
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile deprotection of F-BODIPYs using methylboronic acid†
Craig D. Smith and
Alison Thompson *
*
Department of Chemistry, Dalhousie University, PO BOX 15000, Halifax, NS B3H 4R2, Canada. E-mail: Alison.Thompson@dal.ca
First published on 25th June 2020
Abstract
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacenes (F-BODIPYs) are deprotected through removal of the –BF2 moiety upon treatment with methylboronic acid. The tolerance of various substitution patterns about the dipyrrinato core is demonstrated via the deprotection of thirteen F-BODIPYs and an F-aza-BODIPY. Work-up with aq. HBr affords the desired dipyrin HBr salt in quantitative yield without need for purification.
Introduction
The dipyrrinato framework is a useful ligand for complexation to metals, the most common featuring first-row transition metals and boron.1–4 The photophysical characteristics of –BF2 complexes of dipyrrins, known widely as 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (F-BODIPYs), have enabled applications across many of the chemical sciences.5–8 For example, F-BODIPYs have been used as dyes to label proteins,9 DNA10 and functional materials.11 Furthermore, the potential of F-BODIPYs for use in photo-assisted rechargeable batteries has been investigated.12 Recently, efforts have focused on substituting the boron centre of BODIPYs with alkyl, alkynyl and chloro substituents in order to tune spectroscopic properties and thus potentially extend the performance of these conjugated chemical species.13–25Parallel to the photophysical and spectroscopic advances that have involved BODIPYs, dipyrrins have emerged as capable of providing a useful structural framework for incorporation into catalysts,26–29 supramolecular assembles,30,31 and polypyrroles with desirable biological activity.32–35 As such, there is a desire to substitute, functionalise and manipulate dipyrrins, en route to accessing the dipyrrinato ligand. Given the broad range of strategies by which to functionalise F-BODIPYs, as well as the facile manner by which to purify them using chromatography, the use of the –BF2 moiety has emerged as a useful approach to the protection of dipyrrins.36–44 Reported methods for the removal of the –BF2 moiety from F-BODIPYs, i.e. deprotection to yield the parent dipyrrin, have involved the use of alkoxides and strong Brønstead and Lewis acids. Herein, we report using methylboronic acid to deprotect thirteen F-BODIPYs and an F-aza-BODIPY to generate the corresponding dipyrrins and aza-dipyrrin in quantitative yield.
Results and discussion
Upon treating an F-BODIPY with methyl boronic acid, with anticipation that an appended boronic ester would be hydrolysed,45 a dipyrrin was isolated as a result of removal of the –BF2 unit. Cognisant that these conditions represent a mild alternative to existing methods for the deprotection of F-BODIPYs, and in light of the fact that the volatility of methylboronic acid renders work-up facile, we investigated the scope of the reaction conditions. Treatment of 4,4-difluoro-8-phenyl-4-bora-3a,4a-diaza-s-indacene (1a) with 5 equiv. methylboronic acid at room temperature (Table 1) gave clean conversion, after 24 hours, to provide the corresponding dipyrrin 2a in quantitative yield (entry 1). The incorporation of alkyl groups about the pyrrolic units was well tolerated, as was a methyl substituent in the meso-position (entries 2 and 3, respectively). Work-up using aqueous HBr provided the corresponding dipyrrin salts which are known to be more stable and more crystalline than their HX counterparts, an observation of significant value when storing meso-unsubstituted dipyrrins.4 Using these reaction conditions provided the meso-unsubstituted dipyrrin salts 2d and 2e in quantitative yield (entries 4 and 5, respectively). The preparation of the HBr salt of 2d was equally successful on a one-gram scale (entry 4). Furthermore, the corresponding free-base was isolated in quantitative yield, again on a one-gram scale, upon implementation of a basic work-up procedure. The removal of the –BF2 unit using methylboronic acid was effective for F-BODIPYs bearing unsubstituted positions about the pyrrolic core (entry 6), as well as for unsymmetrical variants (entry 7). Given the previously documented sternutatory properties of the free base of 2f,39 an alternative work-up procedure was implemented so as to ensure successful formation of the corresponding HBr salt. Dipyrrin 2g was also isolated in quantitative yield upon treatment of 4,4-difluoro-1,3-dimethyl-2-ethyl-6-pinacolatoboron-8-H-4-bora-3a,4a-diaza-s-indacene with 5 equiv. methylboronic acid, thereby effecting both deborylation (of the BPin moiety) as well as removal of the –BF2 unit. The new method for the deprotection of F-BODIPYs proceeded in the presence of bromo-substituents, as exemplified by the isolation of quantitative yields of the dipyrrins 2h and 2i (entries 8 and 9). Similarly, the dipyrrin 2j was isolated in quantitative yield upon treatment of the corresponding F-BODIPY with methylboronic acid, thereby demonstrating tolerance of the hydroxy and trifluoromethyl functionalities within these electron-poor constructs. Further demonstration of the ability of methylboronic acid to deprotect F-BODIPYs bearing aryl substituents involved isolation of the two triphenyl-substituted dipyrrins 2k and 2l in quantitative yields (entries 11 and 12, respectively), with 2l being isolated as its free-base on account of the rather poor solubility of the corresponding HBr salt. Expanding the scope further, an aza-F-BODIPY i.e. a bridging nitrogen between the two pyrrolic sub-units, was successfully deprotected to provide the aza-dipyrrin 2m in quantitative yield (entry 13).
