
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and anti-osteoporosis activity of novel Teriparatide glycosylation derivatives†
Nan Wang‡
 a,
Jingyang Li‡b,
Hui Songa,
Chao Liua,
Honggang Hua,
Hongli Liao*c and
Wei Cong*a
a,
Jingyang Li‡b,
Hui Songa,
Chao Liua,
Honggang Hua,
Hongli Liao*c and
Wei Cong*a
aInstitute of Translational Medicine, Shanghai University, Shanghai, China. E-mail: viviencong@shu.edu.cn
bDepartment of Pediatric Respiratory Medicine, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
cSchool of Pharmacy, Chengdu Medical College, Chengdu, China. E-mail: liaohl213@126.com
First published on 7th July 2020
Abstract
Osteoporosis is a metabolic bone disease that is characterized by low bone mass and micro-architectural deterioration of bones. The mechanism underlying this disease implicates an imbalance between bone resorption and bone remodeling. In 2002, the US Food and Drug Administration (FDA) approved Teriparatide for the treatment of osteoporosis, and so far, this compound is the only permitted osteoanabolic. However, as a structurally flexible linear peptide, this drug may be further optimized. In this study, we develop a series of novel N-acetyl glucosamine glycosylation derivatives of Teriparatide and examine their characteristics. Of the analyzed compounds, PTHG-9 exhibits enhanced helicity, greater protease stability, and increased osteoblast differentiation promoting ability compared with the original Teriparatide. Accordingly, PTHG-9 is suggested as a therapeutic candidate for postmenopausal osteoporosis (PMOP) and other related diseases. The successful development of an enhanced osteoporosis drug proves that the method proposed herein can be used to effectively enhance the chemical and biological properties of linear peptides with various biological functions.
Introduction
Osteoporosis is defined as the systemic degradation of bone micro-architecture due to low bone mineral density (BMD) and poor bone quality. As such, this disease is associated with a high risk of fragility fractures.1–3 Indeed, more than 8.9 million cases of osteoporosis-stimulated fractures are reported yearly on a global level. Considering the great influence and serious consequences of osteoporosis, this disease is classified by the World Health Organization (WHO) as a major public health concern.4–6To restore proper bone structure and treat osteoporosis, it is important to re-establish the balance between bone resorption and bone formation. Numerous studies have investigated this issue, and many therapeutic agents have been proposed, including antiresorptives such as bisphosphonates, selective estrogen receptor modulator (SERM), and salmon calcitonin.7–9
The parathyroid hormone (PTH1–84) secreted by the parathyroid glands has been suggested as an effective agent for the treatment of osteoporosis, as it is implicated in the regulation of phosphorous and calcium metabolisms, as well as in neuromuscular excitation and blood clotting. In a previous study, it was shown that PTH1–84 has osteoanabolic activity, and that daily, subcutaneous, low-dose injections of this hormone could promote bone formation.10–12
Teriparatide (PTH1–34) is a human parathyroid hormone N-terminal analogue of PTH1–84 that is created using genetic engineering recombination technology. This analogue retains the same properties as the natural hormone, and thus, it is capable of binding to PTH/PTHrP receptors and activating downstream osteoblast signalling, which leads to enhanced bone formation and increased bone density in osteoporosis patients.10,11 Today, PTH1–34 is the only osteoanabolic approved by the U.S. Food and Drug Administration (FDA).13–15 However, the osteoanabolic effectiveness of PTH1–34 may be further improved, especially considering that linear peptides such as PTH1–34 are generally characterized by low oral bioavailability, poor metabolic stability, poor membrane permeability, and rapid clearance.16 Moreover, linear peptides are usually highly unstructured, resulting in a low intrinsic secondary structure in solution, which constitutes a major problem for target binding.17
Previously, we had reported that glycosylation can improve the properties and functions of peptides. Specifically, glycosylation increases peptide hydrophilicity and oral bioavailability, forces peptides to adopt certain conformations and/or stabilizes the existing conformations, and enhances peptide resistance to proteolytic cleavage.18,19 The osteoprotegerin (OPG) peptidomimetic and N-acetyl glucosamine glycosylation strategy designed by our group leads to more stable secondary structures and stronger chymotrypsin resistance, resulting in greater osteoclast inhibitory activity.20 Based on these results, we believe that glycosylation can also be an effective strategy for improving the chemical and biological properties of Teriparatide. In this study, by replacing all the asparagine and serine residues of Teriparatide one by one with the pre-made O-link and N-link N-acetyl glucosamine amino acid building blocks, we synthesize N-acetyl glucosamine glycosylation derivatives of Teriparatide (Fig. 1B, 4), and we demonstrate that these derivatives exhibit a more stable secondary structure conformation, increased protease resistance, and higher anti-osteoporosis activity, compared with the original compound.
