
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically driven, cobalt–carbon bond-mediated direct intramolecular cyclic and acyclic perfluoroalkylation of (hetero)arenes using X(CF2)4X†
Luxia Cuia,
Toshikazu Ono *ab,
Md. Jakir Hossain‡
a and
Yoshio Hisaeda*ab
*ab,
Md. Jakir Hossain‡
a and
Yoshio Hisaeda*ab
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: tono@mail.cstm.kyushu-u.ac.jp; yhisatcm@mail.cstm.kyushu-u.ac.jp
bCenter for Molecular Systems (CMS), Kyushu University 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 30th June 2020
Abstract
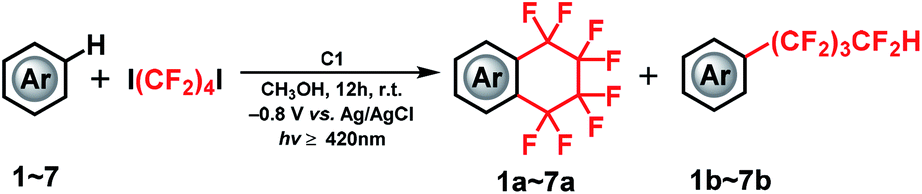
A proof-of-concept for the one-step, synthetically challenging cyclic and acyclic perfluoroalkylation of (hetero)arenes driven by the valence change of a vitamin B12 derivative as a cobalt catalyst in the presence of fluoroalkylating reagents (X(CF2)4X) is presented. The consecutive formation of cobalt–carbon bonds and generation of fluoroalkyl radicals by homolysis are the key steps for the reaction to proceed.
The cobalt–carbon (Co–C) bond is recognized as a crucial catalytic intermediate, and has been extensively studied for radical-mediated reactions in the fields of bioinorganic and organometallic chemistry.1–7 In particular, fluorine substitution of aromatic compounds is an interesting research topic due to the dramatic impact of this reaction on the physical, chemical, and biological properties of the substrates.8–11 In this context, methods for the stoichiometric or catalytic mono-, di-, and trifluoromethylation, and other perfluoroalkylations of (hetero)arenes have been extensively studied.12–20 Nonetheless, catalytic radical fluoroalkylation mediated by Co–C intermediates is still less explored. Recently, our group has investigated electrochemically driven, radical fluoroalkylation reactions using the vitamin B12 derivative, heptamethyl cobyrinate perchlorate [Cob(II)7C1ester]ClO4 (C1), as a cobalt catalyst and fluoroalkylating reagents such as BrCF2COOEt, CF3I, and RfI (Scheme 1(a and b)).21,22 These reactions proceed as follows: first, the Co(I) species is generated from C1 by controlled-potential electrolysis at −0.8 V vs. Ag/AgCl, and it quickly reacts with the fluoroalkylating reagent, e.g., RfI, to form a Co(III)–Rf complex. The Co(III)–Rf complex releases a Rf radical under visible-light irradiation (≥420 nm). Finally, the generated Rf radical reacts with nonactivated (hetero)arenes to afford the fluoroalkylated product. Despite these advances, the radical fluoroalkylation via homolysis of a Co–C bond cannot be clearly distinguished from the reaction obtained by directly reducing fluoroalkylating agents with conventional photoredox catalysts. This situation motivated us to explore in more detail the radical-generating ability from the homolysis of a Co–C bond. Herein, we investigated the intramolecular fluoroalkylating cyclization of (hetero)arenes promoted by formation of a Co–C bond and subsequent generation of fluoroalkyl radicals by homolysis in the presence of the dihalogenated fluoroalkylating reagents X(CF2)4X (Scheme 1(c)).
