
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn expeditious click approach towards the synthesis of galactose coated novel glyco-dendrimers and dentromers utilizing a double stage convergent method†‡
Anand K. Agrahari,
Anoop S. Singh.,
Rishav Mukherjee and
Vinod K. Tiwari *
*
Department of Chemistry, Centre of Advanced Study, Institute of Science, Banaras Hindu University, Varanasi-221005, India. E-mail: Tiwari_chem@yahoo.co.in; Vinod.tiwari@bhu.ac.in
First published on 26th August 2020
Abstract
The primary motive behind this article is to bring to the forefront a unique kind of dendrimer which has remained a dark horse since its discovery, namely dentromer. We herein report the synthesis of glycodendrimers and glycodentromers crowned with galactose units by harnessing an expeditious synthesis of dendrimer core 18 and dentromer core 19, divergently with branching directionality (1 → 2) and (1 → 3), respectively. A competent, double stage convergent synthetic path was chosen to facilitate ease of refining and spectroscopic elucidations. By exploiting a Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction strategy, we successfully developed a new series of galactosylated dendrimers 20, 21, 22, and 24 containing 6, 12, 18, and 18 peripheral galactose units, respectively. We are first to report the practical synthesis of 9-peripheral galactose coated glycodentromer 23 (0th generation) and 27-peripheral galactose coated glycodentromer 25 (1st generation). These synthesized scaffolds were characterized by spectral studies such as 1H, 13C NMR, FT-IR, MALDI-TOF MS, HRMS and SEC analysis. Additionally, gel permeation chromatography depicted the regular progression in size from 6 to 27-peripheral galactose coated glycodendrimers along with glycodentromers, with their high monodispersity. Also, the glyco-dendrimers and dentromers synthesized from two different hypercore units i.e. dendrimers core (18) and dentromer core (19), have been supported by their UV-visible absorbance and emission spectroscopy.
1 Introduction
Dendrimers hail from a distinctive family of macromolecules, tailored with a motif to encompass superior branching, three dimensional, high monodispersity and multivalent functional residue, all in one exquisite architecture.1 The aforementioned attributes of dendrimers have enabled them to have innumerable applications in various fields including drug delivery, gene delivery, photodynamic therapy, biosensors, drugs for cardiovascular diseases, anti-cancer vaccines, anti-microbial vaccines, etc.2–7 However, the field of dendrimers have been widely explored and this provide an origin to a fascinating dendrimer analogue which expands divergently in a 1 → 3 branching fashion, namely dentromer.8 A significant advantage of dentromers over dendrimers corresponds to its densely distributed peripheral groups for the same generation and when conjoined to carbohydrate units, glycodentromers or sugar-linked dentromers undergo an enhanced hypercluster effect which prompts an enhancement in biological activity in comparison to a conventional glycodendrimer.9 Carbohydrates are essential scaffolds owing to their diverse role in biological processes, like functioning of the immune system, enhancing inflammatory response, malignancy, metastasis, apoptosis, etc.10–12This unique and effective feature of carbohydrates results from carbohydrate–lectin interactions playing a cardinal role in various biological systems such as biological recognition, protein folding, cell growth, cell-agglutination, cell signalling and interaction with bacteria/viruses.10,13 This mesmerizing unison of carbohydrates and dendrimers have drawn attention owing to its multivalency effect.14 Incorporating sugar units in a scaffold increases its biological activity as glycan–protein interaction plays a major role in various biological phenomena.15 Dendrimer chemistry is quiet useful in order to develop monodisperse multivalent glycoconjugates.16 and it has been excavated from literature that polyester dendrimers are compatible to biological system with an added advantage of easy preparation techniques. In addition, whenever ester linked dendrimers have been tested, they have been found to be non-toxic in comparison to other dendrimers.17 It has been well established, that double stage convergent method is a combination of convergent and divergent methods and in this strategy, the core unit is synthesized divergently followed by a subsequent attachment of a dendritic wedge in a convergent fashion, thereby generating the hypercore (i.e. dendrimer or dentromer core in present manuscript) containing dendrimer.8,18
The CuAAC 1,3-dipolar cycloaddition reaction has propagated extensively ever since its finding by Sharpless19a and Meldal.19b The atom economy and versatility of these reactions make them a powerful tool for a diverse spectrum of organic synthesis.2,20 Particularly, click chemistry involving Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC), has been profusely explored during the last two decades due to an easy synthesis of diverse two or three dimensional motifs, having a plethora of applications ranging from chemical biology to material science.21 In addition, tailored 1,2,3-triazoles, bioisostere of amides, esters and other heterocycles, are frequently used as pharmacophores in several drugs, exhibiting diverse pharmacological properties. Thus, these moieties play an intriguing role as building blocks of dendrimers and impart to them sufficient biological activity, thereby making them medically important.22
The triply branched glycodendrimer construction is less facile than the doubly branched analogue due to steric reasons.23 So, in present manuscript, we have successfully synthesized a dendrimer core (hexa-alkynylated) as well as a dentromer core (nona-alkynylated) to develop a series of glycodendrimers along with the first ever 0th ‘9-peripheral’ and 1st generation ‘27-peripheral’ galactose coated glycodentromers.