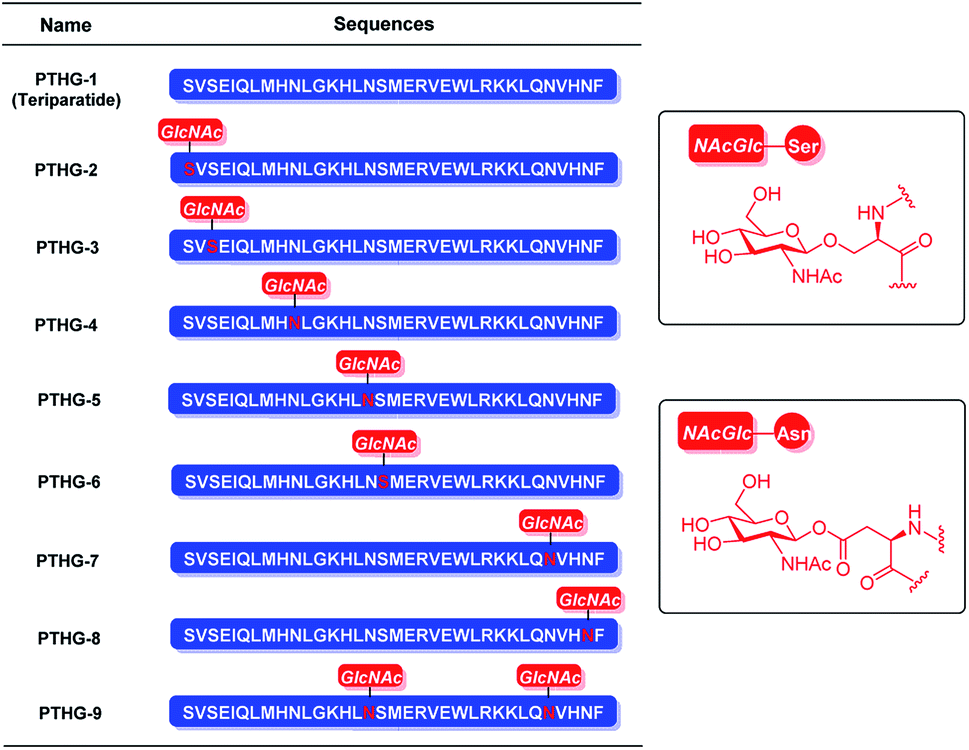 | ||
| Fig. 1 Structures of Teriparatide (PTHG-1) and its N-acetyl glucosamine glycosylation derivatives (PTHG-2–9). | ||
Results and discussion
The Teriparatide glycosylation derivatives were synthesized using the key glycoamino acid building blocks, Fmoc-Asn(Ac3GlcNAc)-OH (4)21–23 and Fmoc-Ser(Ac3GlcNAc)-OH (6).23 To prepare compound 4, N-acetyl-β-D-glucosamine (1) was selected as starting material, and it was reacted with acetyl chloride to obtain the acetyl-protected chloroside intermediate. This intermediate was then reacted with sodium azide, and the two-step reaction was catalyzed by tetrabutylammonium iodide (phase transfer catalyst). The yield of the resulting product (compound 2) was 82.4%. Subsequently, the fully protected glycoamino acid 3 was formed at 71.2% yield by coupling the reduced azide group of compound 2 with Fmoc-Asp-OtBu through HOBt/DIC. Finally, compound 3 was deprotected using a mixture of TFA and DCM, and the resulting building block 4 was obtained in 95.3% yield (Scheme 1A). | ||
Scheme 1 Synthetic route of Fmoc-Asn(Ac3GlcNAc)-OH (4) and Fmoc-Ser(Ac3GlcNAc)-OH (6). Reagents and conditions: (a) (i) acetyl chloride, r.t., 4 days; (ii) NaN3, Bu4NI, DCM/water, r.t., 2 h, 82.4% in 2 steps; (b) (i) H2, Pd/C, MeOH, r.t., overnight; (ii) Fmoc-Asp-OtBu, HOBt, DIC, DMF, r.t., overnight, 71.2% in 2 steps; (c) DCM/TFA (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), r.t., 2 h, 95.3%; (d) (i) 4 Å molecular sieves, BF3·Et2O, DCM, 0 °C, 12 h; (ii) Et3N, Fmoc-Ser-OH, DCM/MeCN (1:2, v/v), r.t., 3 days, 32% in 2 steps. :1, v/v), r.t., 2 h, 95.3%; (d) (i) 4 Å molecular sieves, BF3·Et2O, DCM, 0 °C, 12 h; (ii) Et3N, Fmoc-Ser-OH, DCM/MeCN (1:2, v/v), r.t., 3 days, 32% in 2 steps. | ||
However, building block 6 was generated by direct glycosylation between 1,3,4,6-tetra-O-acetyl-N-acetyl-β-D-glucosamine (5) and Fmoc-Ser-OH, with successive BF3·Et2O and Et3N treatment (Scheme 1B).