|
|||
|---|---|---|---|
| Entry | Product dipyrrin salt yield (%) | Entry | Product dipyrrin salt yield (%) |
| a Work-up A involved quenching with 2 M aq. HBr.b Work-up B involved quenching with 1 M aq. HBr.c Work-up C involved quenching with sat. aq. NaHCO3.d Work-up involved quenching with 2 M aq. HBr performed on a gram scale.e Work-up quenching with sat. aq. NaHCO3 performed on a gram scale.f Using work-up A, dipyrrin 2g was prepared upon treatment of 4,4-difluoro-1,3-dimethyl-2-ethyl-6-pinacolatoboron-8-H-4-bora-3a,4a-diaza-s-indacene with 5 equiv. methylboronic thereby effecting both deborylation (of the BPin moiety) in addition to removal of the –BF2 unit. | |||
| 1 |  |
8 |  |
| 2 |  |
9 |  |
| 3 | 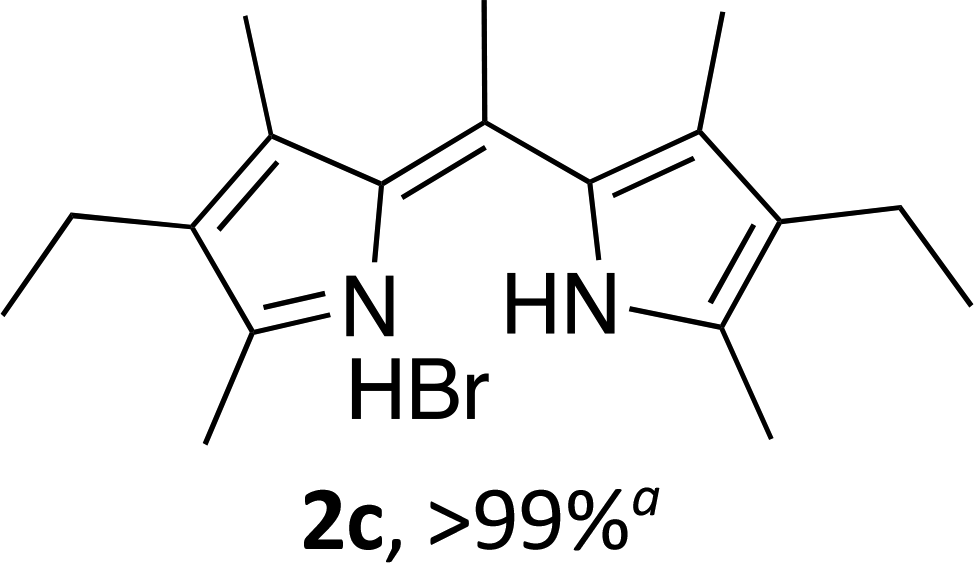 |
10 |  |
| 4 |  |
11 |  |
| 5 |  |
12 |  |
| 6 |  |
13 |  |
| 7 |  |
||

Attempts to reduce the equivalencies of the Lewis acid were briefly explored, resulting in the observation that 2d could be generated in quantitative yield upon treatment of the corresponding F-BODIPY with just 1 equiv. methylboronic acid. However, this achievement did not extend to the deprotection of F-BODIPYs appended with meso-aryl substituents. Despite the successful deprotection of thirteen F-BODIPYs and one F-aza-BODIPY using 5 equiv. methylboronic acid, the bromo-substituted F-BODIPY 3 (Fig. 1) reacted sluggishly to result in decomposition. Equally unsuccessful were attempts to use methylboronic acid to remove the –BF2 unit from F-BODIPYs bearing carboxylate- or azido- functionality around the dipyrrolic core.
