
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn in silico perception for newly isolated flavonoids from peach fruit as privileged avenue for a countermeasure outbreak of COVID-19†
Ahmed E. Allam *a,
Hamdy K. Assafa,
Heba Ali Hassanb,
Kuniyoshi Shimizuc and
Yaseen A. M. M. Elshaier*d
*a,
Hamdy K. Assafa,
Heba Ali Hassanb,
Kuniyoshi Shimizuc and
Yaseen A. M. M. Elshaier*d
aDepartment of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Assiut, 71524, Egypt
bDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, Universities Zone, New Minia City 61111, Egypt
cDepartment of Agro-Environmental Sciences, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, Fukuoka, 819-0395, Japan
dOrganic & Medicinal Chemistry Department, Faculty of Pharmacy, University of Sadat City, Menoufia, Egypt
First published on 13th August 2020
Abstract
3′-Hydroxy-4′-methoxy-chroman-7-O-β-D-glucopyranoside 4 was first isolated from a natural source, together with three known compounds, the ferulic acid heptyl ester 1, naringenin 2, and 4,2′,4′-trihydroxy-6′-methoxychalcone-4′-O-β-D-glucopyranoside 3, which were isolated from peach [Prunus persica (L.) Batsch] fruits. These compounds were subjected to different virtual screening strategies in order to examine their activity to combat the COVID-19 outbreak. The study design composed of some major aspects: (a) docking with main protease (Mpro), (b) docking with spike protein, (c) 3D shape similarity study (Rapid Overlay Chemical Similarity-ROCS) to the clinically used drugs in COVID-19 patients, and finally, (d) the rule of five and the estimated pre-ADMT properties of the separated flavonoids. Docking study with Mpro of SARS-CoV-2 (PDB ID:6LU7, and 6Y2F) showed that compound 3, its aglycone part, and compound 4 have a strong binding mode to a protease receptor with key amino acids, especially Gln:166AA, and having a similar docking pose to co-crystalized ligands. Docking with the spike protein of SARS-CoV-2 illustrated that compounds 3 and 4 have a good binding affinity to PDB ID:6VSB through the formation of HBs with Asp:467A and Asn:422A. According to ROCS analysis, compounds 1, 3, and 4 displayed high similarities to drugs that prevent SARS-Co2 entry to the lung cells or block the inflammatory storm causing lung injury. Compounds 3 and 4 are good candidates for drug development especially because they showed predicted activity against SARS-CoV-2 through different mechanisms either by preventing genome replication or by blocking inflammatory storm that trigger lung injury. These compounds were isolated from peach fruit, and the study supports data and continues with the recommendation of peach fruits in controlling and managing COVID-19 cases.
1. Introduction
Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) also known as 2019-nCoV is the causative microbe for the current pandemic COVID-19.1Coronaviruses (CoV) are a large family of viruses that lead to infection and sickness starting from common cold to severe diseases.2 Middle East Respiratory Syndrome (MERS-CoV) 2012 and Severe Acute Syndrome (SARS-CoV) 2003 are well-known diseases that had arisen from this family. COVID-19 is the third recognized spillover of coronavirus to humans in the last two decades. The International Committee on Taxonomy of Viruses has considered the placement of the human pathogen, tentatively called 2019-nCoV, within the Coronaviridae family,3 Fig. 1.
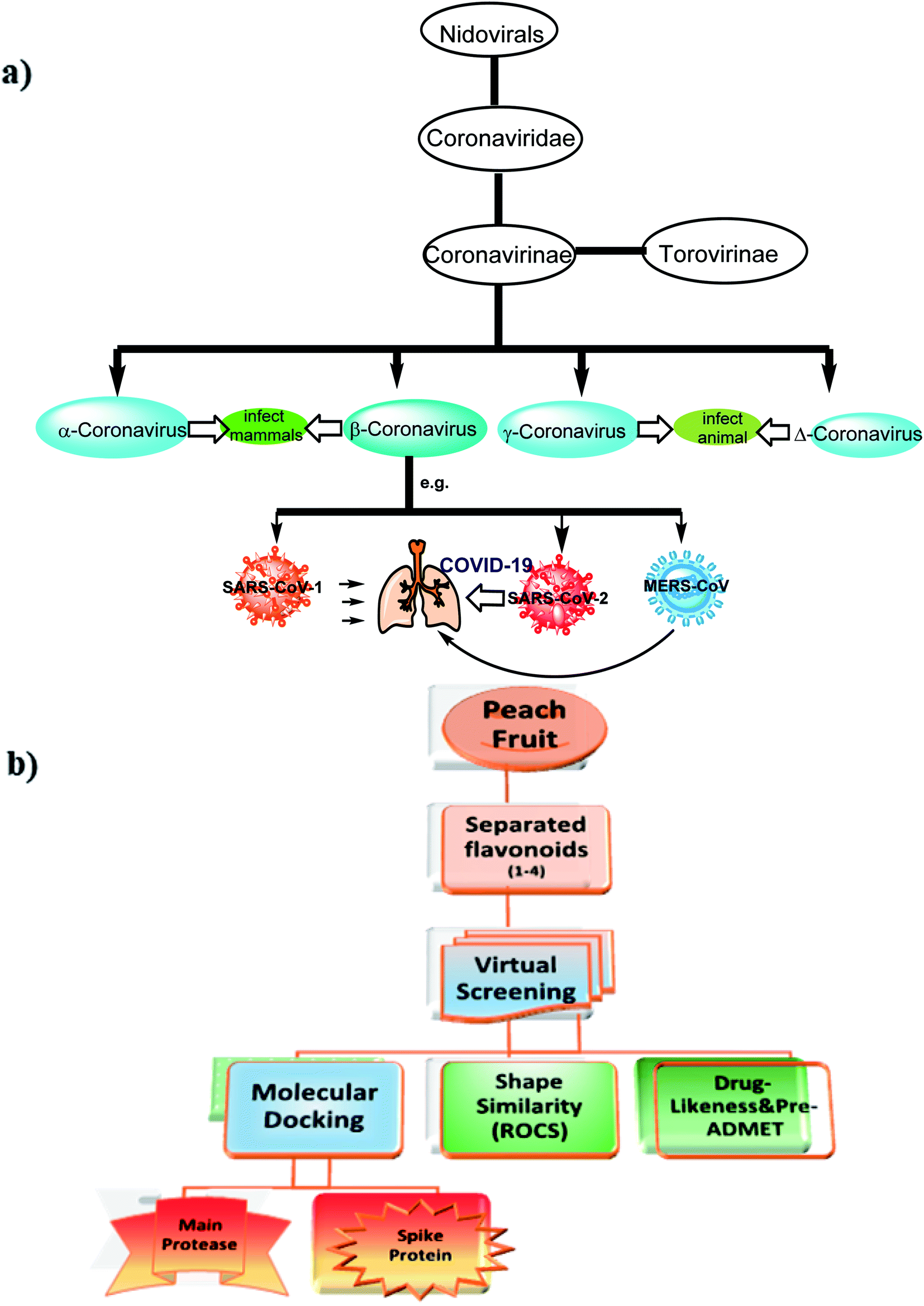 | ||
| Fig. 1 (a) The taxonomy of coronaviridae, (b) Schematic representations of current work design which are composed of two major aspects (1) separation and structural elucidation of new flavonoid from peach fruit and virtual screening approaches (2) structure based approaches. | ||
Although studies on the mechanism of replication as well as the pathogenesis of several coronaviruses have been very active, this family of coronaviruses received much attention because of the new human coronavirus that was responsible for severe acute respiratory syndrome (SARS), a contagious and fatal illness.4
According to the World Health Organization (WHO), from December 2019 to now, about 10 million confirmed cases and about 0.5 million deaths have been reported. The spread of respiratory droplets through sneezing, coughing, or close contact between individuals is the main cause for the transmission of SARS-CoV-2. The symptoms of COVID-19 range from fever, headache, dry coughing, dyspnea, diarrhea, loss of taste, and fatigue to viral pneumonia, acute respiratory distress, and hypoxia. In severe cases, COVID-19 patients require mechanical ventilation.5,6
To date, there is no beneficial therapeutics and limited effective treatment opportunities persist.
1.1. Current therapeutic modalities
Currently, there is insufficient evidence that any of the current antiviral drugs can competently treat COVID-19, and clinical supervision stresses the significance of supportive care and the prevention of complications and avoids social transmission. However, typical precautions, including respiratory and eye protection, are recommended for all healthcare professionals caring for patients with known or suspected COVID-19 pneumonia. Presently, there are numerous clinical trials for targeting SARS-CoV-2 and these drugs, as shown in Fig. 2, are prescribed under what is known as “Drug repurposing approaches or Drug repositioning approaches”.7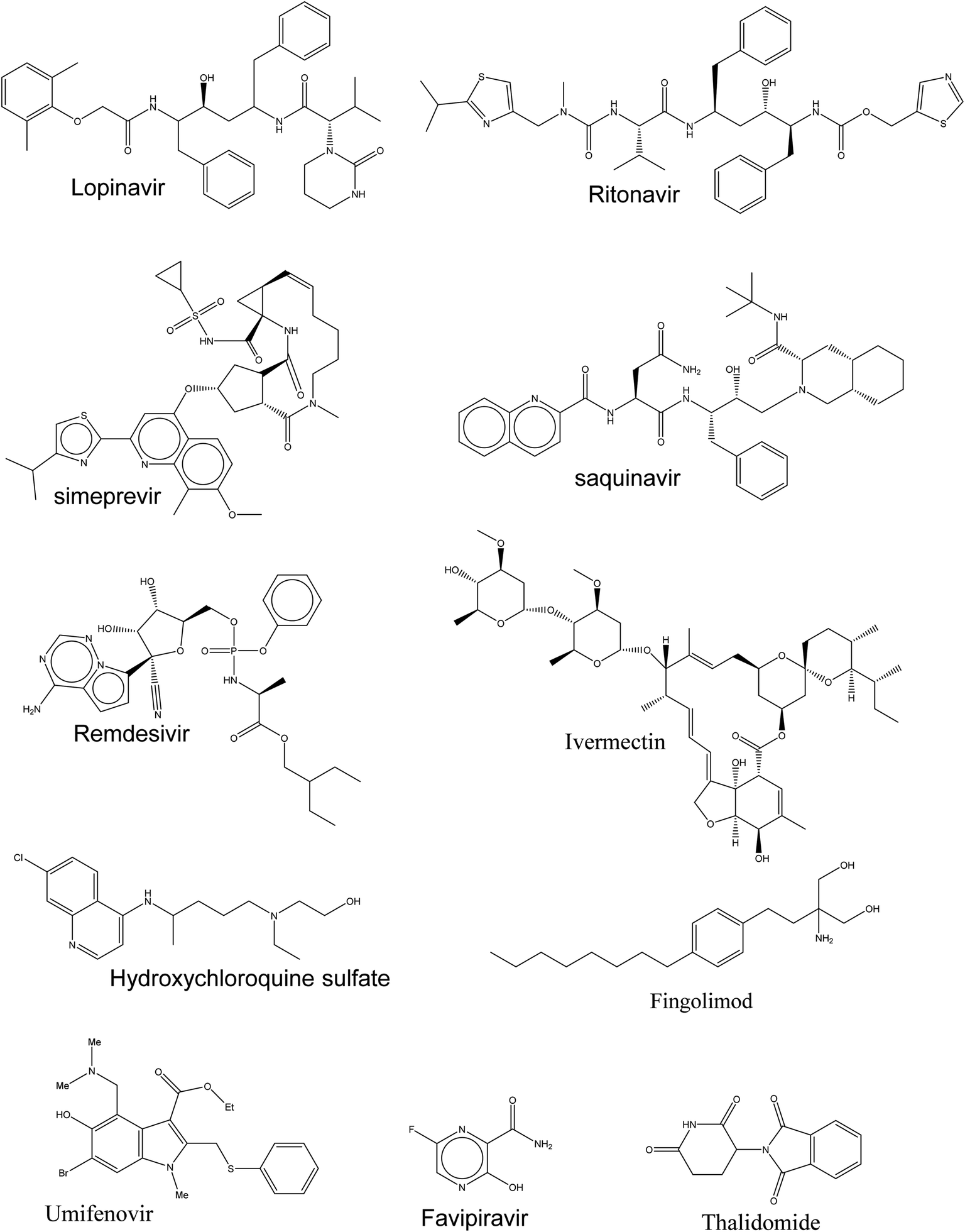 | ||
| Fig. 2 Drugs used in the treatment of COVID-19. | ||
1.2. According to their molecular mechanisms, these drugs could be categorized as follows
(1) Drugs pointing at SARS-CoV-2 directly, these drugs are either;(a) The inhibitors of crucial viral enzymes responsible for genome replication through inhibition of RNA polymerase or viral protease.
