
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A series of four novel alkaline earth metal–organic frameworks constructed of Ca(II), Sr(II), Ba(II) ions and tetrahedral MTB linker: structural diversity, stability study and low/high-pressure gas adsorption properties†‡
Miroslav Almáši *a,
Vladimír Zeleňáka,
Róbert Gyepesb,
Ľuboš Zauškaa and
Sandrine Bourrellyc
*a,
Vladimír Zeleňáka,
Róbert Gyepesb,
Ľuboš Zauškaa and
Sandrine Bourrellyc
aDepartment of Inorganic Chemistry, Faculty of Science, P. J. Šafárik University, Moyzesova 11, SK-041 54 Košice, Slovak Republic. E-mail: miroslav.almasi@upjs.sk
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, CZ-128 43 Prague, Czech Republic
cAix-Marseille University, CNRS, MADIREL, Marseille Cedex 20, F-133 97, France
First published on 1st September 2020
Abstract
A series of four novel microporous alkaline earth metal–organic frameworks (AE-MOFs) containing methanetetrabenzoate linker (MTB) with composition {[Ca4(μ8-MTB)2]·2DMF·4H2O}n (UPJS-6), {[Ca4(μ4-O)(μ8-MTB)3/2(H2O)4]·4DMF·4H2O}n (UPJS-7), {[Sr3(μ7-MTB)3/2]·4DMF·7H2O}n (UPJS-8) and {[Ba3(μ7-MTB)3/2(H2O)6]·2DMF·4H2O}n (UPJS-9) (UPJS = University of Pavol Jozef Safarik) have been successfully prepared and characterized. The framework stability and thermal robustness of prepared materials were investigated using thermogravimetric analysis (TGA) and high-energy powder X-ray diffraction (HE-PXRD). MOFs were tested as adsorbents for different gases at various pressures and temperatures. Nitrogen and argon adsorption showed that the activated samples have moderate BET surface areas: 103 m2 g−1 (N2)/126 m2 g−1 (Ar) for UPJS-7′′, 320 m2 g−1 (N2)/358 m2 g−1 (Ar) for UPJS-9′′ and UPJS-8′′ adsorbs only a limited amount of N2 and Ar. It should be noted that all prepared compounds adsorb carbon dioxide with storage capacities ranging from 3.9 to 2.4 wt% at 20 °C and 1 atm, and 16.4–13.5 wt% at 30 °C and 20 bar. Methane adsorption isotherms show no adsorption at low pressures and with increasing pressure the storage capacity increases to 4.0–2.9 wt% of CH4 at 30 °C and 20 bar. Compounds displayed the highest hydrogen uptake of 3.7–1.8 wt% at −196 °C and 800 Torr among MTB containing MOFs.
1. Introduction
Metal–organic frameworks (MOFs) represent a class of inorganic–organic hybrid materials constructed from metal ions or clusters and organic linkers to form porous polymeric frameworks with interesting properties. Due to their structural and functional diversity, large surface area, porosity and affinity toward various guest molecules, MOFs have been tested as materials for different applications including gas storage and separation,1 heterogeneous catalysis,2 drug delivery,3 magnetic refrigeration4 and additives in batteries.5 However, gas adsorption was and still is one of the most important applications of MOFs. Hydrogen storage in MOF is currently being studied intensively. Hydrogen as a technologically important gas provides an ideal clean energy source, since no detrimental molecules are produced causing an environmental burden. Hydrogen is the smallest and simplest gas molecule with a very low boiling point, and its storage and transportation present a challenge.6 The second technologically important gas is carbon dioxide, which is industrially used to prepare many intermediate or fine chemicals. CO2 is also the main component of greenhouse gases and represents one of the largest anthropogenic impacts on the environment. For this reason, it is necessary to reduce its concentration in the atmosphere and thus limit global warming.7 Physical absorption of hydrogen and carbon dioxide in porous MOFs represents an interesting approach to the storage of the mentioned gases.Alkaline earth metal–organic frameworks (AE-MOFs) are investigated relatively less compared to MOFs that contain transition metals, although they display several unique advantages such as: (a) high abundance in earth's crust, (b) low cost, (c) low toxicity (particularly for calcium), (d) low density (Be, Mg, Ca, Sr) that offers gravimetric benefit for storage applications, (e) stability in air and (f) ionic nature leading to strong interactions with negative functional groups such as carboxylate. Unlike the transition metals, which tend to form largely predictable secondary building units (SBUs), the bonds between alkaline earth metal centres with functional groups are predominantly ionic in nature and the prediction and control over coordination geometry is ambiguous. The nature of organic linker plays an important role in the overall dimensionality and topology of the frameworks. Carboxylate-based linkers are those used most commonly to synthesize AE-MOFs due to the strong interaction between carboxylate oxygen atoms and alkaline earth metals. Based on the HSAB theory, AE metals fall under the hard acids and coordinate well with carboxylate functionalities.8 Carboxylate linkers with tetrahedral symmetry are highly suitable for MOF preparation, because they form the desired porous frameworks with large surface areas. To date, many MOFs have been prepared that contain ligands that possess tetrahedral geometry: methanetetrabenzoate (MTB),9 methanetetrabiphenylcarboxylate,10 methanetetrakis(benzene-4,1-diyl)tetraacrylate,11 methane tetrakis(benzene-4,1-diyl)tetrakis(ethyne-2,1-diyl)tetrabenzoate11 and tetrakis{3,5-bis[(4-carboxylate)phenyl]phenyl}methane.12 The chemical and physical properties of AE-MOFs have found applications in gas storage and separation,13 catalysis,14 sensing15 and proton conduction.16 Numerous AE-MOFs have been explored predominantly for gas storage of carbon dioxide, hydrogen and also for nitric oxide, noble gases and hydrocarbons.13
In our previous studies, we reported several compounds containing tetrahedral methanetetrabenzoate linker (MTB) with Zn(II),17,18 Ni(II)19 and Pb(II)20,21 metal centres and tested them as adsorbents for gases and heterogeneous catalysts. In the course of our continuing investigation into the crystal engineering of MOFs containing MTB linker and their applications, we combined the advantages of AE metals with MTB. Four novel MOFs were successfully prepared, characterized and tested as adsorbents for nitrogen, argon, carbon dioxide, methane and hydrogen at different temperatures and pressures.
2. Experimental
2.1. Materials
All chemicals used in the synthesis of AE-MOFs and H4MTB (see Fig. 1)17 were obtained from Sigma-Aldrich Company or Acros Organics and used without purification.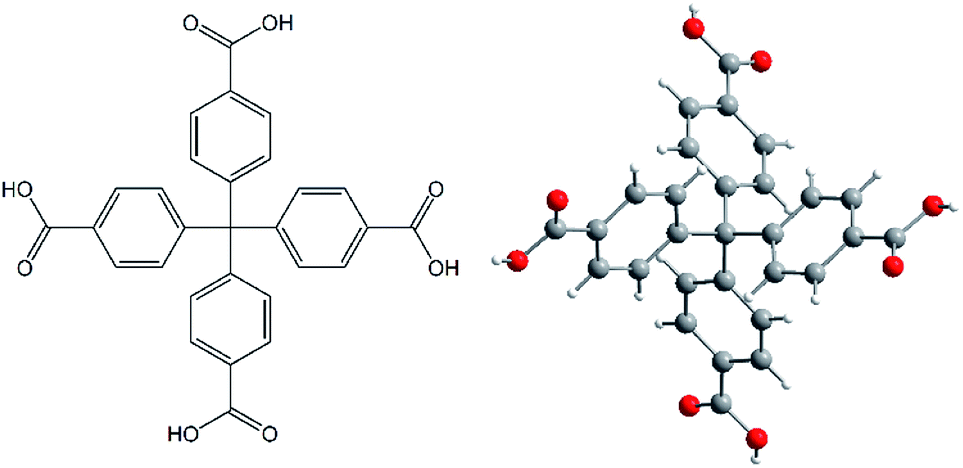 | ||
| Fig. 1 A molecular structure of methanetetrabenzoic acid (H4MTB) as an organic building block used in the synthesis of the final MOF compounds. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.20 mg (0.04 mmol) of methanetetrabenzoic acid and 48 mg (0.20 mmol; Ca(NO3)2·4H2O) for synthesis of compounds UPJS-6 and UPJS-7, 43 mg (0.20 mmol; Sr(NO3)2) for compound UPJS-8 and 53 mg (0.20 mmol, Ba(NO3)2) for synthesis of compound UPJS-9 were used. H4MTB was dissolved in 3 cm3 of N,N′-dimethylformamide (DMF), followed by the addition of the appropriate metal nitrates and then 3 cm3 of ethanol and 2.8 cm3 of water were added while stirring the solutions until the reactants were completely dissolved. Prepared solutions were sealed in glass autoclaves and the temperature programme was controlled by a thermoregulator. The temperature mode was set as follows: the reaction mixtures of compounds UPJS-6, UPJS-8 and UPJS-9 were heated to 80 °C and UPJS-7 to 110 °C with a heating rate of 0.5 °C min−1 for 72 hours. After this time, the reaction vessels were cooled to room temperature with a cooling rate of 0.5 °C min−1. Prepared materials were filtered off, several times washed with mixture of solvents used in the synthesis and dried in the stream of air, yields: 17.8 mg of UPJS-6 (yield 38%), 17.5 mg of UPJS-7 (yield 28%), 18.5 mg of UPJS-8 (yield 35%) and 22.5 mg of UPJS-9 (yield 35%). Elemental analysis for UPJS-6 (C64H54N2O22Ca4; 1363.43 g mol−1) clcd: C, 56.38%; H, 3.99%; N, 2.05%; found: C, 57.14%; H, 4.05%; N 1.98%; for UPJS-7 (C55.5H68N4O25Ca4; 1351.46 g mol−1) clcd: C, 49.32%; H, 5.07%; N, 4.15%; found: C, 48.43%; H, 5.10%; N 4.23%; for UPJS-8 (C55.5H66N4O23Sr3; 1419.99 g mol−1) clcd: C, 46.94%; H, 4.68%; N 3.95%; found: C, 46.51%; H, 4.72%; N, 3.96% and for UPJS-9 (C49.5H58N2O24Ba3; 1476.98 g mol−1) clcd: C, 40.25%; H, 3.96%; N 1.90%; found: C, 40.86%; H, 3.87%; N, 2.01%. Assignment of characteristic absorption bands (in cm−1) in IR spectra for UPJS-6: ν(OH) 3409; ν(CH)ar. 3069, 3038; ν(CH)aliph. 2963, 2932, 2891; ν(C