With the key glycoamino acid building blocks in hand, the Teriparatide glycosylation derivatives were synthesized. For example, PTHG-2 was synthesized by incorporating building block 6 into the peptide backbone via standard Fmoc SPPS, using O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) as a coupling reagent (Scheme 2). The acetyl protection group of the resulting on-resin fully protected peptide 7 was subsequently deprotected using hydrazine hydrate DMF solution, and on-resin intermediate 8 was produced. After acidic cleavage and global deprotection, the crude glycopeptide was obtained (Scheme 2), and then it was purified by preparative high-performance liquid chromatography. The yield of the pure PTHG-2 product was found to be 25.0%.
 | ||
| Scheme 2 Synthetic route of PTHG-2. Reagents and conditions: (a) 10% hydrazine hydrate/DMF, r.t., overnight; (b) TFA/EDT/TIPS/water (95:2:2:1, v/v/v/v), r.t., 2 h, 25%; the resin-bound peptides were protected on side chains at asterisk sites. The following protecting groups for amino acid side chains were used: tert-butyl (tBu; for Glu and Ser), 2,2,4,6,7-pentamethyldihydrobenzo-furane-5-sulfonyl (pbf; for Arg), tert-butyloxycarbonyl (tBoc; for Lys and His) and trityl (Trt; for Asn). | ||
The secondary structures of the synthesized glycopeptides were analysed by circular dichroism (CD). As shown in Fig. 2A, the helicity values of the Teriparatide derivatives ranged between 26.1% and 39.2%, compared with 26.1% for the original Teriparatide (PTHG-1). This confirms that N-acetyl glucosamine glycosylation can improve the helicity of peptides. Among the investigated compounds, PTHG-6, PTHG-8, and PTHG-9 showed the highest degrees of helicity (39.2%, 36.7%, and 36.1%, respectively), with values that are 1.50-, 1.40-, and 1.38-fold greater than that of PTHG-1, respectively.
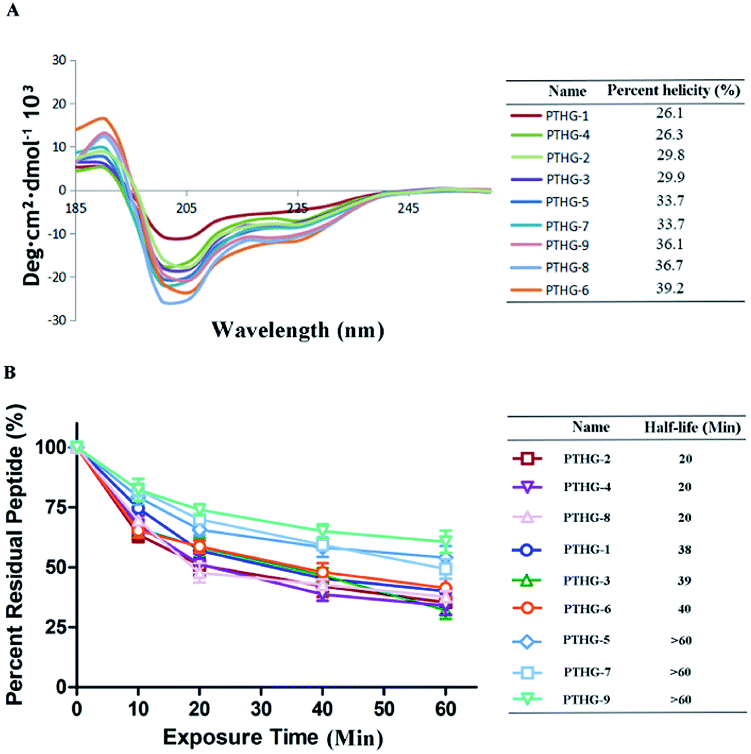 | ||
| Fig. 2 (A) CD spectra of the glycosylation derivatives in 50.0% TFE aqueous solution at 20 °C. (B) Proteolytic stability of glycosylation derivatives under α-chymotrypsin treatment. Data points are displayed as the mean value SEM of duplicate independent experiments. The percent residual peptide was monitored via HPLC. | ||
To assess the protease stability of Teriparatide (PTHG-1) and its derivatives, α-chymotrypsin-mediated degradation tests were performed, and the compounds were monitored by HPLC. As shown in Fig. 2B, 50% of PTHG-1 remained after 60 minutes of protease exposure, compared with 25–75% for the synthesized glycopeptides. Among the investigated compounds, only PTHG-5, PTHG-7, and PTHG-9 had half-lives longer than 60 minutes, and thus, their protease stability is greater than that of the other peptides. These results suggest that the 16- and 30-asparagine residues play extremely significant roles in the process of Teriparatide protease degradation.
