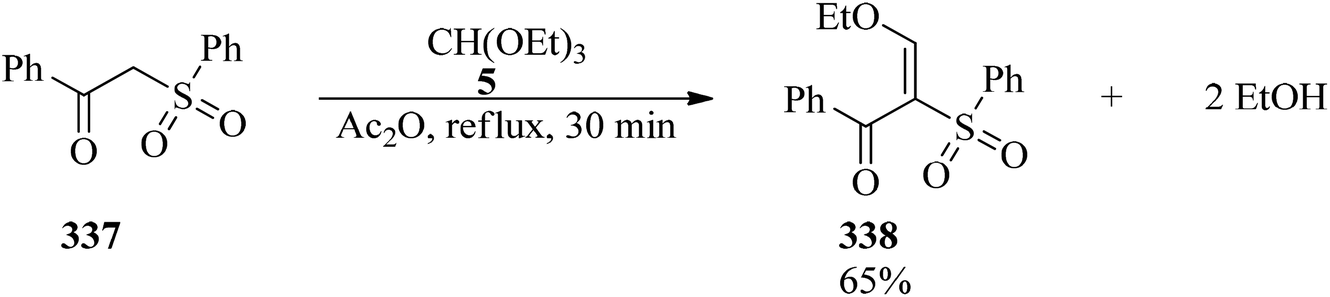
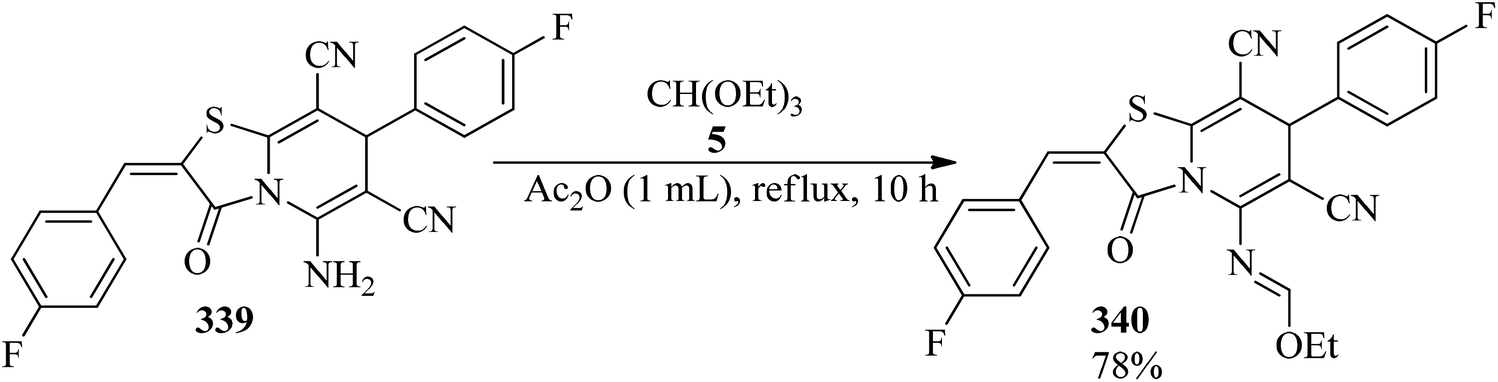
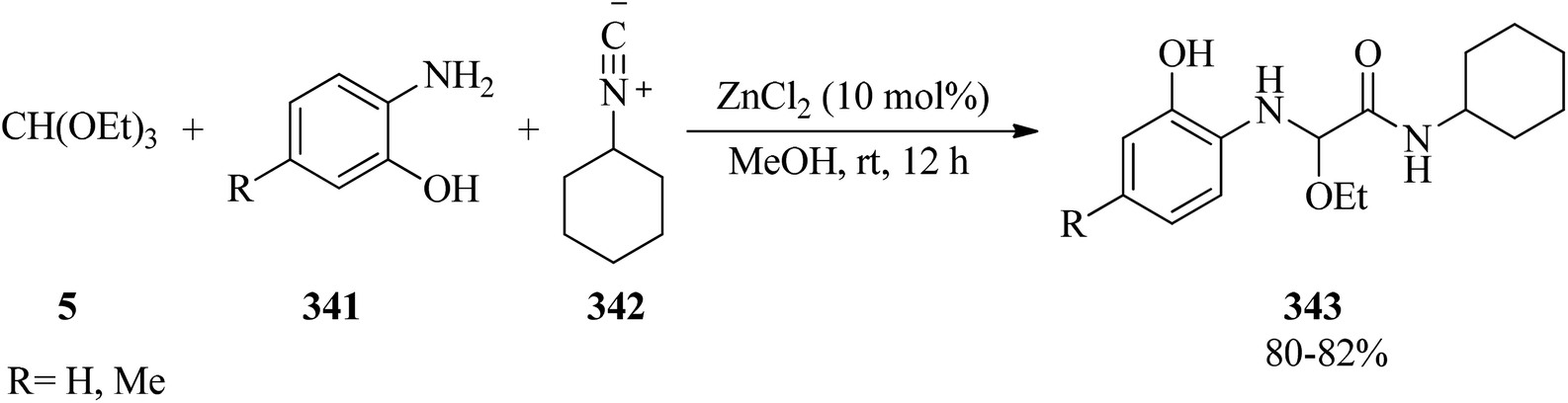
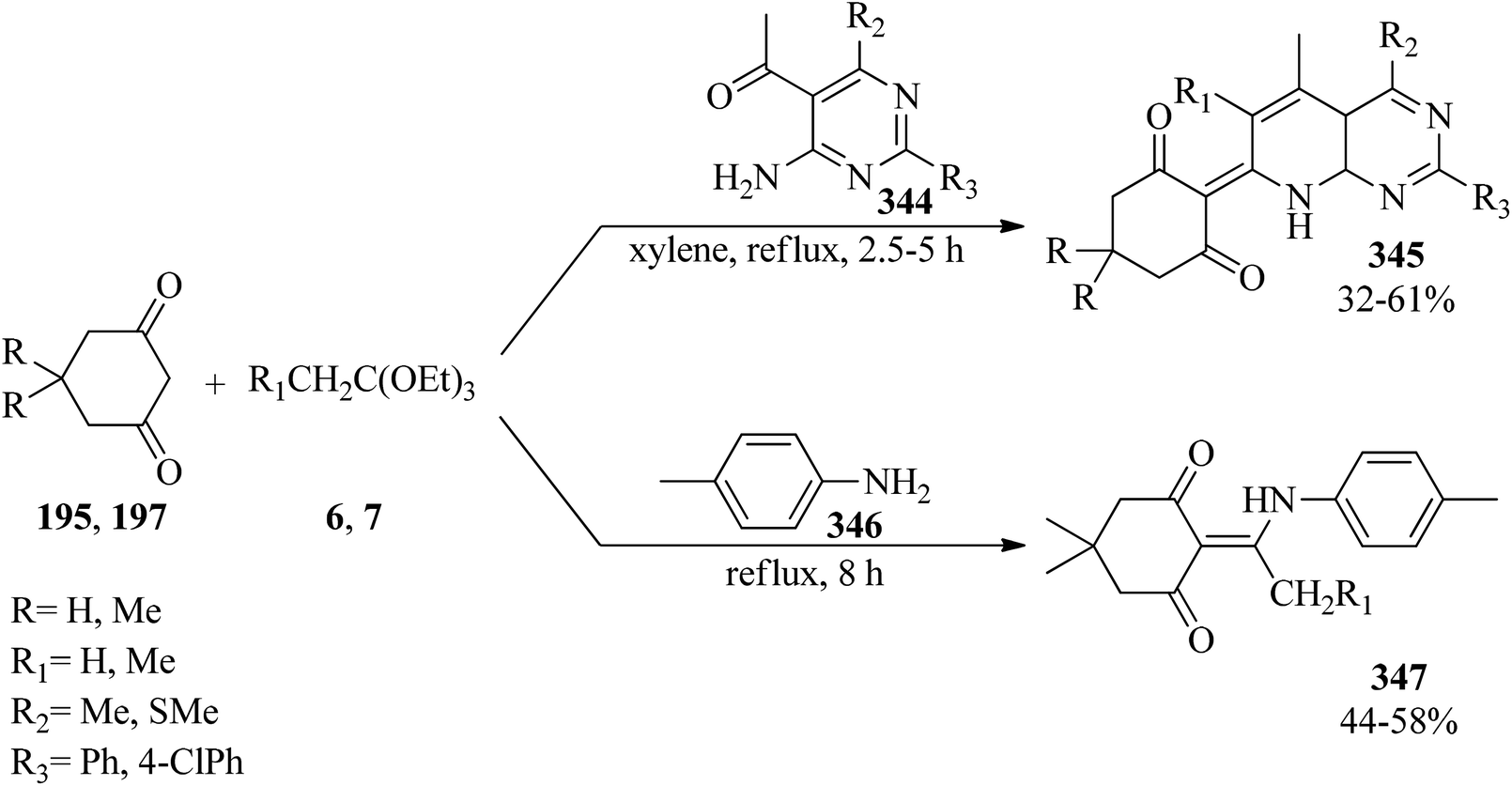
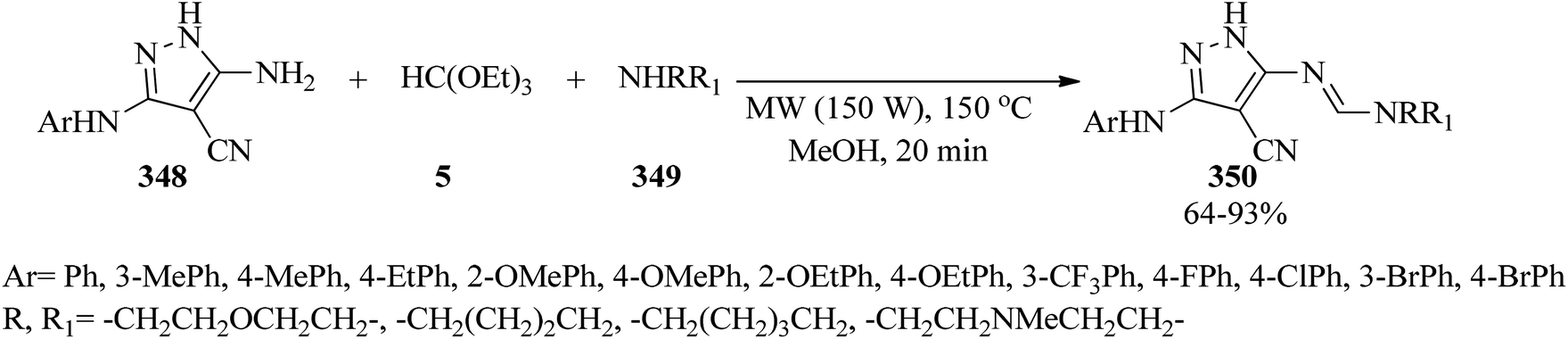
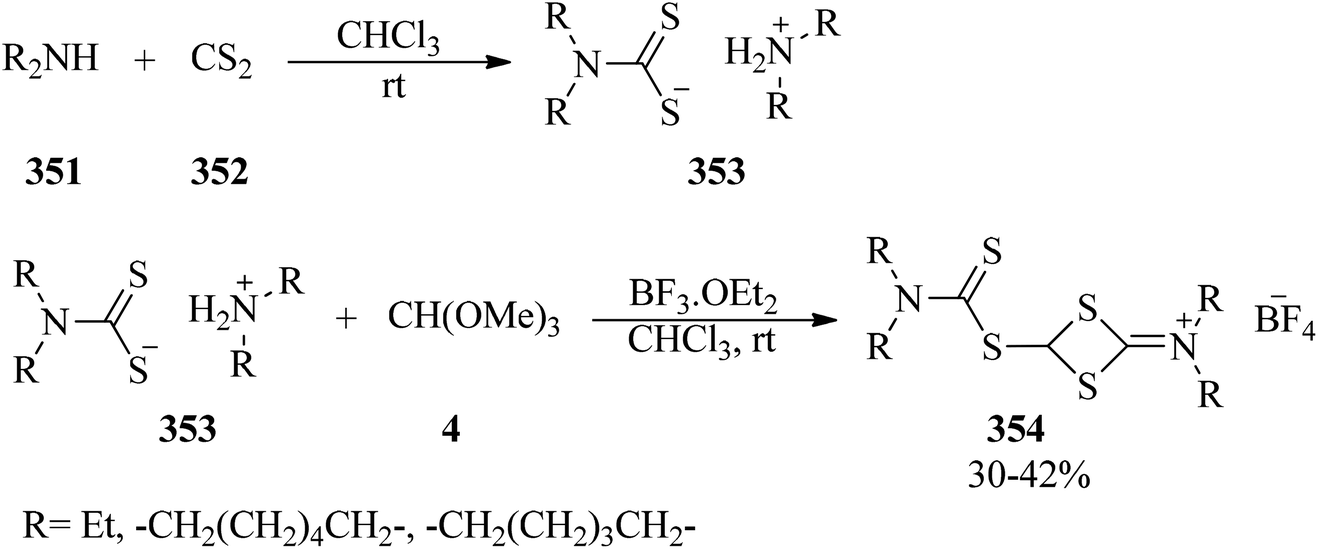
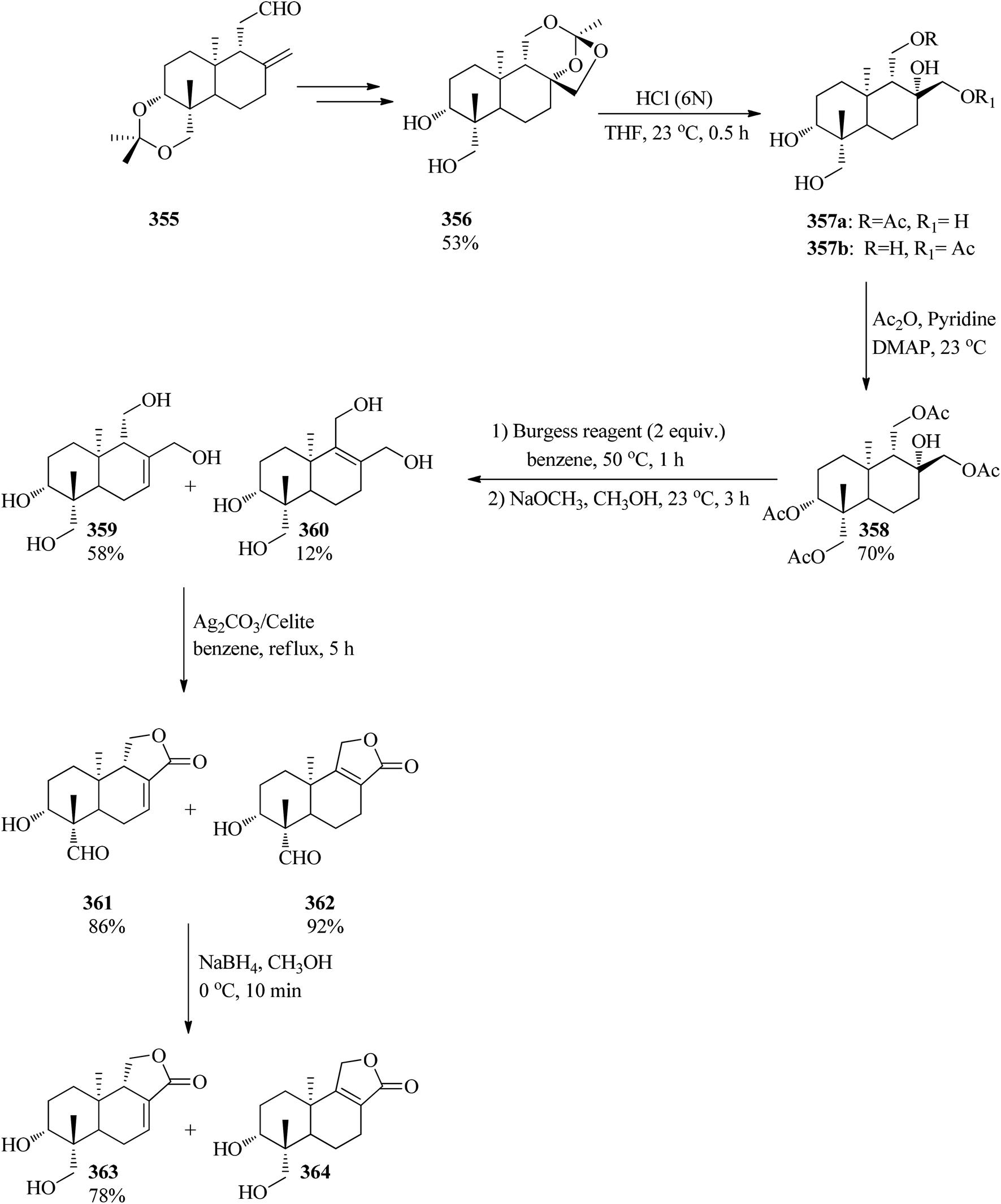
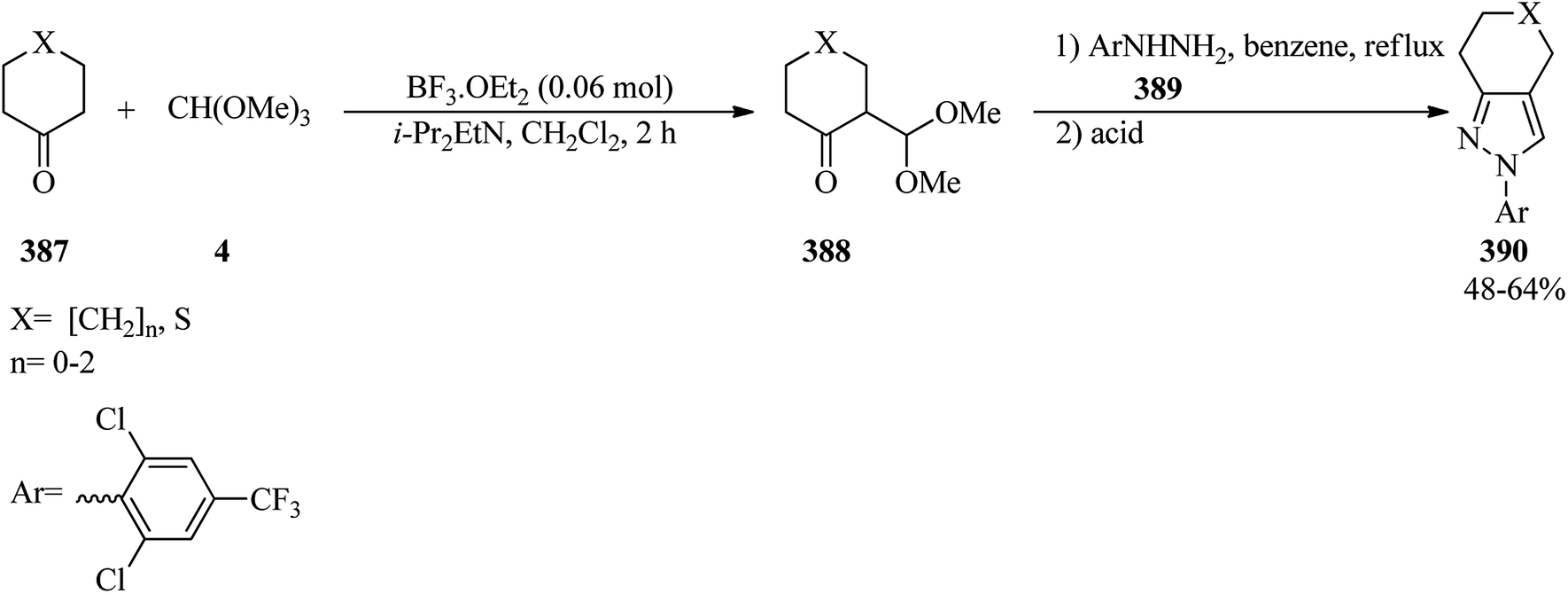
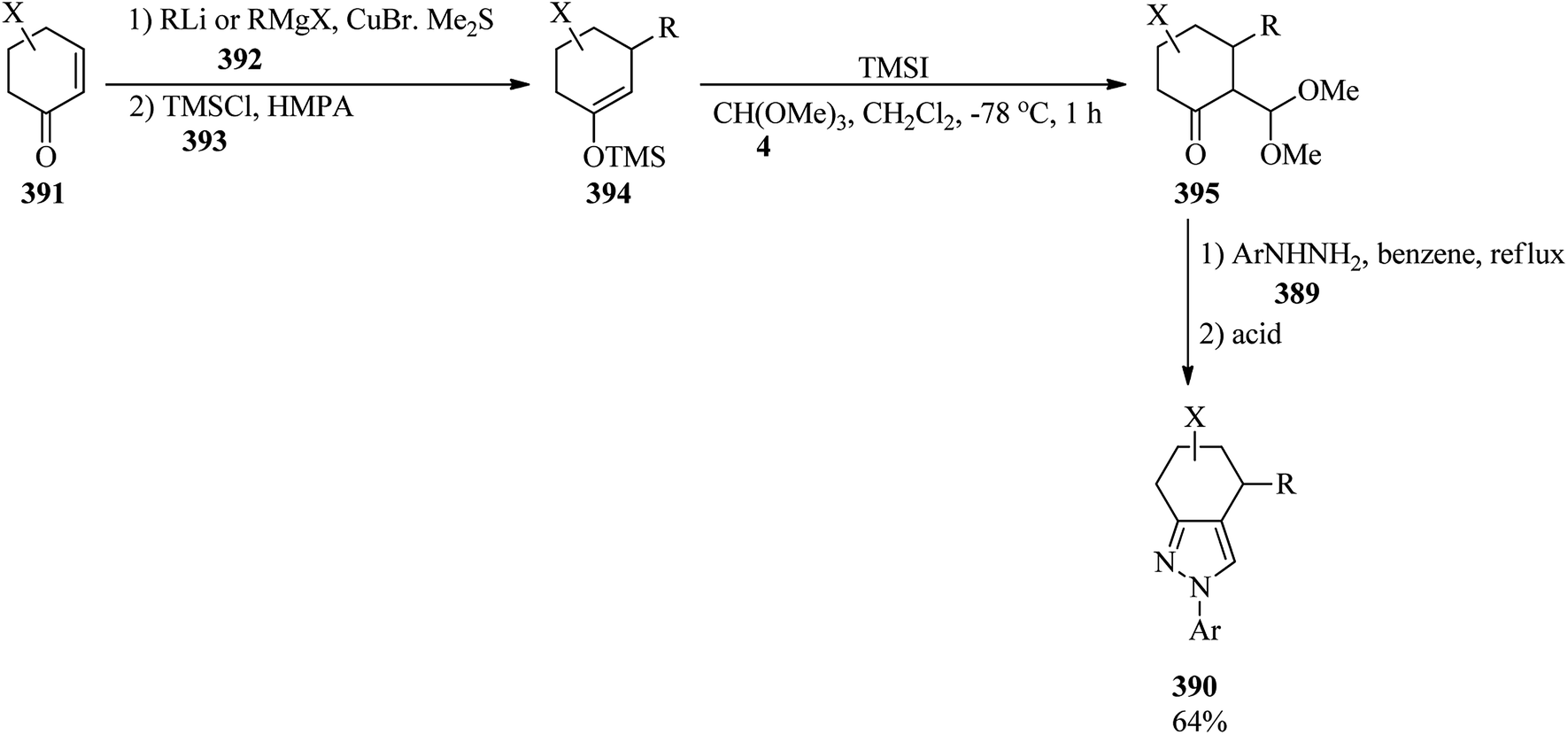
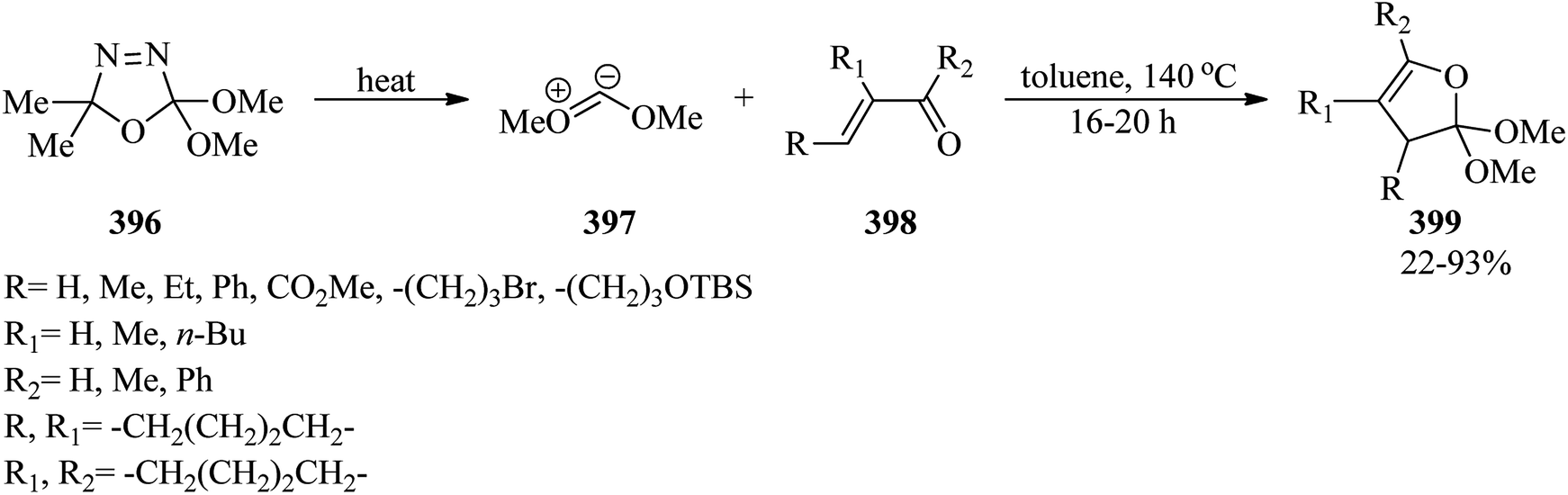
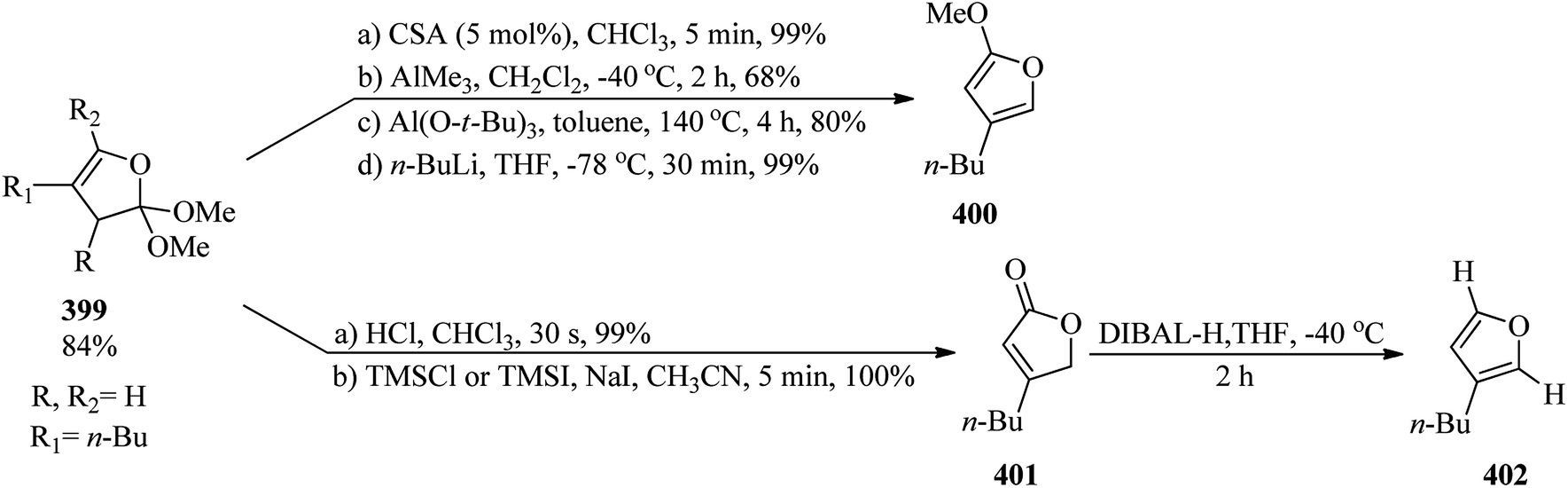
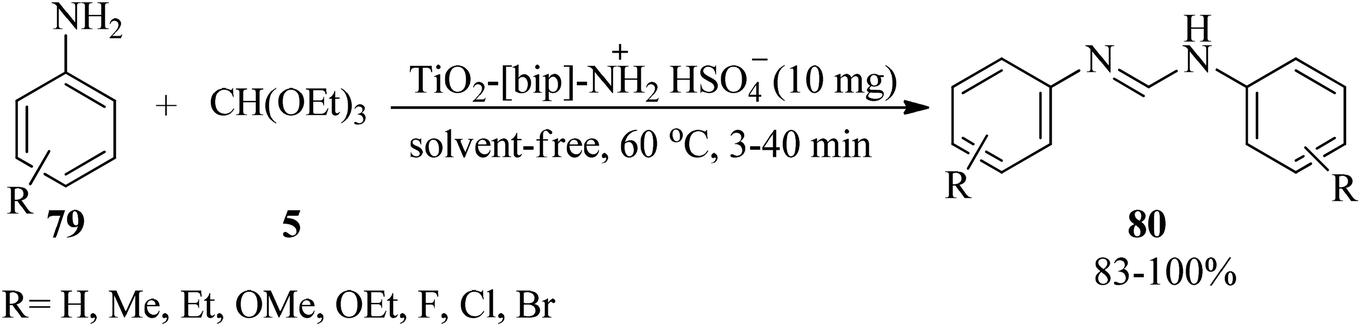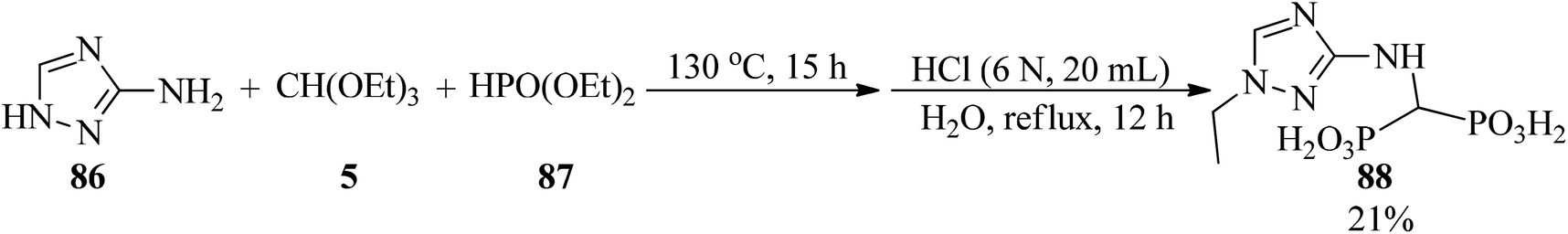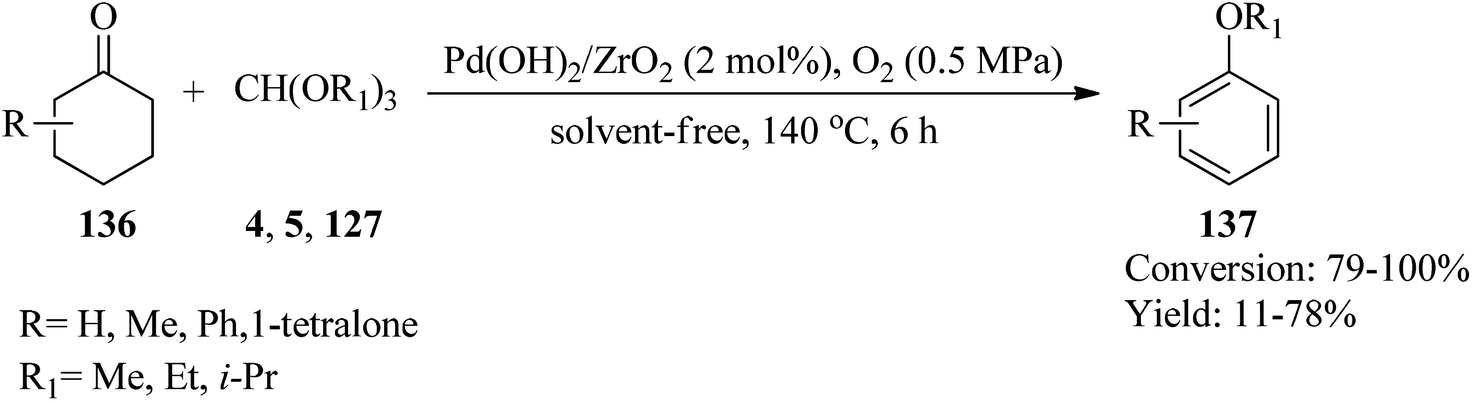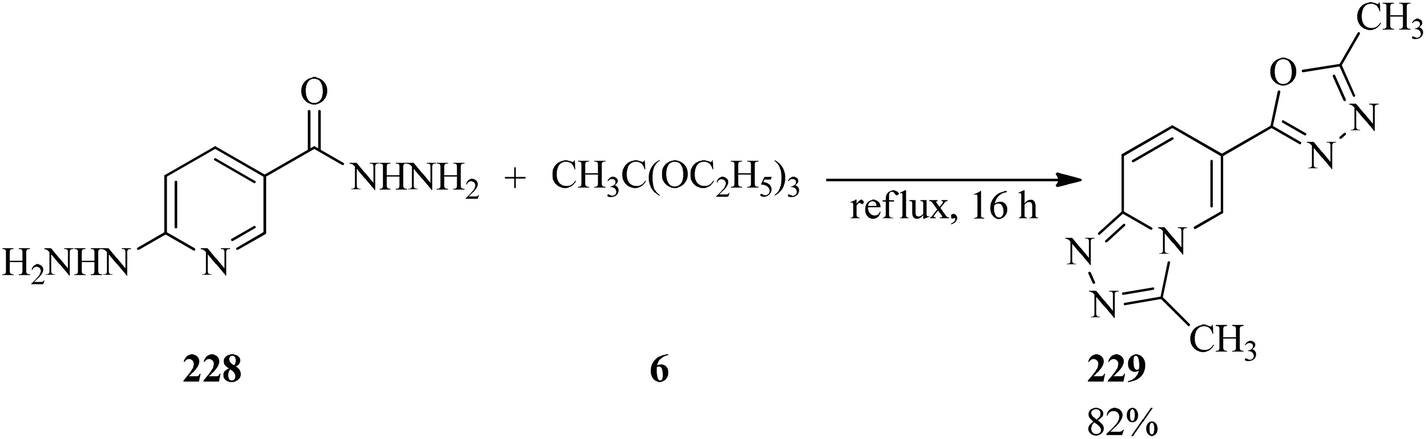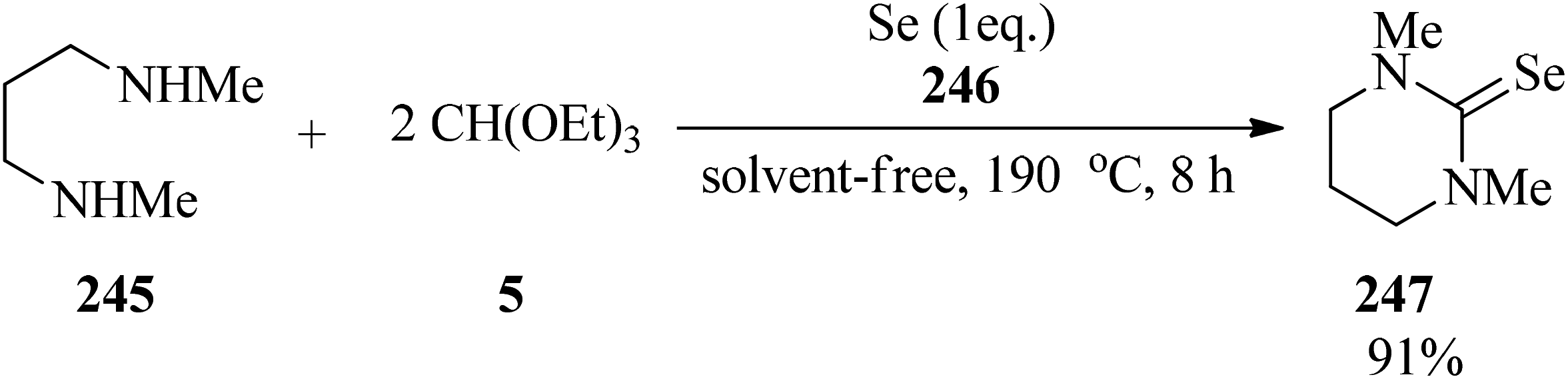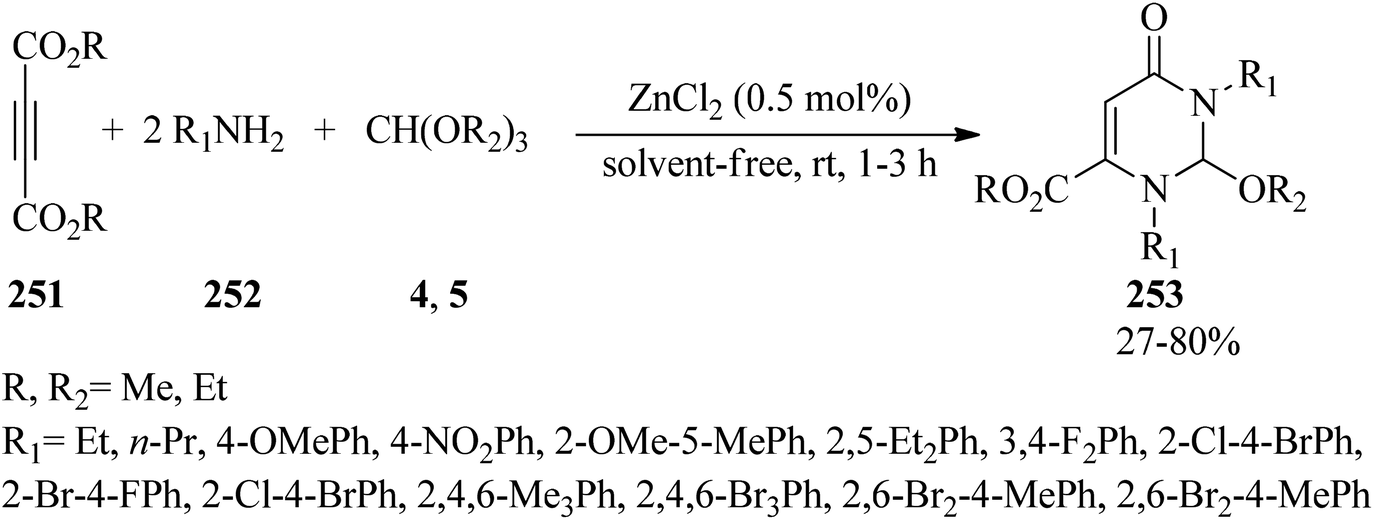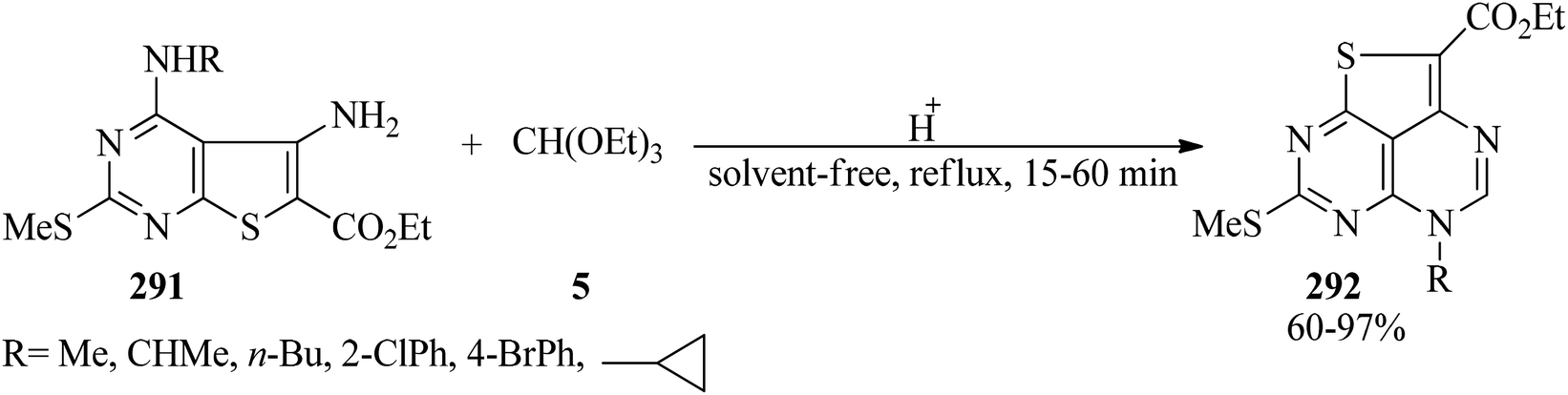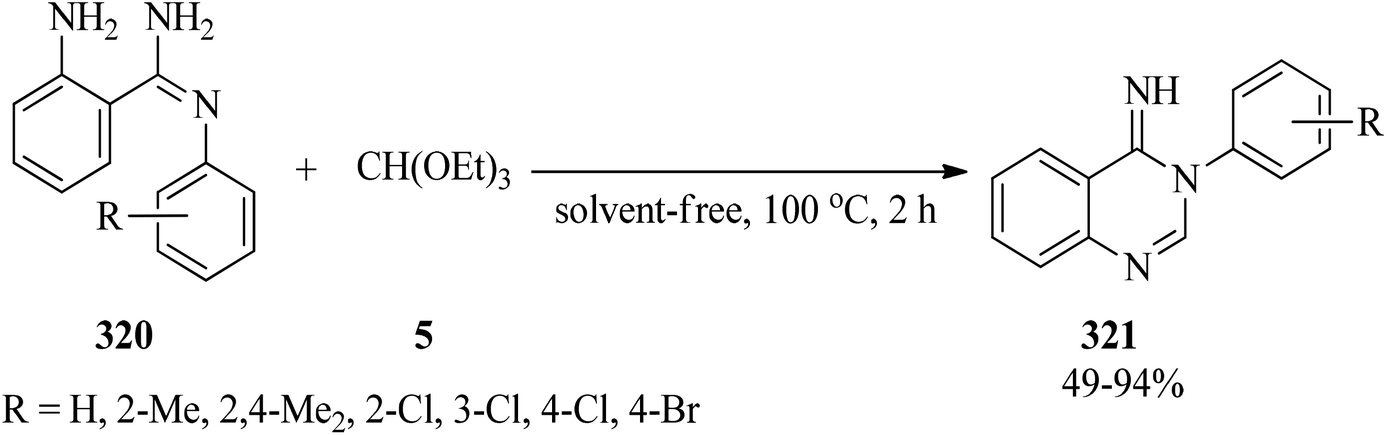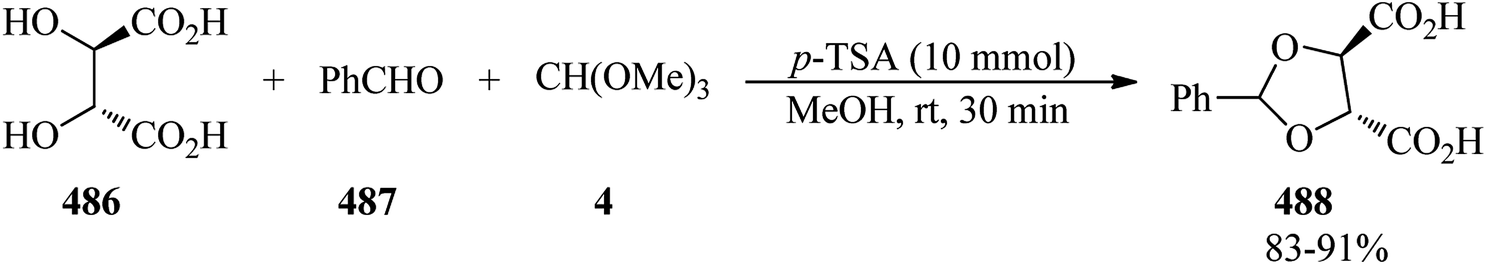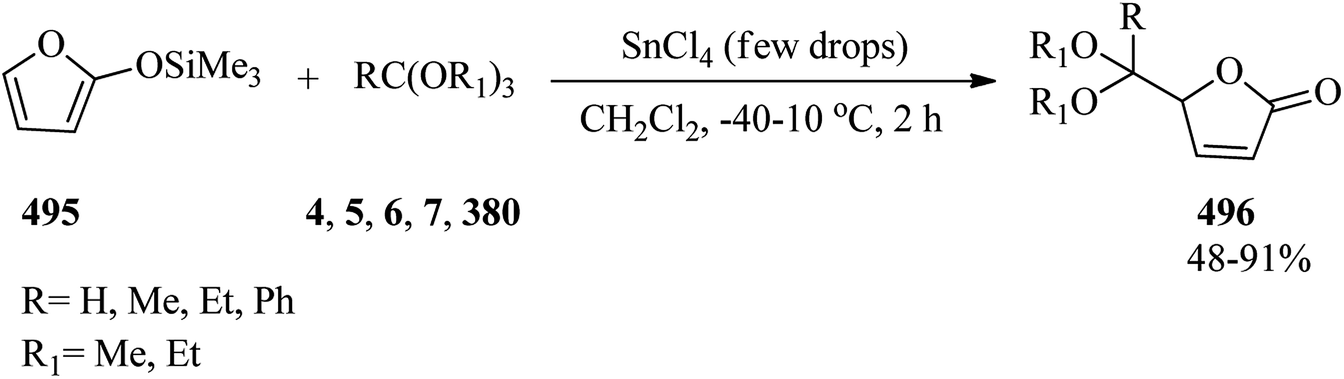DOI:
10.1039/D0RA05276K
(Review Article)
RSC Adv., 2020,
10, 30314-30397
Applications of alkyl orthoesters as valuable substrates in organic transformations, focusing on reaction media
Received
16th June 2020
, Accepted 27th July 2020
First published on 17th August 2020
Abstract
In this review we focus on applications of alkyl orthoesters as valuable and efficient substrates to perform various classes of two-component and multi-component organic reactions. The article has classified them according to two aspects, which are: (i) a focus on the reaction medium (solvent-free conditions, aqueous media, and organic solvents); and (ii) an examination of product structures. Reaction accomplishment under solvent-free conditions is an eco-friendly process with the absence of volatile toxic solvents, which puts it in line with green chemistry goals. Water is an interesting choice in organic transformations due to its inexpensiveness and safety. The authors hope their assessment will help chemists to attain new approaches for utilizing alkyl orthoesters in various organic synthetic methods. The review covers the corresponding literature up to the beginning of 2020.
1. Introduction
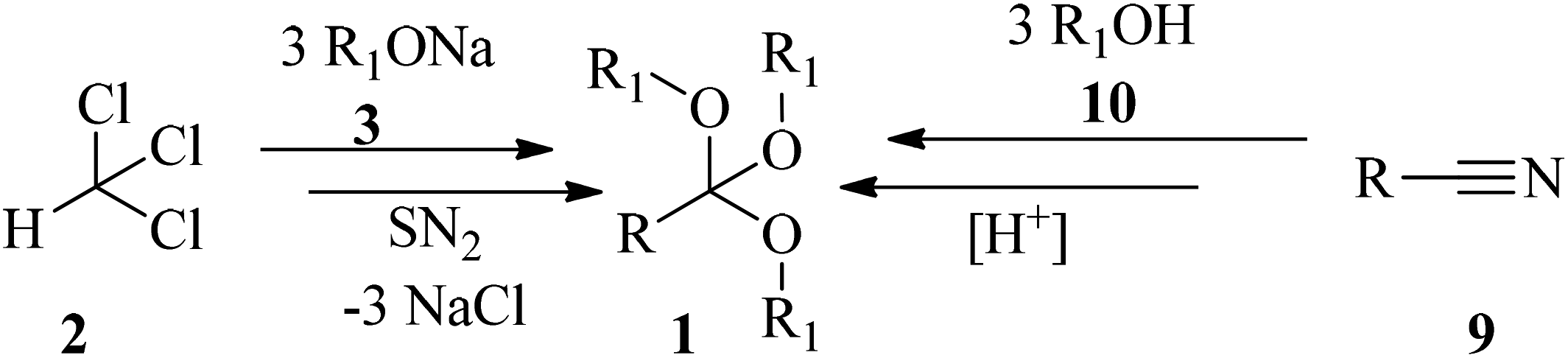
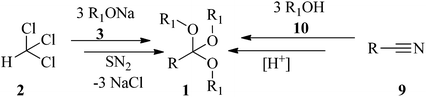
Orthoester (1,1,1-triorganyloxyalkane) (1) is the general name for a functional group containing three alkoxy groups attached to one central carbon atom, with the general formula RC(OR')3. In fact they are the esters of orthoacids which do not exist in a free state.1,2 Orthoesters were first synthesized via nucleophilic the substitution reaction of chloroform (2) with alkoxides (3) by Williamson and Kay in 1854 (Scheme 1).3 There are several examples of acyclic alkylorthoesters such as:4
 |
| | Scheme 1 The Pinner reaction for orthoester synthesis. | |
(a) Trimethyl orthoformate (TMOF) is the simplest orthoester in organic chemistry, which is also called trimethoxymethane, 2-methoxyacetaldehyde dimethyl acetal, methoxymethylal, and methyl orthoformate. This colorless liquid with a pungent odor is soluble in ethanol and ether ((CH(OMe)3, 4), bp = 100.6 °C, and d = 0.9676 g mL−1).
(b) Triethyl orthoformate (TEOF, CH(OEt)3, 5), also called diethoxymethoxyethane, triethoxymethane, and ethyl orthoformate a colorless volatile liquid with bp = 146 °C and d = 0.891 g mL−1, is the orthoester of formic acid.
(c) Triethyl orthoacetate (TEOAc, CH3C(OEt)3, 6) 1,1,1-triethoxyethane, (bp = 142 °C, d = 0.885 g mL−1), is an oily yellow to colorless liquid with a pungent odor.
(d) Triethyl orthopropionate (CH3CH2C(OEt)3, 7), also called 1,1,1-triethoxypropane and orthopropionic acid triethyl ester, is a clear colorless liquid (bp = 155–169 °C, d = 0.886 g mL−1) which is soluble in alcohol, chloroform, ethyl acetate, and ether and hydrolyzes slowly in water.
(e) 3,3,3-Triethoxy-1-propyne (HCC(OEt)3, 8).
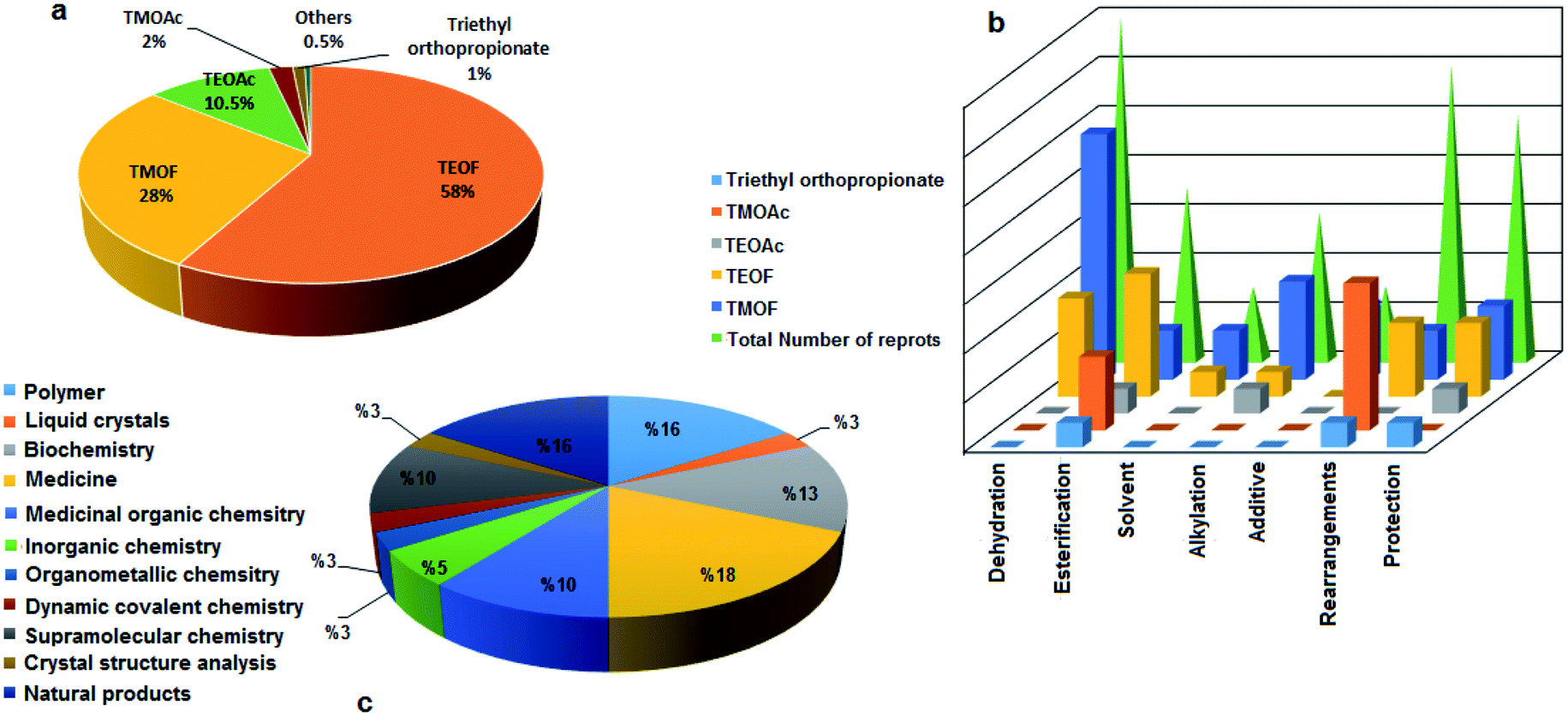
Orthoesters may be considered to be products of the exhaustive alkylation of unstable orthocarboxylic acids and it is from these that the name “orthoester” is derived. Orthoesters are highly reactive owing to the electron-deficient central carbon atom caused by the –I effect of the electronegative groups OR.1 Among the ortheosters introduced above, TEOF involved maximal exploitation in the reactions based on our literature survey (109 reports) followed by TMOF (54 reports), TEOAc (21 cases), TMOAc (4 items), and triethyl orthopropionate (2 cases), and other kinds of alkyl orthoesters (1 case) (Fig. 1a). Several orthoesters were applied in various domains as dehydrating agents,5–18 alkylating agents,19–24 dialkoxymethylation agents,25 esterification agents,26 methoxy sources,27 additives,28–30 solvents,31–33 and protecting groups.34–36 This functional group is also necessary for some conversions such as the Johnson–Claisen rearrangement.37 Fig. 1b demonstrates the assorted applications mentioned vs. the type of orthoester. As specified, TMOF and TEOF are the most commonly used in different processes, except for the case of rearrangements where TMOAc is the most abundant.
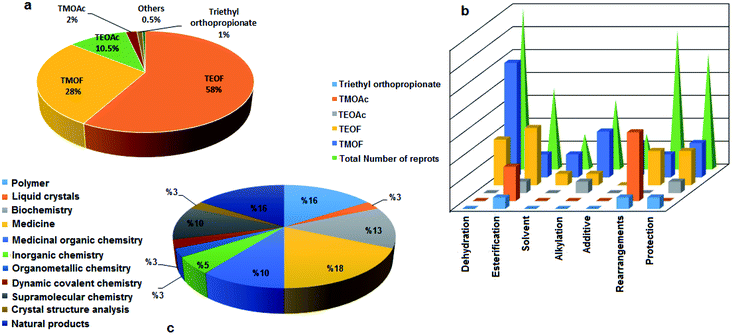 |
| | Fig. 1 The abundances of different orthoesters as substrates in organic transformations (a), assorted applications of different kinds of orthoesters in some processes (b), and the classification of their utilization in various domains of science and technology (c). | |
Different kind of orthoesters also have many applications in several branches of science, such as polymers,38–43 liquid-crystalline compounds,44 biochemistry,45–49 medicine50–56 and medicinal organic chemistry,57–60 inorganic chemistry,61,62 organometallic chemistry,63 dynamic covalent chemistry,64 supramolecular chemistry,65–68 and crystal structure analysis.69 Orthoesters have also been used in the synthesis of natural products, e.g. loganin is a monoterpene glucoside occupying a central position in the generation of secoiridoids and alkaloids, obtained from orthoesters biosynthetically. Because of this, in 1973, Buechi and co-workers described the total synthesis of loganin. It should be noted that trimethyl orthoformate was employed in some of the steps in the preparation of loganin,70 and the total synthesis of (−)-monomorine,71 and (±)-vernolepin.72 It has been reported that TEOF is exploited in the total synthesis of palytoxin, as a marine natural product.73 In 1976, Greenlee and Woodward reported the total synthesis of marasmic acid, one of the many fungal metabolites obtained from the Basidiomycetes, in which trialkyl orthoformates were applied in some of the syntheses.74 Chan and Koide utilized orthoesters for the first total synthesis of the stresgenin B scaffold as a heat shock protein expression inhibitor.75 The application of orthoesters in the synthesis of condensed carbon-cage organics was investigated in the preparation of twistane as a cycloalkane and also the simplest diamondoid. In 1967, Gauthier and Deslongchamps obtained twistane via a multi-step process started by TEOF as one of the substrates.76 The diagram in Fig. 1c illustrates the vast range of applications of different kinds of orthoesters in various sciences. The results indicate the comprehensive use of orthoesters in many domains of science and technology.