2 Results and discussion
The first generation azide functionalized dendrons were synthesized from methyl 3,5-dihydroxybenzoic acid (1) and methyl 3,4,5-tris-hydroxybenzoic acid (2). The crafted first generation dendritic azide was coupled with the alkyne functionalized core unit 18 and 19 to generate a series of six (20–25) peripheral galactosylated glycodendrimers and glycodentromers. In this regard, methyl 3,5-dihydroxybenzoate was derived from esterification of 3,5-dihydroxybenzoic acid using methanol and sulphuric acid as catalyst. Similarly with the same strategy, methyl 3,4,5-tris-hydroxybenzoate was also prepared. Moreover, esterified dendron initiators 3, 4 were propargylated by reacting propargyl bromide, K2CO3, 18-crown-6 ether (co-catalyst) in dry acetone. Furthermore, the propargylated scaffolds 5 and 6 were reduced with LAH in THF followed by bromination using PBr3 in DCM, which furnished 3,5-bis(propargyloxy)benzyl bromide 9 and 3,4,5-tris(propargyloxy)benzyl bromide 10. Owing to their π–π stacking interaction, aromatic ‘aglycones’ have been of substantial interest for dendron synthesis.24 To this end, clickable 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl azide 11 was selected as surface group functionality and this was stitched with aromatic scaffolds 9 and 10 to afford the bromine functionalized first generation 12 and 13 dendrons. The single triazolyl peak at 7.91 ppm in 1H NMR, for compound 12, whereas, for 13, two distinct signals at δ 8.14 and 8.05 ppm were observed with ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which displayed the two chemically non-equivalent triazolyl galactosides. In 13C NMR, the peaks appeared at δ 144.4 and 121.2 ppm for compound 12, and δ 144.9, 144.2 & 123.0, 122.0 ppm for compound 13, confirmed the presence of triazolyl carbons. In continuation, the azidation of the compounds 12 and 13 were executed using NaN3 in DMF, which resulted in azide functionalized dendritic architecture 14 and 15 respectively (Scheme 1). This transformation was illustrated by IR spectra, in which, peak at 2104.5 and 2103.8 cm−1 confirms the formation of corresponding dendritic azides 14 and 15. In addition, there was a down field shifting in 13C NMR, during conversion from –CH2Br (δ 33.2 ppm) to –CH2N3 (δ 54.6 ppm) for moiety 14 and –CH2Br (δ 33.7 ppm) to –CH2N3 (δ 54.6 ppm) for scaffold 15, which supported the formation of respective azide.
:1, which displayed the two chemically non-equivalent triazolyl galactosides. In 13C NMR, the peaks appeared at δ 144.4 and 121.2 ppm for compound 12, and δ 144.9, 144.2 & 123.0, 122.0 ppm for compound 13, confirmed the presence of triazolyl carbons. In continuation, the azidation of the compounds 12 and 13 were executed using NaN3 in DMF, which resulted in azide functionalized dendritic architecture 14 and 15 respectively (Scheme 1). This transformation was illustrated by IR spectra, in which, peak at 2104.5 and 2103.8 cm−1 confirms the formation of corresponding dendritic azides 14 and 15. In addition, there was a down field shifting in 13C NMR, during conversion from –CH2Br (δ 33.2 ppm) to –CH2N3 (δ 54.6 ppm) for moiety 14 and –CH2Br (δ 33.7 ppm) to –CH2N3 (δ 54.6 ppm) for scaffold 15, which supported the formation of respective azide.
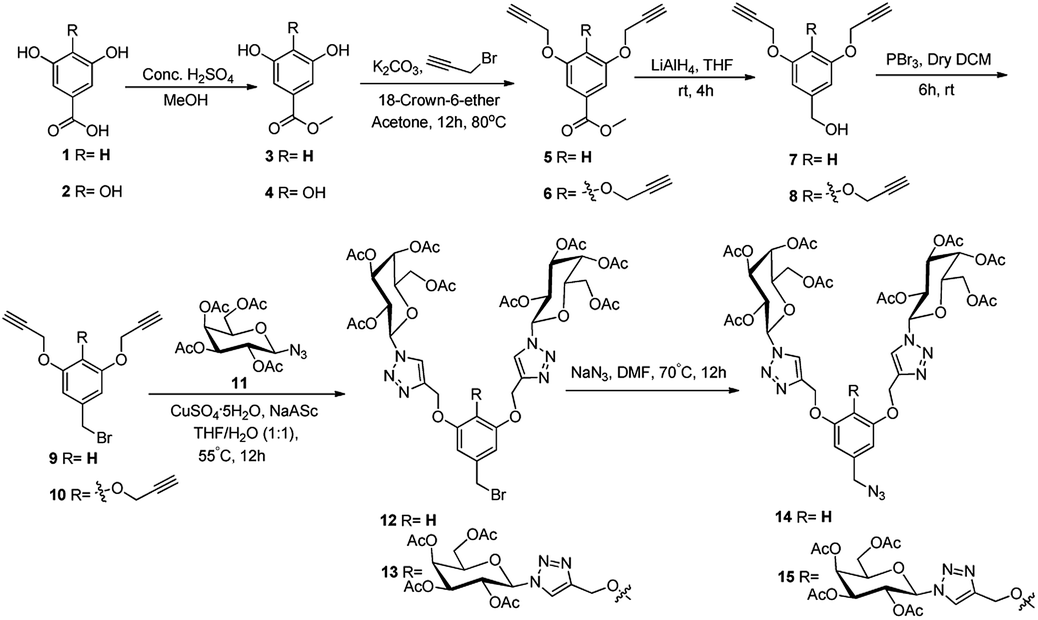 | ||
| Scheme 1 Synthesis of azide functionalized AB2 and AB3 type dendritic structure. | ||
We next, synthesized the other click counterpart of dendritic azides, as depicted in (Scheme 2 and 3). Thus, ester functionalized compound 5 and 6 were separately treated with aqueous NaOH to generate 3,5-bis(propargyloxy)benzoic acid 16 and 3,4,5-tris(propargyloxy)benzoic acid 17, respectively (Scheme 2). The resulted compounds were further divergently condensed with phloroglucinol using DCC, DMAP to afford the hexavalent dendrimer core 18 and nonavalent alkyne dentromer core 19 (Scheme 3).