The alkaline phosphatase (ALP) activity of Teriparatide (PTHG-1) and its N-acetyl glucosamine glycosylation derivatives (PTHG-2–9) was also analysed. For this purpose, MC3T3-E1 cells were treated with different peptides at a concentration of 50 nM, followed by spectrophotometric and optical density measurements (405 nm). As shown in Fig. 3, the PTHG-1 group presented statistically significant ALP activity, which means that PTHG-1 is capable of promoting osteoblast differentiation, as previously reported in the literature. Compared with PTHG-1, the promoting activities of PTHG-2, PTHG-4, and PTHG-9 were 1.18-, 1.12-, and 1.3-times greater, respectively. However, only the stimulation effect of PTHG-9 was statistically significant. Overall, the results indicate that both protease resistance and conformation stability are important factors contributing to an enhanced osteoblast differentiation ability of Teriparatide and its glycosylation derivatives. This explains the improved activity of PTHG-9 compared with PTHG-1.
 | ||
| Fig. 3 Activity of the glycosylation derivatives at 50 nM concentrations as assessed by alkaline phosphatase (ALP) activity in MC3T3-E1 cells. Data represent the mean ± SEM from three independent experiments: *p < 0.01 as compared with PTHG-1. | ||
Conclusions
A novel series of Teriparatide glycosylation derivatives have been designed and successfully synthesized, with reasonable yield, via the standard Fmoc SPPS strategy using pre-made glycoamino acid building blocks. In vitro data suggest that the analogue PTHG-9 has improved helicity, greater protease stability, and increased osteoblast differentiation promoting ability compared with the original Teriparatide. Therefore, PTHG-9 has great potential for use in the development of novel therapeutics for postmenopausal osteoporosis. More importantly, the derivatization method proposed herein constitutes a useful strategy for improving the chemical and biological properties of linear peptides with various biological functions. Further optimization of PTHG-9 and more in-depth biological evaluation are ongoing in our laboratory, and the results will be disclosed in due course.Experimental section
General information
All reagents were purchased from Acros, Sigma-Aldrich, Alfa Aesar and Adamas. Amino acids were commercial available from GL Biochem Shanghai Co. Ltd. All solvents used were bought from Sinopharm Chemical Reagent Co. Ltd. Dichloromethane (DCM) and N,N-dimethylformamide (DMF) were distilled over calcium hydride (CaH2) under argon atmosphere and stored in flask containing 4 Å molecular sieves. All reactions vessels were oven-dried before use. Reactions were monitored by thin-layer chromatography (TLC) and visualized by UV analyzer (254 nm), ninhydrin and/or phosphomolybdic acid. Peptides were analyzed and purified by reverse phase HPLC. A C18 analytic column (Shimadzu Shim-pack VP-ODS, 4.6 × 250 mm, 5 μm particle size, flow rate 1 mL min−1) was used for analytical RP-HPLC, and a C18 column (Shimadzu Shim-pack PRC-ODS, 50 × 250 mm, 15 μm particle size, flow rate 10 mL min−1) was used for preparative RP-HPLC. The solvents systems were buffer A (0.1% TFA in CH3CN) and buffer B (0.1% TFA in water). Data were recorded and analyzed using the software system LC Solution. High resolution mass spectra (HR-MS) were measured on a Waters Xevo G2 QTOF mass spectrometer. 1H- and 13C-NMR spectrum was recorded on a Bruker Avance 300 MHz instrument. Chemical shifts (δ) were reported relative to TMS (0 ppm) for 1H- and 13C-NMR spectra. The coupling constants (J) were displayed in Hertz (Hz) and the splitting patterns were defined as follows: singlet (s); broad singlet (s, br); doublet (d); doublet of doublet (dd); triplet (t); quartet (q); multiplet (m).(2R,3S,4R,5R,6R)-5-Acetamido-2-(acetoxymethyl)-6-azidotetra-hydro-2H-pyran-3,4-diyl diacetate (2)