(b) Hindering viral entrance to human cells through the inhibition of spike proteins.
(2) Drugs as a human immune system modulator, these drugs are either;
(a) Boosting the inherent response, which has a significant role mainly in controlling viruses' replication.
(b) Preventing the inflammatory storm that causes lung injury.
(3) Symptomatic regulator using VEGF inhibitors as Bevacizumab clinical trial (NCT04275414), and Intravenous Immunoglobulin.
Here in Table 1, the ongoing therapeutic choices are represented briefly that may lead to combat this novel pandemic outbreak (Fig. 2).
| Drug | Category | Notes |
|---|---|---|
| Remdesivir | Inhibiting the RNA-dependent RNA polymerase | • It was originally developed by Gilead Sciences (USA) against the Ebola virus as a prodrug because its structure resembles endogenous adenosine8 |
| • It has been previously shown to display antiviral activities against coronaviruses, especially SARS-CoV9 | ||
| • Remdesivir is now being tested in different countries, two randomized phases, III trials10,11 | ||
| Favipiravir | • It was developed by Toyama Chemical (Japan) for the treatment of influenza | |
| • It's a prodrug structurally resembling the endogenous guanine12 | ||
| • Comparing to remdesivir, less preclinical studies have been established for favipiravir to kill SARS-CoV-2 | ||
| • Currently, it was approved by the National Medical Products Administration of China as the first anti-COVID-19 drug in China, as the clinical trial had demonstrated efficacy with minimal side effects | ||
| Ivermectin | Inhibiting the viral Protease | • Anti-parasitic drug and was reported to inhibit both human immunodeficiency virus (HIV) and dengue virus13,14 |
| • A recent in vitro study has proven its capability to reduce the viral RNA of SARS-CoV-2 (ref. 1) | ||
| • Currently, establishing a safety profile is the next step to verify Ivermectin's ability for curing patients diagnosed with COVID-19 (ref. 15) | ||
| Lopinavir/ritonavir | • Both are used in a combination protocol for the treatment of HIV | |
| • Although coronaviruses encode a different enzymatic class of protease, a number of clinical and in vitro model studies have been completed using this combination on SARS and MERS viruses.16,17 | ||
| • Currently, both drugs are prescribed in a clinical trial in early-stage COVID-19; however, no benefit was observed beyond standard care18 | ||
| Hydroxychloroquine (HCQ) | Blocking virus–cell membrane fusion | • A well-known antimalarial and anti-autoimmune agent with basic character |
| • It blocks the viral infection of SARS-CoV-2 by altering the endosomal pH required for the membrane fusion between the virus and the host cell19 or by interfering in the glycosylation of its cellular receptor, ACE2 (ref. 20) | ||
| • It was approved in different countries to control the spread of COVID-19.21 However, larger randomized controlled trials are required for further evaluation22 | ||
| • On 17 June 2020, WHO announced that the use of HCQ in COVID-19 treatment was ceased | ||
| Umifenovir | • It is a fully-functionalized indole with an antiviral activity against influenza infection. It directs hemagglutinin glycoprotein on the surface of the influenza virus and subsequently prevents its fusion with endosome after endocytosis | |
| • Currently, it is undergoing trials for COVID-19 (ref. 23) | ||
| Lactoferrin (LTF) | • It is a globular glycoprotein found in mammalian milk | |
| • It acquires a wide spectrum of biological activities especially immunological properties | ||
| • In some countries e.g. Egypt, it has been prescribed in the protocol for the treatment of COVID-19 patients | ||
| • LF prevents coronavirus to get attached to Heparan Sulfate Proteoglycans (HSPGs) present at the surface of host cells as these viruses are considered to bind to the host cell by binding first to HSPGs24 | ||
| Recombinant human Angiotensin-converting enzyme 2-APN01(rhACE2) | Preventing the inflammatory storm | • It blocks S protein from interacting with cellular ACE2 and so inhibits SARS-CoV-2 replication |
| • The administration of rhACE2 can decrease the serum level of angiotensin II and so there was no further activation of ACE2 receptor | ||
| • Currently, a pilot study is now evaluating the role of rhACE2 in COVID-19 | ||
| Interleukin (IL)-6 inhibitors e.g. sarilumab, siltuximab, and tocilizumab | • Interleukin (IL)-6, IL-1, and TNF-α are the most important pro-inflammatory cytokines in the human body | |
| • Interleukin (IL) inhibitors may ameliorate severe damage to lung tissue caused by cytokine release in patients with serious SARS-CoV-2 infections. Several studies have indicated a “cytokine storm” with the release of IL-6, IL-1, IL-12, and IL-18, along with tumor necrosis factor-alpha (TNFα) and other inflammatory mediators. The increased pulmonary inflammatory response may result in increased alveolar-capillary gas exchange, making oxygenation difficult in patients with severe illness25,26 | ||
| Fingolimod | • It is an![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) immunomodulationdrug, mostly used for treatingmultiple sclerosis27 immunomodulationdrug, mostly used for treatingmultiple sclerosis27 |
|
| • It is a highly potent functional antagonist of S1P1 receptors in lymph node T cells | ||
| • Currently, it is under clinical trials for the treatment of SARS-CoV-2, NCT04280588, MRCTA, and ECFAH of FMU [2020]027 | ||
| Antibiotics as azithromycin | • Known macrolide antibiotics | |
| • Reduce viremia to zero when used in combination with other drugs | ||
| • Approved for hospitalized cases | ||
| Nitazoxanide28 | • Orally active broad-spectrum antiparasitic | |
| • Prodrug transformed rapidly to active metabolites, such as, tizoxanide and tizoxanide | ||
| • Known to potentiate interferon-alfa and interferon-beta assembly | ||
| • Previously revealed an in vitro activity against MERS-CoV and other coronaviruses | ||
| • Recommended in combination with azithromycin | ||
| Thalidomide | • Thalidomide has been repurposed with different pharmacological effects | |
| • It was reported as an anti-inflammatory drug as it prevents the synthesis of TNF-α,29 treating H1N1-infected mice by reducing pro-inflammatory cytokines30 | ||
| • Present studies are based on its immunomodulatory properties to treat COVID-19 (NCT04273529, NCT04273581), phase 2 |
Other therapies, involving mesenchymal stem cells, monoclonal antibody, methylprednisolone, recombinant interferon, and natural killer cells have different functions to boost inherent response. Currently, corticosteroids have been approved in patients with COVID-19 with a hope of preventing lung fibrosis in patients with unresolved acute respiratory distress syndrome.
Drug discovery approaches like virtual screening (VS), drug repositioning, quantitative structure–activity relationship (QSAR), and artificial intelligence (also, called machine learning), are required more as the world is facing unwelcomed and uncontrolled scenario of current pandemic caused by SARS-CoV-2.31,32
Presently, virtual screening libraries, including ligand-based or target-based approaches, are a focal point for medicinal chemist to trigger time and money efficiency.7
Because the discovery of new drugs requires a long time and expense, searching for new compounds from natural sources known for their high safety and applicability will be a good avenue to treat SARS-CoV-2. In order to quickly discovere lead compounds especially from food sources for clinical trials, a virtual screening study was initiated to identify new drugs targeting SARS-CoV-2.
Among these food sources, peach [Prunus persica (L.) Batsch] is one of the nutritionally edible important fruits in the world. In terms of its biological activity, the peach fruit has been reported to have plenty of phenolic acids, flavanones, and chalcones, which have a wide range of activity against viral infections.33 The fruit is also involved in different biological activities, such as growth–inhibition activity against different breast cell lines,34 antioxidant, anti-lipase, and anti-dementia activities.35
Recently, Traditional Chinese Medicine (TCM) has recommended peach, under the Chinese name “Taoren”, as a contributor in the prevention and treatment of COVID-19,36 that evoked us to carry out this study.