 | ||
| Fig. 1 Bromo-substituted F-BODIPY. | ||
Conclusion
In conclusion, we have developed a high yielding and mild method for the deprotection of F-BODIPYs through the use of volatile methylboronic acid using 5% TFA and CH2Cl2 as solvent. The reaction is performed at room temperature, tolerates substrates bearing a range of substituents around the F-BODIPY framework, and requires only a facile work-up procedure in order to isolate quantitative yields of the dipyrrin as either an HBr salt or a free-base.Experimental section
General information
Reactions involving air-sensitive reagents and dry solvents were performed using glassware that had been oven-dried (150 °C) or flame-dried prior to use. NMR spectra were recorded using a 500 MHz spectrometer. 1H chemical shifts are reported in ppm relative to tetramethylsilane using the solvent residual as an internal standard (δ = 7.26 for CDCl3; 5.32 for CD2Cl2; 2.05 for acetone-d6; 2.50 for DMSO-d6) 13C NMR chemical shifts are reported in ppm using the residual solvent as an internal standard (δ = 77.2 for CDCl3; 53.8 for CD2Cl2; 29.8 for acetone-d6; 39.5 for DMSO-d6). Signals in NMR spectra are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (br.), apparent (app.) or a combination of these, referring to the observed spin–spin coupling pattern. Spin–spin coupling constants are reported in hertz (Hz), and are uncorrected. Mass spectrometry was performed using a TOF spectrometer operating in ESI+ or APCI mode, as indicated. Flash chromatography was performed using forced air flow of the indicated solvent system using silica gel 60 as solid support. Reactions were monitored by thin layer chromatography (TLC) on silica gel 60-covered aluminium sheets. TLC plates were developed under UV-light and/or with an acidic ethanolic anisaldehyde solution, vanillin or a KMnO4-solution upon heating. All reagents were purchased from commercial suppliers and used without further purification unless otherwise stated. F-BODIPYs 1a,1 1b,2 1c,3 1d,4 1f,5 1g,6 2h,1 2j![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and 2k8 were prepared according to literature procedures, as was F-aza-BODIPY 1m.9
7 and 2k8 were prepared according to literature procedures, as was F-aza-BODIPY 1m.9
(Z)-4-Bromo-2-((4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide
Following a modified literature procedure,10 to a round-bottom flask was added 3-ethyl-2,4-dimethyl-pyrrole (1.73 g, 14.1 mmol) and 4-bromo-pyrrole-2-carbaldehyde (2.44 g, 14.1 mmol). The pyrroles were dissolved in THF:MeOH (1:1; 85 mL), and the resulting solution was purged with nitrogen before 48% aq. HBr (3.2 mL, 28 mmol) was added drop-wise to the solution. The reaction mixture was stirred at 70 °C for 2 hours, monitoring by TLC analysis until complete consumption of starting material was observed. The reaction mixture was then poured into diethyl ether (100 mL) to induce precipitation. The resulting solid was collected by suction filtration, and dried to afford the title compound as a brown solid (3.60 g, 71%). 1H NMR (500 MHz; CDCl3) δ (ppm): 13.67 (s, 1H), 13.42 (s, 1H), 7.49 (s, 1H), 7.13 (s, 1H), 7.04 (s, 1H), 2.75 (s, 3H), 2.45 (q, J = 7.6 Hz, 2H), 2.30 (s, 3H), 1.09 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ (ppm): 164.9, 146.4, 135.6, 134.7, 130.7, 130.7, 130.3, 127.0, 101.8, 17.4, 14.1, 13.8, 10.4; HRMS-ESI (m/z): [M + H]+ calcd for C13H16BN2, 279.0491; found, 279.0497.
4,4-Difluoro-1,3-dimethyl-2-ethyl-6-bromo-8-H-4-bora-3a,4a-diaza-s-indacene
Following a literature procedure,4 to a solution of (Z)-4-bromo-2-((4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide (3.60 g, 10.0 mmol) in CH2Cl2 (180 mL) under air with stirring at room temperature, was added NEt3 (8.4 mL, 60 mmol) and the resulting solution stirred for 10 minutes. Anhydrous BF3·OEt2 (11 mL, 90 mmol) was then added and the reaction mixture was sealed with a septum and stirred for 1 hour. The septum was then removed, and another aliquot of NEt3 (8.4 mL, 60 mmol) was added. The vessel was resealed and stirred for 10 minutes, after which the septum was again removed and another aliquot of anhydrous BF3·OEt2 (11.1 mL, 90 mmol) was added. The resulting solution was sealed again and stirred for another 1 hour. The reaction mixture was concentrated in vacuo to yield the crude product, which was dissolved in ether (60 mL). The resulting solution washed with 1 M aq. hydrochloric acid (4× 50 mL) and 5 M aq. hydrochloric acid (1× 50 mL). The organic fraction was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude material was purified via column chromatography on silica (hexanes/CH2Cl2; 1:1) to afford the title compound as a red solid (2.30 g, 62%). 1H NMR (500 MHz; CDCl3) δ (ppm): 7.44 (s, 1H), 7.02 (s, 1H), 6.77 (s, 1H), 2.58 (s, 3H), 2.41 (q, J = 7.6 Hz, 2H), 2.19 (s, 3H), 1.09 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ (ppm): 166.2, 142.3, 137.2, 136.1, 136.0, 131.8, 124.4, 122.6, 102.7, 17.4, 14.2, 13.7, 9.7; 11B NMR (160 MHz; CDCl3) δ (ppm): 0.3 (t, J = 31 Hz); 19F NMR (470 MHz; CDCl3) δ (ppm): −146.1 (q, J = 31 Hz); HRMS-APCI (m/z): [M + H]+ calcd for C13H15BBrF2N2, 327.0474; found, 327.0478.