To N-acetyl-β-D-glucosamine (25.0 g, 113.1 mmol) was added acetyl chloride (50 mL, 707.0 mmol), dropwise over 15 min at 0 °C. The reaction mixture was stirred vigorously at room temperature for 4 day. This mixture was diluted with DCM (100.00 mL) and saturated NaHCO3 (100 mL) aqueous solution. The organic phase was separated, washed with saturated NaHCO3 and brine (3 × 100.00 mL), dried over Na2SO4 and concentrated, and used without further purification. A mixture of commercially available NaN3 (8.82 g, 135.70 mmol) and tetrabutylammonium iodide (16.68 g, 45.23 mmol) in DCM/water (1:1, 200.00 mL) was stirred for 2 hours at room temperature. The organic layer was separated, washed with brine (3 × 100.00 mL), dried over Na2SO4, concentrated and purified by column chromatography (20:1–1:1, petro ether/EtOAc) to give 2 as a white powder (13.80 g, 82.4%, 2 steps). 1H-NMR (600 MHz, CDCl3): δ 5.81 (d, J = 6 Hz, 1H), 5.27 (t, J = 12 Hz, 1H), 5.12 (t, J = 12 Hz, 1H), 4.79 (d, J = 6 Hz, 1H), 4.28–4.27 (m, 1H), 4.20 (d, J = 6 Hz, 1H), 3.96–3.91 (m, 1H), 3.83–3.81 (m, 1H), 2.12 (s, 3H), 2.05 (d, J = 6 Hz, 6H), 2.00 (s, 3H). 13C-NMR (600 MHz, CDCl3): δ 170.48, 170.17, 169.94, 168.77, 87.91, 73.48, 71.65, 67.58, 61.36, 53.67, 22.73, 20.21, 20.12, 20.07. ESI-MS m/z calcd for C14H20N4O8 372.1281; found [M + H]+ 373.29.
(2R,3S,4R,5R,6R)-6-(3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(tert-butoxy)-4-oxobutanamido)-5-acetamido-2-(acetoxymethyl)tetrahydro-2H-pyran-3,4-diyl diacetate (3)
To a solution of 4 (10.00 g, 26.88 mmol) in MeOH (200.00 mL) was added Pd/C catalyst (1.00 g). Then the mixture was stirred overnight at room temperature under hydrogen. After the Pd/C catalyst was filtered and the MeOH was removed under vacuum, the residue was used without further purification. To a solution of the intermediate in DMF (50.00 mL) was added Fmoc-Asp-OtBu (12.14 g, 29.56 mmol), HOBt (3.99 g, 29.56 mmol) and DCC (3.99 g, 29.56 mmol) mixture of DCM/DMF (4:1, 200.00 mL) solution. Then DIC (3.72 g, 29.56 mmol) was added to the solution. The reaction was stirred overnight at room temperature. Then, the reaction was filtrated and the filtrate was washed successively with 1 M HCl (3 × 100.00 mL), saturated NaHCO3 (3 × 100.00 mL) and brine (3 × 100.00 mL). The organic phase was dried over Na2SO4, filtered, concentrated and purified by column chromatography (100:1–10:1, DCM/MeOH) to give 3 as a white powder (14.10 g, 71.2%, 2 steps). 1H-NMR (600 MHz, CDCl3): δ 7.77 (d, J = 6 Hz, 2H), 7.62 (d, J = 6 Hz, 2H), 7.41 (t, J = 12 Hz, 2H), 7.32 (t, J = 12 Hz, 2H), 7.23 (d, J = 6 Hz, 1H), 6.18 (d, J = 6 Hz, 1H), 5.98 (d, J = 6 Hz, 1H), 5.14–5.09 (m, 3H), 4.55 (s, 1H), 4.44–4.43 (m, 1H), 4.33–4.31 (m, 2H), 4.24 (t, J = 12 Hz, 1H), 4.16–4.13 (m, 1H), 4.08 (d, J = 6 Hz, 1H), 3.78–3.76 (m, 1H); 2.88–2.85 (m, 1H); 2.74–2.71 (m, 1H); 2.54 (s, 2H); 2.09–2.06 (m, 9H); 1.98 (s, 3H); 1.47 (s, 9H). 13C-NMR (600 MHz, CDCl3): δ 171.91, 171.47, 170.60, 170.15, 169.45, 168.74, 155.64, 143.42, 143.31, 140.78, 127.20, 126.57, 124.69, 119.47, 81.75, 79.73, 73.07, 72.40, 67.11, 66.66, 61.17, 53.00, 50.51, 46.64, 37.48, 27.40, 22.53, 20.19, 20.07. ESI-MS m/z calcd for C37H45N3O13 739.2952; found [M + H]+ 740.19.