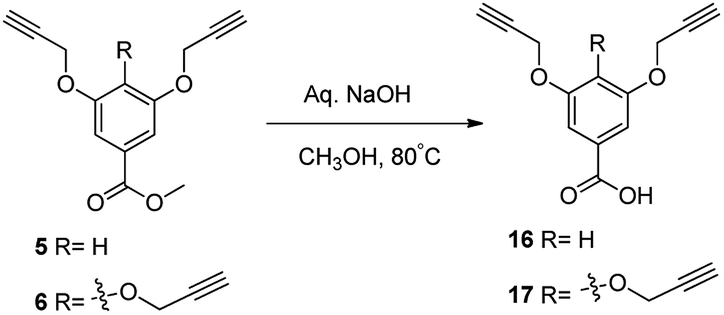 | ||
| Scheme 2 Synthesis of acid functionalized dentritic scaffolds. | ||
 | ||
| Scheme 3 Synthesis of ester-linked dendrimers core and dentromer core by acid–phenol coupling using DCC/DMAP. | ||
At this instant, the synthesized hypercore units were used as starting material for CuAAC coupling with dendritic wedges 14 and 15 to afford a series of glyco-dendrimers and dentromers. In this context, dendrimer core unit 18 was stitched with galactose azide 11, dendritic wedge 14 and 15 using CuSO4·5H2O and sodium-L-ascorbate to prepare a series of 6, 12 and 18 peripheral galactose tethered dendrimers 20, 21 and 22, respectively (Scheme 4). In the same way, dentromer core unit 19 was clicked with galactose-azide 11, dendritic architecture 14 and 15, under similar Cu(I)-catalyzed cycloaddition condition and furnished 9 galactose coated glycodentromer 23, 18 peripheral glycodendrimer 24, and 27 galactose coated glycodentromer 25, respectively with satisfactory to good yield 60–75% (Scheme 5). Interestingly, there was no steric congestion imposed during glycodentromer synthesis. It is to noteworthy to mention that, for the synthesis of higher peripheral galactose tethered dendrimers, elevated equivalents of CuSO4·5H2O was required for amplified yields, because the size of glycodendrimer had been increased with an increase in void number. These voids enable entrapment of the free metal ions thereby delaying the time for completion of reaction and this could be ameliorated by increasing the catalyst amount.25 The symmetrical structure of developed glycodendrimers 20, 21, 22, 24 and glycodentromers 23, 25 was confirmed by their extensive spectral studies (NMR, IR, MALDI-TOF MS, HRMS and size exclusion chromatography) which led to an unequivocal structural illustration. Moreover, the synthesis of glycodendrimer 20, 21, 22, 24 was evidenced by the formation for newly formed triazolyl proton peaks at the desired position (see, ESI‡).