:1.20 mg (0.04 mmol) of methanetetrabenzoic acid and 48 mg (0.20 mmol; Ca(NO3)2·4H2O) for synthesis of compounds UPJS-6 and UPJS-7, 43 mg (0.20 mmol; Sr(NO3)2) for compound UPJS-8 and 53 mg (0.20 mmol, Ba(NO3)2) for synthesis of compound UPJS-9 were used. H4MTB was dissolved in 3 cm3 of N,N′-dimethylformamide (DMF), followed by the addition of the appropriate metal nitrates and then 3 cm3 of ethanol and 2.8 cm3 of water were added while stirring the solutions until the reactants were completely dissolved. Prepared solutions were sealed in glass autoclaves and the temperature programme was controlled by a thermoregulator. The temperature mode was set as follows: the reaction mixtures of compounds UPJS-6, UPJS-8 and UPJS-9 were heated to 80 °C and UPJS-7 to 110 °C with a heating rate of 0.5 °C min−1 for 72 hours. After this time, the reaction vessels were cooled to room temperature with a cooling rate of 0.5 °C min−1. Prepared materials were filtered off, several times washed with mixture of solvents used in the synthesis and dried in the stream of air, yields: 17.8 mg of UPJS-6 (yield 38%), 17.5 mg of UPJS-7 (yield 28%), 18.5 mg of UPJS-8 (yield 35%) and 22.5 mg of UPJS-9 (yield 35%). Elemental analysis for UPJS-6 (C64H54N2O22Ca4; 1363.43 g mol−1) clcd: C, 56.38%; H, 3.99%; N, 2.05%; found: C, 57.14%; H, 4.05%; N 1.98%; for UPJS-7 (C55.5H68N4O25Ca4; 1351.46 g mol−1) clcd: C, 49.32%; H, 5.07%; N, 4.15%; found: C, 48.43%; H, 5.10%; N 4.23%; for UPJS-8 (C55.5H66N4O23Sr3; 1419.99 g mol−1) clcd: C, 46.94%; H, 4.68%; N 3.95%; found: C, 46.51%; H, 4.72%; N, 3.96% and for UPJS-9 (C49.5H58N2O24Ba3; 1476.98 g mol−1) clcd: C, 40.25%; H, 3.96%; N 1.90%; found: C, 40.86%; H, 3.87%; N, 2.01%. Assignment of characteristic absorption bands (in cm−1) in IR spectra for UPJS-6: ν(OH) 3409; ν(CH)ar. 3069, 3038; ν(CH)aliph. 2963, 2932, 2891; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1664; ν(CC) 1600; νas(COO−) 1543; νs(COO−) 1407; UPJS-7: ν(OH) 3423; ν(CH)ar. 3068, 3040; ν(CH)aliph. 2996, 2933, 2890; ν(CO) 1670; ν(CC) 1660; νas(COO−) 1547; νs(COO−) 1408; UPJS-8: ν(OH) 3412; ν(CH)ar. 3067, 3039; ν(CH)aliph. 2959, 2931, 2887; ν(CO) 1662; ν(CC) 1598; νas(COO−) 1541; νs(COO−) 1398; UPJS-9: ν(OH) 3407; ν(CH)ar. 3064, 3037; ν(CH)aliph. 2957, 2929, 2884; ν(CO) 1660; ν(CC) 1590; νas(COO−) 1537; νs(COO−) 1392 cm−1. Corresponding IR spectra of prepared compounds are depicted in Fig. S1 in ESI.‡
O) 1664; ν(CC) 1600; νas(COO−) 1543; νs(COO−) 1407; UPJS-7: ν(OH) 3423; ν(CH)ar. 3068, 3040; ν(CH)aliph. 2996, 2933, 2890; ν(CO) 1670; ν(CC) 1660; νas(COO−) 1547; νs(COO−) 1408; UPJS-8: ν(OH) 3412; ν(CH)ar. 3067, 3039; ν(CH)aliph. 2959, 2931, 2887; ν(CO) 1662; ν(CC) 1598; νas(COO−) 1541; νs(COO−) 1398; UPJS-9: ν(OH) 3407; ν(CH)ar. 3064, 3037; ν(CH)aliph. 2957, 2929, 2884; ν(CO) 1660; ν(CC) 1590; νas(COO−) 1537; νs(COO−) 1392 cm−1. Corresponding IR spectra of prepared compounds are depicted in Fig. S1 in ESI.‡2.2 Characterization of prepared materials
The elemental analysis was performed with a CHNOS Elemental Analyzer vario MICRO from Elementar Analysensysteme GmbH with ±2–3 mg of sample.The infrared spectra were recorded with Avatar FT-IR 6700 spectrometer in the range 4000–400 cm−1. The samples were prepared in the form of a KBr pellet with a complex/KBr mass ratio of 1/100. Before IR measurements KBr was dried at 700 °C for 3 h in an oven and cooled in a desiccator.
Thermal behaviour of as-synthesized and solvent exchanged samples was studied by thermogravimetry analysis (TGA) in the temperature range of 30–800 °C. The sample with a weight of approximately 20 mg was placed in Al2O3 crucible and heated under a dynamic atmosphere of air (60 cm3 min−1) with a heating rate of 10 °C min−1, using the TGA Q500 apparatus.
HE-PXRD measurements were carried out at the P01 wiggler beamline of the PETRA III positron storage ring in DESY (Hamburg, Germany). The patterns were collected during in situ heating with a heating rate of 10 °C min−1 in an atmosphere of argon and beam energy of 60 keV, corresponding to a wavelength of 0.207150 Å. The diffracted photons were collected using an image plate detector PerkinElmer (2300 × 2300 pixels, each pixel having a size of 150 × 150 μm2) carefully aligned orthogonal to the X-ray beam. The sample-to-detector distance and the detector tilt were determined from diffraction patterns obtained from CeO2 standard reference material. The two-dimensional diffraction patterns were integrated using the QXRD software. The observed X-ray scattering intensities were corrected for background, absorption, polarization, incoherent and multiple scattering effects.
2.3 Single-crystal X-ray diffraction (SC-XRD) experiments
Single-crystal diffraction data for UPJS-6 (CCDC: 1978638‡), UPJS-8 (CCDC: 1978639‡), UPJS-9 (CCDC: 1978637‡) were collected on a Nonius Kappa four-circle CCD diffractometer equipped with an Apex II (Bruker) detector at 150 K and for UPJS-7 (CCDC: 1978636‡) on a Bruker D8 VENTURE diffractometer at −153 °C. Data reduction in all cases was carried out by diffractometer software. The phase problem was solved by intrinsic phasing and structure models were refined by full-matrix least-squares on F2 using the Shelx program suite.22 Nonhydrogen atoms were refined anisotropically. Hydrogen atoms were included in idealized positions and refined isotropically. The contribution of guest molecules located in the pores was subtracted by the SQUEEZE procedure in Platon.22 The figures of final crystal structures were drawn using the DIAMOND software. Detailed crystallographic data are given in Table S1 in ESI‡ and selected bond lengths (Å) and angles (°) for all compounds are summarized in Tables S2–S5 in ESI.‡2.4 DFT computations
DFT computations have been carried out using Gaussian 16, Revision A.03. Computations employed the M11 functional and the 6–31 + G(d,p) basis set for all atoms.23 The Hessians for geometry optimizations were estimated before the initial step of optimizations.2.5 Gas adsorption measurements
Nitrogen adsorption isotherms measured at −196 °C were taken using Quantachrome Nova 1200e automatic adsorption analyser. Before the adsorption studies, the solvents located in the channel system of samples were exchanged by ethanol (for procedure see Section 2.1.2 Solvent exchange process) and further, the samples were activated by the evacuation at 100 °C for 24 h to remove the guest molecules. Activated samples were denoted as UPJS-7′′, UPJS-8′′ and UPJS-9′′. The nitrogen adsorption isotherms for desolvated samples were collected in a relative pressure range from p/p0 = 0.005 to 0.95. Based on the nitrogen adsorption isotherm, the BET specific surface area (SBET) and pore volume (Vp) of the samples were calculated.Argon adsorption isotherms were measured at −186 °C, hydrogen adsorption isotherms at −196 °C and carbon dioxide adsorption isotherms were performed at 20 °C using ASAP 2020 apparatus.