N2-(((9H-Fluoren-9-yl)methoxy)carbonyl)-N4-((2R,3R,4R,5S,6R)-3-acetamido-4,5-diacetoxy-6-(acetoxymethyl)tetrahydro-2H-pyran-2-yl)asparagines (4)
3 (10.0 g, 13.53 mmol) was dissolved in TFA/DCM (1:1, 50.00 mL) and stirred for 2 hours at room temperature. The reaction mixture was concentrated in vacuo to yield 4 as a white powder (8.77 g, 95.3%) which was used directly in SPPS without further purification. 1H-NMR (600 MHz, DMSO): δ 8.58 (d, J = 6 Hz, 1H), 7.88 (d, J = 6 Hz, 3H), 7.70 (d, J = 6 Hz, 2H), 7.50 (d, J = 6 Hz, 1H), 7.41 (t, J = 12 Hz, 2H), 7.32 (t, J = 12 Hz, 2H), 5.17 (t, J = 12 Hz, 1H), 5.10 (t, J = 12 Hz, 1H), 4.82 (t, J = 12 Hz, 1H), 4.38–4.37 (m, 1H), 4.28–4.17 (m, 4H), 3.95–3.87 (m, 7H), 2.67–2.64 (m, 1H), 1.99–1.96 (m, 7H), 1.90 (s, 3H), 1.72 (s, 3H). 13C-NMR (600 MHz, DMSO): δ 172.91, 170.00, 169.77, 169.46, 169.28, 155.80, 143.76, 140.59, 127.60, 127.05, 125.22, 120.07, 78.05, 73.33, 72.25, 68.36, 65.67, 61.80, 52.09, 49.96, 46.56, 36.82, 22.55, 20.49, 20.36, 20.33. ESI-MS m/z calcd for C33H37N3O13 683.2326; found [M + Na]+ 706.54, [M − H]− 682.39.
L-Serine,N-[(9H-luoren-9-ylmethoxy)carbonyl]-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl) (6)
To a solution of 1,3,4,6-tetra-O-acetyl-N-acetyl-β-D-glucosamine (5, 1.08 g, 4.74 mmol) and molecular sieve (1.08 g) in DCM (100 mL) was added BF3·Et2O (1 mL, 6.26 mmol) at 0 °C. Then the reaction was stirred at room temperature for 12 h. Et3N (0.4 mL, 5.4 mmol) was added to the mixture and stirred for 10 min. A mixture of commercially available Fmoc-Ser-OH (1.2 g, 3.67 mmol) in DCM/MeCN (1:2, 15 mL) was added to the reaction and stirred for 3 days at room temperature. Then the reaction was filtrated and washed with NaHCO3 (3 × 100.00 mL) and brine (3 × 100.00 mL). The organic phase was dried over Na2SO4, filtered, concentrated and purified by column chromatography (100:1–10:1, DCM/MeOH) to give 6 as a white powder (14.1 g, 32%, 2 step). 1H-NMR (600 MHz, DMSO): δ 9.32–9.31 (m, 3H), 9.16–9.14 (m, 2H), 8.84 (t, J = 12 Hz, 2H), 8.76–8.71 (m, 3H), 6.51 (t, J = 12 Hz, 1H), 6.27 (t, J = 12 Hz, 1H), 6.13 (d, J = 6 Hz, 1H), 5.71–5.61 (m, 6H), 5.44–5.39 (m, 3H), 5.20–5.11 (m, 2H), 3.43 (s, 3H), 3.39 (s, 3H), 3.33 (s, 3H), 3.15 (s, 3H). 13C-NMR (600 MHz, DMSO): δ 171.5, 170.03, 169.61, 169.52, 169.23, 155.91, 143.39, 140.69, 127.63, 127.06, 125.24, 125.19, 120.09, 72.49, 70.82, 68.45, 65.83, 61.75, 54.02, 53.02, 46.56, 22.55, 20.45, 20.37, 20.31. ESI-MS m/z calcd for C32H36N2O13 656.2217; found [M − H]− 654.24.