 | ||
| Scheme 4 Synthesis of galactosylated dendrimer (20, 21 and 22) derived from hexavalent alkyne core 18. | ||
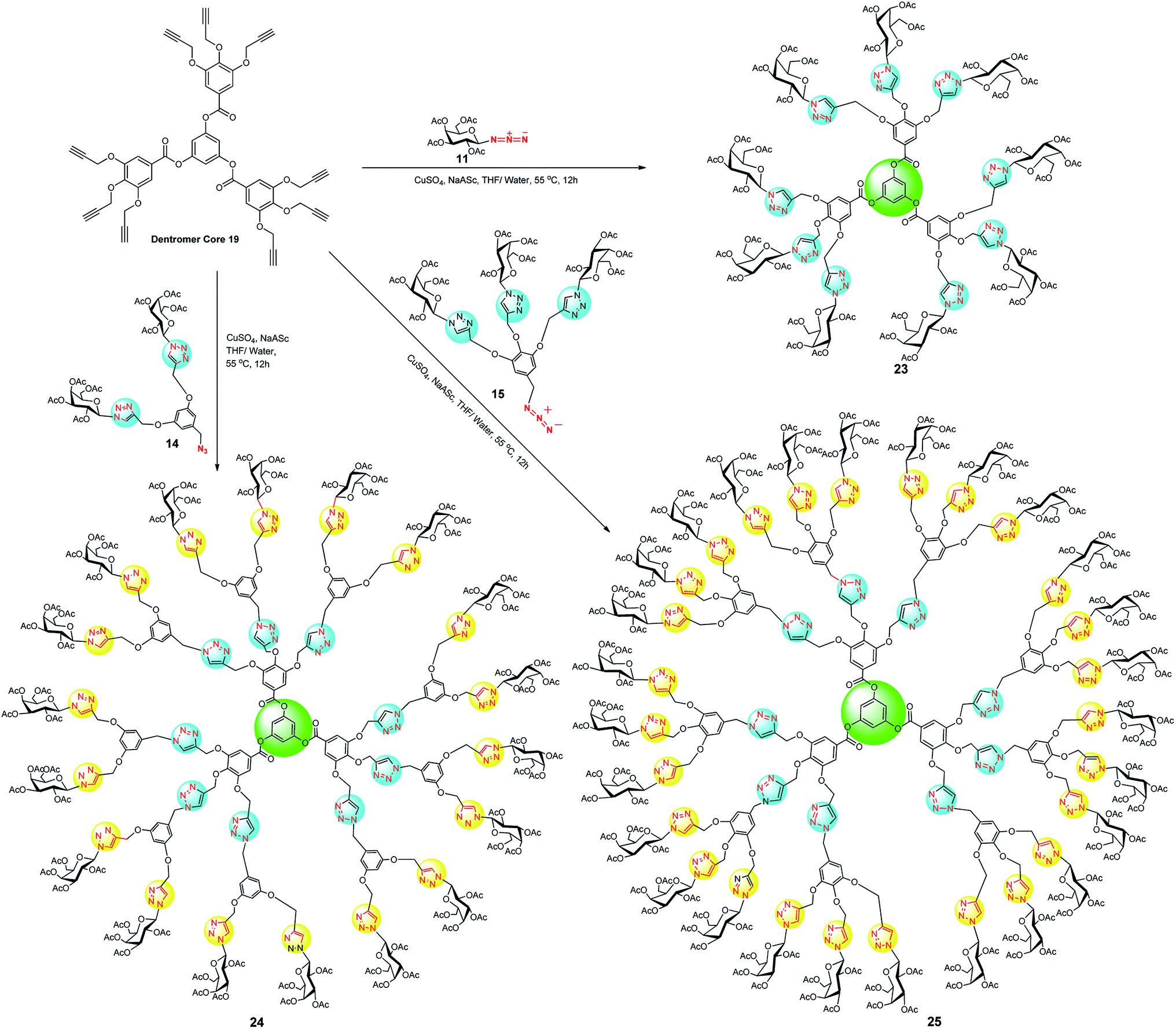 | ||
| Scheme 5 CuAAC mediated synthesis of galactose coated dendrimers (23, 24 and 25) derived from dentromer core 19. | ||
In addition, the formation of 9 and 27 peripheral galactose tethered glycodentromer was established by 1H and 13C NMR, in this regard, two triazolyl proton peaks at 8.22 and 8.11 ppm were obtained, for two different environment of the triazole with ratio 6:3 in the dentromer (23). In contrast to this, 1H NMR of glycodentromer 25 displayed the six peaks i.e. a–f in triazolyl region, each peak for a different environment of the triazole. If, we closely examine only outer triazolyl part of dendritic wedge (Fig. 1), it was noticed that four different triazolyl proton peaks i.e. a–d appeared at δ 8.09, 8.04, 8.13 and 8.11 ppm respectively with ratio a:b:c:d (4:2:2:1) on the periphery of glycodentromer. Next, we observed two inner triazolyl proton peak at 8.17 and 8.18 ppm for e and f with ratio 6:3, which established the unequivocal formation of our targeted glycodentromer 25. Further, the formation of glycodentromers was supported by MALDI-TOF MS.
 | ||
| Fig. 1 ‘1H NMR’ spectral elucidation of triazolyl region of first generation glycodentromer. | ||
There is no vibrating band of alkyne and azide in IR spectra of synthesized dendrimers which validates the absence of free alkyne or azide functionality in the developed glycodendrimers and dentromers, thereby supporting the formation of targeted glycoclusters (see, ESI, Fig. S47–S52‡).
Moreover, size exclusion chromatogram (SEC) depicts the size progression from 6 peripheral glycodendrimer to 27 peripheral glycodentromer, with polydispersity index (PDI) ranging from 1.0–1.03 showing the monodispersity of the synthesized glycoclusters (Fig. 2).
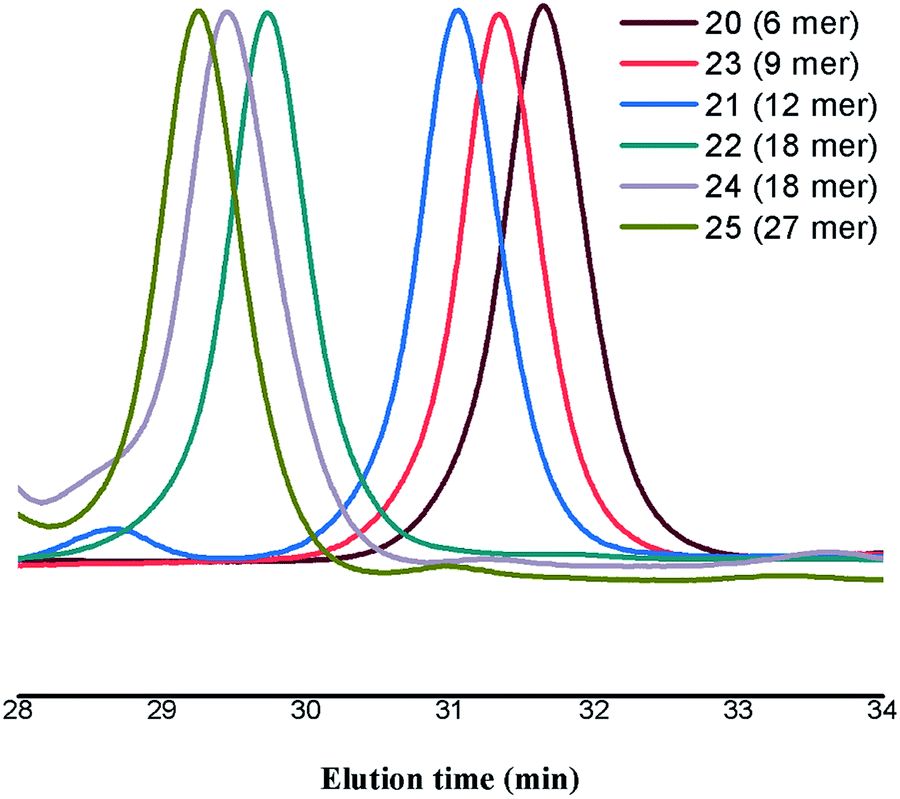 | ||
| Fig. 2 SEC chromatogram of the developed glycodendrimers (20, 21, 22, 24) and glycodentromers (23, 25). | ||
Absorption and emission studies of developed glyco-dendrimer and dentromers were also investigated, glycodendrimer 20, 21, 22 have shown UV-visible absorbance at 309.90, 310.14, 311.38 nm, respectively, whereas for compound 23, 24, 25, peaks were appeared at 274.25, 275.78, 274.61 nm. Thus, compound 20, 21, 22 were excited at the wavelength of ∼309 nm where as scaffold 23, 24, 25, at ∼274 nm, and their emission spectra were also recorded (Fig. 3).
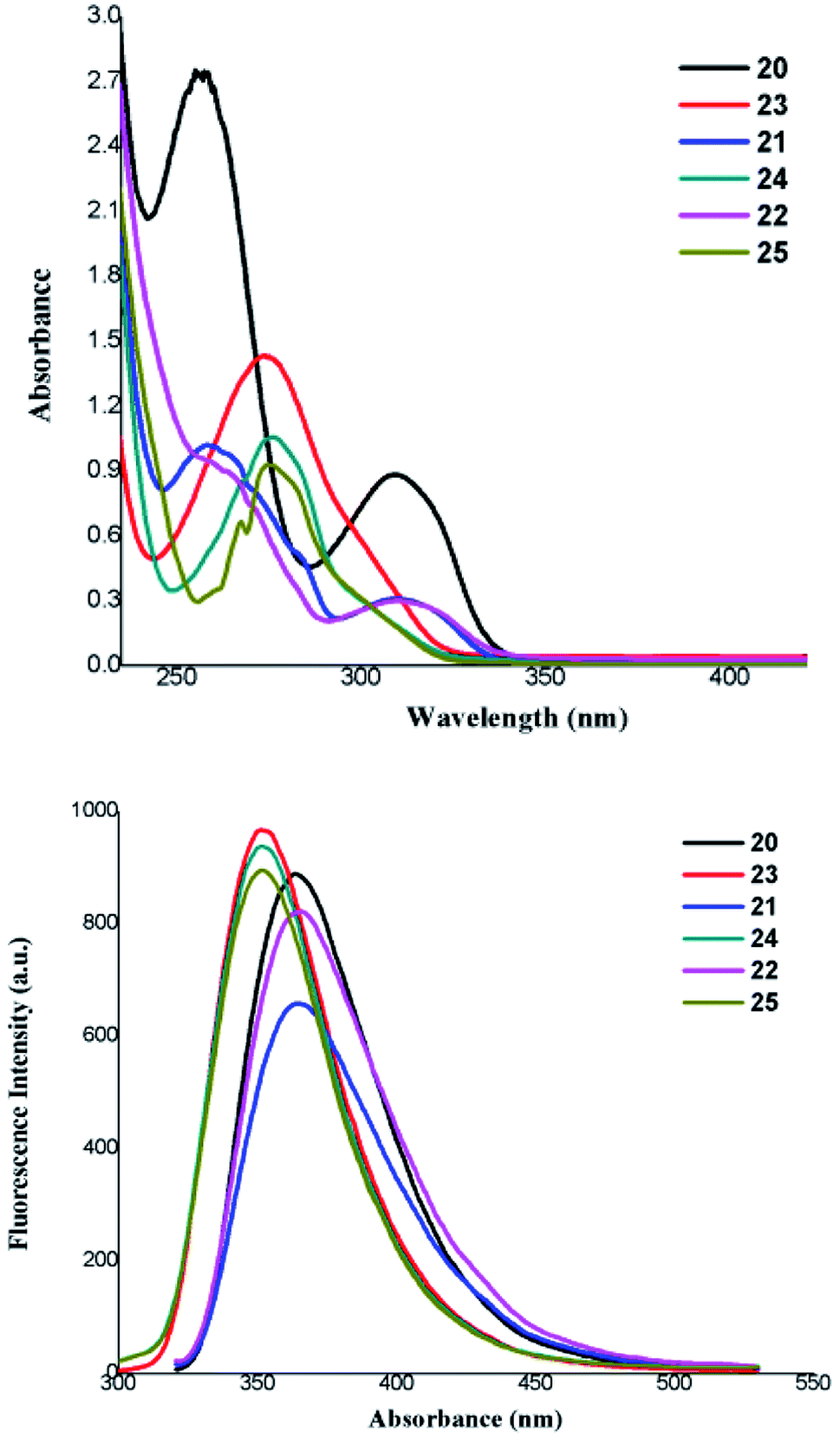 | ||
| Fig. 3 Absorbance and emission spectra of the developed glycodendrimers (20, 21, 22, 24) and glycodentromers (23, 25). | ||
By observing the absorbance and emission pattern of developed glycodendrimer and dentromer, it can be inferred that, two different series of glycoclusters were formed, one with compounds 20, 21 and 22 derived from dendrimers core unit (18) whereas, the second series comprising of compounds 23, 24 and 25 were derived from dentromer core (19). Thus, we reach to a successful end where two different type of cores are involved to construct these glyco-motifs, which was also supported by UV-Vis spectroscopy.
3 Conclusions
In summary, we have fulfilled our objective to synthesize a novel class of dendrimer which was coined as dentromer by Astruc and co-workers. This has highly branched tethers and possesses an unique triple branched architecture which is quite prominent from the scaffolds that we have developed. On this aspect of hyper-branching, dentromers hold superiority over dendrimers and we have portrayed both the development of novel dentromer as well as dendrimer core scaffolds, together with the novel galactose tethered glycodentromer and dendrimers respectively by amalgamation with surface carbohydrate ligands. These hybrid architectures may serve as potential therapeutics owing to the highly biologically active carbohydrate clusters present over the periphery. It has already been disseminated that, glycodendrimers have been employed as anti-adhesives for viral and bacterial adhesion along with anti-biofilms activity but the biological activity of dentromers awaits exploring.4 Experimental
4.1 General methods
All chemicals and solvent were of pure analytical grade. Thin layer chromatography (TLC) was executed on 60 F-254 silica gel, coated on aluminium plates and visualized by UV lamp, 5% H2SO4 methanol solution with subsequent charring by heating. In addition, alkaline KMnO4 solution was used to locate the alkynes with subsequent heating. Flash column chromatography was performed using silica gel (230–400) mesh size. 1H and 13C spectra were recorded in 500 and 125 MHz respectively. Mass spectra were recorded on Sciex-X500R QTOF, MALDI-TOF MS (with 2,5-dihydroxy benzoic acid matrix). IR was recorded in Nujol mulls in KBr pellets. The polydispersity index (Mn/Mw) of the samples was investigated in DMF at 40 °C with a flow rate of 0.5 mL min−1. The calibration of columns was done against polystyrene standard samples. The absorbance and emission spectra were also recorded by using UV-visible spectroscopy.:1), using CuSO4·5H2O (0.3 equiv. per propargyl group) and sodium ascorbate (0.3 equiv. per propargyl), stirred for 12 h at 55 °C, while for glyco-dentromer and dendrimer synthesis (0.8 equiv. of CuSO4·5H2O, NaASc for per propargyl) were used. After completion of reaction (monitored by TLC), it was passed through cellite followed by extraction with ethyl acetate. The organic layer was washed with saturated aq. NH4Cl (2 × 10 mL), water (10 mL) and finally, aq. Brine solution (10 mL). The ethyl acetate layer was collected and dried over anhydrous sodium sulphate followed by evaporation to obtain the crude mass. Moreover, the crude mass was subjected to flash column chromatography (SiO2) to afford the targeted glyco-conjugate with good yield.:1) according to the procedure 1. Yield (6.02 g, 82%); Rf = 0.4 (60% EtOAc/n-hexane); 1H NMR (500 MHz, CDCl3): δ 7.91 (s, 2H), 6.63 (s, 2H), 6.55 (1H), 5.84 (d, J = 9.0 Hz, 2H), 5.54–5.51 (m, 4H), 5.23 (dd, J = 7.0, 3.0 Hz, 2H), 5.18–5.15 (m, 4H), 4.39 (s, 2H), 4.22 (t, J = 6.0 Hz, 2H), 4.17–4.06 (m, 5H), 2.18 (s, 6H), 2.00 (s, 6H), 1.97 (s, 6H), 1.84 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 170.2, 169.8, 169.7, 168.9, 153.3, 144.4, 139.9, 121.2, 108.4, 101.9, 86.1, 73.9, 70.6, 67.8, 66.7, 61.9, 61.1, 60.2, 33.2, 20.57, 20.56, 20.4, and 20.1 ppm.:1) according to the procedure 1. Yield (0.3.44 g, 79%); Rf = 0.4 (80% EtOAc/n-hexane); 1H NMR (500 MHz, CDCl3): δ 8.14 (s, 1H), 8.06 (s, 2H), 6.74 (s, 2H), 5.94–5.88 (m, 3H), 5.69 (t, J = 10.0 Hz, 1H), 5.59–5.42 (m, 5H), 5.31–5.20 (m, 8H), 4.43 (d, J = 4.5 Hz, 2H), 4.30–4.10 (m, 10H), 2.21 (s, 9H), 2.02–1.99 (m, 18H), 1.83 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 170.3, 170.2, 170.1, 170.0, 169.9, 169.8, 168.9, 168.7, 151.9, 144.9, 144.2, 137.7, 133.6, 123.0, 122.0, 108.8, 86.2, 85.7, 73.9, 73.5, 71.0, 70.7, 67.9, 66.7, 66.88, 66.80, 66.4, 62.8, 61.1, 60.9, 60.3, 33.7, 20.6, 20.5, 20.4, and 20.1 ppm.660.1182; found = 13627.0547 (M + H)+.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VKT gratefully acknowledge Science and Engineering Research Board (SERB), New Delhi for the funding (Grant No.: EMR/2016/001123), CISC–BHU for providing spectroscopic studies and Prof. B. Ray for SEC analysis. AKA sincerely acknowledge CSIR, New Delhi for the senior research fellowship (09/013/(0671)/2017-EMR-I). We acknowledge CSIR-National Chemical Laboratory, Pune for providing the facility for MALDI-TOF-MS and thankful to Dr Shaziya Khanam (CSIR-Nehru PDF), NCL Pune, for her help in MALDI-TOF-MS analysis of the developed glycodendrimers.References
