroxyl groups of 5 are derived from atmospheric oxygen (Scheme 1A).14 Tang and co-workers identified the biosynthetic gene cluster (BGC) responsible for the production of 5 in Eupenicillium brefeldianum.12 The bref BGC encodes a highly reducing polyketide synthase (hrPKS, brefPKS), a thiohydrolase (brefTH) and four cytochrome P450 oxygenases (Scheme 1B).12
Reconstitution of BrefPKS and the partnering thiohydrolase BrefTH in yeast and in vitro led to production of an acyclic 2,4,10-triene octaketide as the dominant product, which possesses the same chain length (C16) and expected β-reduction pattern for formation of 5 (ESI Fig. S3†).12 It is notable that in the absence of BrefTH, BrefPKS is a nonaketide synthase and the thiohydrolase plays a critical role in chain length control. Subsequent intramolecular annulation and macrolactonization were proposed to give rise to the bicyclic carbon skeleton of 5.12 However, the mechanism and timing of both macrolactonization and cyclopentane formation are not yet understood and pose intriguing biosynthetic questions. We reasoned that SCH 1 biosynthesis might proceed in a similar fashion. Here, we report the discovery of the sch BGC responsible for formation of SCH 1. Targeted gene knockout experiments together with heterologous expression of key biosynthetic genes in the fungal host Aspergillus oryzae NSAR1 identified a likely minimal gene set needed for production of SCH 1 and shed light on a series of oxidoreductions in the biosynthetic pathway.
Results
Analysis of wild type Phomopsis sp. CMU-LMA
In previous work SCH 1 was co-isolated together with a range of polyketide compounds from the large-scale fermentation of Phomopsis sp. CMU-LMA wildtype (WT, Fig. 1).3,16 Here, repeated small-scale cultivation of the WT strain and analysis of ethyl acetate extracts by liquid-chromatography mass spectrometry (LCMS) confirmed reliable production of SCH 1. SCH 1 was concomitantly produced with LMA P3 6 and DHTO 7 (Fig. 1). LMA-P1 8, a reduced congener of SCH 1, as well as LMA-P2 9, cytosporone B 10 and phomolide C17 11 were observed sporadically across fermentation of WT and knockout strains alike and formation of these metabolites appears to be highly sensitive to small changes in culture conditions. The previously reported compounds benquinol 12, DHTTA 13 and benquoine 4 were never observed in this study, possibly as a result of the small-scale cultivation deployed here.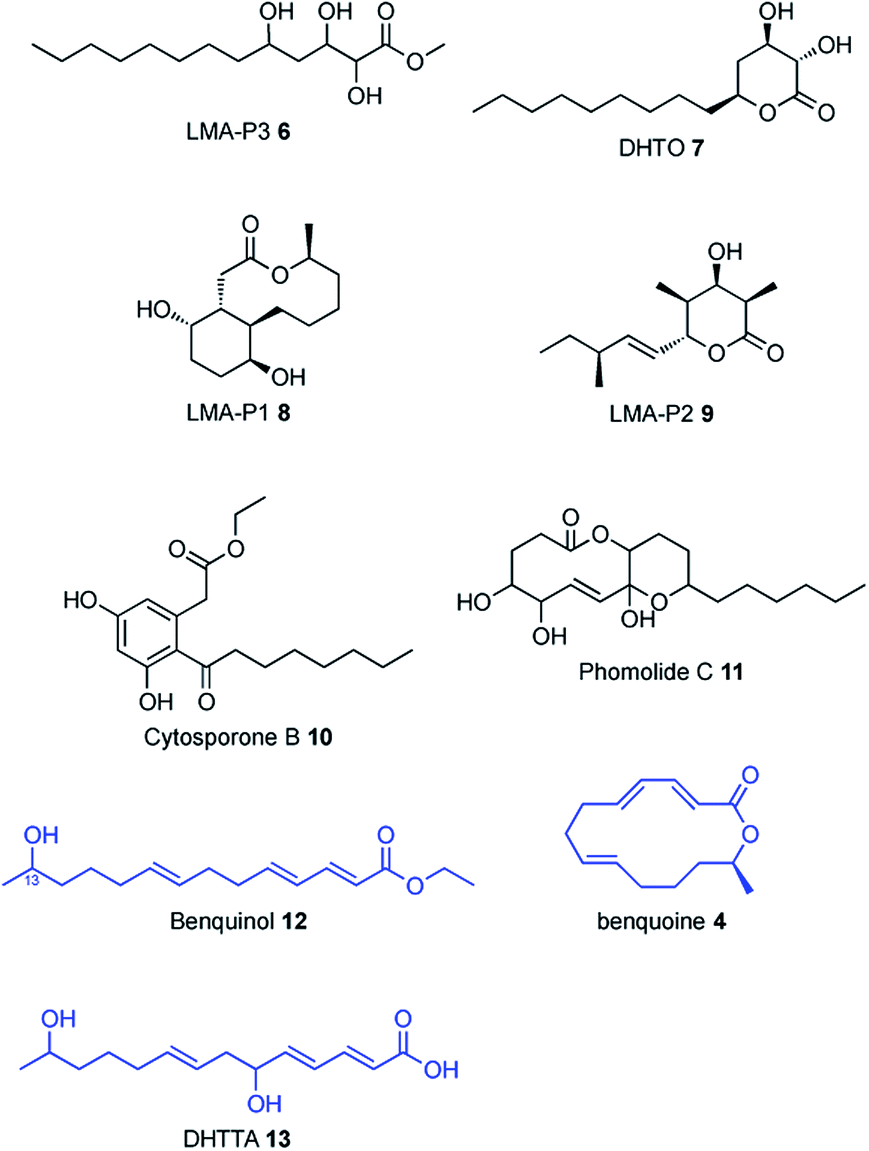 | ||
| Fig. 1 Secondary metabolites concomitantly isolated with SCH 1. Compounds in blue were previously isolated from Phomopsis sp. CMU-LMA but not observed in this study. | ||
In order to discover the biosynthetic origin of 1 we fed sodium [1-13C]-labelled acetate (Scheme 1A) to the WT strain. Labelled SCH 1 (1 mg) was isolated and analysed by 13C nuclear magnetic resonance (NMR). Isotopic labelling resulted in signal enhancement of seven carbons atoms in SCH 1 (C-1, C-3, C-5, C-7, C-9, C-11, C-13; Scheme 1A and ESI Fig. S4†). As expected, the obtained alternating pattern of isotopic labelling indicated that SCH 1 is formed from seven acetate/malonate units and is consistent with a highly reduced heptaketide without rearrangement. Attachment of the hydroxyl group to the Cβ-4 carbon indicates that at least one post-elongation oxidative event occurs during 1 biosynthesis, in analogy to incorporation of both hydroxyl groups in 3 and 9 from atmospheric oxygen.9,14 Notably, the Z-configured C-5/C-6 olefin lies within one of the seven acetate units and it thus cannot have been created by the DH domain of the PKS.18
Genome analysis of Phomopsis sp. CMU-LMA