General procedures for the Fmoc solid phase peptide synthesis
The amino acid residues were attached to the resin with a single coupling procedure. All peptides were synthesized with a scale of 0.15 mmol.(a) Standard pre-activation of resin protocol: the resin was swollen in DCM/DMF mixture solvent for 10 minutes.
(b) Standard Fmoc-deprotection protocol: after treatment with 20% piperidine in DMF solution (15 minutes twice) the resin was washed with DMF (×5), DCM (×5), and DMF (×5).
(c) Standard coupling of amino acids protocol: after pre-activation of 4.0 equivalents of Fmoc-protected amino acid in DMF for 5 minutes using 3.8 equivalents of HCTU and 8.0 equivalents of DIPEA, the solution was added to the resin. After 30 minutes, the resin was washed with DMF (×5), DCM (×5), and DMF (×5). The coupling reaction was monitored with the ninhydrin test.
(d) Standard acetylation protocol: a solution of acetic anhydride and pyridine (1:1, v/v) was added to the resin. After 20 minutes, the resin was washed with DMF (×5), DCM (×5), and DMF (×5).
(e) Standard stapling protocol: after removal of N-terminus Fmoc and acetylation successively, 3.0 equivalents of 1st Grubbs' reagent in dry dichloroethane solution was added to the resin. After overnight reaction, the resin was washed with DMF (×5), DCM (×5), and DMF (×5).
(f) Standard cleavage protocol: the cleavage cocktail (TFA:TIPS:phenol:H2O = 88:5:5:2, v/v/v/v) was added to the resin. After stirring for 2 hours, the cleavage cocktail was collected. The solution was bubbled with argon for concentration and the chilled diethyl ether was added to precipitate the crude peptides. The peptide suspensions were centrifuged for 3 minutes at 3000 rpm and then the clear solution was decanted. The step of precipitation, centrifugation and decantation operations was repeated three times. The resulting white residues were dissolved in CH3CN/water, analyzed and purified by RP-HPLC.
Protease stability experiment
Purified peptides was dissolved in DMSO to a final concentration of 1 mM as solution A. α-chymotrypsin was dissolved in PBS buffer (pH = 7.4, containing 2 mM CaCl2) to a final concentration of 0.5 ng μL−1 as solution B. 50 μL solution A and 1950 μL solution B were mixed. After 0, 1, 2, 4, 8 and 12 hours' incubation at 37 °C, the percent residual peptide was monitored by HPLC.CD spectroscopy study
CD measurements were recorded on a JASCO J-820 spectropolarimeter (JASCO Corp., Ltd). Peptides were dissolved in 50.0% TFE aqueous solution with a concentration of 0.1 mg mL−1. UV spectra were recorded at 20 °C in a quartz cell of 10.0 mm path length. Percent helicity (α) was calculated by the follow eqn (1).24,25
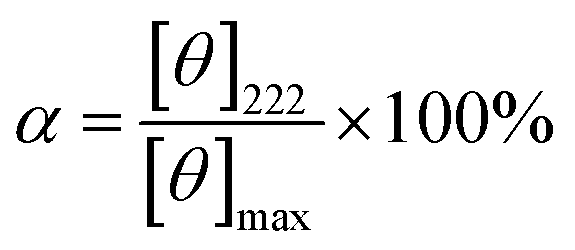 | (1) |
Alkaline phosphatase assay
At 70–80% confluency mice calverial osteoblast were trypsinized and 2 × 103 cells per well were seeded onto 96-well plates for alkaline phosphatase (ALP) measurement. Cells were treated with different concentration of test compound or vehicle for 48 h in α-MEM supplemented with 10% charcoal treated FCS, 10 mM β-glycerophosphate, 50 μg mL−1 ascorbic acid, and 1% penicillin/streptomycin (osteoblast differentiation medium). At the end of incubation period, total ALP activity was measured using p-nitrophenyl phosphate (PNPP) as substrate and absorbance was read at 405 nm.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank LetPub for its linguistic assistance during the preparation of this manuscript. We are grateful to the Key R & D Projects of Science and Technology Department of Sichuan Province (2019YFS0097), the Science and Technology Project of Nantong, China (MS22019026), the National Natural Science Foundation of China (No. 91849129, No. 81703526 and No. 41806088) and instrumental analysis centre of Second Military Medical University for NMR spectroscopic and mass spectrometric analyses.References
- T. D. Rachner, S. Khosla and L. C. Hofbauer, Lancet, 2011, 377, 1276–1287 CrossRef CAS.