4,4-Difluoro-1,3-dimethyl-2-ethyl-6-pinacolatoboron-8-H-4-bora-3a,4a-diaza-s-indacene
A 4 mL screw-top vial was charged with 4,4-difluoro-1,3-dimethyl-2-ethyl-6-bromo-8-H-4-bora-3a,4a-diaza-s-indacene (163 mg, 0.5 mmol), Pd2(dba)3 (5 mg, 5 μmol), XPhos (9 mg, 20 μmol), bis(pinacolato)diboron (380 mg, 1.50 mmol) and oven dried KOAc (147 mg, 1.50 mmol). The vial was capped with a septum and then evacuated and backfilled with N2 (this sequence was carried out two times). 1,4-Dioxane (3 mL) was added via syringe through the septum. Under a flow of nitrogen, the septum was replaced with a screw cap. The reaction mixture was stirred at 110 °C in a preheated aluminium block until consumption of the F-BODIPY starting material was complete, as judged by TLC analysis (approximately 20 minutes). The reaction mixture was allowed to cool to room temperature and then filtered through a thin pad of Celite, eluting with CH2Cl2. The crude material was purified via rapid chromatography on silica (hexanes:EtOAc; 8:2), with minimal residence time ensured in order to minimise decomposition over the solid phase. The product was crystallised from hexanes to give the title compound as a bench- and air-stable brown solid (113 mg, 60%). 1H NMR (500 MHz; CDCl3) δ (ppm): 7.96 (s, 1H), 7.22 (s, 1H), 7.17 (s, 1H), 2.61 (s, 3H), 2.45 (q, J = 7.6 Hz, 2H), 2.24 (s, 3H), 1.36 (s, 12H), 1.12 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ (ppm): 164.2, 145.2, 141.4, 136.9, 135.5, 133.5, 131.8, 123.6, 83.5, 25.0, 17.5, 14.3, 13.5, 9.7, 1× Ar C signal missing; 11B NMR (160 MHz; CDCl3) δ (ppm): 30.4 (br. s) 0.6 (t, J = 32 Hz); 19F NMR (470 MHz; CDCl3) δ (ppm): −146.0 (q, J = 31 Hz); HRMS-APCI (m/z): [M + H]+ calcd for C19H27B2F2N2O2, 375.2221; found, 375.2234.
4,4-Difluoro-1,3,5,7-tetramethyl-2-ethyl-6-bromo-8-H-4-bora-3a,4a-diaza-s-indacene (1i)
To a 100 mL round-bottom flask, charged with a magnetic stirrer bar, was added 4,4-difluoro-1,3,5,7-tetramethyl-2-ethyl-8-H-4-bora-3a,4a-diaza-s-indacene11 (730 mg, 2.64 mmol) and CH2Cl2 (37 mL). To the solution was added N-bromosuccinimide (469 mg, 2.64 mmol). The reaction mixture was stirred at room temperature for 1 hour, then quenched with H2O (30 mL) and extracted with CH2Cl2 (3× 50 mL). The combined organic fractions were dried over Na2SO4, filtered and concentrated in vacuo. The crude material was purified via column chromatography on silica (hexanes/CH2Cl2; 8:2) to afford the title compound as a red solid (906 mg, 97%). 1H NMR (500 MHz; CDCl3) δ (ppm): 6.98 (s, 1H), 2.53 (s, 3H), 2.52 (s, 3H), 2.39 (q, J = 7.6 Hz, 2H), 2.19 (s, 3H), 2.17 (s, 3H), 1.07 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ (ppm): 159.5, 150.8, 139.3, 136.3, 134.1, 133.7, 130.6, 119.5, 107.6, 17.4, 14.5, 13.3, 13.1, 11.0, 9.6; 11B NMR (160 MHz; CDCl3) δ (ppm): 0.7 (t, J = 33 Hz); 19F NMR (470 MHz; CDCl3) δ (ppm): −146.4 (q, J = 33 Hz); HRMS-APCI (m/z): [M + H]+ calcd for C15H19BBrF2N2, 355.0787; found, 355.0781.