The gas adsorption measurements at 30 °C and pressures up to 20 bar were performed with CO2 and CH4 using a homemade high-throughput instrument.24 Gas adsorption experiments were measured via a manometric gas dosing system on three samples in parallel. The amounts of gas adsorbed were calculated by an equation of state using the Reference Fluid Thermodynamic and Transport Properties (REFPROP) software package 8.0 of the National Institute of Standards and Technology (NIST).25 The high pressure adsorption isotherms were measured on the same batch of the sample as nitrogen adsorption. Samples were thermally activated individually in situ under the primary vacuum, at 80 °C overnight.
The regenerability of the samples has been evaluated under mild conditions. For each gas (CO2 and CH4) two measurements with the same parameters were performed on the same sample. Between the first and the second experiment, the samples were submitted to an evacuation step at 30 °C and under the primary vacuum during 80 minutes. In this way, from the second gas adsorption measurement, the regenerability/recovery of the samples was checked under the mentioned conditions.
3. Results and discussion
3.1 Synthesis
All the compounds investigated in the present study were prepared using a solvothermal synthesis under mild synthetic conditions (80–110 °C) and in a molar ratio of 5:1. It is known that the synthesis of MOF may be included in so-called combinatorial synthetic chemistry since the successful preparation of porous coordination polymers depends on many factors such as: reactant concentration, the presence of a co-solvent, solution pH, metal to ligand ratio, properties of the used counterion, reaction temperature, and reaction time, can have a considerable impact on the final composition of the products.26 In the case of the prepared calcium(II) compounds UPJS-6 and UPJS-7, the influence of the reaction temperature was observed. In the synthesis of these materials, the same reaction conditions were used except temperature. Compound UPJS-6 was prepared at 80 °C and represents the low-temperature phase and compound UPJS-7 was synthesized at 110 °C, which can be referred as the high temperature form. The temperature difference of 30 °C had a significant effect on the mutual arrangement of the building components, which led to the formation of different polymeric frameworks with different porosity (see description of crystal structures below).
The exact formula and composition of all prepared samples were established based on the combination of single-crystal X-ray diffraction experiments, elemental analysis and thermogravimetric analysis results.
3.2 Crystal structures
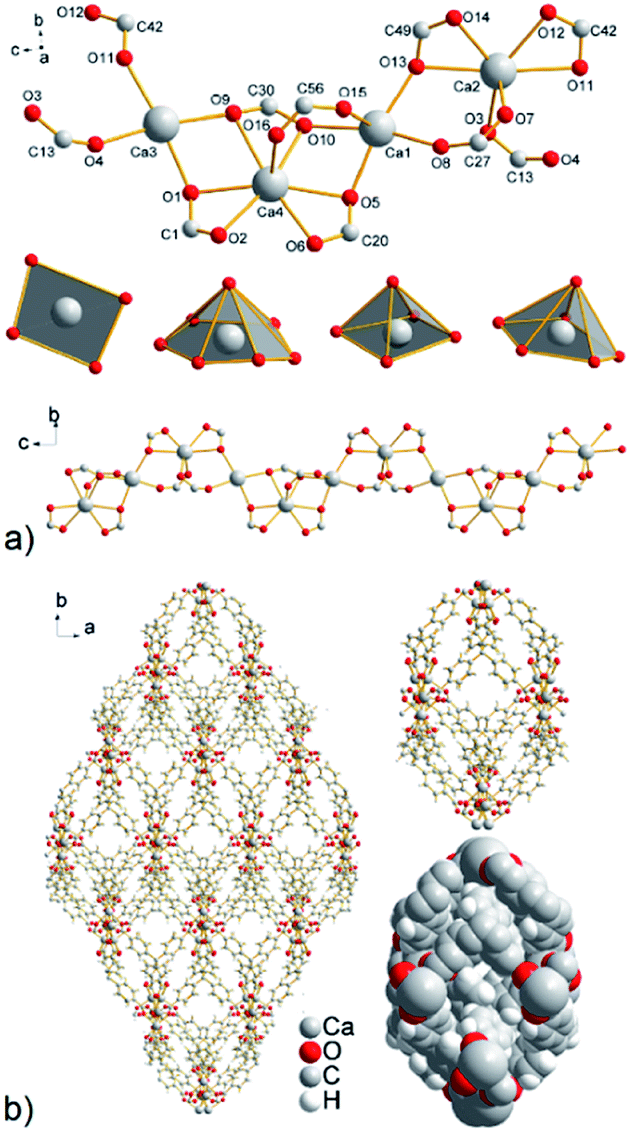 | ||
| Fig. 2 (a) A view of four crystallographically independent calcium ions with the corresponding shape of coordination polyhedra and final 1D sinusoidal calcium chain propagating along the c crystallographic axis. (b) The resulting 3D motif in UPJS-6 with closed/isolated cages. | ||
The SC-XRD analysis showed that in the crystal structure of compound UPJS-6 isolated cages are present. Although the inner diameter of the cage is relatively large with a size of 18.3 × 8.4 Å2, the inlet openings are only 0.7 × 0.5 Å2 in size (see Fig. 2b). The total free volume in the structure of compound UPJS-6 analysed by the PLATON program is 3434.1 Å3 and represents 44.1%, with respect to the total volume of the unit cell (7789.5 Å3). Analysis of the framework using the ToposPro program showed that the compound did not imitate any known mineral and the skeleton topology could be designated as 3,4^2,6,8-c net.27 Since compound UPJS-6 contains isolated pores in the crystal structure, its gas adsorption properties were not studied.
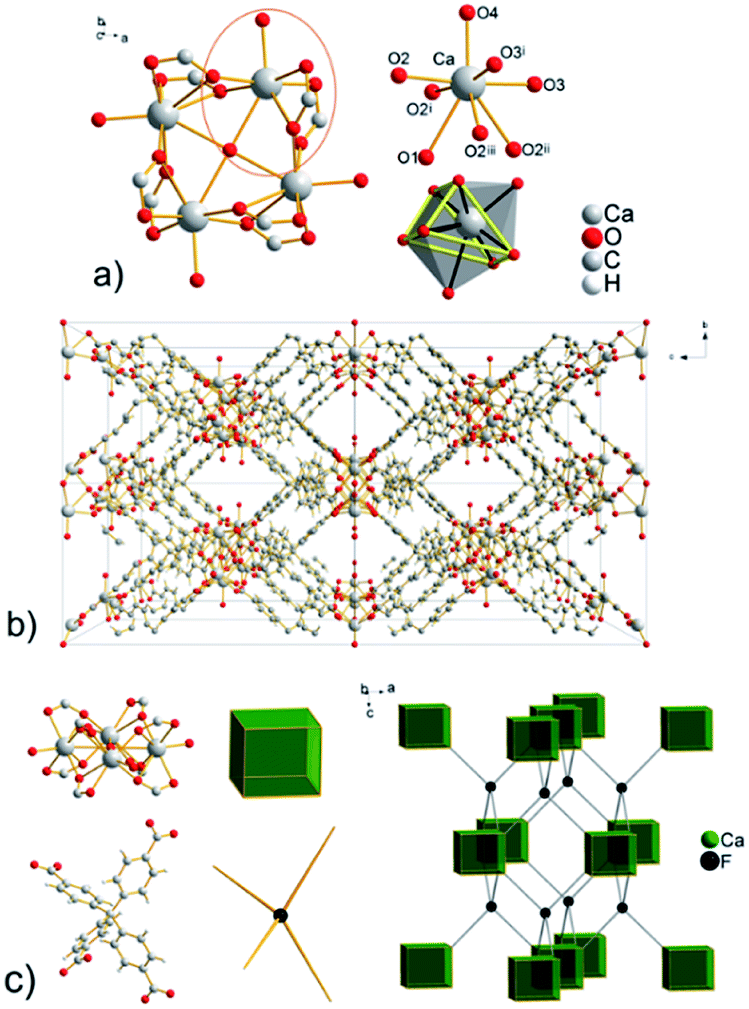 | ||
| Fig. 3 (a) Cyclotranuclear Ca(II) cluster with composition [Ca4(μ4-O)(μ2-COO)8(H2O)4], the coordination environment of Ca(II) ions and a view of bicapped trigonal prismatic polyhedron ((i) x, y, 2 − z; (ii) −x, y, 2 − z; (iii) −x, y, z). (b) View of the framework in UPJS-7 along a = b crystallographic axis. (c) Topological analysis of the framework with a demonstration of the CaF2 structure imitation. | ||
The topological analysis (see Fig. 3b) of the polymeric framework using the ToposPro program27 showed that imitates the fluorite (flu, CaF2) structure. The central carbon atom of the MTB ligand can be considered the F vertex and the [Ca4(μ4-O)(μ2-COO)8(H2O)4] cluster decorates the calcium(II) positions in the CaF2 structure.