Genomic DNA of Phomopsis sp. CMU-LMA was sequenced (4 × 36 cycles) using the v5 Sequencing Kit (FC-104-5001, Illumina) and assembled by the CASAVA-1.8 data analysis pipeline. A draft genome sequence of 64.4 Mb on 2042 scaffolds with scaffold N50 of 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 395 bp was afforded (for assembly data see ESI Table S1†). Analysis of the genome using the Antibiotics & Secondary Metabolite Analysis Shell (AntiSMASH)19 identified more than 150 putative secondary metabolite biosynthetic gene clusters (BGC, ESI Table S2†). Of these, 60 were proposed to encode a PKS as the core gene and manual analysis using the Basic Local Alignment Search Tool (BLAST)20 predicted 36/60 to belong to the class of highly reducing PKS.1 Based on the structural resemblance between SCH 1 and brefeldin A 5 we assumed that the PKS responsible for formation of SCH 1 should share sequence homology to brefPKS. Indeed, homology searches using brefPKS against the Phomopsis draft genome sequence identified a putative sch BGC centred around a highly reducing PKS (65% global identity to brefPKS). Domain analysis of the putative schPKS revealed the expected set of reducing domains and absence of a C-methyl transferase (C-MeT) domain, consistent with the production of a non-methylated highly reduced polyketide. Manual annotation using Softberry FGENESH21 and BLAST of open reading frames within the 50 kbp flanking regions of the putative schPKS revealed several genes putatively involved in secondary metabolism encoding a hydroxylase (schR1); two cytochrome P450 oxygenases (schR2 and schR3); a serine-hydrolase (schR4); an NAD dependent oxidoreductase (schR5); a transporter (schR6) and an FAD dependent oxidase (schR7, Scheme 1B). In the course of this study the genome of Penicillium verrucosum, another producer of SCH 1, became publicly available. Local BLAST analysis against the genome of P. verrucosum using sch PKS as a template revealed a third homologous cluster centred around an hrPKS (here named pvschPKS; 51% global identity to BrefPKS), which contains a similar manifest of genes as observed in the putative sch BGC.
395 bp was afforded (for assembly data see ESI Table S1†). Analysis of the genome using the Antibiotics & Secondary Metabolite Analysis Shell (AntiSMASH)19 identified more than 150 putative secondary metabolite biosynthetic gene clusters (BGC, ESI Table S2†). Of these, 60 were proposed to encode a PKS as the core gene and manual analysis using the Basic Local Alignment Search Tool (BLAST)20 predicted 36/60 to belong to the class of highly reducing PKS.1 Based on the structural resemblance between SCH 1 and brefeldin A 5 we assumed that the PKS responsible for formation of SCH 1 should share sequence homology to brefPKS. Indeed, homology searches using brefPKS against the Phomopsis draft genome sequence identified a putative sch BGC centred around a highly reducing PKS (65% global identity to brefPKS). Domain analysis of the putative schPKS revealed the expected set of reducing domains and absence of a C-methyl transferase (C-MeT) domain, consistent with the production of a non-methylated highly reduced polyketide. Manual annotation using Softberry FGENESH21 and BLAST of open reading frames within the 50 kbp flanking regions of the putative schPKS revealed several genes putatively involved in secondary metabolism encoding a hydroxylase (schR1); two cytochrome P450 oxygenases (schR2 and schR3); a serine-hydrolase (schR4); an NAD dependent oxidoreductase (schR5); a transporter (schR6) and an FAD dependent oxidase (schR7, Scheme 1B). In the course of this study the genome of Penicillium verrucosum, another producer of SCH 1, became publicly available. Local BLAST analysis against the genome of P. verrucosum using sch PKS as a template revealed a third homologous cluster centred around an hrPKS (here named pvschPKS; 51% global identity to BrefPKS), which contains a similar manifest of genes as observed in the putative sch BGC.
A homology analysis was performed between the known bref BGC and the two putative sch BGC from Phomopsis sp. CMU-LMA and Penicillium verrucosum using the Artemis comparison tool (ACT),22 revealing high overall similarity (Scheme 1B). In addition to the hrPKS all clusters share a conserved homologue encoding the thiohydrolase brefTH gene (schR1 and pvschR1). A number of homologous cytochrome P450 oxygenases were identified in the three clusters with SchR2 and SchR3 displaying homology to only one P450 (pvschR3) in P. verrucosum but to three P450s in P. brefeldianum (ORF3, ORF4, ORF6). The presence of an oxidase encoding gene (schR7/pvschR2) is unique to the SCH 1 producing strains and not identified in the bref BGC. In order to link the putative sch BGC to the biosynthesis of 1 we next attempted targeted knockout experiments.