General procedure (GP) for the deprotection of F-BODIPYs using MeB(OH)2
A 4 mL screw-top vial was charged with F-BODIPY (0.2 mmol) and MeB(OH)2 (1.0 mmol). To this was added CH2Cl2 (1.90 mL) and trifluoroacetic acid (0.10 mL). The vial was capped with a screw cap and the mixture was stirred until F-BODIPY had been completely consumed as judged by analysis using TLC (typically stirred for 24 hours).(Z)-2-(Phenyl(2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide (2a)
Following the GP and work-up A, using 4,4-difluoro-8-phenyl-4-bora-3a,4a-diaza-s-indacene (1a),6 provided the title compound as a brown solid (60 mg, >99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 13.43 (br. s, 2H), 8.09 (br. s, 2H), 7.64 (s, 1H), 7.51–7.45 (m, 4H), 6.81 (s, 2H), 6.63 (s, 2H); 13C NMR (125 MHz; CDCl3) δ (ppm): 149.9, 143.9, 136.7, 133.9, 132.1, 131.6, 131.3, 128.2, 117.5; HRMS-ESI (m/z): [M + H]+ calcd for C15H13N2, 221.1073; found, 221.1073.(Z)-3-Ethyl-5-((4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)(phenyl)methyl)-2,4-dimethyl-1H-pyrrole hydrobromide (2b)
Following the GP and work-up A, using 4,4-difluoro-1,3,5,7-tetramethyl-2,6-diethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (1b),2 provided the title compound as a red solid (82 mg, > 99%).1H NMR (500 MHz; CDCl3) δ (ppm): 11.01 (br. s, 2H), 7.59 (t, J = 7.3 Hz, 1H), 7.48 (t, J = 7.3 Hz, 2H), 7.30 (d, J = 7.4 Hz, 2H), 2.70 (br. s, 6H), 2.41–2.35 (m, 4H), 1.36 (br. s, 6H), 1.06–1.03 (m, 6H); 13C NMR (125 MHz; CDCl3) δ (ppm): 152.4, 144.2, 140.1, 137.1, 133.7, 131.8, 128.7, 129.0, 17.5, 14.5, 13.7, 12.3, 1× aryl signal missing; HRMS-ESI (m/z): [M + H]+ calcd for C23H29N2, 333.2325; found, 333.2323.(Z)-3-Ethyl-5-(1-(4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)ethyl)-2,4-dimethyl-1H-pyrrole hydrobromide (2c)
Following the GP and work-up A, using 4,4-difluoro-1,3,5,7,8-pentamethyl-2,6-diethyl-4-bora-3a,4a-diaza-s-indacene (1c),3 provided the title compound as a brown solid (70 mg, > 99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 11.82 (br. s, 2H), 2.87 (s, 3H), 2.54 (s, 6H), 2.39 (q, J = 7.4 Hz, 4H), 2.05 (s, 6H), 1.05 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz; CDCl3) δ (ppm): 149.2, 146.7, 137.1, 132.2, 131.5, 25.0, 17.6, 14.5, 12.7, 12.4. Data correspond to literature values.12(Z)-3-Ethyl-5-((4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-2,4-dimethyl-1H-pyrrole hydrobromide (2d)
Following the GP and work-up A, using 4,4-difluoro-1,3,5,7-tetramethyl-2,6-diethyl-4-bora-3a,4a-diaza-s-indacene (1d),4 provided the title compound as a brown solid (67 mg, > 99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 12.87 (br. s, 2H), 7.01 (s, 1H), 2.64 (s, 6H), 2.40 (q, J = 7.6 Hz, 4H), 2.25 (s, 6H), 1.05 (t, J = 7.6 Hz, 6H); 13C NMR (125 MHz; CDCl3) δ (ppm): 153.7, 141.3, 130.6, 126.1, 118.7, 17.3, 14.5, 12.8, 10.1. Data correspond to literature values.13(Z)-3-(3,3-Dimethylbutyl)-5-((4-(3,3-dimethylbutyl)-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-2,4-dimethyl-1H-pyrrole hydrobromide (2e)
Following the GP and work-up A, using, 4-difluoro-1,3,5,7-tetramethyl-2,6-(3,3-dimethylbutyl)-4-bora-3a,4a-diaza-s-indacene (1e), provided the title compound as a red solid (90 mg, > 99%). 1H NMR (500 MHz; CD2Cl2) δ (ppm): 12.92 (br. s, 2H), 7.06 (s, 1H), 2.59 (s, 6H), 2.36–2.33 (m, 4H), 2.27 (s, 6H), 1.28–1.25 (m, 4H), 0.97 (s, 18H); 13C NMR (125 MHz; CD2Cl2) δ (ppm): 153.4, 141.5, 129.5, 126.0, 118.7, 44.0, 30.3, 28.8, 19.1, 12.5, 10.3; HRMS-ESI (m/z): [M + H]+ calcd for C25H41N2, 369.3264; found, 369.3278.(Z)-2-((3,5-Dimethyl-2H-pyrrol-2-ylidene)methyl)-3,5-dimethyl-1H-pyrrole hydrobromide (2f)
Following the GP and work-up B, using 4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene (1f),2 provided the title compound as a red solid (56 mg, > 99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 13.06 (br. s, 2H), 7.07 (s, 1H), 6.14 (s, 2H), 2.66 (s, 6H), 2.34 (s, 6H); 13C NMR (125 MHz; CDCl3) δ (ppm): 155.6, 146.1, 126.8, 120.1, 117.6, 14.6, 12.3. Data correspond to literature values.14 Caution: the free-base of 2f has been reported to exhibit sternutatory properties.7(Z)-2-((4-Ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide (2g)
Following the GP and work-up A, using 4,4-difluoro-1,3-dimethyl-2-ethyl-4-bora-3a,4a-diaza-s-indacene (1g),15 provided the title compound as a red solid (56 mg, > 99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 13.50 (br. s, 1H), 13.31 (br. s, 1H), 7.65 (s, 1H), 7.21 (s, 1H), 7.09 (s, 1H), 6.47 (s, 1H), 2.73 (s, 3H), 2.43 (q, J = 7.5 Hz, 2H), 2.29 (s, 3H), 1.08 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ (ppm): 162.0, 145.4, 137.6, 133.6, 132.1, 129.3, 127.3, 124.8, 114.6, 17.3, 14.2, 13.6, 10.3. Data correspond to literature values.10Alternatively, following the GP and work-up A using 4,4-difluoro-1,3-dimethyl-2-ethyl-6-pinacolatoboron-8-H-4-bora-3a,4a-diaza-s-indacene provided the title compound as a red solid (56 mg, > 99%). Data as above.