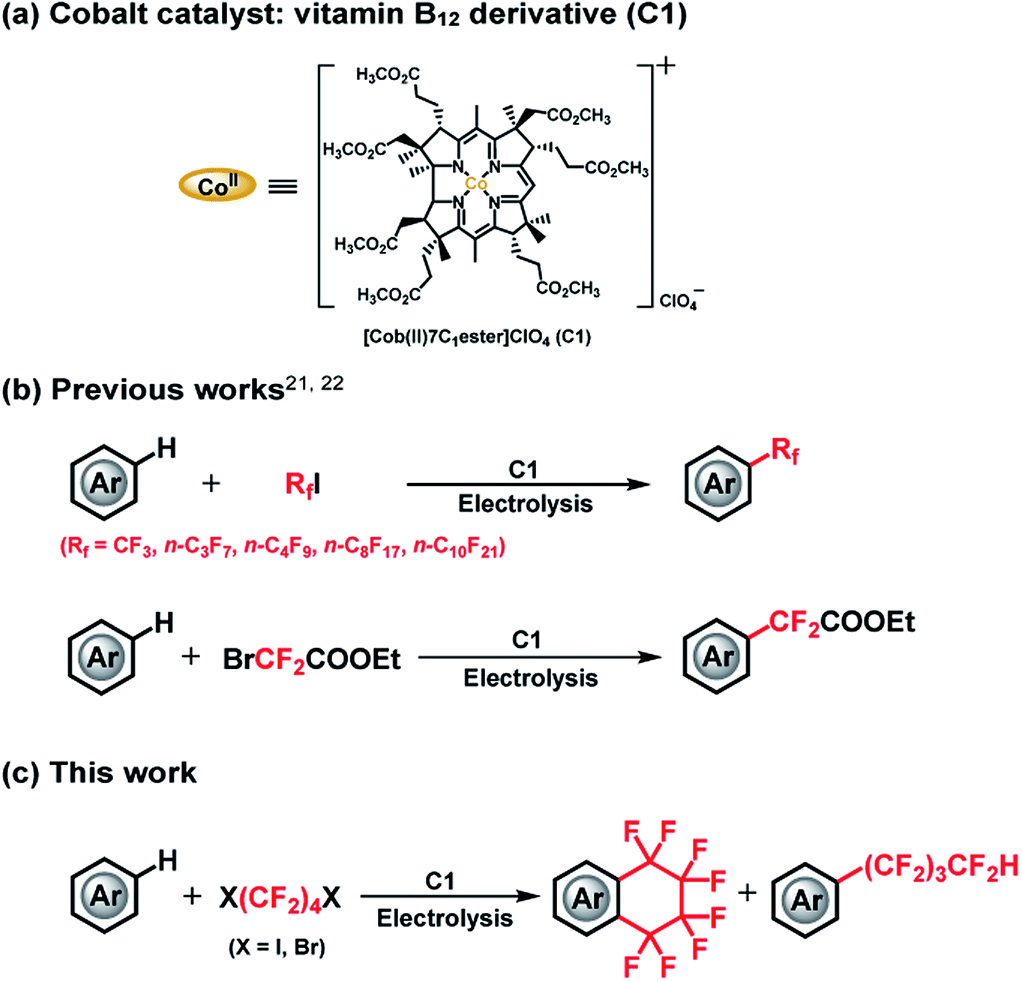 | ||
| Scheme 1 (a) Molecular structure of C1. (b) Trifluoromethylation, perfluoroalkylation and difluoroacylation of (hetero)arenes catalyzed by C1. (c) This work. | ||
We selected X(CF2)nX as alkylating reagents because, although the –(CF2)n– moiety is becoming increasingly important for a diverse array of functional compounds,23–27 methods for the construction of fluoroalkyl-containing rings on aromatic compounds are still scarce. Although a number of studies have been reported in this regard, stoichiometric or harsh conditions are normally required.23,25,28,29 To the best of our knowledge, Co–C bond mediated one-step catalytic C–H intramolecular fluoroalkylating cyclization of unactivated (hetero)arenes through an electrocatalytic method under mild conditions has not been explored yet.
In this study, we demonstrate an electrolysis-driven, intramolecular fluoroalkylating cyclization of (hetero)arenes using dihalogenated fluoroalkylating reagents (X(CF2)4X; X = I, Br) and the vitamin B12 derivative (C1) as cobalt catalyst under mild conditions. We also present that X(CF2)nX (n = 4, 6) can serve as a –(CF2)nH source especially in the presence of methanol (CH3OH) solvent, leading to an acyclic perfluoroalkylated compound containing the –(CF2)nH functional group (Scheme 1(c)). This is the first report on catalytic Co–C bond-mediated intramolecular cyclic and acyclic perfluoroalkylation of (hetero)arenes through an electrochemical method, which provides a new method for the preparation of a large number of synthetically important functional compounds. Although the electrochemically enabled fluoroalkylation strategies30–35 have increasingly emerged in recent years, this work should provide a new insight into various fields.