Targeted gene knockout in Phomopsis sp. CMU-LMA
Cultivation of Phomopsis sp. CMU-LMA on growth media supplemented with different antibiotics showed that hygromycin B effectively inhibited growth of the WT at 50 μg ml−1. A transformation protocol based on generation of Phomopsis protoplasts and insertion of a gene cassette comprising the fungal constitutive A. nidulans gdpA promoter (PgpdA) fused to the hygromycin-resistance conferring gene hph from E. coli was successfully established (ESI Fig. S5†).23 The bipartite knock-out (KO) method developed by Nielsen and co-workers was then deployed to target genes putatively involved in the biosynthesis of SCH 1.24 A total of 137 transformants targeting five biosynthetic genes (schPKS, schR2, schR3, schR5, and schR7) were obtained and polymerase chain reaction (PCR) successfully confirmed 10 transformants to be true KO strains (ESI Fig. S6†). KO of the serine hydrolase schR4 was attempted but not successful. All obtained KO strains were cultivated under producing conditions alongside WT Phomopsis as a positive control and ethyl acetate extracts of fungal cultures were analysed by LCMS.Targeted gene KO of schPKS (ΔschPKS) followed by LCMS analysis showed loss of SCH 1 production and thus linked the sch BGC to the biosynthesis of 1 (Fig. 2A). The ΔschPKS KO strains were also incapable of producing LMA-P1 8, consistent with previous biotransformation experiments that suggested 8 to be a reduced congener of SCH 1 that has undergone both olefin and carbonyl reduction.25 In contrast, the ongoing capability of ΔschPKS KO strains to produce LMA-P3 6 and DHTO 7 showed these to be biosynthetically unrelated.
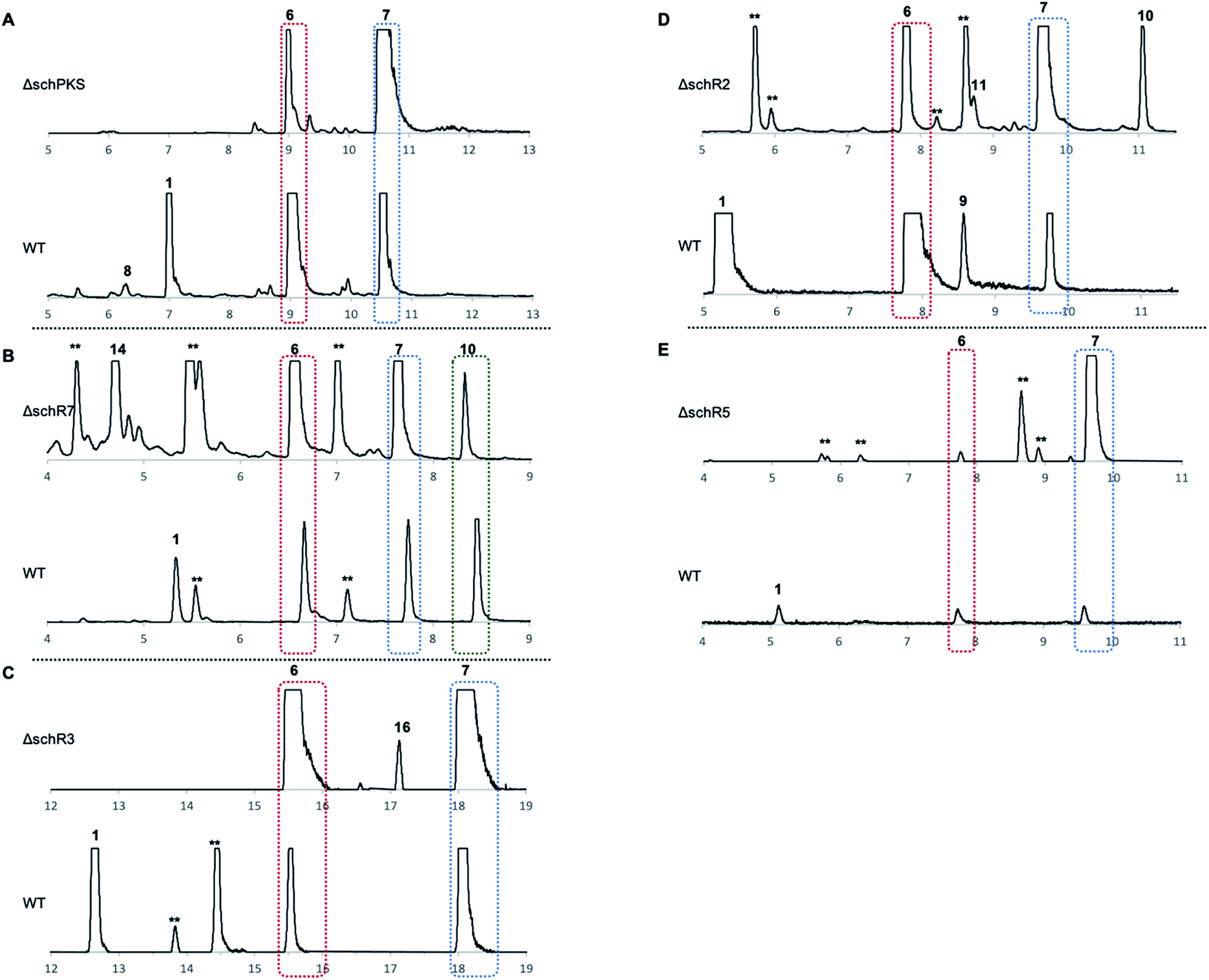 | ||
| Fig. 2 Targeted gene disruption in Phomopsis sp. CMU-LMA. ELSD chromatograms of KO mutants for: (A) schPKS; (B) schR7; (C) schR3; (D) schR2; (E) schR5. ** = unrelated to SCH 1 biosynthesis. X-axis in minutes throughout. | ||
KO of the oxidase encoding gene schR7 (ΔschR7), unique to the sch BGC and not present in the bref BGC, showed loss of SCH 1 production (Fig. 2B). Instead of 1, formation of a new compound 14 (Scheme 2) was observed. This was purified to homogeneity (2.7 mg). High-resolution mass spectrometry (HRMS) data suggested a molecular formula of C14H20O4 ([M]H+ calculated C14H21O4 253.3180, found 253.3178). 1H-NMR confirmed 14 to be a macrolactone as the C-14 methyl showed as a clear doublet at 1.26 ppm. Further analysis using 1H–1H-correlation spectroscopy (COSY) confirmed the carbon skeleton of 14 and showed the same atom connectivity and oxidation pattern as observed in the literature for the known 14-membered lactones mutolide 3 and nigrosporolide 15 (ESI Fig. S7–S10 and ESI Table S3†).9,26 Macrolides 3 and 15 only differ in the configuration of the three olefins, with 3 displaying an E,E,E configuration and 15 an E,Z,Z geometry. However, small differences in chemical shifts between all three compounds suggested 14 to display an alternative olefin configuration. The coupling constant of the coupled protons H-2 and H-3 (JHH 15.6 Hz) indicated E-configuration of this alkene. Coupling constants between the coupled protons H-5 and H-6 were difficult to determine due to signal overlap with proton H-9. Selective decoupling of H-4, however, led to more accurate peak assignment and allowed determination of a smaller coupling constant for H-5/H-6 (JHH 11.3 Hz), corresponding to a Z-configuration (ESI Fig. S11†). The assigned Z-configuration is consistent with literature data for the H-5/H-6 Z-configuration observed in nigrosporolide 15 (JHH 11.4 Hz). Further decoupling of H-10 simplified determination of the coupling constant between the olefin protons H-8 and H-9 (JHH 15.1 Hz), indicating an E-configuration (ESI Fig. S12†). In contrast, the reported Z-configuration in nigrosporolide 15 at the alkene protons H-8 and H-9 is reflected by a significantly smaller coupling constant (JHH 10.6 Hz).25 Further selective decoupling of H-7 did not affect the 1H-signal for H-8, suggesting the dihedral angle between the vicinal protons H-7/H-8 to be around 90° and completing the structural elucidation of 14 (ESI Fig. S12†). Accordingly, disruption of schR7 halted SCH 1 biosynthesis after macrolactonization but prior to annulation.