(Z)-2-((4-Bromophenyl)(2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide (2h)
Following the GP and work-up A, using 4,4-difluoro-8-(phenyl(4-bromo))-4-bora-3a,4a-diaza-s-indacene (1h),7 provided the title compound as a red solid (76 mg, > 99%).1H NMR (500 MHz; CDCl3) δ (ppm): 13.48 (br. s, 2H), 8.10 (br. s, 2H), 7.66 (d, J = 6.3 Hz, 2H), 7.29 (br. s, 2H), 6.81 (br. s, 2H), 6.65 (s, 2H); 13C NMR (125 MHz; CDCl3) δ (ppm): 148.2, 144.4, 136.3, 135.3, 133.5, 131.6, 131.0, 126.6, 117.8.(Z)-3-Bromo-5-((4-ethyl-3,5-dimethyl-2H-pyrrol-2-ylidene)methyl)-2,4-dimethyl-1H-pyrrole hydrobromide (2i)
Following the GP and work-up B, using 4,4-difluoro-1,3,5,7-tetramethyl-2-ethyl-6-bromo-8-H-4-bora-3a,4a-diaza-s-indacene (1i), provided the title compound as a red solid (78 mg, > 99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 13.20 (br. s, 2H), 7.08 (s, 1H), 2.67 (s, 3H), 2.64 (s, 3H), 2.42 (q, J = 7.6 Hz, 2H), 2.32 (s, 3H), 2.29 (s, 3H), 1.07 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz; CDCl3) δ 158.2, 150.3, 143.7, 141.1, 132.3, 127.4, 124.8, 119.8, 106.6, 17.4, 14.3, 13.4, 13.2, 11.9, 10.3; HRMS-ESI (m/z): [M + H]+ calcd for C15H20BrN2, 307.0804; found, 307.0803.(Z)-1-(2-((4-Ethyl-3,5-dimethyl-1H-pyrrol-2-yl)methylene)-3,5-dimethyl-2H-pyrrol-4-yl)-2,2,2-trifluoroethanol hydrobromide (2j)
Following the GP and work-up B, using 4,4-difluoro-1,3,5,7-tetramethyl-2-trifluoroethanol-6-ethyl-4-bora-3a,4a-diaza-s-indacene (1j),7 provided the title compound as a red solid (81 mg, > 99%). 1H NMR (500 MHz; acetone-d6) δ (ppm): 13.75 (br. s, 1H), 13.53 (br. s, 1H), 7.44 (s, 1H), 5.86 (d, J = 5.0 Hz, 1H), 5.39–5.33 (m, 1H), 2.72 (s, 3H), 2.69 (s, 3H), 2.51 (q, J = 7.5 Hz, 2H), 2.46 (s, 3H), 2.39 (s, 3H), 1.11 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz; acetone-d6) δ (ppm): 158.5, 152.5, 145.0, 144.0, 132.8, 128.1, 126.4 (q, J = 281 Hz), 125.9, 121.4, 110.8, 66.8 (q, J = 33 Hz), 17.6, 14.5, 13.3, 12.8, 10.6, 10.1; 19F NMR (470 MHz; acetone-d6) δ (ppm): −78.4 (d, J = 7.6 Hz). Data correspond to literature values.16(Z)-2-Phenyl-5-(phenyl(5-phenyl-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole hydrobromide (2k)
Following the GP and work-up C, using 4,4-difluoro-2,5,8-triphenyl-4-bora-3a,4a-diaza-s-indacene (1k),8 provided the title compound as a red solid (90 mg, > 99%). 1H NMR (500 MHz; acetone-d6) δ (ppm): 13.05 (br. s, 2H), 8.36–8.29 (m, 4H), 7.74 (t, J = 7.2 Hz, 1H), 7.62 (t, J = 7.6 Hz, 2H), 7.57 (d, J = 7.4 Hz, 2H), 7.53–7.50 (m, 6H), 7.33 (s, 2H), 7.20–6.90 (m, 2H); 13C NMR (125 MHz; acetone-d6) δ (ppm): 154.6, 147.2, 137.8, 135.8, 135.1, 134.8, 133.5, 131.7, 130.0, 129.8, 129.5, 129.2, 117.9; HRMS-ESI (m/z): [M + H]+ calcd for C27H21N2, 373.1699; found, 373.1702.(Z)-4-Phenyl-2-(phenyl(4-phenyl-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole (2l)