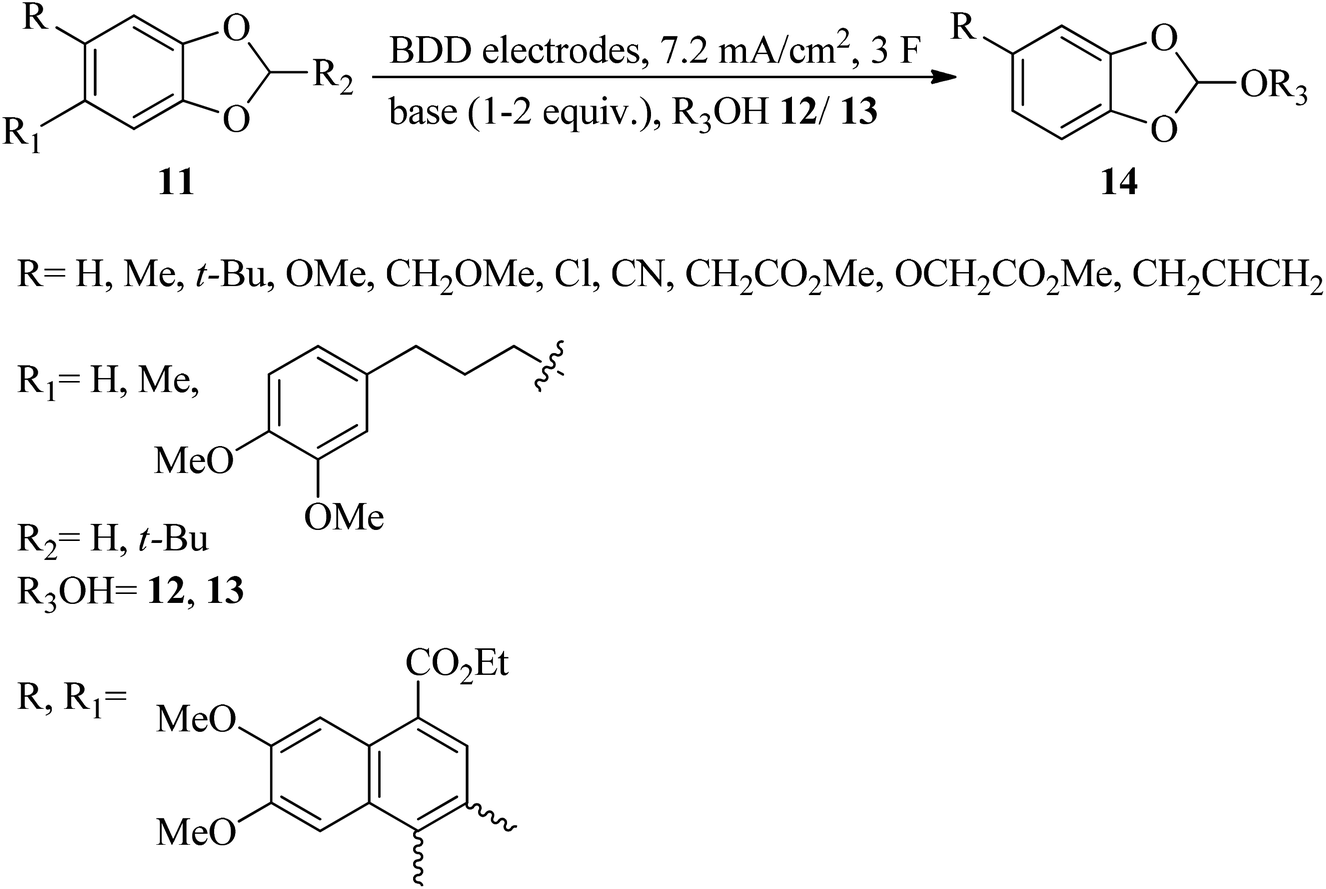
In addition to the Williamson and Kay method, there are various procedures for the preparation of orthoesters. The Pinner reaction of nitriles (9) with alcohols (10) in the presence of strong acids generated orthoesters (Scheme 1).77 Electrochemical approaches78–81 are also employed for the synthesis of these compounds, e.g., new highly fluorinated orthoesters (14) were synthesized through the functionalization of 1,3-benzodioxoles (11) in the 2 position with diverse fluorinated alcohols (such as 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP, 12) or 2,2,2-trifluoroethanol (TFE, 13)) in an undivided cell and in the presence of N,N-diisopropylethylamine or 1,8-diazabicyclo-[5.4.0]undec-7-ene (DIPEA/DBU) as basic electrolytes by boron-doped diamond (BDD) electrodes under a constant current density of 7.2 mA cm−2 until a catalytic quantity of 3.0 F mol−1 of electricity has passed (Scheme 2).82
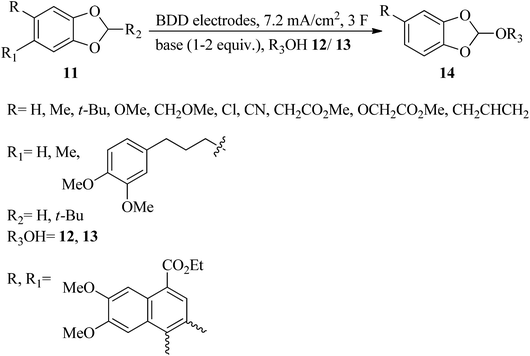 |
| | Scheme 2 The electrochemical synthesis of highly fluorinated orthoesters. | |
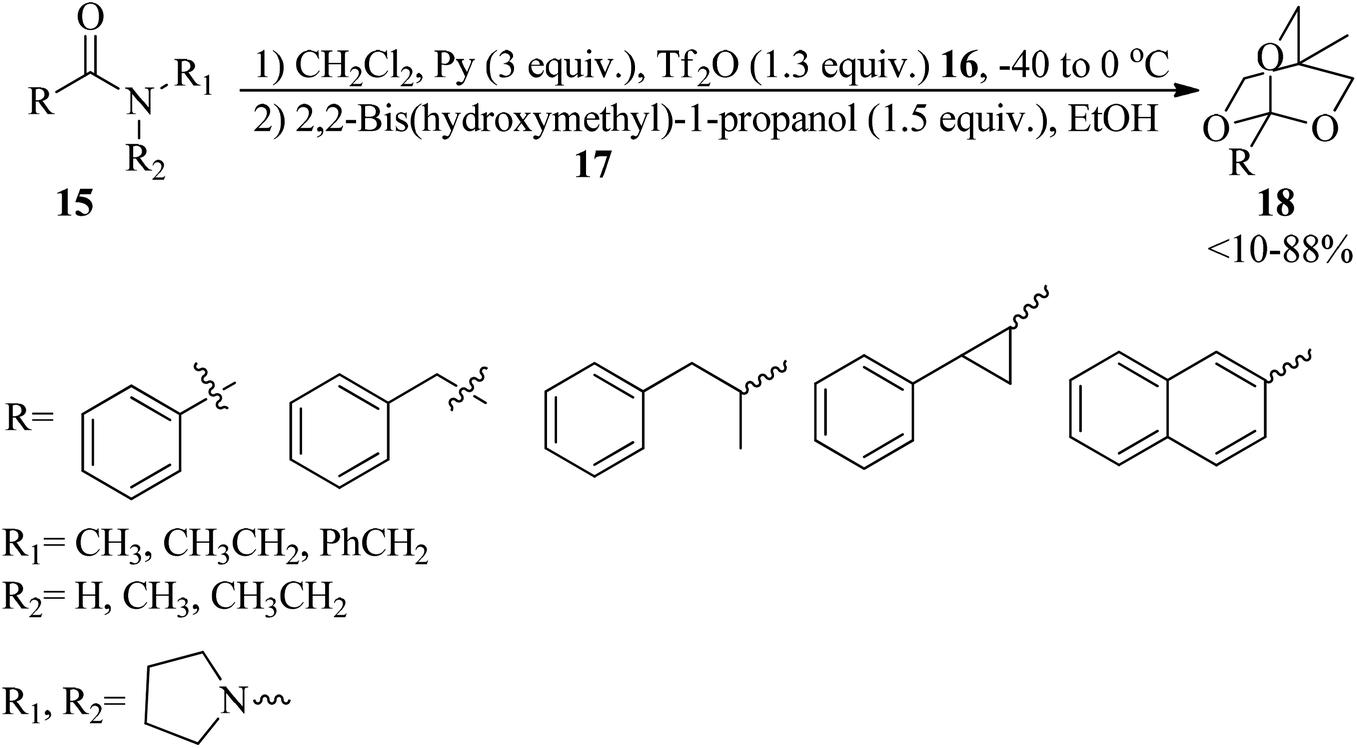
Secondary and tertiary amides (15) were reacted with triflic anhydride (16) by pyridine in CH2Cl2 at low temperatures to form imino and iminium triflates within 4 h, which on treatment with 2,2-bishydroxymethyl-1-propanol (17) utilizing pyridine buffered solution attained 2,6,7-trioxabicyclo[2.2.2]octane orthoesters (18) (Scheme 3).83
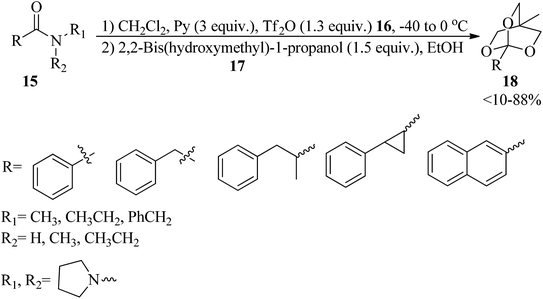 |
| | Scheme 3 A synthetic approach to 2,6,7-trioxabicyclo[2.2.2]octane orthoesters. | |
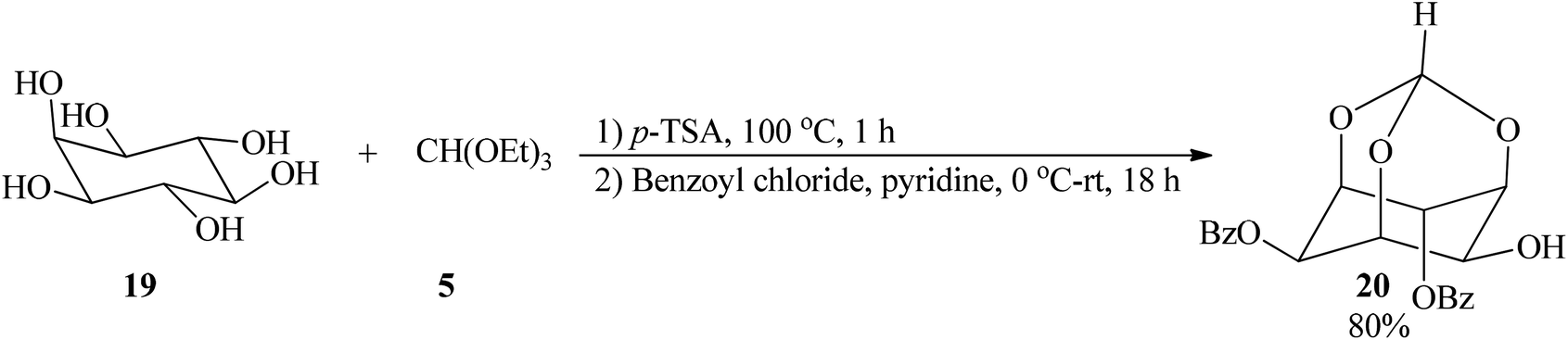
2,4-Di-O-benzoyl-myo-inositol 1,3,5-orthoformate (20), a cyclic alkyl orthoester, was obtained from successive treatment of myo-inositol (19) with TEOF (5) and benzoyl chloride by Shashidhar and Praveen in 2001 (Scheme 4).84
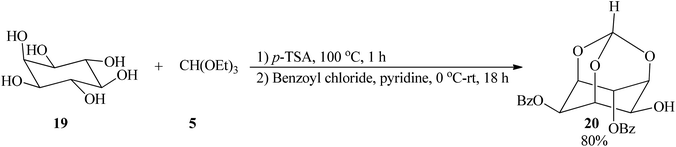 |
| | Scheme 4 The preparation of 2,4-di-O-benzoyl-myo-inositol 1,3,5-orthoformate. | |
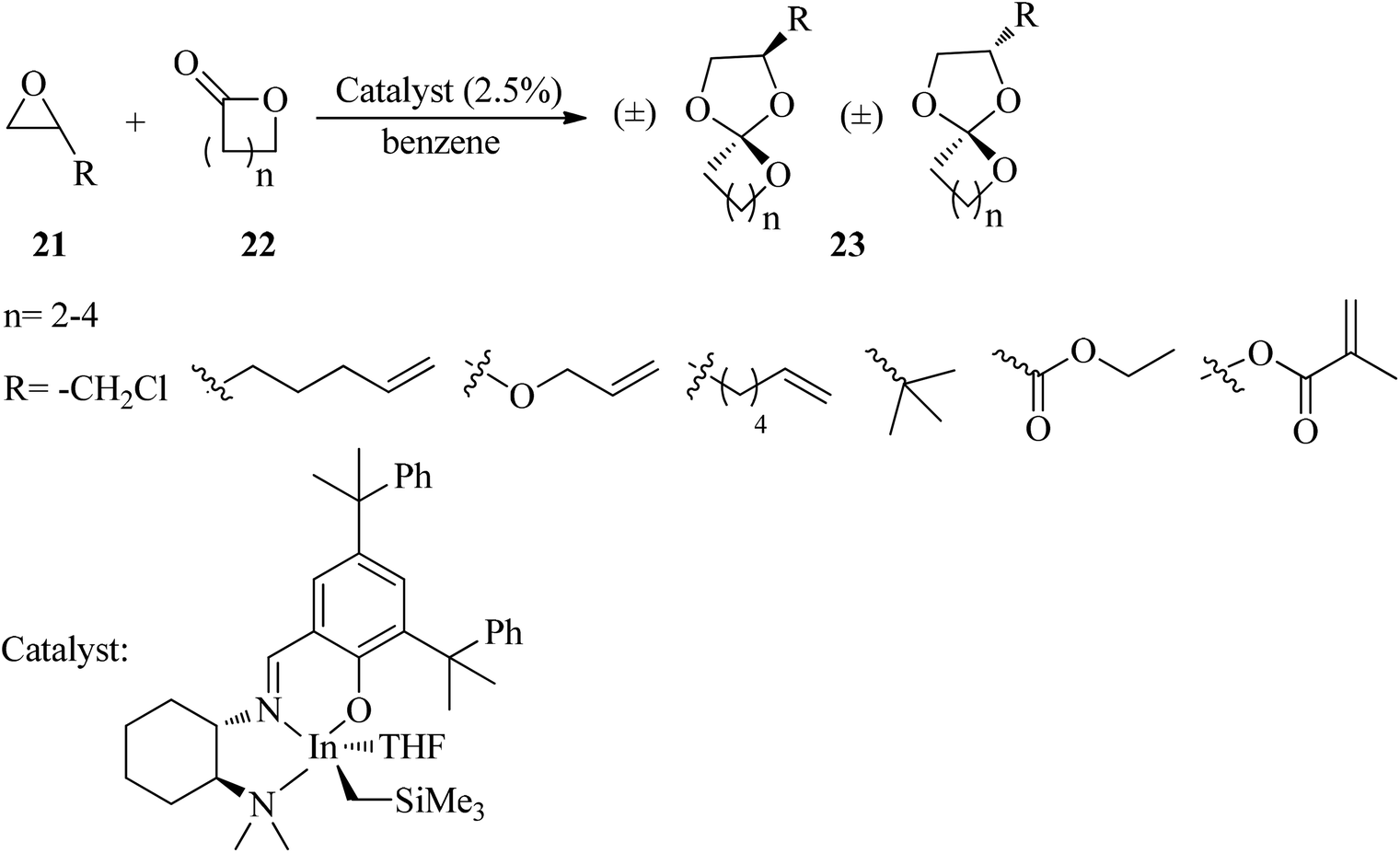
The reaction of epoxides (21) and lactones (22) in the presence of a cationic indium catalyst under heating with benzene resulted in spiro orthoesters (23) (Scheme 5).85
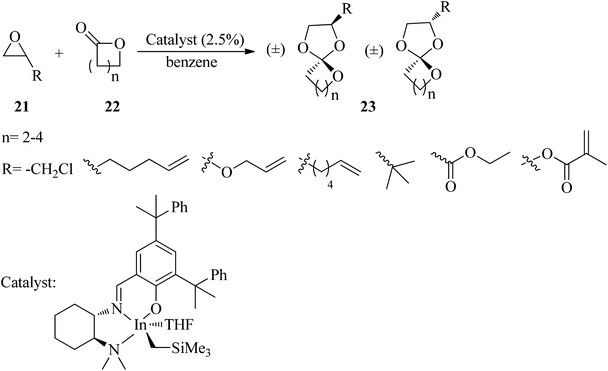 |
| | Scheme 5 The synthesis of spiro orthoesters. | |
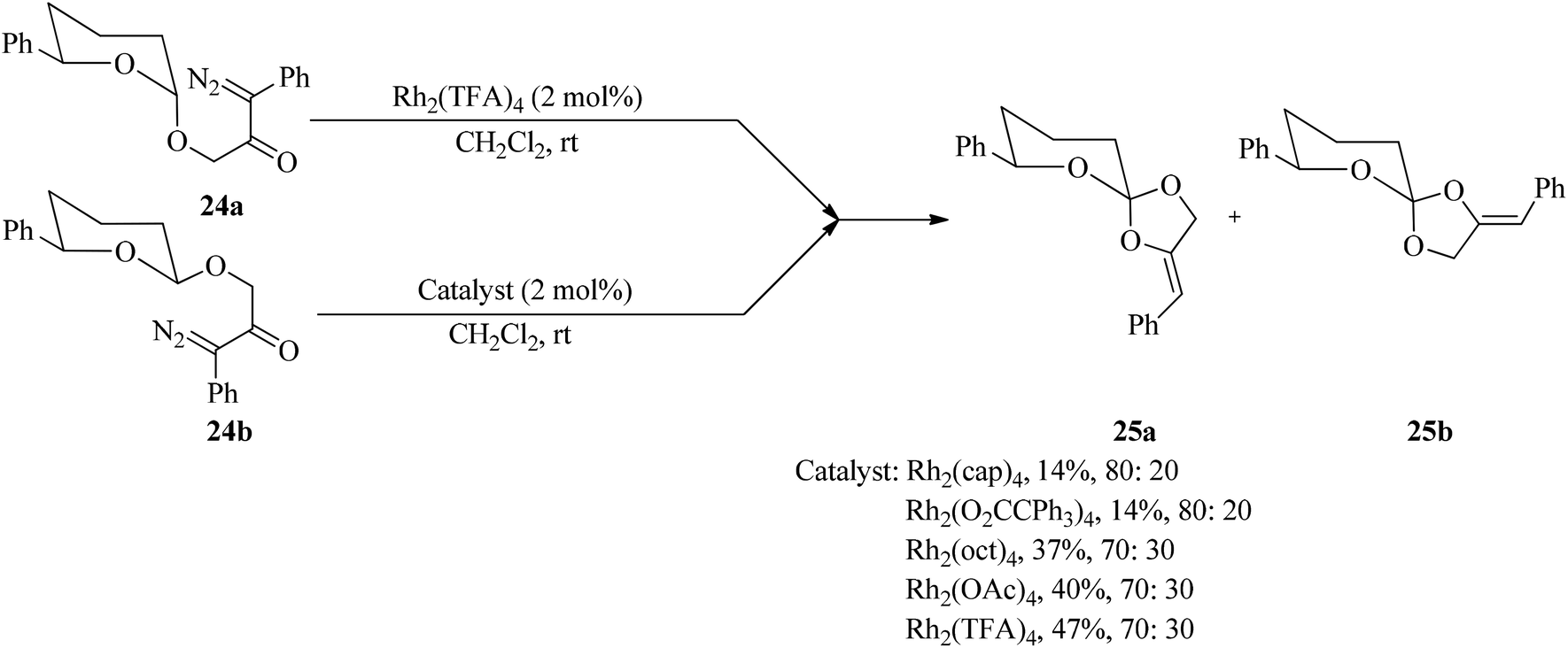
Tetrahydropyranyl acetals containing a phenyl diazoketone substituent (24) underwent Rh(II)-catalyzed C–H insertion through an unusual C–O bond generation to give spirocyclic orthoesters (25) (Scheme 6).86
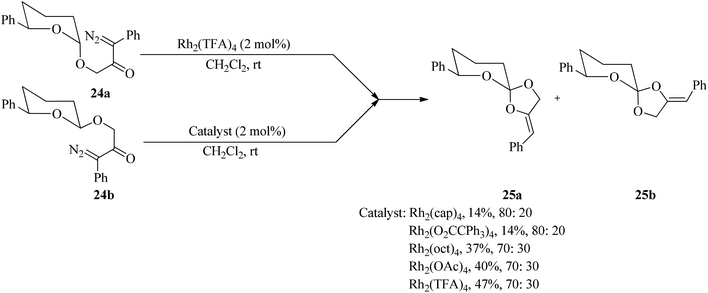 |
| | Scheme 6 The Rh(II)-catalyzed synthesis of spirocyclic orthoesters. | |
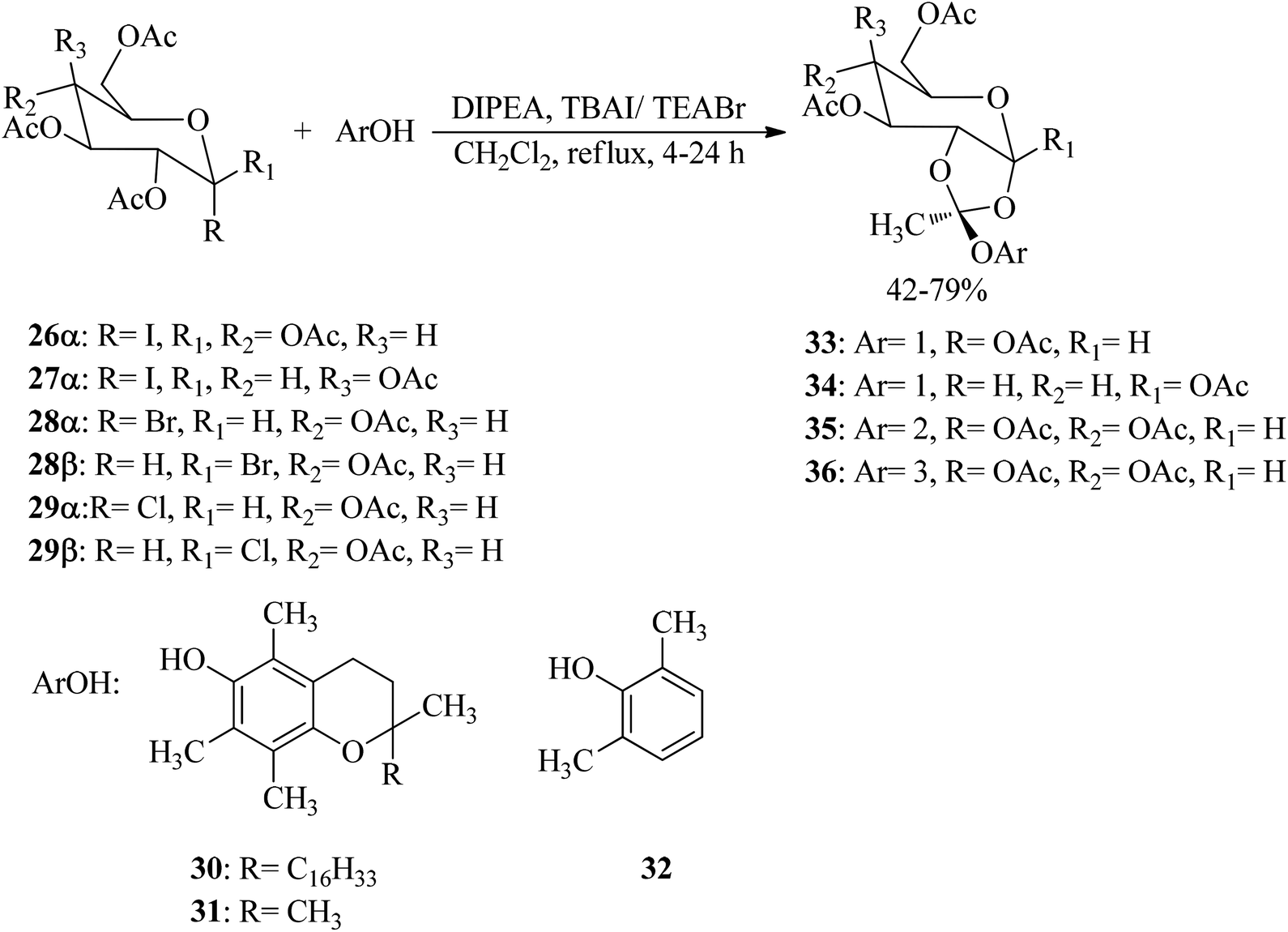
3,4,6-Tri-O-acetyl-1,2-O-(1-tocopheroxyethylidene)-α-D-glucopyranose (vitamin E sugar 1,2-orthoester) (33–36) with exo-type stereoselectivity were synthesized through the reaction of sugar halides (26–29) with various phenols, such as α-tocopherol (30), chroman-6-ol (31), and 2,6-dimethylphenol (32) by N,N-diisopropylethylamine (DIPEA) and tetrabutylammonium iodide (TBAI) or tetraethylammonium bromide (TEABr) in refluxing CH2Cl2 for 4–24 h. The obtained peracetylated orthoesters (33–36) were deprotected by utilizing aminolysis (NH3/MeOH) or by reduction in the presence of DIBAL-H (Scheme 7).87
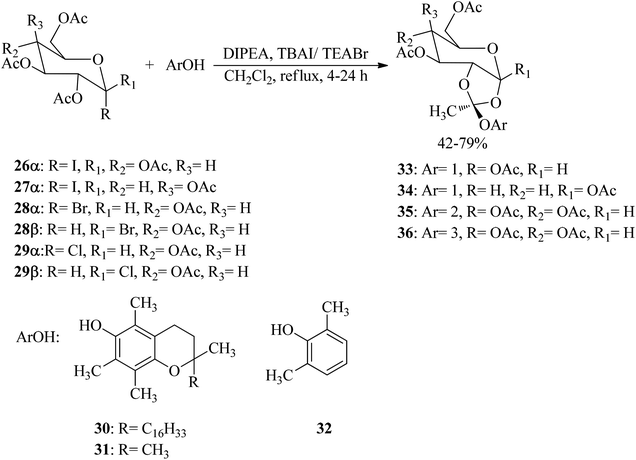 |
| | Scheme 7 Synthetic procedure for peracetylated orthoester. | |
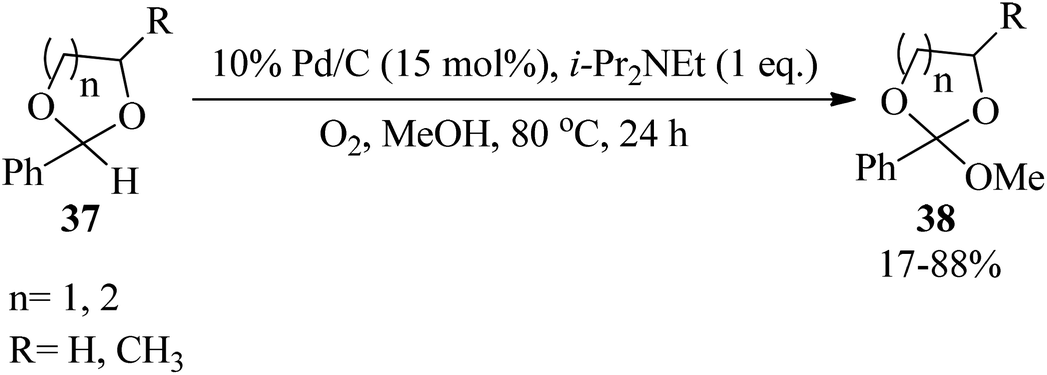
Palladium on carbon (Pd/C) catalyzed the straightforward methoxylation of the benzylic positions of cyclic acetals (37) by i-Pr2NEt in refluxing methanol under atmospheric oxygen as a green oxidant to obtain orthoesters (38) (Scheme 8).88
 |
| | Scheme 8 The synthesis of orthoesters via the methoxylation of cyclic acetals. | |
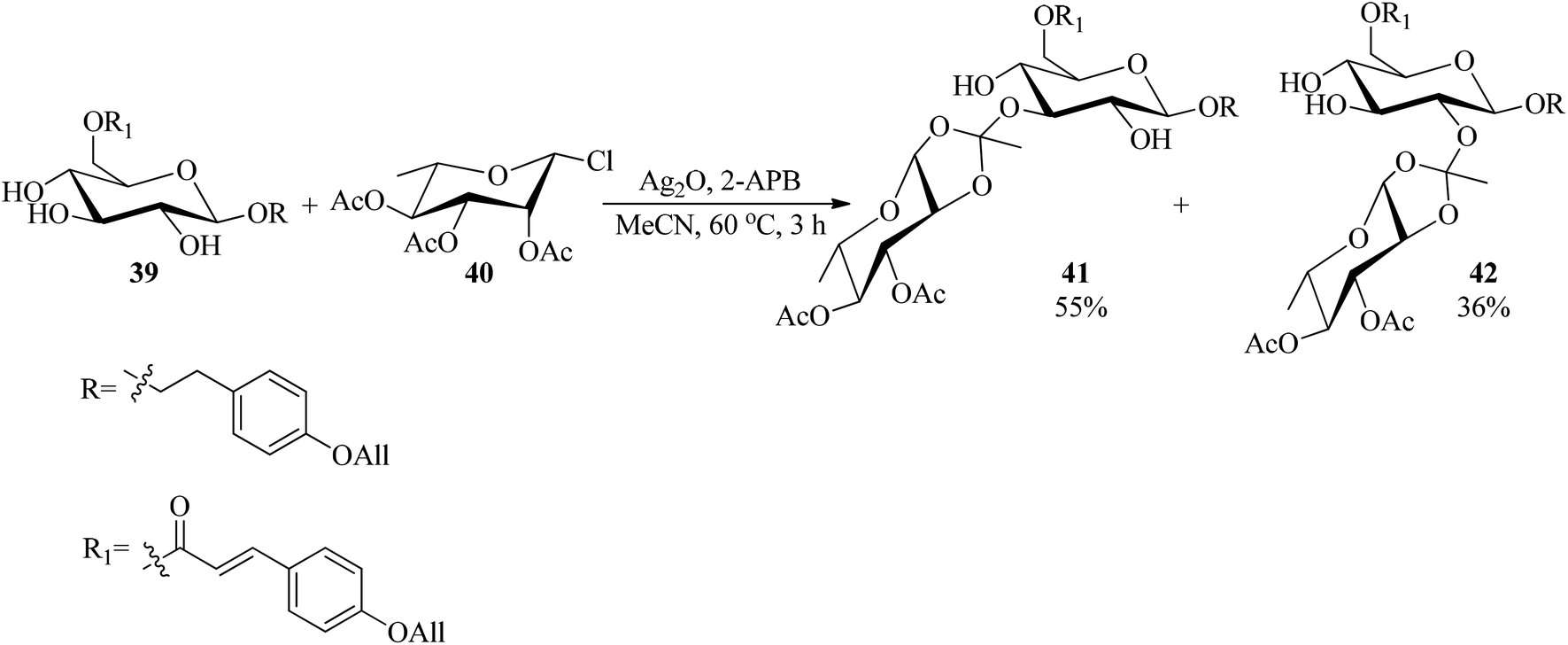
The Koenigs–Knorr glycosylation reaction of glucopyranoside (39) with rhamnosyl chloride (40) as a glycosyl donor was accomplished using a catalytic amount of 2-aminoethyl diphenylborinate (2-APB, 5 mol%)89 and silver(I) oxide (Ag2O) in acetonitrile at 60 °C to obtain the mixture of 3- and 2-orthoesters (41) and (42) in 55% and 36% yields, respectively within 3 h (Scheme 9).90
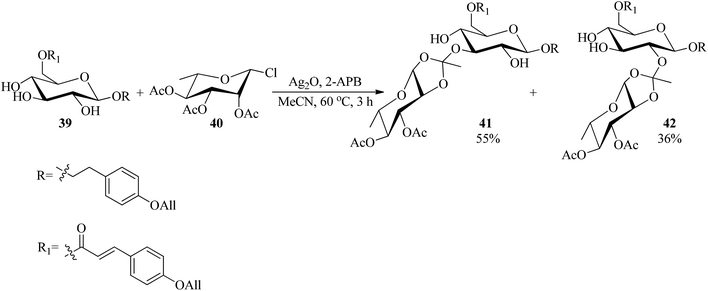 |
| | Scheme 9 The preparation of orthoesters via the Koenigs–Knorr glycosylation reaction. | |
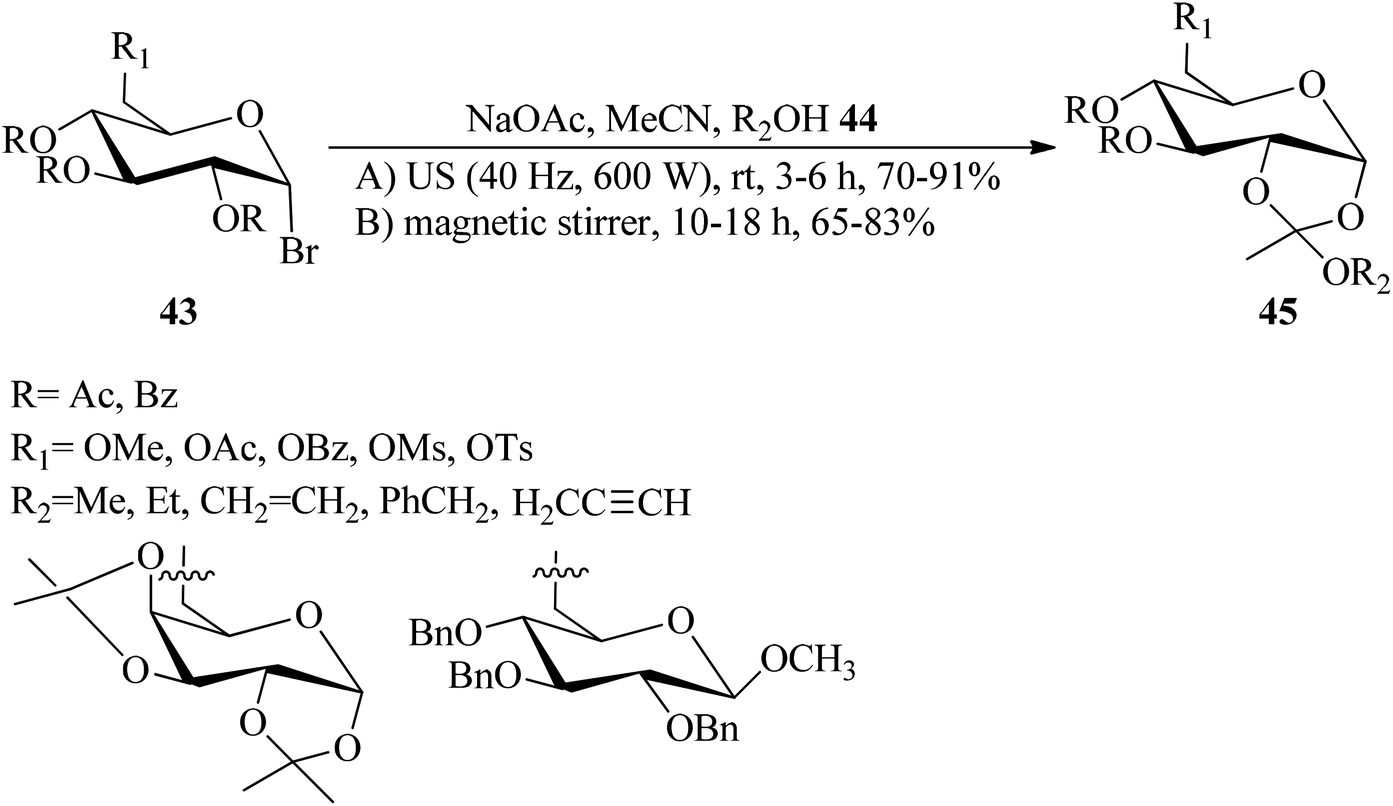
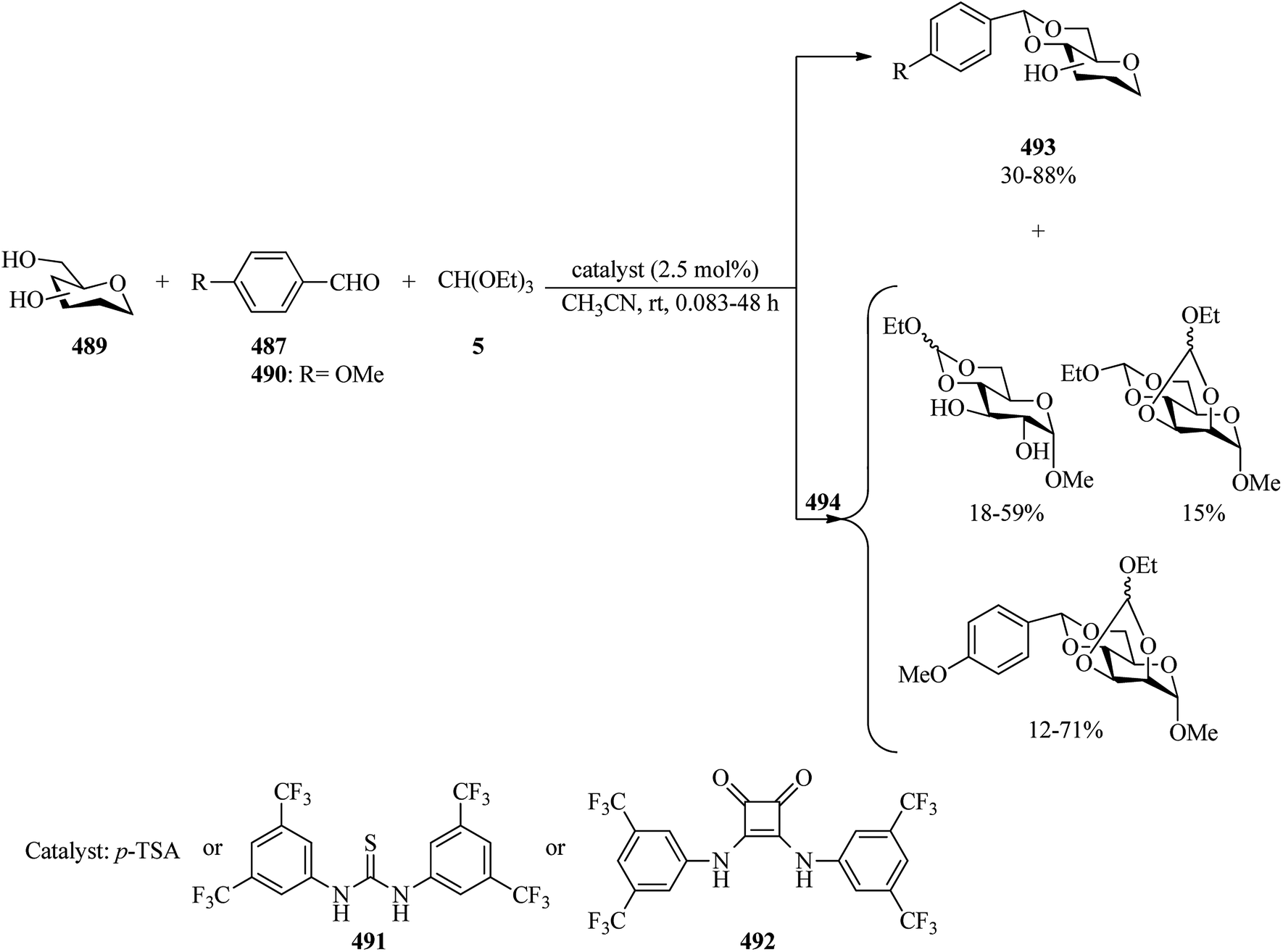
Zhao et al. prepared sugar 1,2-orthoesters (45) via the reaction of peracetylated mannopyranosyl bromides (43) with alcohol derivatives (44) and anhydrous sodium acetate (NaOAc) as a base in acetonitrile at room temperature under ultrasound irradiation (40 KHz and 600 W) (method A), and the traditional reaction under high-speed magnetic stirring (method B). The results confirmed that an ultrasound-assisted reaction proceeded much better than the traditional method (70–91% yields, 3–6 h) (Scheme 10).91
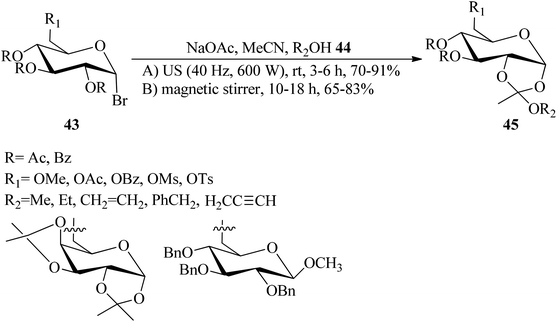 |
| | Scheme 10 The synthetic route to sugar 1,2-orthoesters. | |
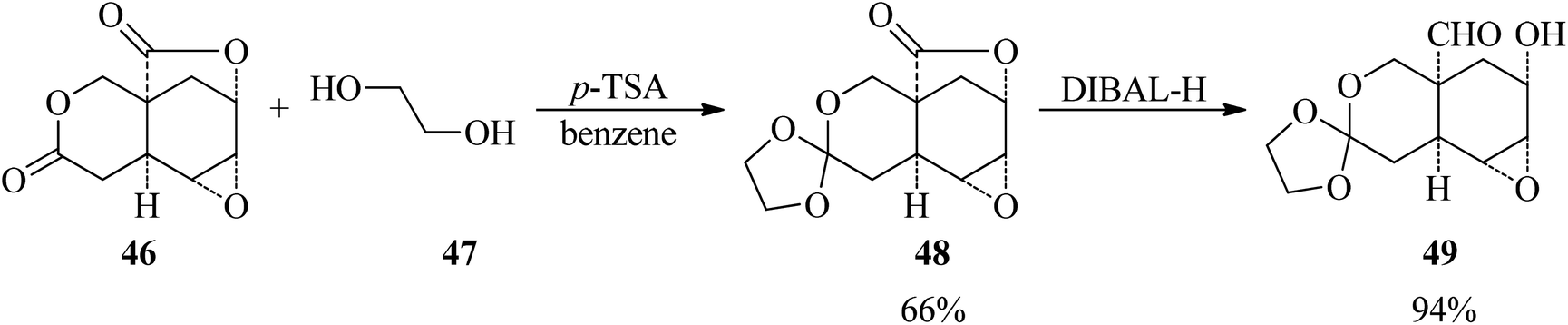
The reaction of dilactone (46) with ethylene glycol (47) by p-toluenesulfonic acid (p-TSA) in the presence of magnesium sulfate in refluxing benzene obtained the crystalline ethylene orthoester (48) in 66% yield within 4.5 h, which underwent a reduction reaction with diisobutylaluminum hydride as a reducing agent in combined dimethoxyethane/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) media at −76 °C to room temperature to afford the hydroxy aldehyde (49) in 94% yield (Scheme 11).92
:2) media at −76 °C to room temperature to afford the hydroxy aldehyde (49) in 94% yield (Scheme 11).92
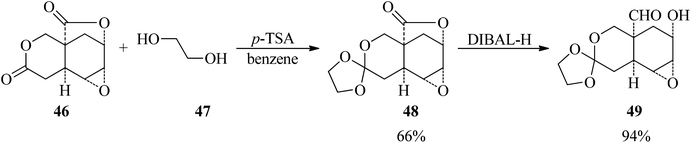 |
| | Scheme 11 The preparation of a crystalline ethylene orthoester possessing an aldehydic moiety. | |
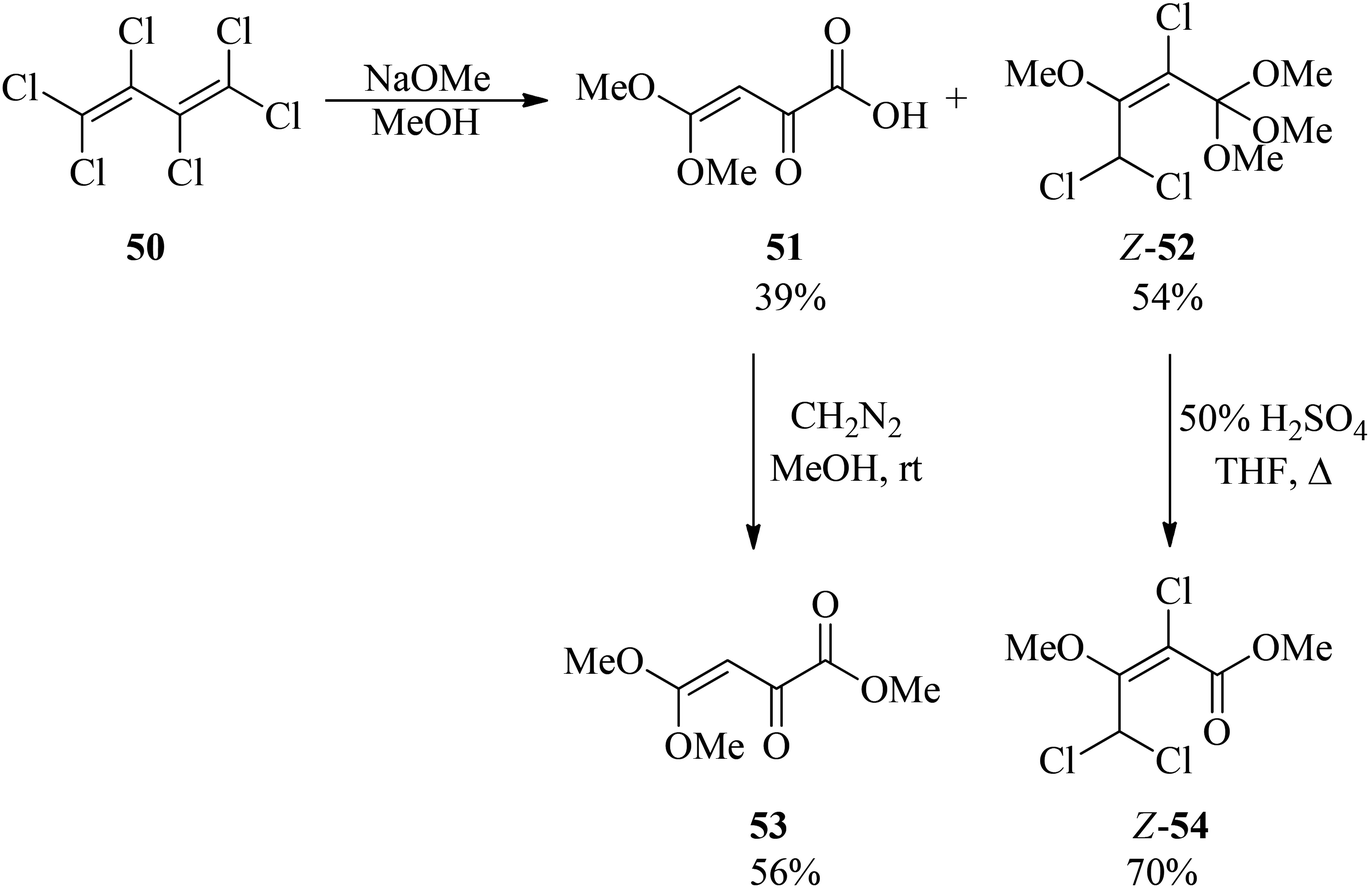
Hexachlorobuta-1,3-diene (50) was refluxed with sodium methoxide in methanol for 12 h to obtain a mixture of 4,4-dimethoxy-2-oxobut-3-enoic acid (51) and (Z)-2,4,4-trichloro-1,1,1,3-tetramethoxybut-2-ene (52) in 39% and 54% yields, respectively in which for easy identification, compound (51) was reacted with diazomethane (CH2N2) in methanol at room temperature to produce methyl (Z)-4,4-dimethoxy-2-oxobut-3-enoate (53) in moderate yield (56%), while orthoester Z-52 was converted into methyl (Z)-2,4,4-trichloro-3-methoxybut-2-enoate (Z-54) in good yield (70%) via acid hydrolysis in THF at 50 °C for 2 h (Scheme 12).93
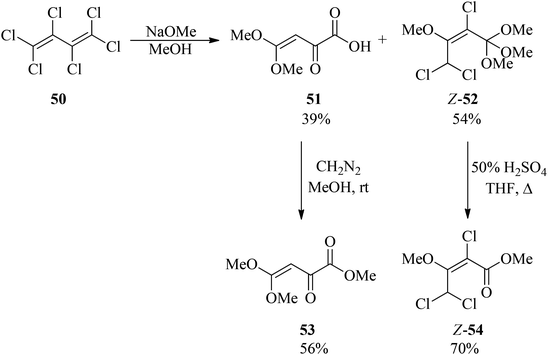 |
| | Scheme 12 The synthesis of some Z-orthoester structures. | |
In 2018, a review entitled “Synthesis and biological activity of oxazolopyrimidines” was published. In some parts of it, orthoesters were used to prepare oxazolo[4,5-d]pyrimidines and 7-aminooxazolo[5,4-d]pyrimidines.94
Perillo et al. presented a review article entitled “Synthesis and properties of seven- to nine-membered ring nitrogen heterocycles. Cyclic amidines and cyclic amidinium salts” which pointed to the application of orthoesters in synthetic methods.95 In 2011, Lavigne's research group published a review article about the route yield from N-heterocyclic carbene precursors. They introduced trialkylorthoesters as the precarbenic unit.96 In 2013, A. Elassar and co-workers published a review article about diaminomaleonitrile (DAMN) in which the mentioned synthon reacted with various orthoesters to prepare different kinds of biologically active heterocycles.97
Numerous review papers have reported on different applications of orthoesters;98,99 for example, a 1986 review article entitled “Alkyl orthoesters and their applications in organic synthesis”, by Pavlova and co-workers, focused on a systematic account of data describing the chemical properties of alkyl esters of orthocarboxylic acids. The proposed reaction mechanisms were discussed and the scope of utility of the orthoesters in the solution of diverse problems in organic synthesis was shown.100 Moreover, a 1988 article entitled “Carbon–carbon bond formation and annulation reactions using trimethyl and triethyl orthoformates”, by Ghosh and Ghatak, focused on the annulation reactions by orthoformates and also the introduction of a formylation–cyclization method for the production of functionalized polycyclic bridged-bicyclo[3.3.1]nonanes, cyclopentenones, and other isolated products to develop the synthetic potential of carbon–carbon bond formation reaction via orthoformates.98 In 2007, a review of “Recent studies on reaction pathways and applications of sugar orthoesters in synthesis of oligosaccharides”, was published by Kong.101 In 2009, Liao and co-workers presented a focused literature survey on plant orthoesters.102 In 2014, a review article with the title “The orthoester Johnson–Claisen rearrangement in the synthesis of bioactive molecules, natural products, and synthetic intermediates-recent advances”, was published with 211 ref. 103. In 2016, another review was also published with the title “Recent development in the orthoester Johnson–Claisen rearrangement and application”, by the Alam group.99 In 2009, a spotlight titled “Alkyl orthoformate: a versatile reagent in organic synthesis”, was compiled by Frizzo.104 It should be noted that, during the preparation of this review article, a paper was published entitled “Orthoesters: multiple role players in organic synthesis”, by Nazarian and Dabiri.105
Due to the central role of orthoesters and their comprehensive applications, the authors considered the literature reports of usage of orthoesters according to several classes of organic transformations. The initial classification is based on the reaction media: solvent-free conditions, aqueous medium and organic solvents. Each part is also subdivided according to the structures of the achieved products. This dual classification demonstrates the importance of the reaction media in progressing the preparation of each class of organics, because the nature of the solvent and its interaction with the reaction components and reagents (orthoesters and other participants) affect the route of the process and may change it seriously. The authors hope the review will highlight the significant role of orthoesters in organic and pharmaceutical chemistry as well as in biosciences and be a proper guide to designing new procedures and approaches to obtain new organics to reach a better life.
2. Orthoester reactions under solvent-free conditions
Performing organic reactions under solvent-free conditions has been notable recently for chemists due to its individual eco-environmental and economical features. In fact, in the absence of solvents the substrates are closer, allowing the right effectual contacts to let the reaction happen. This process, that elevates the reactivity of the substrates, diminishes the reaction time and energy usage, and performs the procedures more selectively in a safe and clean manner. In addition, the absence of hazardous solvents is in line with the rules of green chemistry, turning the transformation into an eco-friendly process. So, because of the importance of the reaction being accomplished under solvent-free conditions, below we focus on reports of utilizing orthoesters in various classes of organic synthesis. The presence of about 150 references in this area confirmed the significance of this method.
2.1. Synthesis of ethoxymethylenes
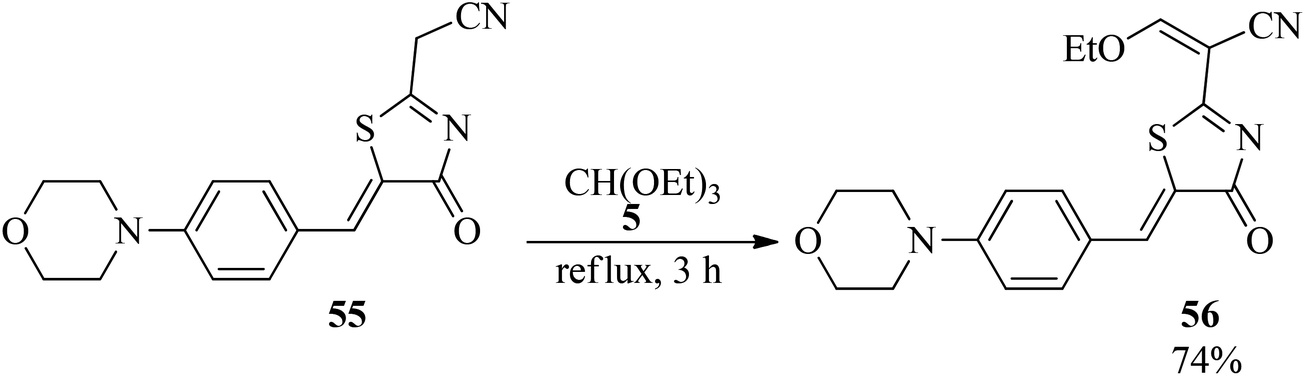
In 2004, Lamphon et al. synthesized 3-ethoxy-2-[5-(4-(morpholin-4-yl)benzylidenyl)-4-oxo-4,5-dihydro-thiaol-2-yl]acrylonitrile containing an ethoxymethylene moiety (56) in 74% yield via the condensation of TEOF (5) with 2-cyanomethyl-5-[4-(morpholin-4-yl)benzylidenyl]-4,5-dihydro-4-thiazolinones (55) under reflux conditions within 3 h (Scheme 13).106
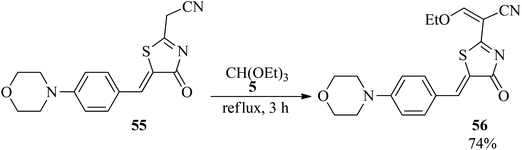 |
| | Scheme 13 The synthesis of 3-ethoxy-2-[5-(4-(morpholin-4-yl)benzylidenyl)-4-oxo-4,5-dihydro-thiaol-2-yl]acrylonitrile by TEOF. | |
2.2. Synthesis of ethoxymethylene amino derivatives
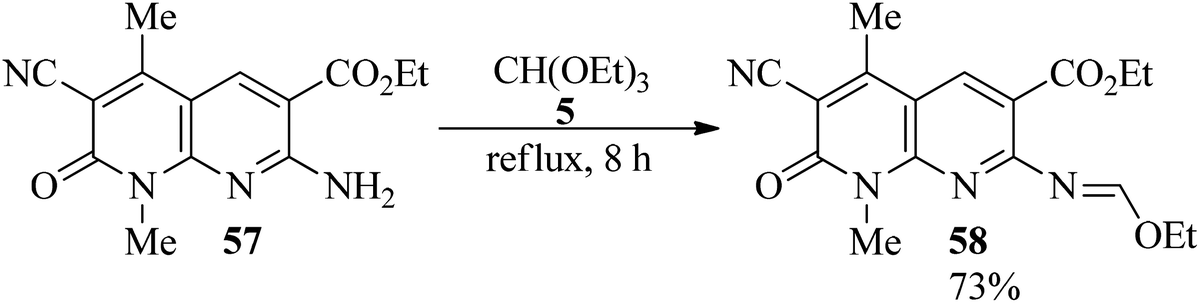
In 2001, Mekheimer presented a novel technique for the synthesis of ethoxymethyleneamino derivative (58)107–110 in high yield (73%) via the condensation of a 1,8-naphthyridin-2-one ring system (57) with TEOF under reflux conditions within 8 h.109 Condensation of TEOF111 with 3-amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carbonitrile112 also generated a Schiff base (Scheme 14).113
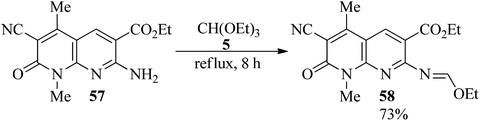 |
| | Scheme 14 The synthesis of ethoxymethyleneamino derivatives. | |
2.3. Synthesis of acetamides
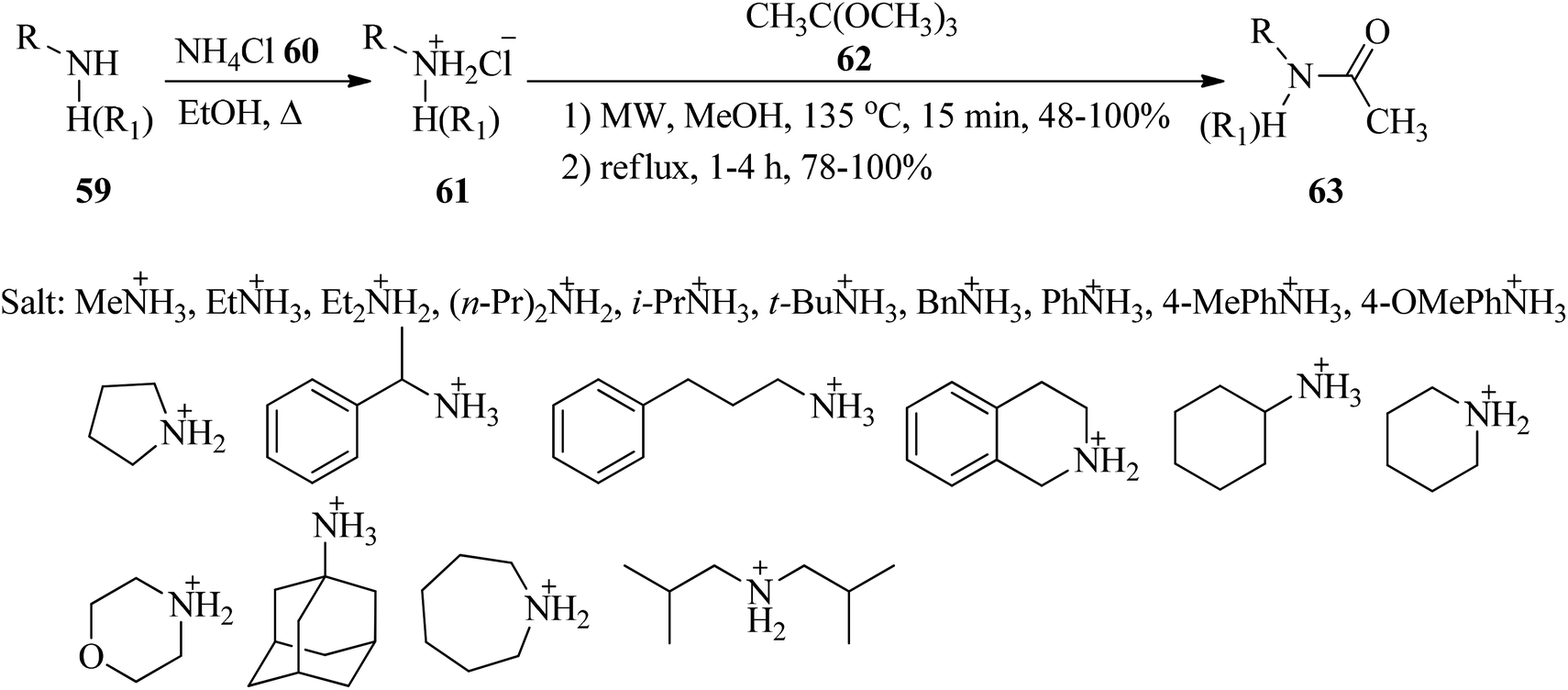
In 2015, Grandi et al. presented an efficacious, fast and high-yielding method for the conversion of amine hydrochloride salts (61) to acetamides (63). Initially, the reaction between equimolar amounts of the primary and secondary amines (59) with ammonium chloride (60) under heating in ethanol obtained amine hydrochloride salts (61) in a 2 h period with excellent yields (>90%)114 which were transformed into their corresponding acetamides (63) via the acetylation reaction with trimethyl orthoacetate (62)115,116 by two different pathways: (a) using microwave irradiation (Biotage Initiator™ Sixty EXP) at 135 °C in methanol, and (b) under conventional heating conditions through refluxing. The results demonstrated that MW-assisted reaction promotion with shorter reaction times (15 min) than conventional heating (1–4 h) (Scheme 15).116
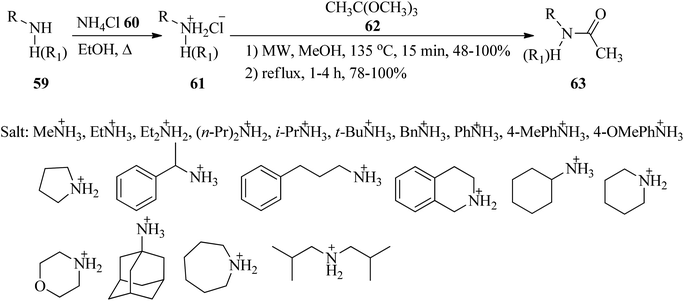 |
| | Scheme 15 The synthesis of acetamides from amine hydrochloride salts and trimethyl orthoacetate. | |
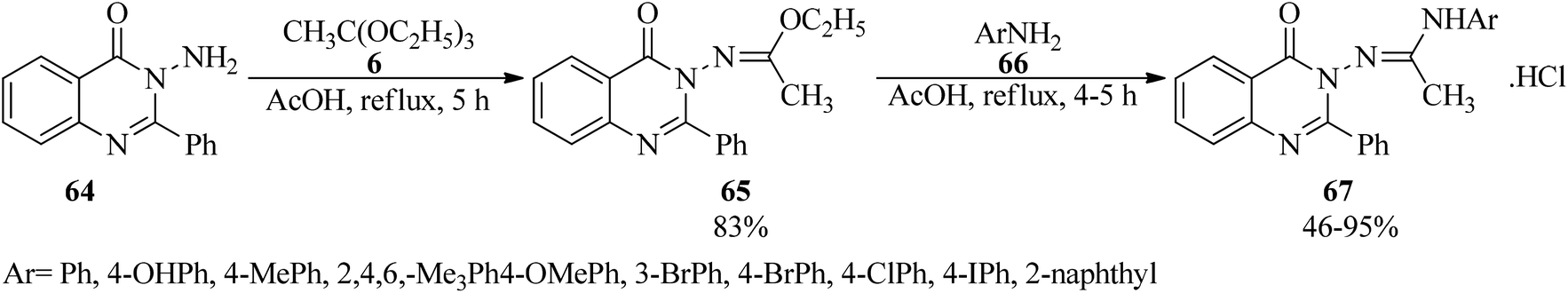
In 2010, Khan et al. presented a simple methodology for the synthesis of unsymmetrical N,N′-disubstituted acetamidines (67).117,118 In this paper, ethyl(1E)-N-(4-oxo-2-phenylquinazolin-3(4H)-yl)ethanimidoate (65) was first prepared via the condensation reaction of 3-amino-2-phenyl-4(3H)-quinazolinone (64) with TEOAc (6) using acetic acid under reflux conditions. After completion of the reaction, the mixture was poured into ice-cold water. The resultant precipitation was filtered, washed with water and recrystallized from ethanol to produce (65) as a white crystalline solid in 83% yield within 5 h.119 In the next step, N-(4-oxo-2-phenyl-3(4H)-quinazolinoyl)-N-(aryl)acetamidines (67) were obtained through the condensation reaction of (65) with substituted aromatic amines (66) by glacial acetic acid at reflux condition in moderate to excellent yields (46–95%) for 4–5 h (Scheme 16). It was found that electron-donating aromatic amines gave the corresponding acetamidines in good yields while the reaction failed with electron-withdrawing aromatic amines. The reaction proceeded selectively with just primary aromatic amines, including electron-donating substituents.118 On the other hand, symmetrical N,N′-disubstituted acetamidines were produced by a microwave-assisted (850 W) condensation reaction of triethyl orthoacetate with the anilines in acetic acid within very short reaction times (5–7 min). High yields and high purity of the product were obtained by the removal of ethanol as a by-product, via placing a receiver between the round-bottom flask and the condenser under heating by microwave irradiation at 850 W for 2–3 min.120
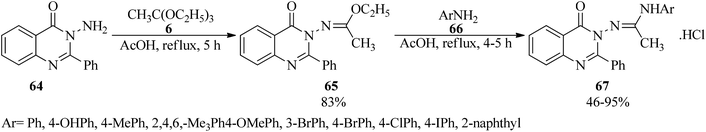 |
| | Scheme 16 The preparation of N-(4-oxo-2-phenyl-3(4H)-quinazolinoyl)-N-(aryl)acetamidines begins by forming amino quinazolinone and TEOAc. | |
2.4. Synthesis of imines
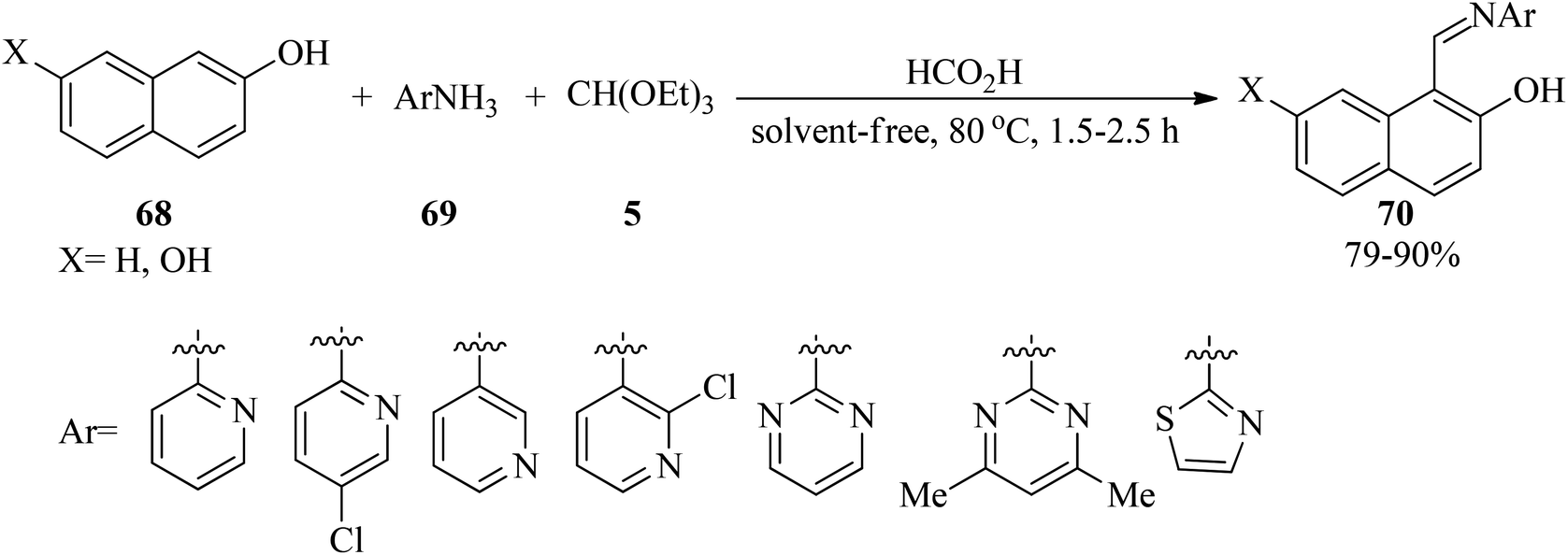
In 2012, Olyaei et al. synthesized novel anil-like products (70)121,122 via the one-pot three-component condensation reaction of naphthols (68), TEOF (5), and heteroaryl amines (69) by formic acid as catalyst under solvent-free conditions at 80 °C in high yields (79–90%) within 1.5–2.5 h (Scheme 17). Investigation of 1H NMR and FT-IR exhibited that the products are in tautomeric equilibrium between the enol–imine and keto–enamine forms. Moreover, intramolecular hydrogen bonds were observed in the synthesized Schiff bases. In this research, 14-(2-hydroxynaphthyl)-14H-dibenzo[a,j]xanthene was also prepared through the treatment of 2-naphthol with TEOF in the absence of heteroaryl amines under solvent-free conditions at 80 °C.122
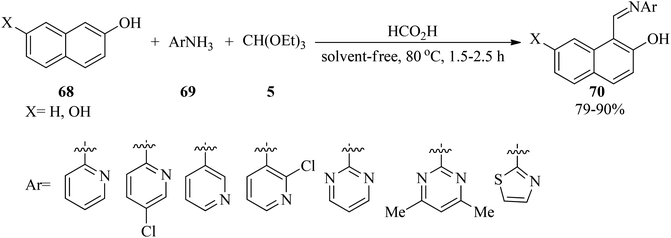 |
| | Scheme 17 Anil-like product formation utilizing TEOF. | |
2.5. Synthesis of imidates
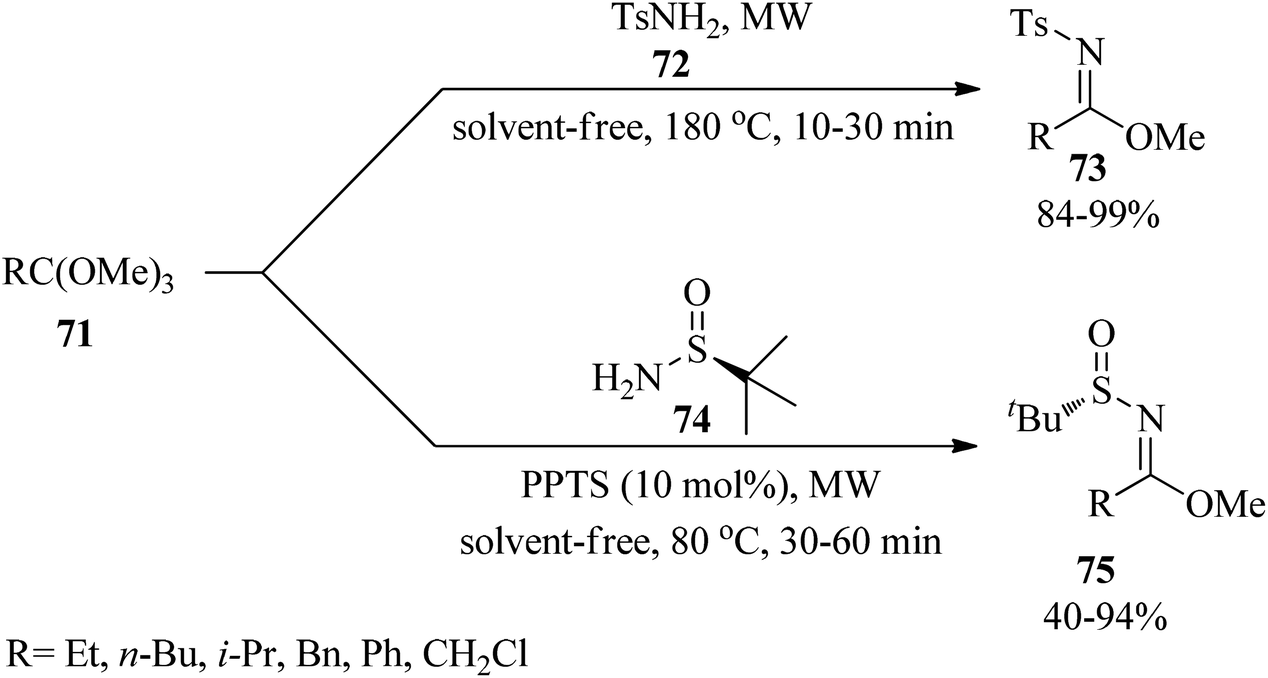
In 2018, Verrier et al. reported metal and solvent-free microwave-assistance for the convenient preparation of N-sulfonyl and N-sulfinyl imidate derivatives by a leaky septum filled up with 4 Å MS which was placed 3 cm above the top of the microwave-tube. The treatment of trimethylorthoesters (71) with 4-methylbenzenesulfonamide (72) under solvent-free conditions at 180 °C afforded N-sulfonylimidates (73) within 10–30 min. Replacing (72) with tert-butanesulfinamide (74) in the presence of pyridinium p-toluenesulfonate (PPTS) (10 mol%) at 80 °C yielded N-sulfinyl imidates (75) within 30–60 min (Scheme 18). The design of an efficient apparatus with the ability to use molecular sieves to trap methanol, short reaction times, easy purification and isolation of the compounds, and excellent conversion and yields are some advantages of this strategy.123
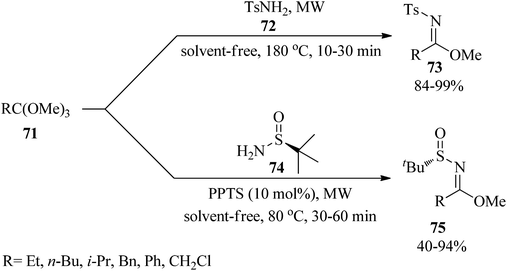 |
| | Scheme 18 Imidate synthetic procedure to form trimethyl orthoesters. | |
2.6. Synthesis of enamines
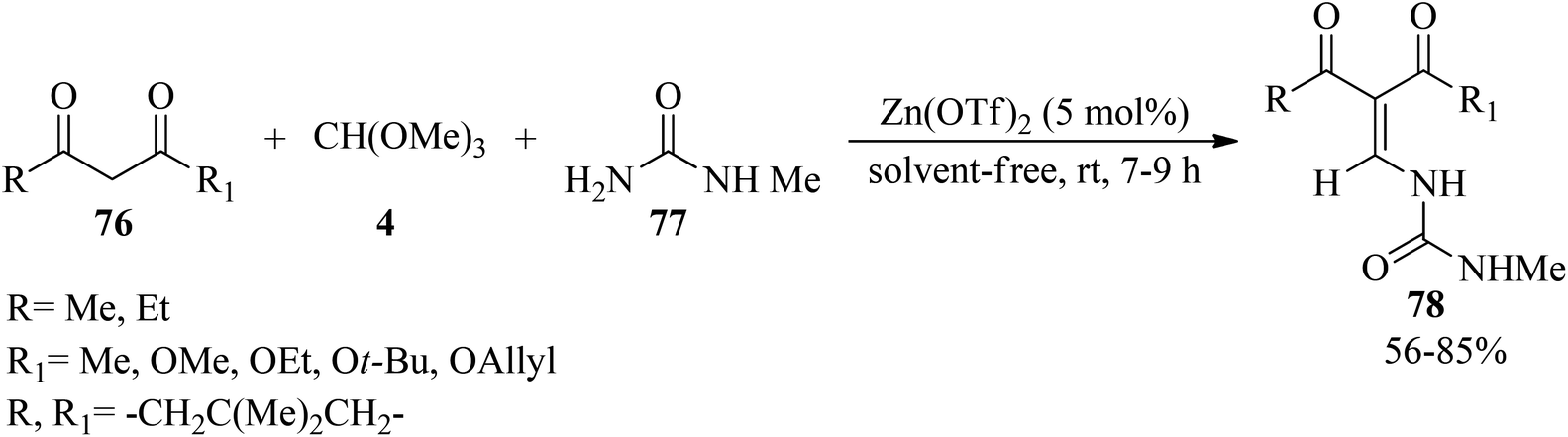
Majee et al. developed an improved method of the Miyashita protocol124 for the synthesis of enamine derivatives (78)125,126 by the three-component condensation reaction of 1,3-dicarbonyls (76), N-methylurea (77) and TMOF (4) using zinc triflate [Zn(OTf)2, (5 mol%)] under solvent-free conditions at room temperature within 7–9 h in good yields (56–85%) (Scheme 19).126
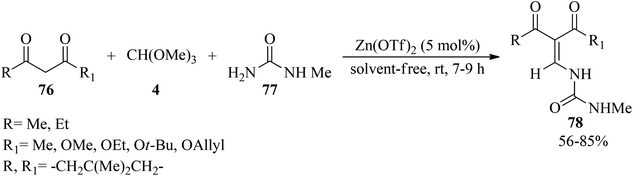 |
| | Scheme 19 Enamine synthesis by TMOF. | |
2.7. Synthesis of formamidines
Mazloumi et al. originated and characterized TiO2-[bip]-NH2+HSO4− as an effective and eco-friendly catalyst through the modification of nanoporous TiO2 with bis-3-(trimethoxysilylpropyl)-ammonium hydrogen sulfate and then its catalytic activity was investigated for the generation of N,N′-diarylformamidines (80)127,128 via the N-formylation reaction of anilines (79) with TEOF (5) under solvent-free conditions at 60 °C in very short reaction times (3–40 min) in high to excellent yields (83–100%) (Scheme 20). In this research, the formation of the unsymmetrical diarylformamidines or aliphatic formamidines was unsuccessful.128
 |
| | Scheme 20 The generation of N,N′-diarylformamidines with TEOF. | |
2.8. Synthesis of lactones
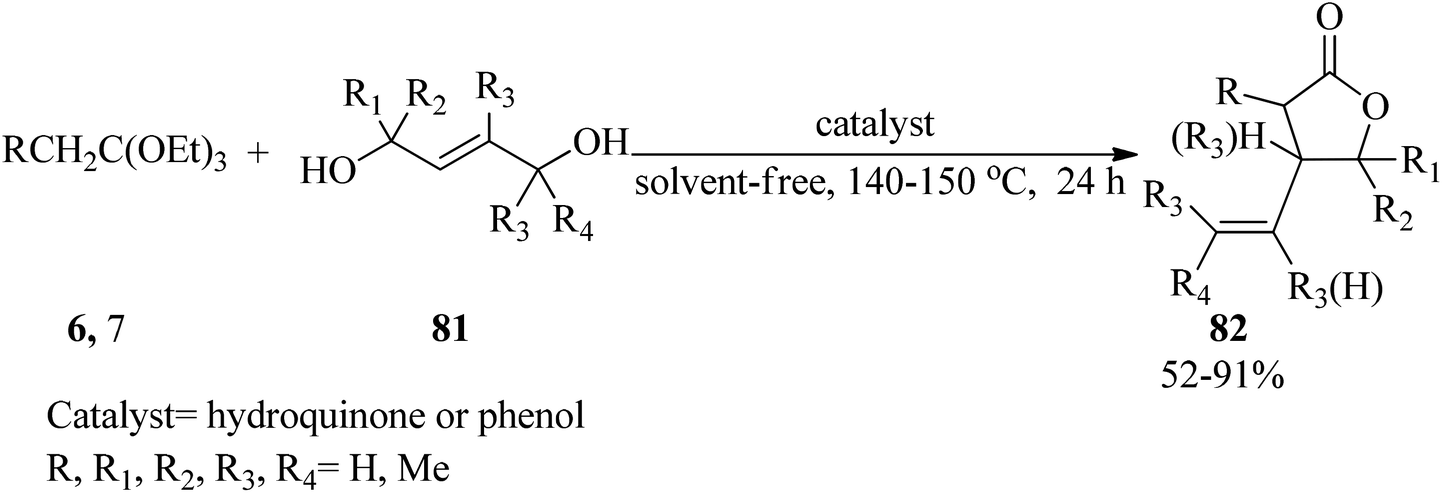
The condensation reaction of trans-2-alkene-1,4-diols (81) with triethyl orthoesters (6, 7) in 1:2 molar ratio was accomplished by utilizing catalytic amounts of hydroquinone or phenol under solvent-free conditions at 140–150 °C to give γ-lactones containing a vinylic substituent on the β-position (82) in 52–91% yields within 24 h (Scheme 21).129