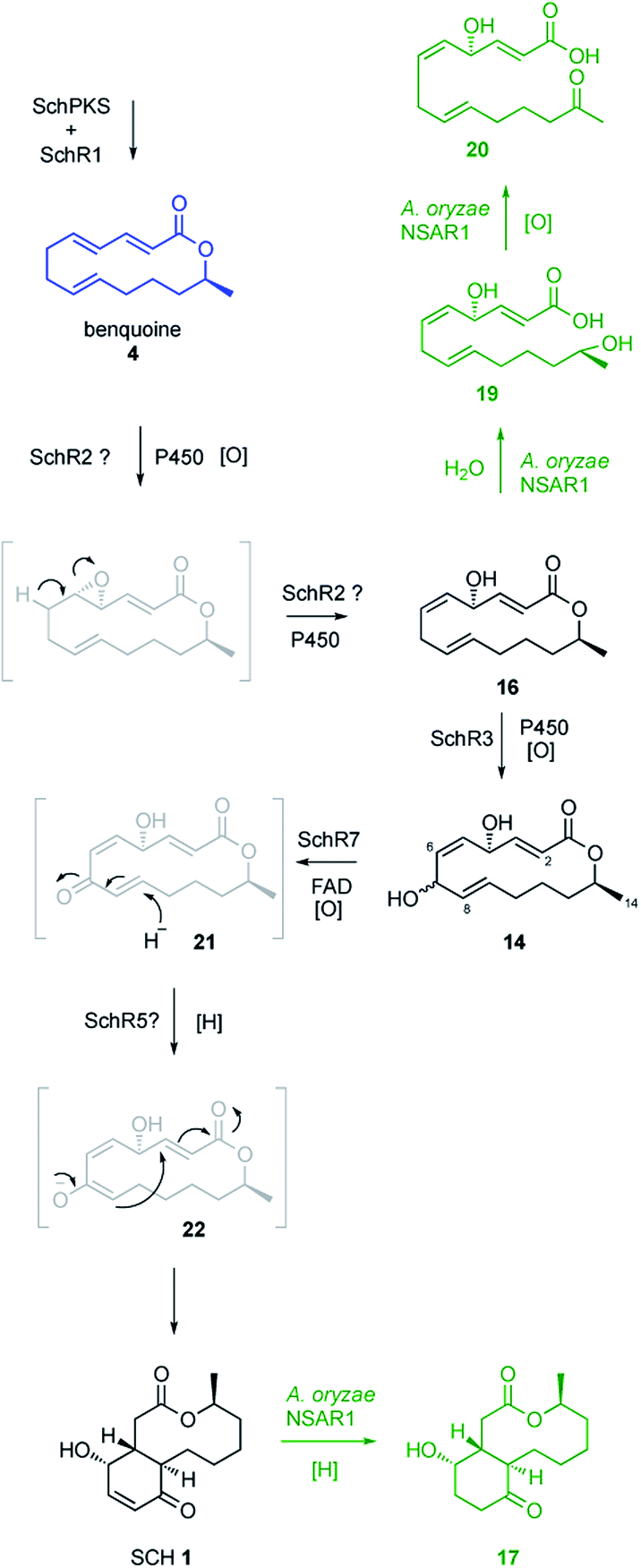 | ||
| Scheme 2 Proposed biosynthesis of SCH 1 and route to shunt metabolites (green). Compounds in black were isolated in this study, compounds in blue were previously isolated from Phomopsis sp. CMU-LMA. Compounds in grey were not observed but suggested as plausible intermediates on the pathway to 1. | ||
Next, we investigated the roles of the two cytochromes P450 (schR2 and schR3) in the biosynthesis of 1, assuming these might be responsible for sequential incorporation of the two hydroxyl groups at the C-4 and C-7 positions. Targeted gene KO of schR3 (ΔschR3) and analysis of ethyl acetate extracts of transformants by LCMS showed that production of SCH 1 was abolished (Fig. 2C). Instead, a less polar compound 16 emerged (Scheme 2), consistent with interrupting the biosynthetic pathway towards 1 at a less oxidized state. HRMS analysis confirmed a molecular formula of C14H20O3 ([M]H+ calculated C14H21O3 237.3185, found 237.3181). Purification to homogeneity afforded 0.8 mg of 16, the structure of which was elucidated by full NMR analysis (ESI Fig. S13–S17 and ESI Table S4†). The obtained NMR data indicated structural similarity to 14 and a 16 amu difference in HRMS data suggested 16 to be the de-hydroxyl analogue of 14. Indeed, replacement of the downfield shifted oxygenated proton in 14 (δH/δC 4.82/70.6 ppm, for H-7/C-7) by two upfield shifted aliphatic protons in 16 (δH/δC 2.82/32.1 ppm, for H2-7/C-7) was clearly observed in the NMR data and together with further COSY and HMBC data confirmed the structure of 16. Observation of 16 conclusively suggests schR3 to incorporate the C-7 hydroxyl group observed in SCH 1 and to act prior to schR7.