Following the GP and work-up C, using 4,4-difluoro-3,6,8-triphenyl-4-bora-3a,4a-diaza-s-indacene (1l), provided the title compound as a dark blue solid (74 mg, >99%). 1H NMR (500 MHz; DMSO-d6) δ (ppm): 8.28 (s, 2H), 7.62–7.57 (m, 9H), 7.36–7.33 (m, 4H), 7.23–7.20 (m, 2H), 6.76 (s, 2H); HRMS-ESI (m/z): [M + H]+ calcd for C27H21N2, 373.1699; found, 373.1699. The title compound was insufficiently soluble to enable the acquisition of 13C NMR data.(Z)-N-(3,5-Diphenyl-2H-pyrrol-2-ylidene)-3,5-diphenyl-1H-pyrrol-2-amine (2m)
Following the GP and work-up C, using 4,4-difluoro-1,3,5,7-tetraphenyl-4-bora-3a,4a,8-triaza-s-indacene (1m),9 provided the title compound as a dark blue solid (90 mg, >99%). 1H NMR (500 MHz; CDCl3) δ (ppm): 8.07 (d, J = 7.3 Hz, 4H), 7.96 (d, J = 7.4 Hz, 4H), 7.54 (t, J = 7.5 Hz, 4H), 7.48 (d, J = 7.4 Hz, 2H), 7.43 (t, J = 7.5 Hz, 4H), 7.36 (t, J = 7.3 Hz, 2H), 7.21 (s, 2H); 13C NMR (125 MHz; CDCl3) δ (ppm): 155.3, 149.8, 142.8, 133.9, 132.4, 130.2, 129.3, 129.2, 128.4, 128.2, 126.7, 115.1. Data correspond to literature values.17Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support for this work was provided by NSERC of Canada via a Discovery Grant and the CREATE Training Program in BioActives (510963). Dr Mike Lumsden and Mr Xiao Feng (both at Dalhousie University) are thanked for sharing their expertise in NMR spectroscopy and mass spectrometry, respectively.References
- R. M. Diaz-Rodriguez, K. N. Robertson and A. Thompson, Chem. Commun., 2018, 54, 13139–13142 RSC.
- R. M. Diaz-Rodriguez, K. N. Robertson and A. Thompson, Dalton Trans., 2019, 48, 7546–7550 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS PubMed.
- M. Benstead, G. H. Mehl and R. W. Boyle, Tetrahedron, 2011, 67, 3573–3601 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- M.-c. Yee, S. C. Fas, M. M. Stohlmeyer, T. J. Wandless and K. A. Cimprich, J. Biol. Chem., 2005, 280, 29053 CrossRef CAS PubMed.
- M. L. Metzker, J. Lu and R. A. Gibbs, Science, 1996, 271, 1420–1422 CrossRef CAS PubMed.
- S. Koleman, O. A. Bozdemir, Y. Cakmak, G. Barin, S. Erten-Ela, M. Marszalek, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Gratzel and E. U. Akkaya, Chem. Sci., 2011, 2, 949–954 RSC.
- T. Godet-Bar, J. C. Leprêtre, P. Poizot, F. Massuyeau, E. Faulques, A. Christen, F. Minassian, J. F. Poisson, F. Loiseau and F. Lafolet, J. Mater. Chem. A, 2017, 5, 1902–1905 RSC.
- L. H. Davies, B. Stewart, R. W. Harrington, W. Clegg and L. J. Higham, Angew. Chem., Int. Ed., 2012, 51, 4921–4924 CrossRef CAS.
- P. Hewavitharanage, P. Nzeata and J. Wiggins, Eur. J. Chem., 2012, 3, 13–16 CrossRef CAS.
- Y. Kubota, J. Uehara, K. Funabiki, M. Ebihara and M. Matsui, Tetrahedron Lett., 2010, 51, 6195–6198 CrossRef CAS.
- K.-M. Liu, M.-S. Tsai, M.-S. Jan, C.-M. Chau and W.-J. Wang, Tetrahedron, 2011, 67, 7919–7922 CrossRef CAS.
- C. Goze, G. Ulrich, L. J. Mallon, B. D. Allen, A. Harriman and R. Ziessel, J. Am. Chem. Soc., 2006, 128, 10231–10239 CrossRef CAS PubMed.
- G. Ulrich and R. Ziessel, J. Org. Chem., 2004, 69, 2070–2083 CrossRef CAS PubMed.
- A. Harriman, G. Izzet and R. Ziessel, J. Am. Chem. Soc., 2006, 128, 10868–10875 CrossRef CAS PubMed.
- Y. Gabe, T. Ueno, Y. Urano, H. Kojima and T. Nagano, Anal. Bioanal. Chem., 2006, 386, 621–626 CrossRef CAS PubMed.
- S. Liu, T.-P. Lin, D. Li, L. Leamer, H. Shan, Z. Li, F. P. Gabbaï and P. S. Conti, Theranostics, 2013, 3, 181–189 CrossRef CAS PubMed.
- Y. Kubo, Y. Minowa, T. Shoda and K. Takeshita, Tetrahedron Lett., 2010, 51, 1600–1602 CrossRef CAS.
- C. Tahtaoui, C. Thomas, F. Rohmer, P. Klotz, G. Duportail, Y. Mely, D. Bonnet and M. Hibert, J. Org. Chem., 2007, 72, 269–272 CrossRef CAS PubMed.
- C. Bonnier, W. E. Piers, A. Al-Sheikh Ali, A. Thompson and M. Parvez, Organometallics, 2009, 28, 4845–4851 CrossRef CAS.
- C. Bonnier, W. E. Piers, M. Parvez and T. S. Sorensen, Chem. Commun., 2008, 4593–4595 RSC.
- M. Yadav, A. K. Ashish Kumar Singh and D. S. Pandey, Organometallics, 2009, 28, 4713–4723 CrossRef CAS.
- Y. Baek and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed.
- S. A. Baudron, Dalton Trans., 2020, 49, 6161–6175 RSC.
- L. Lecarme, A. Kochem, L. Chiang, J. Moutet, F. Berthiol, C. Philouze, N. Leconte, T. Storr and F. Thomas, Inorg. Chem., 2018, 57, 9708–9719 CrossRef CAS PubMed.
- R. Matsuoka and T. Nabeshima, Front. Chem., 2018, 6, 349 CrossRef PubMed.
- A. V. Solomonov, Y. S. Marfin and E. V. Rumyantsev, Dyes Pigm., 2019, 162, 517–542 CrossRef CAS.
- C. S. Gutsche, S. Gräfe, B. Gitter, K. J. Flanagan, M. O. Senge, N. Kulak and A. Wiehe, Dalton Trans., 2018, 47, 12373–12384 RSC.
- S. J. Smalley, M. R. Waterland and S. G. Telfer, Inorg. Chem., 2008, 48, 13–15 CrossRef PubMed.
- R. Sakamoto, T. Iwashima, M. Tsuchiya, R. Toyoda, R. Matsuoka, J. F. Kögel, S. Kusaka, K. Hoshiko, T. Yagi, T. Nagayama and H. Nishihara, J. Mater. Chem. A, 2015, 3, 15357–15371 RSC.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- M. Yu, J. K.-H. Wong, C. Tang, P. Turner, M. H. Todd and P. J. Rutledge, Beilstein J. Org. Chem., 2015, 11, 37–41 CrossRef PubMed.
- S. M. Crawford and A. Thompson, Org. Lett., 2010, 12, 1424–1427 CrossRef CAS PubMed.
- J. A. Melanson, D. A. Smithen, T. S. Cameron and A. Thompson, Can. J. Chem., 2014, 92, 688–694 CrossRef CAS.
- D. A. Smithen, A. E. G. Baker, M. Offman, S. M. Crawford, T. S. Cameron and A. Thompson, J. Org. Chem., 2012, 77, 3439–3453 CrossRef CAS PubMed.
- T. Lundrigan, T. S. Cameron and A. Thompson, Chem. Commun., 2014, 50, 7028–7031 RSC.
- M. Liras, M. Pintado-Sierra, M. Iglesias and F. Sanchez, J. Mater. Chem. A, 2016, 4, 17274–17278 RSC.
- J. Li, Q. Zhang, J. Yin, C. Yu, K. Cheng, Y. Wei, E. Hao and L. Jiao, Org. Lett., 2016, 18, 5696–5699 CrossRef CAS PubMed.
- V. Lakshmi, T. Chatterjee and M. Ravikanth, Eur. J. Org. Chem., 2014, 2014, 2105–2110 CrossRef CAS.
- J. Urieta, B. L. Maroto, F. Moreno, A. R. Agarrabeitia, M. J. Ortiz and S. de la Moya, RSC Adv., 2015, 5, 68676–68680 RSC.
- S. P. A. Hinkes and C. D. P. Klein, Org. Lett., 2019, 21, 3048–3052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Images of NMR spectra. See DOI: 10.1039/d0ra05151a |
| This journal is © The Royal Society of Chemistry 2020 |