The total free space in the crystal structure of UPJS-7, after removal of the guest molecules and analysed by PLATON, is 2529 Å3 (cell volume 4689.0 Å3) and corresponds to 57.7%.
 | ||
| Fig. 4 (a) View of the cyclic triangular strontium cluster with composition [Sr3 (μ3-COO)2(μ2-COO)2(COO)2] and the corresponding coordination polyhedra of crystallographically independent Sr(II) ions. A final crystal packing of the framework with 2D cavities propagating along (b) a and (c) b axis. | ||
Strontium cations are in the structure of compound UPJS-8 hepta- and hexacoordinated. The central atom Sr1 with [SrO7] donor set is coordinated by the seven oxygen atoms derived from the four different carboxylate moieties of the MTB4− ligand and the shape of the coordination polyhedron can be described as a deformed monocapped octahedron. Strontium ion Sr2 is also heptacoordinated by the seven oxygen atoms, but the shape of the coordination polyhedron [SrO7] is a deformed monocaped trigonal prism. The last strontium ion Sr3 is hexacoordinated with [SrO6] donor set and the shape of the coordination polyhedron is deformed octahedron (see Fig. 4a). The Sr–O bond distances ranging from 2.485(2) Å (Sr3–O6) to 2.802(3) Å (Sr3–O2) (see Table S4 in ESI‡). The bond lengths of the tridentate-bridged oxygen atoms Sr2–O2 (2.707(3) Å) and Sr3–O2 (2.802(3) Å) are slightly longer compared to the other Sr–O bonds (see Table S4 in ESI‡), due to the electron-withdrawing from the oxygen atoms as a result of the formation of three coordination bonds. The elongated bond distances are consistent with the bond lengths for other strontium coordination polymers containing μ3-coordinated carboxylate ligand.30
By described coordination of strontium ions and MTB molecules, 3D coordination polymer with the composition of {[Sr3(μ7-MTB)3/2]}n is formed. The polymeric framework contains 2D cavities with sizes of 15.5 × 5.0 Å2 propagating along a crystallographic axis and smaller openings with dimensions of 5.8 and 3.5 Å2 propagating along b axis (see Fig. 4b and c). The total free volume in the crystal structure of compound UPJS-8 is 55.8% (5242.4 Å3 free volume of 9398.2 Å3 unit cell volume). Using the ToposPro program,27 an analysis of the polymeric framework was performed, which showed that the compound does not imitate any known mineral and is unique. The analysis showed that the framework has a 4,5^2,6,8-c net topology.
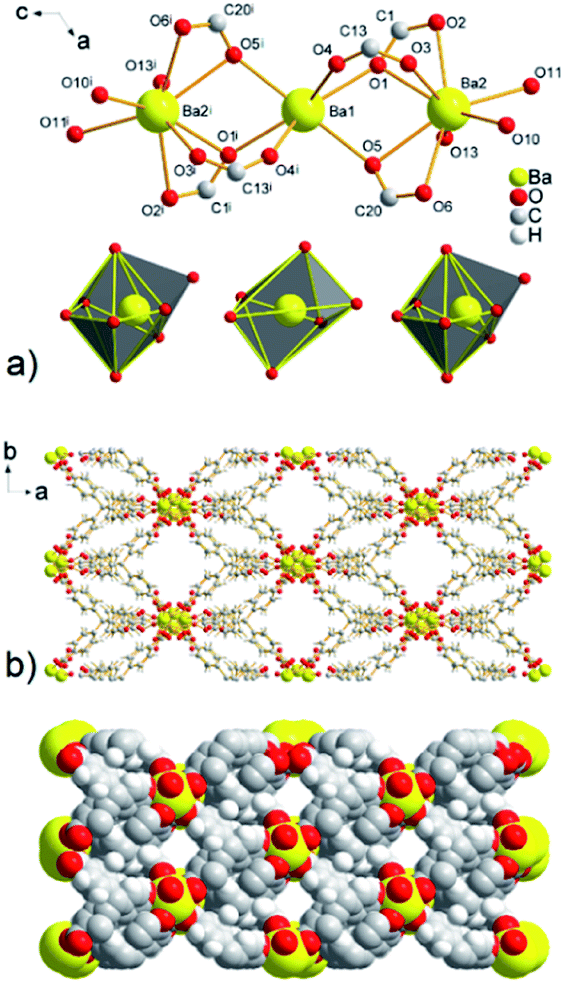 | ||
| Fig. 5 (a) A view of the linear three-nuclear barium cluster with composition [Ba3(μ2-COO)6(H2O)6] in UPJS-9 (hydrogen atoms were omitted for clarity, (i) = −x, y, 1/2 − z). (b) View of a 3D polymeric framework with 1D channels propagating along c crystallographic axis. | ||
The combination of MTB molecules and Ba(II) ions as building blocks results in a three-dimensional framework with 1D channels propagating along the c crystallographic axis (see Fig. 5b). The size of the cavities after the subtraction of the van der Waals radii of the selected atoms is 10.7 × 8.7 Å2. The topological analysis of the UPJS-9 polymeric framework using the ToposPro program27 showed that imitates the fluorite (flu, CaF2) structure similar to UPJS-7. The [Ba3(μ2-COO)6(H2O)6] cluster decorates the calcium(II) positions and the central methane carbon atom of the MTB ligand can be considered the fluoro vertex in fluorite structure.
Using PLATON analysis the total free pore volume of 2973.5 Å3 was found in the structure of compound UPJS-9, which represents 37.1% free space based on the cell volume (8011.1 Å3).
3.3 Thermal behaviour and frameworks stability study
The thermal behaviour and stability of prepared compounds were investigated using thermogravimetry (TG) and obtained TG curves are shown in Fig. S2a in ESI.‡ The thermogravimetric curve of UPJS-6 displayed no significant mass change in the temperature range 30–210 °C. The mass change corresponding to desolvation process was 16.23% and is in good agreement with calculated value of 16.01% (2 DMF and 4H2O molecules). Similar high thermal stability and release of solvent molecules was observed not only for MOF compounds13 but also for common chemicals (CuSO4·5H2O = 50–230 °C; Ca(COO)2·H2O = 180–200 °C). This phenomenon can be explained by the strong interaction between water molecules and the central atoms. We can only speculate that there is such a strong interaction in UPJS-6, but the main reason for the higher thermal stability (210 °C) is the presence of isolated cages/pores in the framework. The water molecules located in them are trapped and are released only after the collapse of the framework. Mentioned explanation is also in good agreement with the results of the HE-PXRD analysis (see below), where the compound is stable at 200 °C and the polymeric skeleton collapses at 250 °C, allowing water molecules to be released, which was reflected by weight loss on the TG curve. TG curve of UPJS-7 showed a solvent weight loss of 32.49% in the 30–400 °C temperature range (expected 32.29% for 4 DMF and 8H2O molecules) and TG curve of UPJS-8 gave a similar solvent weight loss of 29.85% in the 30–300 °C temperature range (expected 29.47% for 4 DMF and 7H2O molecules). For compound UPJS-9 TG analysis gave a distinct weight loss of 22.17% in 30–300 °C temperature range corresponding to the loss of 10 water and 2 DMF molecules (expected 22.09%). After the desolvation process, frameworks exhibit high thermal stability up to 500 °C for UPJS-7 and UPJS-9 and 530 °C for UPJS-8. The high thermal stability of polymeric skeletons can be explained by the strong interactions formed between carboxylate oxygen atoms of MTB linker and AE central ions. The TG curves of ethanol exchanged samples UPJS-7′, UPJS-8′ and UPJS-9′ are shown in Fig. S2b in ESI.‡ As can be seen from obtained TG curves, the solvent exchange process has a significant impact on the activation temperature of the compounds. The activation temperatures decrease from 300–400 °C for as-synthesized samples to 150 °C for ethanol exchanged samples. Solvent exchanged samples were further used for gas adsorption measurements.The framework stability of prepared compounds was studied by high energy powder X-ray diffraction (HE-PXRD) experiments with in situ heating and 50 °C steps. Fig. S3a–S6a in ESI‡ show a comparison of experimental HE-PXRD patterns and the patterns calculated from single-crystal X-ray data, which are in good agreement. The framework of UPJS-6 is stable up to 200 °C (see Fig. S3 in ESI‡). Above this temperature, when the solvent molecules are released (see TG), the structure collapses. At this high temperature, the trapped solvent molecules are transformed into the gas phase, because these molecules cannot leave the isolated cages only if the framework collapses, which causes the destruction of the whole crystal structure. Based on the Kitagawa's MOF classification,31 UPJS-6 can be included in the 1st MOF generation, which is sustained only with guest molecules and shows irreversible framework collapse on the removal of guest molecules. The polymeric skeleton of UPJS-7 (see Fig. S4 in ESI‡) is thermally stable after the release of solvent molecules from the sample and desolvated framework is stable after heating up to 450 °C. Above this temperature, the intensities of the narrow and intense diffraction peaks decrease and at 600 °C formation of amorphous phase was observed. HE-PXRD patterns of UPJS-8 (see Fig. S5b in ESI‡) show similar behaviour to UPJS-7, without changes in the diffraction peak positions during heating. The thermal stability of the polymeric framework of UPJS-8 is higher, up to 500 °C. As the release of the guest molecules located in the cavities (H2O and DMF) in UPJS-7 and UPJS-8 occur in the temperature range 30–300 °C (based on TG) and frameworks stability is up to 450 °C (UPJS-7) and 500 °C (UPJS-8) without phase change (based on HE-PXRD), described compounds could be included into the 2nd MOF generation with stable and permanent polymeric frameworks classified by Kitagawa. Compound UPJS-9 exhibits similar thermal stability compared to previous samples. Moreover, a phase transition was observed (see Fig. S6b in ESI‡). The phase representing the as-synthesized compound is stable up to 250 °C. As this temperature is comparable to the end of desolvation step on TG, their release results in the transformation of the compound into a new phase. Novel formed phase is subsequently thermally stable in a temperature range of 300–500 °C. With increasing temperature, the diffraction peaks of the second phase decrease and the appearance of broad peaks, similar to HE-PXRD patterns of previous compounds. Based on the observed results the framework of compound UPJS-9, could be included in the 3rd MOF generation with a flexible framework.