To investigate the abovementioned fluoroalkylation reactions, we firstly focused on the redox behaviour of C1 as catalyst in the presence or absence of octafluoro-1,4-diiodobutane (1,4-C4F8I2) in CH3OH by cyclic voltammetry (CV) at a scan rate of 100 mV s−1 under nitrogen (Fig. S1†). We observed a reversible Co(II)/Co(I) redox couple of C1 at −0.63 V vs. Ag/AgCl. After adding 2 eq. 1,4-C4F8I2 to C1, the voltammetric pattern was changed, and a new irreversible reduction wave at ca. −0.76 V vs. Ag/AgCl appeared. This can be ascribed to the reduction of the fluoroalkylated derivative of C1, which suggested the suitability of C1 for this molecular transformation. Subsequently, we conducted the fluoroalkylation of (hetero)arenes using 1,4-dimethoxybenzene (1) as the model substrate and 1,4-C4F8I2 as a perfluoroalkylating source in the presence of C1 (1 mol%) catalyst at room temperature employing an electrochemical approach (Fig. S2†). The CV results indicated that the electrolysis potential at −0.8 V vs. Ag/AgCl is suitable for this fluoroalkylation reaction, which was in associating with our previous work.21
The optimized results of the reaction are summarized in Table 1. Firstly, an extensive screening involving the solvent, flow rate and amounts of fluoroalkylating reagent, cathode, other vitamin B12 model complex (C2), and visible-light irradiation afforded the optimal conditions (see the ESI in Table S1, Fig. S3 and S4†). The results initially provided us with some optimal reaction conditions, such as carbon felt as the cathode, C1 as the cobalt catalyst and visible-light irradiation. Subsequently, we firstly continued to perform the reaction with the flow rate of 1,4-C4F8I2 (0.5 eq. of substrate per 1 h, 6 eq. in total) for 12 h, yielding the desired products 1a (13%) and 1b (22%) (Table 1, entry 1). Furthermore, we found that visible-light irradiation condition promoted the better transformation of 1, giving rise to a relatively higher yield of 1a (24%) and 1b (41%) (Table 1, entry 2; Fig. S3†). Using other alcohol solvents and DMSO all led to incomplete conversions and lower yields (Table 1, entries 3–5). To accelerate the catalytic reaction, −1.2 V vs. Ag/AgCl was chosen as the electrolysis potential, which afforded similar yields for both desired products (Table 1, entry 6) compared with the reaction performed under a potential