- (a) D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS; (b) S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3867 CrossRef CAS PubMed; (c) G. R. Newkome and C. Shreiner, Chem. Rev., 2010, 110, 6338–6442 CrossRef CAS PubMed; (d) J.-P. Majoral and A.-M. Caminade, Chem. Rev., 1999, 99, 845–880 CrossRef CAS PubMed; (e) D. Wang, C. Deraedt, J. Ruiz and D. Astruc, Acc. Chem. Res., 2015, 48, 1871–1880 CrossRef CAS PubMed; (f) M. Sowinska and Z. Urbanczyk-Lipkowska, New J. Chem., 2014, 38, 2168–2203 RSC.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- (a) U. Boas and P. M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC; (b) D. Kushwaha and V. K. Tiwari, J. Org. Chem., 2013, 78, 8184–8190 CrossRef CAS PubMed.
- (a) D. C. R. Astruc, Acad. Sci., 1996, 322, 757–766 CAS; (b) A. Archut and F. Vögtle, Chem. Soc. Rev., 1998, 27, 233–240 RSC; (c) D. K. Smith and F. Diederich, Chem.–Eur. J., 1998, 4, 1351–1361 CrossRef.
- (a) M. J. F. Jean and D. A. Tomalia, J. Polym. Sci., Part A: Polym. Chem., 2001, 1, 587–604 Search PubMed; (b) C. Bottcher, B. Schade, C. Ecker, J. P. Rabe, L. J. Shu and A. D. Schlüter, Chem.–Eur. J., 2005, 11, 2923–2928 CrossRef PubMed.
- C. Ornelas, J. Ruiz, C. Belin and D. Astruc, J. Am. Chem. Soc., 2009, 131, 590–601 CrossRef CAS PubMed.
- (a) C. Lee, J. A. Mac Kay, J. M. J. Fréchet and F. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed; (b) M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC.
- (a) D. Astruc, C. Deraedt, R. Djeda and C. Ornelas, Molecules, 2018, 23, 966 CrossRef PubMed; (b) C. Ornelas, Macromol. Chem. Phys., 2016, 217, 149–174 CrossRef CAS.
- (a) K. L. Wooley, C. J. Hawker and J. M. J. Fŕechet, Angew. Chem., Int. Ed. Engl., 1994, 33, 82–85 CrossRef; (b) R. S. Bagul, M. M. Hosseini, T. C. Shiao and R. Roy, Can. J. Chem., 2017, 95, 975–983 CrossRef CAS.
- Carbohydrates in Drug Discovery and Development, ed. V. K. Tiwari, Elsevier, 2020 Search PubMed.
- (a) H. J. Gabius, S. Andre, J. Jimenez-Barbero, A. Romero and D. Solis, Trends Biochem. Sci., 2012, 36, 298–313 CrossRef PubMed; (b) H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed; (c) J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- (a) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef; (b) V. K. Tiwari, A. Kumar and R. R. Schmidt, Eur. J. Org. Chem., 2012, 12, 2945–2956 CrossRef; (c) N. Mishra, A. S. Singh, A. K. Agrahari, S. Singh, M. Singh and V. K. Tiwari, ACS Comb. Sci., 2019, 21, 389–399 CrossRef CAS PubMed; (d) N. Mishra, A. K. Agrahari, P. Bose, S. K. Singh, A. S. Singh and V. K. Tiwari, Sci. Rep., 2020, 10, 3586 CrossRef CAS PubMed.