3.4 Gas adsorption
To characterize the porosity of prepared MOFs, gas adsorption measurements were performed for nitrogen, argon, hydrogen, methane and carbon dioxide. Before adsorption experiments, the samples were immersed in ethanol for one week to exchange the original solvents in the cavities (DMF and water), for easier activation of the compounds. The samples for gas adsorption experiments were prepared by evacuation at 100 °C for 24 h to remove all guest solvent molecules and activated samples were denoted as UPJS-7′′, UPJS-8′′ and UPJS-9′′.Textural properties of materials were initially studied by argon (see Fig. 6a) and nitrogen (see Fig. 6b) adsorption measurements at −196 °C and −186 °C, respectively. Based on the nitrogen and argon adsorption isotherms, corresponding surface areas using the BET equation and pore volumes using the DFT were calculated. Compounds UPJS-7′′ and UPJS-9′′ exhibit typical type I nitrogen and argon adsorption isotherms, consistent with their microporous structures. Compounds exhibit BET surface areas of 126 m2 g−1; pore volume 0.048 cm3 g−1 (based on Ar) and 103 m2 g−1; pore volume 0.045 cm3 g−1 (based on N2) for UPJS-7′′, 358 m2 g−1; pore volume 0.256 cm3 g−1 (based on Ar) and 320 m2 g−1; pore volume 0.243 cm3 g−1 (based on N2) for UPJS-9′′. Compound UPJS-8′′ adsorbs only a limited amount of N2 and Ar and displayed no porosity. The surface areas of the compounds containing MTB linker are summarized in Table 1. By comparing the values of surface areas, it can be concluded that UPJS-7′′ has the lowest surface area and the surface area of UPJS-9′′ is comparable to the nickel(II)33 and cobalt(II)34 analogues.
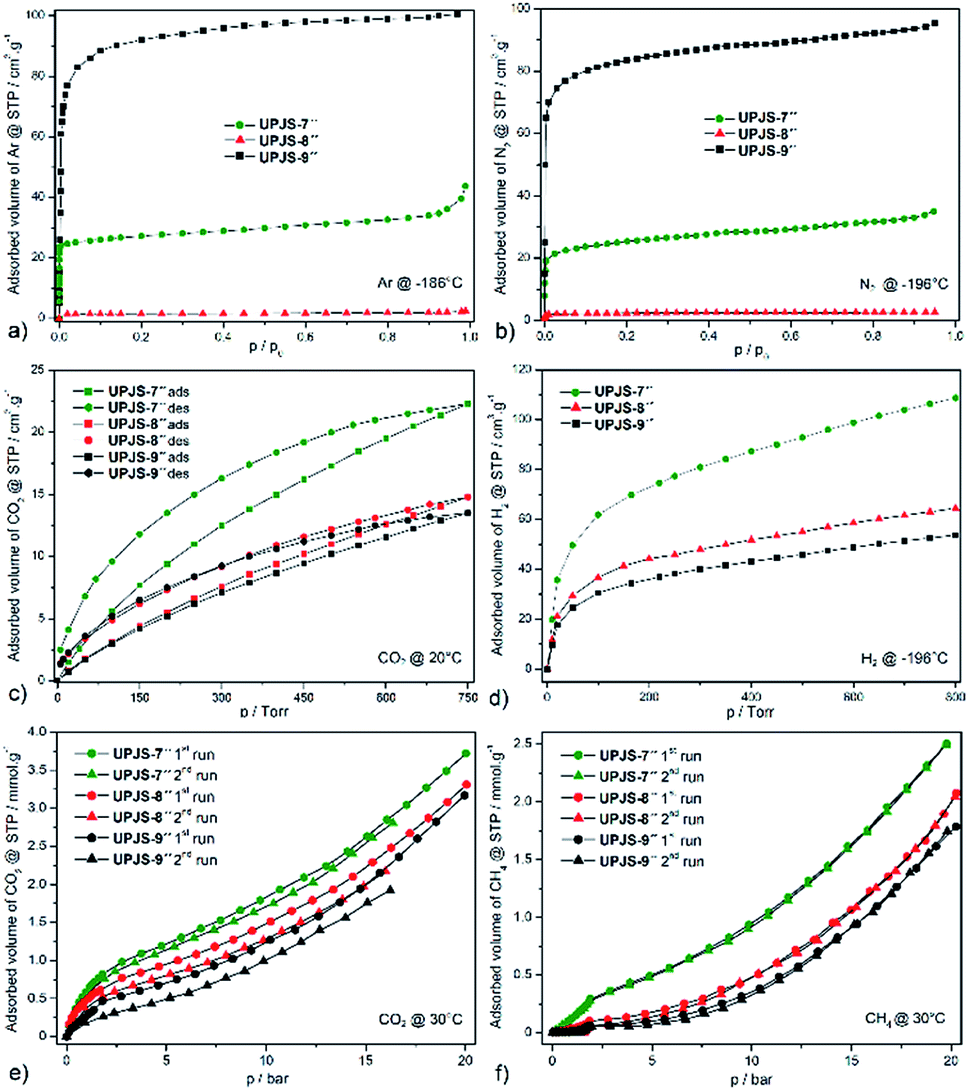 | ||
| Fig. 6 Low pressure adsorption isotherms of (a) argon measured at −186 °C, (b) nitrogen at −196 °C, (c) carbon dioxide at 20 °C and (d) hydrogen at −196 °C. High pressure adsorption isotherm of (e) CO2 and (f) CH4 at measured at 30 °C and up to 20 bar over prepared compounds. | ||
| Compound | Gas | Surface [m2 g−1] | Pore volume [cm3 g−1] | Adsorbed CO2 @ 1 atm [wt%/°C] | Ref. |
|---|---|---|---|---|---|
| a L1 = 1,4,8,11-tetraazacyclotetradecane; L2 = 1,3,6,9,11,14-hexaazatricyclodecane; L3 = hexaazatricycloeicosane; L4 = formate. | |||||
| {[Sr3(μ7-MTB)3/2]·4DMF·7H2O}n (UPJS-8) | N2 | — | — | 2.64/20 | This work |
| {[Ca4(μ4-O)(μ8-MTB)3/2(H2O)4]·4DMF·4H2O}n (UPJS-7) | N2, Ar | 103, 126 | 0.045, 0.048 | 3.92/20 | This work |
| {[Ni2(μ4-MTB)(κ4-L1)2]·4DMF·8H2O}n | CO2 | 141 | 0.055 | — | 32 |
| {[Zn2(μ8-MTB)(H2O)2]·3DMF·3H2O}n (UPJS-2) | N2 | 248 | 0.089 | 5.67/0 | 17 |
| {[Ni2(μ4-MTB)(κ4-L2)2]·8H2O}n | N2 | 288 | 0.230 | — | 33 |
| {[Ba3(μ7-MTB)3/2(H2O)6]·2DMF·4H2O}n (UPJS-9) | N2, Ar | 320, 358 | 0.243, 0.256 | 2.41/20 | This work |
| {[Ni2(μ4-MTB)(κ4-L3)2]·16H2O}n | N2 | 335 | 0.210 | 33 | |
| {[Co4(μ6-MTB)2(μ2-H2O)4(H2O)4]·13DMF·11H2O}n | CO2 | 356 | 0.165 | 7.02/0 | 34 |
| {[Cu2(μ8-MTB)(H2O)2]·6DEF·2H2O}n | N2 | 526 | — | — | 35 |
| {[Zn2(μ4-MTB)(κ4-L1)2]·2DMF·7H2O}n (UPJS-4) | Ar | 644 | 0.246 | 10.50/0 | 18 |
| {[Ni4(μ6-MTB)2(μ2-H2O)4(H2O)4]·10DMF·11H2O}n (UPJS-3) | N2 | 700 | 0.291 | 12.36/0 | 19 |
| {[Pb4(μ8-MTB)2(H2O)4]·5DMF·H2O}n (UPJS-5) | N2 | 980 | 0.666 | 9.30/0 | 20 and 21 |
| {[Zr6(μ3-O)(μ3-OH)(μ8-MTB)2(μ2-L4)4(H2O)4]·xDMF·yMeOH}n | N2 | 1390 | 0.530 | — | 36 |
The carbon dioxide adsorption isotherms of prepared materials measured at 20 °C show type I behaviour with a hysteresis loop (see Fig. 6c). We suppose that this behaviour arises from the kinetic effect, which may be a contributing factor to the hysteresis because the adsorption/desorption kinetics were very slow, due to a strong interaction between the CO2 and framework.37 Compounds adsorb CO2 with maximal storage capacity of 3.92 wt% (0.927 mmol g−1, 22.3 cm3 g−1) for UPJS-7′′, 2.64 wt% (0.615 mmol g−1, 14.8 cm3 g−1) for UPJS-8′′ and 2.41 wt% (0.561 mmol g−1, 13.5 cm3 g−1) for UPJS-9′′ at 20 °C and 1 atm (760 Torr). The storage capacity of prepared compounds decreases in order UPJS-7′′ > UPJS-8′′ > UPJS-9′′. Calcium MOF (UPJS-7′′) adsorbs the highest amount of CO2 from prepared compounds, compared to UPJS-9′′ with the largest surface area (358 m2 g−1). As was mentioned in the introduction, AE-MOFs made of early members of the metal group are thought to be good candidates based on their gravimetric advantages over commonplace first-row transition metal-based MOFs. Surprisingly, the strontium complex UPJS-8′′ which does not adsorb nitrogen or argon, it adsorbs carbon dioxide. Thus, this compound exhibits a high selectivity for CO2 over N2 or Ar and can find its further application as a gas separator. High CO2/N2 selectivity was also observed for Zn-MOF38 and UPJS-1.39 A likely explanation is that nitrogen molecules cannot enter the pores at −196 °C due to large diffusional resistances, but at 20 °C the additional thermal energy allows the CO2 molecules to overcome these resistances. Moreover, higher quadrupole moment of CO2 might induce stronger interaction with the framework of UPJS-8′′.