2. Results and discussion
2.1. Structure elucidation
The total ethanolic extract of Prunus persica L. fruits afforded 3′-hydroxy-4′-methoxy-chroman-7-O-β-D-glucopyranoside 4, and it is the first time it has been isolated from a natural source, together with three known compounds, ferulic acid heptyl ester 1,37 naringenin 2,38 and 4,2′,4′-trihydroxy-6′-methoxychalcone-4′-O-β-D-glucopyranoside 3,35 as shown in Fig. 3.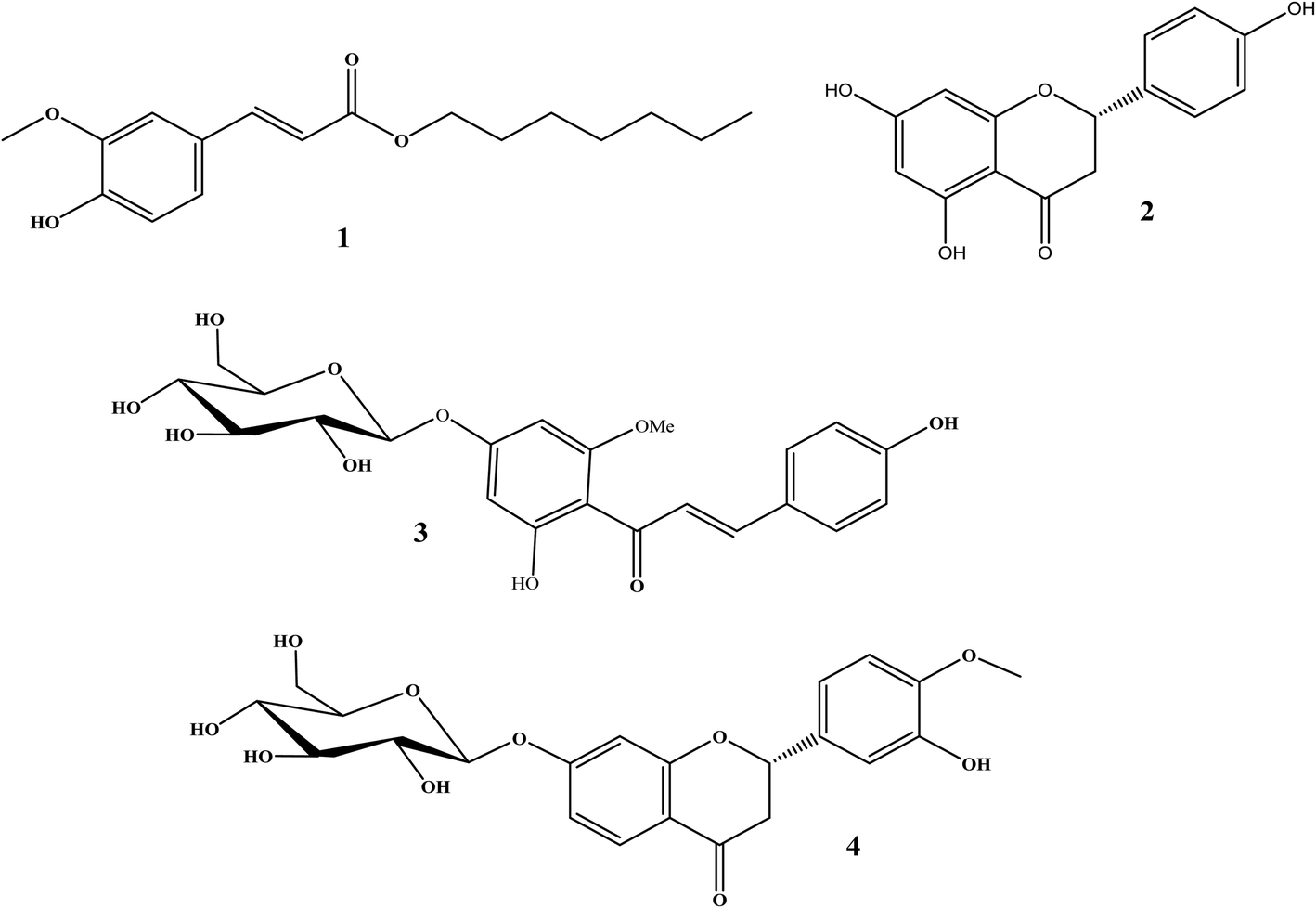 | ||
| Fig. 3 Chemical structures of isolated compounds. | ||
The structures of the known compounds were identified by the comparison of their spectroscopic data with those reported in the literature.
Compound 4 was obtained as a yellow amorphous powder, positive FAB-MS analysis showed [M + Na]+ at m/z 471, corresponding to molecular weight 448 m/z and molecular formula C22H24O10.
The IR spectrum of compound 4 indicated the presence of hydroxyl (3400 cm−1) and carbonyl (1690 cm−1) groups. The UV spectrum of compound 4 exhibited a maximum at 286 and 330 (sh) nm and indicated that 4 is a flavanone.39 The bathochromic shifts induced by AlCl3 (70 nm) and NaOMe (74 nm) are typical of flavanone with two hydroxyl groups at C-3′ and C-4′. No UV bathochromic shift was observed with NaOAc suggesting the absence of free 7-hydroxyl group.39
The 1H-NMR spectrum of 4 reveals the presence of two sets of ABX-spin systems, one reminiscent to the flavanone heterocyclic C ring at δH 5.2 (1H, dd, J = 3, 12.6 Hz, H-2), δH 2.7 (1H, dd, J = 3, 17.4 Hz, H-3eq), and δH 2.9 (1H, dd, J = 13.2, 17.4 Hz, H-3ax); furthermore, the presence of three sets of protons at δH 6.1 (1H, d, J = 2.4 Hz, H-2′), δH 6.7 (1H, d, J = 8.4 Hz, H-5′), and δH 6.9 (1H, dd, J = 2.4, 8.4 Hz, H-6′) indicated the presence of 3′ and 4′ di-substitution in the B ring.35,40 The remaining three aromatic protons at δH 7.3 (1H, d, J = 8.4 Hz), δH 6.8 (1H, dd, J = 2.4, 8.4 Hz), and δH 6.4 (1H, d, J = 2.4 Hz) were assigned to H-5, H-6, and H-8, respectively.41
Also, the 1H-NMR spectrum showed an anomeric proton signal at δH 4.7 (1H, d, J = 7.8 Hz, H-1′′), which indicated the β-configuration of the glucose moiety. Additionally, the 1H-NMR spectrum exhibited a singlet at δH 3.80 (3H), indicating the presence of a methoxy group in the B ring.42
The 13C-NMR spectrum showed the presence of a quaternary carbon C-4 at δC 192.77, an oxymethine carbon C-2 at δC 80.00, an oxymethylene carbon C-3 at δC 46.43, and other carbons of two benzene rings, which indicated the presence of a flavanone moiety.43 Also, the 13C-NMR spectrum showed an anomeric carbon at δC 105.12, which confirmed the presence of glucose moiety.44
The HMBC spectrum showed a significant correlation between the anomeric proton (H-1′′) at δH 4.7 of glucose moiety and the carbon (C-7) at δC 162.52.43,45
The elucidation of the absolute configuration at C-2 was based on the values of coupling constants with methylenic protons H-3 α,β (Jax-ax = 12.6 and Jax-eq = 3 Hz). The close similarity of H-2 chemical shifts, as can be seen in Table 2, with those of the literature,46,47 thus confirmed the S-configuration of C-2.
| No. | Compound 4 | |
|---|---|---|
| δH | δC | |
| 1 | — | — |
| 2 | 5.2 (1H, dd, J = 3, 12.6 Hz) | 80.0 |
| 3 | 2.7 (1H, dd, J = 3, 17.4 Hz), | 46.4 |
| 2.9 (1H, dd, J = 13.2, 17.4 Hz) | ||
| 4 | — | 192.7 |
| 5 | 7.3 (1H, d, J = 8.4 Hz) | 130.2 |
| 6 | 6.8 (1H, dd, J = 2.4, 8.4 Hz) | 101.0 |
| 7 | — | 162.5 |
| 8 | 6.4 (1H, d, J = 2.4 Hz) | 99.8 |
| 9 | — | 166.5 |
| 10 | — | 114.5 |
| 1′ | — | 133.2 |
| 2′ | 6.1 (1H, d, J = 2.4 Hz) | 116.3 |
| 3′ | — | 147.8 |
| 4′ | — | 149.3 |
| 5′ | 6.7 (1H, d, J = 8.4 Hz) | 112.6 |
| 6′ | 6.9 (1H, dd, J = 2.4, 8.4 Hz) | 118.9 |
| 1′′ | 4.7 (1H, d, J = 7.8 Hz) | 105.1 |
| 2′′ | 3.55 (1H, m) | 74.7 |
| 3′′ | 3.42 (1H, m) | 78.6 |
| 4′′ | 3.47 (1H, m) | 71.2 |
| 5′′ | 3.51 (1H, m) | 77.2 |
| 6′′ | 3.7 (1H, dd, J = 4.8, 12.0 Hz), | 62.5 |
| 3.9 (1H, dd, J = 1.8, 12.0 Hz) | ||
| 1′′′ | 3.80 (3H, s) | 56.4 |
From the above-mentioned spectroscopic data, compound 4 was identified as (−)-(2S)-3′-hydroxy-4′-methoxy-chroman-7-O-β-D-glucopyranoside, and it is the first time it has been isolated from a natural source.
2.2. Molecular modeling and virtual screening study
SARS-CoV-2 is a single-stranded positive-sense RNA genome. The virus envelop contains spike protein (S) which regulates viral pass into the host cells. Then, two polyproteins i.e. pp1a and pp1ab are promptly translated upon entry into the host cells, and finally, these are separated by two viral protease enzymes to make viral replication. Thus, these molecular targets can be is considered as druggable targets.49
Numerous molecular dynamic simulation studies were addressed to figure out the active sites of the SARS-CoV-2 main protease (Mpro) or spike protein50 however in this study the docking section was performed with the crystal structures of SARS-CoV-2 reported in PDB. The crystal structure of SARS-Cov-2 main protease (Mpro) forms complex with the inhibitors N3 and α-ketoamide coded with PDB ID 6LU7 (ref. 51) and 6Y2F,52 respectively, Fig. 4. Furthermore, a docking study with the spike protein of SARS-CoV-2 (PDB ID:6vsb53) was represented. By this tactic, we will indicate the possible mechanism of these naturally occurring flavonoids to fight COVID-19.