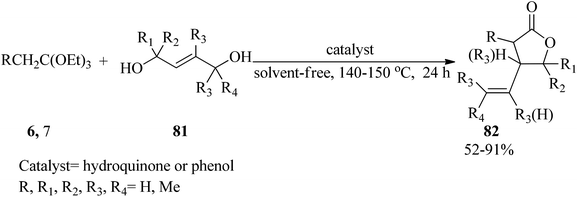 |
| | Scheme 21 The preparation of some classes of γ-lactones. | |
2.9. Synthesis of aminomethylene bisphosphonates
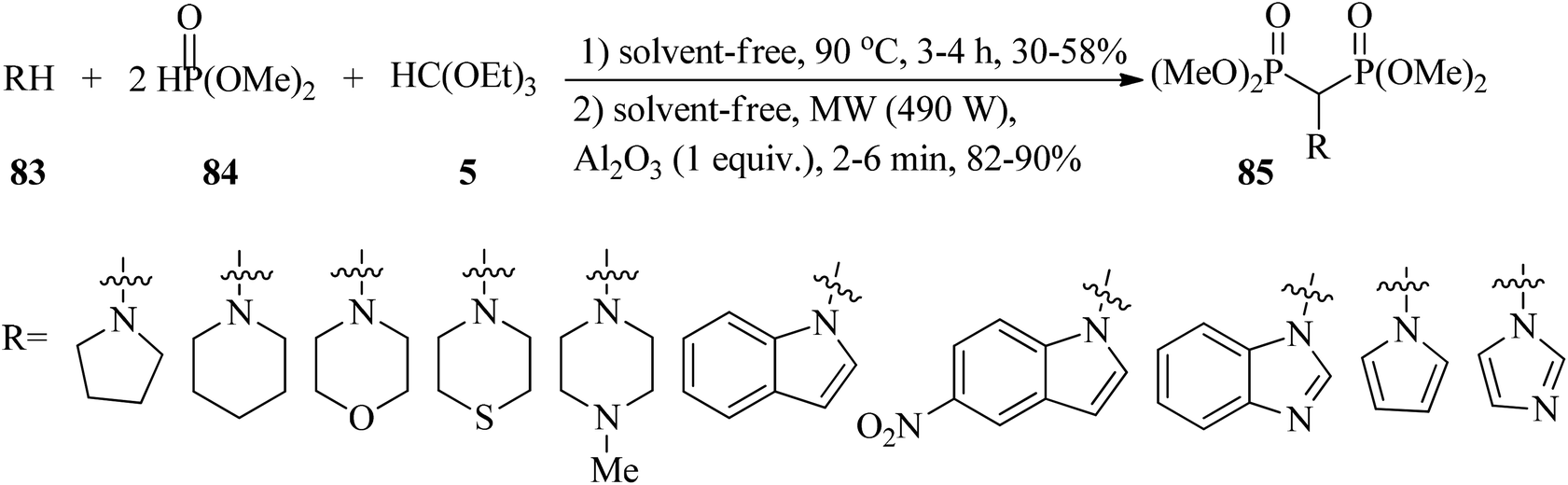
In 2011, Reddy et al. designed an easy, efficient, and green procedure for the synthesis of amino methylene bisphosphonates (85)130–135 via the one-pot three-component condensation reaction of amines (83), TEOF (5), and dimethyl phosphite (84) using an alumina solid support (Al2O3) by microwave irradiation (490 W) in excellent yield (82–90%) for 12 min in two intervals. In addition to the microwave irradiation manner, the reaction was also performed under conventional thermal heating (90 °C, 3–4 h, 30–58%) (Scheme 22). The results revealed shorter reaction times and higher yields using microwave-assistance.135 A similar transformation occurred via the treatment of primary amines, diethyl phosphite, and TEOF using amberlyst-15 under solvent-free conditions at room temperature within 90 min in good to excellent yields (70–95%). All compounds were screened for in vitro antioxidant (by nitric oxide, DPPH radical scavenging, H2O2 methods),136–139 and antimicrobial activities.140
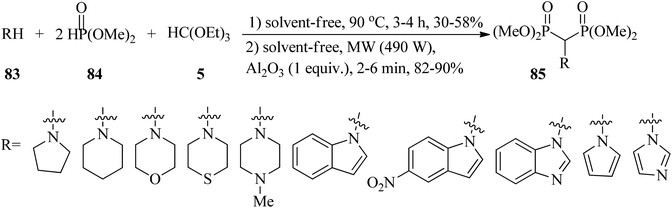 |
| | Scheme 22 A three-component route to obtain amino methylene bisphosphonates. | |
2.10. Synthesis of aminomethylenebisphosphonic acids
In 2017, some aminomethylenebisphosphonic acids (88) were prepared by Miszczyk et al. through the reaction of 3-amino-1,2,4-triazole (86) with diethyl phosphite (87) and TEOF (5), in a 1:2:4 molar ratio (Scheme 23). The products are powerful inhibitors of the activity of J774E cells. So they can be used in antiosteolytic therapy, and are equipotent to the popular drug zoledronate and exhibit higher activity than the drug incadronate.141,142
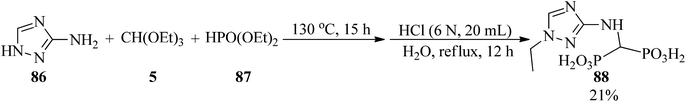 |
| | Scheme 23 The synthesis procedure for biologically active aminomethylenebisphosphonic acid. | |
2.11. Synthesis of indole derivatives
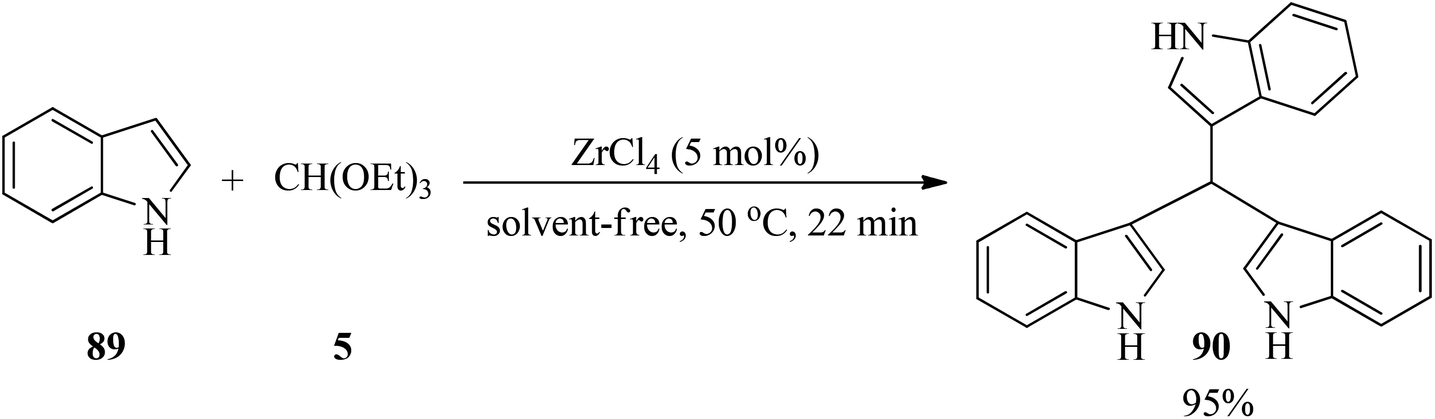
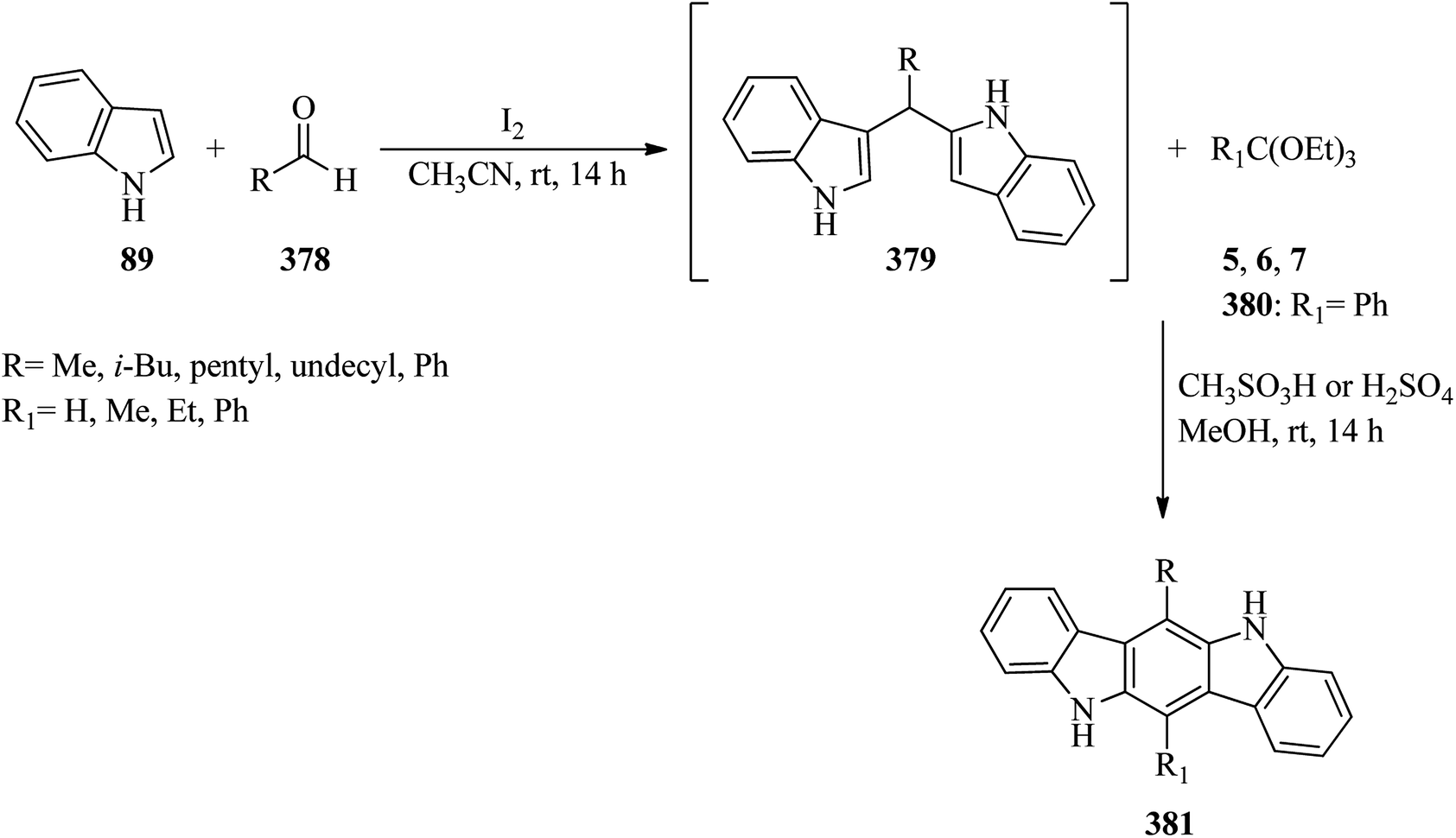
Zirconium(IV) chloride (ZrCl4, 5 mol%) catalyzed the electrophilic substitution reaction of indole (89) with TEOF (5) in a 3:1 molar ratio under solvent-free conditions at 50 °C to obtain tris(indolyl)methane (90)143 in a short reaction time (22 min) in excellent yield (95%) (Scheme 24).144
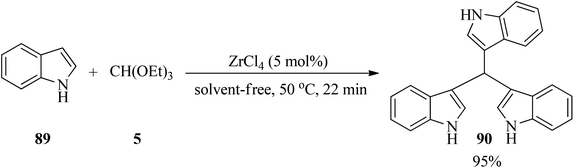 |
| | Scheme 24 Tris(indolyl)methane synthesis. | |
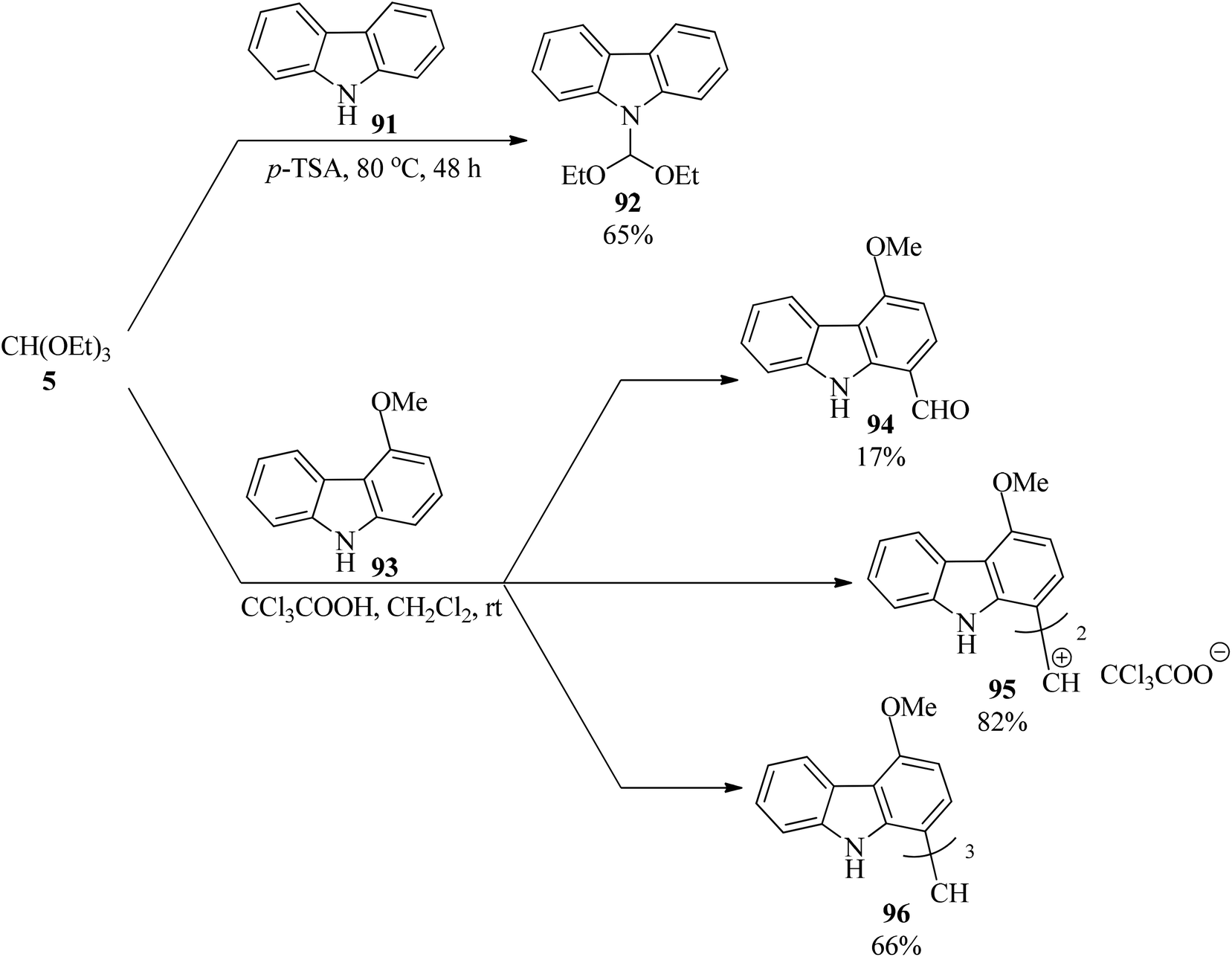
The regioselective reaction of carbazole (91) with TEOF (5) using p-TSA under solvent-free conditions at 80 °C gave 9-(diethoxymethyl)carbazole (92) in 65% yield in 48 h. On the other hand, 4-methoxycarbazole (93) was regioselectively functionalized at the C1 position via reaction with (5) in the presence of trichloroacetic acid in dichloromethane at room temperature to furnish various carbazole derivatives, such as 4-methoxycarbazole-1-carboxaldehyde (94), bis[4-methoxycarbazol-1-yl]methylium trichloroacetate (95), tricarbazolylrnethane (96) according to the reaction conditions (Scheme 25).145
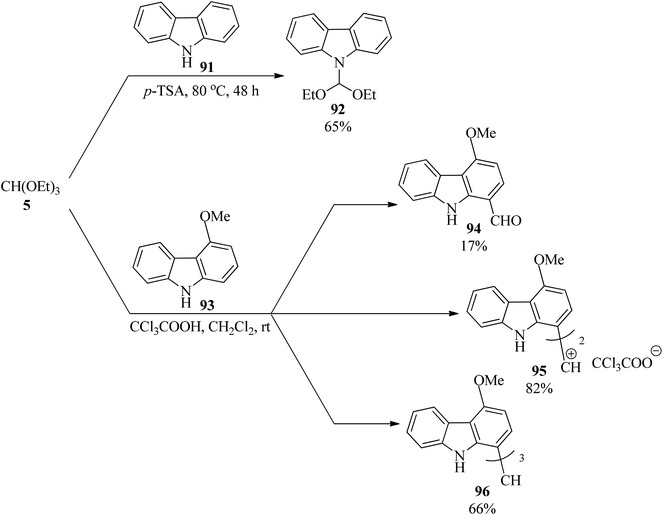 |
| | Scheme 25 The preparation of various heterocycles containing an indole moiety using TEOF. | |
2.12. Synthesis of triaroyl methanes
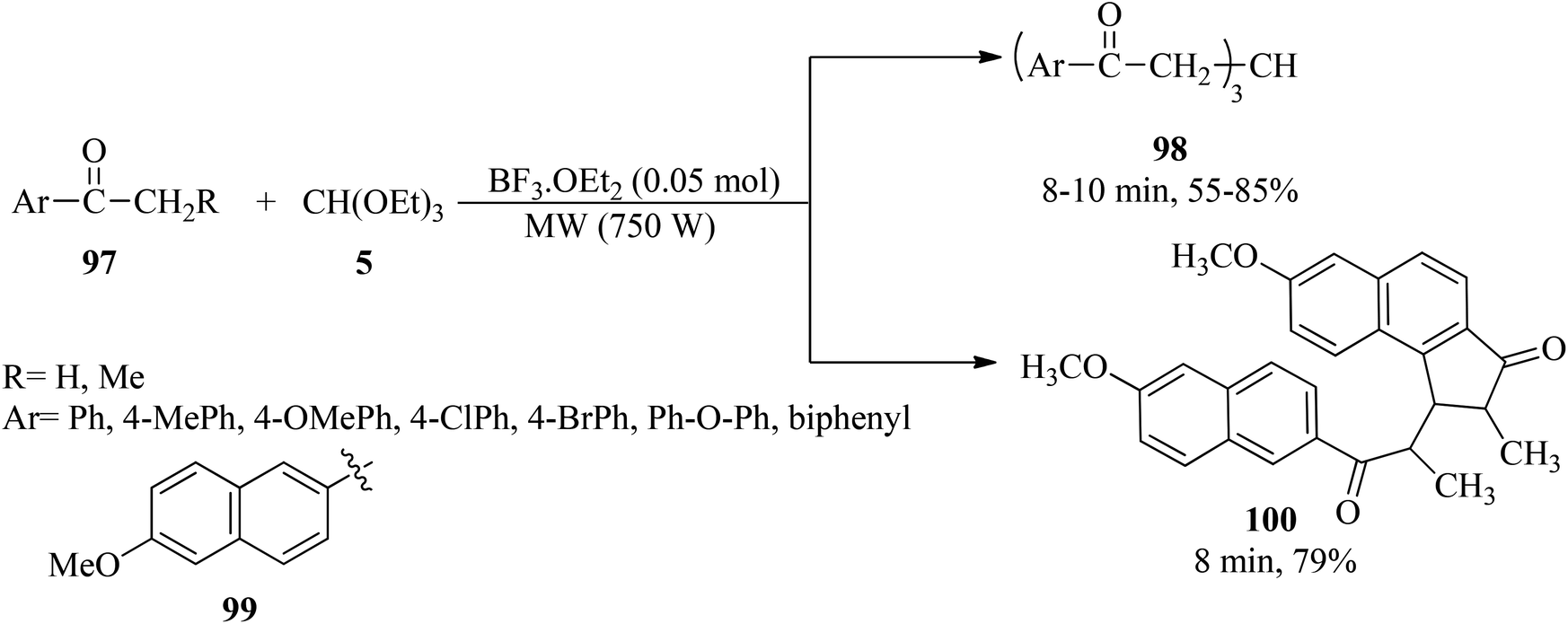
Boron trifluoride etherate liquid (BF3·OEt2, 0.05 mol) catalyzed the condensation reaction of aryl ketones (97) with TEOF (5) under microwave irradiation to obtain triaroyl methanes (98) in 55–85% yields for 8–10 min. It was found that a substrate containing a bulky group such as (99) afforded a fused cyclopentenone ring product (100) in 79% yield within 8 min (Scheme 26).146
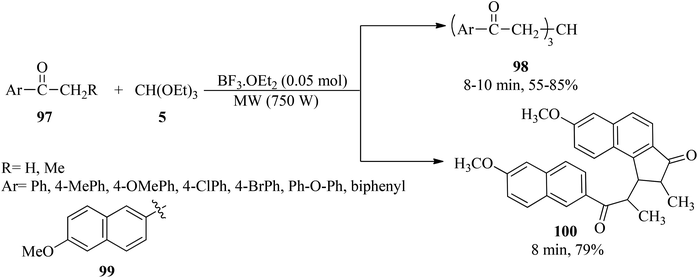 |
| | Scheme 26 The triaroyl methane synthetic procedure. | |
2.13. Synthesis of 5,15-diphenyl-10,20-dimethyl porphyrins
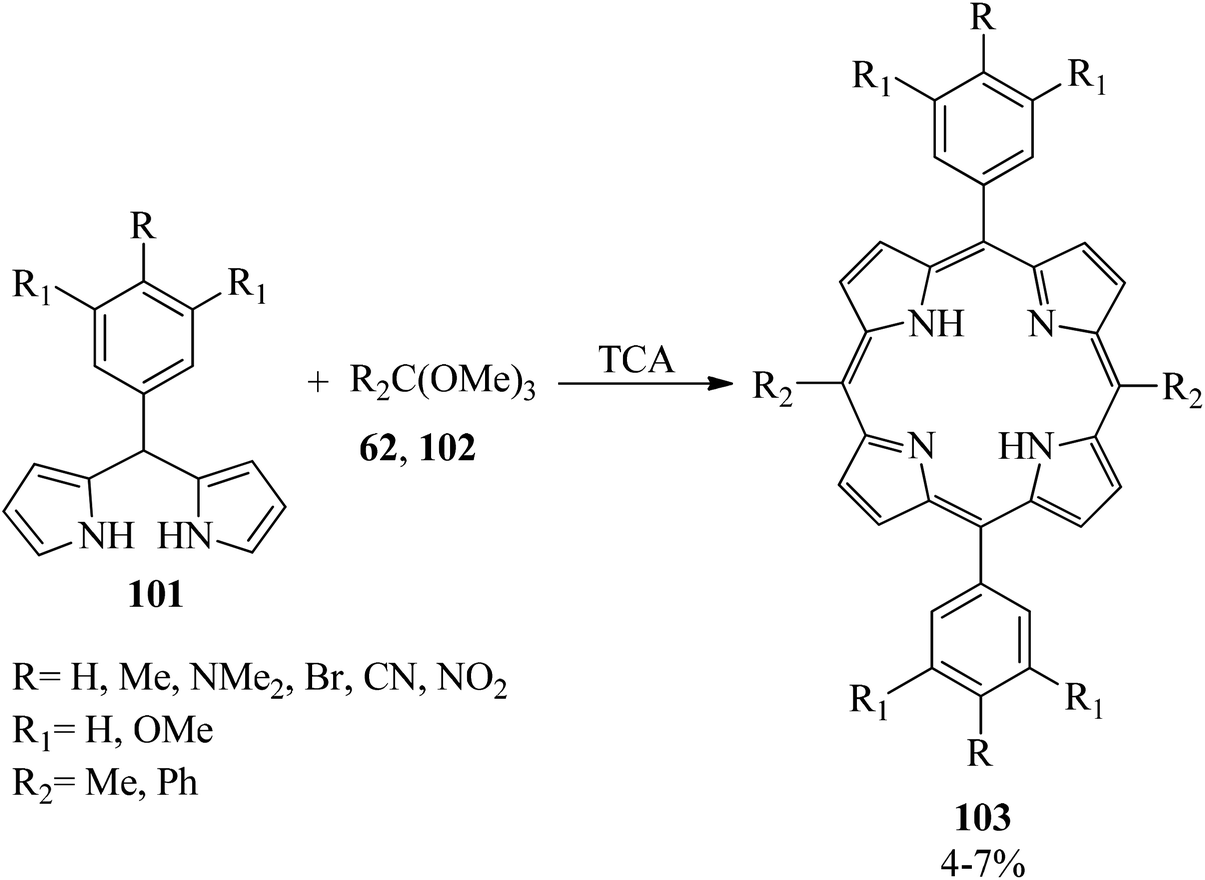
In 2003, the reaction of 5-phenyldipyrromethanes (101) with orthoesters (102) was presented for the generation of 5,15-diphenyl-10,20-dimethyl porphyrins (DPPs) (103) by trichloroacetic acid (TCA) (Scheme 27). Notably, trimethyl orthobenzoates as the bulky orthoesters produced scrambled products. Also, the 5-phenyldipyrromethane, including strong electron-withdrawing substituents on the phenyl ring, improved the scrambled products.147
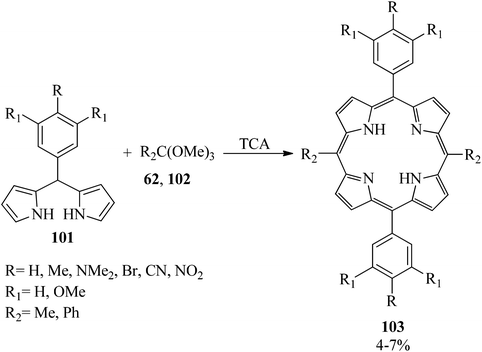 |
| | Scheme 27 Obtaining 5,15-diphenyl-10,20-dimethyl porphyrins (DPPs). | |
2.14. Synthesis of allyl alcohol
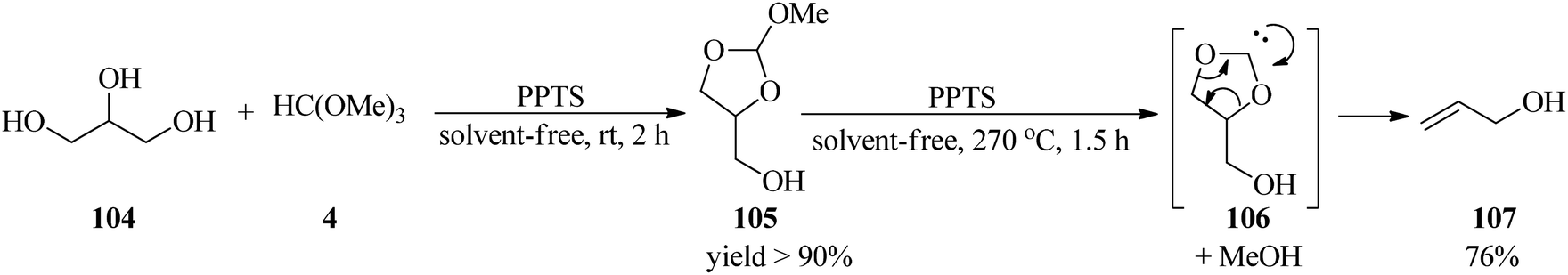
In 2019, Wormann and Maier presented an integrated procedure for the conversion of glycerol (104) to the cyclic orthoester (105) in high yield through the reaction with TMOF (4) by a catalytic amount of PPTS at room temperature for 2 h, which was converted into allyl alcohol (107) under pyrolysis conditions at 270 °C using PPTS under solvent-free conditions. This reaction progressed via the disintegration of the orthoester (105) into carbene (106) and recoverable methanol, which was followed by decarboxylation to produce the mentioned product (107) in high yield (76%) within 1.5 h (Scheme 28).148
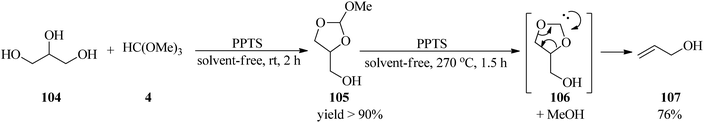 |
| | Scheme 28 Allyl alcohol preparation from glycerol and TMOF. | |
2.15. Orthoester exchange reaction
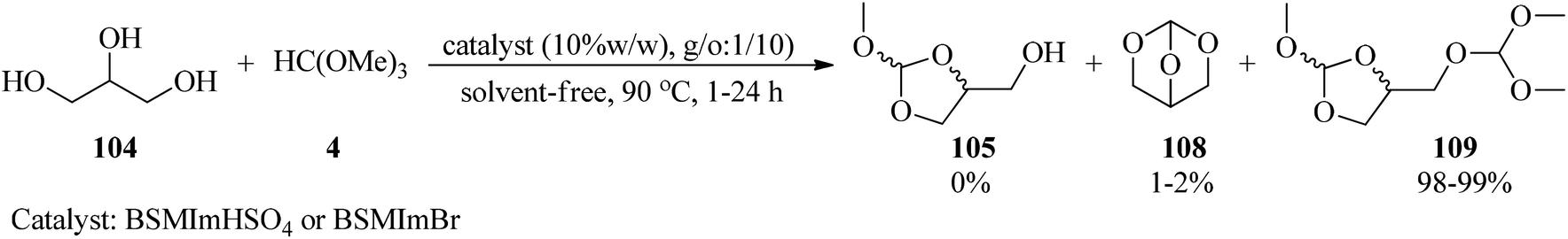
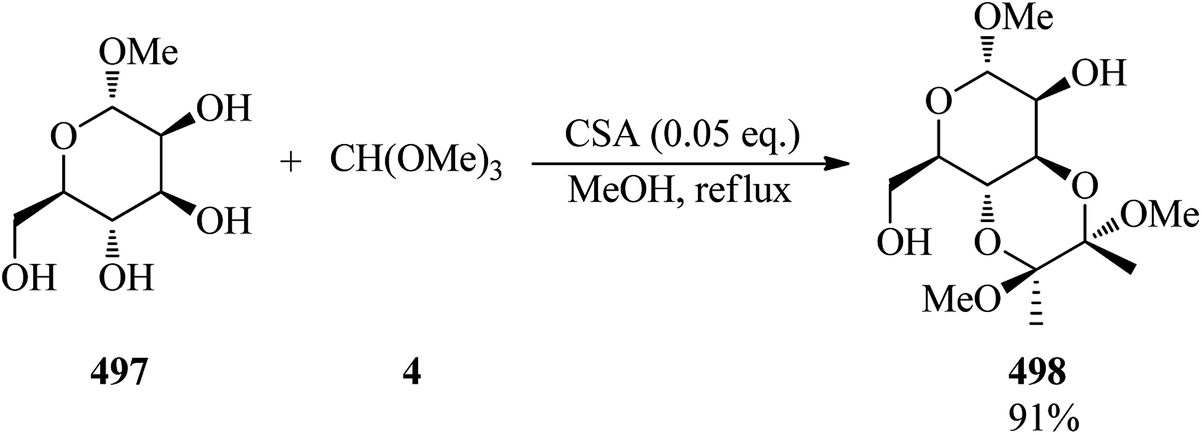
Calmanti et al. investigated the reactivity of glycerol (104) with TMOF (4) with the purpose of developing a credible synthetic strategy for glycerol valorization. It was found that the treatment of glycerol with TMOF under various conditions provided a mixture of compounds (105), (108), (109). Hence, in order to synthesize 4-(dimethoxymethoxy)methyl)-2-methoxy-1,3-dioxolane (109), different parameters, such as different molar ratios of TMOF, temperature, and the absence or presence of various catalysts, were examined. Significantly among different amounts of glycerol and TMOF, the best result was obtained in a molar ratio of 1:10 at 90 °C. Moreover, product (109) was selectively synthesized in the presence of a catalyst as well as in catalyst-free conditions, taking advantage of the thermodynamically controlled equilibrium between intermediates. To probe the effect of a catalyst, various catalysts, such as basic catalysts (K2CO3, ([P1888]CH3OCO2−) or Brønsted and Lewis acid catalysts were employed in which both Brønsted and Lewis acid catalysts progressed this reaction, and specifically Brønsted acidic ionic liquids (BSMImHSO4 and BSMImBr) were the most efficient catalysts (Scheme 29).149
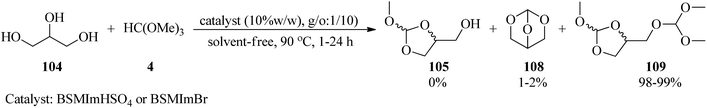 |
| | Scheme 29 The synthesis of 4-(dimethoxymethoxy)methyl-2-methoxy-1,3-dioxolane. | |
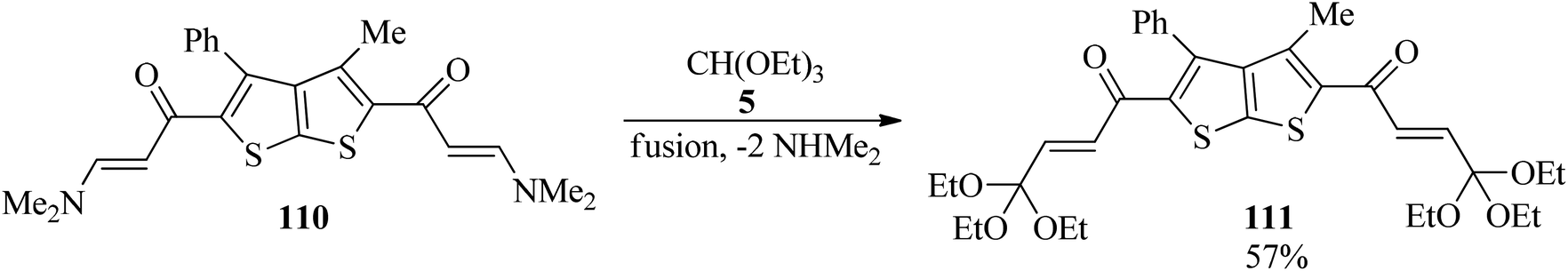
In 2012, the one-pot fusion reaction of bis(enaminone)derivative (110) with TEOF (5) under solvent-free conditions was reported to obtain 1,1′-(3-methyl-4-phenylthieno[2,3-b]thiophene-2,5-diyl)bis(4,4,4-triethoxybut-2-en-1-one) (111) as pale red crystals in relatively high yield (57%) (Scheme 30).150
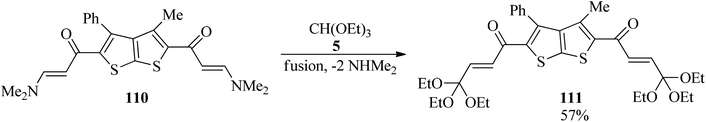 |
| | Scheme 30 Preparing 1,1′-(3-methyl-4-phenylthieno[2,3-b]thiophene-2,5-diyl)bis(4,4,4-triethoxybut-2-en-1-one). | |
2.16. Synthesis of 1,2-dimethyltetramethoxydisilane
The reaction of methyldichlorosilane with TMOF at room temperature afforded dimethoxymethylsilane overnight151,152 in 61% yield. Then, photolysis of dimethoxymethylsilane in the presence of a drop of mercury in the gas phase resulted in 1,2-dimethyltetramethoxydisilane in 81% yield within 8 h.153
2.17. Synthesis of chromanes, chromenes, and thiochromene
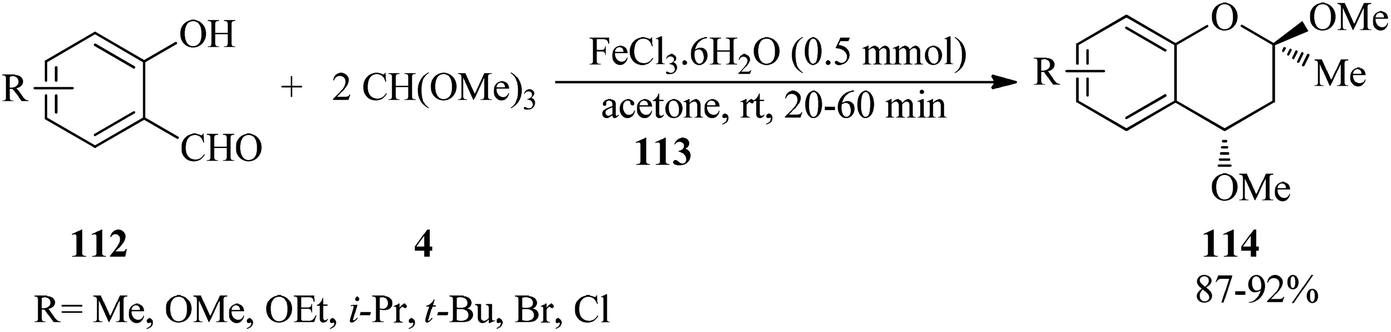
In 2002, Yadav et al. interpreted the highly diastereoselective synthesis of 2,4-dimethoxy-2-methyl-2H-1-benzopyrans (114) via the three-component reaction of o-hydroxybenzaldehydes (112), acetone (113), and trimethyl orthoformate (4) utilizing a catalytic amount of ferric chloride hexahydrate (FeCl3·6H2O, 0.5 mmol) at room temperature in excellent yields (87–92%) for 20–60 min. The only diastereomer bore a trans-relationship between two methoxy groups at the 2- and 4-positions (Scheme 31). Mild reaction conditions, experimental simplicity, easily available substrates, short reaction times, and excellent yields are some of the advantages of this process.154
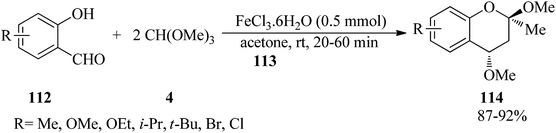 |
| | Scheme 31 The synthesis of 2,4-dimethoxy-2-methyl-2H-1-benzopyrans. | |
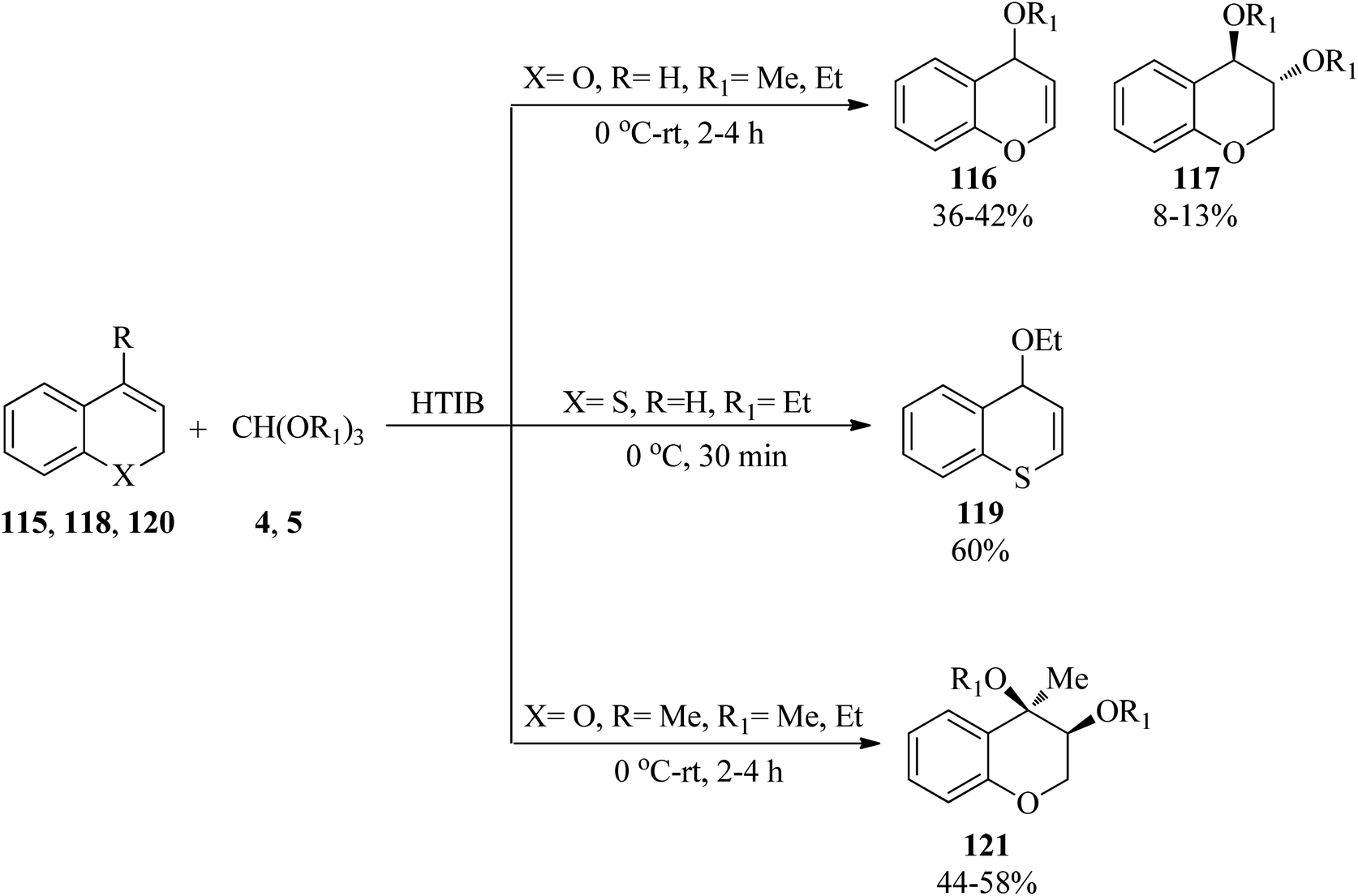
In 2012, Ahmad and Silva Jr investigated the oxidation of different oxygen-containing benzo-fused cycloalkenes with various trialkyl orthoformates (4, 5) in the presence of hypervalent iodine reagent [[hydroxy(tosyloxy)iodo]benzene, HTIB] under solvent-free conditions. It was found that 2H-chromenes (115) generated alkoxy-substituted 4H-chromenes (116) as the main product, along with the trans-addition product (117). On the other hand, 2H-thiochromene (118) and 4-methyl-2H-chromene (120) obtained 4H-thiochromene (119) and cis-3,4-dialkoxy-4-methyl-3,4-dihydro-2H chromenes (121), respectively (Scheme 32).155
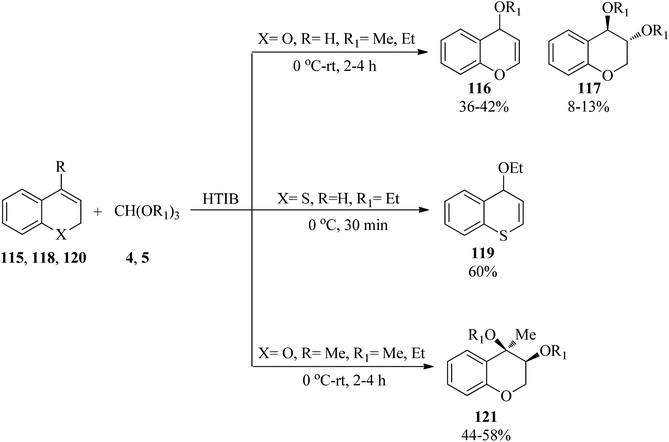 |
| | Scheme 32 The synthesis of different chromenes using trialkyl orthoformates. | |
2.18. Synthesis of coumarin derivatives
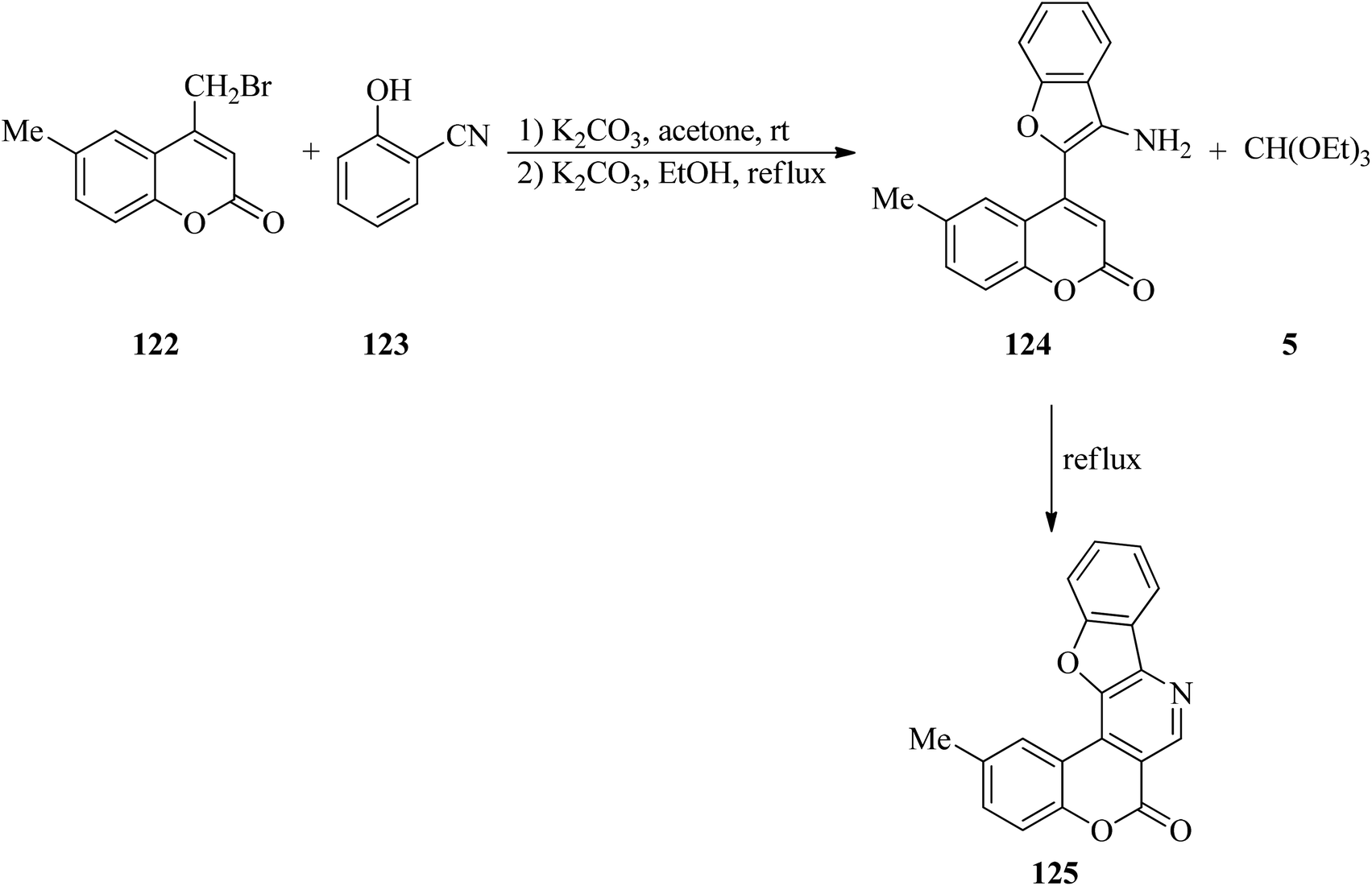
Kulkarni and Sun's research group investigated a three-step sequence, as an array to obtain angularly fused polycyclic heterocycles with coumarin, benzofuran and pyridine rings (125). They started from 4-bromomethylcoumarins (122) and salicylonitrile (123) followed by the addition of TEOF (5) (Scheme 33). Several of these exhibited promising inflammation-inhibiting and anti-microbial properties.156
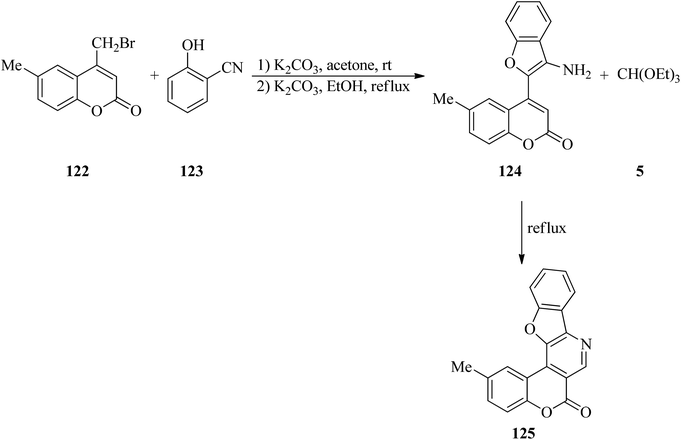 |
| | Scheme 33 The synthesis of fused polycyclic heterocycles utilizing TEOF. | |
2.19. Etherification and esterification reaction, and Johnson–Claisen rearrangement
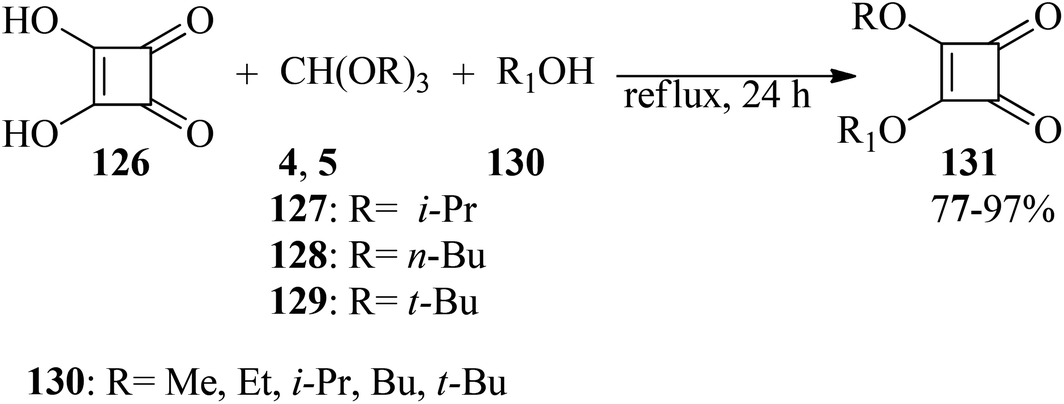
In 1997, Liu and co-workers described a simple and efficient process for the preparation of alkyl squarates (131) through the reaction of squaric acid (126), orthoformates (4, 5, 127–129), and appropriate alcohols (130) under reflux conditions in 77–97% yields (Scheme 34).157
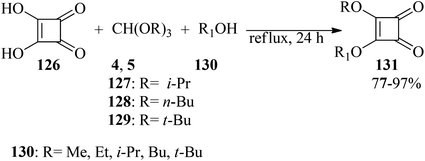 |
| | Scheme 34 Preparation method of alkyl squarates. | |
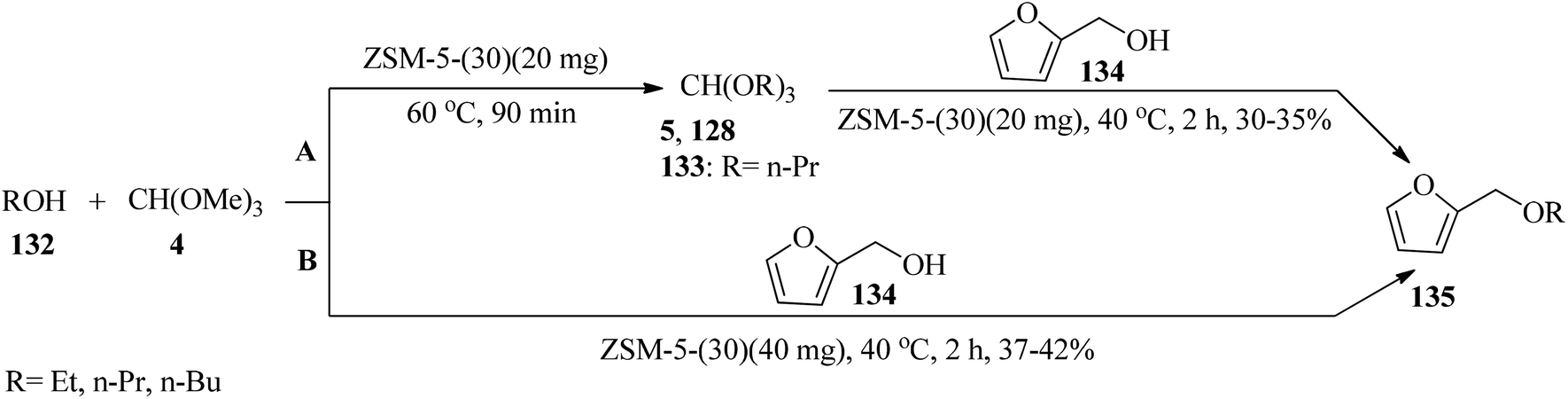
Furfuryl ethers (135) as bio-renewable fuel could be synthesized via two pathways: (a) a sequential reaction method was used in which the appropriate orthoester (5, 128, 133) was synthesized upon treatment with TMOF (4) and an alcohol (132) in the presence of ZSM-5 zeolite (Si/Al ratio = 30:1, ZSM-5-(30)) catalyst (40 mg) at 60–40 °C, followed by the addition of furfuryl alcohol (134) to obtain furfuryl ether (135) bio-fuels. (b) The mentioned products were synthesized through a tandem etherification condensation reaction of (4), an excess amount of alcohol (132), and furfuryl alcohol (134) at 40 °C by ZSM-5-(30) (40 mg) in good yield and good to high selectivity (Scheme 35). It must be mentioned that the low temperature applied improved selectivity by decreasing the production of hydrolysis products, and the competing polymerization reactions resulting in humin by-products.158
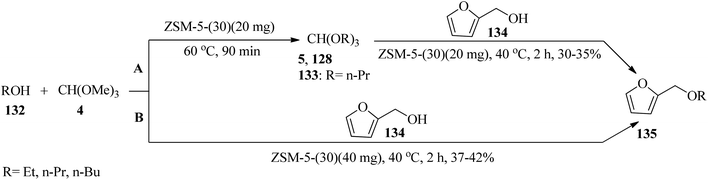 |
| | Scheme 35 The synthesis of furfuryl alcohols. | |
In 2015, Zhang et al. utilized ZrO2 supported Pd(OH)2 (Pd(OH)2/ZrO2) as a new catalytic system for the synthesis of aryl ethers (137) via the reaction of substituted cyclohexanones (136) with orthoesters (4, 5, 127) as both dehydrating and nucleophilic reagents under an atmospheric pressure of oxygen (0.5 MPa) at 140 °C for 6 h (Scheme 36). It was found that in the presence of TMOF (4) reagent, the corresponding aryl ethers were obtained in moderate to good yields. Additionally, replacing other orthoester reagents, such as TEOF (5) and triisopropyl orthoformate (127) with (4) furnished the relevant products in low yields. Comparatively, the weak yields of the products compared to their high conversion were because of the generation of phenols as by-products. Due to the lower reactivity of TIPOF (127) to attain the related product than TMOF (4) or TEOF (5), this orthoester was employed as a dehydrating reagent in the reaction of cyclohexanones with alcohols to provide aryl ethers without forming isopropoxybenzene.159
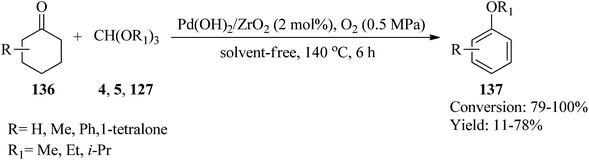 |
| | Scheme 36 The synthesis of aryl ethers. | |
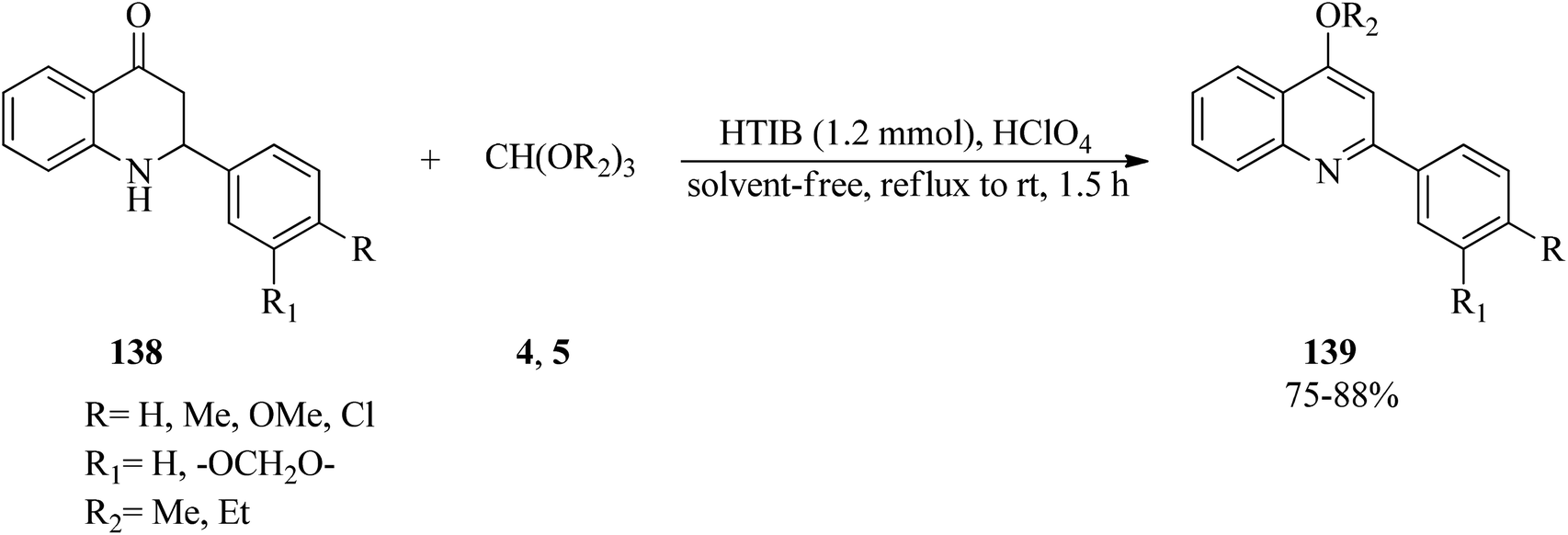
In 1998, Varma and Kumar developed an applicable oxidative method for the synthesis of 4-alkoxy-2-arylquinolines (139) through the reaction of readily available 2-aryl-l,2,3,4-tetrahydro-4-quinolones (138) with orthoformates (4, 5) using the hypervalent iodine oxidative reagent, HTIB, in the presence of a few drops of perchloric acid under solvent-free conditions at reflux to room temperature within 1.5 h in good yields (75–88%) (Scheme 37).160
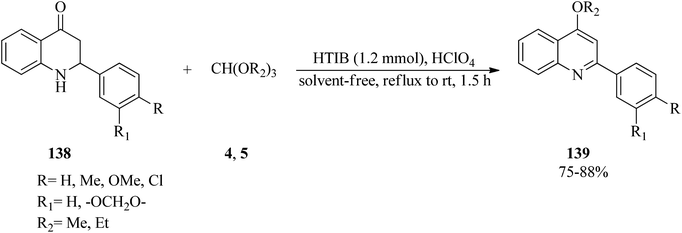 |
| | Scheme 37 The synthesis of 4-alkoxy-2-arylquinolines. | |
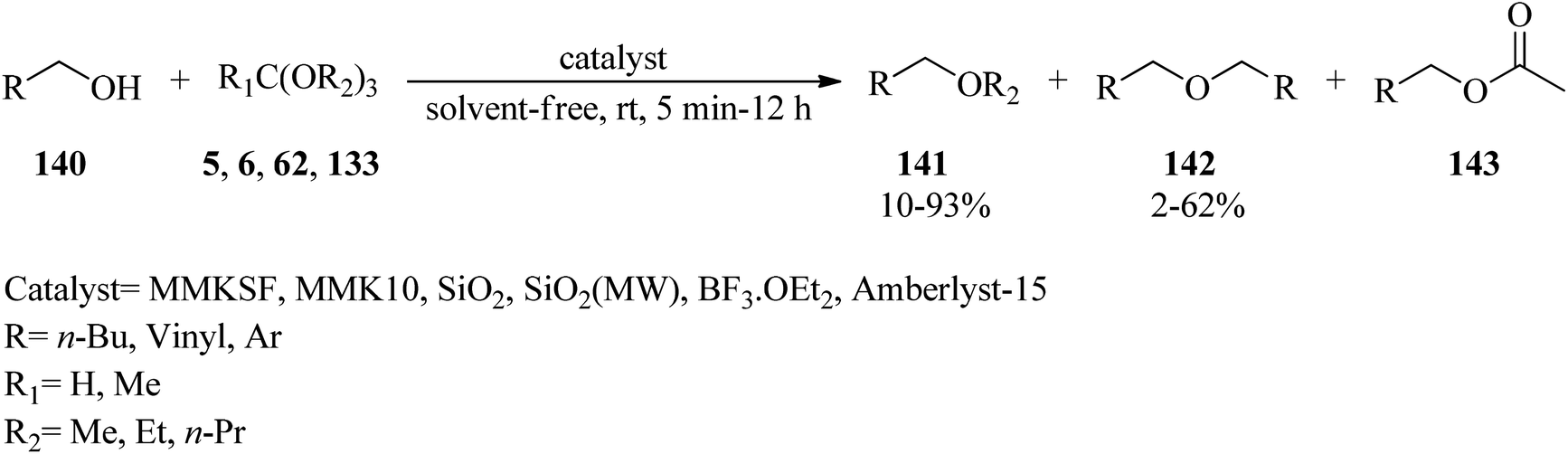
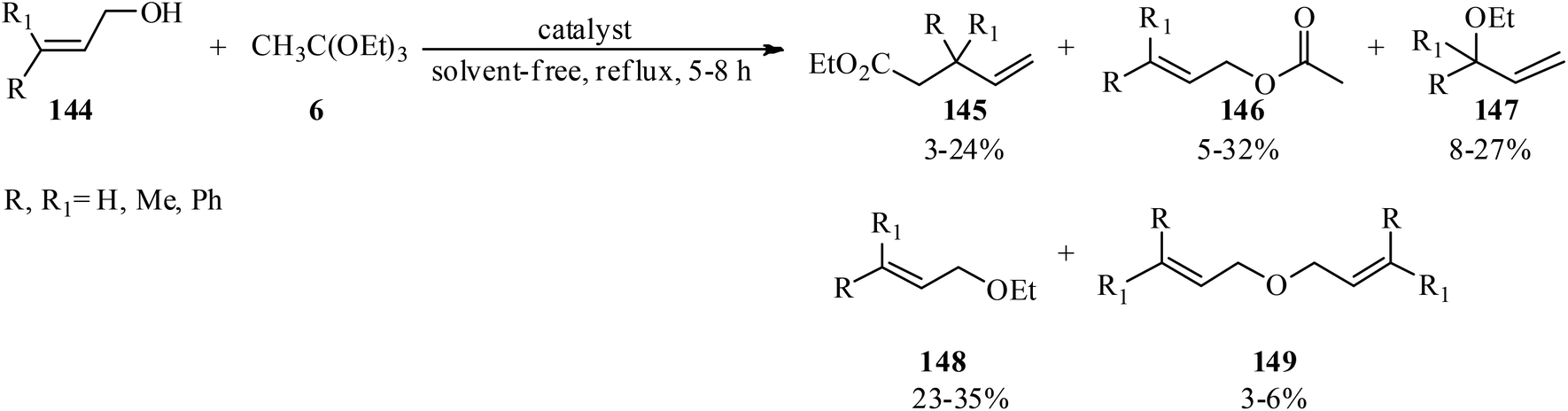
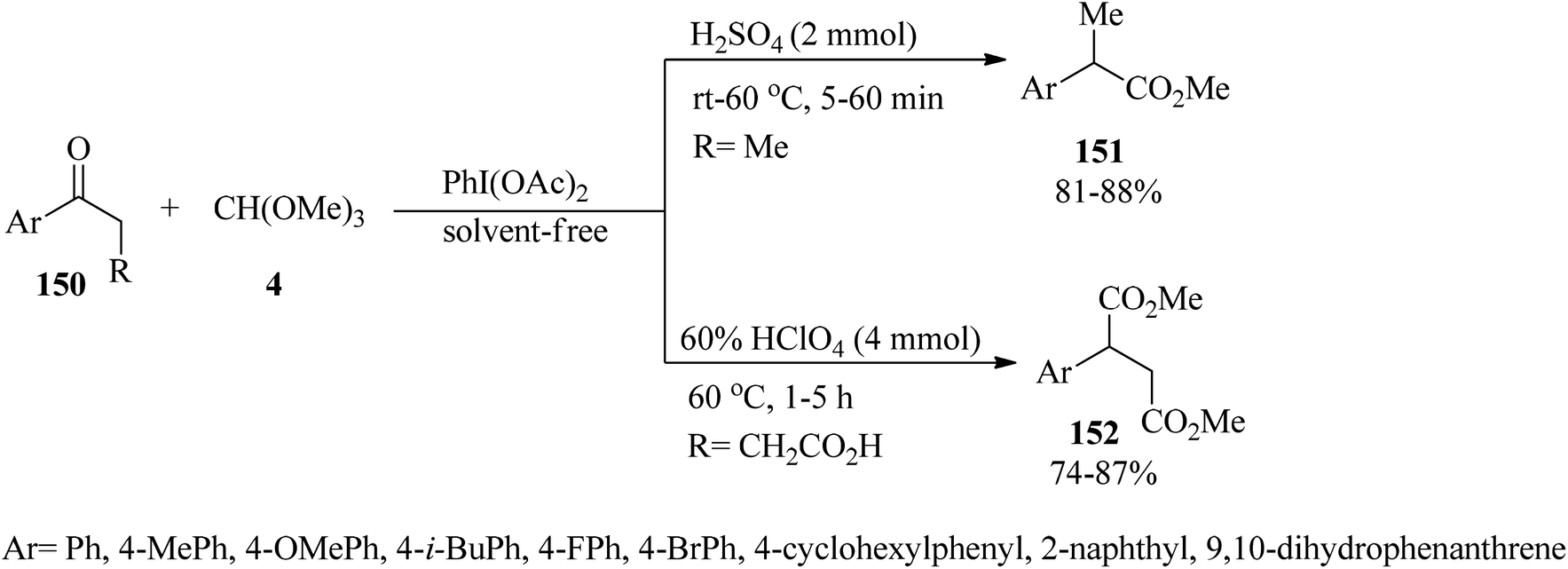
Unsymmetrical ethers (141) were obtained via the reaction of allylic and benzylic alcohols (140) with orthoesters (5, 6, 62, 133) using acidic catalysts such as montmorillonite KSF and K10, BF3·Et2O, SiO2, amberlyst-15 at room temperature under an N2 atmosphere. It is noteworthy that the change in the catalyst was accompanied by an important change in the yields and the type of products obtained, such as dimerized ethers (142) and O-acetylated (143) via competitive reactions (Scheme 38). This reaction was general due to the presence of varied orthoesters, but selective with regard to different allylic and benzylic alcohols. On the other hand, Johnson–Claisen orthoester rearrangement161–164 of allylic alcohols (144) with TEOAc (6) by MMKSF, MMK10, and SiO2 under reflux conditions and N2 atmosphere produced a mixture of Claisen ester (145), acetylated product (146), ethyl ether (147), unsymmetrical terminal ether (148), and dimeric ether (149) in which O-acetylation and O-alkylations were significant side reactions (Scheme 39).164
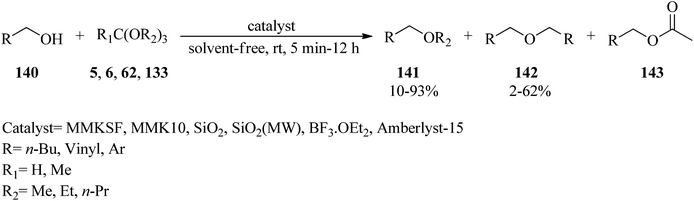 |
| | Scheme 38 The reaction of allylic and benzylic alcohols with orthoesters. | |
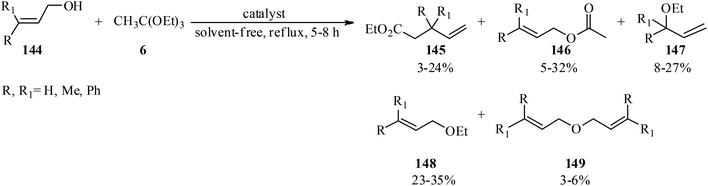 |
| | Scheme 39 The Johnson–Claisen orthoester rearrangement of allylic alcohols. | |
Tamura and co-workers prepared 2-arylalkanoates (151, 152) via oxidative 1,2-aryl migration of alkyl aryl ketones (150) with diacetoxyphenyliodine, as oxidizing reagent, in TMOF (4) under acidic conditions at room temperature or 60 °C in high yields (74–88%) (Scheme 40).165
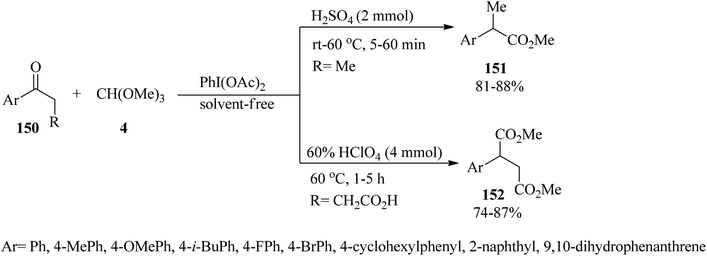 |
| | Scheme 40 2-Arylalkanoate preparation. | |
In 2004, Yoshino and Togo introduced a green, easy, inexpensive, and highly efficient system for the esterification of carboxylic acids (153) with TEOAc (6) by a typical room-temperature ionic liquid, 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF6), at 80–100 °C under neutral and solvent-free conditions in good yields (Scheme 41). The proposed method greatly facilitated the formation of the desired ethyl esters (154), especially for less acidic carboxylic acids, and it could also be used for sterically hindered carboxylic acids, like 2,4,6-triisopropylbenzoic acid or 2,2,2-triphenylacetic acid, and for amino acid without any racemization. Notably, replacement of trimethyl orthoacetate with TEOAc exhibited similar reactivity. On the other hand, the utilization of [bmim]BF4 instead of [bmim]PF6, as an ionic liquid, revealed the same results.166
 |
| | Scheme 41 The esterification of carboxylic acids with TEOAc. | |
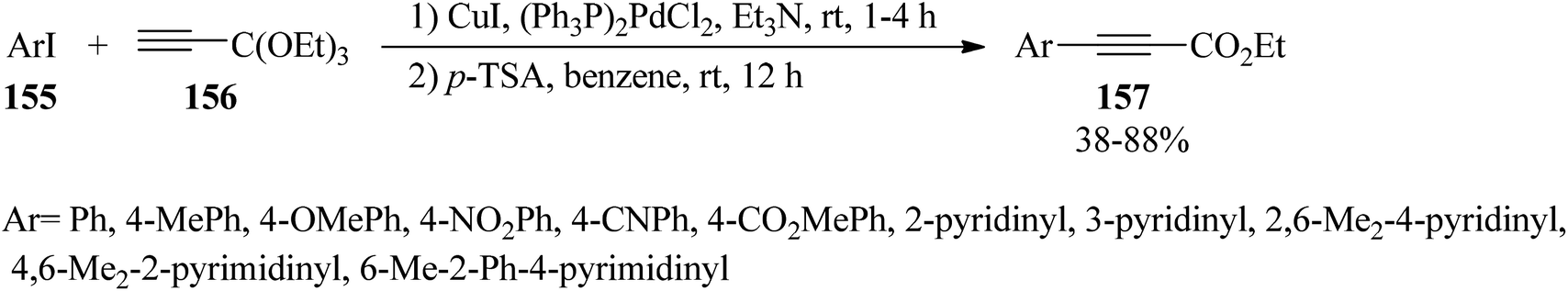
The cross-coupling reaction of aryl iodides (155) with 3,3,3-triethoxy-1-propyne (156) using dichlorobis(triphenylphosphine) palladium [(PPh3)2PdCl2] and cuprous iodide (CuI) in triethylamine (Et3N) at room temperature led to the production of 3,3,3-triethoxy-1-aryl-1-propyne within 1–4 h, which after isolation was subsequently reacted with a catalytic amount of p-TSA in benzene at room temperature for 12 h to yield the ethyl arylpropiolates (157) (Scheme 42).167
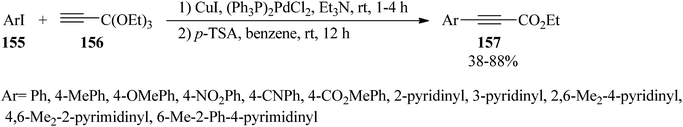 |
| | Scheme 42 The synthesis of ethyl arylpropiolate. | |
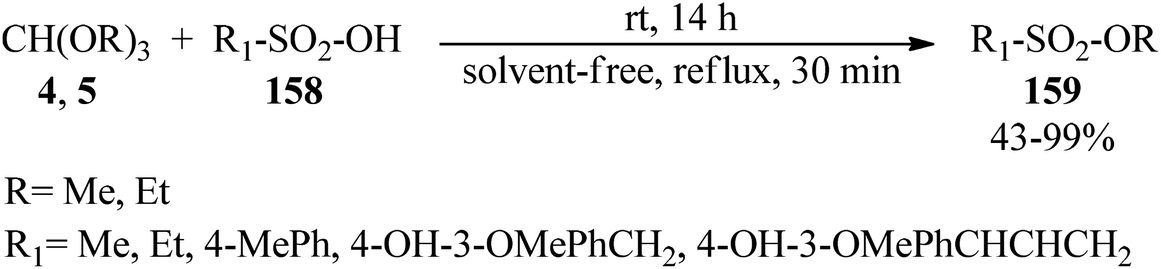
Padmapriya and co-workers reported the alkylation of sulfonic acids (158) by trialkyl orthoformates (4, 5) to generate the alkyl sulfonates (159) under solvent-free conditions at room temperature for 14 h or under reflux conditions in a 30 min period in 43–99% yields (Scheme 43).168
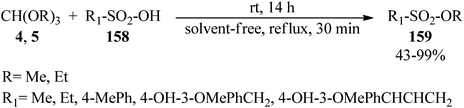 |
| | Scheme 43 The alkylation of sulfonic acids by trialkyl orthoformates. | |
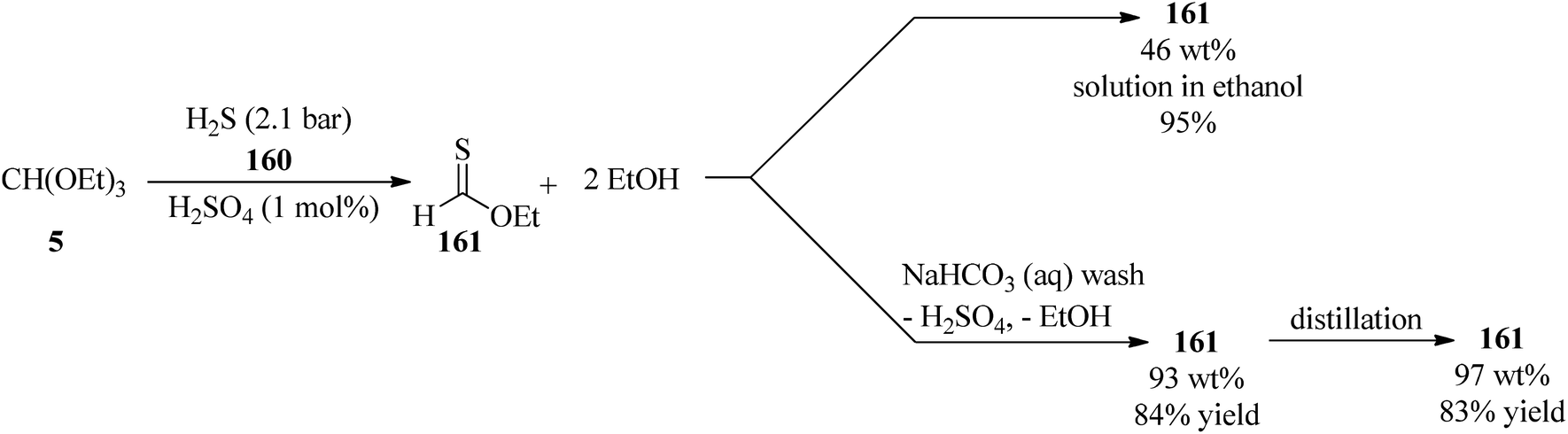
In 2009, Borths et al. synthesized O-ethyl thioformate (161) via the condensation reaction of TEOF (5) with hydrogen sulfide gas (H2S) (160) at 2.1 bar in the presence of sulfuric acid (H2SO4, 98%, 1 mol%) as a Brønsted acid catalyst under solvent-free conditions. The 46 wt% ethanolic O-ethyl thioformate solution was obtained in 95% assay yield and could be purified initially by washing with basic aqueous sodium hydrogen carbonate (NaHCO3) to eliminate the sulfuric acid and ethanol and then by distillation at atmospheric pressure to remove the residual ethanol to provide 97 wt% of O-ethyl thioformate as a yellow oil in 83% yield, which could be utilized as a thioformylating agent for amines (Scheme 44).169
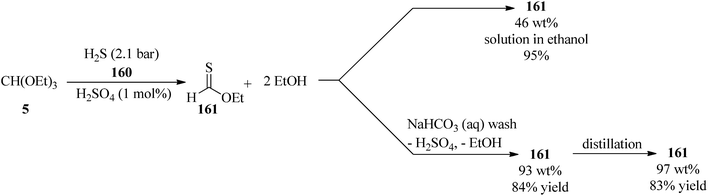 |
| | Scheme 44 The condensation of TEOF with H2S to obtain O-ethyl thioformate. | |
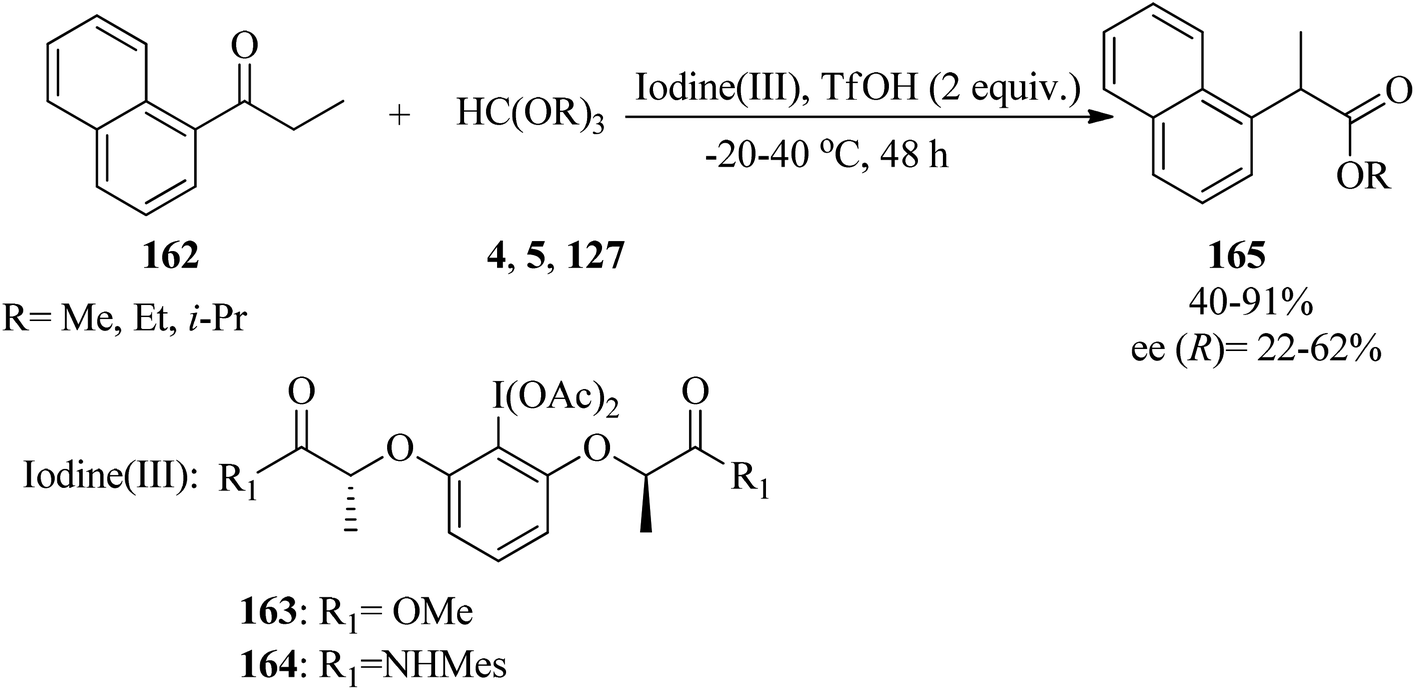
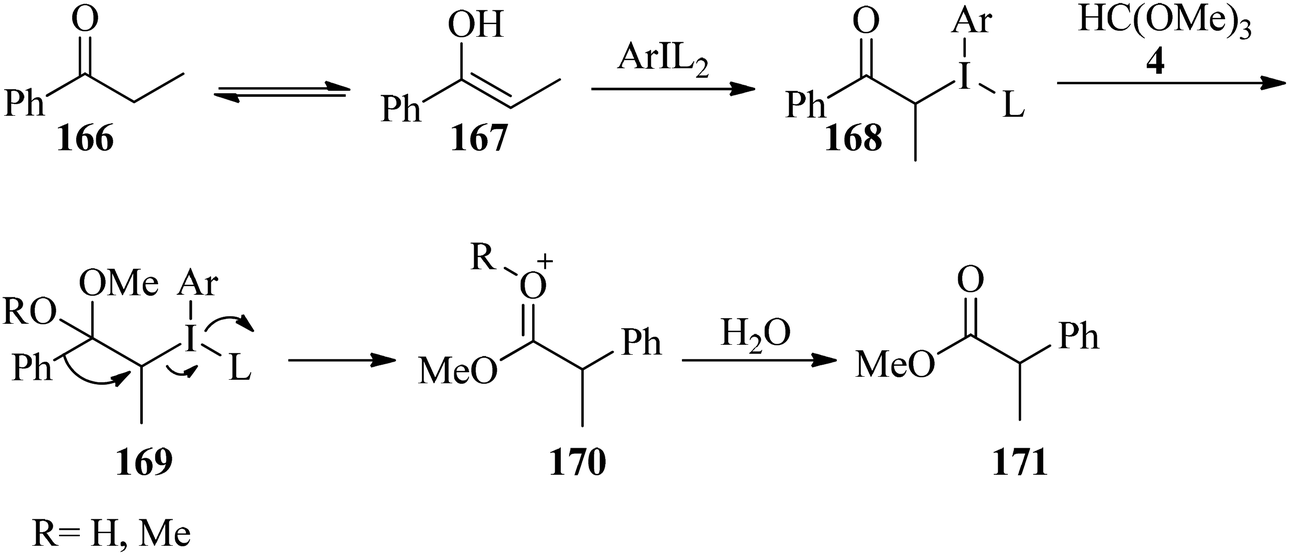
In 2016, Malmedy and Wirth developed a stereoselective reaction to access 2-aryl alkanoates (165) in good enantioselectivity via the rearrangement reaction of 1-(naphthalen-1-yl)propan-1-one (162) with orthoesters (4, 5, 127) by iodine(III) reagent (163, 164) and triflic acid (TfOH) at −20 to 40 °C, yielding the corresponding alkyl 2-(naphthalen-1-yl)propanoate (165) (Scheme 45). In this reaction the effect of different parameters, such as the type of catalyst, catalytic amounts of Lewis acid, the nature of the orthoester, and temperature were investigated. It was found that a reaction with too high a bulkiness of the triisopropyl orthoformate (127) did not result in any rearranged compound. Also, acceptable results were obtained in the presence of 2 equivalents of TfOH. A plausible mechanism for the rearrangement is shown in Scheme 46.170 The reaction was promoted through the reaction of the enolic form (167) of ketone (166) with the iodine(III) moiety to produce the intermediate (168), which provided the ketal (169) by TMOF (4). The elimination of aryliodide was accompanied with 1,2-aryl migration to form the intermediate (170); which was followed by hydrolysis in water to afford the rearranged product (171).171