of −0.8 V vs. Ag/AgCl (Table 1, entry 2). Additionally, the dark condition was optimized at −1.2 V vs. Ag/AgCl and resulted in 1a of 2% yield and 1b of 9% yield, which confirms the efficiency of visible-light irradiation for the fluoroalkylation (Table 1, entry 7). Notably, the reaction was inhibited in the presence of the radical scavenging reagents 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and N-tert-butyl-α-phenylnitrone (PBN), and no formation of the desired products 1a and 1b was observed. Indeed, a signal attributable to the PBN spin adduct was detected by electron spin resonance experiment (Fig. S5†). These results indicate that some radical intermediates were formed during the reaction process. On the basis of these results, the optimized condition for the controlled-potential electrolysis was established at −0.8 V vs. Ag/AgCl in CH3OH with 6 eq. 1,4-C4F8I2 reagent of aromatic substrate in the presence of C1 catalyst (1 mol%) for 12 h at room temperature (Table 1, entry 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Potential (V) vs. Ag/AgCl | Solventb | Conversionc (%) | 1a, Yieldc (%) | 1b, Yieldc (%) | Total yieldc (%) |
| a Reaction conditions: [C1] = 5.0 × 10−4 M; [1,4-dimethoxybenzene (1)] = 5.0 × 10−2 M; [1,4-C4F8I2] = 0.5 eq. of substrate per 1 h, 6 eq. in total; reaction time: 12 h; [n-Bu4NClO4] = 0.1 M; with visible light (≥420 nm); decafluorobiphenyl (C12F10) as the internal standard. Working electrode (WE): carbon felt; counter electrode (CE): Zn plate; reference electrode (RE): Ag/AgCl (3.0 M NaCl aq.).b Abbreviations: CH3OH, methanol; DMSO, dimethyl sulfoxide.c The conversions and yields are based on the initial concentration of 1,4-dimethoxybenzene (1) and were determined by gas chromatography-mass spectrometry (GC-MS).d In the absence of visible-light irradiation.e In the dark. | ||||||
| 1d | −0.8 V | CH3OH | 86 | 13 | 22 | 35 |
| 2 | −0.8 V | CH3OH | >99 | 24 | 41 | 65 |
| 3 | −0.8 V | Ethanol | 38 | 3 | 9 | 12 |
| 4 | −0.8 V | 1-Propanol | 53 | 4 | 12 | 16 |
| 5 | −0.8 V | DMSO | 77 | 3 | 4 | 7 |
| 6 | −1.2 V | CH3OH | >99 | 24 | 38 | 62 |
| 7e | −1.2 V | CH3OH | 75 | 2 | 9 | 11 |
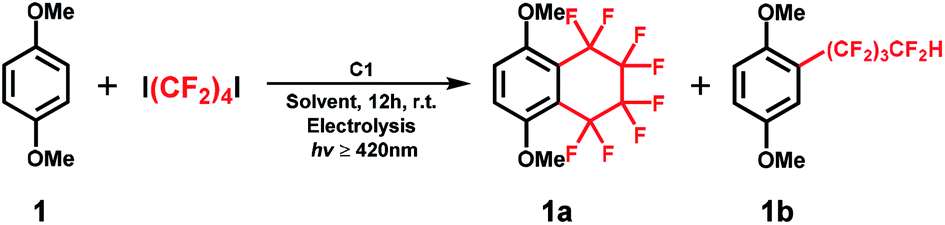