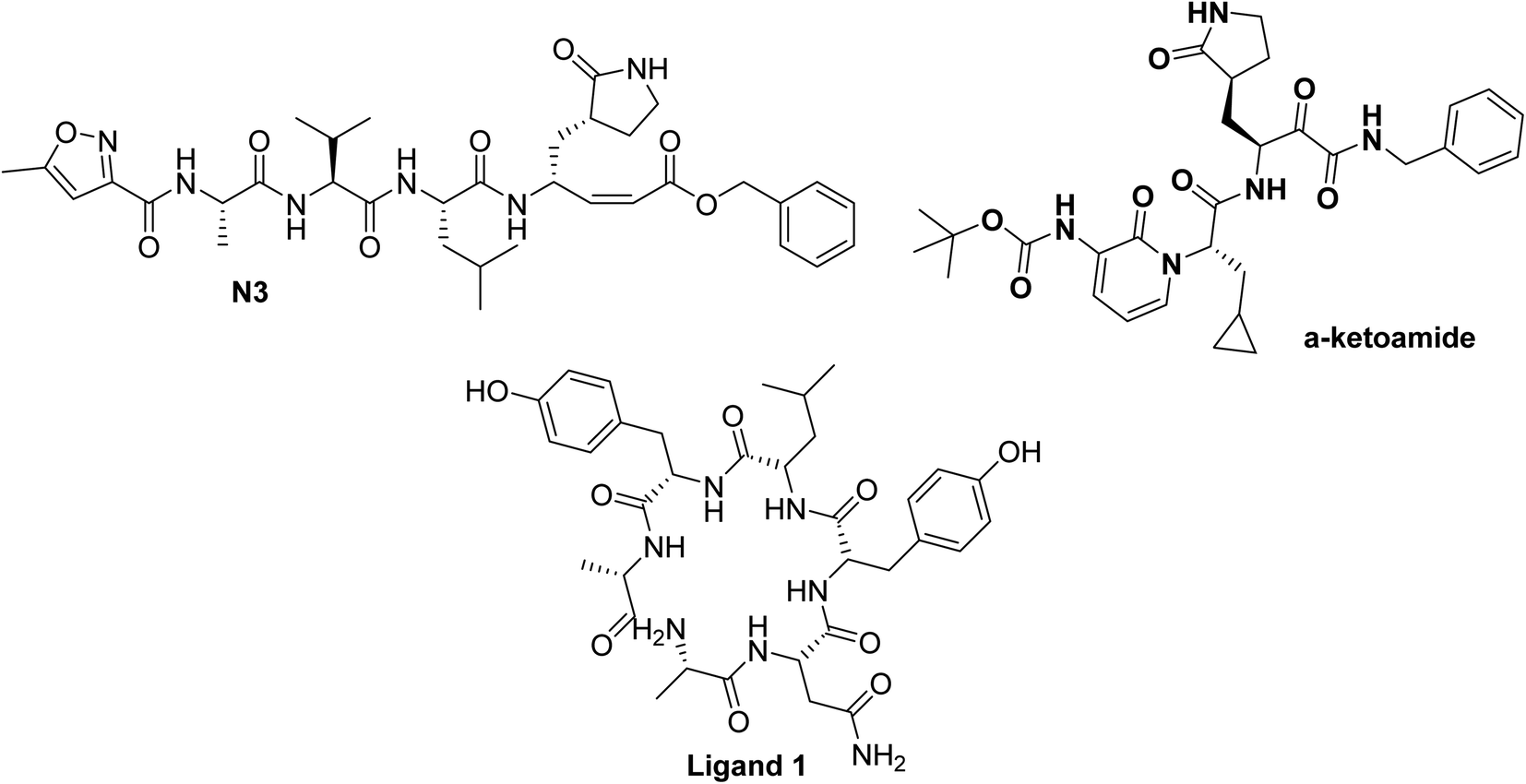 | ||
| Fig. 4 Chemical structure of ligands N3, and α-ketoamide for SARS-CoV-2 Mpro (PDB ID:6LU7,51 and 6Y2F52 respectively) and ligand 1 for SARS-CoV-2 spike protein (PDB ID:6vsb53). | ||
2.2.1.1. Docking study against the structure of Mpro from SARS-CoV-2. The key CoV enzyme which displayed a pivotal role in facilitating viral transcription and replication is called Mpro.51
The standard ligands N3 and α-ketoamide in addition to clinically used protease inhibitor drugs, such as remdisever, lopinavir, and ritonavir were docked with the available co-crystallized main protease (Mpro). The experimental docking of these standards presented similarity in their binding mode inside the receptor especially HBs with the key peptides and amino acids namely Thr:25A–His:41A, Asn:142A–Ser:144A, Glu:166A, Gln:189A, and Ala:191A.
The literature survey displayed that flavonoids were previously reported to inhibit the main protease of SARS-CoV-1.4,54 Recently, there are studies for the activity of phenolic compounds and flavonoids against SARS-CoV-2.26
To well understand the expected activity of the isolated flavonoids from peach fruits, docking analyses were performed using the experimental crystal structure of SARS-CoV-2, which was determined recently. Consensus score for the isolated compounds and some selected antiviral drugs are illustrated in Table 3. Among the isolated compounds and based on the consensus score value, the flavanone 4, chalcone 3, and its aglycone bind strongly with Mpro receptor.
| Compound | PDB ID:6lu7 | PDB ID:6y2f | ||
|---|---|---|---|---|
| Ferulic acid ester 1 | 346 | Hydrophobic interactions only | 279 | HBs with Thr:25A, Cys:44A and hydrophobic interactions |
| S isomer of 2 | 192 | HBs with Glu:166A, Gly:143A, Leu:141A and occupied hydrophobic interaction | 321 | Two HBs with Thr:25A and occupied hydrophobic interaction |
| R isomer of 2 | 167 | HBs with Gly:143A and occupied with Hydrophobic interaction | 333 | Two HBs with Thr:25A, Asp:187A and occupied with hydrophobic interaction |
| Chalcone 3 | 286 | Hydrophobic interactions only | 56 | Two HBs with Gly:189A, Gln:192A and occupied with Hydrophobic interaction |
| Aglycone of 3 | 80 | HBs with Glu:166A, Hist:163A, Ser:144A. Styryl part interacted with hydrophobic interactions | 187 | HBs with Arg:188A. Styryl part interacted with hydrophobic interactions |
| Flavanone 4 | 87 | HBs with Gln:192A, Ser:144A. Benzopyrane architecture interacted with hydrophobic interactions | 133 | HBs with Ala:193A, His:94A. Benzopyrane architecture interacted with hydrophobic interactions |
| Taxifolin | 137 | HB with Glu:166A and occupied deeply with hydrophobic interaction | 151 | HBs with Glu:166A, Thr:190A, Asp:187A and occupied deeply with hydrophobic interaction |
| Saquianiver | 15 | HBs with Glu:166A (two HB), His:164A. Benzyl moiety and quinoline ring occupied with hydrophobic interaction | 123 | HBs with Glu:166A (two HB), His:164A, Gln:189A. Benzyl moiety and quinoline ring occupied with hydrophobic interaction |
| α-ketoamide | 560 | HBs with Glu:166A, Leu:167A, Asn:142A. Benzyl and cyclopropyl moieties participated hydrophobic interactions | 176 | HB with Met:165A. Benzyl and cyclopropyl moieties participated hydrophobic interactions |
| N3 | 347 | HBs with Thr:190A, Glu:166A, Leu:167A, His:164A, Cys:145:A, Met:165A. Benzyl and thiazole parts participated hydrophobic interactions | 258 | HBs with Glu:166A, Gln:189A. Thiazoleo peptide part, isopropyl and benzyl arms participated hydrophobic interactions |
To validate our study, the reported ligand was re-docked with the active pocket. N3 showed interactions with receptor having HB formations with Gln 192: A and Glu 166: A, Fig. 5a. Compound 2, the (S) isomer, displayed HBs by its carbonyl function which is arranged close to the Glu 166: A. The hydroxyl group at C-8 participated in HB interaction with Ser 144: A. The hydroxyl group attached to C2-phenyl moiety at P-position occupied the amino acid clefts through hydrophobic–hydrophobic interactions, Fig. 5b.
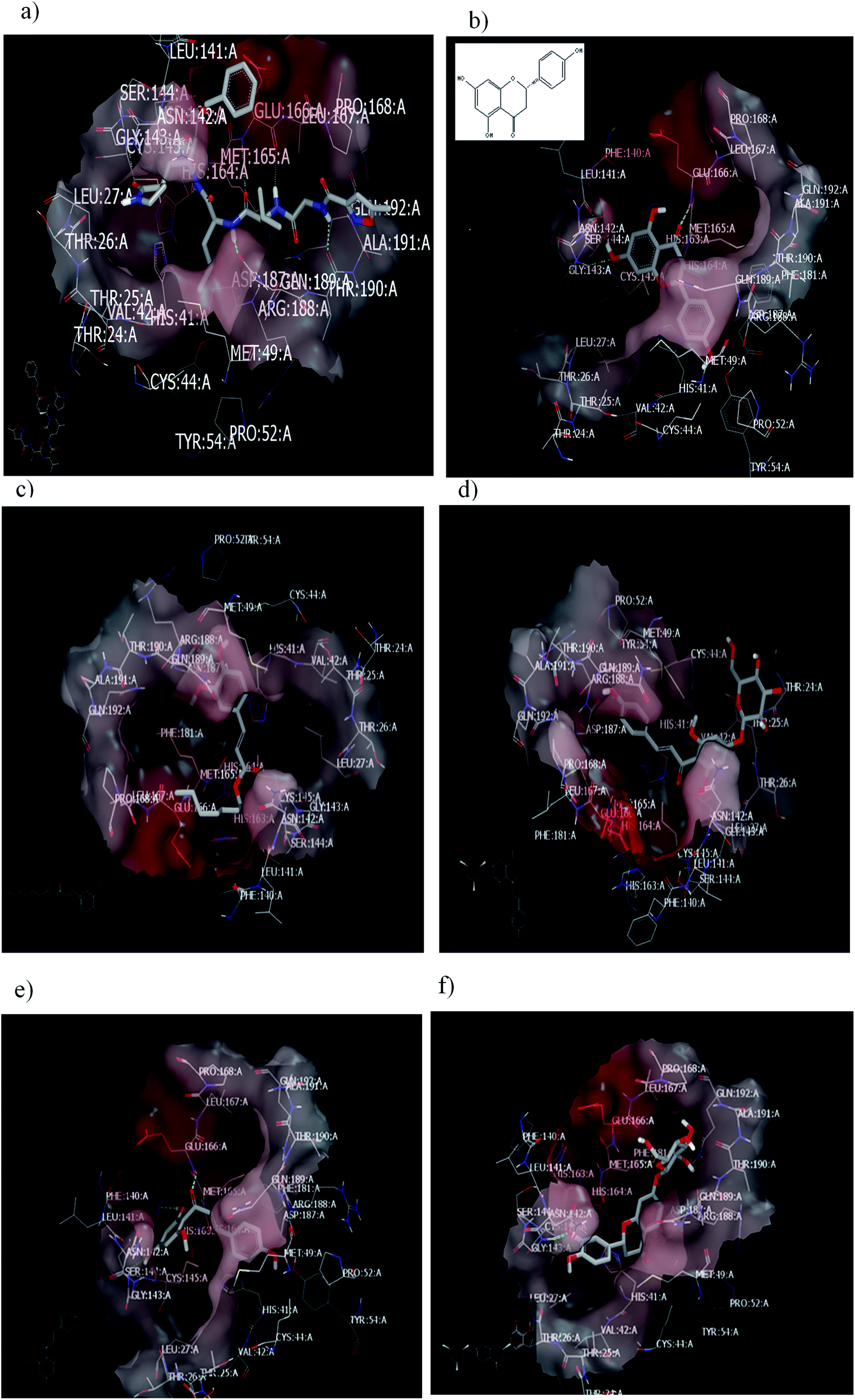 | ||
| Fig. 5 Visual representation by vida for (a) standard ligand N3; (b) compound 2 (S isomer); (c) compound 1; (d) compound 3; (e) Aglycone of compound 3; and (f) compound 4 docked with ID:6lu7.In similar performance, the isolated compounds docked with the PDB ID:6Y2F. Both compounds 3 or its aglycone and compound 4 prioritized with top of consensus score in comparison to other examined compounds, Table 3. | ||
In order to ensure the effect of compound's absolute configuration on docking mode (enantioselective docking relationship) and subsequently explore their structure activity relationship (SAR), the (R) isomer demonstrated different binding pose and mode with the receptor. Docking results clarify that both the enantiomers of compound 2 (Naringenin) displayed dissimilarity in their binding pose inside the active site.
Taxifolin is a dihydro quercetin flavonoid with an interesting spectrum of biological activities.55 Recently, in a virtual screening study, taxifolin showed the lowest binding free energy against Mpro of SARS-CoV-2.56 Our attention was directed to examine the effect of different pharmacophoric features of taxifolin and compound 2 that will support the structure–activity relationship and drug design strategy. Taxifolin presented a binding with the receptor by hydroxyl functionality at C-3, which is arranged close to Glu 166: A with the formation of strong HB. Whereas the phenyl moiety and chroman ring were well-inserted through hydrophobic–hydrophobic interaction in the amino acid clefts. The result showed that this flavonoid (compound 2) can form HB with the same amino acid (Glu 166: A) in spite of devoting from OH at C-3 as in taxifolin.
Regarding the ferulic acid heptyl ester 1, as indicated from its structure, it formed hydrophobic–hydrophobic interactions without the formation of HB, Fig. 5c. This result emphasizes the importance of physicochemical parameters, such as logP, which will be elucidated later. Likewise, compound 3 disclosed strong hydrophobic–hydrophobic interactions, Fig. 5d. To perceive the effect of glycosidation, the 3D shape of compound 3 inside the receptor represented dissimilarity with its aglycone part. The aglycone moiety disclosed strong bonding interaction (consensus score 80). It formed HBs with Glu 166: A, Hist 163: A, and Ser 144: A through the carbonyl at C-2 and OH at C-6 (glycoside linkage), respectively, Fig. 5e.
Concerning compound 4, it facilitated HB formation with Ser. 144: A through the OH group of phenyl at C-2, and the sugar moiety also participated in HB formation with Gln 192: A, Fig. 5f.
2.2.1.2. Docking with PDB ID:6yef. The standard α-ketoamide was re-docked, and it represented different binding modes to our separated flavnoides: HB through the carbonyl amide of cyclopropyl moiety with Glu 166: A and hydrophobic–hydrophobic interactions, Fig. 6a. Flavonoid 3 formed HB with Thr 190:A and hydrophobic–hydrophobic interaction, Fig. 6b. Flavonoid 4 linked the two-terminal clefts of the receptor through the formation of HBs with Ala 133: A and Hist 41:A and through the sugar moiety and 3-hydroxyl group of the phenyl group respectively. The whole part of the molecule displayed hydrophobic–hydrophobic interactions, Fig. 6c.
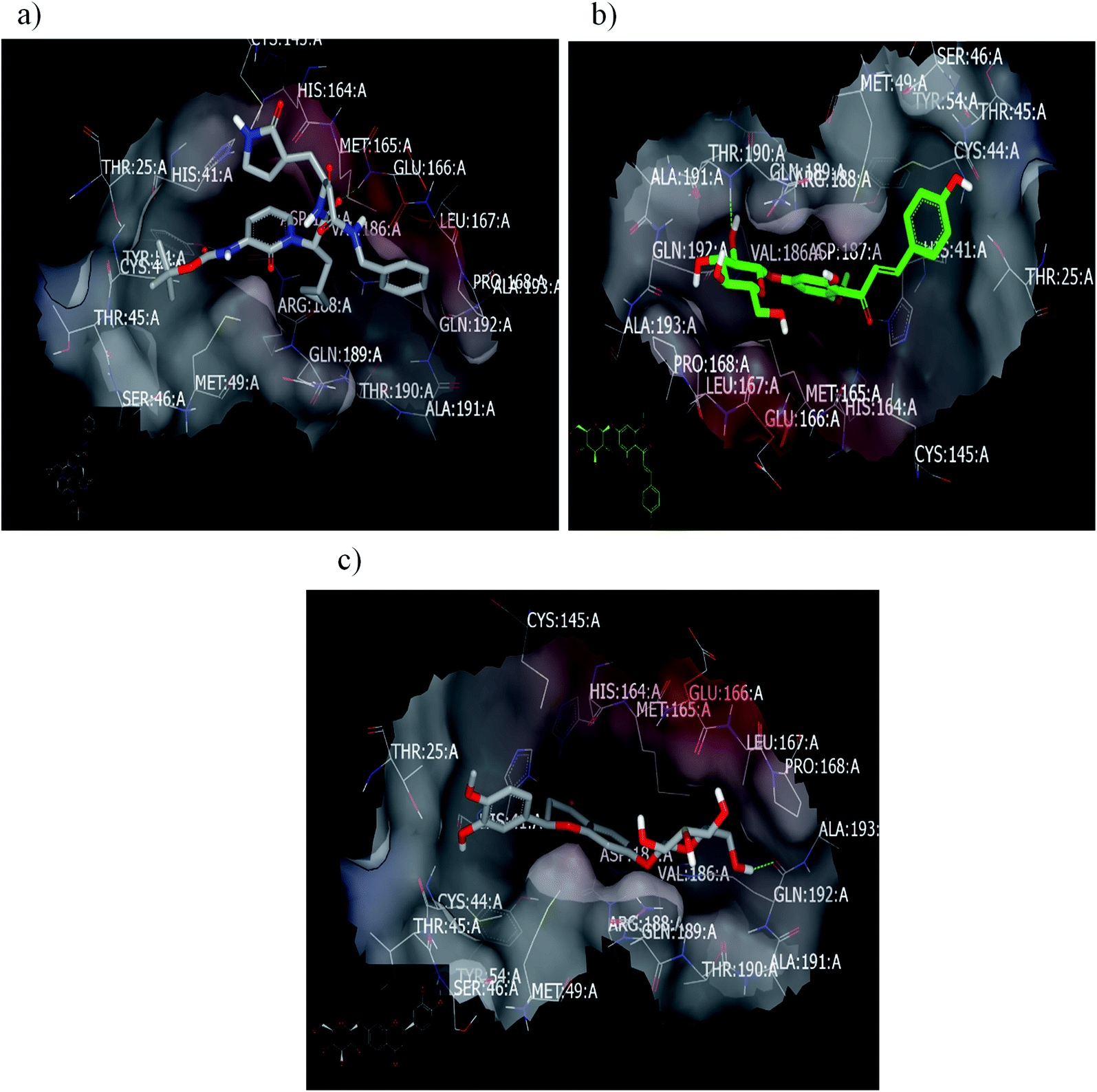 | ||
| Fig. 6 Visual representation by vida for (a) α-ketomide standard; (b) compound 3; and (c) compound 4 docked with ID:6y2f. | ||
2.2.1.3. Docking against the spike protein of SARS-CoV-2. The SARS-CoV-2 spike protein is an important key target for controlling COVID-19 through vaccination, therapeutic treatment, or diagnosis.48 The retrieved receptor for S protein generated by making receptor command/OpenEye represented that the peptide part of Asn:422A–Ser:494A is essential for HB formation with standard ligands. Both the drug candidates, PC786 (ref. 48) (consensus score 70) and ligand 1 (consensus score 204), perceived similar binding mode especially HBs with Pro:491A (strong) and Tyr:351A (weak) with overlay pose to each other, Fig. 7a. The Umifernovir drug exhibited HB with Asn:422A (consensus score 174), ESI.†
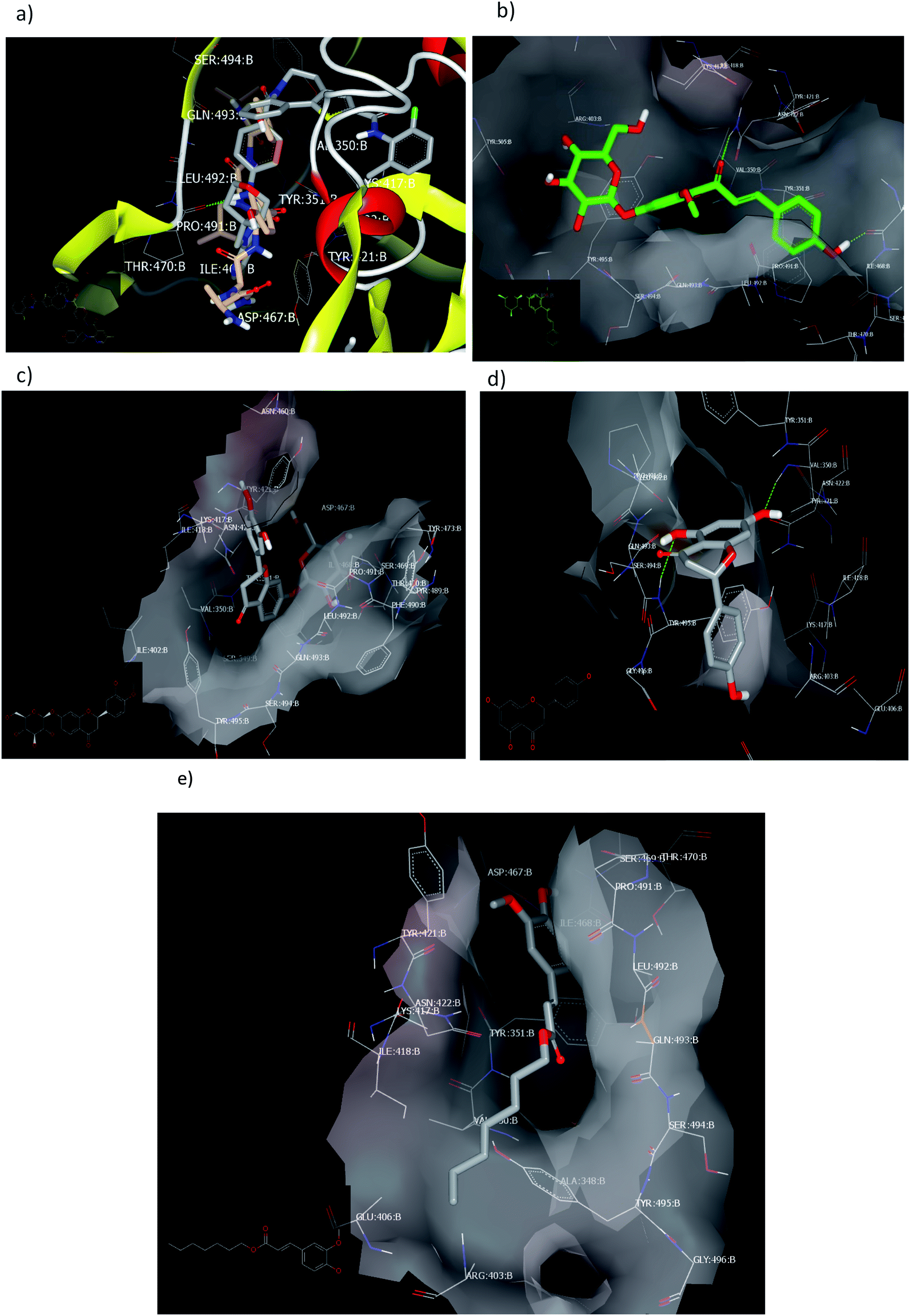 | ||
| Fig. 7 Visual representation by vida for (a) ligand 1 and PC687; (b) compound 3; (c) compound 4; (d) compound 2 docked; and (e) compound 1 with ID:6vsb. | ||
Among the separated flavonoids, compounds 3 and 4 were observed to have a good binding affinity to PDB ID:6vsb. Compound 3 (consensus score 464) showed HB interaction with Asp:467A and Asn:422A, Fig. 7b; while the corresponding aglycone illustrated a docking outside the receptor (consensus score 626), ESI.† This feature indicates that glycosylation is important to provide the compound some polarity essential for binding interactions. Compound 4 formed HBs with Ilu:468A and hydrophobic interactions (consensus score 396), Fig. 7c. Although compound 2 formed HBs with Ser:494A, Val:350A (consensus score 606) but it interacts with the S protein outside the created receptor, Fig. 7d. However, the analog R isomer appeared without docking and was visualized outside the receptor (ESI†), and ferulic ester (1) occupied the receptor with hydrophobic interactions only (consensus score 880), Fig. 7e.
Despite all the prospective approaches of molecular docking, the chemistry of ligands (tautomerism and ionization), the flexibility of the receptor, the availability of receptor crystal structure, and scoring function persisted the challenge. In this regard, although molecular docking is an attractive technique to recognize drug biomolecular interactions for drug design and discovery, many challenges for docking study could occur.
High match in shape reflects a high match in biology while the high match in biology does not reflect the relationship between biological activity and the 3D shape structure. Different applications of ROCS can be applied, such as virtual screening, lead hopping, molecular alignment, pose generation, and structural predictions.34,35,37
The approach of this study is to identify molecules that can adopt shapes extraordinarily similar to reported drugs recommended in COVID-19 disease. ROCS was applied to identify the similarity of our compounds with different drugs or cover other proteins related to SARS-CoV-2 not co-crystallized yet, Table 4.
| Compound | Inhibiting the RNA polymerase | Inhibiting the viral protease | Blocking virus–cell membrane fusion | Prevent cytokine storm | |||
|---|---|---|---|---|---|---|---|
| Remdesivir | Favipiravir | α-ketomide | Lopinavir | Umifenovir | HCQ | Fingolimod | |
| 3; chalcone | 0.58 | 0.57 | 0.64 | 0.56 | 0.76 | 0.66 | 0.78 |
| 4; flavanone | 0.71 | 0.52 | 0.53 | 0.58 (retinover = 0.58) | 0.75 | 0.76 | 0.75 |
| R isomer of 2 | 0.51 | 0.77 | 0.48 | 0.51 | 0.86 | 0.70 | 0.81 |
| S isomer of 2 | 0.50 | 0.78 | 0.48 | 0.47 | 0.86 | 0.69 | 0.80 |
| Aglycone of 3 | 0.53 | 0.78 | 0.52 | 0.57 | 0.87 | 0.85 | |
| 1; ferulic acid-heptyl ester | 0.58 | 0.82 | 0.52 | 0.53 | 0.87 | 0.73 | 0.94 |
| Taxifolin | 0.54 | 0.81 | 0.49 | 0.45 | 0.827 | 0.70 | 0.73 |
| Remdesiver | 2.00 | 0.51 | 0.56 | 0.58 | 0.75 | 0.58 | 0.58 |
| Favipiravir | 0.44 | 2.00 | |||||
| Lopinavir | 0.58 | 0.38 | 0.63 | 2.00 | 0.52 | 0.52 | 0.45 |
| α-ketoamide | 0.53 | 0.45 | 2.00 | 0.56 | 0.52 | 0.51 | 0.51 |
| N3 | 0.51 | 0.30 | 0.54 | 0.59 | 0.45 | 0.48 | 0.47 |
| HQC | 0.56 | 0.77 | 0.54 | 0.54 | 0.83 | 2.00 | 0.84 |
| CQ | 0.52 | 0.83 | 0.53 | 0.59 | 0.73 | 1.5 | 0.71 |
| Umifenovir | 0.45 | 0.66 | 0.44 | 049 | 0.20 | 0.71 | 0.54 |
| Fangiloamide | 0.56 | 0.60 | 0.52 | 0.45 | 0.74 | 0.76 | 2.00 |
ROCS application involves two files,38 which must be in most stable conformer generated by omega algorism: (a) database file, which is a compound collection for this study; (b) query file here is the standard drugs. In this work, we decided to examine our separated compounds with different queries in order to figure out the most possible mechanism in targeting SARS-CoV-2, Table 5. The study depicts a match between the database file and query drug in a separate run. This match is based on the volume overlap of optimally aligned molecules, which are virtually independent of the atom types and bonding patterns of the query. Two main outputs generated from ROCS analysis: (1) shape similarity which includes shape counter, shape atoms, and color atom labels for database set (query compounds) and was visualized by vROCS and VIDA applications. We added this feature which has been written in Fig. 8a–e; and (2) a set of scores expressed in Tanimoto scores. The most imperative score is Tanimoto Combo (TC) that contains both shape fits and color. This has a value between 0 and 2 and the score is used for ranking the hit list, Table 4.
| Compound | Lipinski's rule (rule of five) | Value | PreADMET prediction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | LogP |
HBD | HBA | BBB | PPB | MolPSA | HIA | Caco-2 value | Skin permeability | pKa | Drug-likeness score | ||
| a HBD, hydrogen bond donor; HBA, hydrogen bond acceptor; BBB, blood brain barrier; PPB, plasma protein binding; HIA, percentage human intestinal absorption; MolPSA (molecular polar surface area (PSA); Caco-2 value, permeability to Caco-2 (human colorectal carcinoma) cells in vitro. | |||||||||||||
| 4; flavanone | 448.14 | 0.19 | 5 | 10 | Suitable | 0.036 | 64.54 | 124.24 A2 | 61.51 | 7.17 | −4.56 | <0./9.80 | 0.34 |
| Aglycone of 4 | 386.08 | 2.51 | 2 | 5 | Suitable | 0.122 | 94.84 | 62.00 A2 | 92.75 | 12.82 | −3.42 | <0./8.45 | 0.69 |
| 3; chalcone | 448.14 | 0.55 | 6 | 10 | Suitable | 0.039 | 74.55 | 134.87 A2 | 49.69 | 12.24 | −3.89 | <0./9.72 | 0.56 |
| Aglycone of 3 | 286.08 | 2.86 | 3 | 5 | Suitable | 0.69 | 93.71 | 72.63 A2 | 88.52 | 18.51 | −3.39 | <0./8.06 | 0.10 |
| S isomer of 2 | 272.07 | 2.38 | 3 | 5 | Suitable | 0.95 | 100.00 | 71.98 A2 | 87.31 | 10.52 | −4.18 | <0./8.29 | 0.82 |
| 1 | 292.17 | 5.08 | 1 | 4 | Suitable | 2.35 | 91.71 | 44.87 A2 | 95.44 | 46.70 | −1.33 | <0./9.69 | −0.51 |
Among the separated compounds, compound 4 has the highest similarity to remdesivir, lopinavir, and hydroxychloroquine (HCQ) with TC = 0.71, 0.58, and 0.76 respectively. Table 4 signifies that this compound has high shape similarity to these drugs that prevent the genome of the virus to replicate. Fig. 8a and b illustrated color, volume and shape of remdesivir and lopinavir respectively.