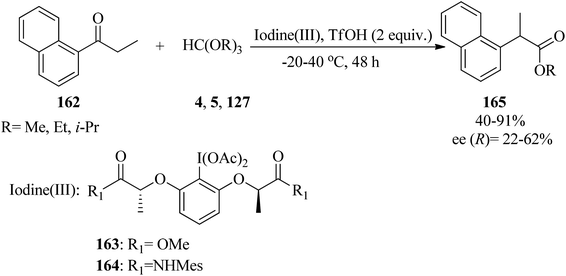 |
| | Scheme 45 The preparation of alkyl 2-(naphthalen-1-yl)propanoates. | |
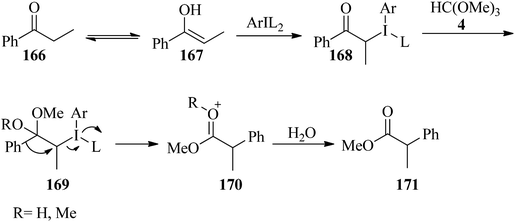 |
| | Scheme 46 Reaction mechanism for the reaction of ketones with TMOF. | |
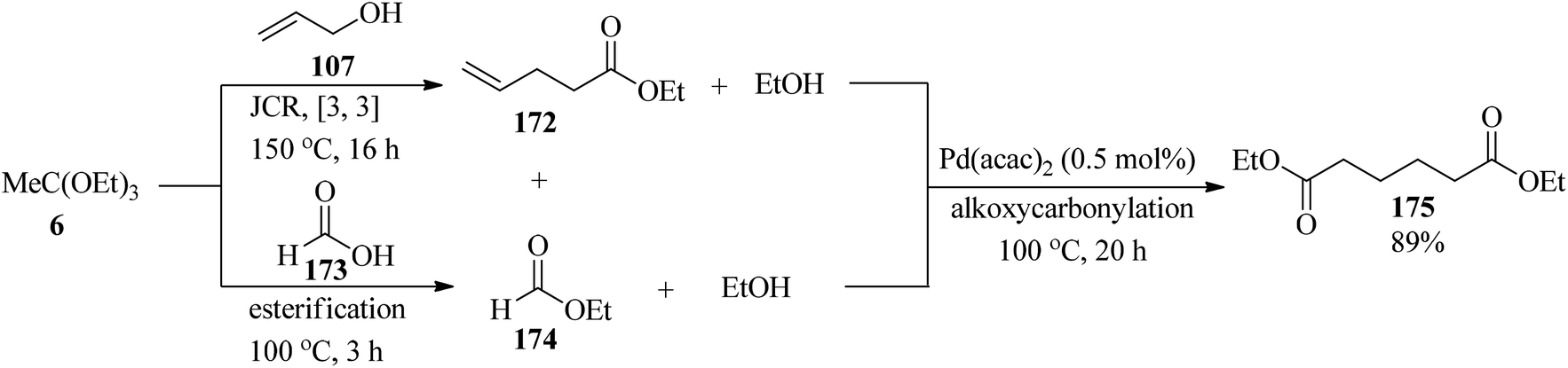
The linear diester diethyl adipate (175) was synthesized by the merger of Johnson–Claisen rearrangement and alkoxycarbonylation via a one-pot procedure. This reaction proceeded through Johnson–Claisen rearrangement (JCR) of allyl alcohol (107) with TEOAc (6, 2 equiv.) to afford ethyl 4-pentenoate (172), which was followed by quenching the excess of (6) by the addition of formic acid (173) to obtain ethyl formate (174). Then, the alkoxycarbonylation of ethyl 4-pentenoate (172) with ethyl formate (174) using the palladium catalytic system such as palladium(II)acetylacetonate (Pd(acac)2, 0.5 mol%) and 1,2-bis((di-tert-butylphosphino)methyl)benzene (2 mol% 1,2-DTBPMB) and methanesulfonic acid (MSA, 8 mol%) produced the linear diester diethyl adipate (175) in good yield (89%) (Scheme 47).172
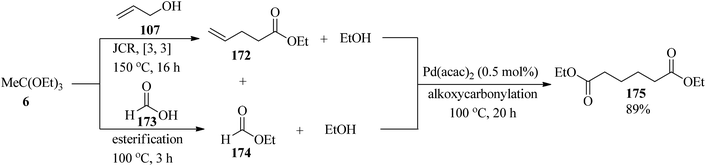 |
| | Scheme 47 The synthesis of linear diester diethyl adipate by the Johnson–Claisen rearrangement and alkoxycarbonylation. | |
2.20. Alkylation reactions
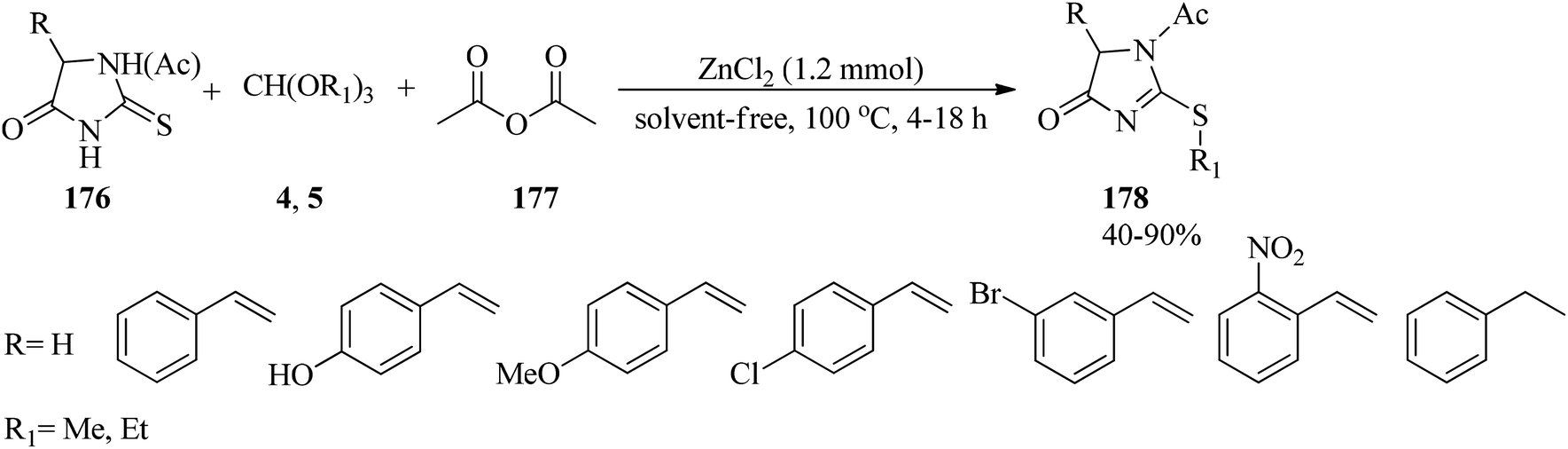
In 2008, the one-pot chemoselective S-alkylation and N-acylation reaction of 2-thiohydantoins (176) utilizing alkyl orthoformates (4, 5) and acetic anhydride (177) was presented for the synthesis of the products (178) by zinc chloride (ZnCl2, 1.2 mmol) at 100 °C within 4–18 h in 40–90% yields (Scheme 48).173
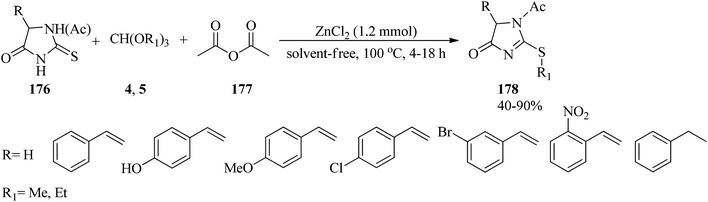 |
| | Scheme 48 S-Alkylation and N-acylation of 2-thiohydantoins. | |
In 1980, Swaringen et al. discovered that the reaction of N-alkylanilines (179) and ortho formats (4, 5), in a 2:1 molar ratio, gave high yields of N-alkylformanilides (180) and N,N-dialkylanilines (181) in the presence of p-TSA (Scheme 49). In addition, aliphatic cyclic amines (such as morpholine or piperidine) and orthoformates gave the corresponding orthoamides.174
 |
| | Scheme 49 The synthesis of N-alkylformanilides and N,N-dialkylanilines. | |
2.21. Acetalization reaction
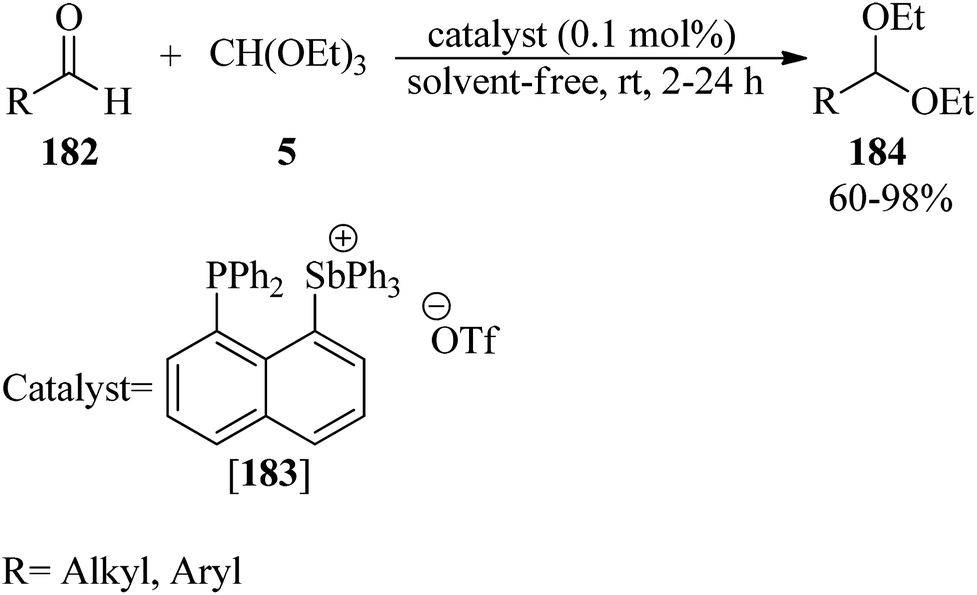
In 2017, Ugarte and Hudnall presented a highly selective, environmentally friendly, rapid, and solvent-free procedure for the protection of carbonyls to acetals (184)175–182 via the reaction of aldehydes (182) with TEOF (5) in equimolar amounts using the tetraarylstibonium salt, 1-diphenylphosphinonaphthyl-8-triphenylstibonium triflate ([183][OTf], 0.1 mol%), at room temperature (Scheme 50). Notably, the rate of the reactions increased in the presence of aliphatic aldehydes. This catalyst was recycled and reused for up to 6 runs without losing activity. Furthermore, the tetraarylstibonium salt could catalyze the deprotection of the acetals into their corresponding aldehydes in aqueous media.182
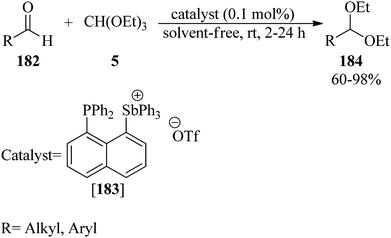 |
| | Scheme 50 Acetalization with TEOF in the presence of tetraarylstibonium salt. | |
In 2009, Kharkongor and Myrboh presented a convenient and efficient one-pot method for the synthesis of α-ketoacetals (186) from substituted acetophenones (185) and TEOF (5) by selenous acid (H2SeO3, 0.7 equiv.) and BF3·Et2O (1.5 mL) as a catalyst under solvent-free conditions at room temperature in moderate to excellent yields (55–90%) within 8–14 h (Scheme 51).183 Acetals could be also synthesized via the reaction of various acidic methines such as cyanoesters and 3,4,5-trimethoxybenzylmalononitrile with orthoformates under heating and solvent-free conditions. The reaction has been shown to be useful for the production of protected aldehydes and 2,4-diamino-5-benzylpyrimidines.184
 |
| | Scheme 51 Acetalization with TEOF in the presence of selenous acid. | |
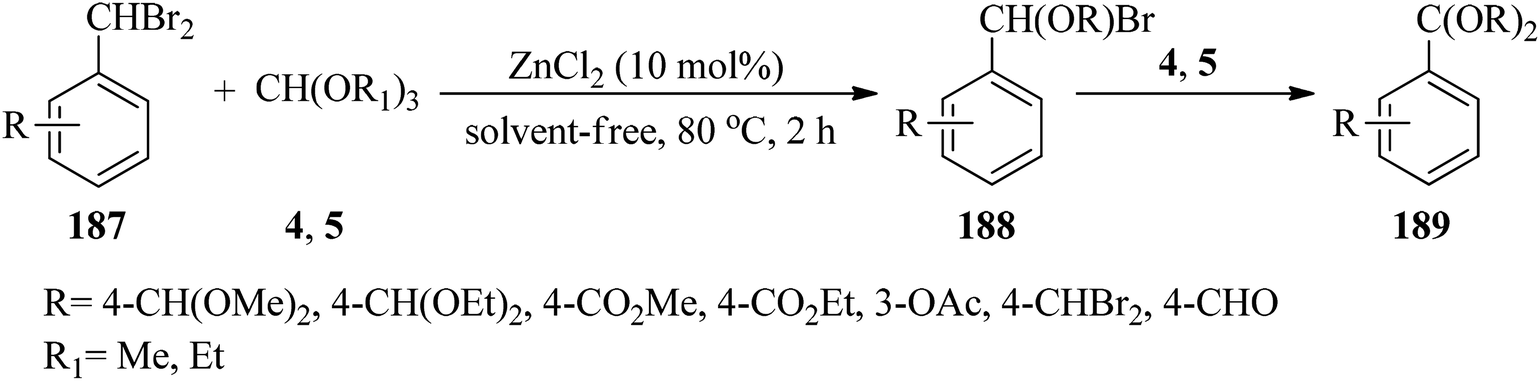
Aromatic aldehyde acetals (189)185,186 were synthesized via double debromoalkoxylation of dibromomethylarenes (187) with trialkyl orthoformate (4, 5) using ZnCl2 (10 mol%) under solvent-free conditions at 80 °C (Scheme 52). This reaction was promoted by the reaction of dibromomethylarenes (187) with an excess of (4, 5) to form α-brominated ether intermediates (188) which underwent a second debromoalkoxylation in the presence of an excess of (4, 5) to provide acetal (189) and the removal of 2 moles of alkyl bromide and 3 moles of formate ester.186
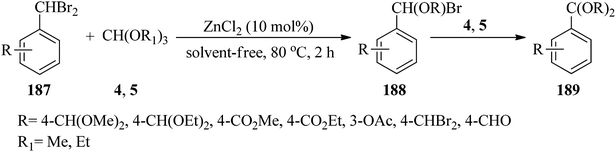 |
| | Scheme 52 Acetalization via the double debromoalkoxylation of dibromomethylarenes. | |
In 2006, Gazizov et al. investigated the reaction of TEOF with 1,1,2,2-tetrabromoethane with different molar ratios at 180 °C under solvent-free conditions within 10 h. It was found that dehydrobromination reaction took place via the reaction of triethyl orthoformate with 1,1,2,2-tetrabromoethane in 1:2 molar ratios to obtain tribromoethylene, ethyl formate, ethyl bromide, and ethanol as the main products along with low amounts of ethyl acetate and dibromoethylene. On the other hand, the debromination reaction was accomplished via the reaction of substrates in a 2:1 molar ratio to give ethyl formate, ethyl bromide, dibromoethylene, ethyl hypobromite, and acetal.187
2.22. Synthesis of α-iodo ketones and α-iodo dimethyl ketals
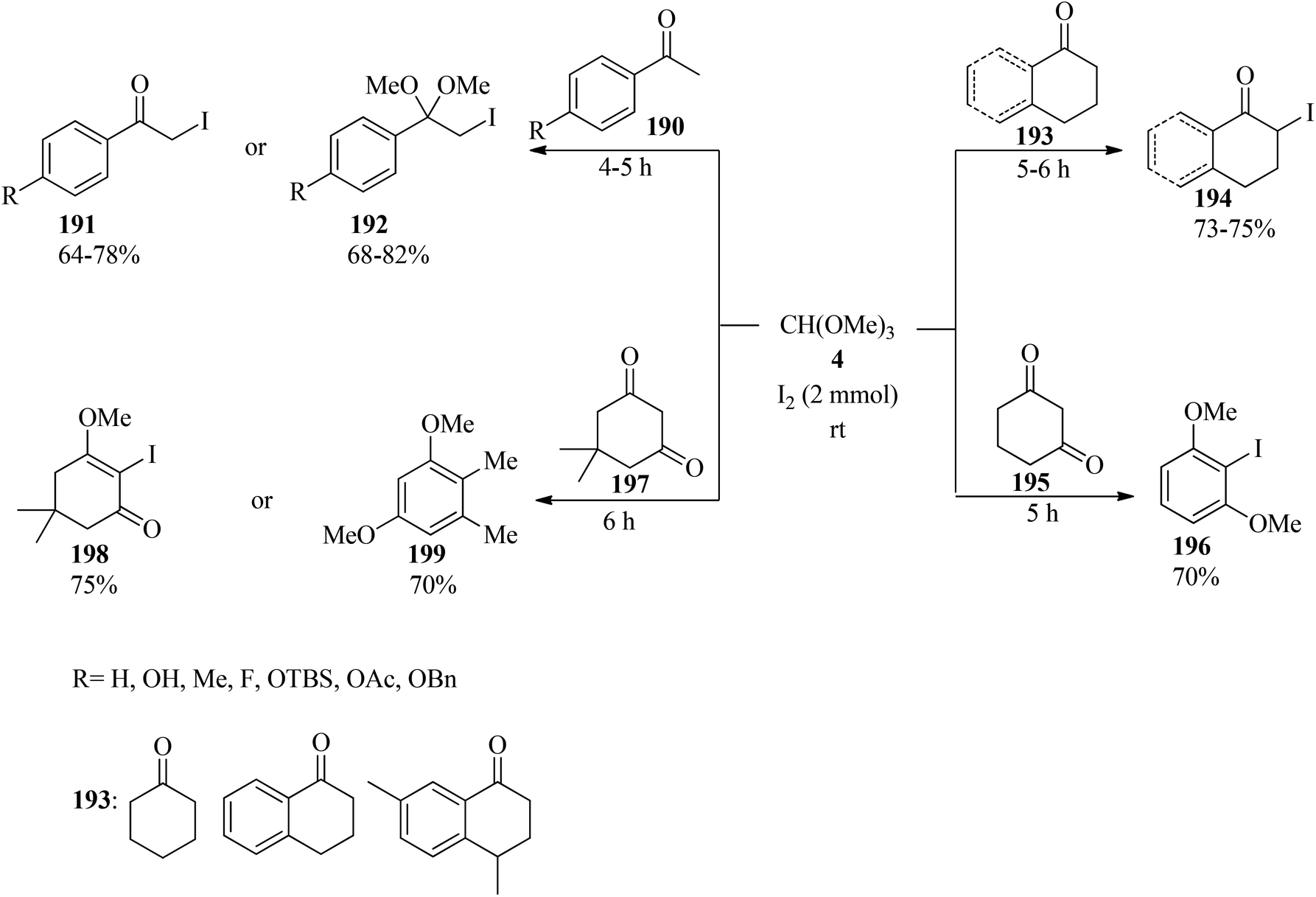
In 2008, Yadav et al. presented a new, effective, and simple protocol for the synthesis of α-iodo ketones (191) and α-iodo dimethyl ketals (192) via the reaction of substituted acetophenones (190), TMOF (4) and iodine at room temperature under solvent-free conditions (Scheme 53). It must be mentioned that the selectivity of the obtained products from acetophenones was attributed to the workup procedures. It was found that, after completion of the reaction, if the reaction mixture was quenched in the presence of aqueous saturated Na2S2O3 and then extracted by dichloromethane, the α-iodinated dimethoxy ketal was obtained as the main product, but when the reaction mixture was stirred in the presence of water before washing with aqueous saturated Na2S2O3, the α-iodo ketones were afforded as the main products. In this conversion, the ring-directed iodination or α,α-diiodination products were never obtained. It is significant that this transformation in the presence of cyclohexanones (193) as a starting material resulted in the α-iodo ketos (194) as the only product, but 1,3-cyclohexadione (195) and substituted 1,3-cyclohexadione such as dimedone (197) yielded the α-iodinated products (196, 198) and the aromatized compound (199)188 in good yields.189
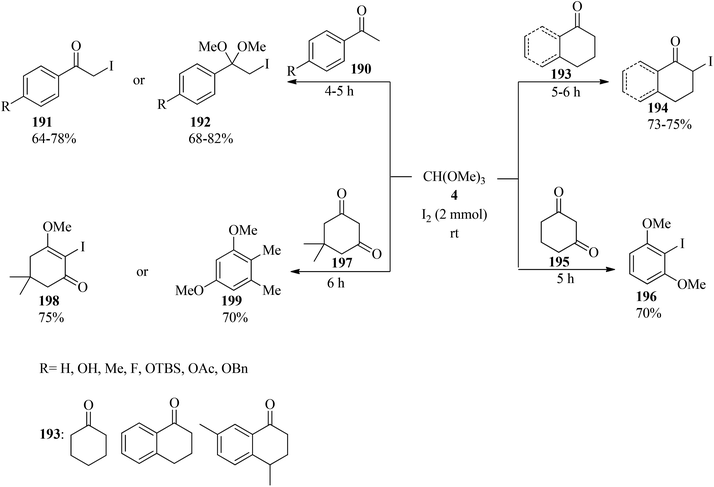 |
| | Scheme 53 The synthesis of iodo ketones and iodo dimethyl ketals. | |
2.23. Ring-contraction reactions
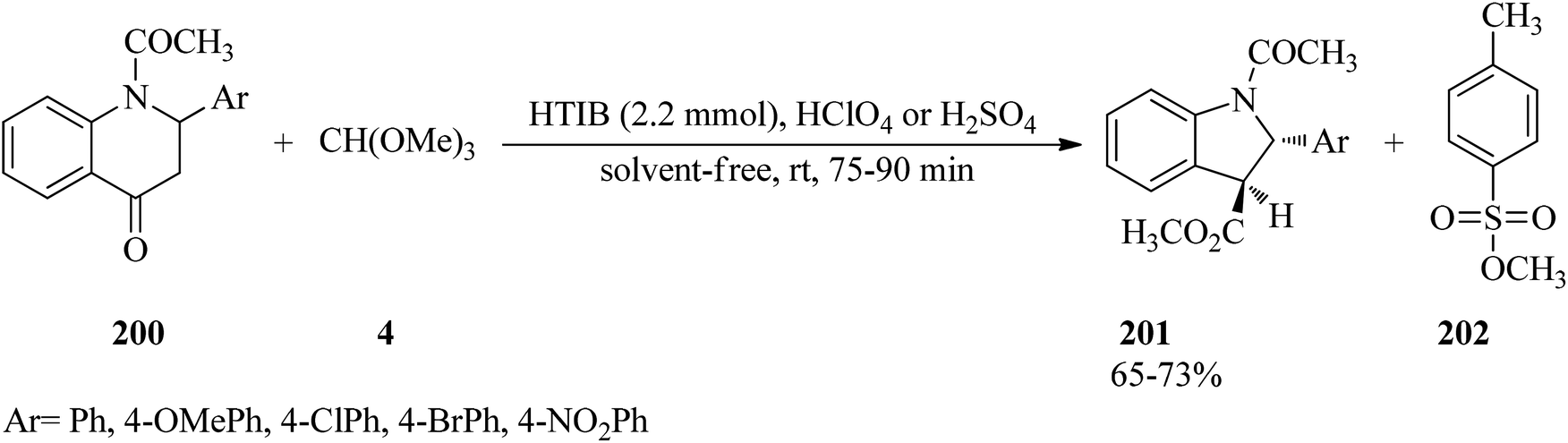
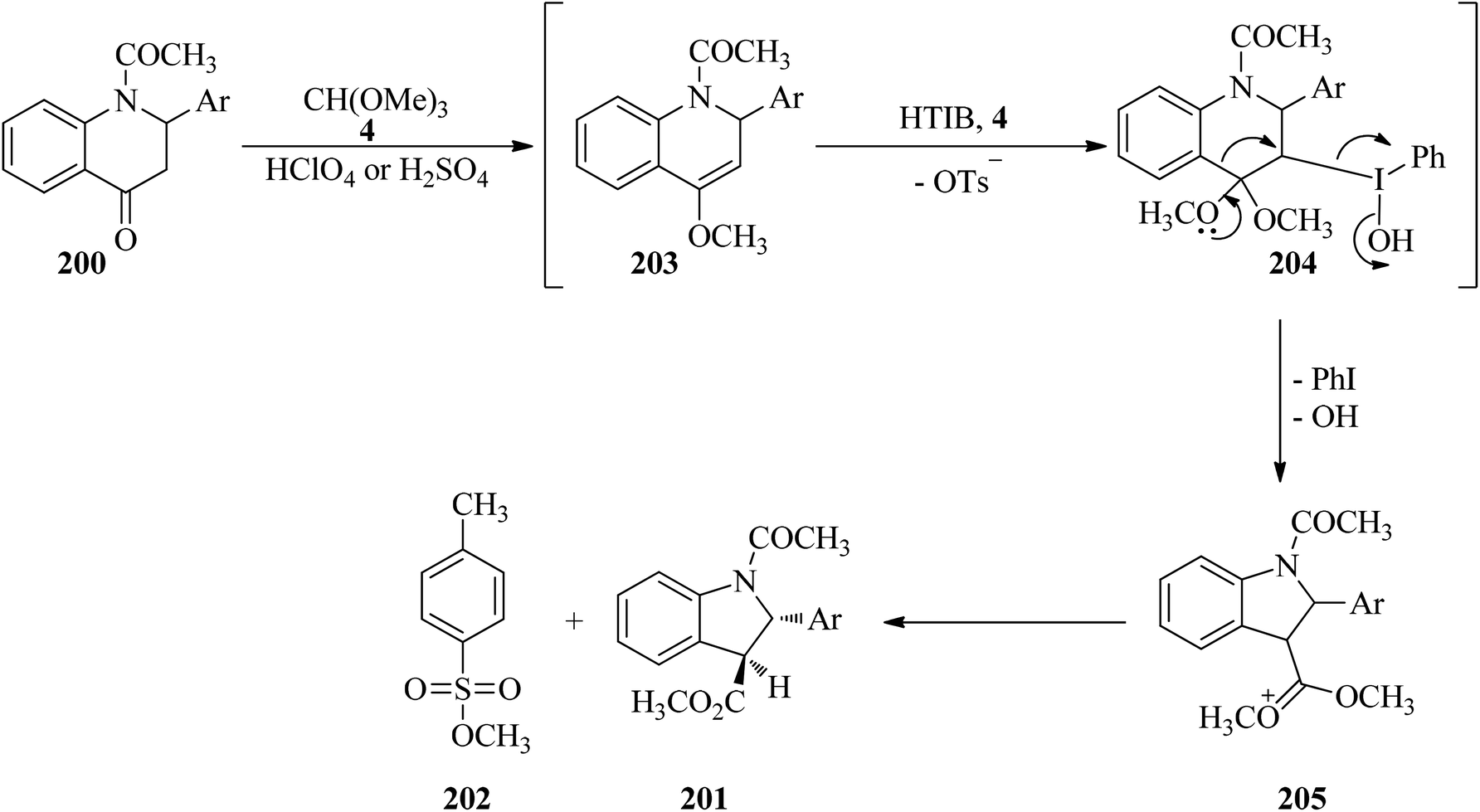
In 2007, Kumar et al. described a stereoselective method for the synthesis of trans-methyl N-acetyl-2-aryl-2,3-dihydroindol-3-carboxylates (201) via the ring contraction of N-acetyl-2-aryl-1,2,3,4-tetrahydro-4-quinolones (200) with TMOF (4) by HTIB as oxidizing agent in the presence of a few drops of either HClO4 or H2SO4 at room temperature in high yields (65–73%) for appropriate times (75–90 min) (Scheme 54). This reaction was accompanied by the formation of methyl p-toluenesulfonate (202) as a by-product in small quantity. The proposed mechanism, shown in Scheme 55, was begun through the ketalisation of (200) with (4) using either HClO4 or H2SO4 to form intermediate enol ether (203). Then the electrophilic attack of HTIB on the double bond of enol ether (203) led to the iodine(III) complex (204), from which the elimination of iodobenzene from (204) with concurrent migration of aryl residue from the C4 to the C3 position gave intermediate carbocation (205), which on hydrolysis obtained the ring-contracted product (201) together with (202). To accomplish greater stability of the resultant carbocation, the migration of aryl residue was preferred over the C2 position aryl ring. Eventually, (201) was afforded by ring contraction of compound (200), and meanwhile by-product (202) was produced, probably through the in situ condensation of methanol with p-TSA.190
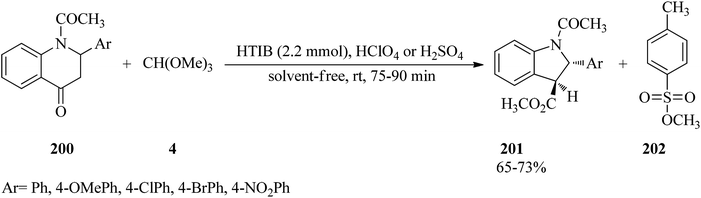 |
| | Scheme 54 The ring contraction of N-acetyl-2-aryl-1,2,3,4-tetrahydro-4-quinolones with TMOF. | |
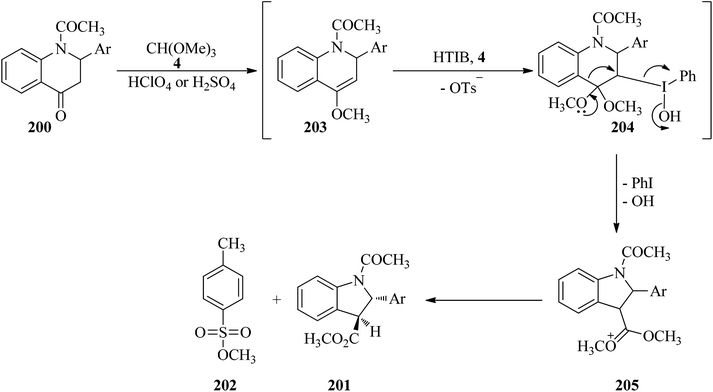 |
| | Scheme 55 A mechanistic approach for the ring contraction of N-acetyl-2-aryl-1,2,3,4-tetrahydro-4-quinolones with TMOF. | |
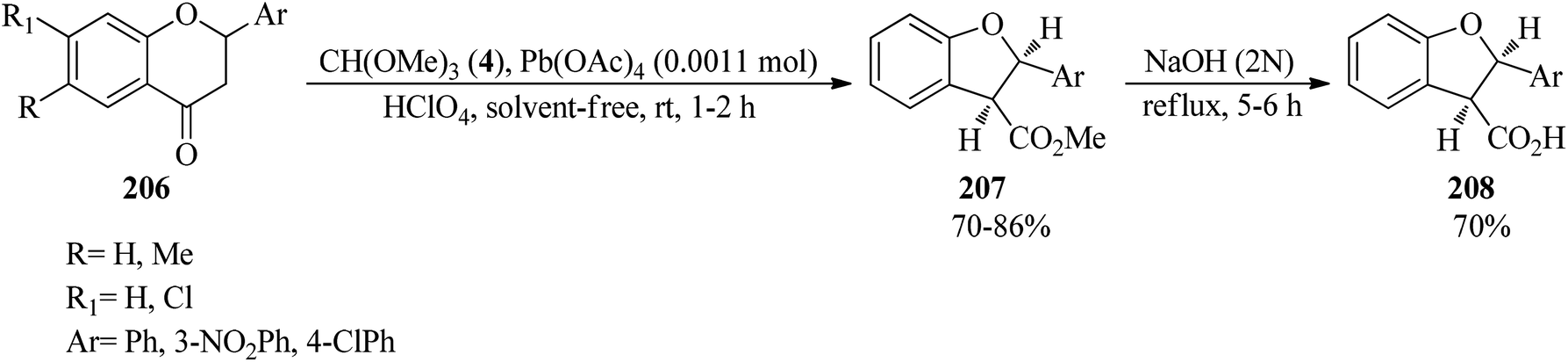
Flavanones (206),191–193 through the oxidative reaction with TMOF (4) using lead tetraacetate (Pb(OAc)4, 0.0011 mol) and 70% perchloric acid under solvent-free conditions at room temperature, were converted into cis-methyl-2,3-dihydro-2-arylbenzofuran-3-carboxylates (207) in high yields (70–86%) within 1–2 h. Subsequent alkaline hydrolysis of the product (207) by aqueous NaOH solution under reflux conditions obtained the corresponding acids (208) within 5–6 h in 70% yields (Scheme 56).193
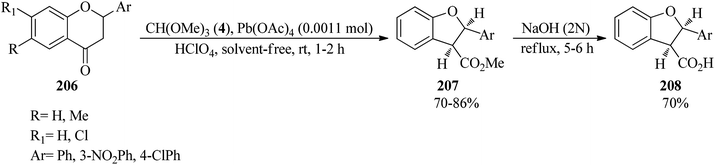 |
| | Scheme 56 The oxidative reaction of flavanones with TMOF using Pb(OAc)4. | |
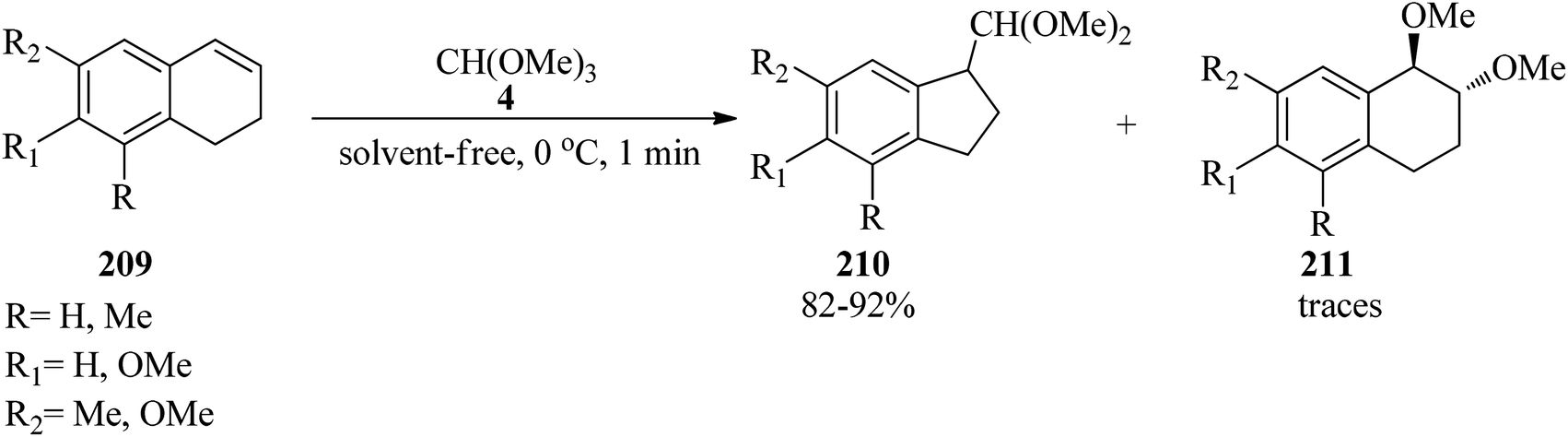
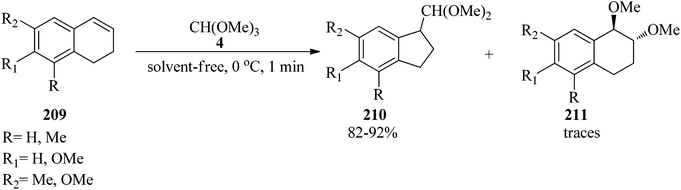
Thallium(III) trinitrate [Tl(NO3)3·3H2O] catalyzed the oxidation of various olefins194 such as 1,2-dihydronaphthalenes (209) with TMOF (4) at 0 °C to obtain indans (210) in good yields (82–92%) within a very short 1 min period (Scheme 57).195 It was found that 1,2-dihydronaphthalenes (209) without an alkyl substituent at the double bond produced mainly the ring-contraction products (210) in high yields,194–204 while the substrates containing an alkyl group because of their lower reactivity gave the addition products (211) in the presence of methanol,197,198,205 although there are some exceptions.200,206
 |
| | Scheme 57 The reaction of 1,2-dihydronaphthalenes with TMOF. | |
2.24. Synthesis of benzimidazoles, benzoxazoles, benzothiazoles, and oxazolo[4,5-b]pyridines
In 2007, the Mohammadpoor-Baltork group presented a green and efficient protocol for the preparation of a new library of benzimidazoles (213), benzoxazoles (215),207,208 benzothiazoles (217),128,209 and oxazolo[4,5-b]pyridines (219)210–212 via the condensation reactions of orthoesters (5, 6, 7) with o-substituted aminoaromatics such as o-phenylenediamine (212), o-aminophenol (214), o-aminothiophenol (216), and 2-amino-3-hydroxypyridine (218), respectively, by catalytic amounts of ZrOCl2·8H2O under solvent-free conditions at room temperature or 85 °C for short reaction times (4–12 min) and moderate to excellent yields (83–97%) (Scheme 58).212
 |
| | Scheme 58 The synthesis of benzimidazoles through the condensation of trialkyl orthoformates with diamines. | |
In 2018, ZrOCl2·8H2O@nano SiO2 as a new catalyst was originated and characterized and subsequently its catalytic activity (0.01 g) has been investigated for the preparation of benzimidazoles213,214 via the condensation reaction of trialkyl orthoformates with diamines under solvent-free condition at 60 °C within reaction times of 1.5–5.5 h and 70–90% yields. High yields (70–90%), recyclability, and reusability of the synthesized catalyst are some of the main advantages of this method.214 Nano-Ni(II)/Y zeolite, as a non-toxic heterogeneous catalyst, at 60 °C under solvent-free conditions also accelerated this transformation within 46–122 min in good to excellent 79–99% yields.215
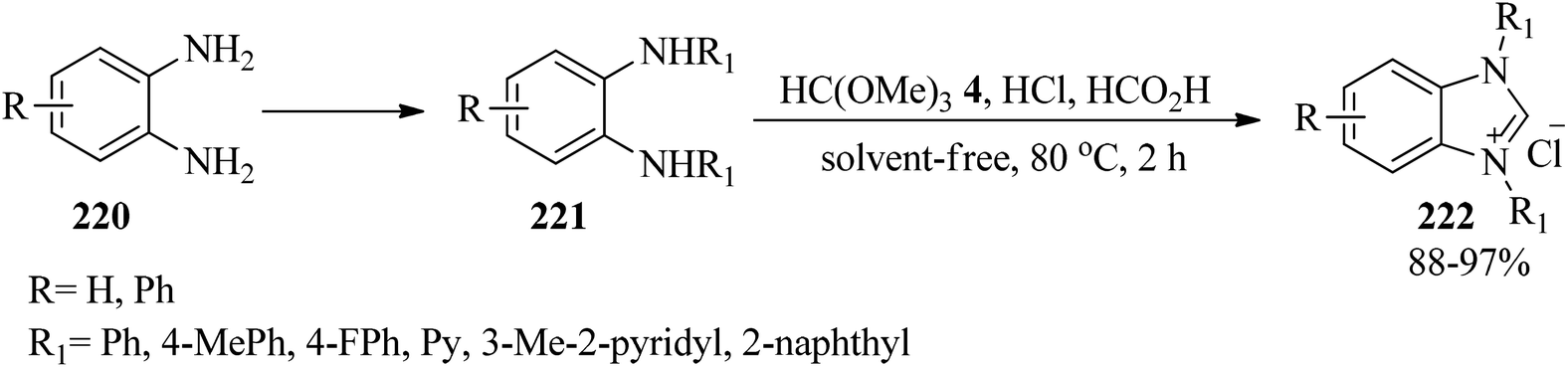
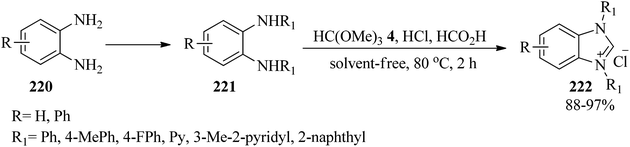
In 2013, Wang and co-workers developed a simple and efficacious process for the synthesis of benzimidazole-based N-heterocyclic carbenes (222) via the cyclization of N,N′-diarylaryldiamines (221), arising from aryldiamines (220), with TMOF (4) using concentrated HCl and formic acid at 80 °C for 2 h in 88–97% yields (Scheme 59). Notably, the reaction was unsuccessful with diamines containing strong electron-withdrawing substitutions.216
 |
| | Scheme 59 The synthesis of benzimidazole-based N-heterocyclic carbenes. | |
2.25. Synthesis of oxadiazoles and thiadiazoles
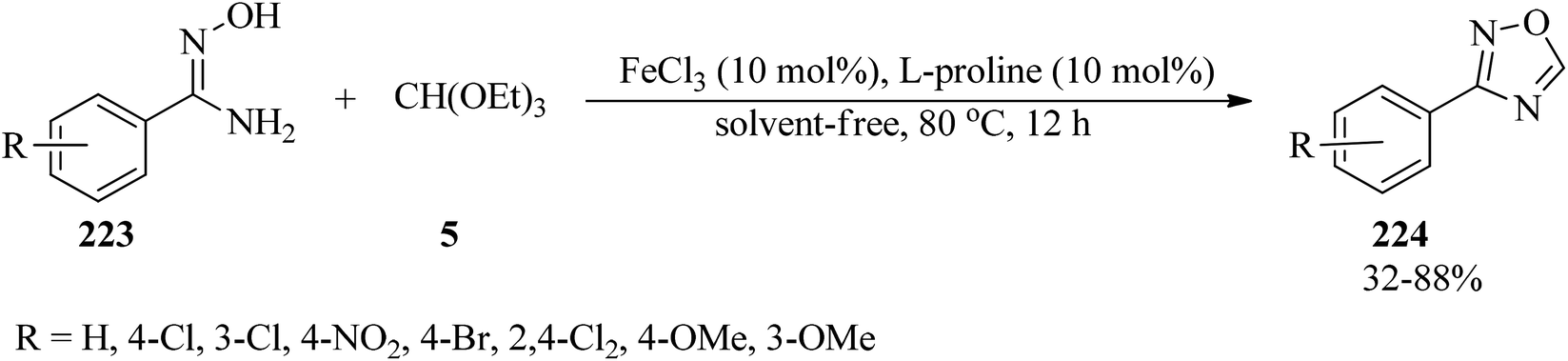
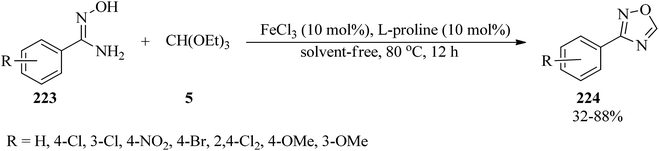
In 2016, Kaboudin et al. investigated iron(III) chloride/L-proline as an efficient catalytic system for the synthesis of 3-substituted 1,2,4-oxadiazoles (224) from amidoximes (223) and TEOF (5) (Scheme 60).217
 |
| | Scheme 60 The preparation of 3-substituted 1,2,4-oxadiazoles. | |
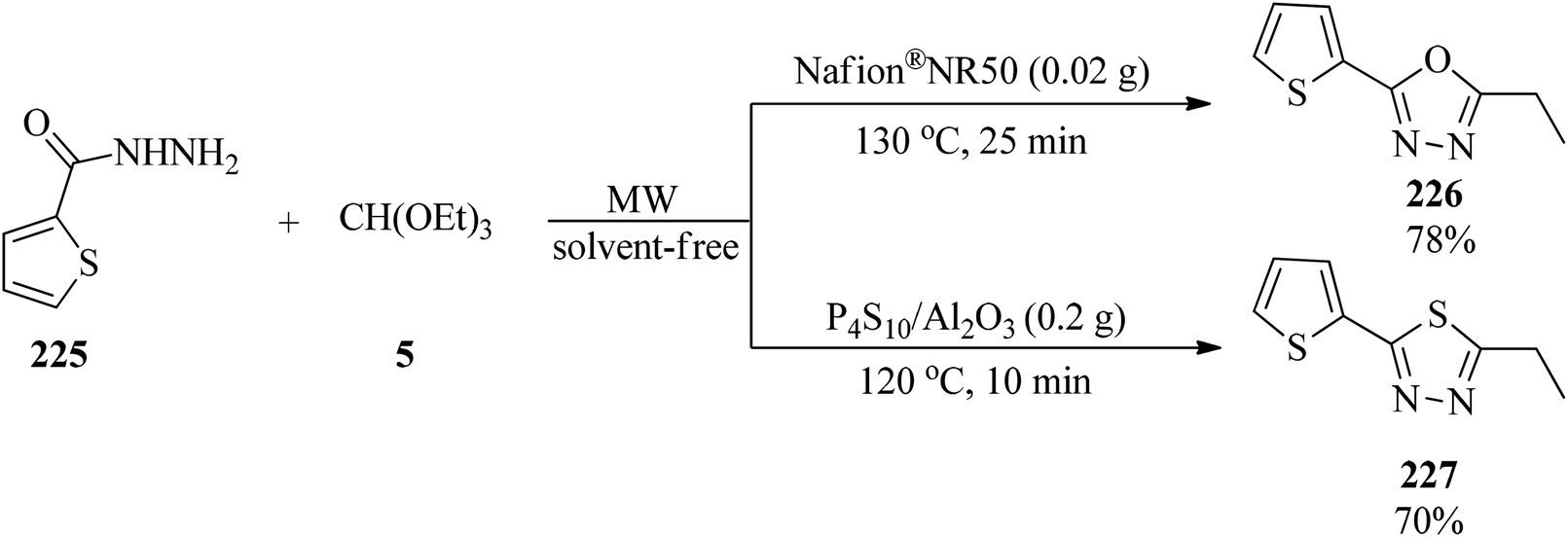
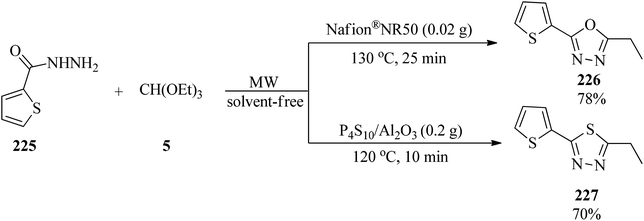
Polshettiwar and Varma performed the solvent-free synthesis of 1,3,4-oxadiazoles (226)218–223 and 1,3,4-thiadiazoles (227) by the condensation of acid hydrazide (225) and triethyl orthoalkanates (5) under microwave irradiation (40–140 W). According to Scheme 61, the condensation of hydrazide and TEOF were successfully undertaken in two catalytic systems which were solid supported Nafion® NR50 and P4S10/Al2O3.223
 |
| | Scheme 61 The microwave-assisted synthesis of 1,3,4-oxadiazoles and 1,3,4-thiadiazoles. | |
The double [4 + 1] cyclocondensation reaction of 6-hydrazinonicotinic acid hydrazide hydrate (228) containing two dinucleophilic centers224 with TEOAc (6) obtained oxadiazolyltriazolopyridine (229) in 82% yield within 16 h under reflux conditions (Scheme 62). In this transformation, the hydrazine and the hydrazide moieties exhibited similar reactivity, whereas in the presence of aldehyde as one other carbon donor moiety the hydrazine and the hydrazide groups were treated chemoselectively and regioselectivly.225
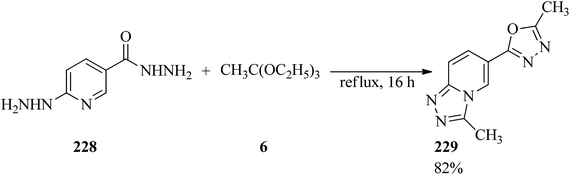 |
| | Scheme 62 Double [4 + 1] cyclocondensation of 6-hydrazinonicotinic acid hydrazide hydrate with TEOAc. | |
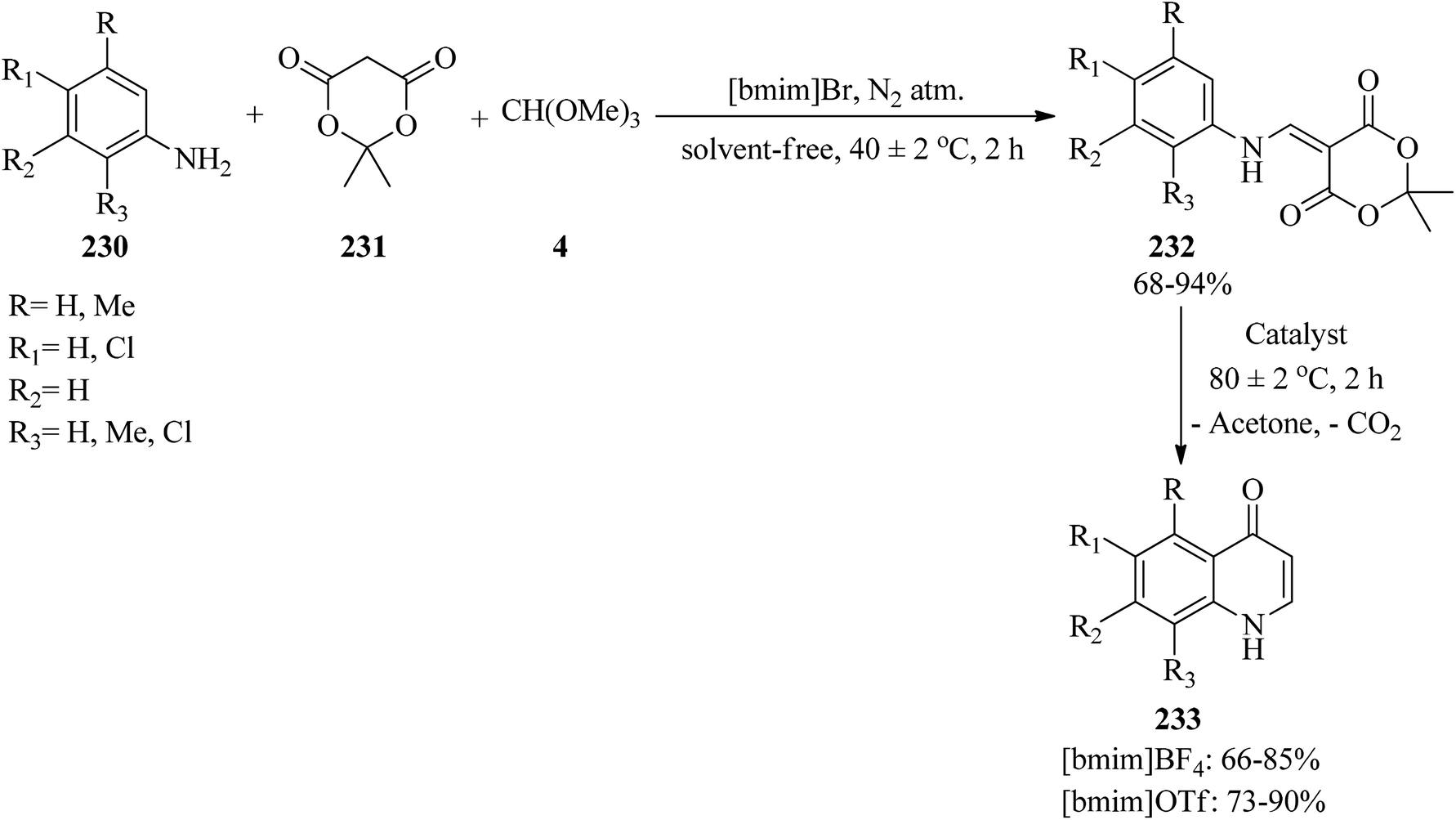
2.26. Synthesis of quinolones
In 2012, Yadav et al. developed a simple and green method for the synthesis of 4(1H)-quinolones via a two-step reaction. At first, the one-pot three-component condensation reaction of arylamines (230), Meldrum's acid (231), and TMOF (4) by 1-butyl-3-methylimidazolium bromide [bmim]Br as ionic liquid catalyst at 40 ± 2 °C obtained arylaminomethylene-1,3-dioxane-4,6-dione (232) in 68–94% yield in 2 h. After purifying, the resulting products underwent a cyclization reaction along with the elimination of acetone and CO2 using [bmim]BF4 or [bmim]OTf at 80 ± 2 °C in a 2 h period to afford 4(1H)-quinolones (233) (Scheme 63). The results revealed the better utility of [bmim]OTf in comparison with [bmim]BF4.226
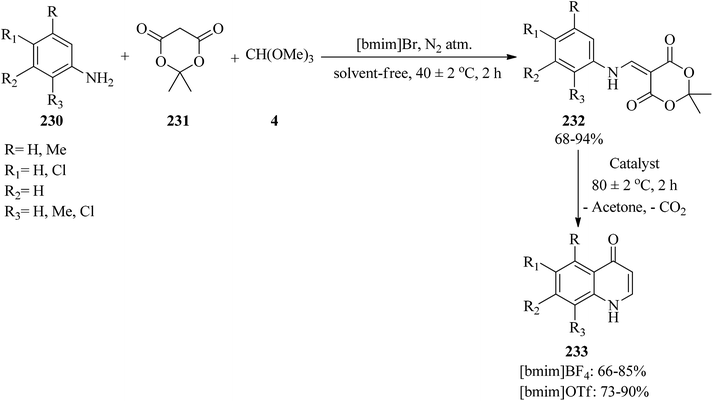 |
| | Scheme 63 The synthesis of 4(1H)-quinolones. | |
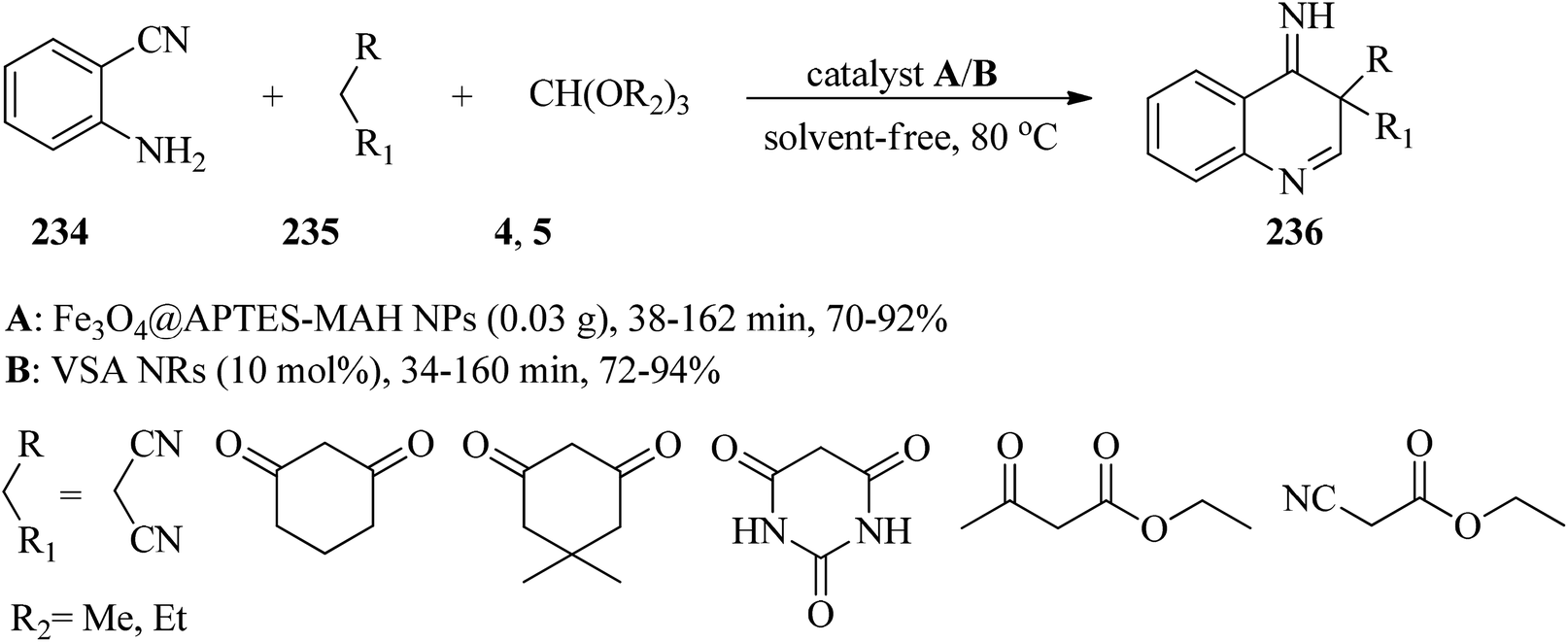
In 2018, Kanaani and Nasr-Esfahani prepared and characterized maleic anhydride coated magnetite nanoparticles (Fe3O4@APTES-MAH NPs) and nanorod vanadatesulfuric acid (VSA NRs) as Brønsted acid nanocatalysts. Subsequently, their catalytic activity was examined for the synthesis of new 4-iminoquinolines (236) via the one-pot three-component reaction of equimolar amounts of 2-aminobenzonitrile (234), orthoesters (4, 5), and active methylenes (235) under solvent-free conditions at 80 °C. In this reaction the influence of different parameters, such as the type of catalyst and orthoesters, was examined. It was found that both VSA NRs and Fe3O4@APTES-MAH NPs catalysts yielded the corresponding 4-iminoquinolines (236) in good to excellent yields. VSA NRs in comparison with Fe3O4@APTES-MAH NPs slightly improved the reaction times and yields (Scheme 64). They discovered that, among different orthoesters, TEOF was more effective. Short reaction times, high yields, simple purification of the products, the inexpensiveness, stability, recyclability and reusability of the catalysts are some of the advantages of this method.227
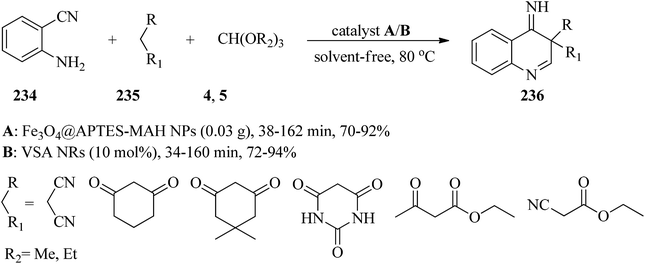 |
| | Scheme 64 The synthesis of new 4-iminoquinolines. | |
2.27. Synthesis of isoquinoline derivatives
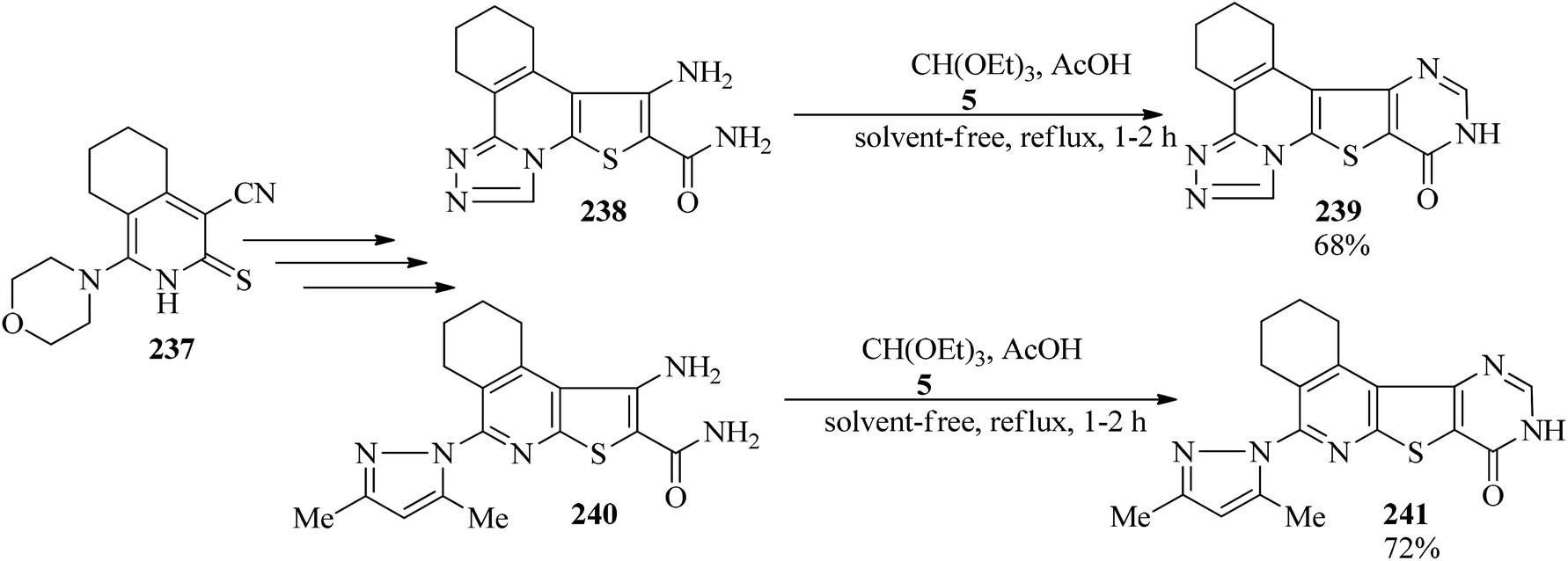
1,2,3,4-Tetrahydro-10-oxopyrimido[4′,5′:4,5]thieno[2,3-c][1,2,4]triazolo[3,4-a]isoquinoline (239, 68%) and 5-(3,5-dimethylpyrazol-1-yl)-1,2,3,4-tetrahydropyrimido[4′,5′:4,5]thieno[2,3-c]isoquinolin-8(9H)-one (241, 72%) were synthesized via the ring closure of triazolo and pyrazolothienotetrahydroisoquinoline (238, 240), derived from key precursors beginning from 4-cyano-1-morpholin-4-yl-5,6,7,8-tetrahydroisoquinoline-3(2H)thione (237), with TEOF (5) using a few drops of glacial acetic acid under reflux conditions for 1–2 h (Scheme 65). The synthesized products were examined and evaluated as antimicrobial agents.228
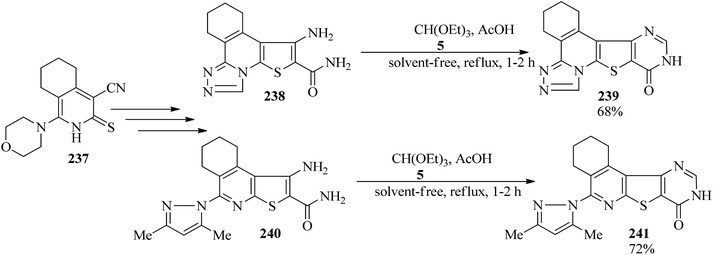 |
| | Scheme 65 The synthesis of isoquinolines with the aid of TEOF. | |
2.28. Synthesis of 1-substituted-1H-1,2,3,4-tetrazoles
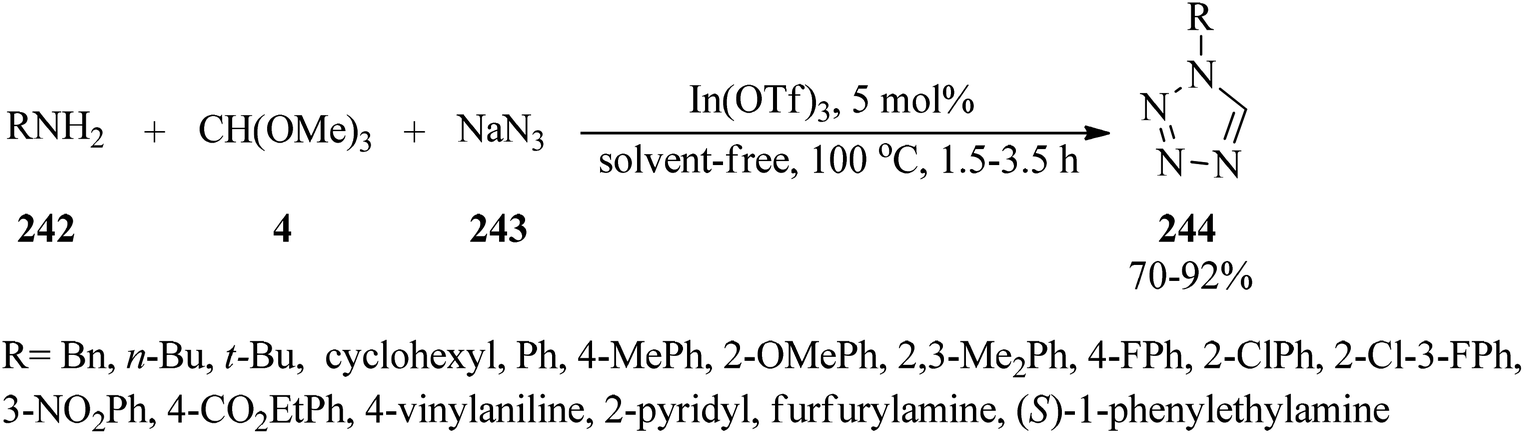
Indium triflate [In(OTf)3, 5 mol%] as a highly efficient Lewis acid catalyst, catalyzed the one-pot three-component condensation reaction of amines (242), TMOF (4), and sodium azide (243) under solvent-free conditions at 100 °C to furnish 1-substituted-1H-1,2,3,4-tetrazoles (244)229–245 in good to excellent yields (70–92%) within 1.5–3.5 h. Both electron-donating and electron-withdrawing anilines yielded the products in high yields. The catalytic system also proceeded well with heterocyclic amine and aliphatic amines (Scheme 66).245 On the other hand, the reaction of substituted anilines, TEOF, and trimethylsilyl azide209,246 also afforded 244 using FeCl3 (20 mmol%) under solvent-free conditions at 70 °C within 5–7 h in 87–95% yields.246
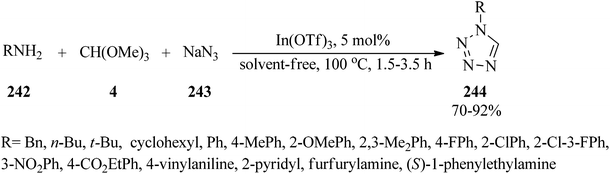 |
| | Scheme 66 The preparation of substituted-1H-1,2,3,4-tetrazoles. | |
2.29. Synthesis of selenoureas
Selenoureas (247) were obtained by a novel three-component condensation reaction from primary or secondary amines (245), TEOF (5) and metallic selenium (246) (Scheme 67).247
 |
| | Scheme 67 Selenourea synthesis by the condensation of amines, TEOF, and Se. | |
2.30. Synthesis of piperazinyl pyridines
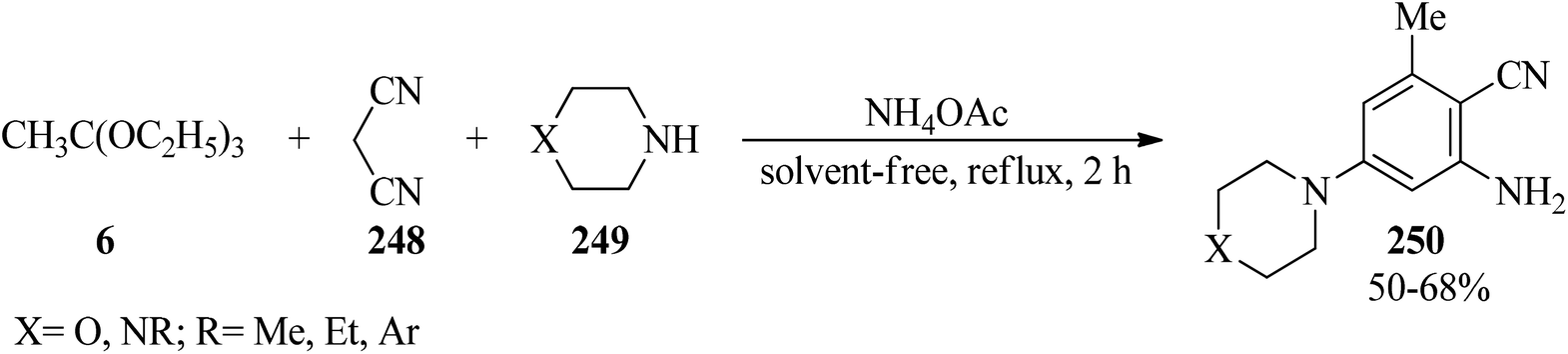
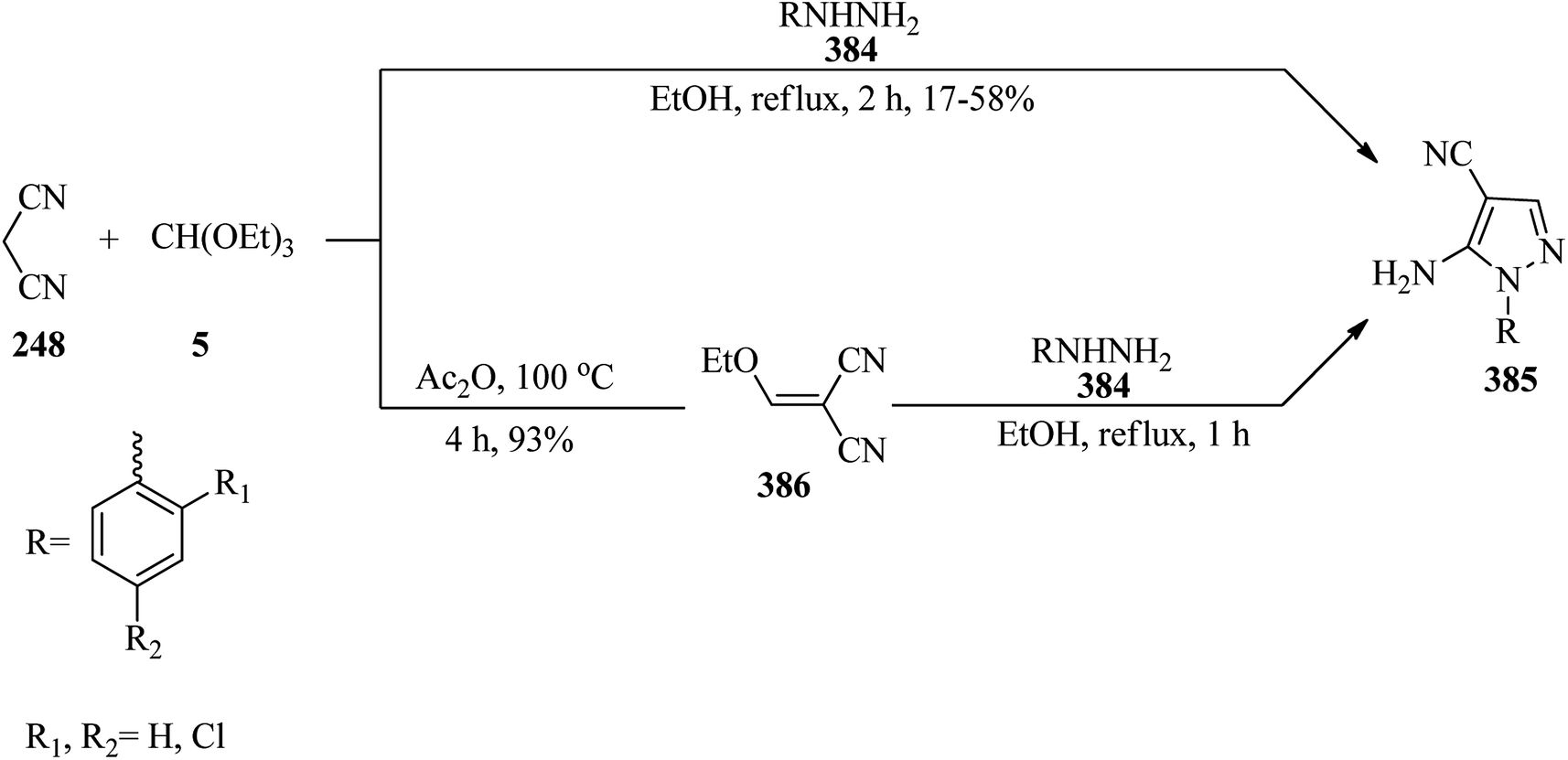
In 2000, Rao et al. described a new simple method for the synthesis of 2-amino-3-cyano-5-N-substitued piperazinyl pyridines (250) via the condensation reaction of TEOAc (6), malononitrile (248), and N-substituted piperazine (249) in a 2:1:1 molar ratio in the presence of excess of ammonium acetate (5 moles) under reflux conditions at 90–92 °C for 2 h in moderate yields (50–68%) (Scheme 68). The products were obtained by a simple filtration and recrystallization from ethylacetate/n-hexane. Elemental and spectral data (FT-IR, PMR) confirmed the structures of the isolated compounds.248
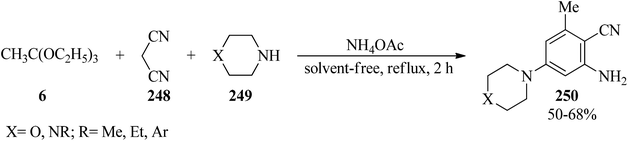 |
| | Scheme 68 Synthetic route to substituted piperazinyl pyridines. | |
2.31. Synthesis of pyrimidine derivatives
In 2010, Nagarajan and Reddy prepared substituted pyrimidine derivatives (253) via the one-pot three-component condensation reaction of acetylenedicarboxylates (251), amines (252), and orthoformates (4, 5) with a molar ratio of 1:2:1 in the presence of ZnCl2 (0.5 mol%) under solvent-free conditions at room temperature in 27–80% yields within 1–3 h (Scheme 69).249
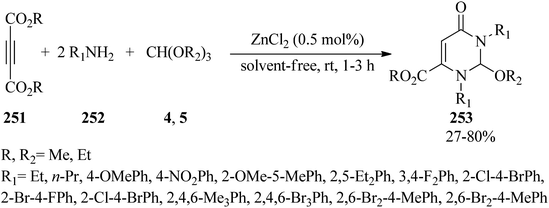 |
| | Scheme 69 The synthesis of substituted pyrimidine derivatives. | |
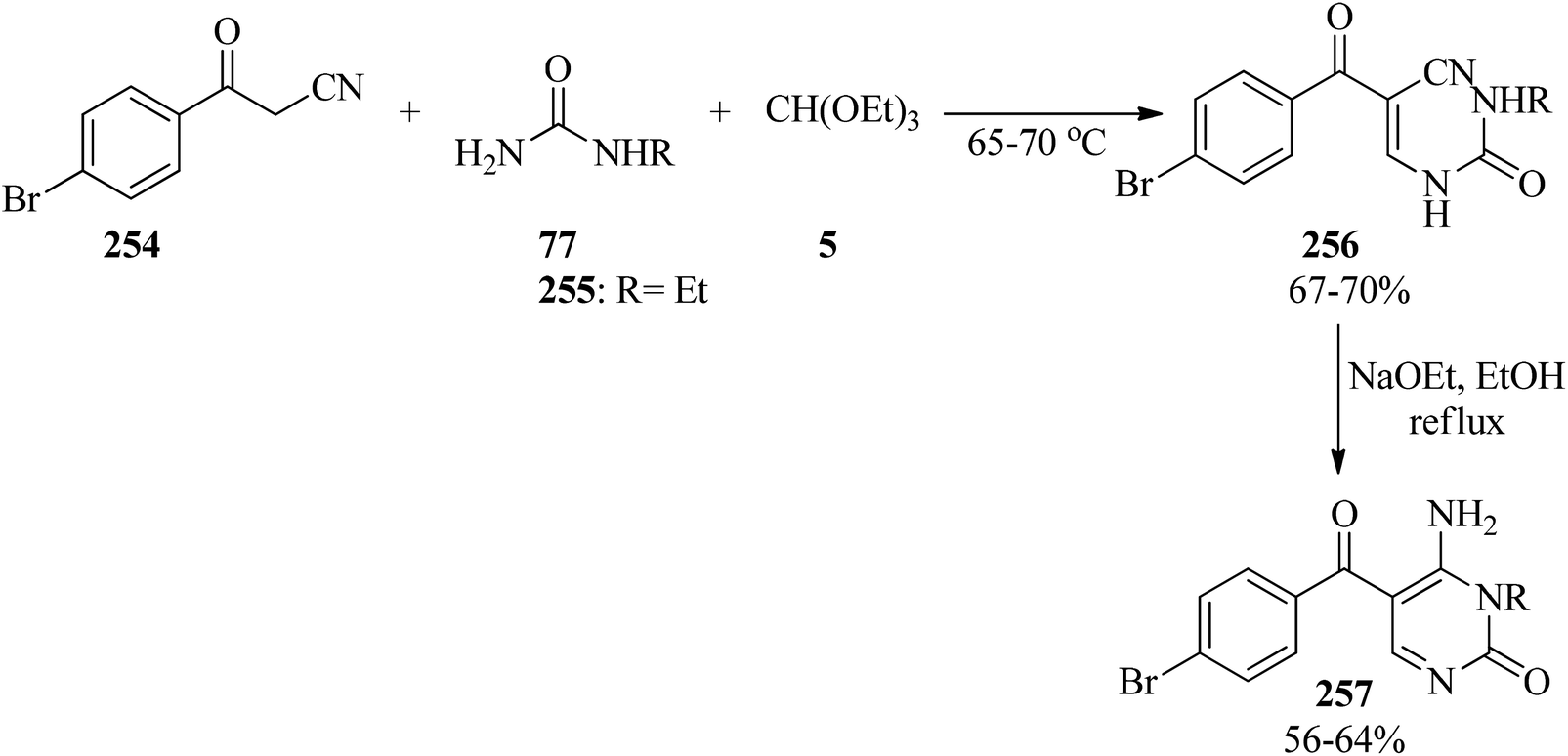
In 2006, Toche et al. presented a two-step procedure for the synthesis of 4-amino-5-(4-bromobenzoyl)-3-ethyl-2(1H)-pyrimidone (257). First, the domino three-component condensation reaction of 4-bromobenzoylacetonitrile (254), mono-alkylated ureas (77, 255), and TEOF (5) at 65–70 °C yielded enamine intermediates (256). Subsequently, the resultant precipitate (which dissolved in EtOH/NaOEt under reflux conditions) underwent an intramolecular cyclization reaction and dissolved in refluxing ethanol to produce 257 in 56–64% yield (Scheme 70). The compounds were evaluated for their insect repellent activity against Sitophilus oryzae (Coleoptera, Curculeonidae).250
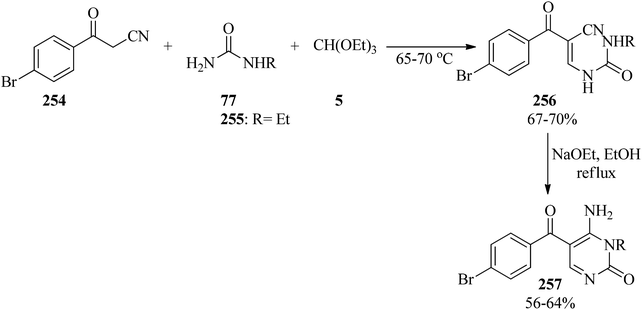 |
| | Scheme 70 The domino synthesis of 4-amino-5-(4-bromobenzoyl)-3-ethyl-2(1H)-pyrimidones. | |
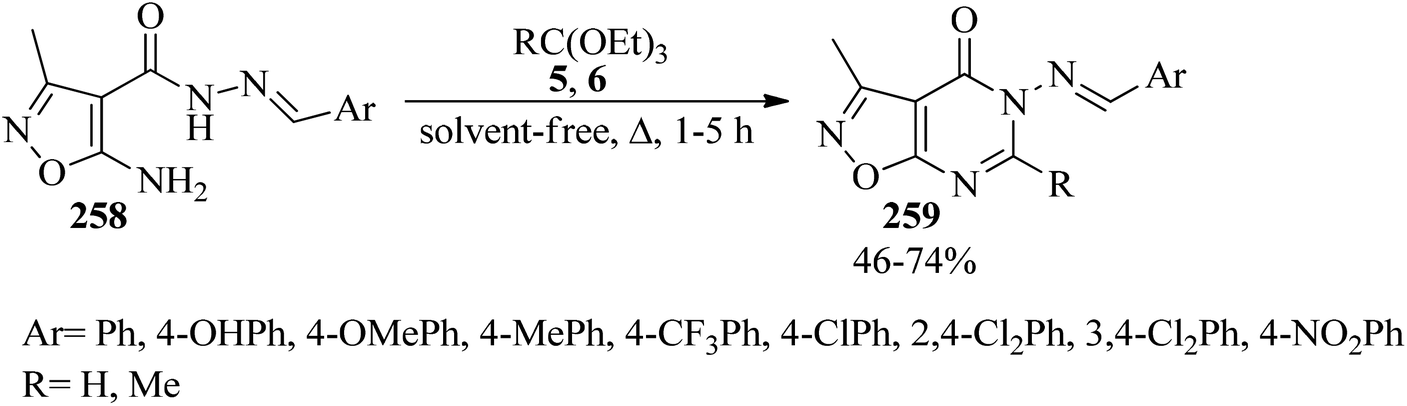
1,2-Oxazolo[5,4-d]pyrimidin-4(5H)-ones (259) were obtained in 46–74% yields via the ring-closure reaction of 1,2-oxazole-4-carbohydrazides (258) with boiling orthoesters (5, 6) under solvent-free conditions within 1–5 h (Scheme 71). The in vitro mycobacterial and cytotoxicity activities of the products were screened against Mycobacterium fortuitum in an MABA test, and lung (A549) and fibroblasts (L929) cell lines, respectively.251
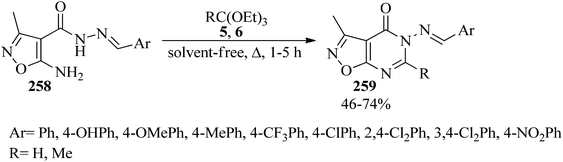 |
| | Scheme 71 Ring closure of 1,2-oxazole-4-carbohydrazides with orthoesters. | |
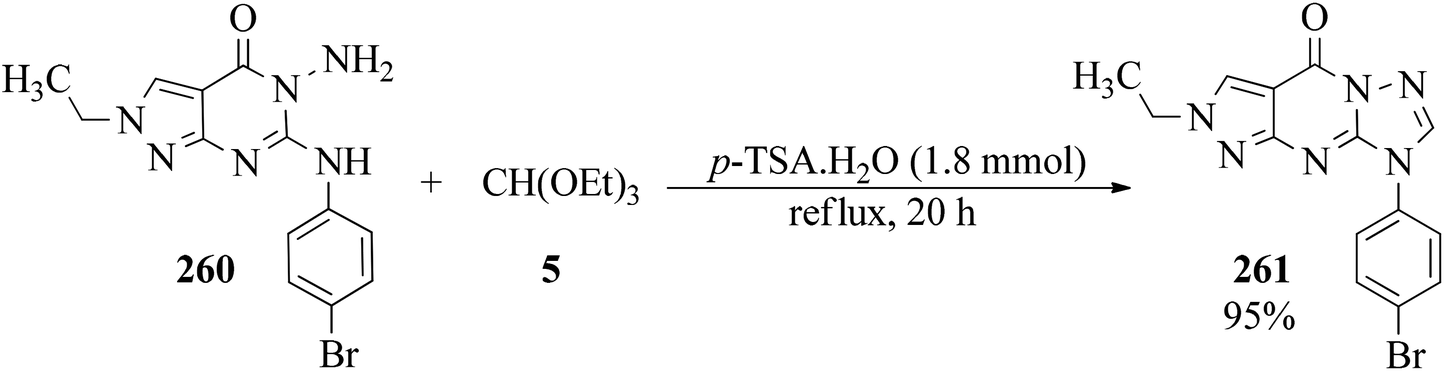
Ethyl-substituted 2-(4-bromo-phenyl)amino-3-aminopyrazolo[3,4-d]pyrimidin-4-one (260) was refluxed in the presence of TEOF (5) and p-TSA, as catalyst, in order to obtain 2-ethyl-8-(4-bromophenyl)-pyrazolo[3,4-d][1,2,4]8H-triazolo[2,3-a]4H-pyrimidin-4-one (261) in 95% yield (Scheme 72).252 The products have previously been reported as potential anti-inflammatory–analgesic agents.253
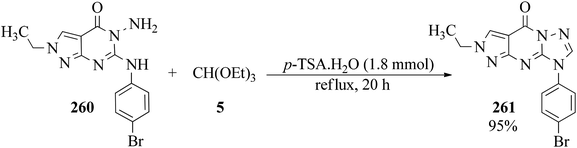 |
| | Scheme 72 The synthesis of 2-ethyl-8-(4-bromophenyl)-pyrazolo[3,4-d][1,2,4]8H-triazolo[2,3-a]4H-pyrimidin-4-ones. | |
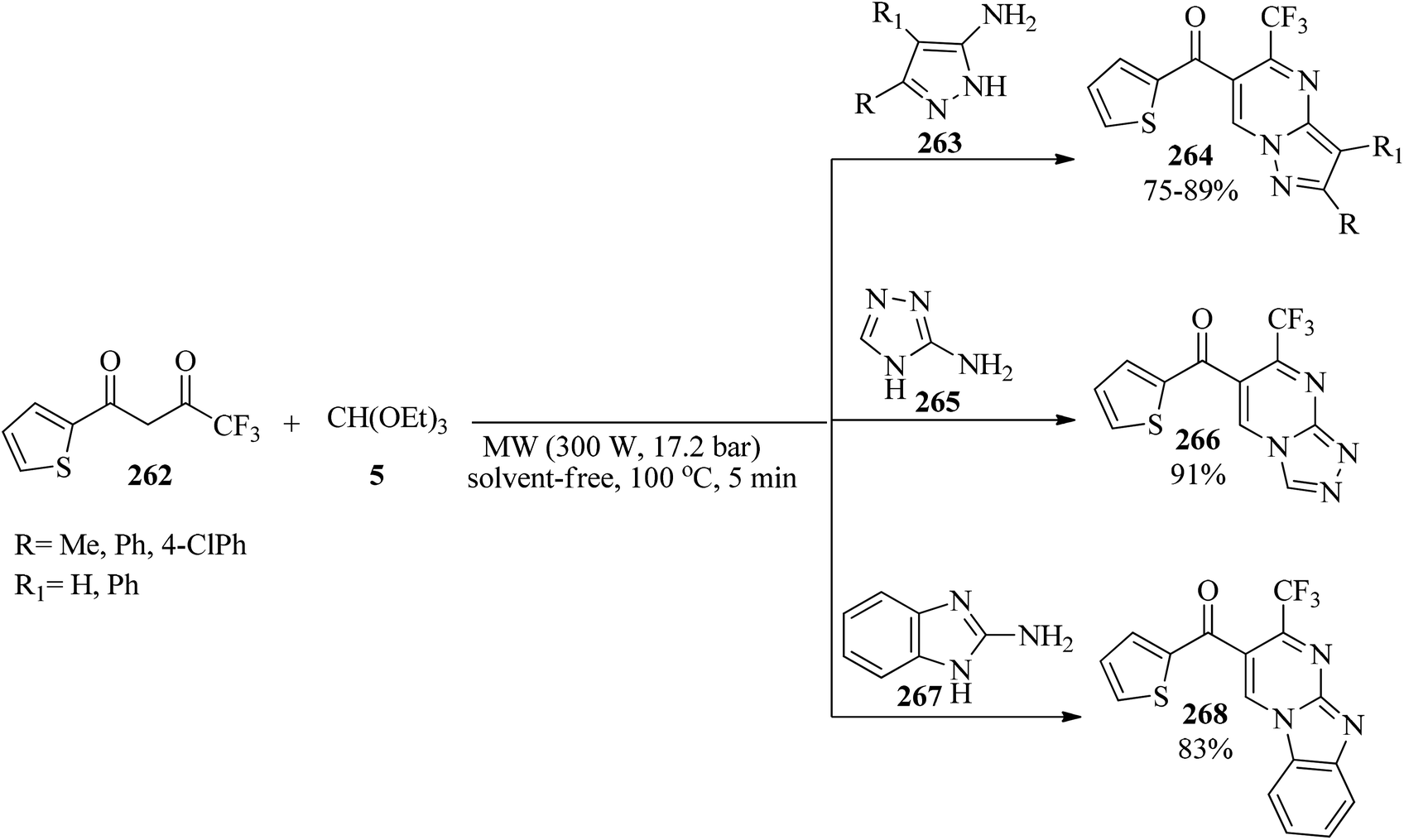
Due to the key role of fluorine-containing heterocyclic products in biological and pharmacological activities,254–256 and also the development of new functional groups,257–263 in 2008, 5-(trifluoromethyl)-pyrazolo[1,5-a]pyrimidines (264), 6-thienoyl-7-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyrimidine (266) and 6-thienoyl-7-(trifluoromethyl)benzimidazo[1,2-a]pyrimidine (268) were synthesized via the condensation reaction of 4,4,4-trifluoro-1-(thien-2-yl)butane-1,3-dione (262), TEOF (5) and the appropriate heterocyclic amines, such as aminopyrazoles (263), 3-amino-l,2,4-triazole (265), and 2-aminobenzimidazole (267), respectively, by pressurized microwave irradiation (300 W, 17.2 bar) at 100 °C under solvent-free conditions for 5 min (Scheme 73).264
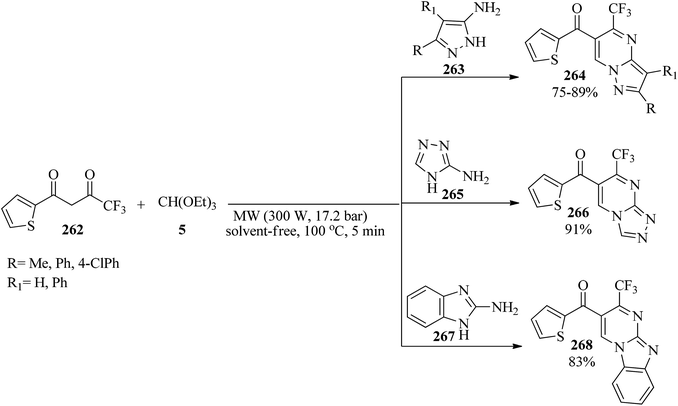 |
| | Scheme 73 The preparation of some fluorinated pyrimidines. | |
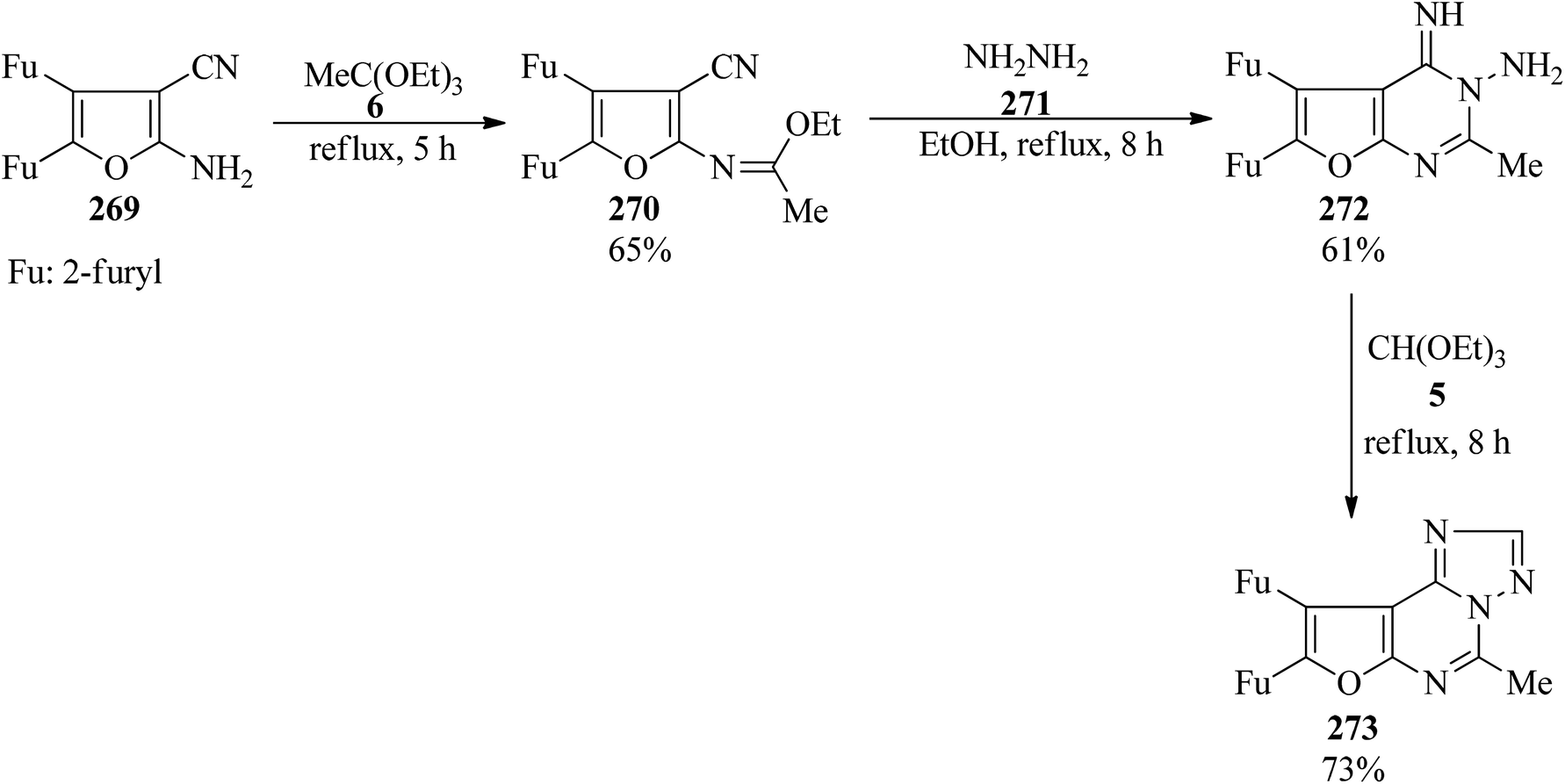
In 2000, 2-amino-4,5-di-(2-furyl)furan-3-carbonitrile (269)265 was utilized as a key starting material for the synthesis of furo[3,2-e][1,2,4]triazolo[1,5-c]pyrimidines (273). At first, the condensation of (269) with TEOAc (6) under reflux conditions for 5 h obtained the corresponding 2-ethoxyimine (270) in good yield (65%) which upon treatment with hydrazine hydrate (271) was converted into (272). Eventually, the cyclocondensation of (272) with TEOF (5) under reflux conditions for 8 h achieved (273) in 73% yield (Scheme 74). The products were purified by simple filtration and crystallization and were finally characterized by their correct analyses and spectroscopic data (IR and 1H NMR).266
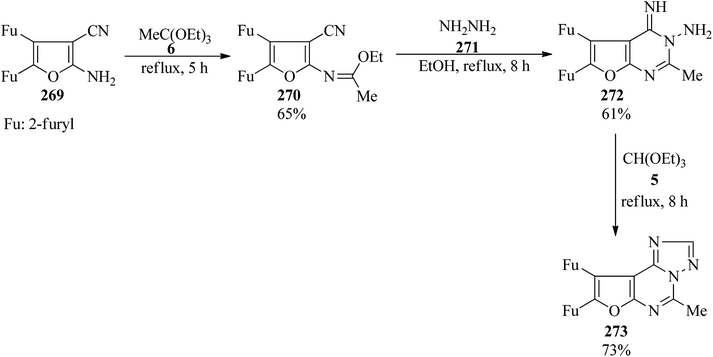 |
| | Scheme 74 Preparation procedure of furo[3,2-e][1,2,4]triazolo[1,5-c]pyrimidines. | |
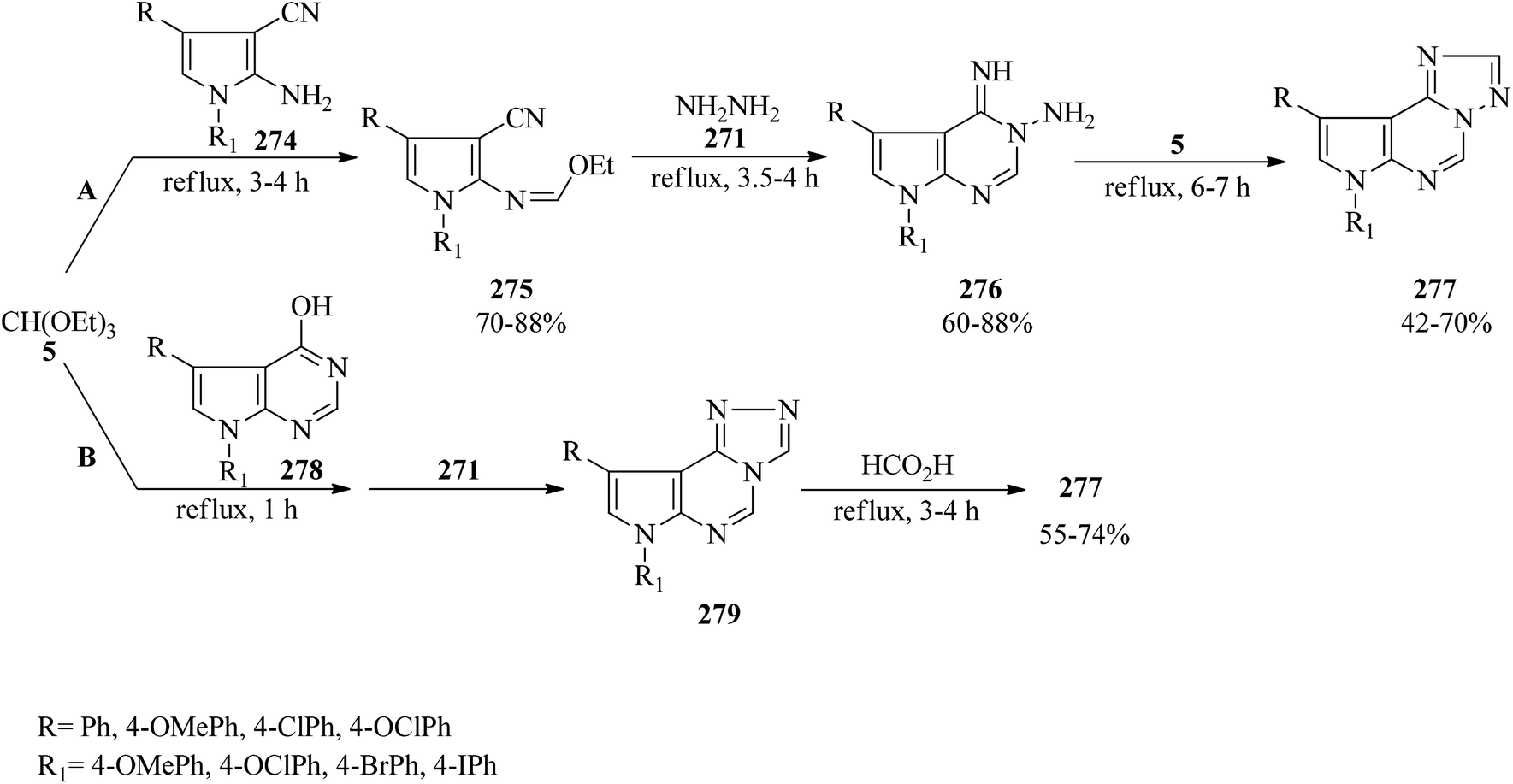
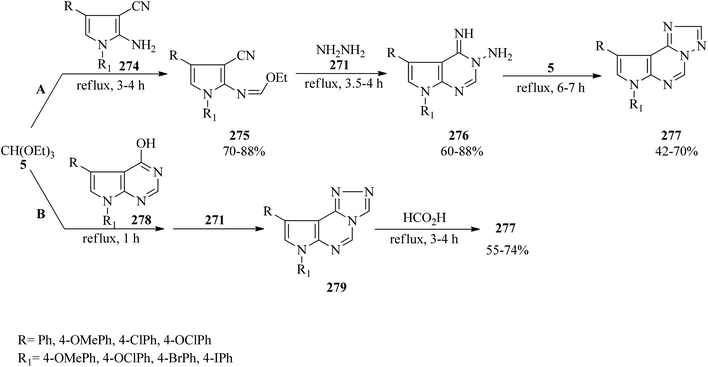
In 2000, Dave and Shah synthesized 7H-1,2,4-triazolo[1,5-c]pyrrolo[3,2-e]pyrimidines (277) via the domino multicomponent reaction of 1,4-disubstituted 2-amino-3-cyanopyrroles (274),267 TEOF (5), and hydrazine hydrate (271) under reflux conditions. This reaction proceeded through the condensation of 274 with 5 under reflux conditions to generate 1,4-disubstituted N-ethoxymethylene-2-amino-3-cyanopyrroles (275) in 3–4 h in good yields (70–88%) which led to 5,7-disubstituted 3-amino-4-imino-7H-pyrrolo[2,3-d]pyrimidines (276) (60–88%) on hydrazinolysis with 271 within 3.5–4 h. Finally, the cyclocondensation reaction of 276 with one carbon donor moiety, such as 5, afforded product (277)268,269 in 6–7 h in weak to high yields (42–70%) (method A). On the other hand, the similar reaction of 4-hydrazino-7H-pyrrolo[2,3-d]pyrimidines (278)270 with 5 and 271 (method B) obtained the [4,3-c] isomers (279), which using formic acid were isomerized to the thermodynamically more stable [1,5-c] isomers (277) through Dimroth rearrangement (Scheme 75). Notably, the latter procedure was faster and more efficient.269
 |
| | Scheme 75 The synthesis of 7H-1,2,4-triazolo[1,5-c]pyrrolo[3,2-e]pyrimidines. | |
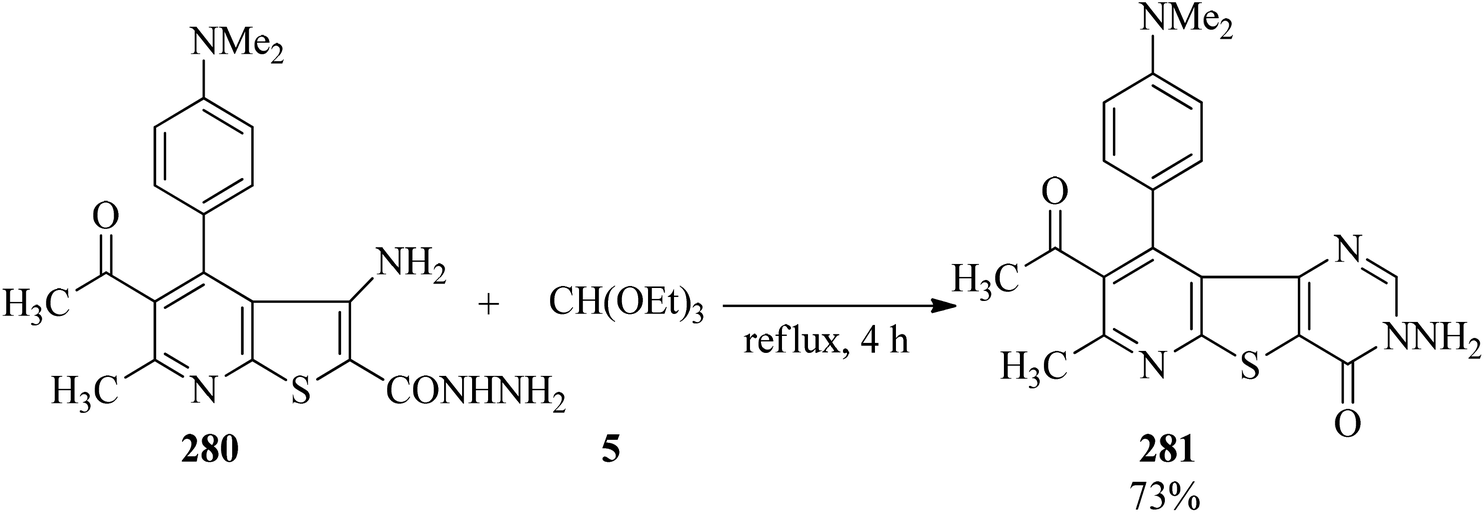
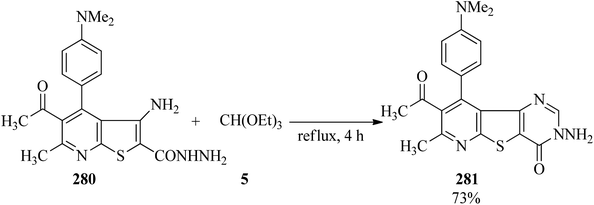
In 2005, Rashad et al. prepared several derivatives containing thieno[2,3-d]pyrimidine systems with potent anti-inflammatory properties: e.g. 11-hydrazino-5,6-dihydronaphtho[1′,2′:4,5]-thieno[2,3-d]pyrimidines were refluxed with triethyl orthoesters for 10 h to obtain 8,9-dihydronaphtho[1′,2′:4,5]thieno[3,2-e][1,2,4]triazolo[4,3-c]pyrimidines271,272 in 72–86% yields; which were isomerized to the thermodynamically more stable triazolo[1,5-c]pyrimidines under heating in either acid or basic media.272 Thieno[2,3-b]-pyridine-2-carbohydrazide (280) was refluxed with TEOF (5) for 4 h to form 3-aminopyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines (281) in 73% yield (Scheme 76).273
 |
| | Scheme 76 Formation of 3-aminopyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines. | |
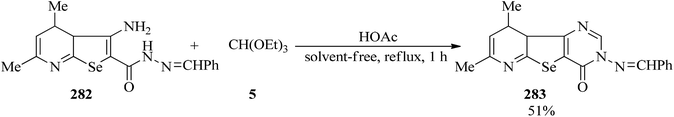
2,4-Dimethyl-7-benzyledineamino-8-oxopyrimido[4′,5′:4,5]-seleno[2,3-b]pyridine (283) was synthesized upon cyclocondensation of benzylidene carbohydrazone (282) with TEOF (5) by a few drops of acetic acid under reflux conditions for 1 h in 51% yield (Scheme 77).274
 |
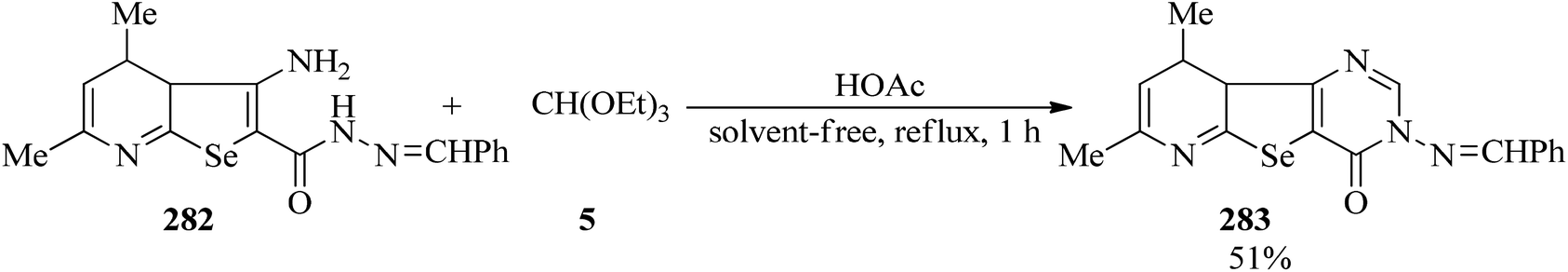
| | Scheme 77 Synthetic approach for 2,4-dimethyl-7-benzyledineamino-8-oxopyrimido[4′,5′:4,5]-seleno[2,3-b]pyridine. | |
In 2004, three pathways were presented for the synthesis of pyrido[2,3-d]pyrimidines (286) through the one-pot three-component condensation reaction of equimolar amounts of 6-aminouracils (284), alkyl nitriles (285), and TEOF (5) in (a) solvent-free conditions, (b) using ethanol as a solvent under heating thermal conditions, and (c) under microwave irradiation with 360 W power at 75 °C utilizing acetic anhydride (Scheme 78), for which the third method had a great effect on decreasing the time and increasing the yield.275 Some compounds have different pharmacological activities, such as antibacterial,276 antitumor,277 cardiotonic, hepatoprotective, and antihypertensive,278 and antibronchitic effects.279
 |
| | Scheme 78 Pyrido[2,3-d]pyrimidine synthetic routes under three sets of conditions. | |
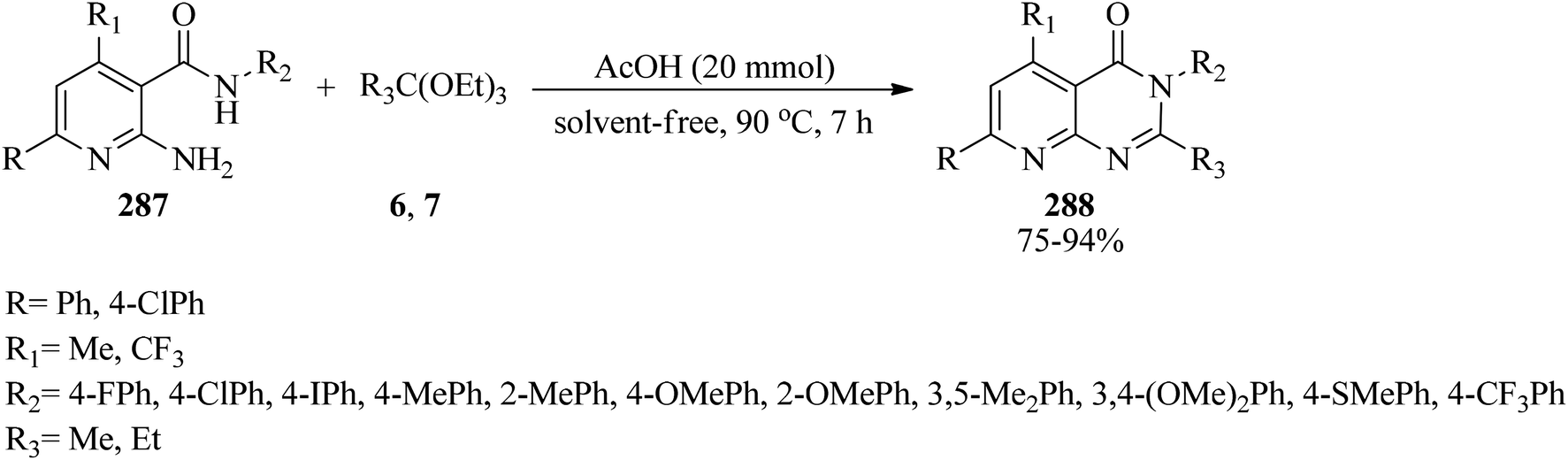
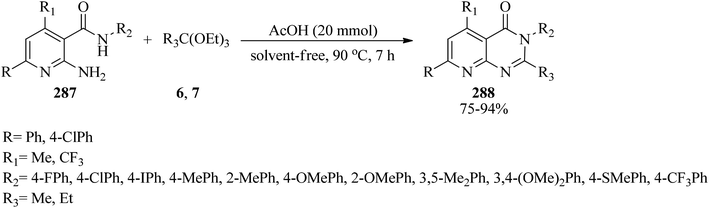
2,3,5,7-Substituted-pyrido[2,3-d]pyrimidin-4(3H)-ones (288) were synthesized in good to excellent yields (75–96%) from the cyclocondensation reaction of 2-amino-N,6-substituted phenyl-4-substituted nicotinamides (287) with orthoesters (6, 7) by catalytic amounts of glacial acetic acid at 90 °C for 7 h (Scheme 79). All the resultant compounds (288) were examined for antibacterial activity against Gram +ve and Gram −ve bacteria.280
 |
| | Scheme 79 The formation of substituted-pyrido[2,3-d]pyrimidin-4(3H)-ones. | |
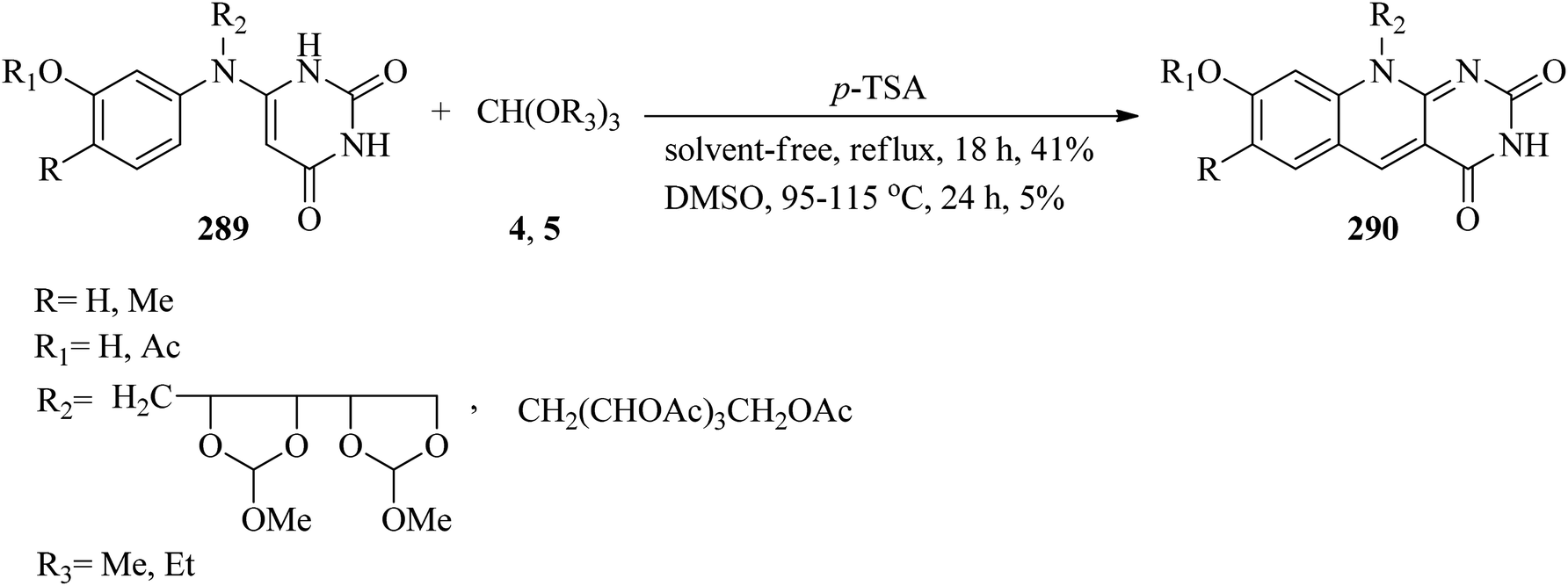
The cyclization of compounds (289) with excess amounts of trialkyl orthoformates (4, 5) resulted in products (290) using catalytic amounts of p-TSA under reflux conditions or in DMSO as solvent at 95–115 °C in 5–41% yields within 18–24 h (Scheme 80).281
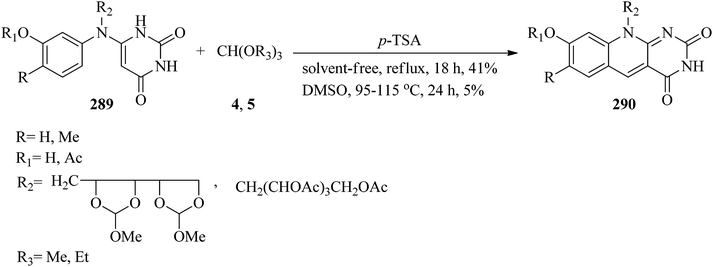 |
| | Scheme 80 Cyclization by TEOF to obtain N-containing heterocycles. | |
Acid-mediated cyclocondensation of ethyl 5-amino-4-(substituted amino)-2-(methylthio)thieno[2,3-d]pyrimidine-6-carboxylates (291) with TEOF (5) resulted in ethyl 5-substituted 7-(methylthio)-5H-1-thia-3,5,6,8-azaacenaphthylene-2-carboxylates (292) in 60–97% yields within 15–60 min (Scheme 81).282
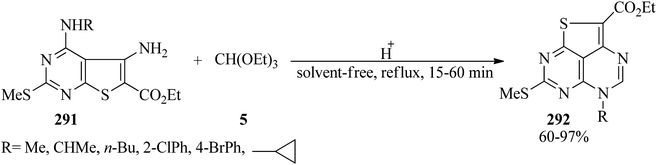 |
| | Scheme 81 The preparation of 7-(methylthio)-5H-1-thia-3,5,6,8-azaacenaphthylene-2-carboxylates. | |
In 2016, Castillo and co-workers developed pyrazolo[5,1-b]purines (294) by a microwave-assisted (300 W) cyclocondensation of pyrazolo[1,5-a]pyrimidine-6,7-diamines (293) with orthoesters (4, 6, 7) at 110–120 °C under solvent-free conditions in good to excellent yields (81–96%) within 5–10 min (Scheme 82).283
 |
| | Scheme 82 The microwave-assisted synthesis of pyrazolo[5,1-b]purines. | |
2.32. Synthesis of triazin derivatives
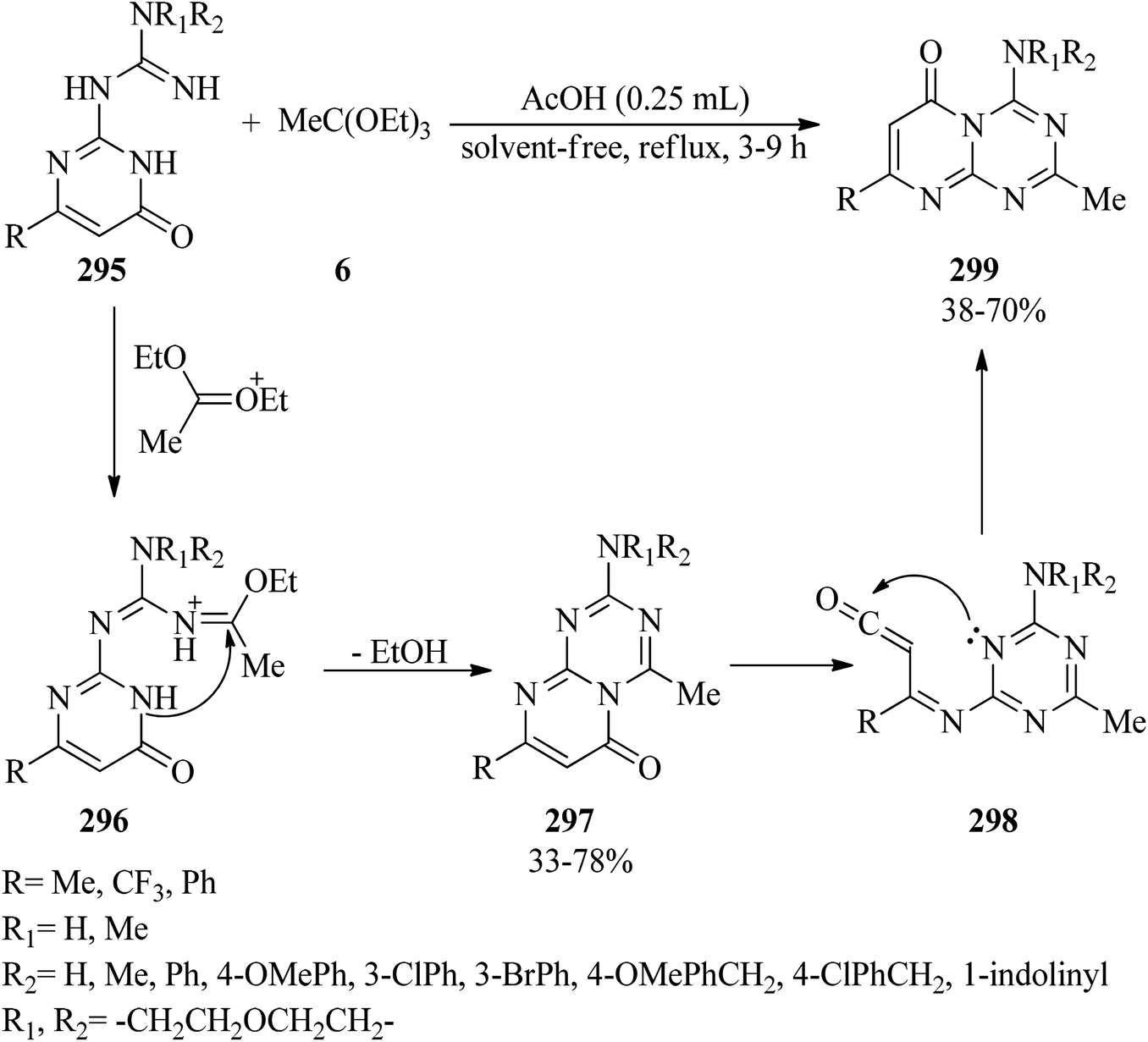
In 2012, Sachdeva and co-workers synthesized 2-amino-4-methylpyrimido[1,2-a][1,3,5]triazin-6-ones (297) and 4-amino-2-methylpyrimido[1,2-a][1,3,5]triazin-6-ones (299) utilizing 1-(6-oxo-1,6-dihydropyrimidin-2-yl)guanidines (295) as starting materials. Firstly, the guanidines (295) were condensed with triethylorthoacetate (6) in glacial acetic acid under reflux conditions to form an iminium ion intermediate (296). Subsequent intramolecular cyclization along with the elimination of ethanol yielded product (297); which was followed by thermal rearrangement for unsubstituted (NH2), alkyl-substituted (NHCH3), and aralkyl guanidines (NHCH2Ph(R)) via ring opening of pyrimidine at the amide moiety to produce the ring-open triazine carbenone (298). Finally, intramolecular nucleophilic attack of the N-1 nitrogen of 1,3,5-triazine on the carbonyl functional group and annulation, respectively, achieved product (299) (Scheme 83). It is noteworthy that the guanidines (295) containing a tertiary amino group such as –N(CH3)2, –N(CH2CH2)2O, indolino and or aryl secondary amino substituent groups, e.g. –NHPh, NHPh(3-Br), NHPh(3-Cl), NHPh(4-OMe) produced mainly the corresponding product 297.284
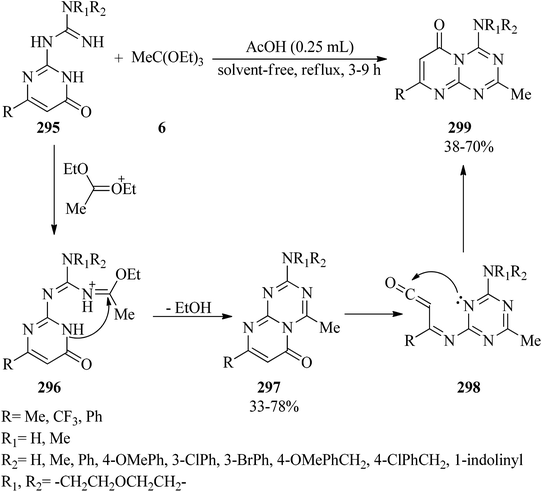 |
| | Scheme 83 The synthesis of 4-amino-2-methylpyrimido[1,2-a][1,3,5]triazin-6-ones. | |
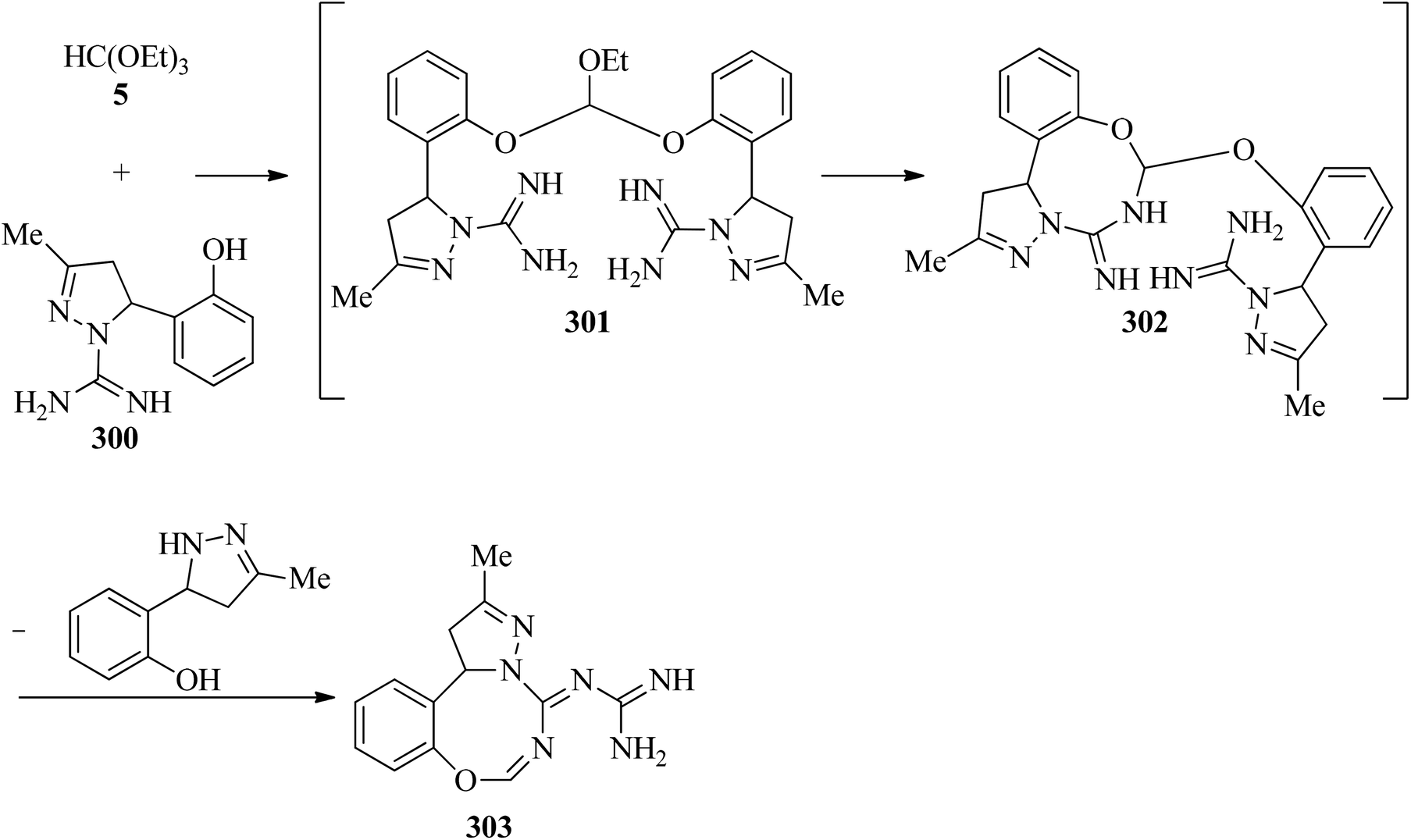
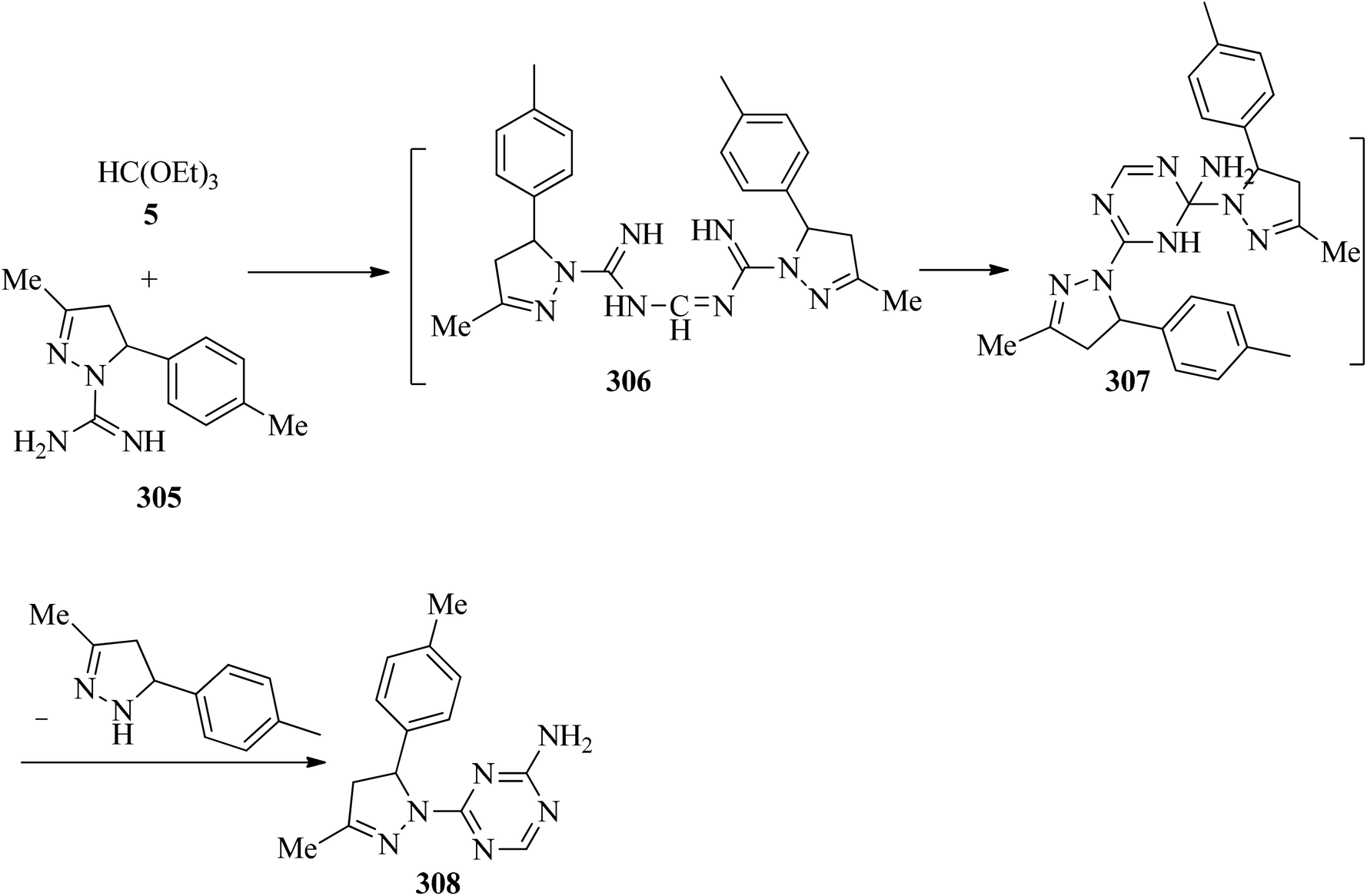
In 2002, Světlík and Liptaj synthesized new heterocyclic ring systems including a pyrazole moiety, such as N-[(5E)-1,12-b-dihydro-substituted-5H-pyrazolo[1,5-e][1,3,5]-benzoxadiazocin-5-ylidene]guanidine (303) or 2-amino-4-[4,5-dihydro-3-methyl-5-(4-methylphenyl)-1H-pyrazol-1-yl]-1,3,5-triazine (308). The products (303) were obtained via the re-esterification of TEOF (5) with phenolic hydroxys of two equivalents of 4,5-dihydro-3-methyl-5-(2-hydroxyphenyl)-1H-pyrazole-1-carboximidamide (300) to produce orthoester (301) which underwent a cyclization reaction with the vicinal amidine moiety to form an oxadiazocine system (302). Then, the nucleophilic attack of the adjoining exocyclic imine nitrogen on the carbon atom of the other amidine group enabled C1-transfer, which was followed by loss of the disubstituted pyrazoline component to obtain (303) (Scheme 84). On the other hand, the condensation reaction of 4,5-dihydro-3-methyl-5-(4-methylphenyl)-1H-pyrazole-1-carboximidamide acetate (305) with 5 under reflux conditions gave intermediate (306) which was followed by a cyclization reaction to get (307). The reaction was completed by the formation of 2-amino-4-[4,5-dihydro-3-methyl-5-(4-methylphenyl)-1H-pyrazol-1-yl]-1,3,5-triazine (308) for 1 h in 65% yield through the elimination of the disubstituted pyrazoline component (Scheme 85).285
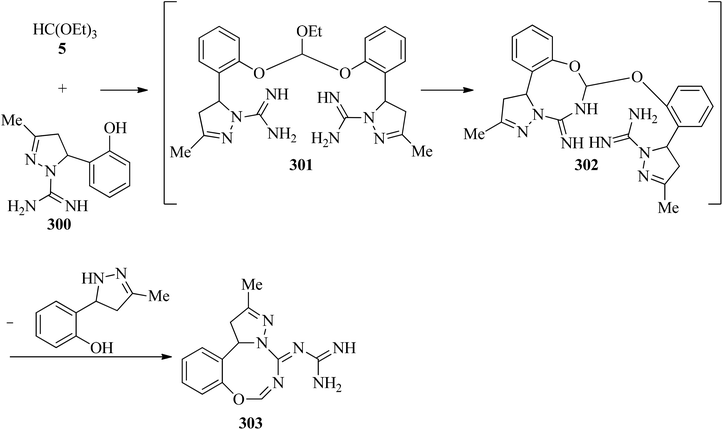 |
| | Scheme 84 The synthesis of dihydro-pyrazolo benzoxadiazocin-5-ylidene guanidines. | |
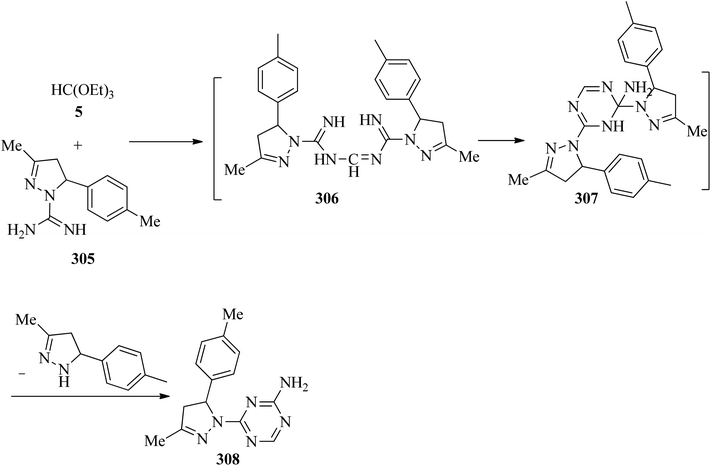 |
| | Scheme 85 Formation procedure for 2-amino-4-[4,5-dihydro-3-methyl-5-(4-methylphenyl)-1H-pyrazol-1-yl]-1,3,5-triazine. | |
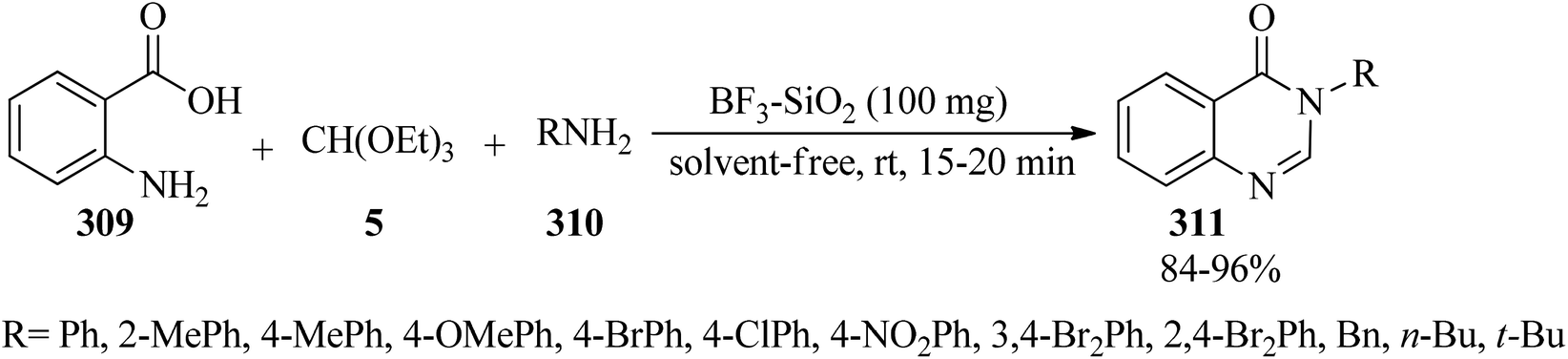
2.33. Synthesis of quinazoline derivatives
In 2010, Wu et al. prepared 4(3H)-quinazolinones (311) in excellent yields (84–96%) and very short reaction times (15–20 min) via the one-pot three-component condensation reaction of anthranilic acid (309),286–293 TEOF (5) and primary amines (310) under solvent-free conditions at room temperature by silica-supported boron trifluoride (BF3–SiO2, 100 mg) as a green, recyclable and reusable heterogeneous catalyst (Scheme 86).293 Microwaves (210 W) accelerated the formation of 4(3H)-quinazolinones in the absence of solvent via the one-pot three-component condensation reaction of anthranilic acid, orthoesters, and ammonium acetate using a catalytic amount of antimony(III) chloride (SbCl3, 1 mol%) in excellent yields (89–93%) within 5 min. According to the results, both aliphatic and aromatic substituents on orthoesters indicated no significant effect on the reaction times and the yields, but generally, the presence of a substituent on orthoesters prolonged the reaction times.294 Quinazolin-4(3H)-ones could also be obtained from the condensation of anthranilic amide with orthoesters using ionic liquids under ultrasound irradiation in 20–25 min in 82–91% yields.209
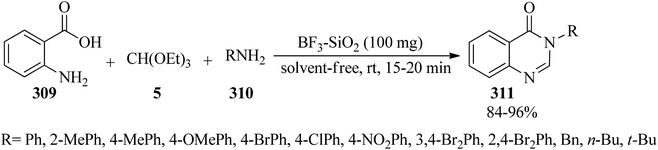 |
| | Scheme 86 The preparation of 4(3H)-quinazolinones. | |
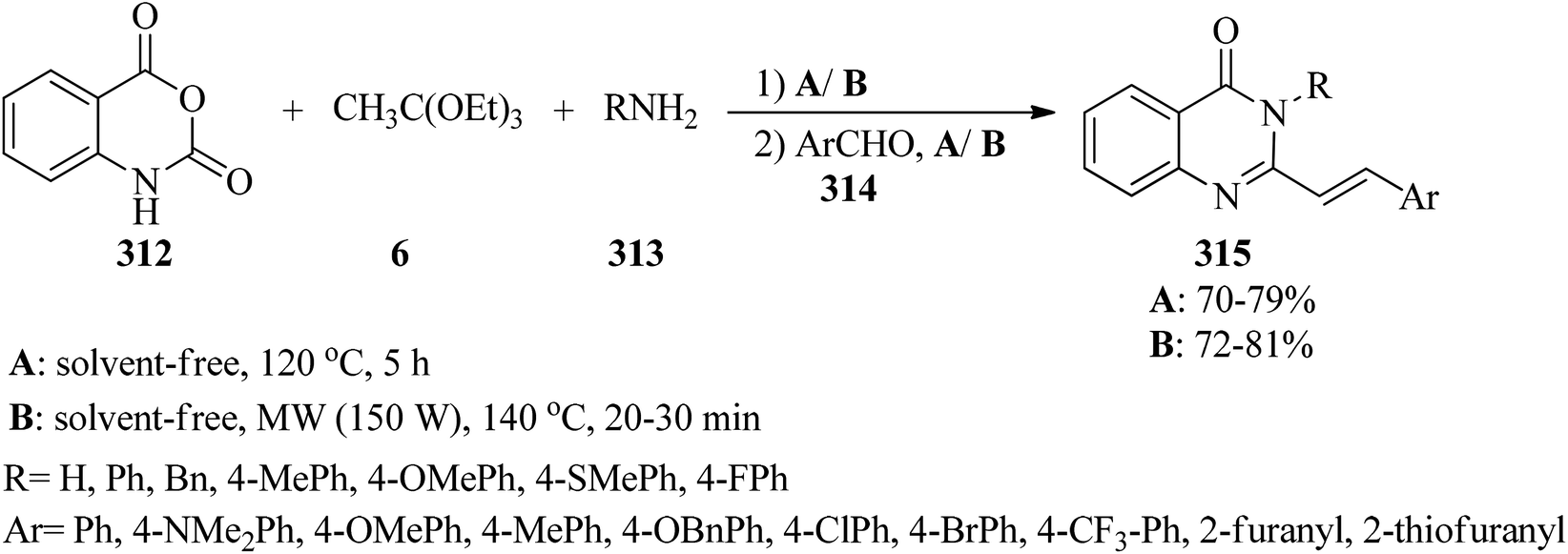
The catalyst/solvent-free one-pot three-component reaction of isatoic anhydrides (312), TEOAc (6), and various amines (313) in equimolar amounts obtained 4(3H)-quinazolinones (311) through two different pathways: (a) utilizing microwave irradiation (150 W) at 140 °C, and (b) under conventional heating at 120 °C. This method developed through the Knoevenagel condensation reaction of 4(3H)-quinazolinones with aromatic aldehydes (314) by a tandem four-component condensation reaction to achieve (E)-3-substituted-2-styrylquinazolin-4(3H)-ones (315).295,296 The results affirmed that MW assisted the reaction progression much better than conventional heating (Scheme 87).296
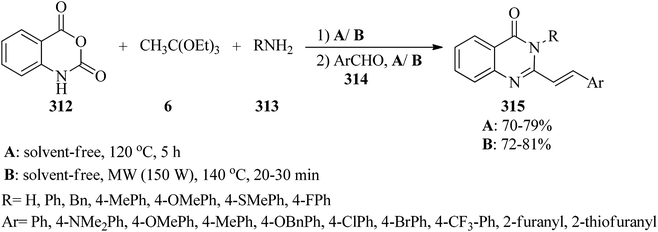 |
| | Scheme 87 Tandem preparation route for (E)-3-substituted-2-styrylquinazolin-4(3H)-ones. | |
In 2006, three pathways were presented for the synthesis of 2-substituted 4-aminoquinazolines (319) via the uncatalyzed one-pot three-component condensation reaction of 2-aminobenzonitrile (234), orthoesters (5, 6, 7, 316, 317), and ammonium acetate (318) in (a) ethanol as solvent under heating conditions, (b) under reflux conditions without a solvent, and (c) microwave irradiation in the absence of a solvent. The results showed that, in comparison with the two premier routes, microwave irradiation has an important influence on decreasing the reaction time (5–7 min) (Scheme 88).297
 |
| | Scheme 88 The synthesis of 2-substituted 4-aminoquinazolines. | |
In 2015, a catalyst and solvent-free one-step method was reported for the synthesis of 3-arylquinazolin-4(3H)imines (321) by heating 2-amino-N′-arylbenzamidines (320) with TEOF (5) (Scheme 89).298
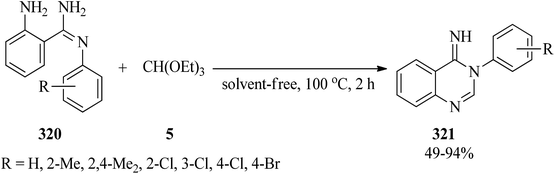 |
| | Scheme 89 The synthesis of 3-arylquinazolin-4(3H)imines. | |
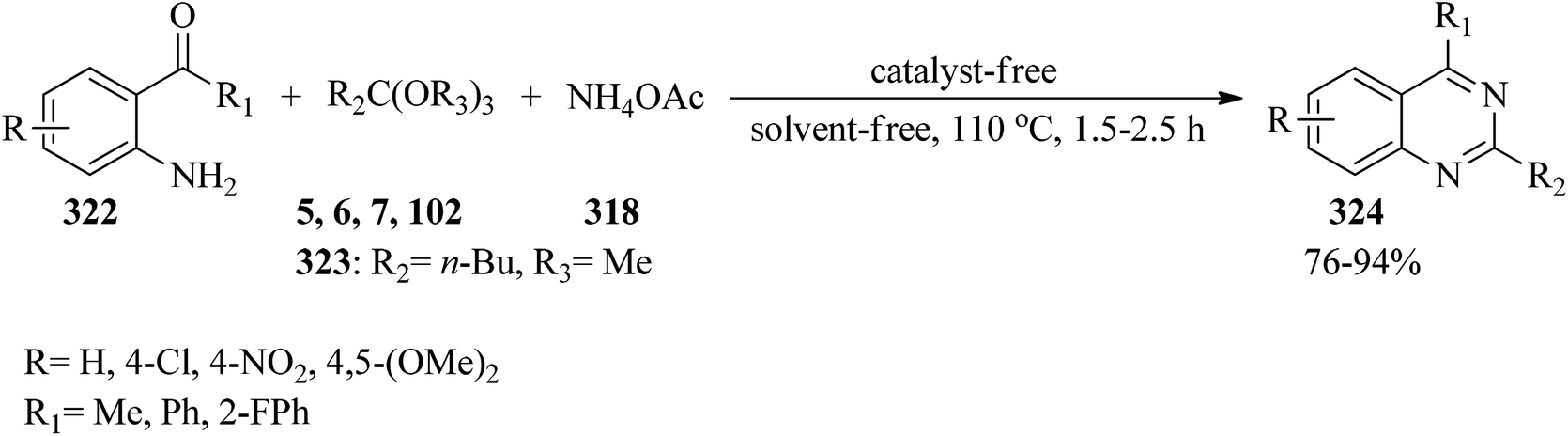
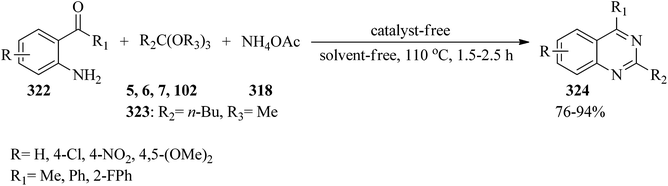
In 2015, Bhat and co-workers, presented an efficient and eco-friendly method for the synthesis of 2,4-disubstituted quinazolines (324) in high yields (76–94%) via the catalyst/solvent-free one-pot three-component reaction of 2-aminoaryl ketones (322), trialkyl orthoesters (5, 6, 7, 102, 323), and ammonium acetate (318) in a 1:1.5:1.5 molar ratio at 110 °C within 1.5–2.5 h (Scheme 90). In this study, in order to investigate the scope and limitations of the presented procedure, the reaction was carried out by diverse substituted 2-aminoaryl ketones and trialkyl orthoesters. The reaction was performed by 2-aminobenzophenones and 2′-aminoacetophenones. It was found that the transformation with 2′-aminoacetophenones proceeded faster, which may be ascribed to the steric effect resulting from the aryl substitution, which decreases the reactivity of the carbonyl group. Remarkably, 2-aminobenzophenones containing both electron-withdrawing and electron-donating groups on the aniline ring provided good results. However, 2′-aminoacetophenones including a strong electron-donating substituent on the aniline ring obtained poor yield. It was also found that the reactivity of aliphatic orthoesters was better than orthobenzoate as an aromatic orthoester, which could probably be attributed to the steric effect of different substituents on the orthoester functional group. It was found that the chain length of the orthoalkylates did not have a significant influence on the reaction results.299
 |
| | Scheme 90 The synthesis of 2,4-disubstituted quinazolines. | |
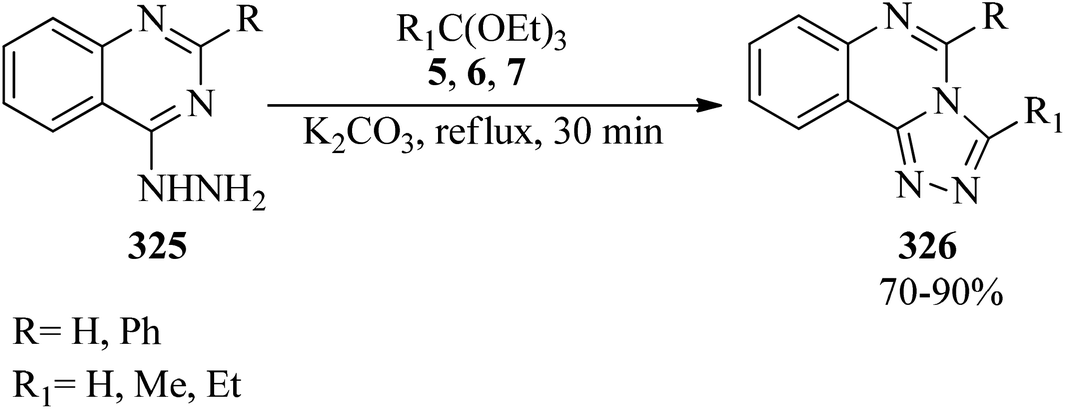
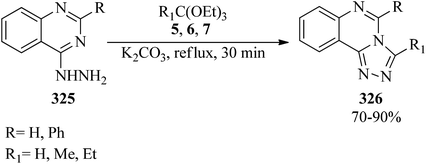
In 1970, Potts and Brugel synthesized 3-substituted-triazolo[4,3-c]quinazolines (326) in good yields (70–90%) via the cyclization reaction of 4-quinazolylhydrazine (325) with orthoesters (5, 6, 7) using potassium carbonate under reflux conditions for 30 min (Scheme 91).300
 |
| | Scheme 91 Formation procedure of 3-substituted-triazolo[4,3-c]quinazolines. | |
2.34. Synthesis of macrocycles
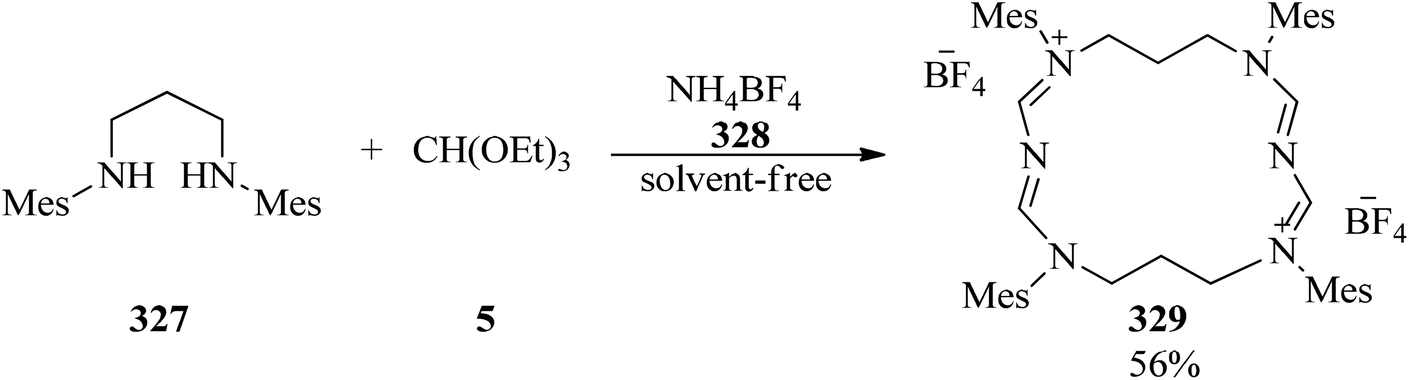
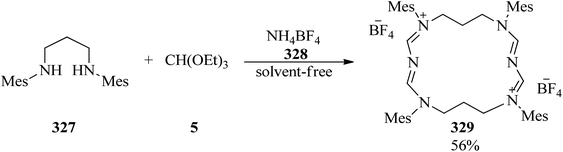
The three-component cyclocondensation of N,N′-dimesityl-propane-1,3-diamine (327), TEOF (5), and ammonium tetrafluoroborate (328) was accomplished to produce the main product macrocycle (329) under solvent-free conditions (Scheme 92) and 1,3-dimesityl-3,4,5,6-tetrahydropyrimidin-1-ium tetrafluoroborate in the presence of ethanol as solvent.301
 |
| | Scheme 92 N-Bearing macrocycle synthesis. | |
3. Orthoester reactions in aqueous media
One of the most interesting challenges to chemists is the reaction performance in water media as a cheap, readily available, safe, and non-toxic solvent. This effective and completely green medium causes the ready hydrolysis of some substrates and/or products, such as orthoesters. In fact, reports of the transformation of orthoesters in aqueous media are very rare (8 references) that are discussed below.