Inactivation of the second cytochrome P450 schR2 (ΔschR2) yielded transformants that were deficient in the production of SCH 1 and LMA-P1 7 (Fig. 2D). However, no new intermediates and neither compounds 14 nor 16 were observed in these strains, indicating that SchR2 acts prior to SchR3 and SchR7 and that the pathway is halted in absence of schR2 at an early biosynthetic stage. Formation of phomolide C 11 and cytosporone B 10 was observed in the ΔschR2 mutant but not assumed to be SCH 1 intermediates due to their structure and unpredictable appearance in WT strains when grown under identical conditions.
Finally, targeted gene disruption of schR5, an oxidoreductase unique to the sch BGC, yielded two true KO transformants. LCMS analysis of these strains under producing conditions showed that production of 1 and 8 was abolished but no new compounds were observed (Fig. 2E). Together, the targeted gene KO experiments identified five genes (schPKS, schR2, schR3, schR5, schR7) that are essential for the biosynthesis of SCH 1.
Reconstitution of SCH biosynthesis in Aspergillus oryzae NSAR1
To further investigate the function of individual genes we attempted reconstitution of the SCH biosynthetic pathway in the fungal host A. oryzae NSAR1.27 As SCH 1 was previously shown to be transformed by Aspergillus ochraceus ATCC 1009 resting cells into several reduced congeners24 we first conducted a biotransformation experiment to assess the stability of SCH 1 in A. oryzae. SCH 1 was fed to a growing culture (2 d) of A. oryzae NSAR1. Extraction at day 5 and analysis of extracts by LCMS showed that 1 was predominantly transformed into compound 17 (Fig. 3A). A minor co-metabolite, compound 18, with identical mass fragmentation pattern and UV absorption as 17 was also observed but eluded characterization due to low production titres (ESI Fig. S18†).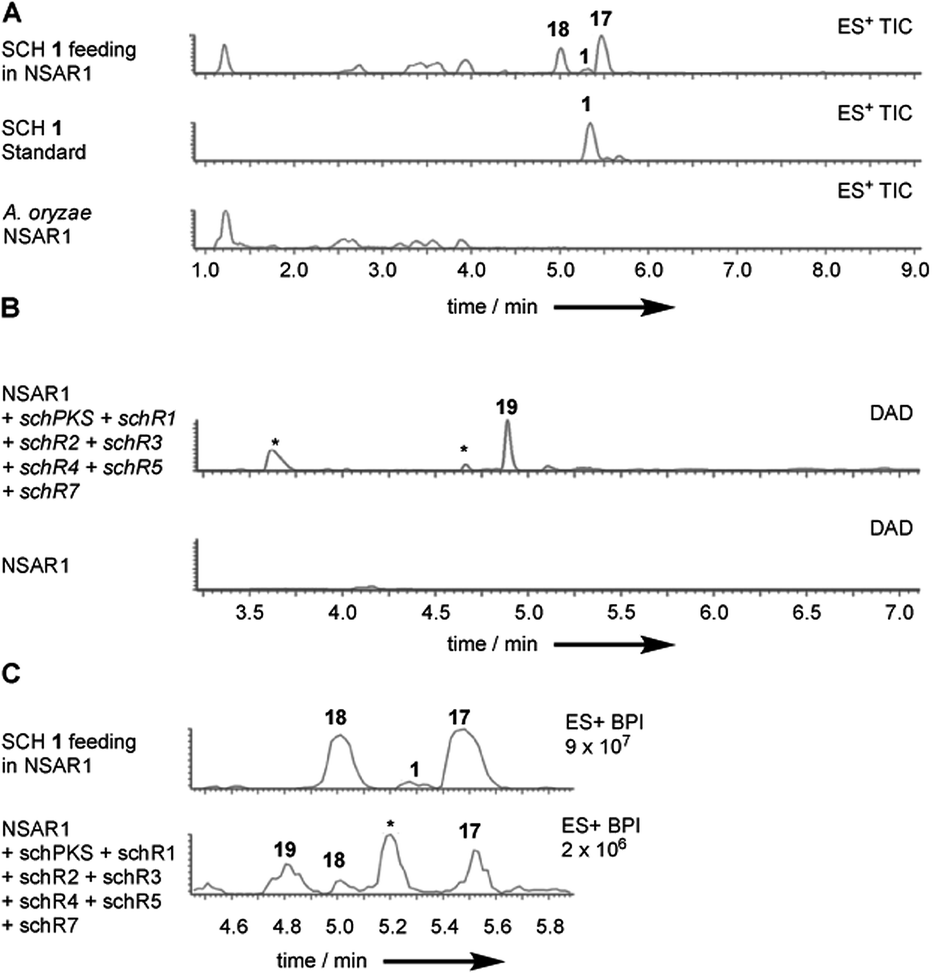 | ||
| Fig. 3 (A) Biotransformation of SCH 1 by A. oryzae NSAR1. ES+ traces of fed 1 to A. oryzae, a standard of 1 and WT control; (B) heterologous expression of the full sch BGC. Diode array detector (DAD) traces of pathway expression and WT control; (C) heterologous expression of the full sch BGC and comparison to fed 1 to A. oryzae. ES+ traces of fed 1 (top) and full sch BGC (bottom). | ||
Isolation of the component 17 and purification to homogeneity afforded 1.2 mg (HRMS C14H22O4 [M − H]− calculated C14H21O4 253.1440, found 253.1440). A 2 amu difference in HRMS compared to SCH 1 suggested 17 to be a reduced congener of 1. Indeed, full NMR analysis identified 17 as the same SCH congener that was previously obtained upon biotransformation of SCH 1 with Aspergillus ochraceus (ESI Fig. S19–S23 and ESI Table S5†).24 Contrary to SCH 1, the C5–C6 olefin in 17 is reduced.