A comparison of the CO2 adsorption capacity with reported compounds containing MTB ligand is summarized in Table 1. The adsorbed values of CO2 for presented AE-MOF appear to be low, but is not suitable to correlate them with other MTB-containing materials because their CO2 adsorption isotherms were measured at 0 °C. A comparison of carbon dioxide capacities can be performed with other MOF compounds that contain alkaline earth metals and measurements were performed under the same conditions, 1 atm and 20 °C. The observed value for UPJS-7′′ is comparable to Ca(SBD) (4.30 wt%; SBET = 145 m2 g−1),40 Ca2(TIA)2(H2O)2 (4.90 wt%)41 but lower than that of compounds Ca5(BTB)2(HBTB)2(H2O)6 (8.70 wt%; SLANG = 914 m2 g−1),42 Mg-MOF-74 (30.1 wt%; SBET = 1416 m2 g−1)7 and Mg-DOBDC (25 wt%; SBET = 1415 m2 g−1).7 Samples UPJS-8′′ and UPJS-9′′ show lower CO2 adsorption capacity values compared to the already published compounds containing strontium or barium ions such as Sr(SBA) (5.10 wt%; SBET = 79 m2 g−1)43 and Ba(HBTB) (9.36 wt%; SLANG = 879 m2 g−1).44
The hydrogen adsorption isotherms of prepared materials measured at −196 °C are shown in Fig. 6d. Compounds adsorb hydrogen with maximal storage capacity of 3.65 wt% (18.11 mmol g−1, 108.7 cm3 g−1) for UPJS-7′′, 2.17 wt% (10.75 mmol g−1, 64.5 cm3 g−1) for UPJS-8′′ and 1.80 wt% (8.95 mmol g−1, 53.7 cm3 g−1) for UPJS-9′′ at −196 °C and 800 Torr. Observed values of hydrogen storage capacities are relatively high and can be explained by the well-known high affinity of hydrogen to AE metal ions.13 Another explanation for the increased affinity of hydrogen for the presented MOFs is the presence of coordinatively unsaturated sites (CUSs) in UPJS-7 and UPJS-9. It is known that MOFs with CUSs offer interesting possibilities for tuning the affinity of these materials towards certain adsorbates, potentially increasing their selectivity and storage capacity. In MOFs with CUSs, at least one available coordination site of the metal centre is occupied by an atom belonging to a solvent molecule after the synthesis. Through activation process using evacuation, or heating, the solvent molecules can be removed, leaving the metal atom with open coordination site. UPJS-7 contains four terminally coordinated water molecules per one cyclotetranuclear cluster [Ca4(μ4-O)(μ2-COO)8(H2O)4], and UPJS-9 contains six water molecules in [Ba3(μ2-COO)6(H2O)6] cluster (see Section 3.2 Crystal structures). Upon activation of the compounds, these solvent molecules are removed, leading to the formation of CUSs. Formed active sites serve as primary adsorption sites for hydrogen where an interaction central atom-hydrogen is present, thereby increasing the adsorption capacity of hydrogen in MOFs. Although UPJS-9 contains a higher number of CUSs compared to UPJS-7, its hydrogen sorption capacity is lower (53.7 cm3 g−1 for UPJS-9; 108.7 cm3 g−1 for UPJS-7). This observation can be explained by the higher relative atomic mass of barium compared to calcium, which is reflected in the resulting values of the volume of adsorbed hydrogen, since in the case of adsorption measurements the amount of gas is given in units of cm3 per one gram of support. In compound UPJS-8, no terminally coordinated solvent molecules are present. However, the DFT modeling23 has shown that there is one unoccupied orbital on each central atom, which can serve as CUSs (see Fig. S7 in ESI‡). Similar results were observed for other coordination compounds containing strontium ions.45
According to the literature data, the H2 adsorption capacities were published only for two MTB-containing compounds, namely {[Zn2(μ4-MTB)(κ4-L1)2]·2DMF·7H2O}n (UPJS-4)18 and {[Ni2(μ4-MTB)(κ4-L1)2]·4DMF·8H2O}n32 (L1 = 1,4,8,11-tetraazacyclotetradecane, cyclam). Their hydrogen adsorption capacities at −196 °C were 1.28 and 0.70 wt%, respectively. It indicates that all novel prepared compounds exhibit significantly larger hydrogen uptake.
Compounds UPJS-7′′, UPJS-8′′ and UPJS-9′′ were also tested as gas adsorbents for carbon dioxide and methane at high pressures. Obtained CO2 and CH4 isotherms measured at 30 °C, pressure up to 20 bar and after two repeated adsorption cycles are depicted in Fig. 6e and f, respectively. The absence of plateaus on the isotherms indicated that the maximal storage capacity for both gases was not achieved and compounds are not saturated by the gases at 20 bar. Both selected probe molecules are nonpolar with different quadrupole moments (CH4 (0 C m2), CO2 (14.3 × 10−40 C m2)), kinetic diameters (CH4 (3.8 Å), CO2 (3.3 Å)) and different boiling points ((CH4 (−161 °C), CO2 (−78 °C))). Described differences may influence the maximal adsorption uptake of gases.
Compounds storage capacities of carbon dioxide at 1 bar and 30 °C were 0.68 mmol g−1 (2.99 wt%) for UPJS-7′′, 0.45 mmol g−1 (1.98 wt%) for UPJS-8′′ and 0.39 mmol g−1 (1.72 wt%) for UPJS-9′′ at 30 °C and 1 bar. A similar trend of adsorbed amounts was also observed for CO2 adsorption isotherms measured at 20 °C and 1 atm, described above. With increasing pressure, an increase in the amount of adsorbed CO2 was observed. The maximal adsorption uptake and corresponding adsorption capacities were 3.72 mmol g−1 (16.4 wt%) for UPJS-7′′, 3.31 mmol g−1 (14.6 wt%) for UPJS-8′′ and 3.07 mmol g−1 (13.5 wt%) for UPJS-9′′ at 30 °C and 20 bar. For repeated measurements, the branches of adsorption isotherms did not overlap and in the second repetition cycle, the samples adsorbed lower amounts of carbon dioxide compared to the first run. As was described in Section 2.5 Gas adsorption measurements above, the samples at repeated measurements were activated only at 30 °C for 8 hours. Observed results from CO2 high pressure adsorption suggest that CO2 molecules interact strongly with the frameworks of prepared compounds. A similar observation was demonstrated by the hysteresis loop on CO2 adsorption isotherms performed at low pressures.
Methane was also used as a probe molecule for high pressure adsorption and measured isotherms are shown in Fig. 6f. Compounds adsorb CH4 with maximal storage capacity of 3.99 wt% (2.49 mmol g−1) for UPJS-7′′, 3.32 wt% (2.07 mmol g−1) for UPJS-8′′ and 2.86 wt% (1.78 mmol g−1) for UPJS-9′′ at 30 °C and 20 bar. The storage capacity of prepared compounds has a similar trend to carbon dioxide and decrease in order UPJS-7′′ > UPJS-8′′ > UPJS-9′′. It could be noted, that the amounts adsorbed for methane are approximately two times lower compared to carbon dioxide. In repeated CH4 adsorption measurements, isotherms do not show a significant change in adsorbed volume. This observation indicates that there is no significant interaction between the methane molecules and the polymeric frameworks. Interestingly, the compound does not adsorb CH4 at low pressures. The compounds start to adsorb methane up to 0.3 bar for UPJS-7′′, UPJS-8′′ and compound UPJS-9′′ at pressure up to 0.8 bar. On the adsorption isotherms, a breakthrough occurs at a pressure of about 2 bar, where the adsorbed amount of methane is significantly increased. The measured isotherms indicate a possible transformation of the polymeric skeletons, which was confirmed for compound UPJS-9 by HE-PXRD measurements. Measured adsorption results indicate the absence of specific adsorption sites for CH4 at the surface of prepared AE-MOFs.
According to the literature data, the high pressure CO2 and CH4 adsorption measurements were performed for two compounds containing MTB linker. Nickel(II) MOF with composition {[Ni4(μ6-MTB)2(μ2-H2O)4(H2O)4]·10DMF·11H2O}n (UPJS-3)20 adsorbed 3.29 wt% (2.05 mmol g−1) of CH4 and 18.05 wt% (4.1 mmol g−1) of CO2 and lead(II) compound with formula {[Pb4(μ8-MTB)2(H2O)4]·5DMF·H2O}n (UPJS-5)20 adsorbed 3.05 wt% (1.9 mmol g−1) of CH4 and 19.81 wt% (4.5 mmol g−1) of CO2 at 30 °C and 21 bar. From the above results, it can be concluded that all prepared AE-MOFs show the same or slightly increased value of methane adsorption capacity and slightly reduced carbon dioxide adsorption capacity.
In the literature, only few MOF compounds containing alkaline earth metals on which the adsorption properties at high pressures and approximately the same conditions have been studied. As an example for comparison could be Mg2(DOBDC) (SBET = 1495 m2 g−1, CO2, 68.9 wt%, 5 °C, 36 bar; CH4, 15.7 wt%, 25 °C, 35 bar);46 Ca2(AZBZ-TC)(H2O)(DMF) (CO2, 5.7 wt%, 25 °C, 20 bar)47 and Ba2(TMA)(NO3)(DMF) (SBET = 190 m2 g−1, CO2, 11.2 wt%, 0 °C, 70 bar; CH4, 1.8 wt%, 0 °C, 70 bar).48
4. Conclusions
We have prepared and characterized four novel alkaline earth metal–organic frameworks. Structural analysis showed that compounds form 3D frameworks consisting of MTB linker and EA ions, which form different clusters with different SBU. Two different calcium(II) compounds UPJS-6 and UPJS-7, were prepared using different reaction temperatures. Compound UPJS-6 represents the low-temperature nonporous phase and compound UPJS-7 as the high temperature form exhibited CaF2 framework topology with an open structure. Compounds UPJS-8 and UPJS-9 also exhibit porosity with different pore dimensionality. The framework stability of prepared materials was investigated using thermogravimetric analysis and high energy powder X-ray diffraction. The thermal stability of prepared compounds ranged at high temperature 500–530 °C (exception UPJS-6), as a result of strong ionic interaction between AE ions and carboxylates of MTB linker. Moreover, HE-PXRD measurements also confirmed the stability of the frameworks and a phase transition was observed in compound UPJS-9. After the solvent exchange process of as-synthesized samples, materials were thermally activated under vacuum and tested as adsorbents for different probe molecules. Activated materials displayed small surface areas and moderate carbon dioxide storage capacities at low and high pressures, comparable with other MTB-containing compounds. Moreover, CO2 adsorption isotherms showed hysteresis at low pressure, respectively reduced adsorbed amount of carbon dioxide at repeated high pressure measurements, indicating an interaction of CO2 molecules with the frameworks. The results of methane adsorption measurements showed that the compounds do not adsorb CH4 at low pressures (0.3 bar for UPJS-7′′, UPJS-8′′ and 0.8 for UPJS-9′′) and with increasing pressure the adsorbed volume increase. From observed results, it can be concluded that the compounds can be used in the separation process of methane and CO2 at low pressures. Moreover, compounds displayed increased adsorbed volume of hydrogen with a sorption capacity of 3.7–1.8 wt% at −196 °C and 800 Torr, which can be explained by the presence of coordinatively unsaturated sites in the MOF's frameworks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the VEGA project no. 1/0745/17 and project from P. J. Safarik University no. VVGS-2019-1033. Authors M. A. and V. Z. thank the Ministry of Education, Science, Research and Sport of the Slovak Republic and the Accreditation commission for the financial support of the TRIANGEL team in the frame of the scheme “Top Research Teams in Slovakia”. M. A. thanks to Dr Arnost Zukal from Heyrovsky Institute of the ASCR (Prague, Czech Republic) for hydrogen adsorption measurements. M. A. thanks to Prof. V. A. Blatov from Samara University in Russia for support with topological analysis of the frameworks. R. G. is grateful for funding provided by OP VVV “Excellent Research Teams”, project CZ.02.1.01/0.0/0.0/15_003/0000417 – CUCAM.References
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC; G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC; M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC; A. Evans, M. Cummings, D. Decarolis, D. Gianolio, S. Shahid, G. Law, M. Attfield, D. Law and C. Petit, RSC Adv., 2020, 10, 5152–5162 RSC.
- R. Fang, A. Dhakshinamoorthy, Y. Li and H. Garcia, Chem. Soc. Rev., 2020, 49, 3638–3687 RSC; P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC; L. Zhao, Y. Zhang, S. Bi and Q. Liu, RSC Adv., 2019, 9, 19236–19242 RSC; J. C. Mansano Willig, G. Granetto, D. Reginato, F. R. Dutra, É. F. Poruczinski, I. M. de Oliveira, H. A. Stefani, S. D. de Campos, É. A. de Campos, F. Manarin and G. V. Botteselle, RSC Adv., 2020, 10, 3407–3415 RSC.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS; R. S. Forgan, Dalton Trans., 2019, 48, 9037–9042 RSC; R. J. Balasamy, V. Ravinayagam, M. Alomari, M. A. Ansari, S. A. Almofty, S. Rehman, H. Dafalla, P. R. Marimuthu, S. Akhtar and M. Al Hamad, RSC Adv., 2019, 9, 42395–42408 RSC; C. Lin, H. He, Y. Zhang, M. Xu, F. Tian, L. Li and Y. Wang, RSC Adv., 2020, 10, 3084–3091 RSC.
- G. M. Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC; S. P. Bera, A. Mondal, S. Roy, B. Dey, A. Santra and S. Konar, Dalton Trans., 2018, 47, 15405–15415 RSC; S. Kuyuldar, D. T. Genna and C. Burda, J. Mater. Chem. A, 2019, 7, 21545–21576 RSC; M. Almáši, V. Zeleňák and A. Zeleňáková, Acta Phys. Pol., A, 2017, 131, 991–993 CrossRef CAS; V. Zeleňák, M. Almáši, A. Zeleňáková, P. Hrubovčák, R. Tarasenko, S. Bourelly and P. Llewellyn, Sci. Rep., 2019, 9, 15572 CrossRef PubMed.
- X. Xiao, L. Zou, H. Pang and Q. Xu, Chem. Soc. Rev., 2020, 49, 301–331 RSC; W. Ye, K. Wang, W. Yin, W. Chai, Y. Rui and B. Tang, Dalton Trans., 2019, 48, 10191–10198 RSC; N. Mohamed and N. K. Allam, RSC Adv., 2020, 10, 21662–21685 RSC; D. Capková, M. Almáši, T. Kazda, O. Čech, N. Király, P. Čudek, A. Straková Fedorková and V. Hornebecq, Electrochim. Acta, 2020, 354, 136640 CrossRef CAS; A. V. Desai, V. Pimenta, C. King, D. B. Cordes, A. M. Z. Slawin, R. E. Morris and A. R. Armstrong, RSC Adv., 2020, 10, 13732–13736 RSC.
- K. K. Gangu, S. Maddila, S. B. Mukkamala and S. B. Jonnalagadd, J. Energy Chem., 2019, 30, 132–144 CrossRef CAS; S. Yu, G. Jing, S. Li, Z. Li and X. Ju, Int. J. Hydrogen Energy, 2020, 45, 6757–6764 CrossRef; D. Pukazhselvan, K. S. Sandhya and D. P. Fagg, Mater. Today, 2020, 97–163 Search PubMed.
- T. K. Pal, D. De and P. K. Bharadwaj, Coord. Chem. Rev., 2020, 408, 213173 CrossRef CAS; T. Ghanbari, F. Abnisa and W. M. A. W. Daud, Sci. Total Environ., 2020, 707, 135090 CrossRef PubMed; Q. Yin, C. Lu, S. Zhang, M. Liu, K. Du, L. Zhang and G. Chang, RSC Adv., 2019, 9, 22604–22608 RSC.
- L. Asgharnejad, A. Abbasi, M. Najafi and J. Janczak, Cryst. Growth Des., 2019, 19, 2679–2686 CrossRef CAS; J. Zhang, Z. Li, Y. Wu, X. Guo, J. Ye, B. Yuan, S. Wang and L. Jiang, RSC Adv., 2019, 9, 408–428 RSC; S. A. Diamantis, A. D. Pournara, A. G. Hatzidimitriou, M. J. Manos, G. S. Papaefstathiou and T. Lazarides, Polyhedron, 2018, 153, 173–180 CrossRef.
- H. Chun, D. Kim, D. N. Dybtsev and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 971 CrossRef CAS PubMed; H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef PubMed; K. Q. Hu, X. Jiang, C. Z. Wang, L. Mei, Z. N. Xie, W. Q. Tao, X. L. Zhang, Z. F. Chai and W. Q. Shi, Chem.–Eur. J., 2017, 23, 529–532 CrossRef PubMed.
- L. Wen, P. Cheng and W. Lin, Chem. Sci., 2012, 3, 2288–2292 RSC; D. Feng, K. Wang, Z. Wei, Y. P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T. F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H. C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed; M. Zhang, Y. P. Chen, M. Bosch, T. Gentle III, K. Wang, D. Feng, Z. U. Wang and H. C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 815–818 CrossRef PubMed; Y. Wang, Q. Liu, Q. Zhang, B. Peng and H. Deng, Angew. Chem., Int. Ed., 2018, 57, 7120–7125 CrossRef PubMed.
- D. Liu, Z. Xie, L. Ma and W. Lin, Inorg. Chem., 2010, 49, 9107–9109 CrossRef CAS PubMed.
- D. Liu, H. Wu, S. Wang, Z. Xie, J. Li and W. Lin, Chem. Sci., 2012, 3, 3032–3037 RSC.
- Q. Zhai, Q. Lin, T. Wu, S. T. Zheng, X. Bu and P. Feng, Dalton Trans., 2012, 41, 2866–2868 RSC , (H2); A. Biswas, M. B. Kim, S. Y. Kim, T. U. Yoon, S. I. Kim and Y. S. Bae, RSC Adv., 2016, 6, 81485–81490 RSC , (H2, CO2); D. Cattaneo, S. J. Warrender, M. J. Duncan, R. Castledine, N. Parkinson, I. Haley and R. E. Morris, Dalton Trans., 2016, 45, 618–629 RSC , (NO); S. Simonato, J. Möllmer, M. Lange, R. Gläser, R. Staudt and C. Feldmann, RSC Adv., 2016, 6, 12446–12452 RSC , (CO2); D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059–14067 RSC , (NO); M. Sagastuy-Breña, P. G. M. Mileo, E. Sánchez-González, J. E. Reynolds, T. Jurado-Vázquez, J. Balmaseda, E. González-Zamora, S. Devautour-Vinot, S. M. Humphrey, G. Maurin and I. A. Ibarra, Dalton Trans., 2018, 47, 15827–15834 RSC , (CO2); M. W. Kadi, H. M. Abd El Salam, T. Zaki and R. M. Mohamed, J. Nanopart. Res., 2020, 22, 143 CrossRef CAS , (CO2); P. Vervoorts, A. Schneemann, I. Hante, J. Pirillo, Y. Hijikata, T. Toyao, K. Kon, K. Shimizu, T. Nakamura, S. Noro and R. A. Fischer, ACS Appl. Mater. Interfaces, 2020, 12, 9448–9456 CrossRef PubMed , (CxHx); J. Qian, G. Chen, S. Xiao, H. Li, Y. Ouyang and Q. Wang, RSC Adv., 2020, 10, 17195–17204 RSC , (Xe/Kr).
- Z. Zhang, Y. Xiao, M. Cui, J. Tang, Z. Fei, Q. Liu, X. Chen and X. Qiao, Dalton Trans., 2019, 48, 14971–14974 RSC; P. Leo, G. Orcajo, D. Briones, A. Rodríguez-Diéguez, D. Choquesillo-Lazarte, G. Calleja and F. Martínez, Dalton Trans., 2019, 48, 11556–11564 RSC.
- Y. Noori and K. Akhbari, RSC Adv., 2017, 7, 1782–1808 RSC; M. Marieeswaran and P. Panneerselvam, RSC Adv., 2020, 10, 3705–3714 RSC.
- X. X. Xie, Y. C. Yang, B. H. Dou, Z. F. Li and G. Li, Coord. Chem. Rev., 2020, 403, 213100 CrossRef CAS; M. K. Sarango-Ramírez, D. W. Lim, D. I. Kolokolov, A. E. Khudozhitkov, A. G. Stepanov and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 6861–6865 CrossRef PubMed.
- M. Almáši, V. Zeleňák, R. Gyepes, A. Zukal and J. Čejka, Colloids Surf., A, 2013, 437, 101–107 CrossRef.
- M. Almáši, V. Zeleňák, A. Zukal, J. Kuchár and J. Čejka, Dalton Trans., 2016, 45, 1233–1242 RSC.
- M. Almáši, V. Zeleňák, M. Opanasenko and J. Čejka, Dalton Trans., 2014, 43, 3730–3738 RSC.
- M. Almáši, V. Zeleňák, R. Gyepes, S. Bourrelly, M. V. Opanasenko, P. L. Llewellyn and J. Čejka, Inorg. Chem., 2018, 57, 1774–1786 CrossRef PubMed.
- M. Almáši, V. Zeleňák, M. Opanasenko and J. Čejka, Catal. Lett., 2018, 148, 2263–2273 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, 3, C71 Search PubMed; A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Wiersum, C. Giovannangeli, D. Vincent, E. Bloch, H. Reinsch, N. Stock, J. S. Lee, J. S. Chang and P. L. Llewellyn, ACS Comb. Sci., 2013, 15, 111–119 CrossRef CAS PubMed.
- E. W. Lemmon and M. H. McLinden, MO Reference Fluid Thermodynamic and Transport Properties, REFPROP 8.0, National Institute of Standards and Technology, Gaithersburg, MD, 2007 Search PubMed.
- S. Khoshhal, A. A. Ghoreyshi, M. Jahanshahi and M. Mohammadi, RSC Adv., 2015, 5, 24758–24768 RSC; M. Almáši, V. Zeleňák, A. Zeleňáková, Z. Vargová and I. Císařová, Inorg. Chem. Commun., 2016, 74, 66–71 CrossRef CAS; S. M. Vornholt, S. E. Henkelis and R. E. Morris, Dalton Trans., 2017, 46, 8298–8303 RSC; R. Seetharaj, P. V. Vandana, P. Arya and S. Mathew, Arabian J. Chem., 2019, 12, 295–315 CrossRef.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; J. Liu, B. Lukose, O. Shekhah, H. K. Arslan, P. Weidler, H. Gliemann, S. Bräse, S. Grosjean, A. Godt, X. Feng, K. Müllen, I. B. Magdau, T. Heine and C. Wöll, Sci. Rep., 2012, 2, 921 CrossRef PubMed.
- H. Lin and P. A. Maggard, Inorg. Chem., 2008, 47, 8044–8052 CrossRef CAS PubMed; D. Sun, Z. H. Wei, C. F. Yang, D. F. Wang, N. Zhang, R. B. Huang and L. S. Zhang, CrystEngComm, 2011, 13, 1591–1601 RSC; S. M. Fang, M. Hu, Q. Zhang, M. Du and C. S. Liu, Dalton Trans., 2011, 40, 4527–4541 RSC.
- I. Stein and U. Ruschewitz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m141–m143 CrossRef CAS; K. Stahl, J. E. T. Andersen and S. Christgau, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m144–m149 CrossRef PubMed; P. C. Cheng, J. X. Zhan, C. Y. Wu and C. H. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1647 CrossRef PubMed; T. Kundu, S. C. Sahoo and R. Banerjee, Chem. Commun., 2012, 48, 4998–5000 RSC.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- Y. E. Cheon and M. P. Suh, Chem.–Eur. J., 2008, 14, 3961–3967 CrossRef CAS PubMed.
- X. Jiang and H. Z. Kou, Chem. Commun., 2016, 52, 2952–2955 RSC.
- Y. E. Cheon and M. P. Suh, Chem. Commun., 2009, 2296–2298 RSC.
- L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905–9908 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- P. Lama, H. Aggarwal, C. X. Bezuidenhout and L. J. Barbour, Angew. Chem., Int. Ed., 2016, 55, 13271–13275 CrossRef CAS PubMed; H. Wu, C. G. Thibault, H. Wang, K. A. Cychosz, M. Thommes and J. Li, Mirop. Mesop. Mater., 2016, 219, 186–189 CrossRef.
- D. Lv, R. Shi, Y. Chen, Y. Chen, H. Wu, X. Zhou, H. Xi, Z. Li and Q. Xia, Chem. Commun., 2018, 57, 12215–12224 CAS.
- V. Zeleňák, Z. Vargová, M. Almáši, A. Zeleňáková and J. Kuchár, Mirop. Mesop. Mater., 2010, 129, 354–359 CrossRef.
- D. Banerjee, Z. Zhang, A. M. Plonka, J. Li and J. B. Parise, Cryst. Growth Des., 2012, 12, 2162–2165 CrossRef CAS.
- A. Mallick, E. M. Schon, T. Panda, K. Sreenivas, D. D. Diaz and R. Banerjee, J. Mater. Chem., 2012, 22, 14951–14963 RSC.
- K. Noh, N. Ko, H. J. Park, S. Y. Park and J. Kim, CrystEngComm, 2014, 16, 8664–8668 RSC.
- C. T. Yeh, W. C. Lin, S. H. Lo, C. C. Kao, C. H. Lin and C. C. Yang, CrystEngComm, 2012, 14, 1219–1222 RSC.
- M. L. Foo, S. Horike and S. Kitagawa, Inorg. Chem., 2011, 50, 11853–11855 CrossRef CAS PubMed.
- Q. Wang, S. Pan, Y. B. Wu, G. Deng, J. H. Bian, G. Wang, L. Zhao, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2019, 58, 17365–17374 CrossRef CAS PubMed; P. Stegner, C. Färber, J. Oetzel, U. Siemeling, M. Wiesinger, J. Langer, S. Pan, N. Holzmann, G. Frenking, U. Albold, B. Sarkar and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 14615–14620 CrossRef PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2017, 5, 32–51 RSC.
- S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun, 2013, 49, 7773–7775 RSC.
- M. L. Foo, S. Horike, Y. Inubushi and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 6107–6111 CrossRef CAS PubMed.
Footnotes |
| † This article is dedicated to the memory of our colleague Dr Arnošt Zukal. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1978636–1978639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05145d |
| This journal is © The Royal Society of Chemistry 2020 |