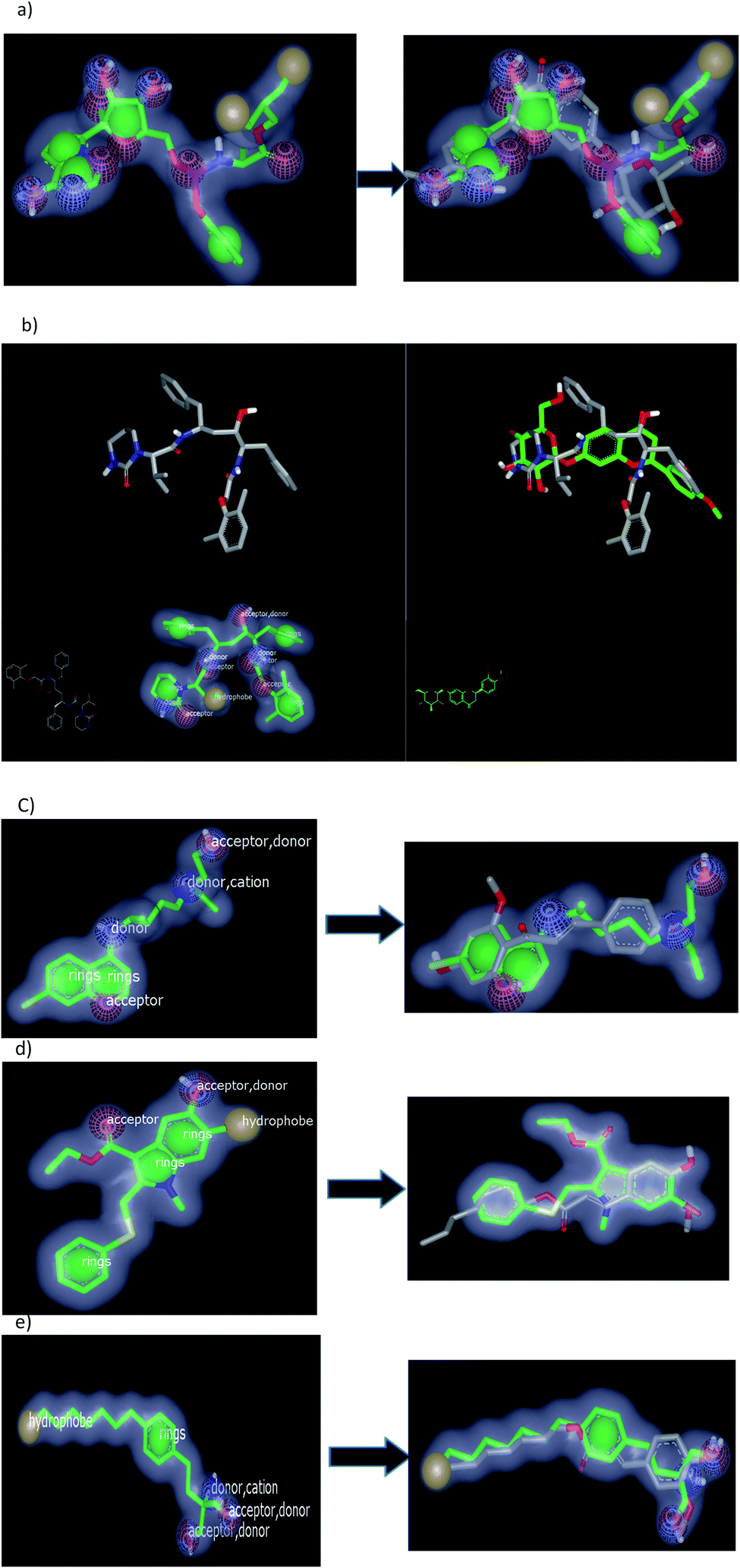 | ||
| Fig. 8 (a) Remidisever ring: 4, donor: 4, acceptor: 8, hydrophobe: 2, cation: 1 (left); and its overly lay with 4 (right); (b) lipinover as ring: 4, donor: 4, acceptor: 5, hydrophobe: 1, cation: 1 (left) and overlay with compound 4 (right); (c) HCQ as ring: 2, donor: 3, acceptor: 2 (left) and HCQ overlay with aglycone of compound 3 (right); (d) umenevir as ring: 3, donor: 1, acceptor: 2, hydrophobe: 1 (left) and overlay with ferrulic ester 1 (right); and (e) fingolamide as ring: 1, donor: 3, acceptor: 2, hydrophobe: 1, cation: 1 (left) and ferrulic ester 1 (right) with fingolamide. | ||
rand their overlay with compound 4 with remdesivir. However, the two benzyl moieties of lopinavir occupied the outside volume of compound 4. Moreover, chalcone 3 has high similarity to protease inhibitor α-ketomide (TC = 0.64). The aglycone part of compound 3 (considered as a metabolite, Fig. 8c) exhibited high similarity to Hydroxychloroquine HCQ (TC 0.85). Additionally, it has also a high similarity to umifenovir (TC 0.87). These results emphasize the hypothesis that this compound (3) has the ability for blocking the entry of virus.
Ferulic ester 1 has the highest similarity to favipiravr, umifenover, and fingolamide, demonstrating that this compound has a high likeliness towards different mechanisms for SARS-CoV-2 treatment, Fig. 8d and e.
In response to their high privilege character but with limited expected drug-likeness properties, these combinations of characters encouraged us to add predicted physicochemical parameter values as will be discussed later to study the drug ability and low toxicity of compounds.
As illustrated in Table 5, compounds 3 and 4 were considered as isomers as both of them have the same molecular weight and are similar in most ADMET and physicochemical parameters (drug-likeness score = 0.56 and 0.34, respectively). The aglycone part of compounds 3 and 4 showed higher acceptable physicochemical and Pr-ADMT parameters. According to their 2D structure (compounds 3 and 4), these compounds originated from two different architecture systems. Compound 1 has a higher lipophilicity value than compound 2, then compound 3 and finally compound 4. Their lipophilicity scores were represented by logP values and BBB.
Compound 1 exhibited high logP value of 5.08 and therefore low hydrophilicity causes poor absorption or permeation with a low drug-likeness score of −0.51, highlighting that lipophilicity is an important parameter in the drug development process. However, compound 2, aglycones of 3, and compound 4 well-exhibited good logP values (2.38–2.81), indicating that the hydrolysis of the glycoside linkage is valuable for compound activity.
3. Experimental
3.1. Reagents
Silica gel and RP-18 silica gel were purchased from Wako (Osaka, Japan). Thin-layer chromatography (TLC) silica gel G60F254 was purchased from Merck (Darmstadt, Germany). All other ingredients used were of the highest grade available.3.2. Preparation of extracts
The fresh materials were freeze-dried and milled below 1 mm. Then, their extracts were prepared by shaking (200 rpm) with solvent (ethanol) at room temperature for 48 h. The extracted solution was filtered and evaporated using ADVANTEC no. 2 filter paper (Toyo Roshi Kaisha, Ltd, Tokyo).3.3. Fractionation and isolation
The ethanol extract was suspended in distilled water and divided between n-hexane, ethyl acetate, and the remaining water to give the n-hexane fraction (12 g), ethyl acetate fraction (5.1 g), and the remaining aqueous fraction (45.5 g). We attempted to isolate the active compounds from the ethanol extract derived from peach fruits. The ethyl acetate fraction from the ethanol extract was sub-fractionated on a silica gel column using chloroform-methanol gradient elution (25%, 50%, 75%, and 100%; 2 L each). The fraction eluted by 25% methanol (1.4 g) was further separated by chromatography on an ODS column (80 × 200 mm; Cosmosil 140 C-18 PREP, Nacalai Tesque, Tokyo, Japan) using six mobile phase systems of methanol–water (10%, 25%, 40%, 50%, 70%, and 90% v/v; elution volume: 1.5 L of each) to give six corresponding fractions. The fraction eluted with 10% methanol (310 mg) was further chromatographed by column chromatography on silica gel and eluted by a stepwise gradient of chloroform-methanol (ratios of 9:1, 85:15, 8:2, 7:3, and 6:4; v/v elution volume: 200 ml each) to give five corresponding fractions. The fraction eluted with 85:15 chloroform-methanol resulted in the elution of compounds 1 and 2 (55 and 40 mg, respectively). The fraction eluted with 8:2 resulted in the elution of compound 3 (35 mg) and that eluted with 7:3 chloroform–methanol resulted in the elution of compound 4 (28 mg). The structures of all compounds were elucidated by 1D and 2D NMR spectroscopy, including 1H, 13C, HSQC, and HMBC experiments, Fig. 3.
The 1H, 13C-NMR, and 2D spectra of the isolated compounds were recorded using a Bruker DRX 600 NMR spectrometer (Bruker Daltonics, Billerica, MA).
3.4. Molecular modeling
The molecular docking and shape similarity known as ROCS studies were carried out using the OpenEye Modeling software [Fast Rigid Exhaustive Docking (FRED) Receptor, version 2.2.5; OpenEye Scientific Software, SantaFe, NM (USA); http://www.eyesopen.com]. A virtual library of target compounds was used and their energies were minimized using the MMFF94 force field, followed by the generation of multi-conformers using the OMEGA application. Data were visualized by vida commands.3.5. ADME prediction
Lipinski's rule (rule of five) and molecular property prediction was calculated by the free accesses to website https://www.molsoft.com/servers.html.PreADMET estimation was determined by utilizing the free access of website https://preadmet.bmdrc.kr/.
4. Conclusion
The isolated compounds from peach fruits showed a high resemblance to drugs that prevent the multiplication of SARS-CoV-2. Although compound 1 is very similar to some of these drugs, such as fingolamide, however, it showed less number of physicochemical parameters and low drug-likeness scores. The aglycones of compounds 3 and 4 illustrated strong binding interactions with Mpro and spike protein of SARS-CoV-2. Compounds originated from flavanone core systems of 2, 4, and those from α,β-unsaturated acetophenone core architecture 3 have high similarity in their 3D structures and their predicted biological activities. This study pointed out the importance of taking peach fruits as a source of naturally occurring compounds isolated from food sources in alleviating SARS-CoV-2, and hence the advice of eating peach fruits to COVID-19 patients. Additionally, it will help us to afford semisynthetic new scaffolds with potential activities against this disease.Our outcomes prove the value of this screening study, which can lead to the rapid discovery of drugs from natural food sources and prioritize them as potential multitargets acting on pandemic COVID-19.
Conflicts of interest
We declare that we have no conflicts of interest.Acknowledgements
Dr Yaseen A. M. M. Elshaier, Organic& Medicinal Chemistry Department, Faculty of Pharmacy, University of Sadat City (USC), Egypt acknowledge OpenEye scientific software for providing the academic license. Also, he acknowledges USC (No. 15) for supporting this project.References
- L. Caly, J. D. Druce, M. G. Catton, D. A. Jans and K. M. Wagstaff, Antivir. Res., 2020, 178, 104787, DOI:10.1016/j.antiviral.2020.104787.
- S. A. Amin and T. Jha, Eur. J. Med. Chem., 2020, 201, 112559, DOI:10.1016/j.ejmech.2020.112559 .
- A. E. Gorbalenya, S. C. Baker, R. S. Baric, R. J. de Groot, C. Drosten, A. A. Gulyaeva, B. L. Haagmans, C. lauber, A. M. leontovich, B. W. Neuman, D. Penzar, S. Perlman, L. L. M. Poon, D. V. Samborskiy, I. A. Sidorov, I. Sola and J. Ziebuhr, Nat. Microbiol., 2020, 5, 536–544 CrossRef PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang and B. Cao, Lancet, 2020, 395, 497–506 CrossRef CAS.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020 DOI:10.1002/cmdc.202000223.
- S. A. Amin, K. Ghosh, S. Gayen and T. Jha, J. Biomol. Struct. Dyn., 2020, 1–10, DOI:10.1080/07391102.2020.1780946.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew and B. Ross, J. Med. Chem., 2017, 60, 1648–1661 CrossRef CAS PubMed.