3.1. Synthesis of formamidines
In 2013, a simple, eco-friendly, and catalyst-free methodology was reported for the N-formylation of amines by TEOF under ultrasound irradiation in water.302 Symmetric formamidines could be also prepared via the N-formylation reaction of amines with TMOF using tin(II) chloride–choline chloride (ChCl/SnCl2, 30 mol%), as catalyst and reaction media, in water at 70 °C for 20–160 min in 30–80% yields.127
3.2. Alkylation reactions
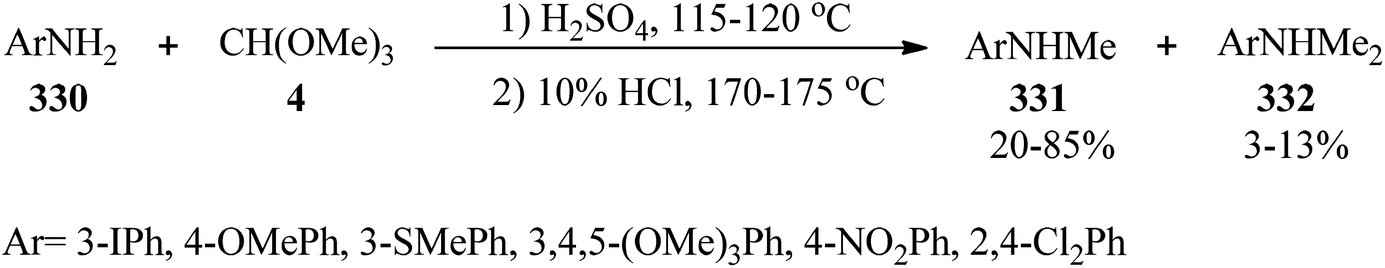
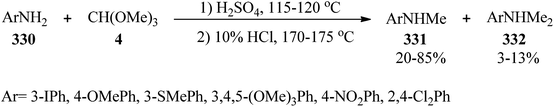
The treatment of anilines (330) with TMOF (4) utilizing sulfuric acid at 115–120 °C resulted in the formation of an N-methylformanilide intermediate; which without isolation was hydrolyzed by aqueous hydrochloric acid (10%) at 170–175 °C to obtain N-methylated anilines (331) in weak to good yields (20–85%) and in some cases along with dimethylated derivatives (332) in minor yields (3–13%) as by-products (Scheme 93).303
 |
| | Scheme 93 The aqueous alkylation of amines. | |
3.3. Synthesis of N-arylureas
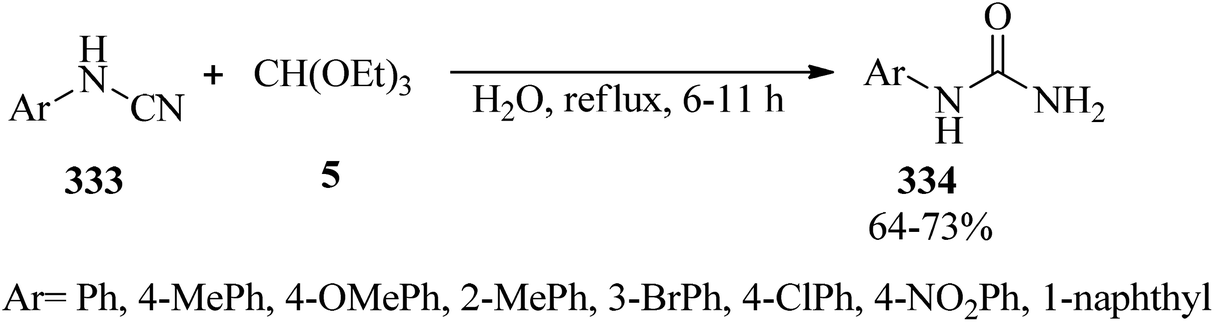
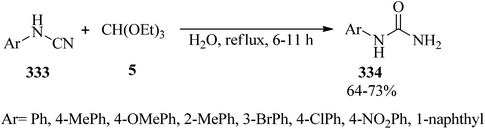
In 2014, a facile and green procedure was presented for the preparation of N-arylureas (334) through the hydration of various aromatic cyanamides (333) with TEOF (5) in refluxing water in 64–73% yields within 6–11 h (Scheme 94). It should be noted that the cyanamides including electron-withdrawing groups prolonged reaction times.304
 |
| | Scheme 94 The water-mediated synthesis of N-arylureas. | |
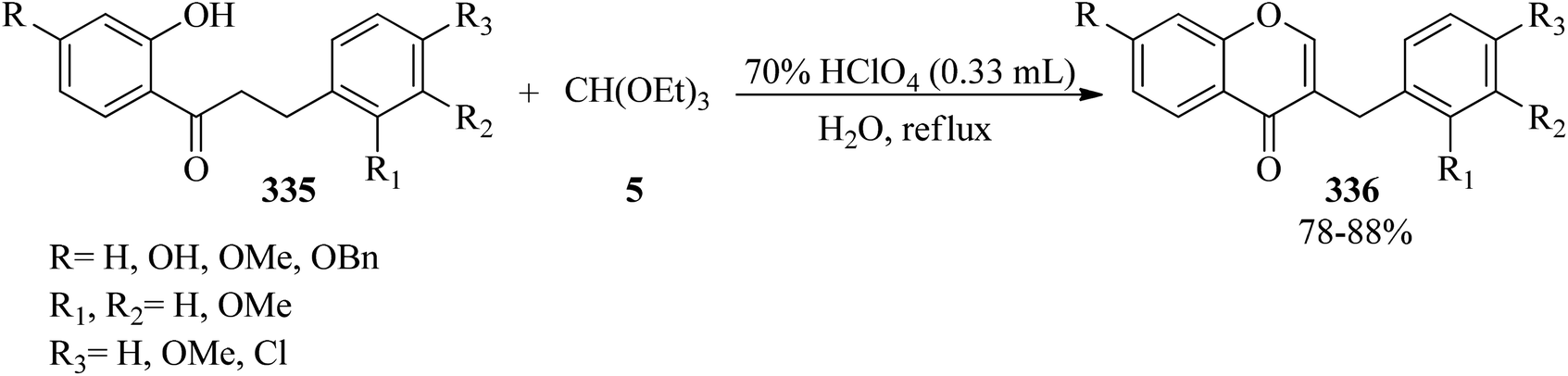
3.4. Synthesis of homoisoflavones
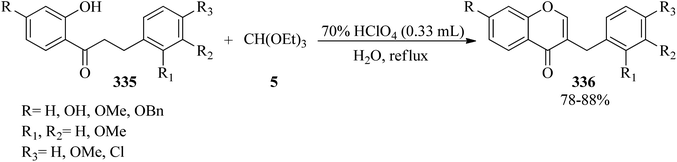
In 2008, Rao et al. described a process for the preparation of homoisoflavones (336)305 in good yields (78–88%) via the ring closure of TEOF (5) with 1-(2-hydroxyphenyl)-3-phenylpropane-1-ones (335) in the presence of 70% perchloric acid, which was followed by aqueous hydrolysis of the intermediate perchlorates (Scheme 95). The in vitro antioxidant and antifungal activities of the products were examined using the superoxide (NBT) and agar cup methods, respectively.306
 |
| | Scheme 95 The preparation of homoisoflavones. | |
3.5. Synthesis of benzoxazoles
Maleki et al. synthesized and characterized Ag (1.5%)@TiO2 (0.1 mL) as a heterogeneous, hazardless, eco-friendly, and reusability nanocatalyst. Then its catalytic activity was examined for the preparation of benzoxazole214,307 via the condensation reaction of orthoesters with 2-amino phenol in water at room temperature within very short reaction times (4–10 min) and at excellent yields (82–93%).307
3.6. Synthesis of 1-substituted-1H-1,2,3,4-tetrazoles
In 2018, Sharghi et al. synthesized iron-doped acidic multi-walled carbon nanotubes (Fe@acidic-MWCNs). The characterization by inductively coupled plasma (ICP), X-ray diffraction (XRD), scanning electron microscopy (SEM), atomic force microscopy (AFM), Raman, and FT-IR analysis confirmed that the iron nanoparticles were supported on the acidic multi-walled carbon nanotubes (MWCNs). Subsequently their catalytic activity was probed for the preparation of 1-substituted-1H-tetrazoles via the one-pot three-component reaction of substituted anilines, TEOf, and sodium azide in water at 50 °C within 1–3.5 h with excellent yields (80–95%). This reaction was carried out faster in the presence of electron-donating anilines than with electron-withdrawing analogs.308 Copper(II) nanoparticles loaded on magnetic core (Fe3O4@SiO2/aza-crown ether–Cu(II)) as a new and highly effective magnetic nanocomposite catalyst in water at 100 °C also facilitated this transformation within 40–70 min in excellent yields (87–98%).309
4. Orthoester reactions in organic solvents
Organic solvents, as carbon-based liquids, are conventional common media to accomplish organic transformations. There are too many organic liquids categorized as solvents. The subdivision of the vast list of solvents could be based on polarity, melting point range, volatility, chemical inertness towards substrates and reagents, or solubility of the existing components in the reaction. In addition to the solvent characteristics, the chemical and physical nature of each reaction member, and the effective relationship between components and solvents determines the choice of reaction media. According to the detailed data below, most solvents utilized in orthoester-participating transformations are alcohols that dissolve many organic compounds as well as most orthoesters (20 reports). The subsequent grades belong to CH2Cl2 (16 cases), acetic anhydride (15 items), benzene and its halogen and alkyl substituted analogs (such as chlorobenzene, xylenes, and toluene, 11 cases), acetic acid (10 items), CH3CN (8 cases), THF (5 points), ether and DMF (each with 3 cases), and dioxane (2 items). The other solvents reported in the literature survey are DMSO, pyridine, CCl4, and ionic liquid that each appeared in just one study.
4.1. Synthesis of ethoxymethylenes
In 2000, Elkholy et al. reported the synthesis of 1-benzoyl-1-phenylsulfonyl-2-ethoxyethene containing an ethoxymethylene group (338),310–315 as yellow crystals, by the reaction of 1-phenyl-2-(phenylsulfonyl)ethanone (337) with TEOF (5) in refluxing acetic anhydride. In this reaction 2 moles of ethanol were removed (Scheme 96).315
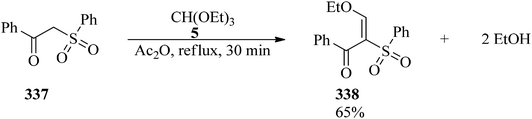 |
| | Scheme 96 The synthesis of ethoxymethylenes. | |
4.2. Synthesis of ethoxymethylene amino derivatives
In 2002, 2-(4-fluorophenyl)methylidene-7-(4-fluorophenyl)-2,3-dihydro-5-ethoxymethyleneamino-7H-3-oxo-thiazolo[3,2-a]-pyrimidin-6,8-dicarbonitrile containing an ethoxymethylene amino motif (340)316–328 was synthesized via the condensation of 5-amino-2-(4-fluorophenyl)methylidene-7-aryl-6,8-dicyano-3-oxo-2,3-dihydro-7H-thiazolo[3,2-a]pyridine (339) with TEOF (5) in refluxing acetic anhydride (10 mL) (as catalyst and solvent) (Scheme 97). Pure (340) was obtained from recrystallization from cold water in 78% yield. Compound (340) exhibited highly antimicrobial activity against Staphylococcus aureus (NCTC-7447).328
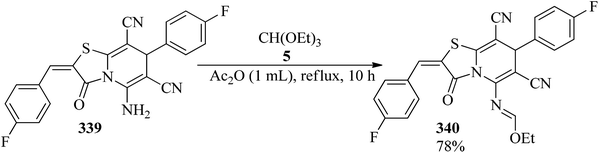 |
| | Scheme 97 The synthesis of an anti-Staphylococcus aureus heterocycle. | |
4.3. Synthesis of acetamides
The three-component Ugi reaction of 2-aminophenols (341), cyclohexyl isocyanide (342), and TEOF (5) in equimolar amounts was accomplished using ZnCl2 (10 mol%) in methanol at room temperature to construct N-cyclohexyl-2-(2-hydroxyphenylamino)-2-ethoxyacetamides (343) for 12 h in high yields (80–82%) (Scheme 98).329
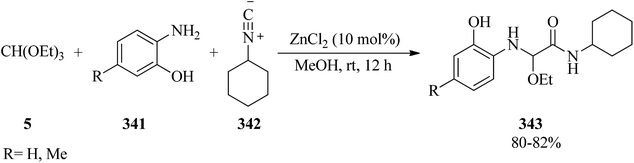 |
| | Scheme 98 The Ugi reaction to obtain acetamides. | |
4.4. Synthesis of enamines
In 2006, Komkov et al. investigated the reaction of cyclohexane-1,3-diones (195, 197) and orthoesters (6, 7) with various amines (344, 346). It was found that the one-pot three-component condensation reaction of cyclohexane-1,3-dione (195) and orthoesters with 5-acetyl-4-aminopyrimidines (344) under reflux in p-xylene obtained a novel series of 7-(1,3-dioxocyclohex-2-ylidene)-7,8-dihydropyrido[2,3-d]pyrimidines (345) in 32–61% yields in 2.5–5 h, while the condensation of dimedone (197) and orthoesters with p-toluidine (346) progressed to generate enamine derivatives (347)330–335 under reflux conditions within 8 h in 44–58% yields (Scheme 99).336
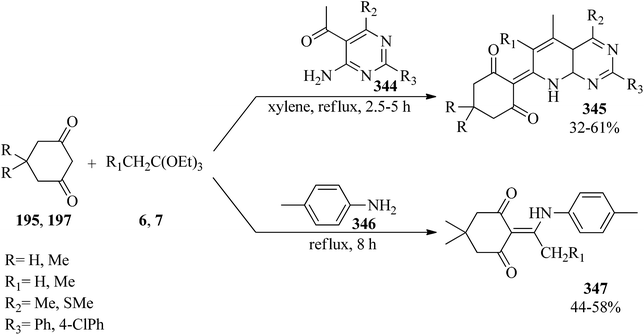 |
| | Scheme 99 The synthesis of enamine-bearing heterocycles. | |
4.5. Synthesis of formamidines
Lim et al. designed the catalyst-free one-pot three-component condensation reaction of polysubstituted 5-aminopyrazoles (348), TEOF (5),337 and cyclic secondary amines (349)338 under microwave irradiation (150 W) at 150 °C in methanol in order to prepare novel N-pyrazolylformamidines (350) in 20 min in 64–93% yields (Scheme 100). Some of the synthesized products were found to inhibit interleukin-17 secretion in phenotypic in vitro evaluations.339
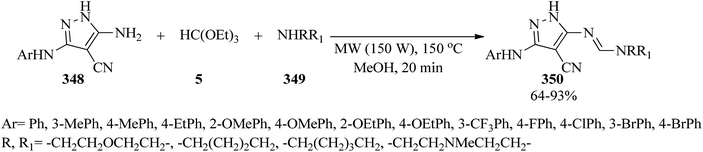 |
| | Scheme 100 Preparing novel N-pyrazolylformamidines. | |
1,3-Bis-arylcarbamylformamidines were prepared via the reaction of N-arylureas with TEOF in boiling acetic anhydride in 85–90% yields.340
4.6. Synthesis of substituted 2-dialkyliminio-1,3-dithietane tetrafluoroborates
In 2015, a facile and straightforward methodology was presented for the synthesis of 4-(N,N-dialkyldithiocarbamato)-2-dialkyliminio-1,3-dithietane tetrafluoroborates (354) in 30–42% yields. In this paper, the condensation reaction of secondary aliphatic amines (351) with carbon disulfide (352) in CHCl3 at room temperature first afforded dithiocarbamic acid salt (353) which upon treatment with TMOF (4) using BF3·OEt2 as catalyst in CHCl3 at room temperature furnished (354) (Scheme 101). This reaction failed with some orthoesters, such as orthovalerate and trimethyl orthobenzoate.341
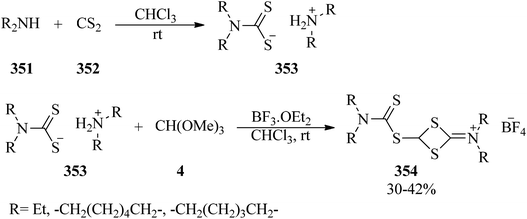 |
| | Scheme 101 Dithiocarbamic acid salt synthetic approach. | |
4.7. Synthesis of lactones
In 2015, Wang and co-workers described the asymmetric total synthesis of (+)-isoiresin (363) and (−)-isoiresin (364) from (3S,5aR,7aS,8R,9R,11aR,11bR)-8-(hydroxymethyl)-3,8,11a-trimethyldecahydro-1H-3,5a-epoxynaphtho[1,2-e][1,3]dioxepin-9-ol (356) derived from readily available aldehyde (355). This transformation occurred via acidic hydrolysis (HCl, 6 N) of the orthoester (356) in THF at 23 °C for 30 min to form a mixture of monoacetate compounds (357a and 357b) which underwent acetylation to produce the tetrakis-acetate (358). Subsequent regioselective dehydration of (358) by various dehydrating agents and then deacetylation resulted in (359) and (360) in different molar ratios, which upon the regioselective oxidative lactonization reaction using Fétizon reagent obtained (361 and 362). Finally, the reduction of the aldehyde functional group by NaBH4 at 0 °C in methanol yielded (363) and (364) (Scheme 102).342
 |
| | Scheme 102 The asymmetric total synthesis of (+)-iresin and (−)-isoiresin. | |
4.8. Synthesis of aminomethylene bisphosphonates
In 2012, Reddy et al. presented an effective, rapid and green method for the synthesis of aminomethylene bisphosphonates via the one-pot three-component reaction of amines, diethyl phosphite and TEOF in a 1:2:1 molar ratio using Yb(PFO)3 (10 mol%) in the presence of ionic liquid [bmim][Cl] at 100 °C for 15–25 min in good to excellent yields (70–93%).343
4.9. Synthesis of 4-dialkylarylamino-1-naphthaldehyderivatives
The reaction of N,N-dialkylarylamines (365) with TMOF (4) was accomplished using TiCl4 (10 mmol) in CH2Cl2 at 0–25 °C to obtain the corresponding formyl derivatives (366) in 75–89% yields (Scheme 103).344
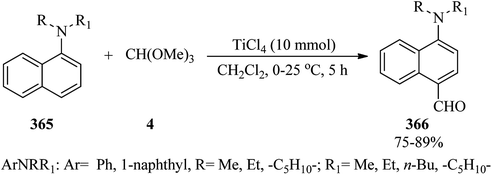 |
| | Scheme 103 The formylation of N,N-dialkylarylamines by TMOF. | |
4.10. Synthesis of pyrrolidines and piperidines
Boron trifluoride diethyl etherate (BF3·OEt2, 0.050 mmol) catalyzed the cyclodehydration of N-carbamate-protected amino alcohols (367) with trimethyl orthobenzoate (102) in CH2Cl2 at room temperature to get azaheterocycles such as pyrrolidines and piperidines within 2–7 h, in high yields (62–87%) (368). Replacing (367) with N-carbamate-protected amino vicinal diols (369) in refluxing CH2Cl2 yielded regio- and stereoselective prolinol derivatives (370) in excellent yields (87–94%) (Scheme 104).345
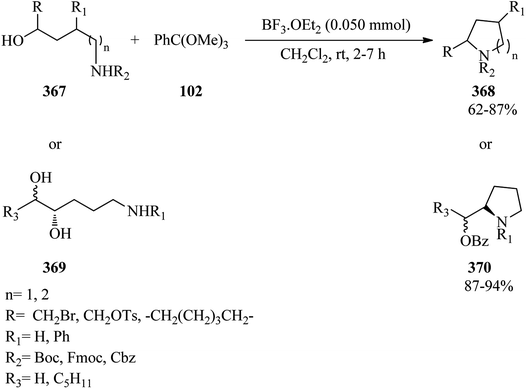 |
| | Scheme 104 The synthesis of pyrrolidines and piperidines. | |
4.11. Synthesis of pyrrole derivatives
In 1998, Tardieux and co-workers presented a simple and mild one-step route for the synthesis of 3,4-disubstituted pyrrole-2,5-dicarbaldehydes (372) via the diformylation346,347 reaction of β-substituted pyrrole-2-carboxylic acids (371) with TEOF (5) in trifluoroacetic acid, in a dual role of catalyst and solvent, at room temperature for 1 h in 22–65% yields (Scheme 105).347
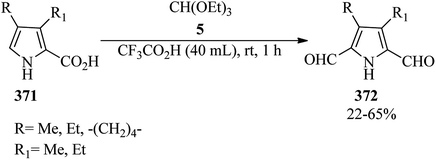 |
| | Scheme 105 The preparation of disubstituted pyrrole-2,5-dicarbaldehydes. | |
Tri-(pyrrol-2-yl)alkanes (374) could be synthesized via the reaction of pyrrole (373) with orthoesters (5, 7, 62, 102) using chloroacetic acid (45 mmol) in CH2Cl2 at 0 °C within 30 min, in 34–38% yields (Scheme 106). 5-Phenyl-4,6-dipyrrin has also been prepared in comparatively good yield (51%) through the reaction of pyrrole with trimethyl orthobenzoate by dichloroacetic acid (38 mmol) under similar conditions.348
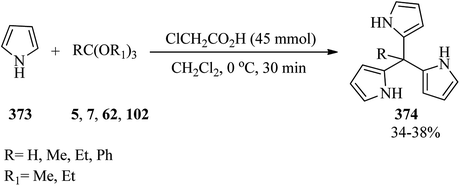 |
| | Scheme 106 Synthetic procedure for tri-(pyrrol-2-yl)alkanes. | |
4.12. Synthesis of indole derivatives
In 1990, indole-3-carbaldehyde (376) as pale yellow crystals was synthesized upon treatment of 4-methoxyindole (375) with TEOF (5) using trichloroacetic acid in CH2Cl2 at room temperature within 20 min in 36% yield. On the other hand, bis-benzo-annellated pentamethine cyanines such as bis(4-methoxyindol-3-yl)methyliumtrichloroacetate (377a) and bis(4-methoxyindol-3-yl)methyliumtetrafluoroborate (377b) were also synthesized in 51 and 76% yields under similar conditions using trichloroacetic acid and tetrafluoroboric acid diethyl ether (HBF4·OEt2), respectively (Scheme 107).349
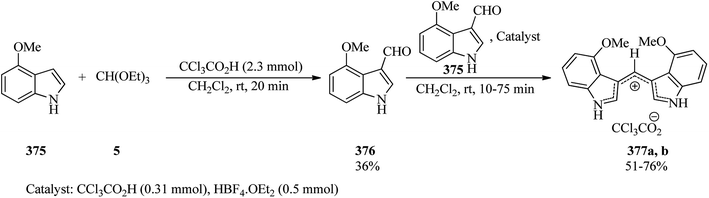 |
| | Scheme 107 The synthesis of bis(indolyl)methane salts. | |
In 2012, Khaksar and co-workers prepared symmetrical tris(indolyl)methanes349,350 via a catalyst-free reaction of indoles with orthoesters in the presence of hexafluoro-2-propanol (HFIP) at room temperature within 15–60 min in excellent yields (88–98%).350
6-Monosubstituted and 6,12-disubstituted 5,11-dihydroindolo[3,2-b]carbazoles (381) were synthesized in 20–50% yields via the one-pot three-stage condensation reaction of indole (89) and aldehydes (378) using a catalytic amount of iodine; which underwent an acid-mediated intramolecular ring-closure reaction by orthoesters (5, 6, 7, 380) in methanol at room temperature for 14 h (Scheme 108).351
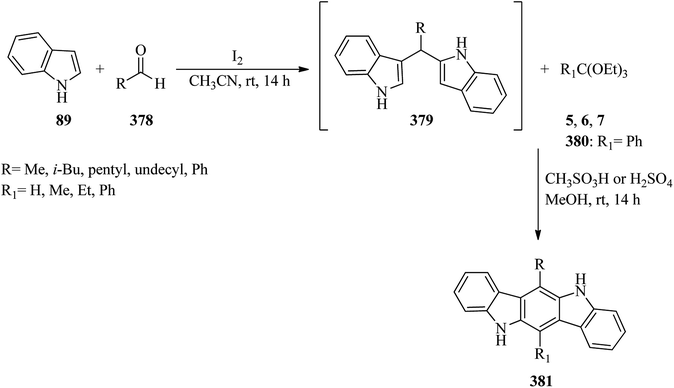 |
| | Scheme 108 The synthesis of substituted dihydroindolo[3,2-b]carbazoles. | |
4.13. Synthesis of pyrazole derivatives
In 2003, Hassen et al. described that N-(2′,2′,2′-trifluoroethyl) phosphonyl β-hydrazones (382) on cyclization with TEOF (5) in the presence of acetic acid in refluxing xylene resulted in novel 1-fluorinated-4-phosphopyrazoles (383) in 46 h, in moderate to excellent yields (60–94%) (Scheme 109).352
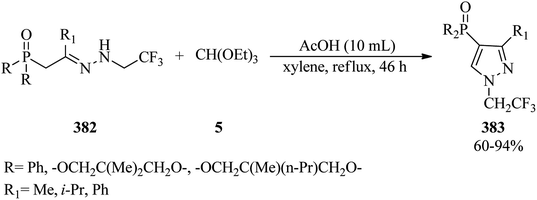 |
| | Scheme 109 The formation of novel 1-fluorinated-4-phosphopyrazoles. | |
1-Substituted 5-aminopyrazole-4-carbonitriles (385) could be prepared via two pathways: (a) The products were produced through the one-pot three-component reaction of 1-substituted hydrazines (384), malononitrile (248), and TEOF (5), in refluxing ethanol within 2 h in low to moderate yields (17–58%). (b) The reaction of malononitrile (248) with TEOF (5) in acetic anhydride at 100 °C which got ethoxymethylenemalononitrile (386) in 93% yield within 4 h. After completion of the reaction, 1-substituted hydrazines (384) were added under similar conditions to afford the desired product (385) in 42–78% within 1 h (Scheme 110).353
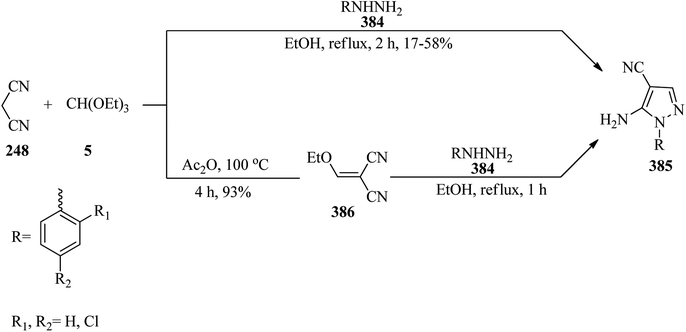 |
| | Scheme 110 The formation of substituted 5-aminopyrazole-4-carbonitriles. | |
In 2002, an effective, useful, and versatile synthetic approach was introduced for the regiospecific synthesis of 3,4-fused-cycloalkyl-1-arylpyrazoles (395) in 48–64% yields via two pathways A and B. In pathway A, BF3-progressed alkylation of symmetric ketones (387) with TMOF (4) in dichloromethane by Hunig's base formed an α-(dimethoxymethyl)ketone intermediate (388), and after isolation, the cyclocondensation of intermediate (388) with arylhydrazine (389) under acidic conditions in refluxing benzene produced product (390) (Scheme 111). In pathway B, the kinetic enolates were first regiospecifically obtained via the condensation of α,β-unsaturated cycloalkanones (391) with organolithium/Grignard reagents (392) by a copper(I) bromide dimethyl sulfide complex (CuBr·Me2S) which was followed by trapping of the above-mentioned enolate utilizing chlorotrimethylsilane (393) as silyl enol–ethers (394). Then, the reaction of the crude enol–ethers (394) with TMOF (4) utilizing catalytic amounts of iodotrimethylsilane (TMSI) in CH2Cl2 at −78 °C for 1 h achieved intermediate (395) regiospecifically, which on cyclocondensation with arylhydrazine (389) resulted in the product (390) (Scheme 112).354
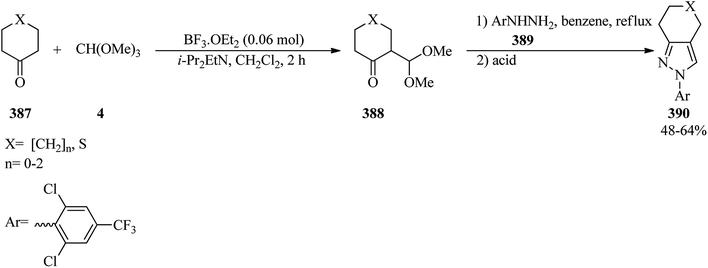 |
| | Scheme 111 The regiospecific synthesis of 3,4-fused-cycloalkyl-1-arylpyrazoles (method A). | |
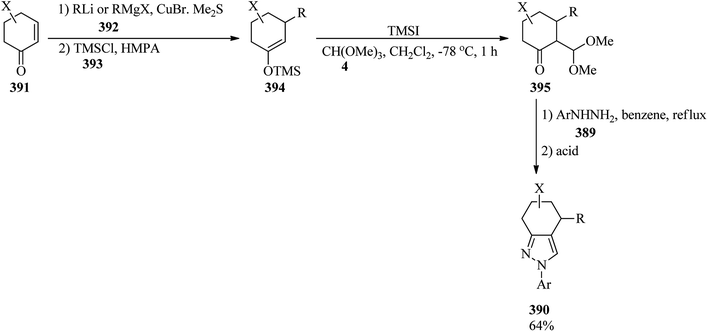 |
| | Scheme 112 The regiospecific synthesis of 3,4-fused-cycloalkyl-1-arylpyrazoles (method B). | |
4.14. Synthesis of methoxyfurans, furanones, and furans
In 2018, Croisetière and Spino devised a rapid and effective sequence of reactions to synthesize O-heterocycles, containing methoxyfurans, furanones, and furans. Initially, the [4 + 1]-annulation reaction of dimethoxycarbene (397),355 arising from Warkentin's oxadiazoline (396), with readily available α,β-unsaturated carbonyls (398) gave cyclic orthoesters (399) (Scheme 113), which were applied as key precursors for the synthesis of methoxyfurans (400), furanones (401), and furans (402). The synthetic methoxyfurans in this report were classified in four parts: first, by a catalytic amount of camphorsulfonic acid in CHCl3; secondly, trimethylaluminum in CH2Cl2 at −40 °C; third, using a weaker Lewis acid aluminum tris(tert-butoxide) in toluene at 140 °C; finally, in the presence of n-BuLi in THF at −78 °C. Furanone (401) could be prepared from the acid hydrolysis of orthoester by concentrated HCl in CHCl3 (method A). Also, utilizing a mild Lewis acid such as TMSCl/TMSI in the presence of NaI iodide in CH3CN furnished furanones (method B). Furan (402) was achieved from the reduction of the resultant furanone by DIBAL-H at −40 °C in THF356 (Scheme 114).357
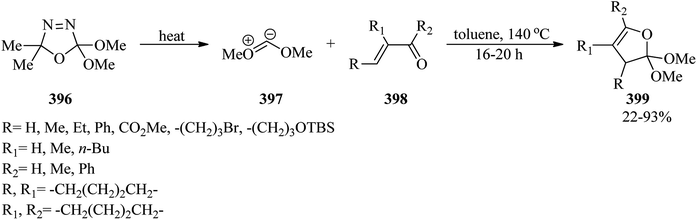 |
| | Scheme 113 Synthesizing O-heterocycles containing methoxyfurans, furanones, and furans. | |
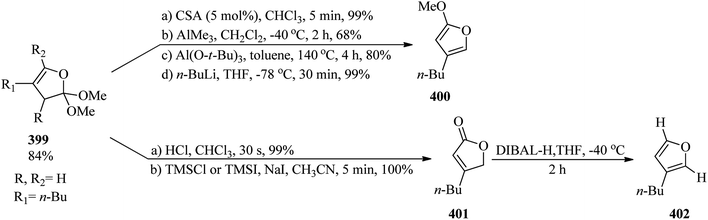 |
| | Scheme 114 The synthesis of some novel methoxyfurans and furans. | |
4.15. Synthesis of dihydropyran-4-ones
In 2015, Clarke et al. utilized a novel modification Maitland–Japp cyclization reaction for the synthesis of highly functionalized dihydropyran-4-ones (404) via the reaction of δ-hydroxy-β-ketoesters (403) with two easily available orthoesters (62, 323) in the presence of acetic anhydride as a dehydrating agent in dry toluene under heating conditions through refluxing and microwave irradiation. It was found that replacing conventional heating with microwave irradiation reduced the large excess amount of orthoesters from 10 equivalents to only 2 equivalents and also the reaction progressed in a few minutes (Scheme 115).358
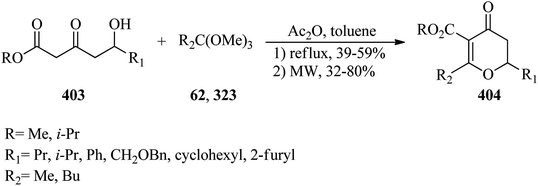 |
| | Scheme 115 The synthesis of highly functionalized dihydropyran-4-ones. | |
4.16. Synthesis of pyrano[2,3-c]pyrazoles
3-Substituted pyrazolin-5-ones (405) were condensed with TEOF (5) in refluxing acetic anhydride to give the intermediate (406) which was then cyclized via Michael addition with hippuric acids (407) together with ethanol elimination and acetylation of the ring nitrogen to achieve pyrano[2,3-c]pyrazoles (408) for 10–30 min in good to excellent yields (75–94%) (Scheme 116).359
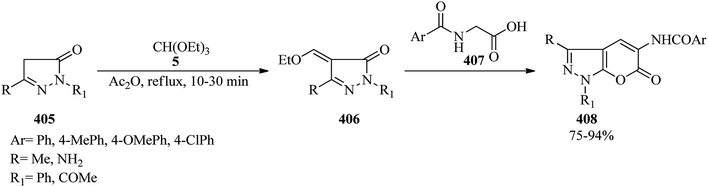 |
| | Scheme 116 Pyrano[2,3-c]pyrazoles achievement route. | |
4.17. Synthesis of chromane derivatives
Scandium triflate [Sc(OTf)3, 3 mol%] catalyzed the diastereoselective reaction of o-hydroxybenzaldehydes (409), glycals (410), and TMOF (4) in dichloromethane at room temperature to furnish cis-fused pyranobenzopyrans (411) within 0.5–1.5 h in good yields (74–83%) with high diastereoselectivity (Scheme 117).360 This transformation was also accomplished with various other alkenes, such as indene, styrene and allyltrimethylsilane under similar conditions to give substituted benzopyrans within 2–6 h in 78–90% yields with high selectivity.361
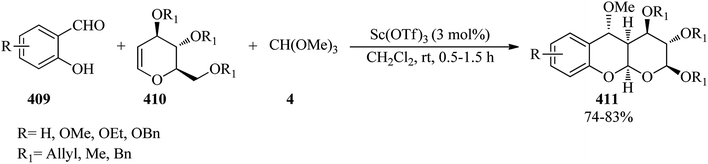 |
| | Scheme 117 The formation of cis-fused pyranobenzopyrans. | |
In 1999, Miyazaki et al. reported a novel, effective, and stereoselective procedure for the synthesis of trans-tetrahydropyrano[3,2-c]benzopyrans (414) in 8–96% yields in 1–24 h via the one-pot three-component reaction of o-hydroxybenzaldehydes (412), TMOF (4), and unsaturated alcohols (413) utilizing a catalytic amount of p-TSA (0.2 mmol) in benzene at room temperature (Scheme 118).362
 |
| | Scheme 118 The stereoselective synthesis of trans-tetrahydropyrano[3,2-c]benzopyrans. | |
In 2003, the one-pot three-component reaction of 4-hydroxy coumarins (415),363,364 carbamates (416), and TEOF (5) was presented for the synthesis of 2-[(substituted 2,4-dioxochromane-3-ylidene)methyl]amino acetic acids (417)365 in refluxing 2-propanol within 2 h in moderate yields (40.5–55%) (Scheme 119). All the products were screened for their biological activity but only a few molecules were active against B. subtilis, S. aureus, E. coli and P. aeruginosa at minimum inhibitory concentrations (MIC) of 100, 300, 500 μg mL−1 compared to streptomycin (CAS 57-92-1) and kanamycin (CAS 59-01-8).366
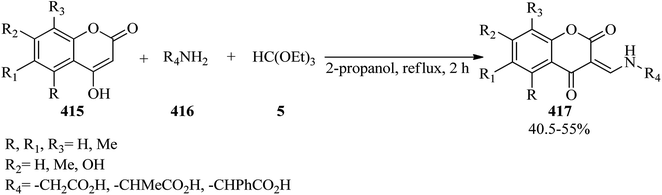 |
| | Scheme 119 The synthesis of 2-[(substituted-2,4-dioxochromane-3-ylidene)methyl]amino acetic acids. | |
Pd(II) catalyzed the one-pot cascade three-component coupling reaction of o-hydroxybenzaldehydes (418), 4-pentyn-1-ols (419), and orthoesters (4, 5) in CH3CN at −30 °C to room temperature achieved a direct diastereoselective synthetic method for chromane spiroacetals (420).367–369 It was found that replacing 4-pentyn-1-ols (419) with the 5α- and 5β-epimers of 4,5-secocholestan-5-ol (421a, b) yielded the corresponding steroid chroman spiroketals (422a, b and 423a, b) (Scheme 120).368
 |
| | Scheme 120 A synthetic method for chromane spiroacetals. | |
4.18. Synthesis of chromene derivatives
Various 2,2-dimethyl-2H-chromenes (426)370–374 could be prepared in 42–67% yields through a two-step Yb(OTf)3-catalyzed reaction of o-hydroxybenzaldehydes (424), TMOF (4), and 2-methylpropene (425) in CH2Cl2 at room temperature for 60 h, which was followed by refluxing toluene in the presence of a catalytic amount of p-TSA (0.03 g) for 1 h to eliminate the methanol from the reaction mixture (Scheme 121). Notably, the reaction failed with salicylaldehydes containing methoxy substituents at the 2- and 4-positions. On the other hand, aldehydes including hydroxyl functional groups at the 3- and 5-positions could not be fully isolated because other unidentified compounds were formed with similar polarity that prevented a complete purification.374
 |
| | Scheme 121 A synthetic approach to various 2,2-dimethyl-2H-chromenes. | |
4.19. Synthesis of coumarin derivatives
The synthesis of substituted 3-benzoylamino-5-oxo-5,6,7,8-tetrahydrocoumarins (427) from 1,3-cyclohexanediones (195, 197), hippuric acid (407), and TEOF (5) (or other one-carbon synthetic equivalents, such as diethoxymethyl acetate or N,N-dimethylformamide dimethyl acetal) in acetic anhydride has been described (Scheme 122).375
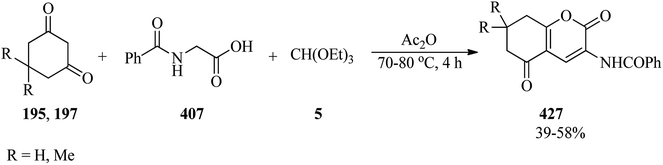 |
| | Scheme 122 The synthesis of substituted 3-benzoylamino-5-oxo-5,6,7,8-tetrahydrocoumarins. | |
In 2003, 8-amino-7-(p-bromophenyl)-10-hydroxypyridino[3′,2′-6,5]4H-pyrano[3,2-c][1]benzopyran-6-one (429) was prepared via the cyclocondensation of 2-amino-4-(4′-bromophenyl)-3-cyano-4H,5H-pyrano[3,2-c][1]benzopyran-5-one (428) with TEOF (5) in refluxing acetic anhydride within 2 h in 55% yield (Scheme 123). The resultant product exhibited antibacterial and antifungal activities.376
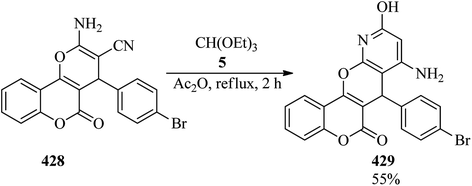 |
| | Scheme 123 The formation of amino-7-(p-bromophenyl)-10-hydroxypyridino[3′,2′-6,5]4H-pyrano[3,2-c][1]benzopyran-6-ones. | |
In 2000, the El-Agrody group synthesized 14-methyl-13,14H-[1]benzopyrano[3′,4′:5,6]-pyrano[3,2-e][1,2,4]triazolo[1,5-c]-pyrimidine-13-one as colorless crystals in 87% yield via the ring-closure reaction of 9-amino-8,9-dihydro-8-imino-7-methyl-6H,7H-[1]benzopyrano[3′,4′:5,6]-pyrano[2,3-d]-pyrimidine-6-one with TEOF in refluxing benzene within 3 h.377 In 2001, they also reported the preparation of antibacterial and antifungal active 11-bromo-14-(p-methoxyphenyl)-14H-naphtho[1′,2′:5,6]pyrano[3,2-e][1,2,4]triazolo[2,3-c] pyrimidine through the cyclocondensation of 10-amino-3 -bromo-12-(p-methoxyphenyl)-11-imino-10,11-dihydro-12H-naphtho[1′,2′:5,6]-pyrano[2,3-d]pyrimidine with TEOF under similar condition for 6 h in 79% yield.326
4.20. Etherification and esterification reactions
Bakos et al. reported that a single frustrated Lewis pair catalyst B(C6Cl3H2)(C6F4H)2 could progress the tandem reductive etherification reaction of aldehydes/ketones (430) with trialkyl orthoformate (4, 5), as alkylating agent, to produce ethers (431) under 20 bar H2 pressure in THF at 55–100 °C, for 16–84 h with high selectivity (98–99%) and weak to good yields (35–92%) (Scheme 124).378
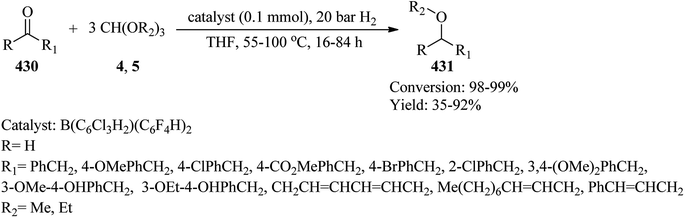 |
| | Scheme 124 Tandem reductive etherification reaction of aldehydes/ketones. | |
In 2016, estrone 3-secondary ethers (435) were made through the eco-friendly etherification/aromatization reaction of readily accessible dienone (432) with trialkyl orthoformates (127, 433, 434), as alkylating agent, in a 1:5 molar ratio by triflic acid (TfOH, 0.5 mmol) in the corresponding alcohol (5 mL) at 100 °C for 2 h in 80–87% yields (Scheme 125). The above-mentioned procedure was applied for the generation of three marketed 3-etherified estrogen drugs, quinestrol, quinestradol, and nilestriol, utilizing 19-hydroxyandrost-4-ene-3,17-dione as a commercially available precursor.379
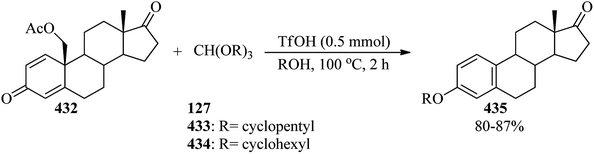 |
| | Scheme 125 Etherification/aromatization reaction of dienone. | |
Scandium triflate [Sc(OTf)3, 10 mol%] catalyzed the one-pot three-component condensation of aldehydes, allylsilane and TMOF in dichloromethane to produce homoallyl ethers380,381 at room temperature. Mild reaction conditions, good yields (70–90%), high selectivity, recyclability, and reusability of the catalyst are some important advantages of this procedure.381 On the other hand, the reaction of equimolar amounts of homoallyl alcohol (436) with TMOF (4) using Lewis acids such as InCl3 (3 mmol) in CH2Cl2 at room temperature obtained homoallyl ethers (437) and homoallyl chlorides (439) in good yields. Notably, homoallyl alcohols with aryl moieties with electron-donating substituents furnished exclusively homoallyl ethers (437) in 84–92% yields. Homoallyl alcohol including halogen atoms as the substituents resulted in homoallyl ethers (437, 61–72%) along with bishomoallyl ethers (438) (9–14%) as by-products. The homoallyl alcohols containing several halogen atoms or an electron-withdrawing substituent produced only the homoallyl chlorides (439) in 67–88% yields (Scheme 126). Similarly, this transformation was accomplished using BiCl3 at room temperature and BF3·OEt2 at −78 °C, while treatment utilizing TiCl4 at −78 °C afforded exclusively the homoallyl chloride (439) in 92% yield. Hence, the type of product could be controlled through the selection of the desired Lewis acid.382
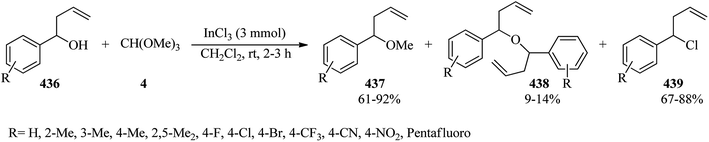 |
| | Scheme 126 The preparation of some homoallylic compounds. | |
Maulide and Markó synthesized spirocyclic ethers (442) in 35–63% yields via the Mukaiyama aldol reaction of the silylated acyloins (440) with functionalized orthoesters (441) by ZnCl2 in dichloromethane at room temperature, which was followed by desilylation of the resultant adducts using TBAF in THF, and finally annulation in the presence of t-BuOK (Scheme 127).383
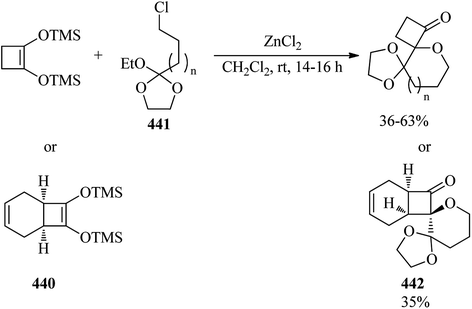 |
| | Scheme 127 Mukaiyama aldol reaction of silylated acyloins. | |
In 2013, Lee and Kraus presented a new technique for the synthesis of substituted benzoates (446) in 72–94% yields via the one-step one-pot inverse electron-demand Diels–Alder reaction of methyl coumalate (443) with orthoesters (62, 444, 445) as an electron-rich dienophile in toluene at 200 °C for 16 h (Scheme 128).384
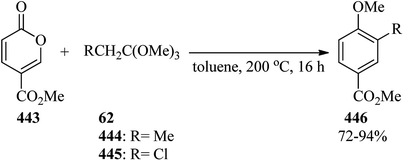 |
| | Scheme 128 The synthesis of substituted benzoates. | |
In 1998, Fletcher co-workers reported a facile and inexpensive process for the formation of alkoxy/aryloxycyclopropanes (448) with preference for the generation of the more hindered cis-isomers through the reaction of TMOF (4) with alkenes (447) in the presence of zinc amalgam and Me3SiCl in refluxing diethyl ether for 24 h (Scheme 129).385
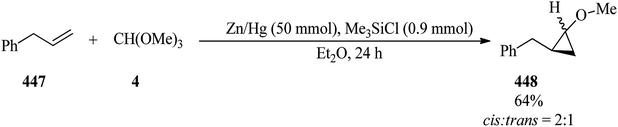 |
| | Scheme 129 The formation of alkoxy/aryloxycyclopropanes. | |
In 2018, Bunse et al. synthesized ethoxycarbonyl-substituted 1-alkylidenephthalanes (451) from the reaction of orthoester (449) arising from triethyl orthopropanoate (7). This reaction was prompted via halogen–metal exchange of the orthoester (449) with n-butyllithium (n-BuLi) in THF at −78 °C for 15 min which was followed by the nucleophilic addition reaction with diverse carbonyl compounds (450) along with regioselective 5-exo-dig cyclization of the corresponding intermediate lithium alcoholate and eventually hydrolysis to produce the products (451) as mixtures of (Z)- and (E)-configured diastereomers with an approximate range of 90:10 to 95:5 (Scheme 130). Notably, the aromatic aldehyde benzaldehyde was unsuccessful in getting the phenyl-substituted derivative.386
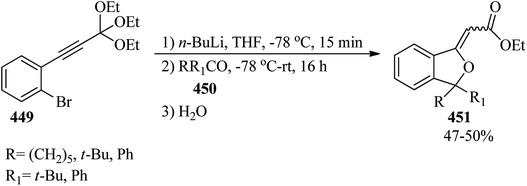 |
| | Scheme 130 The preparation of ethoxycarbonyl-substituted 1-alkylidenephthalanes. | |
In 2018, Sempere and Carreira prepared β-substituted γ,δ-unsaturated esters (453) via an iridium-catalyzed enantioselective allylic alkylation reaction of trimethyl orthoacetate (62), as an acetate enolate source, with racemic allylic carbonates (452), utilizing ZnBr2 as an additive in 1,4-dioxane at room temperature (Scheme 131). Significantly, halogenated and electron-rich substrates yielded the relative products with high yields and excellent enantioselectivity. Notably, the meroterpenoid (+)-conicol has been synthesized through employment of this new procedure in a formal enantioselective synthesis.387
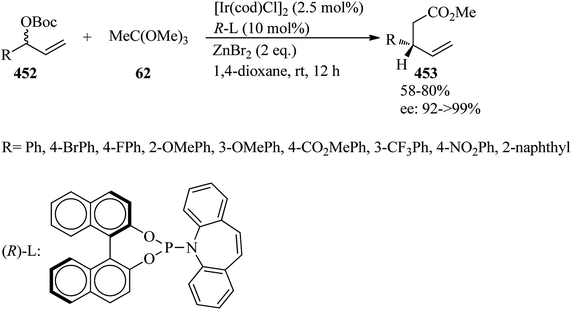 |
| | Scheme 131 The preparation of β-substituted γ,δ-unsaturated esters. | |
Brodzka and co-workers probed the enzymatic kinetic resolution (EKR) of Novozym 435 as a biocatalyst for the synthesis of enantiomerically pure carboxylic acid esters (456) via the esterification of racemic 3-phenyl-4-pentenoic acid (454) with trialkyl orthoesters (4, 5, 6, 62, 102, 323, 380, 444, 455) as alkoxy group donors in toluene at 40 °C (Scheme 132). Based on the resulting data, trialkyl orthoesters including bulky alkyl groups, such as trialkyl orthobenzoate, obtained excellent results for both yield and enantioselectivity (50% yield, >99% ee). It was found that the enantioselectivity of EKR is highly dependent on the structure of an alkoxy group donor.388
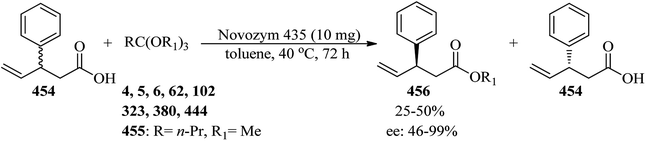 |
| | Scheme 132 The synthesis of enantiomerically pure carboxylic acid esters. | |
In 2015, Koszelewski et al. described a novel method of kinetic resolution based on irreversible enzymatic esterification of rac-3-hydroxy-3-(aryl)propanoic acids (457) with triethyl orthobenzoate (380) using Novozym 435 tandem metal-enzyme dynamic kinetic resolution (DKR) in toluene at 40 °C to access enantiomerically active β-hydroxy ester (458) with low enantioselectivities in low yields (<1–45%) for 48 h (Scheme 133).389
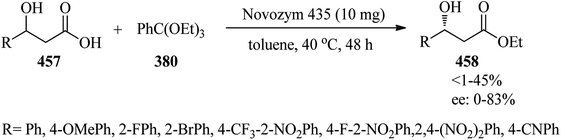 |
| | Scheme 133 The synthesis of enantiomerically active β-hydroxy ester. | |
In 1993, Trujillo and Gopalan used triethylorthoacetate as an ideal reagent for the transformation of sulfonic acids into their ethyl esters under a catalyst-free mode in dichloromethane (CH2Cl2) at room temperature in high to excellent yields (69–97%) for 30 min. The uncatalyzed esterification reaction of carboxylic acids using this reagent was also conveniently performed in refluxing toluene within 24 h in high yields (81–92%). Replacing trimethylorthoacetate with triethylorthoacetate resulted in the desired methyl esters. According to the resulting data, the utilization of TEOF instead of triethylorthoacetate was unsuccessful.390
In 1996, Schwabacher and Stefanescu designed a useful and convenient method for the generation of methyl phosphinate (460) together with the elimination of methyl formate and methanol via the reaction of phosphinic acid (459) with TMOF (4) in a combined dry THF/toluene (1:1) medium at 5 °C to room temperature. This reaction was accompanied by the formation of a by-product (461), which was assumed to form through the attack of the dimethoxymethyl cation on the phosphorus(III) tautomer of (459) or (460) (Scheme 134).391
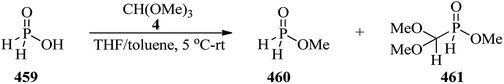 |
| | Scheme 134 The generation of methyl phosphinate. | |
In 1994, King introduced a convenient method for the formation of ester–ethers (463) in 61–95% yields via the reaction of lactones (462) with orthoformates (4, 5, 133) using sulfuric acid in the desired alcohol as solvent at 50 °C within 3–8 h (Scheme 135).392
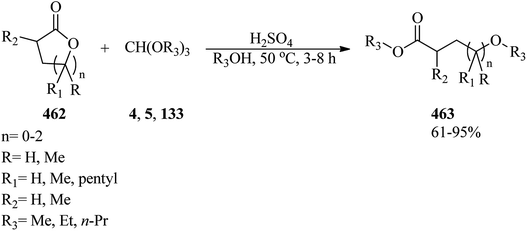 |
| | Scheme 135 The formation of ester–ethers from lactones and orthoformates. | |
4.21. Johnson–Claisen rearrangement
In 2001, Takao et al. showed that the Claisen rearrangement of the substrate (464)393,394 derived from 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose, with triethyl orthopropionate (7) by a catalytic amount of propanoic acid in DMF at 130 °C for 4 h obtained an inseparable mixture of the rearrangement products (465, 466, and 467) as a 1:2.5:10 diastereomeric mixture in a combined yield of 87% (Scheme 136). Under similar conditions, the reaction of (464) with trimethyl orthobutyrate (455) by powdered molecular sieves 4A (MS-4 Å) afforded an inseparable mixture of the rearrangement products (468 and 469) as a 1:3 diastereomeric mixture in a combined yield of 46% (Scheme 137). In both reactions, the σ-bond formation progressed prevalently from the β-side. This resultant stereochemical was opposite to the observed rearrangement of (464) with TEOAc.395
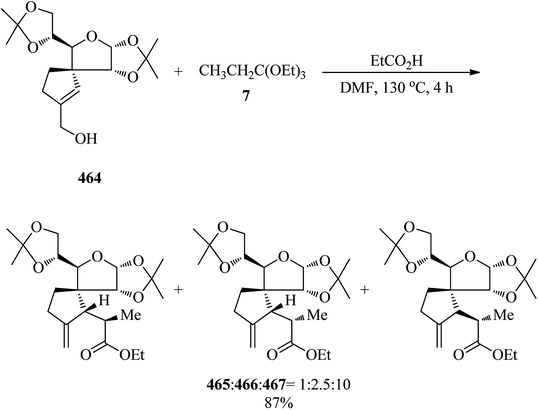 |
| | Scheme 136 The Claisen rearrangement of 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose with triethyl orthopropionate. | |
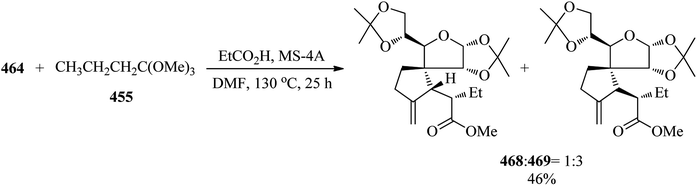 |
| | Scheme 137 The Claisen rearrangement of 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose with trimethyl orthobutyrate. | |
4.22. Acetalization reaction
In 2003, Ma et al. presented an efficient and chemoselective method for the preparation of diethyl acetals in good to excellent yields (63–96%) via the acetalization178,396–408 of aldehydes/ketones with TEOF using TiO2/SO42− in refluxing ethanol for 25 min to 3 h. It was found that, in competitive reactions, aldehydes and cyclic aliphatic ketones revealed excellent chemoselectivity.408 The reaction of enamines (470) with trialkyl orthoformates (4, 5) was also accomplished in the presence of BF3·OEt2 in CH2Cl2 to give α-dialkoxymethyl carbonyl compounds (471), after hydrolysis, in 61–85% yields within 1 h (Scheme 138).409
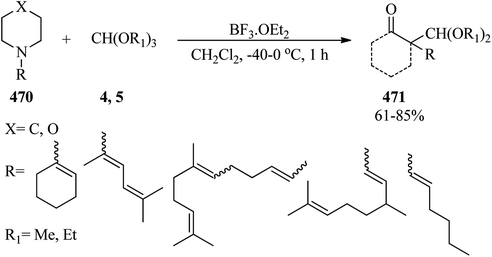 |
| | Scheme 138 The chemoselective preparation of diethyl acetals. | |
In 1999, Martins et al. reported the regioselective reaction of 2-acylcycloalkanones (472) with TMOF (4) using p-TSA (0.78 mmol) in methanol at room temperature to afford the corresponding acetals within 24 h (Scheme 139). It was found that the reverse Claisen condensation of electron-donating substituted 2-acylcyclohexanones, where R was Me, Et or CH2Ph with TMOF led to 7,7-dimethoxyalkanoate methyl esters (473) in 80–93% yields. On the other hand, 2-acyl cyclohexanone dimethyl acetal derivatives (474) in 80–85% yields were produced when R was Ph, CF3 or OMe. Similar to the products (474), 2-acetylcyclooctanone derivative (R = Me) resulted in acetal (475) in 86% yield. Finally, 2-acetylcyclopentanone (R = Me) and 2-acetyl cycloheptanone (R = Me) gave rise to acetals (476) and (477) in 75 and 74% yields, respectively.410
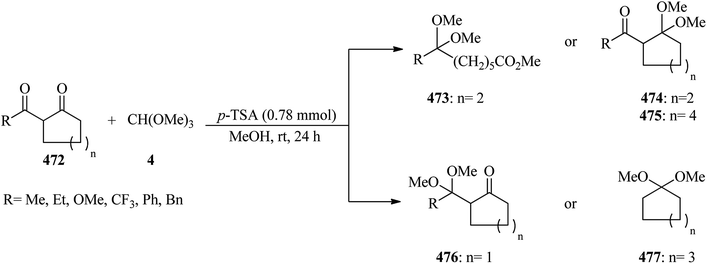 |
| | Scheme 139 The regioselective reaction of 2-acylcycloalkanones with TMOF. | |
In 1991, the highly enantioselective hydroformylation of vinyl aromatics (478) was reported by the Hegedus group in the presence of TEOF (5). They discovered that TEOF plays a special role in stereoselectivity. As can be seen in Scheme 140, the reaction proceeded with (PtIIBPPM)/SnCl2 (PtIIBPPM: l-(tert-butoxycarbonyl)-(2S,4S)-4-(dibenzophospholyl)-2-[(dibenzophospholyl)methyl]pyrrolidine) in o-dichlorobenzene as solvent.411
 |
| | Scheme 140 The enantioselective hydroformylation of vinyl aromatics. | |
In 2015, Pospech and co-workers introduced an intermolecular one-step methoxylation/acetalization reaction of donor–acceptor substituted diazo compounds (480) with TMOF (4) utilizing Rh2(S-pttl)4 (1.0 mol%) in CH2Cl2 at room temperature to synthesize functionalized tertiary α-alkoxy-β-oxo-esters (481) in 1 h in 40–91% yields (Scheme 141). The concurrent C–O/C–C bond generation produces products with exclusive quaternary carbon centers containing substituent groups with various oxidation levels, such as esters, protected aldehydes and alkoxidse.412
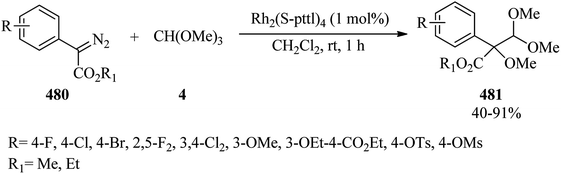 |
| | Scheme 141 Methoxylation/acetalization of diazo compounds with TMOF. | |
In 2015, the cyanation of orthoesters (5, 102, 317, 380),413 with sterically congested α-cyanoamines, like dicyclohexylcyanoamine (482), as effective cyanating reagents, was described for the formation of cyanoacetals (483) by trichlorosilyl triflate (SiCl3OTf) in CH2Cl2 at 0 °C within 0.5 h, in excellent yields (73–97%) (Scheme 142).414
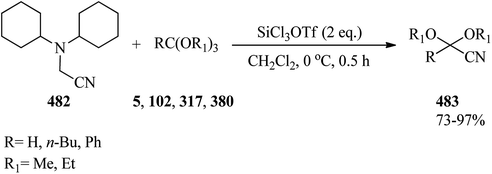 |
| | Scheme 142 The cyanation of orthoesters. | |
Casiraghi and co-workers prepared 2-hydroxyaryl-1-carboxyaldehyde diethylacetals (485) via the reaction of special phenol salts like magnesium halides (484) with TEOF (5) in benzene or diethylether at 20–80 °C (Scheme 143). This reaction was promoted by a C-regiospecific attack at the ortho positron of the phenoxy moiety.415
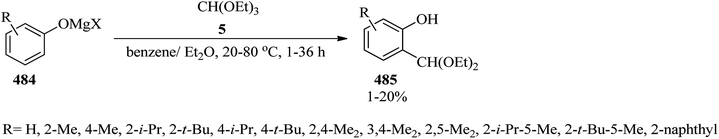 |
| | Scheme 143 The regioselective preparation of 2-hydroxyaryl-1-carboxyaldehyde diethylacetals. | |
In 1993, Byun and Bittman synthesized (−)-dimethyl 2,3-O-benzylidene-L-tartrate and its enantiomer (488) in 83–91% yields via the reaction of L-tartaric acid (486), benzaldehyde (487), and TMOF (4) by p-TSA (10 mmol) in methanol at room temperature (Scheme 144).416
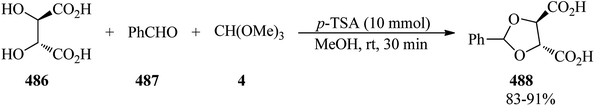 |
| | Scheme 144 The formation of (−)-dimethyl 2,3-O-benzylidene-L-tartrate. | |
In 2013, Geng et al. prepared 4,6-O-arylidenated derivatives (493) and orthoesters (494), regioselectively via the protected/unprotected sugars (489), benzaldehydes (487, 490), and TEOF (5) by organocatalysts such as thiourea (491)/squaramide derivatives (492) in anhydrous CH3CN at room temperature (Scheme 145). In this procedure the influence of various parameters, such as the amount and type of aldehyde, catalyst and orthoester, was investigated on the time and yield of reaction. It was found that a strong acid-catalyst like p-TSA has a great effect in reducing the reaction time.417
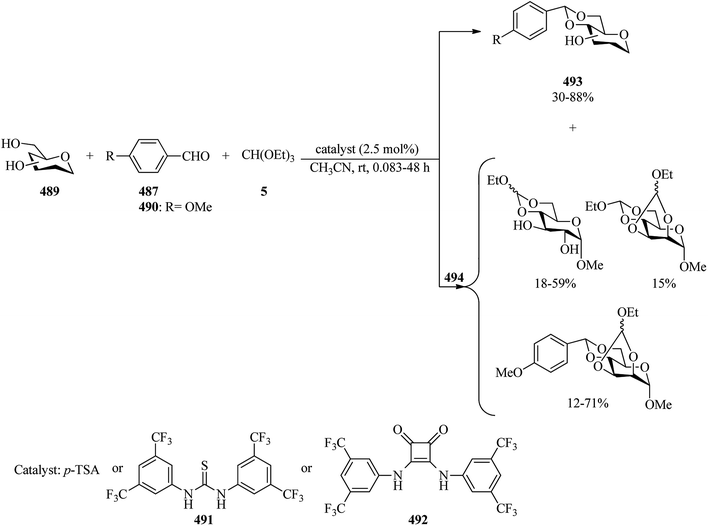 |
| | Scheme 145 The regioselective preparation of 4,6-O-arylidenated derivatives of sugars. | |
5-Substituted 5,5-dialkoxy-2-penten-4-olides (496) were made through the reaction of 2-(trimethylsiloxy)furan (495) with orthoesters (4, 5, 6, 7, 380) in the presence of a few drops of tin(IV)chloride (SnCl4) in CH2Cl2 at −40 to 10 °C in 48–91% yields in a 2 h period (Scheme 146).418
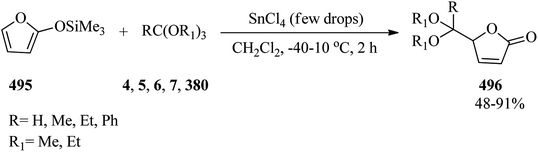 |
| | Scheme 146 The synthesis of 5-substituted 5,5-dialkoxy-2-penten-4-olides. | |
The vicinal diequatorial diols reaction of 2,2,3,3-tetramethoxybutane (497) with TMOF (4) using camphorsulfonic acid in refluxing methanol for 12–18 h yielded butane 2,3-bisacetal (498) in moderate to excellent yields (54–91%) to protect trans-1,2-diols (Scheme 147).419
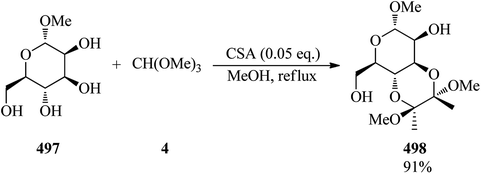 |
| | Scheme 147 The reaction of 2,2,3,3-tetramethoxybutane with TMOF. | |
In 1991, Hoffman and Salvador described a one-pot conversion of cyclic ketones to N-methyl lactams. The reaction of ketones (499) with TEOF (5) by p-TSA in ethanol gave acetal (500) which was treated in situ with N-(((p-nitrobenzene)sulfonyl)oxy)methylamine (501) to furnish the N-methyl group of the imidate salt (502). Finally, dealkylation of the resulting O-ethyl imidate with sodium iodide in refluxing acetonitrile achieved lactams (503) in moderate to high yields (Scheme 148).420
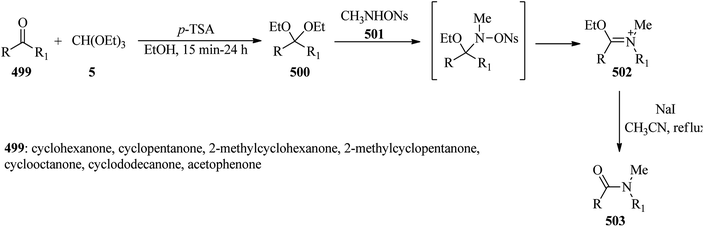 |
| | Scheme 148 The conversion of cyclic ketones to N-methyl lactams with TEOF. | |
DeMong and Williams suggested an asymmetric synthesis of 2-amino-3,3-dimethoxypropanoic acid (−)-506. The key step is the quenching of a chiral glycine (504) titanium enolate with TMOF (4) in CH2Cl2 at −78 °C to form the appropriate α-dimethylacetal421,422 (+)-505 in 94% yield. The (−)-506 was obtained in 98% yield through the hydrogenolysis of the lactone moiety by 20 mol% of Pd(OH)2 in a combined THF/MeOH (3:1) medium; followed by utilizing trituration with ether to eliminate the dibenzyl by-product (Scheme 149).422
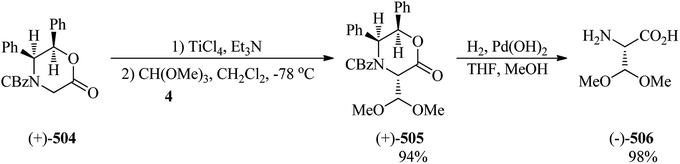 |
| | Scheme 149 The asymmetric synthesis of 2-amino-3,3-dimethoxypropanoic acid. | |
The reaction of chalcones (507) with TMOF (4) was accomplished using thallium(III) trinitrate (TTN) in acidic methanol to afford a 50:50 mixture of 3,3-dimethoxy-1,2-diphenylpropan-1-one (508, Ar = Ar1 = C6H5) and methyl 2,3-diphenyl-3-methoxypropanoate (512, Ar = Ar1 = C6H5). The keto acetal (508)423 was obtained through normal Ar ring migration, while the ester (512) was obviously achieved by migration of the Ar1 ring. As shown in Scheme 150, owing to the low rate of the treatment of chalcone with TTN and also the faster reaction of aldehydes and ketones with TMOF by TTN to form acetals and ketals,424 the ketalization reaction of chalcone apparently competes with oxidative rearrangement. Hence, the oxidative rearrangement resulted in product (508) and the ketalization reaction formed intermediate (510) which upon treatment with TTN was converted into intermediate (511). Finally, the 1,2-migration of the Ar1 group and the respective elimination of TTN furnished product (512) (Scheme 150).425