A series of fungal expression plasmids were constructed to express different combinations of sch biosynthetic genes in A. oryzae. For this, cDNA was prepared from Phomopsis WT under producing conditions and yeast homologous recombination was used to clone biosynthetic genes into the modular expression system described by Lazarus and co-workers.28
Plasmids corresponding to the “full” SCH biosynthetic gene cluster (schPKS, schR1, schR2, schR3, schR4, schR5, schR7, but omitting the transporter schR6) were successfully transformed into A. oryzae. Biotransformation of 1 by A. oryzae WT indicated that successful reconstitution of the sch BGC in the heterologous host should yield the reduced SCH analogue 17 instead of 1. Indeed, growth under inducing conditions and LCMS analysis of transformants carrying the full cluster showed production of trace amounts of 17 and the minor co-metabolite 18 previously observed in biotransformation experiments (Fig. 3C). SCH 1 was not observed.
Observation of trace amounts of 17 upon expressing the “full” sch BGC shows that the transformed set of genes is sufficient for production of 1 and that no other gene is needed for SCH 1 biosynthesis. However, transformation of the full cluster also yielded 19 as the dominant product and 20 as a minor co-metabolite (Fig. 3B and ESI Fig. S24†). Isolation of 19 and purification to homogeneity (1 mg; HRMS [M − H]− calculated C14H21O4 253.1440, found 253.1440) followed by NMR characterization revealed a high similarity to data previously obtained for compound 16 (ESI Fig. S25–S29 and ESI Table S6†), obtained upon KO of schR3. An 18 amu difference observed in HRMS indicated addition of water in the case of 19, consistent with an opened lactone. Indeed, significant differences in chemical shifts of the methyl group protons at C-14 (δH3–14 1.02 ppm in 19, δH3–14 1.27 ppm in 16) and upfield shift of the proton attached to C-13 (δH-13 3.56 ppm in 19, δH-13 4.82 ppm in 16) supported 19 to be the open acid form of 16. Bromocresol staining of 16 and 19 supported 19 to be the corresponding free acid derivative of 16 (ESI Fig. S30†). The coupling constant (JHH 15.4 Hz) of the alkene protons H-2/H-3 indicated 2E-configuration. Determination of the coupling constant between olefin protons H-5 and H-6 was difficult due to overlapping 1H-signals with H-8 and H-9. 1H-decoupling of H-7 facilitated determination of the coupling constant of the olefin protons H-5/H-6 (JHH 10.8 Hz) and suggested a Z-configuration (ESI Fig. S31†). Further decoupling of H-10 allowed determination of the coupling constant for the alkene protons H-8/H-9 (JHH 15.4 Hz) and suggesting an E-olefin (ESI Fig. S32†). The observed double bond configurations were consistent with the configurations determined for compound 16.
Purification of compound 20 to homogeneity (1.9 mg) and subsequent NMR analysis revealed almost identical chemical shifts compared to 19 (ESI Fig. S33–S37 and ESI Table S7†). However, C-13 was identified as a quaternary position as observed by heteronuclear single quantum coherence spectroscopy (HSQC) and the corresponding chemical shift of C-13 (208.3 ppm) indicated a carbonyl group instead of an alcohol, agreeing with HRMS data obtained for 20 ([M − H]− calculated C14H19O4 251.1283, found 251.1286). Compound 20 thus constitutes the 13-keto-congener of 19. Since gene disruption of schR3 and schR7 yielded the intact lactones 14 and 16, observation of 19 and 20 suggests these to be early shunt products, probably derived by early hydrolysis of the lactone moiety in 16.
Expression of early genes in A. oryzae
To investigate early steps in SCH 1 biosynthesis we attempted to assemble the pathway stepwise, starting with expression of the standalone schPKS. But neither expression of schPKS alone nor co-expression with thiohydrolase encoding gene schR1 revealed any early biosynthetic intermediates (ESI Fig. S38†) in A. oryzae. Full cluster expression and observation of compound 17 however had previously demonstrated that SchPKS is active in A. oryzae. We thus assumed that the PKS product is likely to be shunted in this fungal host. This is consistent with the previous finding that upon KO of schR2 no intermediates are observed, suggesting that the early intermediates of SCH 1 biosynthesis are quickly degraded. This is consistent with previous observations of rapid degradation of linear (non-methylated) polyketides in Aspergillus oryzae, probably by β− or ω-oxidation.29Inclusion of the P450 schR2 in the expression affords acyclic compounds 19 and 20 again, but no trace amounts of 17 were identified (Fig. 4). This is consistent with SchR2 introducing the C-4 hydroxyl group prior to the pathway being redirected into the shunt pathway. Addition of oxidase encoding gene schR7 and the second P450 encoded by schR3 gave 19 and 20 but at lower titre. Together these results show that SchPKS, the partnering hydrolase SchR1 and one of the two P450s are sufficient to produce the 16 analogue 19 which is probably a shunt product in A. oryzae. Further oxidation by the heterologous host results in 20.