- M. L. Agostini, E. L. Andres, A. C. Sims, R. L. Graham, T. P. Sheahan, X. Lu, E. C. Smith, J. B. Case, J. Y. Feng and R. Jordan, MBio, 2018, 9, e00221, DOI:10.1128/mBio.00221-18.
- M. L. Holshue, C. DeBolt, S. Lindquist, K. H. Lofy, J. Wiesman, H. Bruce, C. Spitters, K. Ericson, S. Wilkerson and A. Tural, N. Engl. J. Med., 2020, 382, 929–936 CrossRef CAS PubMed.
- M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong and G. Xiao, Cell Res, 2020, 30, 269–271 CrossRef CAS PubMed.
- Y. Furuta, T. Komeno and T. Nakamura, Proc. Jpn. Acad., Ser. B, 2017, 93, 449–463 CrossRef CAS PubMed.
- K. M. Wagstaff, H. Sivakumaran, S. M. Heaton, D. Harrich and D. A. Jans, Biochem. J., 2012, 443, 851–856 CrossRef CAS PubMed.
- S. N. Yang, S. C. Atkinson, C. Wang, A. Lee, M. A. Bogoyevitch, N. A. Borg and D. A. Jans, Antivir. Res., 2020, 177, 104760, DOI:10.1016/j.antiviral.2020.104760.
- M. Bray, C. Rayner, F. Noël, D. Jans and K. Wagstaff, Antivir. Res., 2020, 178, 104805, DOI:10.1016/j.antiviral.2020.104805.
- A. H. de Wilde, D. Jochmans, C. C. Posthuma, J. C. Zevenhoven-Dobbe, S. van Nieuwkoop, T. M. Bestebroer, B. G. van den Hoogen, J. Neyts and E. J. Snijder, Antimicrob. Agents Ch., 2014, 58, 4875–4884 CrossRef PubMed.
- J. F. W. Chan, Y. Yao, M. L. Yeung, W. Deng, L. Bao, L. Jia, F. Li, C. Xiao, H. Gao and P. Yu, J. Infect. Dis., 2015, 212, 1904–1913 CrossRef CAS PubMed.
- B. Cao, Y. Wang, D. Wen, W. Liu, J. Wang, G. Fan, L. Ruan, B. Song, Y. Cai and M. Wei, N. Engl. J. Med., 2020, 382, 1787–1799 CrossRef PubMed.
- A. Savarino, J. R. Boelaert, A. Cassone, G. Majori and R. Cauda, Lancet Infect. Dis., 2003, 3, 722–727 CrossRef CAS PubMed.
- M. J. Vincent, E. Bergeron, S. Benjannet, B. R. Erickson, P. E. Rollin, T. G. Ksiazek, N. G. Seidah and S. T. Nichol, Virol. J., 2005, 2, 1–10 CrossRef PubMed.
- P. Gautret, J. C. Lagier, P. Parola, L. Meddeb, M. Mailhe, B. Doudier, J. Courjon, V. Giordanengo, V. E. Vieira and H. T. Dupont, Int. J. Antimicrob. Ag., 2020, 105949, DOI:10.1016/j.ijantimicag.2020.105949.
- J. M. Molina, C. Delaugerre, J. Le Goff, B. Mela-Lima, D. Ponscarme, L. Goldwirt and N. de Castro, Med. Mal. Infect., 2020, 50, 30085–30088 Search PubMed.
- C. Chen, J. Huang, Z. Cheng, J. Wu, S. Chen, Y. Zhang, B. Chen, M. Lu, Y. Luo and J. Zhang, MedRxiv, 2020 DOI:10.1101/2020.03.17.20037432.
- D. B. Kell, E. L. Heyden and E. Pretorius, Front. Immunol., 2020 DOI:10.3389/fimmu.2020.01221.
- Y. N. Lamb and E. D. Deeks, Drugs, 2018, 78, 929–940 CrossRef CAS PubMed.
- A. M. Sayed, A. R. Khattab, A. M. AboulMagd, H. M. Hassan, M. E. Rateb, H. Zaid and U. R. Abdelmohsen, RSC Adv., 2020, 10, 19790–19802 RSC.
- D. Pelletier and D. A. Hafler, N. Engl. J. Med., 2012, 366, 339–347 CrossRef CAS PubMed.
- M. Kelleni, Preprints, 2020, 2020040432, DOI:10.20944/preprints202004.0432.v1.
- N. Vargesson, Birth Defects Research Part C: Embryo Today: Reviews, 2015, 105, 140–156 CrossRef CAS PubMed.
- H. Zhu, X. Shi, D. Ju, H. Huang, W. Wei and X. Dong, Inflammation, 2014, 37, 2091–2098 CrossRef CAS PubMed.
- A. O. Adeoye, B. J. Oso, I. F. Olaoye, H. Tijjani and A. I. Adebayo, J. Biomol. Struct. Dyn., 2020, 1–14, DOI:10.1080/07391102.2020.1765876.
- N. Adhikari, A. K. Halder, S. Mallick, A. Saha, K. D. Saha and T. Jha, Bioorgan. Med. Chem., 2016, 24, 4291–4309 CrossRef CAS PubMed.
- H. Zakaryan, E. Arabyan, A. Oo and K. Zandi, Arch. Virol., 2017, 162, 2539–2551 CrossRef CAS PubMed.
- G. Noratto, W. Porter, D. Byrne and L. Cisneros-Zevallos, J. Agric. Food Chem., 2009, 57, 5219–5226 CrossRef CAS PubMed.
- T. Nakagawa, A. E. Allam, K. Ohnuki and K. Shimizu, Nat. Prod. Commun., 2018, 13, 1293–1296 CrossRef.
- S. X. Wang, Y. Wang, Y. B. Lu, J. Y. Li, Y. J. Song, M. Nyamgerelt and X. X. Wang, J. Integr. Med., 2020 DOI:10.1016/j.joim.2020.04.001.
- G. C. L. Ee, C. K. Lim, Y. H. Taufiq-Yap and R. Go, Malays. J. Chem., 2005, 7, 45–48 Search PubMed.
- K. Patel, G. K. Singh and D. K. Patel, Chin. J. Integr. Med., 2018, 24, 551–560 CrossRef CAS PubMed.
- K. R. Markham, Techniques of flavonoid identification, Academic press London, 1982 Search PubMed.
- E. A. Ragab, M. Hosny, H. A. Kadry and H. A. Ammar, J. Nat. Prod., 2010, 3, 35–46 CAS.
- C. Ganbaatar, M. Gruner, D. Mishig, R. Duger, A. W. Schmidt and H. J. Knölker, Nat. Prod. J., 2015, 8, 1–7 CrossRef CAS.
- A. B. Nouga, J. C. Ndom, E. M. Mpondo, J. C. N. Nyobe, A. Njoya, L. M. Meva'a, P. B. Cranwell, J. A. Howell, L. M. Harwood and J. D. Wansi, Nat. Prod. Res., 2016, 30, 305–310 CrossRef CAS PubMed.
- S. Diao, M. Jin, C. S. Jin, C. X. Wei, J. Sun, W. Zhou and G. Li, Nat. Prod. Res., 2019, 33, 3021–3024 CrossRef CAS PubMed.
- N. Sultana, K. Akter, N. Nahar, M. S. H. Khan, M. Mosihuzzaman, M. H. Sohrab and K. Krohn, Nat. Prod. Res., 2010, 24, 1018–1026 CrossRef CAS PubMed.
- H. L. Li, J. Tang, R. H. Liu, C. Zhang and W. D. Zhang, Nat. Prod. Res., 2009, 23, 122–126 CrossRef CAS PubMed.
- T. H. Kang, S. J. Jeong, W. G. Ko, N. Y. Kim, B. H. Lee, M. Inagaki, T. Miyamoto, R. Higuchi and Y. C. Kim, J. Nat. Prod., 2000, 63, 680–681 CrossRef CAS PubMed.
- V. Singh, B. Yadav and V. Pandey, Phytochemistry, 1999, 51, 587–590 CrossRef CAS PubMed.
- P. K. Panda, M. N. Arul, P. Patel, S. K. Verma, W. Luo, H. G. Rubahn, Y. K. Mishra, M. Suar and R. Ahuja, Sci. Adv., 2020 DOI:10.1126/sciadv.abb8097.
- A. K. Ghosh, J. Takayama, Y. Aubin, K. Ratia, R. Chaudhuri, Y. Baez, K. Sleeman, M. Coughlin, D. B. Nichols, D. C. Mulhearn, B. S. Prabhakar, S. C. Baker, M. E. Johnson and A. D. Mesecar, J. Med. Chem., 2009, 52, 5228–5240 CrossRef CAS PubMed.
- M. F. Sk, R. Roy, N. A. Jonniya, S. Poddar and P. Kar, J. Biomol. Struct. Dyn., 2020 DOI:10.1080/07391102.2020.1768149.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang and C. Peng, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CAS.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Grahamand and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- T. T. H. Nguyen, H. J. Woo, H. K. Kang, Y. M. Kim, D. W. Kim, S. A. Ahn, Y. Xia and D. Kim, Biotechnol. Lett., 2012, 34, 831–838 CrossRef CAS PubMed.
- Y. A. Vladimirov, E. Proskurnina, E. Demin, N. Matveeva, O. Lubitskiy, A. Novikov, D. Y. Izmailov, A. Osipov, V. Tikhonov and V. Kagan, Biochemistry (Moscow), 2009, 74, 301–307 CrossRef CAS PubMed.
- A. Fischer, M. Sellner, S. Neranjan, M. Smiesko and M. A. Lill, Int. J. Mol. Sci., 2020, 21, 3626, DOI:10.3390/ijms21103626.
- P. C. Hawkins, A. G. Skillman and A. Nicholls, J. Med. Chem., 2007, 50, 74–82 CrossRef CAS PubMed.
- K. R. Abdellatif, W. A. Fadaly, G. M. Kamel, Y. A. Elshaier and M. A. El-Magd, Bioorg. Chem., 2019, 82, 86–99 CrossRef CAS PubMed.
- M. A. Elbastawesy, Y. A. El-Shaier, M. Ramadan, A. B. Brown, A. A. Aly and G. E.-D. A. Abuo-Rahma, Mol. Divers., 2020 DOI:10.1007/s11030-019-10021-0.
- M. S. Islam, A. M. Al-Majid, F. F. El-Senduny, F. A. Badria, A. Rahman, A. Barakat and Y. A. Elshaier, Appl. Sci., 2020, 10, 2170, DOI:10.3390/app10062170.
- T. J. Ritchie and S. J. Macdonald, Drug Discov. Today, 2014, 19, 489–495 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05265e |
| This journal is © The Royal Society of Chemistry 2020 |