With the optimized reaction conditions in hand, we evaluated the scope of the C1-mediated direct fluoroalkylation of (hetero)arenes (Fig. 1 and Table 2). As shown in Fig. 1, other two fluoroalkylating reagents, 1,4-dibromooctafluorobutane (1,4-C4F8Br2) and dodecafluoro-1,6-diiodohexane (1,6-C6F12I2), were examined for these attractive fluoroalkylations. The two desired fluoroalkylated products 1a and 1b were obtained in lower yields using 1,4-C4F8Br2 (5% and 16%, respectively), indicating that the latter exhibited lower reactivity than 1,4-C4F8I2. Moreover, after examining the reactivity of long-chain fluoroalkylating reagent (1,6-C6F12I2), a moderate yield of 51% was obtained for the selective formation of acyclic perfluoroalkylated arene 1c, whereas the product of the intramolecular fluoroalkylating cyclization of 1 was not detected. This suggests that the intramolecular cyclization depends on the number of –CF2– units of the (CF2)n group. These results indicated that 1,4-C4F8I2 was the best fluoroalkylating reagent, and it was used for further expanding the substrate scope of the reaction.
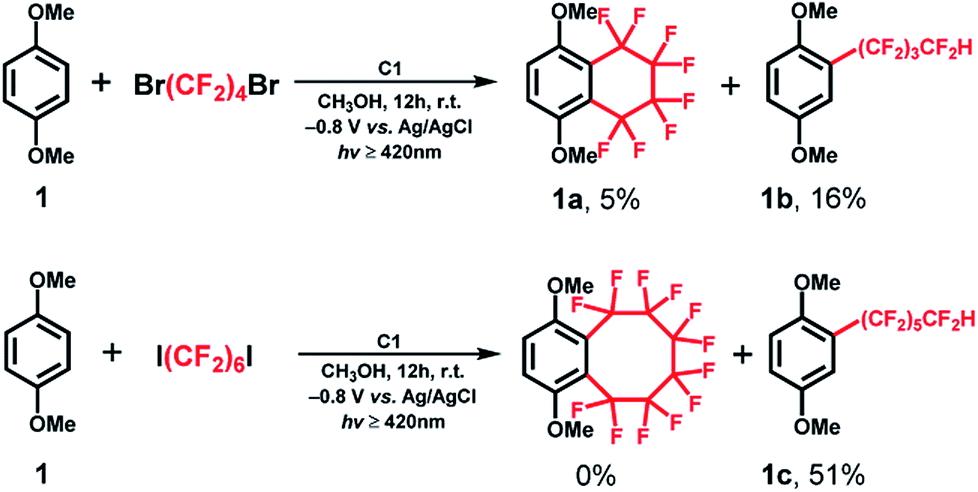 | ||
| Fig. 1 Optimization of the reaction conditions with other fluoroalkylating reagents such as 1,4-C4F8Br2 and 1,6-C6F12I2. | ||
|
|---|
| a Reaction conditions: [C1] = 5.0 × 10−4 M; [substrate (1–7)] = 5.0 × 10−2 M; [1,4-C4F8I2] = 0.5 eq. of substrate per 1 h, 6 eq. in total; reaction time: 12 h; [n-Bu4NClO4] = 0.1 M; decafluorobiphenyl (C12F10) as the internal standard. WE: carbon felt; CE: Zn plate; RE: Ag/AgCl (3.0 M NaCl aq.). The yields are based on the initial concentration of aromatic substrate and were determined by gas chromatography-mass spectrometry (GC-MS). |
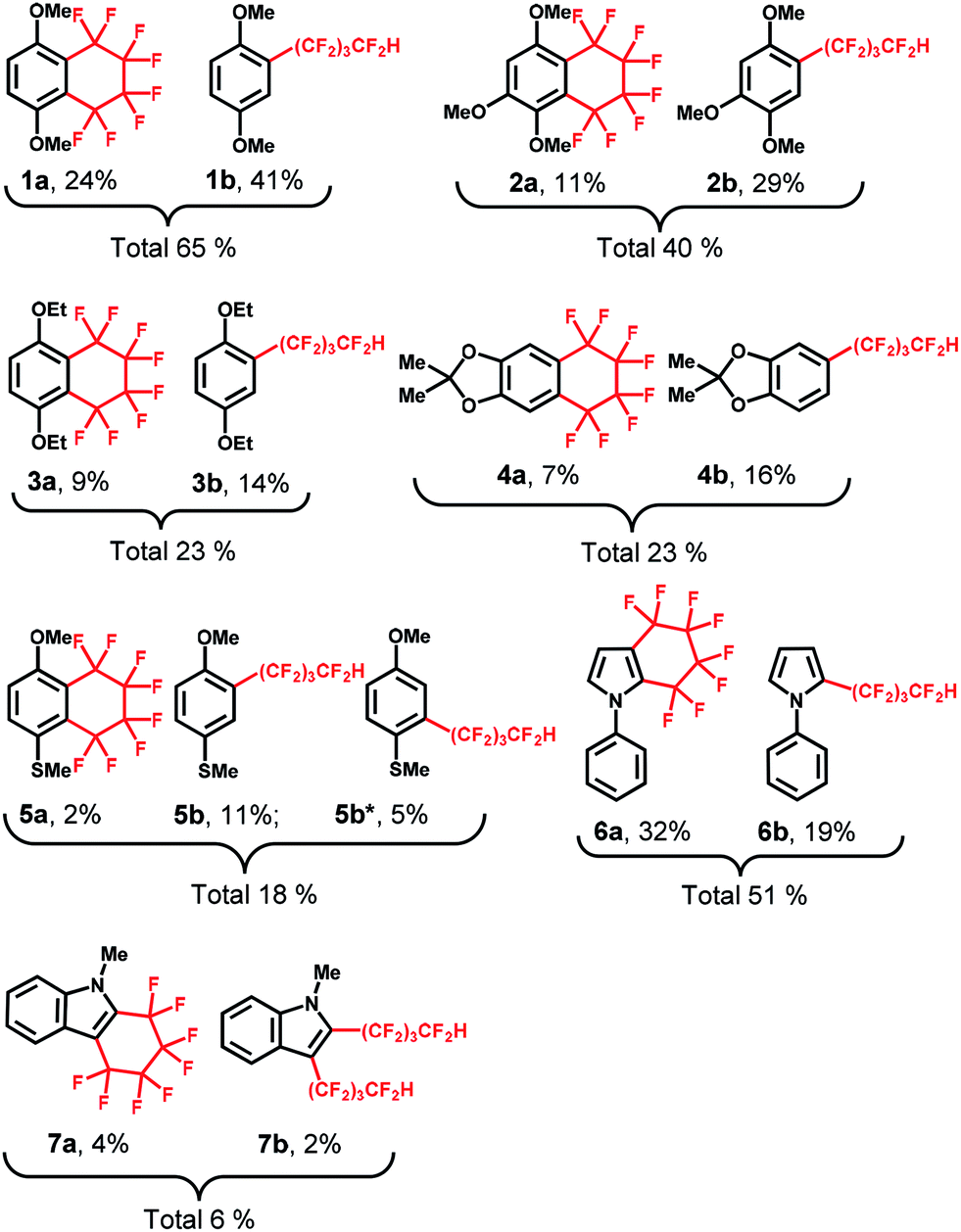 |
Thus, several (hetero)arenes (2–7) were evaluated for these transformations under the standard conditions (Table 2). The substrate 1,2,4-trimethoxybenzene (2) underwent this transformation, affording 2a and 2b in 11% and 29% yield, respectively. Other aromatic compounds, such as 1,4-diethoxybenzene (3) and 2,2-dimethyl-1,3-benzodioxole (4) also showed good tolerance to the reaction conditions, yielding the desired products 3a and 3b in 9% and 14% yield and 4a and 4b in 7% and 16% yield, respectively. In addition, 4-methoxythioanisole (5) was examined, and the cyclic perfluoroalkylating product 5a was obtained in 2% yield, along with acyclic products 5b and 5b* in 11% and 5% yield, respectively. Fortunately, the substrate 1-phenylpyrrole (6) reacted completely