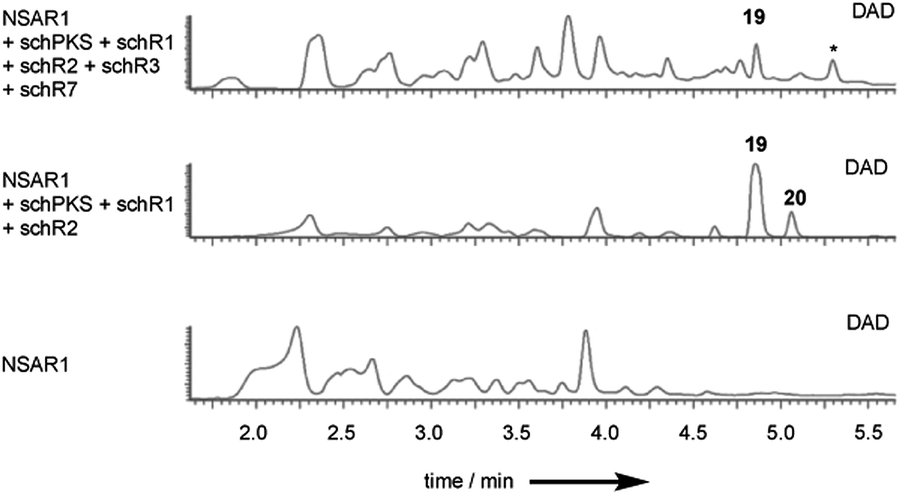 | ||
| Fig. 4 DAD chromatograms of fungal extracts from the heterologous expression of early biosynthetic genes in A. oryzae NSAR1. Top, co-expression of schPKS, schR1, schR2, schR3, schR7; middle, co-expression of schPKS, schR1, schR2; bottom, NSAR1 WT. | ||
Finally, omission of schR1, the thiohydrolase encoding gene, from the full cluster expression abolishes production of any SCH 1 congener (ESI Fig. S39†) as expected from the brefeldin A pathway.12
Discussion and conclusion
In this study the Sch-642305 BGC (sch BGC) was discovered and validated. Reconstitution of the entire SCH 1 biosynthetic gene cluster in A. oryzae NSAR1 successfully afforded the biotransformed SCH 1 congener 17 and demonstrated that a minimal set of 7 genes is sufficient to produce 1. Early steps in SCH 1 biosynthesis eluded characterization as gene disruptions in the native producer did not lead to formation of early stage biosynthetic intermediates. Likewise, investigation of early steps in A. oryzae repeatedly led to formation of the acyclic SCH 1 congeners 19 and 20. We assume that 19 and 20 constitute shunt products derived by hydrolysis of the lactone 16 by the heterologous host. Unexpected and undesired side reactions upon heterologous expression in A. oryzae are well-known. Oikawa and coworkers e.g. previously reported undesired oxidative shunting upon expressing key biosynthetic genes involved in cytocholasin K and solanapyrone formation.30,31 Similar oxidations have been described for the heterologous expression of desmethylbassianin and for the avirulence gene ACE1.32,33 All observed oxidations occurred at the terminus of linear polyketide precursors such as the observed oxidation of compound 19 to 20.Given the high homology between the sch BGC and the bref BGC we believe it is reasonable to assume that early biosynthetic steps are similar. Tang and co-workers showed that reconstitution of the hrPKS BrefPKS and the thiohydrolase BrefTH led to formation of an acyclic octaketide likely to be the precursor of 5, with formation of the correct octaketide being dependent on the presence of the thiohydrolase BrefTH.12 They proposed a mechanism where the thiohydrolase BrefTH performs both off-loading of the PKS chain and lactonization by using its catalytic His-276 to deprotonate the terminal hydroxyl group of the linear octaketide and thus yielding a suitable nucleophile for lactonization. It is easy to imagine how small changes in PKS programming lead to formation of a heptaketide in 1 biosynthesis in contrast to the octaketide observed in 5 biosynthesis and we propose that a similar interplay between SchPKS and SchR1 gives rise to such early intermediates. Co-isolation of benquinol 12 and benquoine 4 from the SCH 1 producing strain Phomopsis sp. CMU-LMA indeed identified such likely early biosynthetic intermediates. Deprotonation of the C-13 hydroxyl group of 12 would indeed give rise to a suitable nucleophile for 4 formation. A similar mechanism was recently proposed for the fungal macrolides phaeospelide A and phaseolide A where macrolactone formation was solely dependent on the associative work of a hrPKS and a standalone trans acting thiolesterase.34
Furthermore, omission of schR1 from the heterologous expression in A. oryzae abolished production of any SCH congeners and thus demonstrates the crucial role it plays in SCH biosynthesis. The obtained data from KO experiments suggest that the two cytochrome P450 oxygenases SchR2 and SchR3 subsequently introduce the C-4 and C-7 hydroxyl groups. SchR2 most likely acts on the C-4/C-5 E-configured olefin inserting an epoxide which can open to the isolated compound 16 with consequent double bond migration. Subsequent action of SchR3 inserts the C-7 hydroxyl and gives rise to the observed intermediate 14 (Scheme 2).
The key structural feature of SCH 1 is the presence of the 4-hydroxycyclohexenone ring, formation of which is not yet understood. Based on the results obtained from KO experiments and heterologous expression experiments functions have been assigned to all genes except schR5 and schR7, suggesting one of these to be responsible for formation of the six-membered ring. Bioinformatic analysis of schR7 shows highest homology (28% sequence identity) to the flavoprotein 6-hydroxy-D-nicotine oxidase from Neurospora crassa, an enzyme known to convert hydroxyl groups to ketones and we thus propose SchR7 to catalyse conversion of the C-7 hydroxyl group in 14 to the corresponding ketone 21 (Scheme 2). Putative reduction of the C-5/C-6 olefin by oxidoreductase SchR5 would lead to formation of the enolate 22 which could undergo subsequent Michael-type cyclization to generate the 4-hydroxycyclohexenone ring observed in SCH 1. Disruption of the schR5 gene abolished production of 1 and thus it has to play a crucial role in 1 formation.
It is notable that the timing of formation of the 4-cyclohexenone moiety in SCH 1 must differ from formation of the cyclopentane ring observed in brefeldin A 5. Observations by Tang and co-workers that reconstitution of BrefPKS and BrefTH affords acyclic PKS products only suggests that cyclopentane formation needs to occur prior to macrolactonization, thus generating a configuration more favourable for lactone formation.12 On the contrary, in SCH 1 biosynthesis lactone formation occurs prior to formation of the six-membered ring, as indicated by observation of the lactones 14 and 16 which lack the cyclohexenone moiety. Additionally, during SCH 1 biosynthesis we conclusively demonstrated that both C-4 and the C-7 hydroxylation occur prior to formation of the cyclohexenone moiety whereas labelling studies by Hutchinson and co-workers suggest that ring closure in 3 is not dependent on C-7 hydroxylation.13
Overall, we have identified the sch BGC in Phomopsis sp. CMU-LMA, and shown key steps of the biosynthesis are similar to those proposed for brefeldin A 5, although the transannulation steps differ. This is consistent with differences in the respective gene clusters. Combined data from gene disruptions and reconstitution of SCH 1 biosynthesis in A. oryzae suggest a minimal gene set needed for formation of 1 and a plausible pathway towards 1 was suggested for the first time. Further work will be required to conclusively demonstrate the mechanism of ring formation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FT thanks the HSBDR Doctoral Training Centre for funding. KEL and CS thank the Leibniz Universität Hannover for funding. Christopher Lambert is thanked for technical assistance. Genome data assembly were performed by CeBiTeC at the University of Bielefeld, and Prof. J. Kalinowski and Dr. D. Wibberg are thanked for data assembly work. DFG is thanked for the provision of NMR equipment (INST 187/686-1). J. O. thanks the IMAGIF High throughput sequencing platform at CNRS, Gif-sur-Yvette. The publication of this article was funded by the Open Access Fund of the Leibniz Universität Hannover.Notes and references
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC
.