- N. K. Saadeh, R. Roy, R. S. Bagul, T. C. Shiao and M. Hosseini, Polym. Chem., 2017, 8, 5354–5366 RSC.
- (a) R. Sharma, I. Zhang, L. Abbassi, R. Rej, D. Maysinger and R. Roy, Polym. Chem., 2015, 33, 490–498 Search PubMed; (b) J. Camponovo, C. Hadad, J. Ruiz, E. Cloutet, S. Gatard, J. Muzart, S. Bouquillon and D. Astruc, J. Org. Chem., 2009, 74, 5071–5074 CrossRef CAS PubMed.
- Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC.
- J.-d' A. K. Twibanire and T. B. Grindley, Polymers, 2012, 4, 794–879 CrossRef.
- Y. M. Chabre, P. P. Brisebois, L. Abbassi, S. C. Kerr, J. V. Fahy, I. Marcotte and R. Roy, J. Org. Chem., 2011, 76, 724–727 CrossRef CAS PubMed.
- (a) K. L. Wooley, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4252–4261 CrossRef CAS; (b) R. Sharma, K. Naresh, Y. M. Chabre, R. Rej and N. K. Saadeh, Polym. Chem., 2014, 4321–4331 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- A. K. Agrahari, A. S. Singh, A. K. Singh, N. Mishra, M. Singh, P. Prakas and V. K. Tiwari, New J. Chem., 2019, 43, 12475–12482 RSC.
- (a) K. B. Mishra and V. K. Tiwari, J. Org. Chem., 2014, 79, 5752–5762 CrossRef CAS PubMed; (b) P. Dwivedi, K. B. Mishra, B. B. Mishra, N. Singh, R. K. Singh and V. K. Tiwari, Glycoconjugate J., 2015, 32, 127–140 CrossRef CAS PubMed; (c) K. B. Mishra, R. C. Mishra and V. K. Tiwari, RSC Adv., 2015, 5, 51779–51789 RSC; (d) A. Mishra and V. K. Tiwari, ChemistrySelect, 2017, 2, 9466–9471 CrossRef CAS; (e) D. Kushwaha, R. Singh and V. K. Tiwari, Tetrahedron Lett., 2014, 55, 4532–4536 CrossRef CAS; (f) A. Kushwaha, A. K. Agrahari, K. Manar, C. Yadav, V. K. Tiwari, M. G. B. Drew and N. Singh, New J. Chem., 2019, 43, 8939–8949 RSC; (g) A. K. Singh, A. K. Agrahari, A. S. Singh, V. K. Tiwari and P. Prakash, Sci. Rep., 2020 DOI:10.1038/s41598-020-70921-2.
- (a) E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discov. Today, 2017, 22, 1572–1581 CrossRef CAS PubMed; (b) B. zhang, Eur. J. Med. Chem., 2019, 168, 357–372 CrossRef CAS PubMed.
- (a) M. Touaibia and R. Roy, Mini-Rev. Med. Chem., 2007, 7, 1270–1283 CrossRef CAS PubMed; (b) A. Imberty, Y. A. Chabre and R. Roy, Chem.–Eur. J., 2008, 14, 7490–7499 CrossRef CAS PubMed.
- (a) F. Casoni, L. Dupin, G. Vergoten, A. Meyer, C. Ligeour, T. Gehin, O. Vidal, E. Souteyr, J. J. Vasseur, Y. Chevolot and F. Morvan, Org. Biomol. Chem., 2014, 12, 9166–9179 RSC; (b) R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Camara, A. Stocker, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- (a) M. Arseneault, C. Wafer and J.-F. Morin, Molecules, 2015, 20, 9263–9294 CrossRef CAS PubMed; (b) M. Arseneault, P. Dufour, I. Levesque and J.-F. Morin, Polym. Chem., 2011, 2, 2293–2298 RSC; (c) A. Papadopoulos, T. C. Shiao and R. Roy, Mol. Pharm., 2012, 9, 394–403 CrossRef CAS PubMed; (d) C.-B. Yim, I. Dijkgraaf, R. Merkx, C. Versluis, A. Eek, G. E. Mulder, D. T. S. Rijkers, O. C. Boerman and R. M. J. Liskamp, J. Med. Chem., 2010, 53, 3944–3953 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, B. P. Bode, C. T. Reidl, J. W. Sawicki, A. J. Mason, J. A. Maguire and N. S. Hosmane, Organometallics, 2012, 31, 2931–2935 CrossRef CAS.
- C. J. Ruiz, E. Cloutet and D. Astruc, Chem.–Eur. J., 2009, 15, 2990–3002 CrossRef PubMed.
- C. Yang, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2018, 57, 16194–16199 CrossRef CAS PubMed.
- X. Liu, Q. Ling, L. Zhao, G. Qiu, Y. Wang, L. Song, Y. Zhang, J. Ruiz, D. Astruc and H. Gu, Macromol. Rapid Commun., 2017, 1700448, 1–11 Search PubMed.
Footnotes |
| † This manuscript is dedicated to Prof. Shiv P. Singh, emeritus Professor at Kurukshetra University, Kurukshetra, India. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05289b |
| This journal is © The Royal Society of Chemistry 2020 |