with 1,4-C4F8I2 under the standard conditions, providing fluoroalkylated products 6a in 32% yield and 6b in 19% yield. Moreover, this C–H fluoroalkylation could be extended to 1-methylindole (7), yielding the cyclic fluoroalkylated product 7a in 4% yield. Interestingly, the –(CF2)4H group could be introduced at two positions of the indole ring, yielding the 7b product in 2% yield. Although moderate to low yields and selectivity are obtained mainly due to other by-products formation, these results demonstrate that this method for the electrochemically-driven C1-catalyzed intramolecular cyclic and acyclic perfluoroalkylation of aromatic compounds containing the –(CF2)n– group provides a useful tool for the modification of multifunctionalized molecules. The crystal structures of cyclic perfluoroalkylated compounds (1a, 3a, 5a and 7a) are shown in Fig. 2(a)–(d). On the basis of the above experimental results and the previously reported studies,21,22 we propose the preliminary reaction mechanism illustrated in Fig. 2(e).
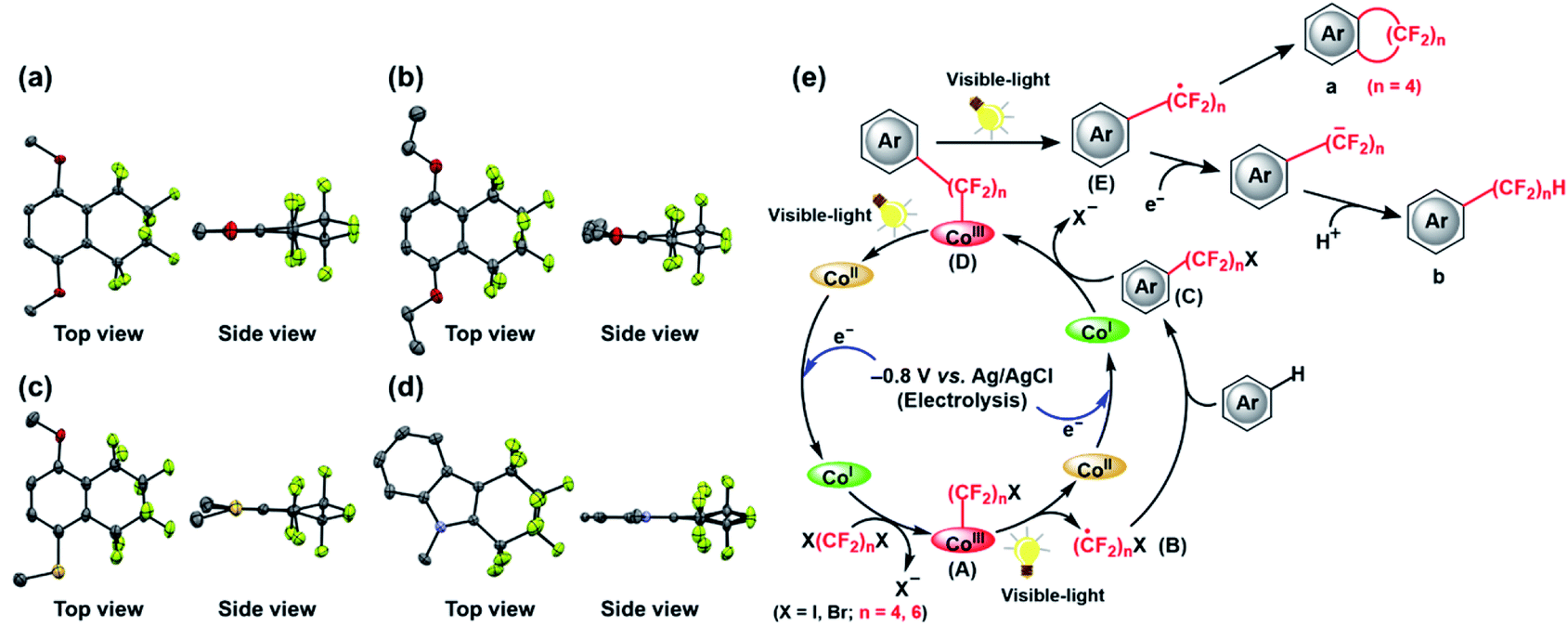 | ||
| Fig. 2 Crystal structures of (a) 1a, (b) 3a, (c) 5a, and (d) 7a, showing displacement ellipsoids at the 50% probability level. Hydrogen atoms have been omitted for clarity. Color code: C, gray; N, light blue; O, red; F, light green; S, yellow. (e) Mechanistic study of radical fluoroalkylation mediated by C1. | ||
Firstly, the potential at −0.8 V vs. Ag/AgCl gives rise to a supernucleophilic Co(I) species as the key intermediate during the whole catalytic process. Subsequently, the Co(I) species reacts with X(CF2)nX (X = I, Br; n = 4, 6), yielding the intermediate Co(III)–(CF2)nX complex (A) with the concomitant release of a halogen ion X−.36 Formation of the key perfluoroalkylated radical intermediate ·(CF2)nX (B) is then accomplished via photodynamically driven homolytic Co(III)–(CF2)nX (A) cleavage, and (B) then reacts with the nonactivated (hetero)arenes to afford another crucial intermediate (hetero)arene–(CF2)nX (C). Similarly, this complex reacts with the Co(I) species to form the Co(III)–(CF2)n–(hetero)arene (D), along with the release of another X−. The homolysis of this Co(III)–C bond provides a radical adduct ·(CF2)n–(hetero)arene (E) that undergoes intramolecular addition to the aromatic substrate to furnish the cyclic fluoalkylated product a. This intramolecular