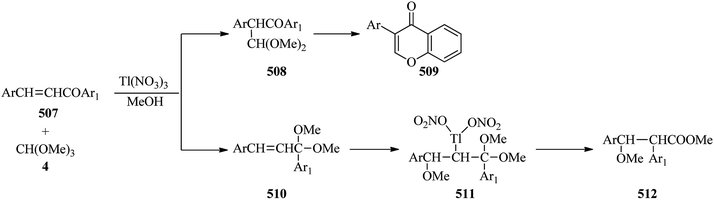 |
| | Scheme 150 The reaction of chalcones with TMOF. | |
2-Dimethoxymethyl-4,4-dimethoxycyclohexa-2,5-dienone (514) could be prepared via oxidation of 2-formyl-4-methoxyphenol (513) with TMOF (4), utilizing thallium trinitrate426,427 in methanol at −20 °C while the oxidation of 2-methoxycarbonyl-4-methoxyphenol furnished 2-methoxycarbonyl-3,4,4-trimethoxycyclohexa-2,5-dienone (Scheme 151).427
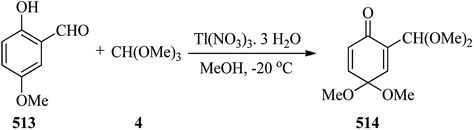 |
| | Scheme 151 The oxidation of 2-formyl-4-methoxyphenol with TMOF. | |
In 2007, Graham and Smith demonstrated that alcohols could be transformed directly into either acyclic or cyclic acetals in both tandem and sequential oxidation/acetalization processes using manganese dioxide, trialkyl orthoformates and an indium triflate catalyst in dichloromethane at ambient temperature.428 In 2015, Manojveer and Balamurugan developed a convenient method for the synthesis of naphthalenes (517) via a domino three-component condensation reaction of o-alkynylbenzaldehydes (515), ketones (516), and TMOF (4) in the presence of TfOH (20 mol%) in acetonitrile at room temperature (Scheme 152). This transformation was initiated via the condensation of o-alkynylbenzaldehyde (515) with TMOF (4) by TfOH to obtain the in situ formed acetal (518), which was condensed with enol ether (519), arising from the reaction of ketones with TMOF, to form chalcone (520) which underwent intramolecular cyclization reaction to afford naphthyl ketones (517).429
 |
| | Scheme 152 The synthesis of naphthalenes via domino three-component condensation. | |
Manojveer and Balamurugan presented a facile and efficient process to get substituted benzo[a]fluorenes (524), with regioselectivity via a domino three-component reaction of o-alkynylbenzaldehydes, alkynes, and TMOF by TfOH (20 mol%) in CH3CN or CH2Cl2 at room temperature. This reaction proceeded through the reaction of aldehydes (515) with TMOF (4) by TfOH to obtain the in situ formed acetal (521). Subsequent [2 + 2] cycloaddition between acetal (521) and alkynes (522) generated the cis-isomer (523) which on intramolecular annulation reaction achieved substituted benzo[a]fluorenes (524) in 45–98% yields in 0.5–24 h (Scheme 153).430
 |
| | Scheme 153 The regioselective synthesis of substituted benzo[a]fluorenes. | |
TfOH catalyzed the one-pot three-component reaction of ketones, such as acetophenones and ethyl phenyl ketones (525), chalcones (526), and TMOF (4) in carbon tetrachloride (CCl4) at room temperature to obtain a series of symmetrical and unsymmetrical 1,5-diketones (527) within 6–8 h. This reaction progressed through the in situ formation of acetals (528) from the reaction of ketones with TMOF which set up an equilibrium with the enol ether (529), as an Michael acceptor, which was followed by Michael addition with the activated chalcones by an acidic catalyst (530) to afford 1,5-diketones (527). Replacing acetophenones with ethyl phenyl ketones yielded a mixture of diastereomeric 1,5-diketones (where the syn diastereomers were the major product) in 55–95% yields (Scheme 154).431
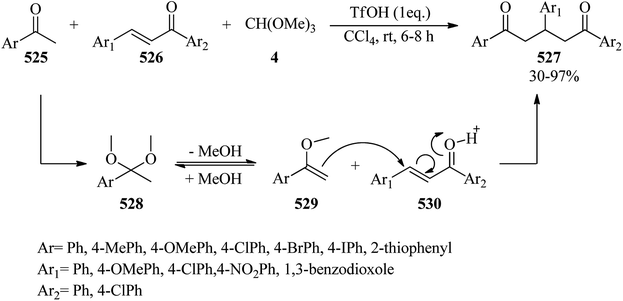 |
| | Scheme 154 The preparation of 1,5-diketones. | |
4.23. Synthesis of benzoxazoles, benzothiazoles, benzimidazoles, oxazoles, oxadiazoles, and imidazoles
In 2009, Lee et al. reported a simple and effective procedure for the generation of benzoxazoles and oxazoles via the one-pot reduction-triggered heterocyclization reaction. Indium/acetic acid promoted the reaction of 2-nitrophenols with trimethyl orthoesters in refluxing benzene to produce benzoxazoles432–434 in 50–98% yields in a 1 h period. In a similar manner, 1-aryl-2-nitroethanones upon treatment with trimethyl orthobenzoate using In/AcOH in refluxing acetonitrile afforded oxazoles in 1 h in 46–72% yields.434 In 2014, Gnanasekaran and co-workers reported arylhydrazides (531) on reaction with orthoesters (5, 6, 7, 380) by NH4Cl (30 mol%) as catalyst in a polar solvent like ethanol under reflux conditions yielding 2-substituted and 2,5-disubstituted 1,3,4-oxadiazoles (532)435–437 within 0.5–10 h, in 22–98% yields (Scheme 155). The reaction was successful with electron-releasing and electron-withdrawing groups on the arylhydrazide precursor and progressed gently for both non-aromatic and aromatic orthoesters.438
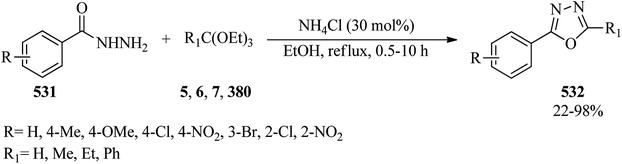 |
| | Scheme 155 Synthetic approach for substituted 1,3,4-oxadiazoles. | |
In 2014, Al-Majidi obtained 1,2,4-triazolo[3,4-b]benzothiazole (534) as pale yellow crystals on treatment of 2-hydrazinobenzothiazole (533) with TEOF (5) in the presence of a few drops of acetic acid in refluxing methanol for 3 h in 65% yield (Scheme 156). The synthesized compound showed moderate antimicrobial activity against Serratia but was inactive against Staphylococcus aureus.439
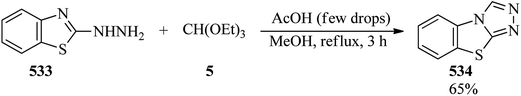 |
| | Scheme 156 The formation of 1,2,4-triazolo[3,4-b]benzothiazole. | |
2-Oxo-2H-pyrimido[2,1-b]benzothiazole-3-carbonitriles were prepared through the cyclocondensation of cyanoacetamides with TEOF in nitrobenzene at 190–200 °C in 77–79% yields within 3 h.440 In 2010, the Jeffries-EL group prepared some classes of synthetically useful 2,6-disubstituted benzobisthiazoles (539) based on the Lewis acid-catalyzed ring-closing reaction between diamino benzene dithiol (535) and substituted orthoesters (5, 6, 536, 537, 538) in dimethylacetamide (DMA) as organic solvent (Scheme 157). These products are of interest for the development of new organic semiconductors.441
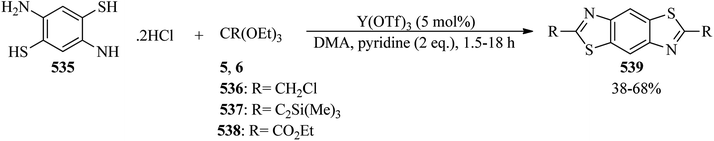 |
| | Scheme 157 The formation of disubstituted benzobisthiazoles. | |
Wang et al. developed a mild and facile two-step one-pot method for the synthesis of benzo and heteroaromatic fused imidazoles.442–445 In first step, ortho-nitroanilines were converted into ortho-diamines by the reduction of the nitro group using iron powder. In the next step, cyclization occurred between ortho-diamines and TEOF by catalytic amounts of ytterbium triflate (0.005 equiv.) in acetic acid (1.0 M) at 75 °C to obtain the corresponding imidazoles within 3 h.445 Benzimidazoles308,446,447 were also prepared by Zhang et al. from o-phenylenediamines with orthoesters using sulfamic acid (0.05 mmol) in methanol at room temperature in 0.5–5 h in excellent yields (85–98%).447 ZrCl4 in ethanol at room temperature also accelerated this transformation within 0.8–6 h in excellent yields (81–95%).448 In 2004, the one-pot three-component reaction of 2-cyanobenzimidazole (540), TEOF (5), and hippuric acid (407) was presented for the synthesis of pyrido[1,2-a]benzimidazole derivatives (541)359,449 in refluxing acetic anhydride within 10 min, in excellent yields (84–91%) (Scheme 158).359
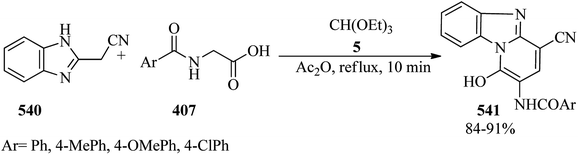 |
| | Scheme 158 The synthesis of pyrido[1,2-a]benzimidazole derivatives. | |
Due to the noteworthy biological activities of the natural nucleoside antibiotic, bredinin, such as antiviral and antihepatitis C effects, and its versatile biological activities, Nair and Zhang synthesized a new carbocyclic analog of bredinin (545) using (3aS,4R,6R,6aR)-6-((methoxymethoxy)methyl)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-amine (542) as a key precursor. In the first step, (542) was converted into the malondiamide intermediate (543) via side-chain functional group conversions. Subsequently, (543) on reaction with TEOF (5) by acetic acid as catalyst under heating in ethanol at 90 °C underwent cyclization to afford the appropriate 4-carbamoyl-imidazolium-5-olate ring (544) within 1.5 h in moderate yield (55%). In the final step, the deprotection reaction of the recent compound (544) resulted in (545) (Scheme 159). The resultant product displayed antiviral activity, although it was low, against a number of RNA viruses. More biological properties are under investigation.450
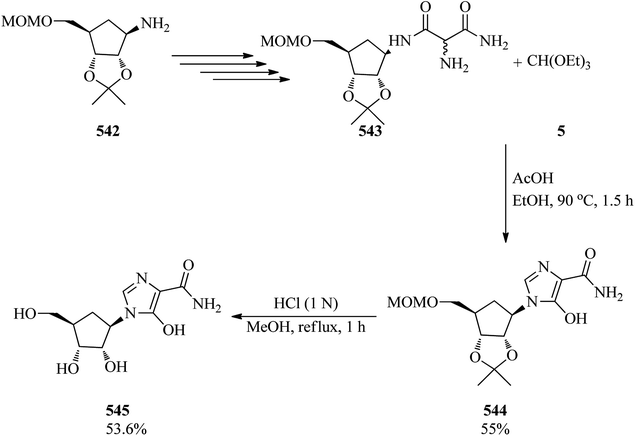 |
| | Scheme 159 Synthesizing a carbocyclic analog of bredinin. | |
The cyclodehydration of urea derivatives (546) with TMOF (4) was accomplished in the presence of boronic acid (10–20 mol%) under refluxing toluene to attain compounds (547) in 75–95% yields in 12–24 h which as key intermediates were converted into a 1H-imidazo[1,2-a]imidazol-2-one LFA-1 antagonist (548) (Scheme 160).451
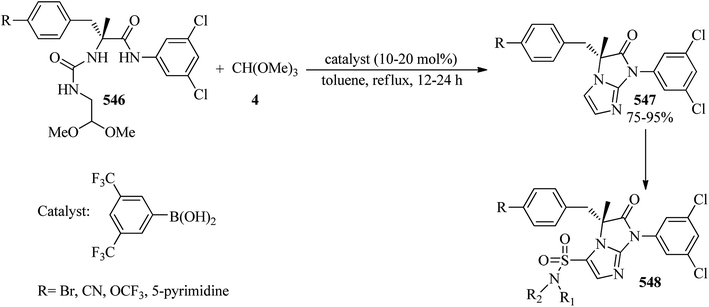 |
| | Scheme 160 The cyclodehydration of ureas with TMOF. | |
4.24. Synthesis of thiadiazoles, triazoles, and 1H-tetrazoles
2-Alkylamino-1,3,4-thiadiazoles have been synthesized via the ethanol-mediated reaction of 4-alkylthiosemicarbazide and TEOF, in a 1:2 molar ratio, through the accelerated role of concentrated hydrochloric acid by Tomalia and Pmge in 1973.452 In 2015, Aouali et al. prepared 5-amino-1-phenyl-1,2,4-triazole derivatives (552) through the one-pot three-component reaction of orthoester (6, 7, 549), phenylhydrazine (550) and cyanamide (551) by acetic acid (5 mol%) in methanol through two different pathways: (a) utilizing microwave irradiation (200 W) at 80 °C, and (b) under classical heating conditions. The results indicated shorter reaction times and higher yields by the microwave method (87–95%, 40 min) (Scheme 161).453
 |
| | Scheme 161 The synthesis of 5-amino-1-phenyl-1,2,4-triazoles. | |
Kerr and co-workers synthesized various chiral and achiral 1,2,4-triazolium salts as key precursors for the generation of N-heterocyclic carbine. In this paper, various 1,2,4-triazolium salts were obtained under different conditions, e.g. pentafluorophenyl triazolium salt (555) as a light tan solid was produced through the reaction of hydrazinium tetrafluoroborate (554), arising from morpholinone derivative (553), with TEOF (5) in chlorobenzene at 110 °C in 24 h in 66% yield (Scheme 162).454
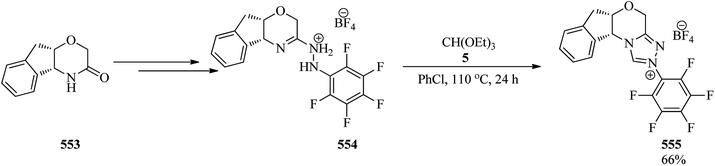 |
| | Scheme 162 The preparation of pentafluorophenyl triazolium salt. | |
Chuprun et al. synthesized tetrazoles via the enantioselective reaction of α-amino acids with sodium azide and TEOF in the presence of glacial acetic acid both as catalyst and solvent at 55 °C within 4 h in weak to good yields (23–83%). The product structure was characterized by 1H and 13C NMR and mass spectra, and their stereoisomeric excess amounts were assigned by chiral HPLC (20–100%).455 1-(6,7,9,10,17,18,20,21-Octahydrodibenzo[b,k]-[1,4,7,10,13,16]hexaoxacyclooctadecen-2-yl)-1H-tetrazole (2) could be synthesized via the domino three-component reaction of an arylamino derivative of dibenzo-18-crown-6 (1), TEOF, and sodium azide under heating in glacial acetic acid at 105 °C for 3 h in 73% yield.456 Yb(OTf)3 also promoted the reaction of amines, TEOF, and sodium azide in 2-methoxyethanol at 100 °C to prepare the 1-susbstituted 1H-1,2,3,4-tetrazoles457–462 in 6–9 h in 71–91% yields. Some of the products revealed phytocidal activity.462 Muralidharan et al. also synthesized a library of nitrogen-rich heterocycles, such as 2-(1H-tetrazol-1-yl)-1H-imidazole-4,5-dicarbonitrile (557), 1-(1H-1,2,4-triazol-3-yl)-1H-tetrazole (559), and 5-(1H-tetrazol-1-yl)-1H-1,2,4-triazol-3-amine (561) via the critical role of TEOF. As could be seen, the above-mentioned compounds were obtained via the reaction of TEOF and also NaN3 (243) with 2-amino-1H-imidazole-4,5-dicarbonitrile (556), 1H-1,2,4-triazol-3-amine (558), and 1H-1,2,4-triazole-3,5-diamine (560), as starting materials, respectively (Scheme 163).463
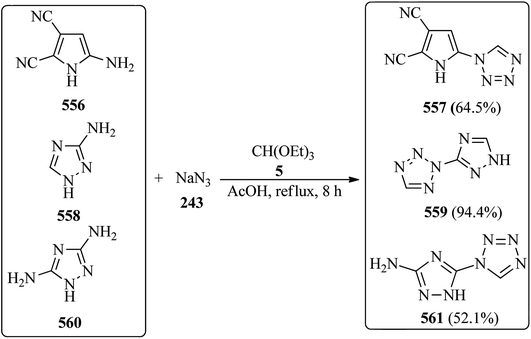 |
| | Scheme 163 Synthesizing a library of nitrogen-rich heterocycles. | |
4.25. Synthesis of pyridines
5-Nitropyridines (564) were obtained by Sagitullina et al. via the cyclocondensation of nitroacetone (562), TEOF (5) and enamines (563) in acetic acid media (Scheme 164).464
 |
| | Scheme 164 The cyclocondensation of nitroacetone, TEOF, and enamines. | |
4.26. Synthesis of pyridazine derivatives
6-Methyl-3,4-diphenylpyrimido[4′,5′:4,5]selenolo[2,3-c]pyridazine-8(7H)-one was synthesized via the cyclocondensation reaction of 5-amino-3,4-diphenyl-6-substitutedselenolo[2,3-c]pyridazine with TEOF in acetic anhydride under reflux conditions within 8 h in 72% yield.465 In 2015, Arghiani and co-workers reported a simple procedure for the synthesis of new [1,2,4]triazolo[4,3-b]pyridazines (567) on treatment of 6-choloro-5-(4-substituted phenyl)-3-pyridazinyl hydrazines (565) with orthoesters (5, 6, 7, 566) in refluxing acetic acid for 5 h (Scheme 165). The products were filtered off and crystallized from methanol.466
 |
| | Scheme 165 The synthesis of some new [1,2,4]triazolo[4,3-b]pyridazines. | |
4.27. Synthesis of tetrahydropyrimidinium salts
The cyclization of N,N′-dialkyl or diaryl ethane-1,2-diamines or propane-1,3-diamines (568) with inorganic ammonium salts (328) and TEOF (5) proceeds under microwave irradiation to afford the corresponding imidazolinium or tetrahydropyrimidinium salts (569). As can be seen in the Scheme 166, the microwave-assisted preparation of some 1,3-diaryl-3,4,5,6-tetrahydropyrimidium tetrafluoroborates was performed in ethanolic media. The TEOF is in 2–6 excess to the other substrates.467
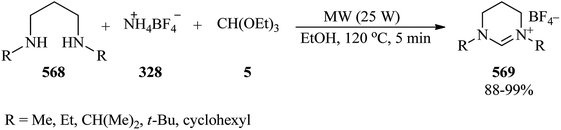 |
| | Scheme 166 The cyclization of N,N′-dialkyl ethane-1,2-diamines with ammonium salts and TEOF. | |
4.28. Synthesis of pyrimidine derivatives
In the synthetic route to obtain 1,3,4-oxadiazole tagged thieno[2,3-d]pyrimidine derivatives (571), the first step was reported via the reaction of 5-amino-4-cyano-3-methylthiophene-2-carboxylate (570) with TEOF, in refluxing toluene through the catalytic activity of acetic acid (Scheme 167).468
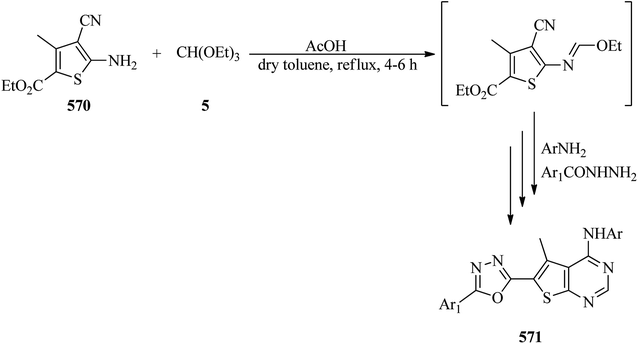 |
| | Scheme 167 Obtaining 1,3,4-oxadiazole tagged thieno[2,3-d]pyrimidine derivatives. | |
In 2003, Abdel-Rahman and coworkers have reported that pyridine-2-carboxamide derivatives (572) were reacted with TEOF in refluxing acetic anhydride to furnish white crystals of 9-aryl-7-(2′-thienyl)-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-4(3H)-ones (573) (Scheme 168).469–471
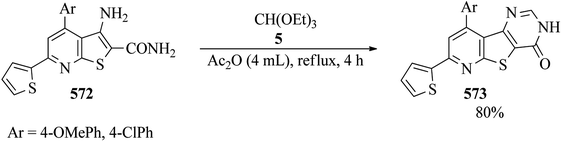 |
| | Scheme 168 The formation of 9-aryl-7-(2′-thienyl)-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine-4(3H)-ones. | |
The cyclocondensation of 5-acetyl-3-amino-4-aryl-2-(N-aryl)carbamoyl-6-methyl-thieno[2,3-b]pyridines (574) with TEOF was accomplished in refluxing acetic anhydride, in a dual role of catalyst and solvent, to achieve 8-acetyl-3,9-diaryl-7-methylpyrido[3′,2′:4,5]thieno[2,3-d]pyrimidine-4(3H)-ones (575)325,472 in excellent yields (87–94%) in 2 h (Scheme 169).325
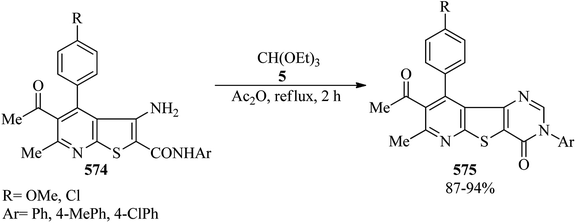 |
| | Scheme 169 The preparation of 8-acetyl-3,9-diaryl-7-methylpyrido[3′,2′:4,5]thieno[2,3-d]pyrimidine-4(3H)-ones. | |
Pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]pyrimidines (577) were produced via the cyclocondensation reaction of pyrazolo[3,4-d]pyrimidine (576) with triethyl orthoesters (5, 6, 7) in a 1:2 molar ratio in acetic acid, in a dual role of solvent and catalyst, under reflux conditions within 3 h in 63–83% yields (Scheme 170).473
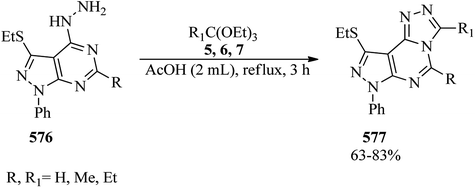 |
| | Scheme 170 Obtaining pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]pyrimidines. | |
Pyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidin-6(7H)-ones (579) were made through cyclocondensation of 7-hydrazinyl-substituted pyrimido[4,5-d]pyrimidin-4(3H)-ones (578) with triethyl orthoesters (5, 6, 7) in equimolar amounts in refluxing AcOH in a 6 h period in 40–90% yields (Scheme 171). The products showed fluorescent and photophysical properties.474
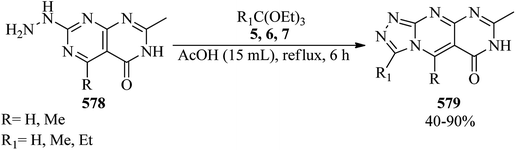 |
| | Scheme 171 The formation of pyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidin-6(7H)-ones. | |
In 2016, 2,4-dichloro-5-amino-6-methylpyrimidine (580) was applied as a starting material for the synthesis of 5-hydrazinyl-7-methyl[1,3]thiazolo[5,4-d]pyrimidines (581) via a multi-step reaction which, on a cyclization reaction with orthoesters (5, 6) in refluxing acetonitrile using catalytic amounts of acetic acid along with the elimination of 3 moles of ethanol, furnished novel thiazolo[5,4-d][1,2,4]triazolo[4,3-a]pyrimidines (582) for 2–4 h in high yields (62–89%) (Scheme 172).475
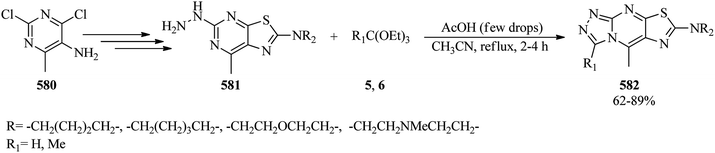 |
| | Scheme 172 A novel thiazolo[5,4-d][1,2,4]triazolo[4,3-a]pyrimidine preparation approach. | |
Benzo[4,5]imidazo[1,2-c]-[1,2,5]oxadiazolo[3,4-e]pyrimidine (584) was obtained via the cyclocondensation of 3-amino-4-(1H-benzo[d]imidazol-2-yl)-1,2,5-oxadiazole (583) with TEOF in boiling acetic anhydride for 12 h, in 60% yield (Scheme 173).476
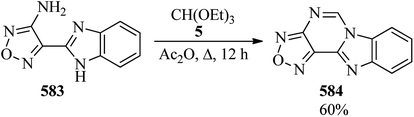 |
| | Scheme 173 The synthesis of benzo[4,5]imidazo[1,2-c]-[1,2,5]oxadiazolo[3,4-e]pyrimidine. | |
6-{[(5-Nitrothiophen-2-yl)methylene]amino}-3-phenyl-2-thioxo-2,3-dihydrothiazolo-[4,5-d]-pyrimidin-7(6H)-one (587)477,478 as a brown compound was prepared via the cyclocondensation of thiazoline derivative (586), arising from 5-nitro-2-thiophenecarboxaldehyde (585), with TEOF in the presence of acetic anhydride under reflux conditions within 3 h in 61% yield (Scheme 174). The product (587) revealed good anti-inflammatory activity.478
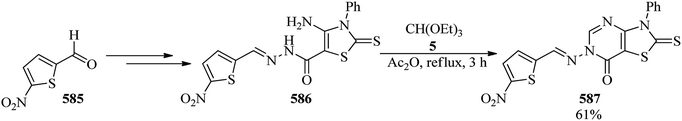 |
| | Scheme 174 The synthesis of 6-{[(5-nitrothiophen-2-yl)methylene]amino}-3-phenyl-2-thioxo-2,3-dihydrothiazolo-[4,5-d]-pyrimidin-7(6H)-one. | |
Due to the growing biological importance of fused thiazoles, in 2007, Bondock et al. employed 1-chloro-3,4-dihydronaphthalene-2-carboxaldehyde (588) as a key precursor for the formation of thiazoline derivative (589) via a multi-step reaction. Subsequent cyclization with TEOF in acetic anhydride under reflux conditions yielded the thiazolo[5,4-d]pyrimidine derivative (590) in 60% yield in 3 h (Scheme 175). The compounds were screened and evaluated as antimicrobial agents.479
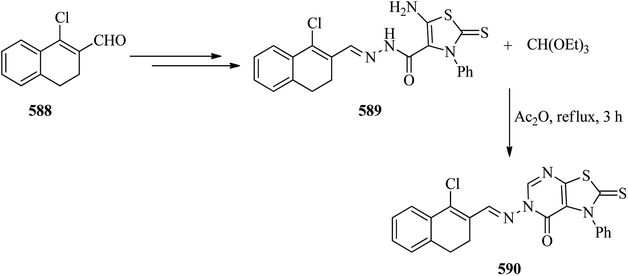 |
| | Scheme 175 Achieving thiazoline derivatives. | |
In 1998, Porcari and Townsend developed a fast and efficient method for the preparation of 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine (594) from 2-amino-5-bromo-3,4-dicyanopyrrole (591), TEOF (5), and ethanolic ammonia (593). In the first step the reaction of 2-amino-5-bromo-3,4-dicyanopyrrole (591) with triethylorthoformate (5) in dry acetonitrile at reflux temperature, for 1 h formed 2-bromo-5-(ethoxymethylene)iminopyrrole-3,4-dicarbonitrile (592) which was used without further purification. Ring annulation to obtain 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine (594) was accomplished by dissolving the unpurified intermediate (592) in saturated ethanolic ammonia and heating this mixture at 105 °C in a sealed steel reaction vessel for 6 h (Scheme 176).480
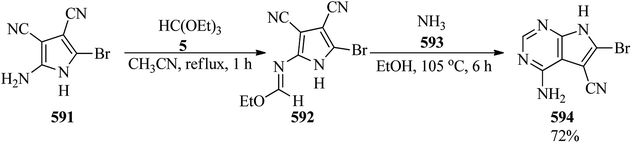 |
| | Scheme 176 The preparation of 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine. | |
In 2015, Abdel Hameed et al. demonstrated a regioselective procedure for the synthesis of pyrazolo[1,5-a]pyrimdines-7(4H)-ones (598) via the reaction of 5-amino pyrazoles (595), Meldrum's acid (231), and TEOF (5) in dioxane at room temperature. This transformation took place via a Knoevenagel condensation of Meldrum's acid (231) with TEOF (5) to produce intermediate (596), as a Michael acceptor, followed by the regioselective Michael addition with the exocyclic amino moiety of 5-amino pyrazoles (595) to obtain the corresponding acyclic adducts (597) in dioxane at room temperature; which after isolation underwent an intramolecular cyclization and then the elimination of acetone and CO2 to get pyrazolo[1,5-a]pyrimdines-7(4H)-ones (598) in refluxing dioxane for 12 h. The compound (598) could also be directly obtained at once via the one-step condensation reaction of substrates in refluxing dioxane for 15 h (Scheme 177).481
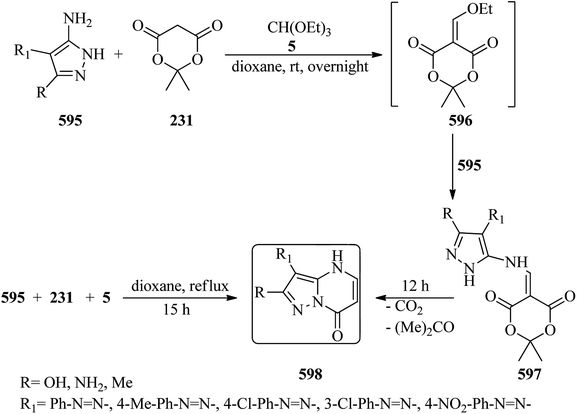 |
| | Scheme 177 The regioselective synthesis of pyrazolo[1,5-a]pyrimdines-7(4H)-ones. | |
In 2008, a facile and high-yielding process was developed for the synthesis of pyrazolo[3,4-d]pyrimidin-4-ones (601) in excellent yields (82–92%). First, aminopyrazoles (599) were condensed with TEOF (5) in glacial acetic acid under reflux conditions for 3 h to form the intermediate (600) which was then followed by cyclization and removal of an equivalent ethanol to achieve (601) (Scheme 178).482
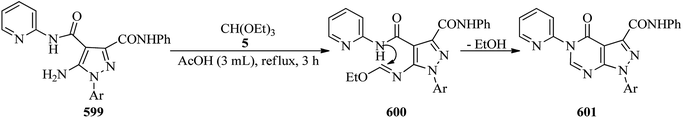 |
| | Scheme 178 The synthesis of pyrazolo[3,4-d]pyrimidin-4-ones. | |
In 2015, 6-amino-3-methyl-4-fluorophenyl-1,4-dihydro-pyrano[2,3-c]pyrazole-5-carbonitrile (602) was employed as a starting material for the preparation of 4-(4-fluorophenyl)-5-imino-3-methyl-1,4-dihydropyrazolo[4′,3′:5,6]pyrano[2,3-d]pyrimidin-6(5H)-amine (603)483,484 as yellow powder. Hence, in the first step, (602) was condensed with TEOF (5) in refluxing acetic anhydride for 5 h; which was followed by the reaction of the isolated solid with hydrazine hydrate (271) in ethanol under reflux conditions for 2 h to produce (603) in 60% yield (Scheme 179). The product was screened for antibacterial and antifungal activities compared with norfloxacin and fluconazole as standard drugs.484
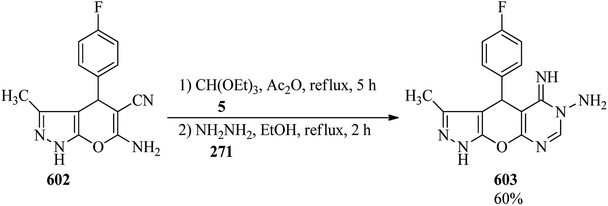 |
| | Scheme 179 The preparation of 4-(4-fluorophenyl)-5-imino-3-methyl-1,4-dihydropyrazolo[4′,3′:5,6]pyrano[2,3-d]pyrimidin-6(5H)-amine. | |
4.29. Synthesis of triazine derivatives
Montazeri et al. established a simple and catalyst-free method for the preparation of new bis[1,2,4]triazolo[4,3-b:1′,5′-d][1,2,4]triazines (606). This reaction progressed through the nucleophilic substitution reaction of 8-methyl-5-methylthio-2-aryl-6H-[1,2,4]triazolo[1,5-d][1,2,4]triazines (604) with hydrazine hydrate (271) in refluxing ethanol for 3 h to generate 5-hydrazino compounds (605); which was followed a cyclocondensation by triethyl orthoesters (5, 6) under refluxing in ethanol for 8 h to produce products (606) in 51–67% yields (Scheme 180).485
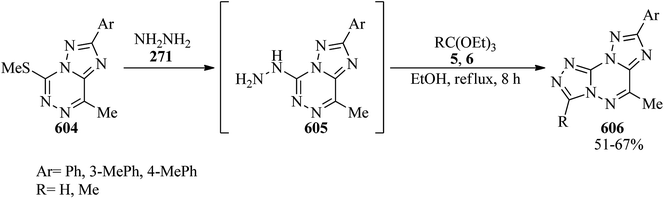 |
| | Scheme 180 The preparation of new bis[1,2,4]triazolo[4,3-b:1′,5′-d][1,2,4]triazines. | |
In 2017, the Zemanová group reported that 4H-furo[3,2-b]pyrrole-5-carbohydrazide (607) on reaction with orthoesters (5, 6, 536) in a polar solvent like DMF under reflux conditions yielded furo[2′,3′:4,5]pyrrolo[1,2-d][1,2,4]triazin-8(7H)-ones (608) within 3.5–24 h, in high yields (69–75%) (Scheme 181). The products were screened for their antibacterial activity on Gram-negative bacterial species Escherichia coli CCM 7929, Pseudomonas syringae CCM 2114 and Gram-positive bacterial species Micrococcus luteus CCM 732, Bacillus pumilus CCM 2218 and it was found to have good activity.486
 |
| | Scheme 181 The synthesis of furo[2′,3′:4,5]pyrrolo[1,2-d][1,2,4]triazin-8(7H)-ones. | |
In 1989, some new 8-substituted-10-methyl-l,2-dihydro-1-oxo-l,2,4-triazino[4,5-a]indoles (610) were prepared in 70–78% yields from the reaction of various 3-methylindole-2-carbohydrazides (609) with TEOF (5) in refluxing dimethylformamide within 8 h (Scheme 182). These derivatives were evaluated for their antimicrobial activity. Most of the compounds showed good activity against Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa. Chloro-substituted derivatives showed the highest activity against all of the organisms tested.487
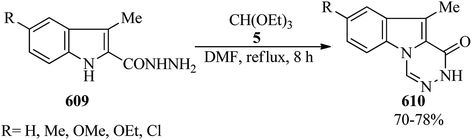 |
| | Scheme 182 The formation of new 8-substituted-10-methyl-l,2-dihydro-1-oxo-l,2,4-triazino[4,5-a]indoles. | |
In 2018, Lim and et al. described a chemo- and regioselective procedure for the synthesis of new 2-alkyl substituted 4-aminoimidazo[1,2-a][1,3,5]triazines (612) in weak to good yields (11–61%) via the catalyst-free one-pot three-component reaction of 2-aminoimidazoles (611), trialkylorthoesters (6, 7, 316, 317), and cyanamide (551) under microwave irradiation (Discover SP CEM) at 160 °C in ethyl acetate for 35 min (Scheme 183). It was found that a growth in length of the alkyl chain group on compound (612) resulted in a reduction in isolated yields. For example, the products originating from TEOAc (6) and triethyl orthopropionate, (7) obtained higher isolated yields than triethyl orthobutyrate (316) or trimethyl orthovalerate (317), which could be ascribed to the proportionately lower reactivity of long-chain orthoesters for the preparation of compounds (612). Molecular and crystal building blocks of resultant molecules were examined using X-ray crystallography.488
 |
| | Scheme 183 The synthesis of new 2-alkyl substituted 4-aminoimidazo[1,2-a][1,3,5]triazines. | |
4.30. Synthesis of tetrazine derivatives
Preyssler heteropolyacid (H14[NaP5W30O110], 0.1 mmol) catalyzed the green one-pot reaction of 4-amino-3-hydrazino-6-methyl-1,2,4-triazin-5(2H)-one with orthoesters in acetic acid under reflux conditions to produce [1,2,4]triazino[4,3-b][1,2,4,5]tetrazines within 1–6 h in excellent yields (79–95%).489 [1,2,4]Triazino[4,5-b][1,2,4,5]tetrazines were also prepared from the reaction of 6-methyl-4-amino-5-hydrazino-1,2,4-triazin-3-one with orthoesters.490 [1,2,4]Triazino[3,4-b][1,2,4,5]-tetrazine-6-thiones (614) were synthesized in 58–78% yields upon treatment of 4-amino-6-methyl-3-hydrazino-4,5-dihydro-1,2,4-triazine-5-thione (613) with excess amounts of triethyl orthoesters (5, 6) in refluxing ethanol for 1 h (Scheme 184).491
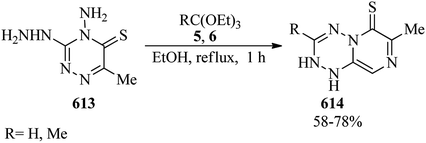 |
| | Scheme 184 The preparation of [1,2,4]triazino[3,4-b][1,2,4,5]-tetrazine-6-thiones. | |
4.31. Synthesis of cinnoline derivative
The cyclocondensation of 10-amino-7-(p-tolyl)pyrimido[4′′,5′′:4′,5]thieno[3′,2′:5,6]pyrido[3,2-c]cinnoline-11-imine (615) with TEOF (5) was accomplished in refluxing acetic acid for 5 h to give 7-(p-tolyl)-1,2,4-triazolo[4′′′,3′′′:1′′,6′′]pyrimido[4′′,5′′:4′,5′]thieno[3′,2′:5,6]pyrido[3,2-c]cinnoline (616) in 70% yield (Scheme 185).492
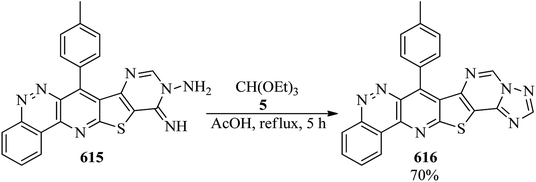 |
| | Scheme 185 The synthesis of 7-(p-tolyl)-1,2,4-triazolo[4′′′,3′′′:1′′,6′′]pyrimido[4′′,5′′:4′,5′]thieno[3′,2′:5,6]pyrido[3,2-c]cinnoline. | |
4.32. Synthesis of quinazoline derivatives
Cobalt(II) chloride (CoCl2, 10 mol%) catalyzed the one-pot three-component reaction of anthranilic acid, TEOF, and anilines in CH3CN at 70 °C to obtain 3-substituted-quinazolin-4(3H)-ones.493–495 Both electron-donating and electron-withdrawing anilines provided the products in good yields (75–95%).495 Quinazolin-4(3H)-ones (618) could also be obtained in good yields via the novel reductive cyclization of o-nitrobenzamides (617) and TEOF (5), promoted by TiCl4/Zn in refluxing THF (Scheme 186).496
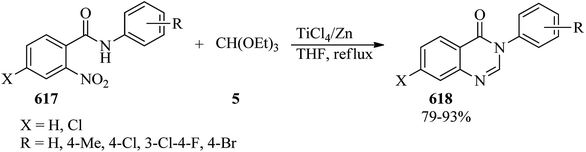 |
| | Scheme 186 The synthesis of quinazolin-4(3H)-ones. | |
The cyclocondensation of 6-hydrazino-substituted benzimidazo[1,2-c]quinazolines (619) with orthoesters (5, 6, 7) was accomplished in refluxing ethanol to obtain new benzimidazo[1,2-c][1,2,4]triazolo[4,3-a]quinazolines (620) fin 1 h in high yields (70–81%) (Scheme 187).497
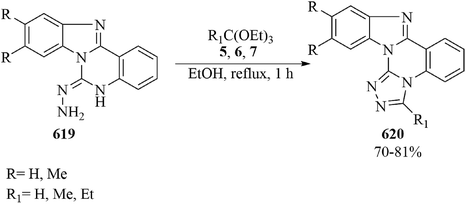 |
| | Scheme 187 Obtaining new benzimidazo[1,2-c][1,2,4]triazolo[4,3-a]quinazolines. | |
Bis(quinazolinon-4(1H)-one) derivatives (622) were also synthesized via the regioselective one-pot pseudo five-component cyclocondensation reaction of isatoic anhydride (312), orthoesters (5, 6, 7, 102, 323, 455), and diamines (621) using KAl(SO4)2·12H2O (alum, 20 mol%) as an inexpensive, eco-friendly, readily available, easily separable, and recyclable catalyst in refluxing DMF within 30–50 min in good to excellent yields (79–90%) (Scheme 188).498 4-Arylaminoquinazolines499 were synthesized in excellent yields (84–96%) within 2 h via the one-pot three-component condensation reaction of 2-aminobenzamide, anilines, and orthoesters by a catalytic amount of Keggin-type heteropolyacid (H6[PMo9V3O40], 0.03 mmol) in refluxing CH3CN. Significantly, anilines containing electron-donating groups were obtained in slightly better yield. In this reaction, 3-quinazolin-4-one (2) was formed as a by-product in low yield.500 Preyssler heteropolyacid H14 [NaP5W30O110] also progressed this transformation in refluxing CH3CN within 2 h.501
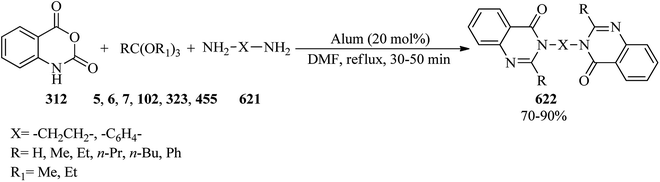 |
| | Scheme 188 The formation of bis(quinazolinon-4(1H)-one) derivatives. | |
4.33. Synthesis of quinoxalines
Novel 3-substitued [1,2,4]triazolo[3′,4′;2,3][1,3]triazolo[4,5-b]quinoxaline derivatives (624) were achieved through the cyclocondensation of [1,3]thiazolo[4,5-b]quinoxaline-2(3H)-one hydrazone (623) with orthoesters (4, 6) in acetic acid under reflux conditions within 4–8 h (Scheme 189).502
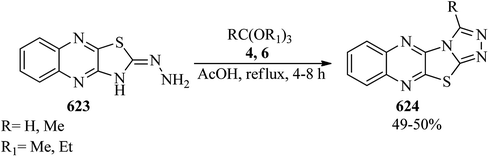 |
| | Scheme 189 The synthesis of novel 3-substitued [1,2,4]triazolo[3′,4′;2,3][1,3]triazolo[4,5-b]quinoxalines. | |
4.34. Glycosidation, glycosylation, and oligomerization reactions
In 2015, Rao et al. employed propargyl 1,2-orthoesters (625) as key synthons and glycosyl donors for the synthesis of thiosaccharides and 1-thiotrehaloses (627) in a 1,2-trans diastereoselective form only via the glycosidation reaction503–509 with glycosyl mercaptans (626), as glycosyl acceptor, using gold(III) bromide (AuBr3, 7 mol%) as catalyst in CH2Cl2 at room temperature under argon atmosphere in excellent yields (83–94%) within 6 h (Scheme 190).509
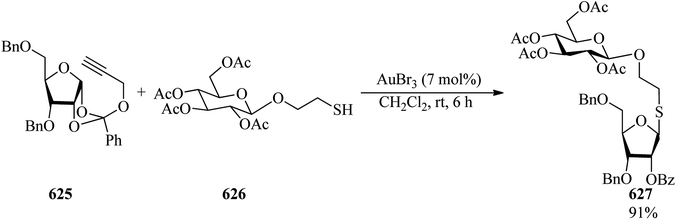 |
| | Scheme 190 The synthesis of thiosaccharides and 1-thiotrehaloses. | |
In 2015, Vadhadiya and Ramana reported the first total synthesis of sinenside A. In this survey, the glycosidation reaction of orthoester (628)510 with allylic alcohol (629), as a glycosyl acceptor, by BF3·Et2O (0.15 equiv.) as catalyst in dichloromethane at 0 °C in the presence of 4 Å molecular sieves obtained β-glucoside (630) in good yield (76%) in a 1 h period, which was employed as a key precursor for the synthesis of sinenside A (631) via a multi-step reaction (Scheme 191).511
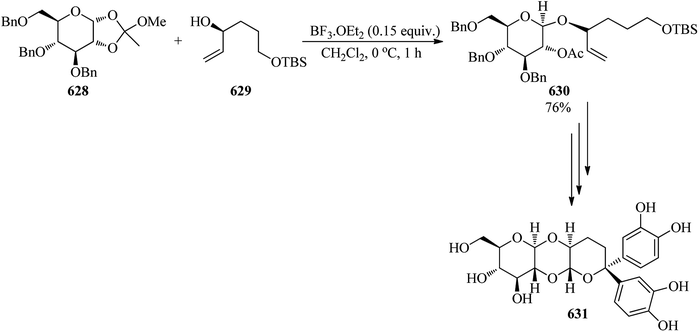 |
| | Scheme 191 The total synthesis of sinenside A. | |
The stereoselective O-glycosylation of glycals (632) and glycosylbromides (634) by orthoformates (4, 5, 127) in the presence of InCl3 in corresponding solvents (CH3CN and CH2Cl2) yielded O-glycopyranosides (633, 635) at room temperature (Scheme 192). It must be mentioned that both perbenzyl and peracetyl glycals resulted in 2,3-unsaturated-O-glycosides with major α-selectivity while glycosylbromides furnished alkoxy glycosides with high β-selectivity in most cases.512
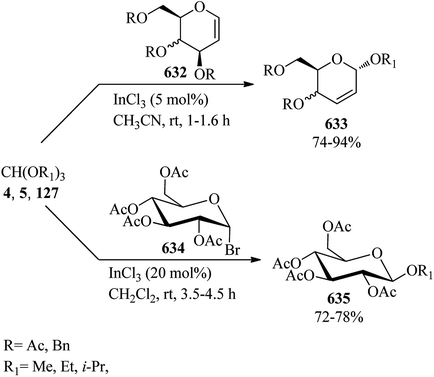 |
| | Scheme 192 The formation of O-glycopyranosides. | |
SnCl4 catalyzed the oligomerization reaction of 3-O-benzoyl-β-D-arabinofuranose 1,2,5-orthobenzoate (636)513,514 with 4-(2-chloroethoxy) phenol (637a) or (4-(2-chloroethoxy)phenoxy)trimethylsilane (CEP-OTMS, 637b) as a terminating agent in CH2Cl2 at −25 °C to form selectively α(1 → 5) linked arabinofuranose oligomers, including 4-(2-chloroethoxy)phenyl (CEP) aglycon with a deprotected 5-OH group at the non-reducing end (638) (Scheme 193).515 The resultant glycosyl acceptor (638) could be used in glycosylation reactions.514,516
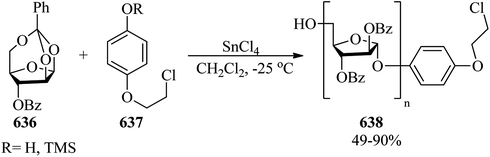 |
| | Scheme 193 Glycosyl acceptor synthetic procedure. | |
4.35. Synthesis of (±)-1,2-O-ethylidene-myo-inositol
The reaction of myo-inositol (639) with TEOF (5) using p-TSA (2g) under heating in dimethyl sulfoxide at 100 °C has been used to obtain myo-inositol monoorthoformate (640) and (±)-1,2-O-ethylidene-myo-inositol (641) within a 24 h period in 42% and 33% yields, respectively (Scheme 194). It was found that the presence of the chiral carbon of ethylidene in the compound (641) formed a mixtures of the four diastereomers which were not isolated by silica gel column chromatography but were characterized by NMR spectra.517
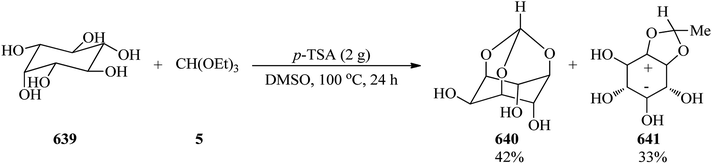 |
| | Scheme 194 The synthesis of myo-inositol monoorthoformate and (±)-1,2-O-ethylidene-myo-inositol. | |
4.36. α-Arylation and alkylation of protected pyruvate esters
In 2017, Esteves et al. interpreted a novel process for the enolate arylation and alkylation of protected pyruvate esters. Hence, in the first step, the orthoester-masked pyruvate equivalent (oxabicyclo[2.2.2]octyl orthoester, OBO)518–521 was prepared through the reaction of pyruvic acid (642) with oxalyl chloride (643) to form pyruvoyl chloride (644) which underwent an esterification reaction with 3-methyl-3-oxetanemethanol (645) in the presence of pyridine to afford the ester (646) via a two-step reaction which on reaction with BF3·OEt2 in CH2Cl2 at 0 °C yielded the OBO orthoester (647) (Scheme 195). Then, the mono α-arylation (649) reaction was accomplished via the reaction of (647) with aryl bromide (648) by Pd(dtbpf)Cl2 (5 mol%), utilizing at least two equivalents of base in THF at 50 °C for 24 h. Due to the greater acidity of the product than the starting material,522 the resultant aryl ketone was deprotonated in situ by the extra base. Subsequent alkylation by different equivalents of electrophile at 50–80 °C obtained α-arylation and alkylation products (Scheme 196). In the final step, the OBO protective group was conveniently removed using acidic conditions such as p-TSA in ethanol at 90 °C for 3–8 h. The method also enabled mono- and diarylation reactions to happen on a readily available OBO-protected precursor.521
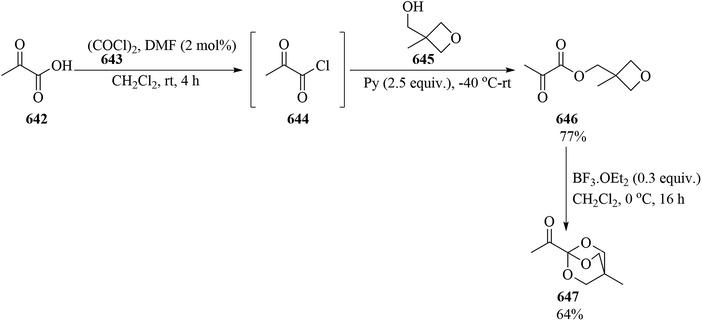 |
| | Scheme 195 The preparation of OBO orthoester. | |
 |
| | Scheme 196 The mono α-arylation of OBO orthoester. | |
5. Conclusions
In this review, several types of orthoesters have been reported as applicable substrates in classified organic transformations. These treatments have been subdivided according to the reaction medium. This focus is because of the significance of economical and green reactions in organic syntheses. Based on the overall results, the reaction could happen in solvent-free media, in the presence of aqueous conditions, or in organic solvents. Detailed investigations in the form of a literature survey affirmed that the processes containing orthoesters progressed well in the absence of solvents. This important attainment has let researchers design new procedures to achieve compounds with multi-functionalized motifs in the absence of hazardous solvents, utilizing the greater activity potential of substrates and reagents, as they could come into contact effectively within short periods. The aqua-mediated reactions were not extensive, which may be due to the hydrolysis affinity of orthoesters in water. Traditional organic solvents were also mentioned a great deal in reports. The greatest usage involved alcohols, dichloromethane, acetic anhydride, acetonitrile, acetic acid, benzene and its derivatives, and THF. Other common solvents used in lower abundance were DMF, ether, DMSO, dioxane, and CCl4. Some transformations were done under heating conditions and some others required microwave or ultrasound assistance for promotion. Based on the many mentions, TEOF was the most commonly used alkyl orthoester in a vast range of reactions. The authors hope that this article will guide general readers to gain an intense interest in using orthoesters, as versatile and easily-handled synthons, in a great range of reactions with a focus on the environment.
Conflicts of interest
There are no conflicts to declare.
References
- V. V. Mezheritskii, E. P. Olekhnovich and G. N. Dorofeenko, Russ. Chem. Rev., 1973, 42, 392 CrossRef.
- R. H. Dewolfe, Synthesis, 1974, 1974, 153 CrossRef.
- A. W. Williamson and G. Kay, Ann. Chem., 1854, 92, 346 CrossRef.
- R. H. Dewolfe, Carboxylic Ortho Acid Derivatives: Preparation and Synthetic Applications, organic chemistry a series of monographs, Academic Press, New York and London, 1970, ch. 1, pp. 54–122 Search PubMed.
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2008, 634, 267 CrossRef CAS.
- S. L. Repetto, R. Patel, T. Johnson, J. F. Costello, J. K.-W. Lam and C. J. Chuck, Energy Fuels, 2016, 30, 9080 CrossRef CAS.
- J. C. Jansen and J. Reedijk, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1974, 29, 527 CAS.
- T. Wagner, C. G. Hrib, V. Lorenz, F. T. Edelmann, D. S. Amenta, C. J. Burnside and J. W. Gilje, Z. Anorg. Allg. Chem., 2012, 638, 2129 CrossRef CAS.
- G. C. Look, M. M. Murphy, D. A. Campbell and M. A. Gallop, Tetrahedron Lett., 1995, 36, 2937 CrossRef CAS.
- Y. F. Song, G. A. van Albada, J. Tang, I. Mutikainen, U. Turpeinen, C. Massera, O. Roubeau, J. S. Costa, P. Gamez and J. Reedijk, Inorg. Chem., 2007, 46, 4944 CrossRef CAS PubMed.
- S. K. Shannon and G. Barany, J. Org. Chem., 2004, 69, 4586 CrossRef CAS PubMed.
- N. Yu, R. Poulain and J.-C. Gesquiere, Synlett, 2000, 2000, 355 CrossRef.
- M. Schöttner, J. Reiner and F. S. K. Tayman, Phytochemistry, 1997, 46, 1107 CrossRef.
- D. M. Ketcha, L. J. Wilson and D. E. Portlock, Tetrahedron Lett., 2000, 41, 6253 CrossRef CAS.
- R. T. Stibrany, D. N. Schulz, S. Kacker, A. O. Patil, L. S. Baugh, S. P. Rucker, S. Zushma, E. Berluche and J. A. Sissano, Macromolecules, 2003, 36, 8584 CrossRef CAS.
- I. Abrunhosa-Thomas, O. Roy, M. Barra, T. Besset, P. Chalard and Y. Troin, Synlett, 2007, 2007, 1613 CrossRef.
- F. Zaragoza and S. V. Petersen, Tetrahedron, 1996, 52, 10823 CrossRef CAS.
- M. O. Frederick and D. P. Kjell, Tetrahedron Lett., 2015, 56, 949 CrossRef CAS.
- Y. L. Janin, J. Chiki, M. Legraverend, C. Huel and E. Bisagni, Tetrahedron, 1999, 55, 12797 CrossRef CAS.
- Y. Chen, Chem.–Eur. J., 2019, 25, 3405 CrossRef CAS PubMed.
- Y. C. Mei, B. W. Yang, W. Chen, D. D. Huang, Y. Li, X. Deng, B. M. Liu, J. J. Wang, H. Qian and W. L. Huang, Lett. Org. Chem., 2012, 9, 276–279 CrossRef CAS.
- R. S. Hosmane and C. M. Bertha, Synth. Commun., 1990, 20, 2921–2935 CrossRef CAS.
- N. N. Balaneva, O. P. Shestak and V. L. Novikov, Russ. Chem. Bull., Int. Ed., 2019, 68, 55 CrossRef CAS.
- M. Nabati and M. Mahkam, Iran. Chem. Commun., 2014, 2, 162–167 Search PubMed.
- M. J. Gallagher and H. Honegger, Tetrahedron Lett., 1977, 18, 2987 CrossRef.
- J. Gloede, S. Ozegowski, D. Matt and A. De Cian, Tetrahedron Lett., 2001, 42, 9139–9142 CrossRef CAS.
- W. Noll, Chemistry and Technology of Silicones, Academic Press, New York, 1968, pp. 68–115 Search PubMed.
- Y. Kita, Y. Yoshida, S. Mihara, D.-F. Fang, K. Higuchi, A. Furukawa and H. Fujioka, Tetrahedron Lett., 1997, 38, 8315 CrossRef CAS.
- G. Dujardin, S. Leconte, A. Bénard and E. Brown, Synlett, 2001, 2001, 147 CrossRef.
- H. Yanai, T. Oguchi and T. Taguchi, J. Org. Chem., 2009, 74, 3927 CrossRef CAS PubMed.
- M. Cheong, M.-N. Kim and J. Y. Shim, J. Organomet. Chem., 1996, 520, 253 CrossRef CAS.
- A. D. Morley, Tetrahedron Lett., 2000, 41, 7405 CrossRef CAS.
- D. A. Wacker and P. Kasireddy, Tetrahedron Lett., 2002, 43, 5189 CrossRef CAS.
- J. J. Reina, A. D. Maio, J. Ramos-Soriano, R. C. Figueiredo and J. Rojoa, Org. Biomol. Chem., 2016, 14, 2873 RSC.
- A. Gambacorta, D. Tofani and A. Migliorini, Molecules, 2007, 12, 1762 CrossRef CAS PubMed.
- J. L. Giner, Org. Lett., 2005, 7, 499 CrossRef CAS PubMed.
- W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T.-T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741 CrossRef CAS.
- M. P. Dreyfuss and P. Dreyfuss, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2179 CrossRef CAS.
- S. Okamoto, K. Tanaka and A. Sudo, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1743 CrossRef CAS.
- T. Haider, O. Shyshov, O. Suraeva, I. Lieberwirth, M. von Delius and F. R. Wurm, Macromolecules, 2019, 52, 2411 CrossRef CAS PubMed.
- L. Li, K. Knickelbein, L. Zhang, J. Wang, M. Obrinske, G. Z. Ma, L.-M. Zhang, L. Bitterman and W. Du, Chem. Commun., 2015, 51, 13078 RSC.
- S. Maiti, S. Manna, J. Shen, A. P. Esser-Kahn and W. Du, J. Am. Chem. Soc., 2019, 141, 4510 CrossRef CAS PubMed.
- Y. Furusho and T. Endo, Polym. Bull., 2017, 74, 1207 CrossRef CAS.
- M. Rillich, D. Jehnichen, H. Komber and F. Bohme, Macromol. Chem. Phys., 1995, 196, 1635–1644 CrossRef CAS.
- C. Adamson, R. J. Pengelly, S. S. K. Abadi, S. Chakladar, J. Draper, R. Britton, T. M. Gloster and A. J. Bennet, Angew. Chem., 2016, 128, 15202 CrossRef.
- J. L. Darlix, P. Fromageot and E. Reich, Biochemistry, 1973, 12, 914 CrossRef CAS PubMed.
- B. E. Griffin, M. Jarman, C. B. Reese and J. E. Sulston, Tetrahedron, 1967, 23, 2301 CrossRef CAS PubMed.
- B. S. Ross, R. H. Springer, Z. Tortorici and S. Dimock, Nucleos. Nucleot. Nucl., 1997, 16, 1641 CrossRef CAS.
- L. J. Lawrence, C. J. Palmer, K. W. Gee, T. Wang, H. I. Yamarnura and J. E. Casida, J. Neurochem., 1985, 45, 798 CrossRef CAS PubMed.
- G. Leriche, M. Nothisen, N. Baumlin, C. D. Muller, D. Bagnard, J.-S. Remy, S. A. Jacques and A. Wagner, Bioconjugate Chem., 2015, 26, 1461 CrossRef CAS PubMed.
- B. Li, B. Duan, J. Lia, M. Zhang, Y. Yuan, Y. Ding and A. Hu, Tetrahedron, 2018, 74, 6419–6425 CrossRef CAS.
- D. Song, S. Sun, Y. Tian, S. Huang, Y. Ding, Y. Yuan and A. Hu, J. Mater. Chem. B, 2015, 3, 3195–3200 RSC.
- R. Cai, L. Zhu, W. Shen and Y. Zhao, Fitoterapia, 2019, 137, 104198 CrossRef CAS PubMed.
- H. J. Schaeffer and C. F. Schwender, J. Med. Chem., 1974, 17, 6–8 CrossRef CAS PubMed.
- X. Huang, F. Du, J. Cheng, Y. Dong, D. Liang, S. Ji, Sh. Lin and Z. Li, Macromolecules, 2009, 42, 783–790 CrossRef CAS.
- A. H. Shamroukh, M. E. A. Zaki, E. M. H. Morsy, F. M. Abdel-Motti and F. M. E. Abdel-Megeid, Arch. Pharm., 2007, 340, 345–351 CrossRef CAS PubMed.
- A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., 2009, 48, 8547 CrossRef CAS PubMed.
- A. Jackson and O. Meth-Cohnt, J. Chem. Soc., Chem. Commun., 1995, 1319 RSC.
- A. Alsughayer, A. A. Elassar, S. Mustafa and F. Al Sagheer, J. Biomater. Nanobiotechnol., 2011, 2, 144–149 CAS.
- L. Shahzadi, M. Yar, A. Jamal, S. A. Siddiqi, A. A. Chaudhry, S. Zahid, M. Tariq, I. Rehman and Sh. MacNeil, J. Biomater. Appl., 2016, 31, 582–593 CrossRef CAS PubMed.
- J.-R. Li, R.-H. Zhang and X.-H. Bu, Eur. J. Inorg. Chem., 2005, 2005, 1913 CrossRef.
- G. A. van Albada, S. Amani Komaei, H. Kooijman, A. L. Spek and J. Reedijk, Inorg. Chim. Acta, 1999, 287, 226 CrossRef CAS.