- M. Chu, R. Mierzwa, L. Xu, L. He, J. Terracciano, M. Patel, V. Gullo, T. Black, W. Zhao, T.-M. Chan and A. T. McPhail, J. Nat. Prod., 2003, 66, 1527–1530 CrossRef CAS PubMed
- E. Adelin, C. Servy, S. Cortial, H. Lévaique, M.-T. Martin, P. Retailleau, G. Goff, B. Bussaban, S. Lumyong and J. Ouazzani, Phytochemistry, 2011, 72, 2406–2412 CrossRef CAS
- H. Jayasuriya, D. L. Zink, J. D. Polishook, G. F. Bills, A. W. Dombrowski, O. Genilloud, F. F. Pelaez, L. Herranz, D. Quamina, R. B. Lingham, R. Danzeizen, P. L. Graham, J. E. Tomassini and S. B. Singh, Chem. Biodiversity, 2005, 2, 112–122 CrossRef CAS PubMed
- G. Mehta and H. M. Shinde, Tetrahedron Lett., 2005, 46, 6633–6636 CrossRef CAS
- G. Mehta and H. M. Shinde, Chem. Commun., 2005, 3703 RSC
- E. M. Wilson and D. Trauner, Concise Synthesis of the Bacterial DNA Primase Inhibitor (+)-Sch 642305, Org. Lett., 2007, 9, 1327–1329 CrossRef CAS
- B. B. Snider and J. Zhou, Org. Lett., 2006, 8, 1283–1286 CrossRef CAS PubMed
- H. B. Bode, M. Walker and A. Zeeck, Eur. J. Org. Chem., 2000, 2000, 1451–1456 CrossRef
- V. L. Singleton, N. Bohonos and A. J. Ullstrup, Nature, 1958, 181, 1072–1073 CrossRef CAS PubMed
- Y. Suzuki, H. Tanaka, H. Aoki and T. Tamura, Agric. Biol. Chem., 1970, 34, 395–413 CAS
- A. O. Zabala, Y.-H. Chooi, M. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed
- Y. Yamamoto, A. Hori and C. R. Hutchinson, J. Am. Chem. Soc., 1985, 107, 2471–2474 CrossRef CAS
- C. T. Mabuni, L. Garlaschelli, R. A. Ellison and C. R. Hutchinson, J. Am. Chem. Soc., 1979, 101, 707–714 CrossRef CAS
- B. E. Cross and P. Hendley, J. Chem. Soc., Chem. Commun., 1975, 124b–1125 RSC
- E. Adelin, M.-T. Martin, S. Cortial, P. Retailleau, S. Lumyong and J. Ouazzani, Phytochemistry, 2013, 93, 170–175 CrossRef CAS PubMed
- A. Ito, H. Maeda, A. Tonouchi and M. Hashimoto, Biosci., Biotechnol., Biochem., 2015, 79, 1067–1069 CrossRef CAS PubMed
- D. H. Kwan and F. Schulz, Molecules, 2011, 16, 6092–6115 CrossRef CAS PubMed
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed
- A. A. Salamov and V. V. Solovyev, Genome Res., 2000, 10, 516–522 CrossRef CAS PubMed
- T. J. Carver, K. M. Rutherford, M. Berriman, M.-A. Rajandream, B. G. Barrell and J. Parkhill, Bioinformatics, 2005, 21, 3422–3423 CrossRef CAS
- P. J. Punt, R. P. Oliver, M. A. Dingemanse, P. H. Pouwels and C. A. M. J. J. van den Hondel, Gene, 1987, 56, 117–124 CrossRef CAS
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed
- E. Adelin, C. Servy, S. Cortial, H. Lévaique, J. Gallard, M.-T. Martin, P. Retailleau, B. Bussaban, S. Lumyong and J. Ouazzani, Bioorg. Med. Chem. Lett., 2011, 21, 2456–2459 CrossRef CAS
- J. S. Harwooda, H. G. Cutler and J. M. Jacyno, Nigrosporolide, Nat. Prod. Lett., 2006, 6, 181–185 CrossRef
- F. J. Jin, J. Maruyama, P. R. Juvvadi, M. Arioka and K. Kitamoto, FEMS Microbiol. Lett., 2004, 239, 79–85 CrossRef CAS PubMed
- M. N. Heneghan, A. A. Yakasai, L. M. Halo, Z. Song, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, ChemBioChem, 2010, 11, 1508–1512 CrossRef CAS PubMed
- E. J. Skellam, D. Hurley, J. Davison, C. M. Lazarus, T. J. Simpson and R. J. Cox, Mol. BioSyst., 2010, 6, 680–682 RSC
- R. Fujii, A. Minami, K. Gomi and H. Oikawa, Tetrahedron Lett., 2013, 54, 2999–3002 CrossRef CAS
- R. Fujii, T. Ugai, H. Ichinose, M. Hatakeyama, T. Kosaki, K. Gomi, I. Fujii, A. Minami and H. Oikawa, Biosci., Biotechnol., Biochem., 2015, 80, 426–431 CrossRef
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05311b |
| ‡ Current address: Max Planck Institute for Chemical Ecology, Department of Natural Product Biosynthesis, Jena. |
| This journal is © The Royal Society of Chemistry 2020 |