cyclization proceeds in the case of n = 4. At the same time, a single-electron transfer to ·(CF2)n–(hetero)arene (E) proceeds to give the corresponding radical anion, which is then protonated to form the final acyclic perfluoroalkylated product b. Overall, the reactions are driven by the consecutive formation of Co–C bonds and the formation of a fluoroalkyl radical species by homolysis, which is a very rare example up to date. A relatively high turnover number (TON) of 130 was observed due to the inherent high stability of the vitamin B12 framework, indicating the significant factor of Co–C bond for these molecular transformations. At this stage, isolation of the Co(III)–Rf species (A), (D) and (C) intermediate under the described conditions was not attempted.
In conclusion, we have developed an electrochemically driven, cobalt(III)–carbon bond-mediated direct C–H radical intramolecular fluoroalkylating cyclization and acyclic perfluoroalkylation of (hetero)arenes using X(CF2)4X as fluoroalkylating reagent and a vitamin B12 derivative as a cobalt catalyst. This protocol provides a new direction for one-step synthesis of cyclic perfluoroalkylated (hetero)arenes with a C4F8-containing six-membered ring and acyclic perfluoroalkylated compounds containing –C4F8H group, which cannot be obtained by the stoichiometric reactions. Although yields and selectivity still have room for improvement, and electron-rich compounds are required as substrates, these new reactions proceeding under mild conditions are valuable as a proof-of-concept from the viewpoint of bioinorganic and organometallic chemistry. Mechanistic studies, substrate variations, and improvement of selectivity for these perfluoroalkylated compounds are still underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant numbers JP17H04875, JP16H06514, JP18H04265 and JP19K22204. This work was also supported by the Nissan Chemical Corporation.Notes and references
- G. Pattenden, Chem. Soc. Rev., 1988, 17, 361 RSC.
- B. Giese, J. Hartung, J. He, O. Hüter and A. Koch, Angew. Chem., Int. Ed. Engl., 1989, 28, 325 CrossRef.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391 RSC.
- K. Tahara, L. Pan, T. Ono and Y. Hisaeda, Beilstein J. Org. Chem., 2018, 14, 2553 CrossRef CAS PubMed.
- J. Demarteau, A. Debuigne and C. Detrembleur, Chem. Rev., 2019, 119, 6906 CrossRef CAS PubMed.
- M. Ociepa and D. Gryko, in Cobalt Catalysis in Organic Synthesis: Methods and Reactions, 2020, p. 417 Search PubMed.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- M. Bassetto, S. Ferla and F. Pertusati, Future Med. Chem., 2015, 7, 527 CrossRef CAS PubMed.
- S. Caron, Org. Process Res. Dev., 2020, 24, 470 CrossRef CAS.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC.
- S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287 CrossRef CAS.
- X. Li, X. Shi, X. Li and D. Shi, Beilstein J. Org. Chem., 2019, 15, 2213 CrossRef CAS PubMed.
- S. Barata-Vallejo and A. Postigo, Molecules, 2019, 24, 4483 CrossRef CAS PubMed.
- J. Zhou, F. Wang, Z. Lin, C. Cheng, Q. Zhang and J. Li, Org. Lett., 2020, 22, 68 CrossRef CAS PubMed.
- W. Zhang, X.-X. Xiang, J. Chen, C. Yang, Y.-L. Pan, J.-P. Cheng, Q. Meng and X. Li, Nat. Commun., 2020, 11, 638 CrossRef CAS PubMed.
- T. Mao, M.-J. Ma, L. Zhao, D.-P. Xue, Y. Yu, J. Gu and C.-Y. He, Chem. Commun., 2020, 56, 1815 RSC.
- M. J. Hossain, T. Ono, K. Wakiya and Y. Hisaeda, Chem. Commun., 2017, 53, 10878 RSC.
- M. J. Hossain, T. Ono, Y. Yano and Y. Hisaeda, ChemElectroChem, 2019, 6, 4199 CrossRef CAS.
- P. T. Kaplan, L. Xu, B. Chen, K. R. McGarry, S. Yu, H. Wang and D. A. Vicic, Organometallics, 2013, 32, 7552 CrossRef CAS.
- J. Charpentier, N. Früh, S. Foser and A. Togni, Org. Lett., 2016, 18, 756 CrossRef CAS PubMed.
- K. C. Rippy, E. V. Bukovsky, T. T. Clikeman, Y.-S. Chen, G.-L. Hou, X.-B. Wang, A. A. Popov, O. V. Boltalina and S. H. Strauss, Chem.–Eur. J., 2016, 22, 874 CrossRef CAS PubMed.
- L. Li, C. Ni, Q. Xie, M. Hu, F. Wang and J. Hu, Angew. Chem., Int. Ed., 2017, 56, 9971 CrossRef CAS PubMed.
- X.-P. Fu, X.-S. Xue, X.-Y. Zhang, Y.-L. Xiao, S. Zhang, Y.-L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948 CrossRef CAS PubMed.
- H.-P. Cao, J.-C. Xiao and Q.-Y. Chen, J. Fluorine Chem., 2006, 127, 1079 CrossRef CAS.
- I. V. Kuvychko, C. Dubceac, S. H. M. Deng, X.-B. Wang, A. A. Granovsky, A. A. Popov, M. A. Petrukhina, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2013, 52, 7505 CrossRef CAS PubMed.
- H. Mei, R. Pajkert, L. Wang, Z. Li, G.-V. Röschenthaler and J. Han, Green Chem., 2020, 22, 3028 RSC.
- A. Claraz, T. Courant and G. Masson, Org. Lett., 2020, 22, 1580 CrossRef CAS PubMed.
- Z. Zou, W. Zhang, Y. Wang, L. Kong, G. Karotsis, Y. Wang and Y. Pan, Org. Lett., 2019, 21, 1857 CrossRef CAS PubMed.
- S. Zhang, L. Li, J. Zhang, J. Zhang, M. Xue and K. Xu, Chem. Sci., 2019, 10, 3181 RSC.
- P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460 CrossRef CAS PubMed.
- G.-Y. Dou, Y.-Y. Jiang, K. Xu and C.-C. Zeng, Org. Chem. Front., 2019, 6, 2392 RSC.
- T. Ono, K. Wakiya, M. J. Hossain, H. Shimakoshi and Y. Hisaeda, Chem. Lett., 2018, 47, 979 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2004379–2004382. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05295g |
| ‡ Current address: Department of Chemistry, Begum Rokeya University, Park Mor, Modern, Rangpur-5404, Bangladesh. |
| This journal is © The Royal Society of Chemistry 2020 |