- Y. Sasaki and C. Pittman, J. Org. Chem., 1973, 38, 3723–3726 CrossRef CAS.
- R.-C. Brachvogel and M. von Delius, Chem. Sci., 2015, 6, 1399 RSC.
- R.-C. Brachvogel, H. Maid and M. von Delius, Int. J. Mol. Sci., 2015, 16, 20641 CrossRef CAS PubMed.
- R.-C. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 CrossRef PubMed.
- M. von Delius, Synlett, 2016, 27, 177 CrossRef CAS.
- H. Löw, E. Mena-Osteritz and M. von Delius, Chem. Sci., 2018, 9, 4785 RSC.
- N. T. Patil, M. S. Shashidhar, M. I. Tamboli and R. G. Gonnade, Cryst. Growth Des., 2017, 17, 5432 CrossRef CAS.
- G. Buechi, J. A. Carlson, J. E. Powell and L. F. Tietze, J. Am. Chem. Soc., 1973, 95, 540 CrossRef CAS.
- S. Zhang, L. Xu, L. Miao, H. Shu and M. L. Trudell, J. Org. Chem., 2007, 72, 3133 CrossRef CAS PubMed.
- G. R. Kieczykowski and R. H. Schlessinger, J. Am. Chem. Soc., 1978, 100, 1938 CrossRef CAS.
- J. Uenishi, J. M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS.
- W. J. Greenlee and R. B. Woodward, J. Am. Chem. Soc., 1976, 98, 6075 CrossRef CAS.
- W. C. Chan and K. Koide, Org. Lett., 2018, 20, 7798–7802 CrossRef CAS PubMed.
- J. Gauthier and P. Deslongchamps, Can. J. Chem., 1967, 45, 297–300 CrossRef CAS.
- A. Pinner, Chem. Ber., 1883, 16, 1643 CrossRef.
- A. Fischer and H. Puetter, Eur. Pat., BASF SE, DE10043789A12000, 2000.
-
(a) H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257 CrossRef;
(b) F. Lebreux, F. Buzzo and I. E. Markó, J. Electrochem. Soc., 2008, 13, 1 CAS.
- H. G. Thomas and A. Schmitz, Synthesis, 1985, 31 CrossRef CAS.
- R. Fardel, U. Griesbach, H. Pütter and C. Comninellis, J. Appl. Electrochem., 2006, 36, 249 CrossRef CAS.
- J. L. Röckl, A. V. Hauck, D. Schollmeyer and S. R. Waldvogel, ChemistryOpen, 2019, 8, 1167 CrossRef PubMed.
- A. B. Charette and P. Chua, Tetrahedron Lett., 1997, 38, 8499 CrossRef CAS.
- T. Praveen and M. S. Shashidhar, Carbohydr. Res., 2001, 330, 409–411 CrossRef CAS PubMed.
- H.-J. Jung, C. Chang, I. Yu, D. C. Aluthge, T. Ebrahimi and P. Mehrkhodavandi, ChemCatChem, 2018, 10, 3219 CrossRef CAS.
- F. J. Lombard, R. J. Lepage, B. D. Schwartz, R. C. Johnston, P. C. Healy, E. H. Krenske and M. J. Coster, Org. Biomol. Chem., 2018, 16, 256 RSC.
- P. Wałejko and A. Baj, Monatsh. Chem., 2019, 150, 275 CrossRef.
- N. Yasukawa, T. Kanie, M. Kuwata, Y. Monguchi, H. Sajiki and Y. Sawama, Chem.–Eur. J., 2017, 23, 10974 CrossRef CAS PubMed.
- C. Gouliaras, D. Lee, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 13926 CrossRef CAS PubMed.
- D. T. Khong and Z. M. A. Judeh, Org. Biomol. Chem., 2017, 15, 2638 RSC.
- J. Zhao, S. Wei, A. Yue and H. Shao, Chin. J. Chem., 2012, 30, 627 CrossRef CAS.
- S. Danishefsky, P. F. Schuda, T. Kitahara and S. J. Etheredge, J. Am. Chem. Soc., 1977, 99, 6066 CrossRef CAS.
- V. A. Egorov, F. A. Gimalova, L. S. Khasanova and M. S. Miftakhov, Russ. Chem. Bull., Int. Ed., 2015, 64, 355–358 CrossRef CAS.
- L. M. De Coen, B. I. Roman, M. Movsisyan, T. S. A. Heugebaert and C. V. Stevens, Eur. J. Org. Chem., 2018, 2018, 2148 CrossRef CAS.
- I. A. Perillo, M. C. Caterina and A. Salerno, Arkivoc, 2018, i, 288 Search PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Ceesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- A. Al-Azmi, A. A. Elassar and B. L. Booth, Tetrahedron, 2003, 59, 2749–2763 CrossRef CAS.
- S. Ghosh and U. R. Ghatak, Proc. - Indian Acad. Sci., Chem. Sci., 1988, 100, 235 CAS.
- S. Alam, Int. J. Chem. Res., 2016, 8, 173 Search PubMed.
- L. A. Pavlova, Y. A. Davidovich and S. V. Rogozhin, Russ. Chem. Rev., 1986, 55, 1026 CrossRef.
- F. Kong, Carbohydr. Res., 2007, 342, 345 CrossRef CAS PubMed.
- S.-G. Liao, H.-D. Chen and J.-M. Yue, Chem. Rev., 2009, 109, 1092 CrossRef CAS PubMed.
- R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, Eur. J. Org. Chem., 2014, 2014, 2833 CrossRef CAS.
- C. P. Frizzo, Synlett, 2009, 2009, 1019 CrossRef.
- Z. Nazarian and M. Dabiri, ChemistrySelect, 2020, 5, 4394 CrossRef CAS.
- R. Q. Lamphon, M. S. A. El-Gaby, M. M. Khafagy, G. A. M. El-Hag Ali, A. A. El-Maghraby, H. A. Eyada and M. H. M. Helal, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1279–1292 CrossRef CAS.
- E. K. Ahmed, F. F. Abdel-latif and M. A. Ameen, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 95–107 CrossRef CAS.
- M. A. El-Hashash, M. H. A. Soliman, I. I. Abd El-Gwad, S. S. El-Sakka and M. A. Morsy, Afinidad, 2010, 67, 367–373 CAS.
- R. A. Mekheimer, Synthesis, 2001, 2001, 103–107 CrossRef.
- M. Khashi, A. Davoodnia and V. S. P. R. Lingam, Res. Chem. Intermed., 2015, 41, 5731–5742 CrossRef CAS.
- A. S. Oganisyan, A. S. Noravyan and M. Z. Grigoryan, Chem. Heterocycl. Compd., 2004, 40, 75–78 CrossRef CAS.
- A. Krauze, Z. Bomika, A. M. Shestopalov, L. A. Rodinovskaya, J. Pelcers, G. Duburs, Y. A. Sharanin and V. K. Promonenkov, Khim. Geterotsikl. Soedin., 1981, 3, 377 (Chem. Abstr., 1981, 95, 80662) Search PubMed.
- H. M. F. Madkour, A. A. E. Afify, A. A. Abdalha, G. A. Elsayed and M. S. Salem, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 719–732 CrossRef CAS.
- S. Saba, R. Hernandez, C. Chin Choy, K. Carta, Y. Bennett, S. Bondi, S. Kolaj and C. Bennett, J. Fluorine Chem., 2013, 153, 168–171 CrossRef CAS.
- S. Saba and J. A. Ciaccio, J. Chem. Educ., 2016, 93, 945–948 CrossRef CAS.
- M. J. D. Grandi, C. Bennett, K. Cagino, A. Muccini, C. Suraci and S. Saba, Synth. Commun., 2015, 45, 2601–2607 CrossRef.
- N. L. Reddy, J. B. Fischer, K. J. Burke-Howie, P. Barmettler, M. R Rhodes, E. Weber and J. F. W. Keana, Bioorg. Med. Chem. Lett., 1993, 3, 2113–2116 CrossRef CAS.
- R. Khan, M. Arfan, J. Mahmood, S. Anjum and M. I. Choudhary, Chin. Chem. Lett., 2010, 21, 905–910 CrossRef CAS.
- R. N. Laxma, J. B. Fisher and K. J. Burke-Howie, Bioorg. Med. Chem. Lett., 1993, 3, 2113 CrossRef.
- P. Harding and D. J. Harding, Synth. Commun., 2007, 37, 2655–2661 CrossRef CAS.
- S. M. Nair and I. Bhattacharya, Asian J. Chem., 2009, 21, 504–510 CAS.
- A. Olyaei, F. Gesmati, M. Sadeghpour, B. Shams and M. Alizadeh, Synth. Commun., 2012, 42, 1650–1660 CrossRef CAS.
- C. Verrier, S. Carret and J.-F. Poisson, ACS Sustainable Chem. Eng., 2018, 6, 8563–8569 CrossRef CAS.
- W. D. Jones Jr, E. W. Huber, J. M. Grisar and R. A. Schnettler, J. Heterocycl. Chem., 1987, 24, 1221–1223 CrossRef.
- G. I. Graf, D. Hastreiter, L. E. da Silva, R. A. Rebelo, A. G. Montalban and A. McKillop, Tetrahedron, 2002, 58, 9095–9100 CrossRef CAS.
- A. Majee, S. K. Kundu, S. Santra and A. Hajra, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2014, 53, 124–126 Search PubMed.
- N. Azizi, E. Gholibeglo, M. Babapour, H. Ghafuri and S. M. Bolourtchian, C. R. Chim., 2012, 15, 768–773 CrossRef CAS.
- M. Mazloumi, F. Shirini, O. Goli-Jolodar and M. Seddighi, New J. Chem., 2018, 42, 5742–5752 RSC.
- K. Kondo and F. Mori, Chem. Lett., 1974, 3, 741–742 CrossRef.
- M. B. Martin, J. S. Grimley, J. C. Lewis, H. T. Heath, B. N. Bailey, H. Kendrick, V. Yardley, A. Caldera, R. Lira, J. A. Urbina, S. N. J. Moreno, R. Docampo, S. L. Croft and E. Oldfield, J. Med. Chem., 2001, 44, 909–916 CrossRef CAS PubMed.
- A. Balakrishna, M. V. N. Reddy, P. V. Rao, M. A. Kumar, B. S. Kumar, S. K. Nayak and C. S. Reddy, Eur. J. Med. Chem., 2011, 46, 1798–1802 CrossRef CAS PubMed.
- B. Kaboudin and S. Alipour, Tetrahedron Lett., 2009, 50, 4243–4245 CrossRef CAS.
- V. I. Krutikov, A. V. Erkin, P. A. Pautov and M. M. Zolotukhina, Russ. J. Gen. Chem., 2003, 73, 205–210 CrossRef.
- E. Dąbrowska, A. Burzyńska, A. Mucha, E. Matczak-Jon, W. Sawka-Dobrowolska, Ł. Berlicki and P. Kafarski, J. Organomet. Chem., 2009, 694, 3806–3813 CrossRef.
- G. C. S. Reddy, M. V. N. Reddy, N. B. Reddy and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 74–80 CrossRef CAS.
- A. Shirwaiker, K. Rajendran and C. Dinesh Kumar, Indian J. Exp. Biol., 2004, 42, 803–807 Search PubMed.
- B. H. Babu, B. S. Shailesh and J. Paddikala, Fitotherapia, 2001, 72, 272–277 CrossRef CAS.
- K. Kato, S. Terao, N. Shimamoto and M. Hirata, J. Med. Chem., 1988, 31, 793–798 CrossRef CAS PubMed.
- Y. Gow-Chin and C. Hui-Yin, J. Agric. Food Chem., 1995, 43, 27–32 CrossRef.
- U. M. R. Kunda, S. K. Balam, B. R. Nemallapudi, S. S. Chereddy, S. K. Nayak and S. R. Cirandur, Chem. Pharm. Bull., 2012, 60, 104–109 CrossRef CAS PubMed.
- P. Miszczyk, D. Wieczorek, J. Gałęzowska, B. Dziuk, J. Wietrzyk and E. Chmielewska, Molecules, 2017, 22, 254–265 CrossRef PubMed.
- P. Miszczyk, I. Turowska-tyrk, P. Kafarski and E. Chmielewska, Molecules, 2017, 22, 450–461 CrossRef PubMed.
- N. G. Singh, C. Kathing, J. W. S. Rani and R. L. Nongkhlaw, J. Chin. Chem. Soc., 2014, 61, 442–446 CrossRef CAS.
- S. F. Hojati and A. S. Kaheh, Jordan J. Chem., 2019, 14, 17–27 CAS.
- H. Witzel and U. Pindur, J. Heterocycl. Chem., 1988, 25, 907–910 CrossRef CAS.
- J. Yu, Y. Hu, Q. Huang, R. Ma and S. Yang, Synth. Commun., 2000, 30, 2801–2806 CrossRef CAS.
- S. Fox, R. Hudson and R. W. Boyle, Tetrahedron Lett., 2003, 44, 1183–1185 CrossRef CAS.
- M. Wormann and M. E. Maier, RSC Adv., 2019, 9, 15314–15317 RSC.
- R. Calmanti, E. Amadio, A. Perosa and M. Selva, Catalysts, 2019, 9, 534 CrossRef.
- Y. N. Mabkhot, A. Barakat and S. Alshahrani, J. Mol. Struct., 2012, 1027, 15–19 CrossRef CAS.
- L. M. Shorr, J. Am. Chem. Soc., 1954, 76, 1390 CrossRef CAS.
- O. J. Klejnot, Inorg. Chem., 1963, 2, 825 CrossRef CAS.
- M. E. Childs and W. P. Weber, J. Organomet. Chem., 1975, 86, 169 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy and V. Geetha, Synth. Commun., 2002, 32, 763–769 CrossRef CAS.
- A. Ahmad and L. F. Silva Jr, Synthesis, 2012, 44, 3671–3677 CrossRef CAS.
- I. A. Khan, M. V. Kulkarni, M. Gopal, M. S. Shahabuddin and C. M. Sun, Bioorg. Med. Chem. Lett., 2005, 15, 3584–3587 CrossRef CAS PubMed.
- H. Liu, C. S. Tomooka and H. W. Moore, Synth. Commun., 1997, 27, 2177–2180 CrossRef CAS.
- D. R. Chaffey, T. E. Davies, S. H. Taylor and A. E. Graham, ACS Sustainable Chem. Eng., 2018, 6, 4996–5002 CrossRef CAS.
- Z. Zhang, T. Hashiguchi, T. Ishida, A. Hamasaki, T. Honma, H. Ohashi, T. Yokoyama and M. Tokunaga, Org. Chem. Front., 2015, 2, 654–660 RSC.
- R. S. Varma and D. Kumar, Tetrahedron Lett., 1998, 39, 9113–9116 CrossRef CAS.
- H. Egami, S. Tamaoki, M. Abe, N. Ohneda, T. Yoshimura, T. Okamoto, H. Odajima, N. Mase, K. Takeda and Y. Hamashima, Org. Process Res. Dev., 2018, 22, 1029–1033 CrossRef CAS.
- G. V. Kryshtal, G. M. Zhdankina, N. V. Ignat'ev, M. Schulte and S. G. Zlotin, J. Fluorine Chem., 2016, 183, 23–29 CrossRef CAS.
- E. Brenna, C. Fuganti, F. G. Gatti, M. Passoni and S. Serra, Tetrahedron: Asymmetry, 2003, 14, 2401–2406 CrossRef CAS.
- H. M. Kumar, S. Joyasawal, B. V. Reddy, P. P. Chakravarthy, A. D. Krishna and J. S. Yadav, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44, 1686–1692 Search PubMed.
- Y. Tamura, T. Yakura, Y. Shirouchi and J.-I. Haruta, Chem. Pharm. Bull., 1985, 33, 1097 CrossRef CAS.
- T. Yoshino and H. Togo, Synlett, 2004, 2004, 1604–1606 Search PubMed.
- T. Sakamoto, F. Shiga, A. Yasuhara, D. Uchiyama, Y. Kondo and H. Yamanaka, Synthesis, 1992, 746–748 CrossRef CAS.
- A. A. Padmapriya, G. Just and N. G. Lewis, Synth. Commun., 1985, 15, 1057–1062 CrossRef CAS.
- C. J. Borths, J. Chan, B. J. Burke and R. D. Larsen, Synlett, 2009, 2009, 3139–3142 CrossRef.
- Y. Tamura, T. Yakura, Y. Shirouchi and J.-I. Haruta, Chem. Pharm. Bull., 1985, 33, 1097–1103 CrossRef CAS.
- F. Malmedy and T. Wirth, Chem.–Eur. J., 2016, 22, 16072–16077 CrossRef CAS PubMed.
- T. Seidensticker, D. Möller and A. J. Vorholt, Tetrahedron Lett., 2016, 57, 371–374 CrossRef CAS.
- R. Kumar and P. M. S. Chauhan, Tetrahedron Lett., 2008, 49, 5475–5479 CrossRef CAS.
- R. A. Swaringen Jr, J. F. Eaddy and T. R. Henderson, J. Org. Chem., 1980, 45, 3986–3989 CrossRef.
- H. G. Bonacorso, C. A. Cechinel, M. R. Oliveira, M. B. Costa, M. A. Martins, N. Zanatta and A. F. Flores, J. Heterocycl. Chem., 2005, 42, 1055–1061 CrossRef CAS.
- A. Ladépêche, E. Tam, J.-E. Ancel and L. Ghosez, Synthesis, 2004, 2004, 1375–1380 CrossRef.
- E. Pérez-Mayoral, R. M. Martín-Aranda, A. J. López-Peinado, P. Ballesteros, A. Zukal and J. Čejka, Top. Catal., 2009, 52, 148–152 CrossRef.
- R. Kumar and A. K. Chakraborti, Tetrahedron Lett., 2005, 46, 8319–8323 CrossRef CAS.
- Y. Du and F. Tian, Synth. Commun., 2005, 35, 2703–2708 CrossRef CAS.
- W. Gong, B. Wang, Y. Gu, L. Yan, L. Yang and J. Suo, Synth. Commun., 2004, 34, 4243–4247 CrossRef CAS.
- H. Mansilla and M. M. Afonso, Synth. Commun., 2008, 38, 2607–2618 CrossRef CAS.
- R. A. Ugarte and T. W. Hudnall, Green Chem., 2017, 19, 1990–1998 RSC.
- I. Kharkongor and B. Myrboh, Tetrahedron Lett., 2015, 56, 4359–4362 CrossRef CAS.
- R. A. Swaringen Jr, D. A. Yeowell, J. C. Wisowaty, H. A. El-Sayad, E. L. Stewart and M. E. Darnofall, J. Org. Chem., 1979, 44, 4825–4829 CrossRef.
- M. B. Gazizov, S. Y. Ivanova, S. N. Ibragimov, K. S. Gazizova, R. A. Khairullin and K. A. Medvedeva, Russ. J. Gen. Chem., 2016, 86, 2132–2134 CrossRef CAS.
- M. B. Gazizov, S. Y. Ivanova, N. Y. Bashkirtseva, O. D. Khairullina, R. A. Khairullin and O. V. Gazizova, Russ. Chem. Bull., Int. Ed., 2017, 66, 1230–1233 CrossRef CAS.
- M. B. Gazizov, R. F. Karimova, O. M. Chernova, K. M. Gazizov, E. R. Khazeeva and O. G. Sinyashin, Russ. J. Gen. Chem., 2006, 76, 1172–1174 CrossRef CAS.
- P. H. Nelson and J. T. Nelson, Synthesis, 1992, 1992, 1287–1291 CrossRef.
- J. S. Yadav, G. Kondaji, M. S. R. Reddy and P. Srihari, Tetrahedron Lett., 2008, 49, 3810–3813 CrossRef CAS.
- A. Kumar, S. Kumar, R. K. Gupta and D. Kumar, J. Chem. Res., 2007, 2007, 680–682 CrossRef.
- M. S. Khanna, O. V. Singh, C. P. Garg and R. P. Kapoor, Synth. Commun., 1993, 23, 585–590 CrossRef CAS.
- I. Németh, A. Kiss-Szikszai, T. Z. Illyés, A. Mándi, I. Komáromi, T. Kurtán and S. Antus, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 1289–1296 Search PubMed.
- M. S. Khanna, Org. Prep. Proced. Int., 1994, 26, 125–127 CrossRef CAS.
- H. M. C. Ferraz, T. O. Vieira and L. F. Silva Jr, Synthesis, 2006, 2006, 2748–2752 CrossRef.
- L. F. Silva Jr, R. M. F. Sousa, H. M. C. Ferraz and A. M. Aguilar, J. Braz. Chem. Soc., 2005, 16, 1160–1173 Search PubMed.
- H.-J. Kabbe, Justus Liebigs Ann. Chem., 1962, 656, 204 CrossRef CAS.
- H. M. C. Ferraz, L. F. Silva Jr and T. O. Vieira, Tetrahedron, 2001, 57, 1709–1713 CrossRef CAS.
- C.-H. Liao, F.-N. Ko, T.-S. Wu and C.-M. Teng, J. Pharm. Pharmacol., 1997, 49, 1248 CrossRef CAS PubMed.
-
(a) H. M. C. Ferraz, A. M. Aguilar and L. F. Silva Jr, Synthesis, 2003, 2003, 1031–1034 CrossRef;
(b) H. M. C. Ferraz, A. M. Aguilar and L. F. Silva Jr, Tetrahedron, 2003, 59, 5817–5821 CrossRef CAS.
- A. McKillop, J. D. Hunt, F. Kienzle, E. Bigham and E. C. Taylor, J. Am. Chem. Soc., 1973, 95, 3635–3640 CrossRef CAS.
- H. M. C. Ferraz, L. F. Silva Jr and T. O. Vieira, Tetrahedron, 2001, 57, 1709 CrossRef CAS.
- H. M. C. Ferraz, A. M. Aguilar and L. F. Silva Jr, Synthesis, 2003, 2003, 1031 CrossRef.
- H. M. C. Ferraz, A. M. Aguilar and L. F. Silva Jr, Tetrahedron, 2003, 59, 5817 CrossRef CAS.
- G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva Jr, J. Org. Chem., 2009, 74, 2561–2566 CrossRef CAS PubMed.
- P. Abley, J. E. Byrd and J. Halpern, J. Am. Chem. Soc., 1973, 95, 2591 CrossRef CAS.
- H. Sekizaki, M. Ito and S. Inoue, Bull. Chem. Soc. Jpn., 1978, 51, 2439 CrossRef CAS.
- L. Wang, J. Sheng, H. Tian and C. Qian, Synth. Commun., 2004, 34, 4265–4272 CrossRef CAS.
- A. R. Katritzky, R. P. Musgrave, B. Rachwal and C. Zaklika, Heterocycles, 1995, 41, 345–352 CrossRef CAS.
- G. Aridoss and K. K. Laali, Eur. J. Org. Chem., 2011, 2011, 2827–2835 CrossRef.
- S. F. Hojati, B. Maleki and Z. Beykzadeh, Monatsh. Chem., 2011, 142, 87–91 CrossRef CAS.
- I. Mohammadpoor-Baltork, A. R. Khosropour and S. F. Hojati, Monatsh. Chem., 2007, 138, 663–667 CrossRef CAS.
- I. Mohammadpoor-Baltork, A. R. Khosropour and S. F. Hojati, Catal. Commun., 2007, 8, 1865–1870 CrossRef CAS.
- B. Karami, S. Nikoseresht and S. Khodabakhshi, Chin. J. Catal., 2012, 33, 298–301 CrossRef CAS.
- K. Nikoofar and S. M. Dizgarani, Iran. Chem. Commun., 2018, 6, 62–69 Search PubMed.
- A. Mobinikhaledi, M. Zendehdel, F. Goudarzi and G. R. Bardajee, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1526–1531 CrossRef CAS.
- H. Wang, Y. Xia, S. Lv, J. Xu and Z. Sun, Tetrahedron Lett., 2013, 54, 2124–2127 CrossRef CAS.
- B. Kaboudin, F. Kazemia, M. Pirouza, A. Baharian Khoshkhoo, J. Kato and T. Yokomatsu, Synthesis, 2016, 48, 3597–3602 CrossRef CAS.
- F. Fouad, D. R. Davis and R. Twieg, Liq. Cryst., 2018, 45, 1508–1517 CrossRef CAS.
- J. L. Malavolta, L. M. Frigo, S. Moura, D. C. Flores and A. F. C. Flores, J. Braz. Chem. Soc., 2017, 28, 2106–2116 CAS.
- M. M. Ghorab, F. A. Ragab, S. I. Alqasoumi, A. M. Alafeefy and S. A. Aboulmagd, Eur. J. Med. Chem., 2010, 45, 171–178 CrossRef CAS PubMed.
- G. S. Gadaginamath and S. R. Pujar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2003, 42, 2896–2900 Search PubMed.
- M. R. Mahmoud, W. S. I. Abou-Elmagd, H. A. Derbala and M. H. Hekal, J. Chem. Res., 2012, 36, 75–82 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Tetrahedron Lett., 2008, 49, 879–883 CrossRef CAS.
- C. Ainsworth, J. Am. Chem. Soc., 1955, 77, 1148 CrossRef CAS.
- H. G. Bonacorso, G. R. Paim, L. M. F. Porte, E. P. Pittaluga, M. A. P. Martins and N. Zanatta, Arkivoc, 2012, 8, 214–225 Search PubMed.
- A. K. Yadav, G. R. Sharma, P. Dhakad and T. Yadav, Tetrahedron Lett., 2012, 53, 859–862 CrossRef CAS.
- E. Kanaani and M. Nasr-Esfahani, Monatsh. Chem., 2018, 149, 543–550 CrossRef CAS.
- A. M. Kamal, S. M. Radwan and R. M. Zaki, Eur. J. Med. Chem., 2011, 46, 567–578 CrossRef CAS PubMed.
- T. M. Potewar, S. A. Siddiqui, R. J. Lahoti and K. V. Srinivasan, Tetrahedron Lett., 2007, 48, 1721–1724 CrossRef CAS.
- M. Esmaeilpour, J. Javidi, F. N. Dodeji and M. M. Abarghoui, J. Mol. Catal. A: Chem., 2014, 393, 18–29 CrossRef CAS.
- C. N. S. S. P. Kumar, D. K. Parida, A. Santhoshi, A. K. Kota, B. Sridhar and V. J. Rao, MedChemComm, 2011, 2, 486–492 RSC.
- D. Habibi, M. Nasrollahzadeh, L. Mehrabi and S. Mostafaee, Monatsh. Chem., 2013, 144, 725–728 CrossRef CAS.
- H. Naeimi and S. Mohamadabadi, Dalton Trans., 2014, 43, 12967–12973 RSC.
- H. Naeimi, F. Kiani and M. Moradian, Green Chem. Lett. Rev., 2018, 11, 361–369 CrossRef CAS.
- M. Maham and M. Nasrollahzadeh, Appl. Organomet. Chem., 2019, 33, e5097 Search PubMed.
- H. Naeimi, F. Kiani and M. Moradian, Iran. J. Catal., 2013, 3, 243–247 Search PubMed.
- M. Esmaeilpour, J. Javidi and S. Zahmatkesh, Appl. Organomet. Chem., 2016, 30, 897–904 CrossRef CAS.
- M. Nasrollahzadeh, M. Sajjadi, M. R. Tahsili, M. Shokouhimehr and R. S. Varma, ACS Omega, 2019, 4, 8985–9000 CrossRef CAS PubMed.
- D. Habibi, H. Nabavi and M. Nasrollahzadeh, J. Chem., 2012, 2013, 1 Search PubMed.
- T. M. Potewar, S. A. Siddiqui, R. J. Lahoti and K. V. Srinivasan, Tetrahedron Lett., 2007, 48, 1721–1724 CrossRef CAS.
- A. K. Gupta, C. H. Song and C. H. Oh, Tetrahedron Lett., 2004, 45, 4113–4116 CrossRef CAS.
- D. Habibi, M. Nasrollahzadeh and T. A. Kamali, Green Chem., 2011, 13, 3499–3504 RSC.
- D. Habibi, M. Nasrollahzadeh and T. A. Kamali, Green Chem., 2011, 13, 3499–3504 RSC.
- S. N. Dighe, K. S. Jain and K. V. Srinivasan, Tetrahedron Lett., 2009, 50, 6139–6142 CrossRef CAS.
- D. Kundu, A. Majee and A. Hajra, Tetrahedron Lett., 2009, 50, 2668–2670 CrossRef CAS.
- F. Darvish and S. Khazraee, Int. J. Org. Chem., 2015, 5, 75–80 CrossRef CAS.
- Y. Zhou and M. K. Denk, Tetrahedron Lett., 2003, 44, 1295–1299 CrossRef CAS.
- V. S. Rao, A. K. Gupta, P. Giridhar and B. S. Reddy, Synth. Commun., 2000, 30, 2625–2628 CrossRef CAS.
- A. S. Nagarajan and B. S. R. Reddy, Synlett, 2010, 2010, 2311 CrossRef.
- R. Toche, V. Medhane, A. Desai, A. Avhale, D. Kendre and M. Jachak, J. Indian Chem. Soc., 2006, 83, 1039–1040 CAS.
- P. Ploszaj, A. Junka, B. Szponar, M. Maczynski, S. Ryng, M. Bartoszewicz and A. Piwowar, Acta Pol. Pharm., 2018, 75, 637–647 CAS.
- S. Guccione, A. Raffaelli, G. U. Barretta, L. M. Scolaro, S. Pucci and F. Russo, Eur. J. Med. Chem., 1995, 30, 333–337 CAS.
- F. Russo, S. Guccione, G. Romeo, L. Monsu'Scolaro, S. Pucci, A. Caruso, V. Cutuli and M. A. Roxas, Eur. J. Med. Chem., 1992, 27, 73–80 CrossRef CAS.
- G. Resnati, Tetrahedron, 1993, 49, 9385–9445 CrossRef CAS.
- K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS.
- G. Lal, S. Pez and G. P. Syvret, Chem. Rev., 1996, 96, 1737–1756 CrossRef CAS PubMed.
- J. Mann, Chem. Soc. Rev., 1987, 16, 381–436 CAS.
- K. Burger, U. Wucherpfennig and E. Brunner, Adv. Heterocycl. Chem., 1994, 60, 1–23 CrossRef CAS.
- Biomedicinal Aspects of Fluorine Chemistry, ed. R. Filler and Y. Kobayashi, Kondansha and Elsevier Biomedical, Tokyo, 1982 Search PubMed.
- J. T. Welch and S. Eswarakrishnan, Fluorine in Bioorganic Chemistry, Wiley, New York, 1991 Search PubMed.
- Biomedical Frontiers of Fluorine Chemistry, ed. I. Ojima, J. R. McCarthy and J. T. Welch, American Chemical Society, Washington, DC, 1996, ACS Symposium Series 639 Search PubMed.
- S. Narizuka and T. Fuchigami, Bioorg. Med. Chem. Lett., 1995, 5, 1293–1294 CrossRef CAS.
- T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer, Berlin, 2000 Search PubMed.
- M. R. Shaaban, J. Fluorine Chem., 2008, 129, 1156–1161 CrossRef CAS.
- K. Gewald, Chem. Ber., 1966, 99, 1002–1007 CrossRef CAS.
- N. A. Hassan, Molecules, 2000, 5, 826–834 CrossRef CAS.
- K. Gewald and M. Henschel, J. Prakt. Chem., 1976, 318, 663 CrossRef CAS.
- C. G. Dave and R. D. Shah, Molecules, 2002, 7, 554–565 CrossRef CAS.
- C. G. Dave and R. D. Shah, J. Heterocycl. Chem., 2000, 37, 757–761 CrossRef CAS.
- C. G. Dave and R. D. Shah, J. Heterocycl. Chem., 1998, 35, 1295 CrossRef CAS.
- C. J. Shishoo, M. B. Devani, G. V. Ullas, S. Ananthan and V. S. Bhadti, J. Heterocycl. Chem., 1981, 18, 43–46 CrossRef CAS.
- A. E. Rashad, O. A. Heikal, A. O. H. El-Nezhawy and F. M. E. Abdel-Megeid, Heteroat. Chem., 2005, 16, 226–234 CrossRef CAS.
- M. A. A. Elneairy, M. A. M. Gad-Elkareem and A. M. Abdel-Fattah, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 1451–1466 CrossRef CAS.
- S. H. Abdel-Hafez, S. A. Abdel-Mohsen and Y. A. El-Ossaily, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 2297–2305 CrossRef CAS.
- I. Devi and P. J. Bhuyan, Synlett, 2004, 2004, 283–286 Search PubMed.
-
(a) M. Y. Gavrilov, G. N. Novoseleva, M. I. Vakhrin and M. E. Konshin, Khim.-Farm. Zh., 1996, 30, 39 Search PubMed;
(b) M. M. Ghorab and A. Y. Hassan, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 141, 257 CrossRef.
-
(a) G. L. Anderson, J. L. Shim and A. D. Broom, J. Org. Chem., 1976, 41, 1095 CrossRef PubMed;
(b) E. M. Grivaky, S. Lee, C. W. Siyal, D. S. Duch and C. A. Nichol, J. Med. Chem., 1980, 23, 327 CrossRef PubMed.
-
(a) S. Furuya and T. Ohtaki, Eur. Pat., EP608565, 1994, Chem. Abstr., 1994, 121, 205395;
(b) D. Heber, C. Heers and U. Ravens, Pharmazie, 1993, 48, 537–541 CAS.
- R. H. Bradbury, G. A. Breault, P. J. Jewbury and J. E. Pease, US Pat., US6593326B1, 1998.
- L. N. Bheemanapalli, R. R. Akkinepally and S. R. Pamulaparthy, Chem. Pharm. Bull., 2008, 56, 1342–1348 CrossRef CAS PubMed.
- W. T. Ashton, R. D. Brown, F. Jacobson and C. Walsh, J. Am. Chem. Soc., 1979, 101, 4419 CrossRef CAS.
- S. TumkeviCius, Liebigs Ann., 1995, 1703–1705 CrossRef CAS.
- J.-C. Castillo, D. Estupiñan, M. Nogueras, J. Cobo and J. Portilla, J. Org. Chem., 2016, 81, 12364–12373 CrossRef CAS PubMed.
- N. Sachdeva, A. V. Dolzhenko and W. K. Chui, Org. Biomol. Chem., 2012, 10, 4586–4596 RSC.
- J. Světlík and T. Liptaj, J. Chem. Soc., Perkin Trans. 1, 2002, 1260–1265 RSC.
- K. Ighilahriz, B. Boutemeur, F. Chami, C. Rabia, M. Hamdi and S. M. Hamdi, Molecules, 2008, 13, 779–789 CrossRef CAS PubMed.
- K. Laxminarayana, C. Rajendiran and K. Mukkanti, Der Pharma Chem., 2012, 4, 517–522 Search PubMed.
- B. P. Bandgar, P. E. More and V. T. Kamble, Chin. J. Chem., 2009, 27, 1123–1126 CrossRef CAS.
- A. R. Khosropour, I. Mohammadpoor-Baltork and H. Ghorbankhani, Tetrahedron Lett., 2006, 47, 3561–3564 CrossRef CAS.
- M. Wang, Z. Song and T. Zhang, Synth. Commun., 2011, 41, 385–391 CrossRef CAS.
- M. Wang, Z. G. Song and T. T. Zhang, Chem. Heterocycl. Compd., 2010, 46, 581–584 CrossRef CAS.
- M. M. Heravi, N. Javanmardi, H. A. Oskooie and B. Baghernejad, Bull. Chem. Soc. Ethiop., 2011, 25, 305–308 CAS.
- L. Wu, W. Ma, L. Yang and F. Yan, Asian J. Chem., 2010, 22, 6053–6058 CAS.
- W.-L. Wang, X.-X. Liu, T. Zhang, J.-M. Zhang and J.-H. Zhou, J. Chem. Soc. Pak., 2016, 38, 1196–1202 CAS.
- M. Dabiri, M. Baghbanzadeh and A. S. Delbari, J. Comb. Chem., 2008, 10, 700–703 CrossRef CAS PubMed.
- D. Kumar, P. S. Jadhavar, M. Nautiyal, H. Sharma, P. K. Meena, L. Adane, S. Pancholia and A. K. Chakraborti, RSC Adv., 2015, 5, 30819–30825 RSC.
- K. Rad-Moghadam and L. Samavi, J. Heterocycl. Chem., 2006, 43, 913–916 CrossRef CAS.
- W. Szczepankiewicz and N. Kuźnik, Tetrahedron Lett., 2015, 56, 1198–1199 CrossRef CAS.
- S. I. Bhat, U. K. Das and D. R. Trivedi, J. Heterocycl. Chem., 2015, 52, 1253–1259 CrossRef CAS.
- K. T. Potts and G. Brugel, J. Org. Chem., 1970, 35, 3448–3451 CrossRef CAS.
- A. L. Paulsen and R. Madsen, Heterocycles, 2004, 63, 2051 CrossRef CAS.
- D. Habibi, H. Sahebekhtiari, M. Nasrollahzadeh and A. Taghipour, Lett. Org. Chem., 2013, 10, 209–212 CrossRef CAS.
- S. Padmanabhan, N. L. Reddy and G. J. Durant, Synth. Commun., 1997, 27, 691–699 CrossRef CAS.
- M. Nasrollahzadeh, M. Enayatia and M. Khalaj, RSC Adv., 2014, 4, 26264–26270 RSC.
- J. C. Jaen, L. D. Wise, T. G. Heffner, T. A. Pugsley and L. T. Meltzer, J. Med. Chem., 1991, 34, 248–256 CrossRef CAS PubMed.
- V. M. Rao, G. L. V. Damu, D. Sudhakar, V. Siddaiah and C. V. Rao, Arkivoc, 2008, 11, 285–294 Search PubMed.
- B. Maleki, M. Baghayeri, S. M. Vahdat, A. Mohammadzadeh and S. Akhoondi, RSC Adv., 2015, 5, 46545–46551 RSC.
- H. Sharghi, M. Mozaffari, J. Aboonajmi, M. M. Doroodmand, P. Shiri and M. Aberi, ChemistrySelect, 2018, 3, 13534–13540 CrossRef CAS.
- F. Rezaei, M. A. Amrollahi and R. Khalifeh, Inorg. Chim. Acta, 2019, 489, 8–18 CrossRef CAS.
- P. Černuchová, G. Vo-Thanh, V. Milata and A. Loupy, Heterocycles, 2004, 64, 177–191 CrossRef.
- F. A. Abu-Shanab, J. Sulfur Chem., 2006, 27, 433–440 CrossRef CAS.
- M. F. Farhat, A. M. M. EL-Saghier, M. A. Makhlouf, K. M. Kreddan and A. B. Elmezoughi, J. Sulfur Chem., 2007, 28, 563–572 CrossRef CAS.
- M. M. Heravi, N. Nami, N. Seifi, H. A. Oskooie and R. Hekmatshoar, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 591–599 CrossRef CAS.
- A. M. Hussein, A. A. Harb and I. A. Mousa, J. Heterocycl. Chem., 2008, 45, 1819–1823 CrossRef CAS.
- Y. M. Elkholy, F. A. Abu-Shanab and A. W. Erian, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 167, 151–159 CrossRef CAS.
- N. M. Elwan, J. Heterocycl. Chem., 2004, 41, 281–284 CrossRef CAS.
- H. Abdel-Ghany and A. Khodairy, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 166, 45–56 CrossRef CAS.
- E. I. Al-Afaleq and S. A. Abubshait, Molecules, 2001, 6, 621–638 CrossRef CAS.
- A. A. Amer, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 2330–2343 CrossRef CAS.
- W. M. Basyouni and K. A. H. M. El-Bayouki, J. Chem. Res., 2005, 2005, 356–360 CrossRef.
- A. M. S. El-Sharief, J. A. A. Micky, N. A. M. M. Shmeiss and G. El-Gharieb, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 439–451 CrossRef CAS.
- A. A. Fadda, H. A. Etman, M. Y. El-Seidy and K. M. Elattar, J. Heterocycl. Chem., 2012, 49, 774–781 CrossRef CAS.
- S. A. Al-Issa, Asian J. Chem., 2006, 18, 2145–2150 CAS.
- N. M. Nahas and A. A. A. Hafez, Heterocycl. Commun., 2007, 13, 381–386 CAS.
- A. E. Abdel-Rahman, E. A. Bakhite and E. A. Al-Taifi, Pharmazie, 2003, 58, 372–377 CAS.
- A. H. Bedair, H. A. Emam, N. A. El-Hady, K. A. R. Ahmed and A. M. El-Agrody, Il Farmaco, 2001, 56, 965–973 CrossRef CAS PubMed.
- S. A. El-Assiery, G. H. Sayed and A. Fouda, Acta Pharm., 2004, 54, 143–150 CAS.
- A. A. El-Maghraby, G. A. M. El-Hag Ali, A. H. A. Ahmed and M. S. A. El-Gaby, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 293–302 CrossRef CAS.
- A. Shaabani, S. Keshipour, S. Shaabani and M. Mahyari, Tetrahedron Lett., 2012, 53, 1641–1644 CrossRef CAS.
- K. A. Nazish, A. Rauf, A. Sharif, E. Ahmed, F.-U. H. Nasim, A. Yaqoob and A. M. Qureshi, J. Chem. Soc. Pak., 2015, 37, 1047–1055 CAS.
- A. Yousefi, R. Yousefi, F. Panahi, S. Sarikhani, A. R. Zolghadr, A. Bahaoddini and A. Khalafi-Nezhad, Int. J. Biol. Macromol., 2015, 78, 46–55 CrossRef CAS PubMed.
- A. M. Salaheldin and M. K. Alphy, J. Heterocycl. Chem., 2008, 45, 307–310 CrossRef CAS.
- A. F. A. Khattab, D. V. Tinh and W. Stadlbauer, J. Prakt. Chem., 1996, 338, 151–156 CrossRef CAS.
- M. Seidel, G. C. Van Tuyle and W. D. Weir, J. Org. Chem., 1970, 35, 1662–1664 CrossRef CAS.
- A. K. Osipov, A. A. Anis'kov and A. Y. Yegorova, Russ. J. Org. Chem., 2017, 53, 210–214 CrossRef CAS.
- A. V. Komkov, M. A. Prezent, A. V. Ignatenko, I. P. Yakovlev and V. A. Dorokhov, Russ. Chem. Bull., Int. Ed., 2006, 55, 2085–2090 CrossRef CAS.
- B. Holló, V. M. Leovac, P. Bombicz, A. Kovács, L. S. Jovanović, G. Bogdanović, V. Kojić, V. Divjaković, M. D. Joksović and K. M. Szécsényi, Aust. J. Chem., 2010, 63, 1557–1564 CrossRef.
- J. Balog, Z. Riedl and G. Hajós, Tetrahedron Lett., 2013, 54, 5338–5340 CrossRef CAS.
- F. P. L. Lim, G. Luna and A. V. Dolzhenko, Synthesis, 2016, 48, 2423–2428 CrossRef CAS.
- C. W. Whitehead and J. J. Traverso, J. Am. Chem. Soc., 1955, 77, 5872–5877 CrossRef CAS.
- A. Z. Halimehjani, M. H. Shayegan, S. S. Poshteh, V. Amani, B. Notash and M. M. Hashemi, Tetrahedron Lett., 2015, 56, 7124–7127 CrossRef.
- B.-L. Wang, H.-T. Gao and W.-D. Z. Li, J. Org. Chem., 2015, 80, 5296–5301 CrossRef CAS PubMed.
- M. V. N. Reddy, J. Kim and Y. T. Jeong, J. Fluorine Chem., 2012, 135, 155–158 CrossRef CAS.
- M. Periasamy, N. Kishorebabu and K. N. Jayakumar, Tetrahedron Lett., 2007, 48, 1955–1958 CrossRef CAS.
- H. Park, Y. Kwon, J. E. Shin, W.-J. Kim, S. Hwang, S. Lee and S. Kim, Synthesis, 2017, 49, 2761–2767 CrossRef CAS.
- R. Paolesse, L. Jaquinod, M. O. Senge and K. M. Smith, J. Org. Chem., 1997, 62, 6193–6198 CrossRef CAS.
- C. Tardieux, F. Bolze, C. P. Gros and R. Guilard, Synthesis, 1998, 1998, 267–268 CrossRef.
- C. B. Reese and H. Yan, Tetrahedron Lett., 2001, 42, 5545–5547 CrossRef CAS.
- U. Pindur and H. Witzel, Arch. Pharm., 1990, 323, 427–432 CrossRef CAS PubMed.
- S. Khaksar, S. M. Vahdat, M. Gholizadeh and S. M. Talesh, J. Fluorine Chem., 2012, 136, 8–11 CrossRef CAS.
- R. Gu, A. Hameurlaine and W. Dehaen, J. Org. Chem., 2007, 72, 7207–7213 CrossRef CAS.
- Z. Hassen, A. B. Akacha and B. Hajjem, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 2349–2356 CrossRef CAS.
- M. J. Dooley, R. J. Quinn and P. J. Scammells, Aust. J. Chem., 1989, 42, 747–750 CrossRef CAS.
- S. K. Meegalla, D. Doller, R. Liu, D. Sha, R. M. Soll and D. S. Dhanoa, Tetrahedron Lett., 2002, 43, 8639–8642 CrossRef CAS.
- J. Warkentin, Acc. Chem. Res., 2009, 42, 205–212 CrossRef CAS PubMed.
-
(a) H. Minato and T. Nagasaki, J. Chem. Soc. C, 1966, 377–379 RSC;
(b) C. W. Jefford, A. W. Sledeski, J.-C. Rossier and T. Boukouvalas, Tetrahedron Lett., 1990, 31, 5741–5744 CrossRef CAS.
- J.-P. Croisetière and C. Spino, J. Org. Chem., 2018, 83, 5609–5618 CrossRef PubMed.
- P. A. Clarke, P. B. Sellars and N. M. Nasir, Org. Biomol. Chem., 2015, 13, 4743–4750 RSC.
- G. H. Elgemeie, A. H. Elghandour and A. M. Hussein, Synth. Commun., 2004, 34, 3293–3302 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, L. Chandraiah, B. Jagannadh, S. K. Kumar and A. C. Kunwar, Tetrahedron Lett., 2002, 43, 4527–4530 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, C. Parisse, P. Carvalho and T. P. Rao, Tetrahedron Lett., 2002, 43, 2999–3002 CrossRef CAS.
- H. Miyazaki, K. Honda, M. Asami and S. Inoue, J. Org. Chem., 1999, 64, 9507–9511 CrossRef CAS.
- V. R. Shah, J. L. Bose and R. C. Shah, J. Org. Chem., 1960, 25, 677 CrossRef CAS.
- N. S. Bhatt, A. K. Shah, R. V. Raval and V. Thakor, Curr. Sci., 1984, 53, 1289–1290 CAS.
- H. Maamar, J. Heterocycl. Chem., 1993, 30, 1155 CrossRef.
- M. Chavda, A. Shah, S. Bhatt, K. Deo and P. Kundu, Arzneim.-Forsch./Drug Res., 2003, 53, 196–200 CAS.
- J. Barluenga, A. Mendoza, F. Rodríguez and F. J. Fañanás, Angew. Chem., Int. Ed., 2009, 48, 1644–1647 CrossRef CAS PubMed.
- M. C. Mayorquín-Torres, M. Flores-Álamo and M. A. Iglesias-Arteaga, Tetrahedron Lett., 2017, 58, 3500–3504 CrossRef.
- C. Alarcón-Manjarrez, R. Arcos-Ramos, M. Flores-Álamo and M. A. Iglesias-Arteaga, Steroids, 2016, 109, 66–72 CrossRef.
- H. Miyazaki, K. Honda, M. Asami and S. Inoue, J. Org. Chem., 1999, 64, 9507–9511 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, M. Aruna, C. Venugopal, T. Ramalingam, S. K. Kumar and A. C. Kunwar, J. Chem. Soc., Perkin Trans. 1, 2002, 165–171 CAS.
- J. S. Yadav, B. V. S. Reddy, C. V. Rao and K. V. Rao, J. Chem. Soc., Perkin Trans. 1, 2002, 1401 RSC.
- R. Merten and G. Müller, Chem. Ber., 1964, 97, 682–694 CrossRef CAS.
- S. Prado, Y. L. Janin and P.-E. Bost, J. Heterocycl. Chem., 2006, 43, 1605–1608 CrossRef CAS.
- M. Kočevar, S. Polanc, M. Tišler and B. Verček, Synth. Commun., 1989, 19, 1713–1719 CrossRef.
- M. A. Al-Haiza, M. S. Mostafa and M. Y. El-Kady, Molecules, 2003, 8, 275–286 CrossRef CAS.
- A. H. Bedair, N. A. El-Hady, M. S. Abd El-Latif, A. H. Fakery and A. M. El-Agrody, Il Farmaco, 2000, 55, 708–714 CrossRef CAS PubMed.
- M. Bakos, Á. Gyömöre, A. Domján and T. Soós, Angew. Chem., Int. Ed., 2017, 56, 5217–5221 CrossRef CAS PubMed.
- D.-Q. Zheng, Y. Jing, B.-Y. Zheng, Y.-F. Ye, S. Xu, W.-S. Tian, H.-Y. Ma and K. Ding, Tetrahedron, 2016, 72, 2164–2169 CrossRef CAS.
- P. W. Anzalone, A. R. Baru, E. M. Danielson, P. D. Hayes, M. P. Nguyen, A. F. Panico, R. C. Smith and R. S. Mohan, J. Org. Chem., 2005, 70, 2091–2096 CrossRef CAS PubMed.
- J. S. Yadav, B. V. S. Reddy and P. Srihari, Synlett, 2001, 2001, 673–675 CrossRef.
- E. Merişor, J. Conrad, C. C. Malakar and U. Beifuss, Synlett, 2008, 2008, 903–907 CrossRef.
- N. Maulide and I. E. Markó, Org. Lett., 2007, 9, 3757–3760 CrossRef CAS.
- J. J. Lee and G. A. Kraus, Tetrahedron Lett., 2013, 54, 2366–2368 CrossRef CAS.
- R. J. Fletcher, W. B. Motherwell and M. E. Popkin, Chem. Commun., 1998, 2191–2192 RSC.
- P. Bunse, E.-U. Wurthwein and B. Wunsch, Eur. J. Org. Chem., 2018, 2018, 1806–1812 CrossRef CAS.
- Y. Sempere and E. M. Carreira, Angew. Chem., 2018, 130, 7780–7784 CrossRef.
- A. Brodzka, D. Koszelewski, M. Zysk and R. Ostaszewski, Catal. Commun., 2018, 106, 82–86 CrossRef CAS.
- D. Koszelewski, M. Zysk, A. Brodzka, A. Żądło, D. Paprocki and R. Ostaszewski, Org. Biomol. Chem., 2015, 13, 11014–11020 RSC.
- J. I. Trujillo and A. S. Gopalan, Tetrahedron Lett., 1993, 34, 7355–7358 CrossRef CAS.
- A. W. Schwabacher and A. D. Stefanescu, Tetrahedron Lett., 1996, 37, 425–428 CrossRef CAS.
- S. A. King, J. Org. Chem., 1994, 59, 2253–2256 CrossRef CAS.
- K.-I. Tadano, T. Murata, T. Kumagai, Y. Isshiki and S. Ogawa, J. Carbohydr. Chem., 1993, 12, 1187–1202 CrossRef CAS.
- K.-I. Takao, H. Saegusa, G. Watanabe and K.-I. Tadano, Tetrahedron: Asymmetry, 2000, 11, 453–464 CrossRef CAS.
- K.-I. Takao, H. Saegusa and K.-I. Tadano, J. Carbohydr. Chem., 2001, 20, 57–69 CrossRef CAS.
- T. Okano, T. Michihashi and J. Kiji, Appl. Organomet. Chem., 1995, 9, 473–478 CrossRef CAS.
- A. Roy, M. Rahman, S. Das, D. Kundu, S. K. Kundu, A. Majee and A. Hajra, Synth. Commun., 2009, 39, 590–595 CrossRef CAS.
- H. Mansilla, Synth. Commun., 2006, 36, 2195–2201 CrossRef.
- M. J. Climent, A. Corma, S. Iborra, M. C. Navarro and J. Primo, J. Catal., 1996, 161, 783–789 CrossRef CAS.
- A. Ziyaei-Halimjani, N. Azizi and M. R. Saidi, J. Sci., Islamic Repub. Iran, 2005, 16, 37–41 CAS.
- C. C. Silveira, S. R. Mendes, F. I. Ziembowicz, E. J. Lenardão and G. Perin, J. Braz. Chem. Soc., 2010, 21, 371–374 CrossRef CAS.
- S. H. Lee, J. H. Lee and C. M. Yoon, Tetrahedron Lett., 2002, 43, 2699–2703 CrossRef CAS.
- R. Chen and L. Wang, RSC Adv., 2015, 5, 20155–20159 RSC.
- D. J. M. Lyons, R. D. Crocker, D. Enders and T. V. Nguyen, Green Chem., 2017, 19, 3993–3996 RSC.
- S. V. Ley and H. W. M. Priepke, Angew. Chem., Int. Ed., 1994, 33, 2292–2294 CrossRef.
- R. Gopinath, Sk. J. Haque and B. K. Patel, J. Org. Chem., 2002, 67, 5842–5845 CrossRef CAS PubMed.
- N. M. Leonard, M. C. Oswald, D. A. Freiberg, B. A. Nattier, R. C. Smith and R. S. Mohan, J. Org. Chem., 2002, 67, 5202–5207 CrossRef CAS PubMed.
- Y.-R. Ma, T.-S. Jin, S.-X. Shi and T.-S. Li, Synth. Commun., 2003, 33, 2103–2108 CrossRef CAS.
- O. Takazawa, K. Kogami and K. Hayashi, Bull. Chem. Soc. Jpn., 1984, 57, 1876–1881 CrossRef CAS.
- M. A. P. Martins, G. P. Bastos, A. P. Sinhorin, A. F. C. Flores, H. G. Bonacorso and N. Zanatta, Synlett, 1999, 6, 789–791 CrossRef.
- J. K. Stille, H. Su, P. Brechot, G. Parrinello and L. S. Hegedus, Organometallics, 1991, 10, 1183–1189 CrossRef CAS.
- J. Pospech, A. J. J. Lennox and M. Beller, Chem. Commun., 2015, 51, 14505–14508 RSC.
-
(a) W. Kantlehner, T. Maier and J. J. Kapassakalidis, Liebigs Ann. Chem., 1981, 1981, 70–84 CrossRef;
(b) K. Utimoto, Y. Wakabayashi, Y. Shishiyama, M. Inoue and H. Nozaki, Tetrahedron Lett., 1981, 22, 4279–4280 CrossRef CAS;
(c) S. Kirchmeyer, A. Mertens, M. Arvanaghi and G. A. Olah, Synthesis, 1983, 1983, 498–500 CrossRef;
(d) J. H. Babler and C. J. Marcuccilli, Tetrahedron Lett., 1987, 28, 4657–4660 CrossRef CAS;
(e) T. Hiyama, H. Oishi, Y. Suetsugu, K. Nishide and H. Saimoto, Bull. Chem. Soc. Jpn., 1987, 60, 2139–2150 CrossRef CAS.
- S. Kotani, M. Sakamoto, K. Osakama and M. Nakajima, Eur. J. Org. Chem., 2015, 2015, 6606–6609 CrossRef CAS.
- G. Casiraghi, G. Casnati and M. Cornia, Tetrahedron Lett., 1973, 14, 679–682 CrossRef.
- H.-S. Byun and R. Bittman, Synth. Commun., 1993, 23, 3201–3204 CrossRef CAS.
- Y. Geng, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Eur. J. Org. Chem., 2013, 2013, 7035–7040 CrossRef CAS.
- M. Asaoka, N. Sugimura and H. Takei, Bull. Chem. Soc. Jpn., 1979, 52, 1953–1956 CrossRef CAS.
- J.-L. Montchamp, F. Tian, M. E. Hart and J. W. Frost, J. Org. Chem., 1996, 61, 3897–3899 CrossRef CAS PubMed.
- R. V. Hoffman and J. M. Salvador, Tetrahedron Lett., 1991, 32, 2429–2432 CrossRef CAS.
- J. Morris, D. G. Wishka, G. P. Luke, T. M. Judge and R. B. Gammill, Tetrahedron, 1997, 53, 11211–11222 CrossRef CAS.
- D. E. DeMong and R. M. Williams, Tetrahedron Lett., 2002, 43, 2355–2357 CrossRef CAS.
- A. McKillop, B. P. Swann, M. E. Ford and E. C. Taylor, J. Am. Chem. Soc., 1973, 95, 3641–3645 CrossRef CAS.
- E. C. Taylor, R. L. Robey, K.-T. Liu, B. Favre, H. T. Bozimo, R. A. Conley, C.-S. Chiang, A. McKillop and M. E. Ford, J. Am. Chem. Soc., 1976, 98, 3037–3038 CrossRef CAS.
- E. C. Taylor, R. A. Conley, D. K. Johnson and A. McKillop, J. Org. Chem., 1977, 42, 4167–4169 CrossRef CAS.
- D. J. Crouse and D. M. S. Wheeler, Tetrahedron Lett., 1979, 20, 4797–4798 CrossRef.
- T. W. Hart and F. Scheinmann, Tetrahedron Lett., 1980, 21, 2295–2296 CrossRef CAS.
- B. M. Smith and A. E. Graham, Tetrahedron Lett., 2007, 48, 4891–4894 CrossRef CAS.
- S. Manojveer and R. Balamurugan, Eur. J. Org. Chem., 2015, 2015, 4254–4260 CrossRef CAS.
- S. Manojveer and R. Balamurugan, Chem. Commun., 2014, 50, 9925–9928 RSC.
- S. R. Koppolu and R. Balamurugan, New J. Chem., 2017, 41, 1186–1192 RSC.
- K. R. Kunz, E. W. Taylor, H. M. Hutton and B. J. Blackburn, Org. Prep. Proced. Int., 1990, 22, 613–618 CrossRef CAS.
- B. C. Tlach, A. L. Tomlinson, A. G. Ryno, D. D. Knoble, D. L. Drochner, K. J. Krager and M. Jeffries-EL, J. Org. Chem., 2013, 78, 6570–6581 CrossRef CAS PubMed.
- J. J. Lee, J. Kim, Y. M. Jun, B. M. Lee and B. H. Kim, Tetrahedron, 2009, 65, 8821–8831 CrossRef CAS.
- S. J. Maddirala, V. S. Gokak and L. D. Basanagoudar, J. Heterocycl. Chem., 2004, 41, 7–11 CrossRef CAS.
- A. Kudelko, K. Jasiak and K. Ejsmont, Monatsh. Chem., 2015, 146, 303–311 CrossRef CAS PubMed.
- M. S. Khajavi, S. S. S. Hosseini and F. Sefidkon, Iran. J. Chem. Chem. Eng., 1997, 16, 68–71 CAS.
- K. K. Gnanasekaran, B. Nammalwar, M. Murie and R. A. Bunce, Tetrahedron Lett., 2014, 55, 6776–6778 CrossRef CAS.
- S. M. H. Al-Majidi, J. Saudi Chem. Soc., 2014, 18, 893–901 CrossRef.
- J. Štetinová, R. Kada and J. Leško, Molecules, 1996, 1, 251–254 Search PubMed.
- J. F. Mike, J. J. Inteman, A. Ellern and M. Jeffries-EL, J. Org. Chem., 2010, 75, 495–497 CrossRef CAS PubMed.
- S. Khaksar, A. Heydari, M. Tajbakhsh and S. M. Vahdat, J. Fluorine Chem., 2010, 131, 1377–1381 CrossRef CAS.
- M. M. Heravi, N. Montazeri, M. Rahmizadeh, M. Bakavoli and M. Ghassemzadeh, J. Chem. Res., 2000, 2000, 584–585 CrossRef.
- Y.-C. Chi and C.-M. Sun, Synlett, 2000, 2000, 591–594 CrossRef.
- F. Wang, M. Tran-Dubé, S. Scales, S. Johnson, I. McAlpine and S. Ninkovic, Tetrahedron Lett., 2013, 54, 4054–4057 CrossRef CAS.
- W. Huang and R. M. Scarborough, Tetrahedron Lett., 1999, 40, 2665–2668 CrossRef CAS.
- Z.-H. Zhang, T.-S. Li and J.-J. Li, Monatsh. Chem., 2007, 138, 89–94 CrossRef CAS.
- Z.-H. Zhang, L. Yin and Y.-M. Wang, Catal. Commun., 2007, 8, 1126–1131 CrossRef CAS.
- I. B. Dzvinchuk, A. V. Turov and M. O. Lozinskii, Chem. Heterocycl. Compd., 2006, 42, 929–934 CrossRef CAS.
- V. Nair and F. Zhang, Molecules, 2013, 18, 11576–11585 CrossRef CAS PubMed.
- R. P. Frutos, T. Tampone, J. A. Mulder, Y. Xu, D. Reeves, X.-J. Wang, L. Zhang, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2011, 52, 2465–2467 CrossRef CAS.
- D. Tomalia and J. N. Pmge, J. Org. Chem., 1973, 38, 3949–3951 CrossRef CAS.
- M. Aouali, D. Mhalla, F. Allouche, L. El Kaim, S. Tounsi, M. Trigui and F. Chabchoub, Med. Chem. Res., 2015, 24, 2732–2741 CrossRef CAS.
- M. S. Kerr, J. R. de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725–5728 CrossRef CAS PubMed.
- S. S. Chuprun, A. V. Protas, O. S. Fedorova, D. D. Vaulina, R. N. Krasikova, E. A. Popova and R. E. Trifonov, Russ. J. Org. Chem., 2016, 52, 1863–1865 CrossRef CAS.
- V. A. Ostrovskii, M. S. Mazur, V. V. Mikhailenko, N. A. Aksenov and A. V. Aksenov, Chem. Heterocycl. Compd., 2016, 52, 849–851 CrossRef CAS.
- S. V. Voitekhovich, Y. V. Grigoriev, A. S. Lyakhov, L. S. Ivashkevich and O. A. Ivashkevich, Dalton Trans., 2016, 45, 13406–13414 RSC.
- S. N. Dighe, K. S. Jain and K. V. Srinivasan, Tetrahedron Lett., 2009, 50, 6139–6142 CrossRef CAS.
- S. V. Voitekhovich, A. N. Vorob'ev, P. N. Gaponik and O. A. Ivashkevich, Chem. Heterocycl. Compd., 2005, 41, 999–1004 CrossRef CAS.
- H. Wang, F. Wei, Q. Chen, X. Hu and X. Niu, J. Chem. Res., 2016, 40, 570–572 CrossRef CAS.
- Y. Satoh and N. Marcopulos, Tetrahedron Lett., 1995, 36, 1759–1762 CrossRef CAS.
- W.-K. Su, Z. Hong, W.-G. Shan and X.-X. Zhang, Eur. J. Org. Chem., 2006, 2006, 2723–2726 CrossRef.
- D. Srinivas, V. D. Ghule and K. Muralidharan, RSC Adv., 2014, 4, 7041–7051 RSC.
- G. P. Sagitullina, A. K. Garkushenko, E. G. Atavin and R. S. Sagitullin, Mendeleev Commun., 2009, 19, 155–156 CrossRef CAS.
- Sh. H. Abdel-Hafez, Eur. J. Med. Chem., 2008, 43, 1971–1977 CrossRef CAS PubMed.
- Z. Arghiani, S. M. Seyedi, M. Bakavoli and H. Eshghi, J. Heterocycl. Chem., 2015, 52, 1099–1107 CrossRef CAS.
- A. Aidouni, S. Bendahou, A. Demonceau and L. Delaude, J. Comb. Chem., 2008, 10, 886–892 CrossRef CAS PubMed.
- Y. Kotaiah, N. Harikrishna, K. Nagaraju and C. V. Rao, Eur. J. Med. Chem., 2012, 58, 340–345 CrossRef CAS PubMed.
- A. E. Abdel-Rahman, E. A. Bakhite, O. S. Mohamed and E. A. Thabet, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 89–106 CrossRef CAS.
- A. A. Harb, A. M. Hussein and I. A. Mousa, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 2247–2261 CrossRef CAS.
- A. H. M. Hussein, M. A. M. Gad-Elkareem, A. B. A. A. M. El-Adasy and I. M. Othman, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 2263–2280 CrossRef CAS.
- Z. A. Hozien, A. A. Abdel-Wahab, K. M. Hassan, F. M. Atta and S. A. Ahmed, Pharmazie, 1997, 52, 753–758 CAS.
- S. Sheikhi-Mohammareh, A. Shiri and M. Bakavoli, J. Chem. Res., 2015, 39, 403–406 CrossRef CAS.
- M. Mohadeszadeh, M. Rahimizadeh, H. Eshghi, A. Shiri, M. Gholizadeh and A. Shams, Helv. Chim. Acta, 2015, 98, 474–481 CrossRef CAS.
- H. Moradi, H. Eshghi and M. Bakavoli, J. Chem. Res., 2016, 40, 276–279 CrossRef CAS.
- A. V. Sergievskii, O. A. Krasnoshek, S. F. Mel'nikova and I. V. Tselinskii, Russ. J. Org. Chem., 2002, 38, 915–917 CrossRef.
- J. A. Bekhit, H. T. Y. Fahmy, S. A. F. Rostom and A. M. Baraka, Eur. J. Med. Chem., 2003, 38, 27–36 CrossRef PubMed.
- S. M. I. Badr, Turk. J. Chem., 2011, 35, 131–143 CAS.
- S. Bondock, W. Khalifa and A. A. Fadda, Eur. J. Med. Chem., 2007, 42, 948–954 CrossRef CAS PubMed.
- A. R. Porcari and L. B. Townsend, Synth. Commun., 1998, 28, 3835–3843 CrossRef CAS.
- A. M. A. Hameed, A. M. Nour-Eldin, M. M. Ibrahim and K. U. Sadek, Chem. Sci. Int. J., 2015, 8, 1–5 CrossRef.
- K. M. Dawood, A. M. Farag and N. A. Khedr, Arkivoc, 2008, xv, 166–175 Search PubMed.
- M. E. A. Zaki, Molecules, 1998, 3, 71–79 Search PubMed.
- H. N. Hafez, A. G. Alshammari and A.-R. B. A. El-Gazzar, Acta Pharm., 2015, 65, 399–412 CAS.
- N. Montazeri, M. M. Heravi, F. Tajfirooz and N. Nazari, Monatsh. Chem., 2017, 148, 691–694 CrossRef CAS.
- I. Zemanová, R. Gašparová, F. Kraic, D. Kružlicová, T. Maliar, A. Boháč and G. Addová, Arkivoc, 2017, 2017, 184–193 Search PubMed.
- S. B. Rajur, A. Y. Merwade, L. D. Basanagoudar and P. V. Kulkarni, J. Pharm. Sci., 1989, 78, 780–782 CrossRef CAS PubMed.
- F. P. L. Lim, L. Y. Tan, E. R. T. Tiekink and A. V. Dolzhenko, RSC Adv., 2018, 8, 21495–21504 RSC.
- M. M. Heravi, F. F. Bamoharram, G. Rajabzadeh, N. Seifi and M. Khatami, J. Mol. Catal. A: Chem., 2006, 259, 213–217 CrossRef CAS.
- M. M. Heravi, M. Rahimizadeh and M. Ghassemzadeh, Indian J. Heterocycl. Chem., 2006, 15, 387–388 CAS.
- M. M. Heravi, M. Bakavoli, M. Rahimizadeh, G. Rajabzadeh and M. Ghassemzadeh, Russ. J. Org. Chem., 2008, 44, 1233–1234 CrossRef CAS.
- M. Z. A. Badr, A. A. Geies, M. S. Abbady and A. A. Dahy, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 2581–2593 CrossRef CAS.
- S.-L. Cao, M. Zhang, Y.-P. Feng, Y.-Y. Jiang and N. Zhang, Synth. Commun., 2008, 38, 2227–2236 CrossRef CAS.
- M. Adib, E. Sheikhi and H. R. Bijanzadeh, Synlett, 2012, 2012, 85–88 CrossRef.
- A. Nasreen and R. M. Borik, Orient. J. Chem., 2014, 30, 761–768 CrossRef CAS.
- D. Shi, L. Rong, J. Wang, Q. Zhuang, X. Wang and H. Hu, Tetrahedron Lett., 2003, 44, 3199–3201 CrossRef CAS.
- A. Davoodnia, M. M. Heravi, E. Golshani, M. Bakavoli and L. Dehabadi, J. Chem. Res., 2007, 2007, 257–258 CrossRef.
- A. A. Mohammadi, S. Taheri and S. Askari, J. Heterocycl. Chem., 2017, 54, 484–488 CrossRef CAS.
- E. Marinho, R. Araújo and F. Proença, Tetrahedron, 2010, 66, 8681–8689 CrossRef CAS.
- M. M. Heravi, S. Sadjadi, N. M. Haj, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Tetrahedron Lett., 2009, 50, 943–945 CrossRef CAS.
- F. F. Bamoharram, M. M. Heravi, M. Roshani and M. Monabati, Asian J. Org. Chem., 2011, 23, 511–514 CAS.
- M. Bakavoli, B. Reihani, M. Rahimizadeh and M. Nikpour, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 99–104 CrossRef CAS.
- B. V. Rao, S. Manmode and S. Hotha, J. Org. Chem., 2015, 80, 1499–1505 CrossRef CAS PubMed.
- S. A. Thadke, M. Neralkar and S. Hotha, Carbohydr. Res., 2016, 430, 16–23 CrossRef CAS PubMed.
- C. Uriel, P. Rijo, A. S. Fernandes, A. M. Gómez, B. Fraser-Reid and J. C López, ChemistrySelect, 2016, 1, 6011–6015 CrossRef CAS.
- A. Mallick, Y. Mallikharjunarao, P. Rajasekaran, R. Roy and Y. D. Vankar, Eur. J. Org. Chem., 2016, 2016, 579–588 CrossRef CAS.
- S. A. Thadke, B. Mishra, M. Islam, S. Pasari, S. Manmode, B. V. Rao, M. Neralkar, G. P. Shinde, G. Walke and S. Hotha, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- A. Y. Shaikh, G. Sureshkumar, D. Pati, S. S. Gupta and S. Hotha, Org. Biomol. Chem., 2011, 9, 5951–5959 RSC.
- B. V. Rao, S. Manmode and S. Hotha, Carbohydr. Res., 2015, 417, 103–108 CrossRef PubMed.
- M. L. Wolfrom, J. W. Spoors and R. A. Gibbons, J. Org. Chem., 1957, 22, 1513–1514 CrossRef CAS.
- P. M. Vadhadiya and C. V. Ramana, Org. Lett., 2015, 17, 1724–1727 CrossRef CAS PubMed.
- D. Mukherjee, S. K. Yousuf and S. C. Taneja, Tetrahedron Lett., 2008, 49, 4944–4948 CrossRef CAS.
- M. V. Panova, N. M. Podvalnyy, E. L. Okun, P. I. Abronina, A. O. Chizhov and L. O. Kononov, Carbohydr. Res., 2018, 456, 35–44 CrossRef CAS PubMed.
- N. M. Podvalnyy, A. O. Chizhov, A. I. Zinin and L. O. Kononov, Carbohydr. Res., 2016, 431, 25–32 CrossRef CAS PubMed.
- N. M. Podvalnyy, P. I. Abronina, E. L. Zdorovenko, A. O. Chizhov, A. I. Zinin, V. I. Torgov and L. O. Kononov, Russ. Chem. Bull., 2014, 63, 497–500 CrossRef CAS.
- T. Uchino, Y. Tomabechi, A. Fukumoto and H. Yamada, Carbohydr. Res., 2015, 402, 118–123 CrossRef CAS PubMed.
- W. Zhu and Z. Li, Synth. Commun., 2000, 30, 3823–3828 CrossRef CAS.
- E. J. Corey and N. Raju, Tetrahedron Lett., 1983, 24, 5571–5574 CrossRef CAS.
- Y. Norimura, D. Yamamoto and K. Makino, Org. Biomol. Chem., 2017, 15, 640–648 RSC.
- H. J. Shirley, M. Koyioni, F. Muncan and T. J. Donohoe, Chem. Sci., 2019, 10, 4334–4338 RSC.
- C. H. Alves Esteves, C. J. J. Hall, P. D. Smith and T. J. Donohoe, Org. Lett., 2017, 19, 5248–5251 CrossRef CAS PubMed.
-
(a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef CAS;
(b) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed;
(c) G. A. Grasa and T. J. Colacot, Org. Lett., 2007, 9, 5489–5492 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*