- R. Baron, Nat. Rev. Endocrinol., 2012, 8, 76–78 CrossRef CAS PubMed.
- R. Diaz, Nat. Rev. Endocrinol., 2012, 8, 255–262 CrossRef PubMed.
- L. L. Tegola, M. Mattera, S. Cornacchia, X. Cheng and G. Guglielmi, J. Frailty Sarcopenia Falls, 2018, 3, 138–147 Search PubMed.
- Q. Wu, X. Xiao and Y. Xu, J. Clin. Med., 2020, 9, 1–14 Search PubMed.
- M. Zamani, V. Zamani, B. Heidari, H. Parsian and S. M. Esmaeilnejad-Ganji, Archives of Osteoporosis, 2018, 13, 129 CrossRef PubMed.
- J. S. Chen and P. N. Sambrook, Nat. Rev. Endocrinol., 2012, 8, 81–91 CrossRef CAS PubMed.
- B. C. Silva and J. P. Bilezikian, Annu. Rev. Med., 2011, 62, 307–322 CrossRef CAS PubMed.
- A. Wysocki, M. Butler, T. Shamliyan and R. L. Kane, Ann. Intern. Med., 2011, 155, 680–W213 CrossRef PubMed.
- P. Sun, M. Wang and G.-Y. Yin, Biochem. Biophys. Res. Commun., 2020, 525, 850–856 CrossRef CAS PubMed.
- L. L. Ding, F. Wen, H. Wang, D. H. Wang, Q. Liu, Y. X. Mo, X. Tan, M. Qiu and J. X. Hu, Osteoporosis Int., 2020, 31, 961–971 CrossRef CAS PubMed.
- M. E. Kraenzlin and C. Meier, Nat. Rev. Endocrinol., 2011, 7, 647–656 CrossRef CAS PubMed.
- H. Chtioui, F. Lamine and R. Daghfous, N. Engl. J. Med., 2011, 364, 1081–1082 CrossRef CAS PubMed.
- V. Heath, N. Engl. J. Med., 2010, 363, 2396–2405 CrossRef PubMed.
- A. Farooki, M. Fornier and M. Girotra, N. Engl. J. Med., 2007, 357, 2410–2411 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37, 1186–1197 CrossRef CAS PubMed.
- Y. S. Tan, D. P. Lane and C. S. Verma, Drug Discovery Today, 2016, 21, 1642–1653 CrossRef CAS PubMed.
- Y. Zou, Q. Zhao, C. Zhang, L. Wang, W. Li, X. Li, Q. Wu and H. Hu, J. Pept. Sci., 2015, 21, 586–592 CrossRef CAS PubMed.
- H. Hu, J. Xue, B. M. Swarts, Q. Wang, Q. Wu and Z. Guo, J. Med. Chem., 2009, 52, 2052–2059 CrossRef CAS PubMed.
- C. Liu, X. Chen, X. Zhi, W. Weng, Q. Li, X. Li, Y. Zou, J. Su and H.-G. Hu, Eur. J. Med. Chem., 2018, 145, 661–672 CrossRef CAS PubMed.
- B. Premdjee, A. L. Adams and D. Macmillan, Bioorg. Med. Chem. Lett., 2011, 21, 4973–4975 CrossRef CAS PubMed.
- C. Toonstra, M. N. Amin and L. X. Wang, J. Org. Chem., 2016, 81, 6176–6185 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, M. Wu, Q. Chang, H. Hu and X. Zhao, ACS Chem. Biol., 2019, 14, 516–525 CrossRef CAS PubMed.
- D. Wang, K. Chen, J. L. Kulp III and P. S. Arora, J. Am. Chem. Soc., 2006, 128, 9248–9256 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05136e |
| ‡ Nan Wang and Jingyang Li contributes equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |