
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReporter-recruiting bifunctional aptasensor for bioluminescent analytical assays†
Anna Davydovaa,
Vasilisa Krasitskayab,
Pavel Vorobjevac,
Valentina Timoshenkoa,
Alexey Tupikina,
Marsel Kabilov a,
Ludmila Frankbd,
Alya Venyaminovaa and
Mariya Vorobyeva*a
a,
Ludmila Frankbd,
Alya Venyaminovaa and
Mariya Vorobyeva*a
aInstitute of Chemical Biology and Fundamental Medicine SB RAS, Novosibirsk 630090, Russia. E-mail: maria.vorobjeva@gmail.com
bInstitute of Biophysics SB RAS, Federal Research Center “Krasnoyarsk Science Center SB RAS”, Krasnoyarsk 660036, Russia
cNovosibirsk State University, Pirogova St., 2, 630090 Novosibirsk, Russia
dSiberian Federal University, Krasnoyarsk 660041, Russia
First published on 1st September 2020
Abstract
We report a novel bioluminescent aptasensor, which consists of 2′-F-RNA aptamer modules joined into a bi-specific aptamer construct. One aptamer module binds the analyte, then after structural rearrangement the second module recruits non-covalently Ca2+-dependent photoprotein obelin from the solution, thus providing a bioluminescent signal. This concept allows using free protein as a reporter, which brings such advantages as no need for aptamer–protein conjugation, a possibility of thermal re-folding of aptamer component with no harm to a protein, and simpler detection protocol. We developed the new 2′-F-RNA aptamer for obelin, and proposed the strategy for engineering structure-switching bi-modular aptamer constructs which bind the analyte and the obelin in a sequential manner. With the use of hemoglobin as a model analyte, we showed the feasibility of utilizing the aptasensor in a fast and straightforward bioluminescent microplate assay. With a proper design of a secondary structure, this strategy of aptasensor engineering might be further extended to bi-specific aptamer-based bioluminescent sensors for other analytes of interest.
Introduction
Nucleic acid aptamers have been known for more than 25 years as highly specific and affine binders. Almost any molecule of interest can serve as a target for developing aptamers by a SELEX (Systematic Evolution of Ligands by EXponential Enrichment) technology. Nowadays, aptamers represent the most promising alternative to monoclonal antibodies for analytical and therapeutic applications.1,2 Their target binding affinity and specificity are comparable to those of antibodies, while aptamers possess a lot of unique advantages, such as generation by in vitro selection on a lab bench; relatively easy, reproducible, and cost-effective chemical synthesis; long shelf-life; minimal batch-to-batch variation; and a broad spectrum of chemical modifications.3Aptamers continue to find numerous applications in molecular engineering for bioanalysis, medical therapeutics, and diagnostics. By their very nature, aptamers are made of nucleic acids, which brings several advantages to the smart design of aptamer-based molecular constructs.
First, the functional activity of an aptamer is determined by its spatial structure. The latter, in turn, relies on intermolecular complementary base-pairing and thus can be regulated by altering the aptamer's length and nucleotide sequence. The ability of aptamers to change their conformation after target binding gives ample opportunities to design biosensing platforms. For instance, a large variety of nanomaterial-assisted aptasensors have been developed, detecting the binding event after a conformational change (see the review4).
Second, aptamers as building blocks are very compatible, so one can combine them in a “Lego-like” manner to obtain complex molecules with tuneable properties depending on the certain research task. More to the point, the design of such joint molecules hangs upon well-established algorithms of nucleic acids' secondary structure prediction.5 So-called multivalent aptamer constructs can comprise several copies of the same aptamer to increase the binding avidity. Otherwise, joining aptamers to different targets opens the widest possibilities to obtain a desired repertoire of functionalities.6 Specifically, bioanalytical systems of this type can combine two aptamer modules: one for the analyte recognition, and the other for recruiting a reporter molecule which, in turn, provides an analytical signal.7–10 Once generated, the aptamer module that non-covalently immobilizes the reporting group could then be used for the engineering of bi-aptameric constructs for different analytes.
In the present study, we aimed to create bioanalytical bi-modular aptamer constructs that recruit Ca2+-regulated photoprotein obelin as a reporter. Obelin is an extraordinarily sensitive, triggerable reporter suitable for different bioluminescent assays.11 Particularly, we have previously demonstrated that obelin covalently attached to the specific aptamer gives a very sensitive and selective reporter for the bioluminescent aptasensor.12–14 So far, bioluminescent proteins are quite rarely used as reporter groups for aptamer-based assays, although they provide excellent analytical response and signal-to-noise ratio. For example, Moutsiopoulou et al. recently reported the aptamer beacon biosensing system comprising structure-switching anti-IFNγ DNA aptamer covalently joined with Gaussia luciferase and its inhibitor.15 However, the covalent joining of the aptamer and the protein, may narrow the possibilities for thermal denaturation and refolding of the aptamer due to the risk of protein deactivation. Here, we propose a fundamentally different approach to the engineering of bioluminescent aptasensors: the non-covalent recruitment of obelin, which does not require chemical conjugation or engineering of a fused protein to incorporate the reporter into the bioanalytical system. Moreover, prior to the obelin addition, the analytical system comprises only the nucleic acid and may undergo denaturation/renaturation steps with no risk of deactivation for the reporter protein. A number of aptamers capable of binding small fluorogens and enhancing their fluorescence have been described to the moment, and successfully employed for RNA imaging.16 Nevertheless, to the best of our knowledge, the only example of an aptamer for a reporter protein is the GFP-binding one.17 We selected and rationally truncated the novel 2′-F-RNA aptamer for obelin, and proposed a strategy for its integration into bi-modular structure-switching construct. The proof of concept for this strategy was demonstrated by the example of bi-functional constructs built of obelin-binding and hemoglobin-binding 2′-F-RNA aptamers which were successfully employed in the microplate bioluminescent assay.
Experimental
Materials and reagents
Streptavidin, human hemoglobin (Hb), bovine serum albumin (BSA), SYBR Green I, Tween 20 and other reagents were purchased from Sigma-Aldrich (St. Louis, USA). Ni Sepharose 6 FF resin and His Mag Sepharose Ni magnetic beads (MB) were purchased from GE Healthcare (United Kingdom). “UltraMild” protected nucleoside β-cyanoethyl phosphoramidites and modified polymer supports for the synthesis of oligonucleotides were purchased from Glen Research (Sterling, USA). 2′-F-Modified pyrimidine triphosphates were purchased from Nanotech-C (Novosibirsk, Russia). Dulbecco's phosphate buffered saline (DPBS), T7 RNA polymerase, inorganic pyrophosphatase, DNase I, RevertAid reverse polymerase, FastAP thermosensitive alkaline phosphatase, T4 polynucleotide kinase and unmodified ribonucleoside triphosphates were purchased from Thermo Fisher Scientific (Waltham, USA). Taq DNA polymerase, and deoxyribonucleoside triphosphates were purchased from Biosan (Novosibirsk, Russia).Target proteins
His-Obe carrying His6 sequence at the N-terminus was obtained as described in ref. 18. Wild type obelin (wt-Obe), aequorin and clytin were obtained according to the methods, described in ref. 19 and 20.Synthesis of nucleic acid libraries, primers, and 2′-F-RNA aptamers
Single-stranded DNA library, primers, and individual 2′-F pyrimidine (C and U) modified RNA aptamers were synthesized by the solid phase phosphoramidite method on 0.4 μmol scale on an automated DNA/RNA synthesizer ASM-800 (Biosset, Novosibirsk, Russia) using protocols optimized for the instrument. The ssDNA random library was 5′-GCCTGTTGTGAGCCTCCTGTCGAAN40TTGAGCGTTTATTCTTGTCTCCC-3′ (where N represents a deoxynucleotide (A, T, G, or C)). The PCR primers were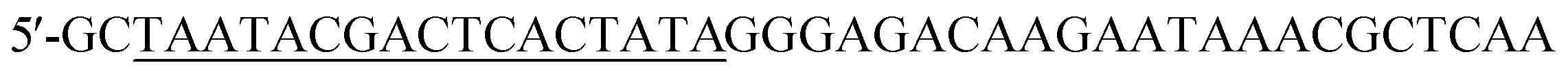 (forward primer, T7 promoter sequence underlined) and 5′-GCCTGTTGTGAGCCTCCTGTCGAA (reverse primer). To obtain the ssDNA library with a smooth distribution of all four nucleotides in the randomized region, we optimized the molar ratio of nucleoside phosphoramidites for the automated chemical synthesis of the N40 fragment.21
(forward primer, T7 promoter sequence underlined) and 5′-GCCTGTTGTGAGCCTCCTGTCGAA (reverse primer). To obtain the ssDNA library with a smooth distribution of all four nucleotides in the randomized region, we optimized the molar ratio of nucleoside phosphoramidites for the automated chemical synthesis of the N40 fragment.21
Biotinylated aptamers were synthesized from 3′-aminohexyl containing 2′-F-RNA aptamers by incubation in 0.1 M Na2B4O7 with a 100-fold molar excess of biotin N-hydroxysuccinimide ester (Merck, Germany) for 2.5 h at 25 °C. The product was precipitated by 2% NaClO4 in acetone, washed twice with acetone and diluted in water. The excess reagent was removed by centrifugation using an Amicon Ultra-0.5 mL Centrifugal Filters 3 K device (Merck, Germany).
The dsDNA library was obtained by the primer extension using the Klenow fragment of E. coli DNA polymerase I. The product of the extension reaction was purified with the MinElute Reaction Cleanup kit (Qiagen, Germany). The purified dsDNA library was then used as a template for in vitro transcription of the 2′-F-modified RNA library. The transcription was performed using 1 mM ATP, GTP and 3 mM 2′-F-UTP, 2′-F-CTP, and T7 RNA polymerase. The resulting 2′-F-modified RNA library was purified by gel filtration and analyzed by denaturing PAGE.
Aptamer selection
For every selection round, the His-Obe protein was immobilized onto Ni Sepharose 6 FF resin or His Mag Sepharose Ni magnetic beads. An aliquot of Ni Sepharose resin (5 μL) was washed three times with 0.5 mL of DPBS buffer containing 1 mM EDTA (DPBSE). His-Obe in DPBSE (20 μg mL−1, 150 μL) was vortexed with the resin at room temperature for 1 h, and then the resin was washed three times with 150 μL of DPBSE.2′-F-Modified RNA library was folded in DPBSE by heating at 90 °C for 5 min and cooled down for 10 min at 25 °C. Then Tween 20, BSA and E. coli tRNA (rounds 1–5) or polyA (rounds 6–12) were added as nonspecific competitors for final concentrations of 0.05%, 0.01%, and 0.1%, respectively. To remove nonspecifically binding RNAs, the initial library was incubated with 5 μL of Ni Sepharose resin at room temperature with mixing for 1 h. Unbound RNAs were collected by centrifugation and mixed with 5 μL of Ni Sepharose with pre-immobilized His-Obe at room temperature for 1 h. Unbound RNAs were washed out with 0.05% Tween 20 in DPBSE. After several washes, RNA–protein complexes were eluted with 20 mM Tris–HCl buffer (pH 7.0) containing 100 mM imidazole. The recovered RNAs were subjected to reverse transcription by RevertAid reverse transcriptase followed by the cDNA amplification and T7 RNA transcription to regenerate an enriched RNA pool. The selection pressure was progressively increased by decreasing the amount of protein and the time of incubation and adding more washes (see details at Table S1†).
High-throughput sequencing and bioinformatic analysis of the data
Double-stranded DNA libraries after the 6th, 10th and 12th SELEX round (1 μg) were re-amplified with the primers containing adapters and barcodes for high-throughput Illumina sequencing. The libraries were sequenced on a MiSeq platform using the 2 × 300 bp paired-ends sequencing kit (Illumina) in Genomics Core Facility (ICBFM SB RAS, Novosibirsk, Russia). Raw sequences were analyzed with UPARSE pipeline22 using Usearch v10.0.240. The UPARSE pipeline included the merging of paired reads, read quality filtering, length trimming, merging of identical reads (dereplication), discarding singleton reads, removing chimeras, and OTU clustering using the UNOISE23 algorithm. Nucleotide sequences of the most represented aptamers are listed in Table S2†.Differential radial capillary action of ligand assay (DRaCALA)
For 5′-[32P]-labeling, 2′-F-RNAs were phosphorylated using γ-[32P]-ATP (Biosan, Novosibirsk, Russia) and T4 polynucleotide kinase (see ESI† for details). The 5′-[32P]-labeled 2′-F-RNA probes (≥5 pmol) in 100 μL of DPBS containing 1 mM MgCl2 (DPBSM) were incubated at 90 °C for 5 min and then cooled down for 10 min at 25 °C. Tween 20, BSA and polyA were added to the reaction mixture for final concentrations of 0.05%, 0.01% and 0.1 mg mL−1, respectively. To start binding, 5 μL of the RNA solution were mixed with 5 μL of protein solution (His-Obe, wt-Obe, aequorin, or clytin) in DPBSM. After 40 min of incubation at 25 °C, aliquots were withdrawn and analyzed by the DRaCALA assay according to Donaldson et al.24 Briefly, 5 μL of the reaction mixture were spotted on a dry nitrocellulose filter (Millipore, USA). Spots were allowed to dry for approx. 20 min and then exposed to the Bio-Rad phosphorimager screen overnight. For each spot, the intensities of the inner and outer circles were quantitated using Molecular Imager FX and Quantity One 4.5.1 software (Bio-Rad, USA). The fraction of bound 2′-F-RNA (FB) was calculated from the intensities (Iinner, Itotal) and areas (Ainner, Atotal) of the inner and outer circles using the eqn (1), as described in:21
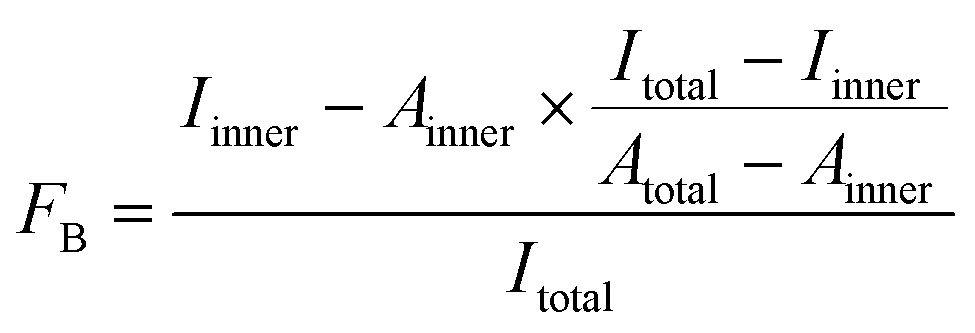 | (1) |
Each binding assay was performed in triplicate. The values of the equilibrium dissociation constants (KD) were determined by approximation of experimental data using a standard equation for bimolecular ligand–receptor binding (2) in the GraphPad Prism software.
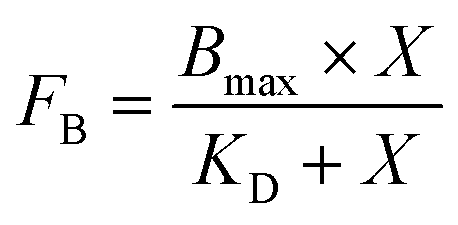 | (2) |
Solid-phase bioluminescent assays with bi-functional 2′-F-RNA aptamers
Before measurement biotinylated bi-functional 2′-F-RNA aptamers (O79t1-H9t11, H9t11-L-O79t1 or O79t1-L-H9t11) were folded in DPBSM by heating at 90 °C for 5 min and cooling down for 10 min at 25 °C. Then Tween 20, BSA and E. coli tRNA were added as nonspecific competitors for final concentrations of 0.05%, 0.01%, and 0.1%, respectively. The 25 nM solution of biotinylated bi-functional 2′-F-RNA aptamers (50 μL) were added into streptavidin activated wells (10 μg mL−1 in PBS, pH 7.5, 50 μL in each well, overnight at 4 °C) in binding buffer (DPBSM, 0.05% Tween 20, 0.01% BSA, 0.01% E. coli tRNA) and incubated with shaking for 30 min at room temperature.Then, the wells were washed, and 50 μL aliquots of the mixed solution of human hemoglobin (final concentration from 100 to 1.6 nM) and His-Obe (final concentration of 100 nM) in binding buffer were added into the wells and incubated for 40 min at room temperature.
Aliquots of 100 nM His-Obe in binding buffer were placed into the control wells. After washing the wells, bioluminescence of bound obelin was initiated by injection of 0.1 M CaCl2 in 0.1 M Tris–HCl, pH 8.8 (50 μL) and measured with Mithras LB 940 plate luminometer (Berthold, Germany).
The signal was integrated for 5 s. Signals from the control wells were subtracted from those obtained from the respective aptamer-containing wells.
Results and discussion
In vitro selection of 2′-F-RNA aptamers to obelin
In our study, we chose a uniform 2′-fluoro modification of all pyrimidine residues within RNA library. This type of modification is quite widespread for in vitro selection of RNA aptamers since it provides nuclease resistance and stable secondary structures, and is well compatible with all enzymatic steps of the SELEX cycle (see, e.g., the reviews25,26).The Ca2+-regulated photoprotein obelin elongated from the N-terminus by the hexahistidine fragment (His-Obe)18 was used as a SELEX target, according to commonly used selection technique27–29 (see ESI† for Experimental details of the selection). To monitor the course of the enrichment, we assessed the diversity of the obtained libraries in the dsDNA form by analyzing remelting profiles of dsDNA pools (DiStRO assay) (Fig. S1†). The progressive increase in remelting temperature with the number of the round pointed to the loss of diversity, which corresponds to the enrichment of the library After 12 rounds of selection and high-throughput sequencing of the final and intermediate selection pools, the most represented aptamers (Table S2 and Fig. S3†) were chemically synthesized and tested for their ability to bind His-tagged and tag-free obelins.
Binding affinities of candidate aptamers
In this study, we applied for the first time the DRaCALA (Differential Radial Capillary Action of Ligand Assay) technique proposed by Donaldson et al.24 to examine the aptamer–protein complex formation. The method is based on different mobilities of free nucleic acid and NA–protein complexes in the probe spotted on the dry nitrocellulose filter. In our case, aptamer–protein complexes were easily identified as the spots of smaller diameter inside the larger spots corresponding to the free [32P]-labelled aptamers (see, e.g., Fig. 1B). Even the complexes hardly detectable by the electrophoretic mobility shift assay (EMSA) (e.g., Fig. 4C) were clearly registered by the DRaCALA (Fig. S5†). The method also enables the quantitative measurement of binding affinity in terms of KD (Table 1). Rather surprisingly, the results revealed that some of the SELEX leaders bound only the His-tagged obelin and show no affinity to the wild-type one.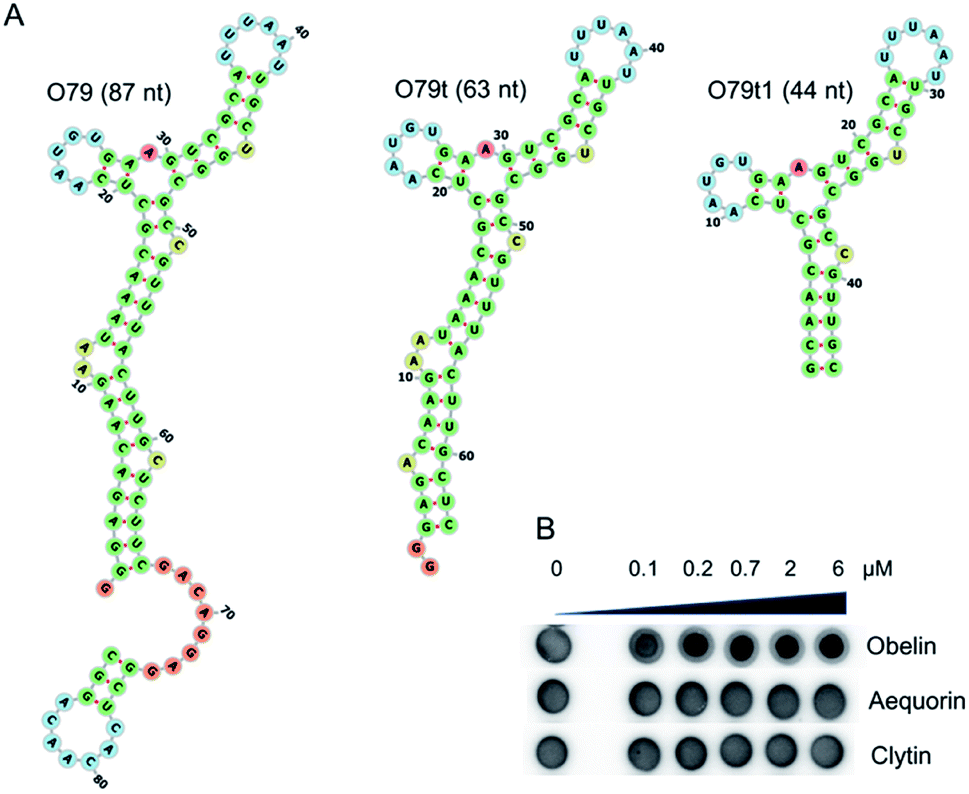 | ||
| Fig. 1 Obelin-binding aptamer O79 and its truncated versions. (A) Predicted secondary structures for O79, O79t, O79t1. (B) Example of binding of O79t to close related photoproteins obelin, aequorin, and clytin examined by the DRaCALA assay. | ||
| Name | Sequence (5′–3′) | KD, μM (His-Obe) | KD, μM (wt-Obe) |
|---|---|---|---|
| a NB, no binding detected. | |||
| O79 | gggagacaagaauaaacgcucaaugugaagucgcauuuaauugcuggcgccguuuacuugcucuucgacaggaggcucacaacaggc | 0.28 | 0.69 |
| O79t | gggagacaagaauaaacgcucaaugugaagucgcauuuaauugcuggcgccguuuacuugcuc | 0.34 | 0.72 |
| O3t | gggagacaagaauaaacgcucaaugugaagucgcacuuaguugcuggcgucguuuacuugcuc | 0.23 | 1.37 |
| O4t | gggagacaagaauaaacgcucaacuaggcugugcgcggugcccuaucuuauccgcgccucuccu | 0.58 | NBa |
| O6t | gggagacaagaauaaacgcucaagacgugcgcgggaaagaccgacgcucuaccccuacaagcu | 0.94 | NBa |
| O35 | gggagacaagaauaaacgcucaauaggguacgcggacagcgaugggaccgcguugccagccccuucgacaggaggcucacaacaggc | 0.12 | NBa |
| O5 | gggagacaagaauaaacgcucaaguuguacgcgguuggcaauccgcguugcuuuacggguuccuucgacaggaggcucacaacaggc | 0.45 | NBa |
As the most promising candidate for the obelin-recruiting module, we chose the aptamer O79, which demonstrated sufficient binding affinity to both His-tagged and tag-free targets. Of note, the bioluminescent assay with microplate-immobilized aptamers proved that obelin retains its bioluminescent properties being bound to the aptamers (Fig. S7†).
Rational design of O79 aptamer
For incorporation of O79 into the single oligonucleotide chain of a bi-modular aptamer, it was rationally truncated from 87 to 44 nt (Fig. 1A) to minimize the overall length of the construct. In the aptamer O79t1, the longest stem was shortened to 6 bp, and two terminal AU pairs were replaced by GC pairs to stabilize the structure (see ESI† for details).Further truncation of O79t1 aptamer led to the complete loss of binding affinity (Fig. S6, ESI†). We therefore considered this sequence as a minimal binding motif. Of note, the aptamers of O79 series demonstrated an excellent selectivity for obelin. We have not registered any complex formation with closely related Ca2+-dependent photoproteins aequorin and clytin (e.g., Fig. 1B), even though these three proteins exhibit high structural homology.20
Engineering of bi-specific 2′-F-RNA aptamers
We then examined the possibility to use the optimized aptamer O79t1 for non-covalent recruiting of obelin into a bi-specific aptamer construct. The bi-specific aptamers proposed in this study consist of two structural modules: (1) the molecular recognition module, i.e., an aptamer binding the target analyte, and (2) the obelin-binding aptamer for a reporter-recruiting module.Since the analytical signal should turn-on only in the presence of the target analyte, we aimed to engineer a structure-switching construct capable of strictly sequential activation. To this point, we employed the rational modular design relying on the secondary structure prediction. Within the bi-specific construct, the analyte-binding aptamer mostly retains its structure, while the obelin-binding part is hybridized with a complementary fragment of the analyte aptamer or with a short additional linker sequence. It is, therefore, ‘hidden’ and unable to recognize the obelin. Binding of the analyte induces the structural rearrangement of the whole construct and ‘unmasking’ of the obelin-binding aptamer. Thus, upon the addition of obelin, it binds the cognate aptamer, which, in turn, enables the generation of the specific bioluminescent signal after Ca2+ addition. The principal detection scheme is depicted in Fig. 2.
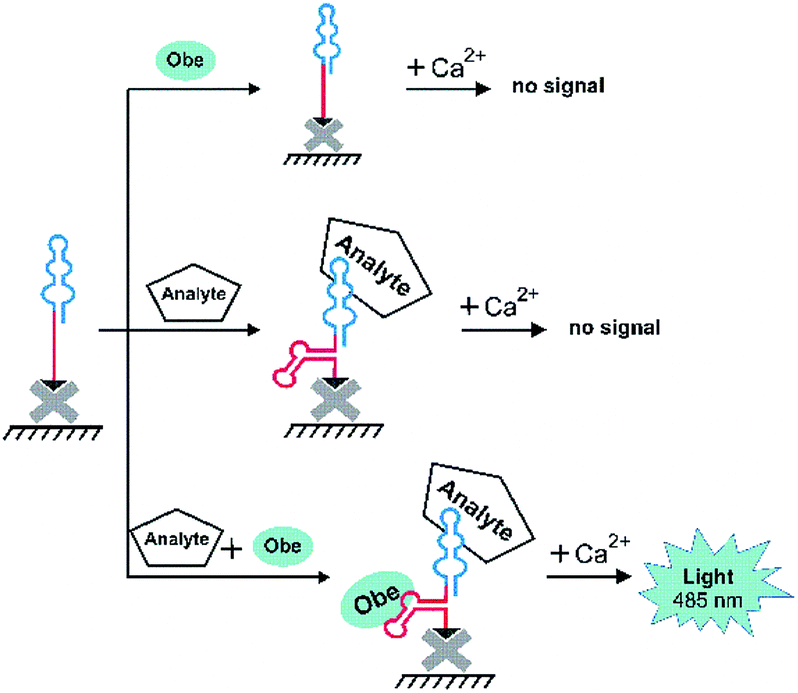 | ||
| Fig. 2 A general scheme of microplate bioluminescent detection of an analyte with bi-specific aptamers. | ||
As a proof of principle for this strategy, we employed the human hemoglobin (Hb) binding 2′-F-RNA aptamer H9t11 as an analyte-binding module.30 This G-quadruplex aptamer is of relatively small size (43 nt), which makes it possible to synthesize the whole 2′-F-RNA molecule built of two aptamer sequences on an automated DNA/RNA synthesizer. Theoretically, two aptamers can be fused into the single construct differently, with or without an additional oligonucleotide linker between the aptamer modules. Generally, linkers of “neutral” nucleotide sequences such as A10 and (dT)10, or non-nucleotide hexaethylene glycol linker are employed to connect different aptamers in bi-functional molecules that expose both aptamers for the simultaneous binding of the targets.5 However, for the constructs that provide the sequential target binding, the particular sequence of the linker requires a more thorough design since it contributes to the overall secondary structure. So far, there are no universal guidelines for engineering structure-switching bifunctional aptamers. In some cases, the fusion of two aptamers without a linker allows hiding one of them in the resulting structure.9 Other constructs require a smart design of the oligonucleotide linker made from the fragment of the one of the aptamers.7
Here, we tested both approaches. For the engineering of structure-switching bimodular aptamers, we either joined two aptamers without a linker or derived the linker sequences from short terminal fragments (6–8 nt) of both aptamers. In the latter case, we chose the length of the linker having in mind that the secondary structure of the bi-functional construct should be stable enough to hide the obelin aptamer, but also capable of rearrangement after hemoglobin binding. A set of possible variants was designed and analyzed for their secondary structures. In doing this, we considered two requirements: (1) the hemoglobin aptamer retains its structure, and (2) the structure of the obelin-binding motif within the whole molecule is significantly changed as compared to O79t1 alone. The following three constructs met the criteria mentioned above (Fig. 3): (1) O79t1 followed by H9t11 directly, without the linker, (2) 5′-H9t11-L-O79t1-3′, and (3) 5′-O79t1-L-H9t11-3′, where L is a hexanucleotide linker sequence corresponding to the 3′-terminal fragment of O79t1.
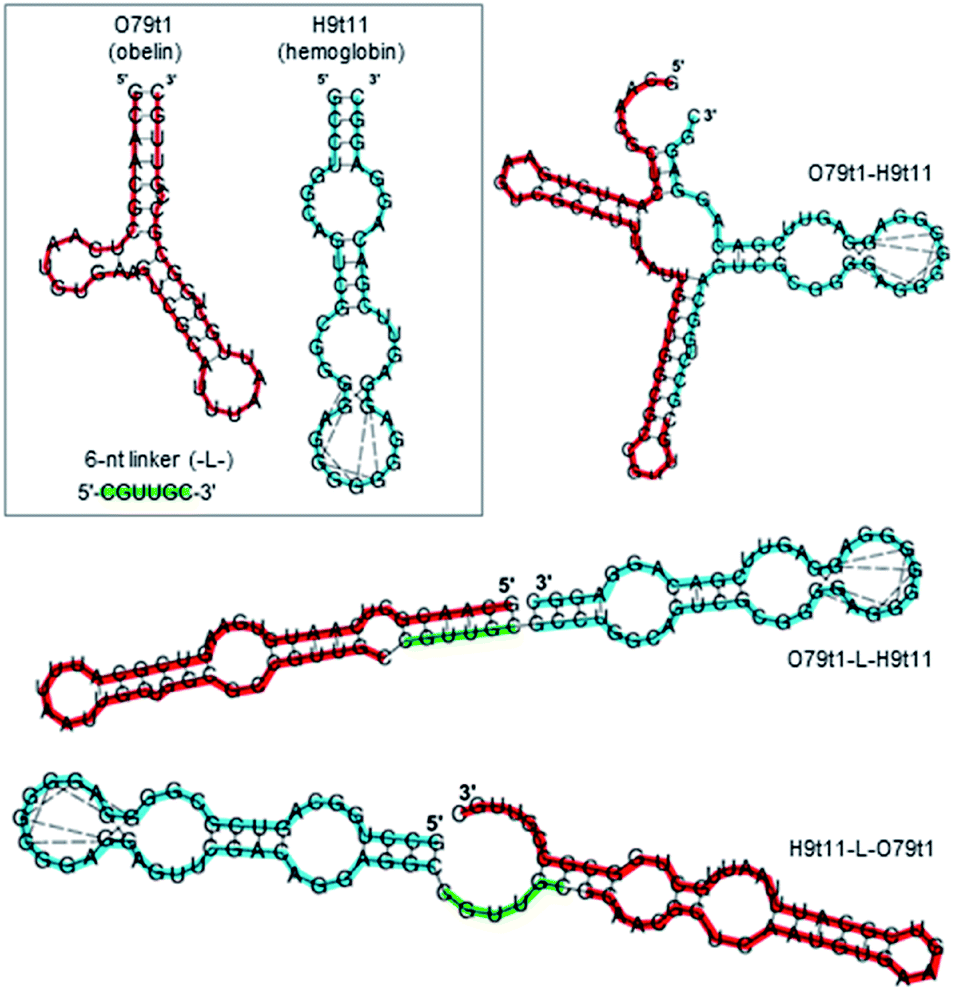 | ||
| Fig. 3 Secondary structures of obelin-binding (red) and hemoglobin-binding (blue) aptamer modules and bi-specific aptamers on their basis (obtained by Vienna RNA fold). Dashed lines connect G residues involved in quadruplex formation. Hexanucleotide linker (green) between the modules is derived from the 3′-terminal fragment of O79t1. | ||
Bioluminescent microplate assay
The obtained bifunctional aptamers with dual hemoglobin/obelin specificity were tested by a bioluminescent microplate assay. 3′-Biotinylated aptamers were immobilized on the streptavidin-coated wells of the microplate and incubated with varying concentrations of hemoglobin in the presence of a constant concentration of obelin. The optimal concentration of obelin (100 nM) was chosen as described in ESI (Fig. S8†). The bioluminescent signals were measured after Ca2+ addition in the concentration which is saturating for all obelin molecules bioluminescence.11 The scheme and the results of the assay are shown in Fig. 4. In all three cases, we registered a low bioluminescent signal of the obelin in the absence of hemoglobin.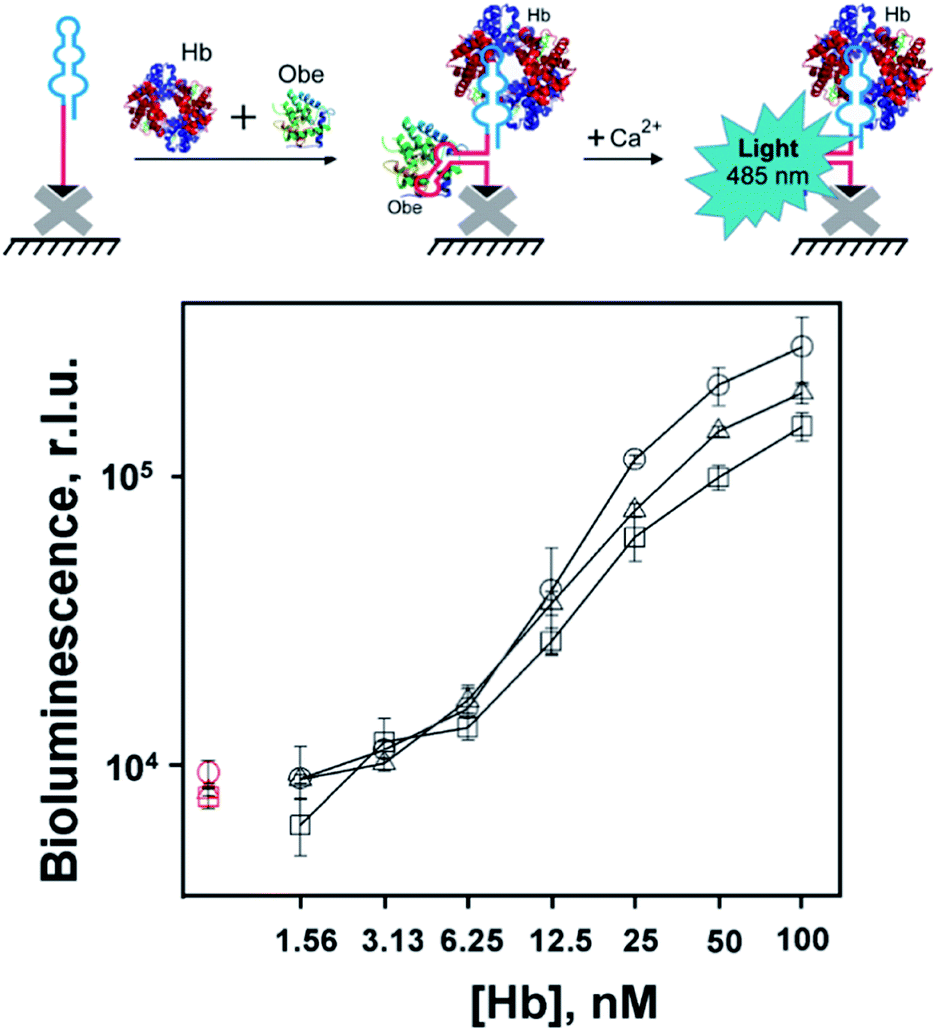 | ||
| Fig. 4 Bioluminescent detection of human hemoglobin with bispecific aptamers Apt79t1-H9t11 (–□–), H9t11-L-Apt79t1 (–○–) or Apt79t1-L-H9t11 (–Δ–) at simultaneous addition of obelin (100 nM) and hemoglobin (1.56–100 nM). Red symbols show the signals from hemoglobin-free (control) samples. Biotinylated aptamers were immobilized in streptavidin-coated microplate wells. Each point is the average ± standard deviation, n = 3. | ||
In contrast, upon the addition of hemoglobin, all constructs provided a specific growth in the bioluminescent signal proportional to the analyte's concentration, with slightly different bioluminescence curves. Namely, in the case of linker-containing aptamers, we observed a linear growth in the bioluminescent signal from 1.6 to 50 nM Hb for O79t1-L-H9t11 and from 3.1 nM Hb for the H9t11-L-O79t1 construct. For linker-free O79t1-H9t11, this growth started from 6.25 nM Hb. Thereby, the designed constructs possessed the ability to derive the specific bioluminescent signal upon the analyte binding. The developed assay allows for the simultaneous addition of hemoglobin and obelin to the microplate well with immobilized aptamer, followed by washing step and measurement of Ca2+ bioluminescence. This makes the detection protocol very easy and fast to perform. What is also important, the bi-functional aptamer is re-folded by the common heating/cooling procedure before plate immobilization, without jeopardizing the reporter photoprotein.
As compared to other aptasensing systems for hemoglobin detection, our assay surpasses the analytical performance of fluorescent aptasensor based on fluorescein-labeled aptamer and graphene oxide (range of detection 77–770 nM).31 Electrochemical hemoglobin-specific aptasensors provide lower detection limits of 10−12 M (ref. 32) and even 10−20 M.33 It should be noted, however, that physiological blood hemoglobin concentration is about 2 mM (13.2 g dL−1). All reported aptasensors provide at least nanomolar sensitivity and therefore perfectly suit for Hb detection.
Conclusions
To sum up, we generated, characterized, and truncated 2′-F-RNA aptamer affine and selective to the Ca2+-regulated photoprotein obelin. The aptamer was applied for non-covalent recruiting of the photoprotein into novel bimodular aptamer constructs with double hemoglobin/obelin functionality, which were engineered with a particular focus on the linker sequence between two modules. A model bioluminescent microplate assay showed that the constructs comprising hemoglobin-specific and obelin-specific 2′-F-RNA aptamers bind their targets in strictly a sequential manner. The bioluminescent signal from the obelin was registered only in the presence of hemoglobin, and its magnitude depended on the hemoglobin concentration.We thus established a proof-of-principle for non-covalent integration of the reporter protein into the aptamer-based bioanalytical system using the specific aptamer. The proposed novel approach offers the advantages of using a free protein as a reporter group, wide possibilities for re-folding of the aptamer part without any harm for the protein component, and fast and straightforward protocol of detection. With a proper design of a secondary structure, this strategy of aptasensor engineering might be further extended to the bi-specific aptamer-based bioluminescent sensors for other analytes of interest.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Russian Science Foundation (grant # 16-14-10296), Russian State funded budget projects # АААА-А17-117020210021-7 to ICBFM SB RAS and # АААА-А19-119031890015-0 to IBP SB RAS.Notes and references
- S. Poolsup and C. Y. Kim, Curr. Opin. Biotechnol., 2017, 48, 180–186 CrossRef CAS.
- H. Zhang, L. Zhou, Z. Zhu and C. Yang, Chem.–Eur. J., 2016, 22, 9886–9900 CrossRef CAS.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1683 CrossRef.
- G. Wang, Y. Wang, L. Chen and J. Choo, Biosens. Bioelectron., 2010, 25, 1859–1868 CrossRef CAS.
- J. Fallmann, S. Will, J. Engelhardt, B. Grüning, R. Backofen and P. F. Stadler, J. Biotechnol., 2017, 261, 97–104 CrossRef CAS.
- M. Vorobyeva, P. Vorobjev and A. Venyaminova, Molecules, 2016, 21, 14–16 CrossRef.
- T. Bing, X. Liu, X. Cheng, Z. Cao and D. Shangguan, Biosens. Bioelectron., 2010, 25, 1487–1492 CrossRef CAS.
- T. Kato, I. Shimada, R. Kimura and M. Hyuga, Chem. Commun., 2016, 52, 4041–4044 RSC.
- C. C. Chang, S. Lin, C. H. Lee, T. L. Chuang, P. R. Hsueh, H. C. Lai and C. W. Lin, Biosens. Bioelectron., 2012, 37, 68–74 CrossRef CAS.
- T.-L. Chuang, C.-C. Chang, Y. Chu-Su, S.-C. Wei, X.-H. Zhao, P.-R. Hsueh and C.-W. Lin, Lab Chip, 2014, 2968–2977 RSC.
- L. A. Frank, Sensors, 2010, 10, 11287–11300 CrossRef CAS.
- M. A. Vorobjeva, V. V. Krasitskaya, A. A. Fokina, V. V. Timoshenko, G. A. Nevinsky, A. G. Venyaminova and L. A. Frank, Anal. Chem., 2014, 86, 2590–2594 CrossRef CAS.
- V. V. Krasitskaya, V. V. Chaukina, M. V. Abroskina, M. A. Vorobyeva, A. A. Ilminskaya, M. R. Kabilov, S. V. Prokopenko, G. A. Nevinsky, A. G. Venyaminova and L. A. Frank, Anal. Chim. Acta, 2019, 1064, 112–118 CrossRef CAS.
- E. E. Bashmakova, V. V. Krasitskaya, G. S. Zamay, T. N. Zamay and L. A. Frank, Talanta, 2019, 199, 674–678 CrossRef.
- A. Moutsiopoulou, D. Broyles, H. Joda, E. Dikici, A. Kaur, A. Kaifer, S. Daunert and S. K. Deo, Anal. Chem., 2020, 92, 7393–7398 CrossRef CAS.
- S. Neubacher and S. Hennig, Angew. Chem., Int. Ed., 2018, 1266–1279 Search PubMed.
- B. Shui, A. Ozer, W. Zipfel, N. Sahu, A. Singh, J. T. Lis, H. Shi and M. I. Kotlikoff, Nucleic Acids Res., 2012, 40, e39 CrossRef CAS.
- V. V. Krasitskaya, A. S. Davydova, M. A. Vorobjeva and L. A. Frank, Russ. J. Bioorg. Chem., 2018, 44, 296–301 CrossRef CAS.
- B. A. Illarionov, L. A. Frank, V. A. Illarionova, V. S. Bondar, E. S. Vysotski and J. R. Blinks, Methods Enzymol., 2000, 305, 223–249 CAS.
- S. V. Markova, E. S. Vysotski, J. R. Blinks, L. P. Burakova, B.-C. Wang and J. Lee, Biochemistry, 2002, 41, 2227–2236 CrossRef CAS.
- A. S. Davydova, O. A. Krasheninina, A. E. Tupikin, M. R. Kabilov, A. G. Venyaminova and M. A. Vorobyeva, Russ. J. Bioorg. Chem., 2019, 45, 656–661 CrossRef CAS.
- R. C. Edgar, Nat. Methods, 2013, 10, 996–998 CrossRef CAS.
- R. C. Edgar, bioRxiv, 2016, 081257 Search PubMed.
- G. P. Donaldson, K. G. Roelofs, Y. Luo, H. O. Sintim and V. T. Lee, Nucleic Acids Res., 2012, 40, e48 CrossRef CAS.
- S. Y. Tan, C. Acquah, A. Sidhu, C. M. Ongkudon, L. S. Yon and M. K. Danquah, Crit. Rev. Anal. Chem., 2016, 46, 521–537 CrossRef CAS.
- F. Guo, Q. Li and C. Zhou, Org. Biomol. Chem., 2017, 15, 9552–9565 RSC.
- M. B. Murphy, S. T. Fuller, P. M. Richardson and S. A. Doyle, Nucleic Acids Res., 2003, 31, e110 CrossRef.
- P. Bouvet, Methods Mol. Biol., 2009, 543, 139–150 CrossRef CAS.
- T. K. Sharma, J. G. Bruno and A. Dhiman, Biotechnol. Adv., 2017, 35, 275–301 CrossRef CAS.
- A. Davydova, M. Vorobyeva, E. Bashmakova, P. Vorobjev, O. Krasheninina, A. Tupikin, M. Kabilov, V. Krasitskaya, L. Frank and A. Venyaminova, Anal. Biochem., 2019, 570, 43–50 CrossRef CAS.
- M. Lin, W. Li, Y. Wang, X. Yang, K. Wang, Q. Wang, P. Wang, Y. Chang and Y. Tan, Chem. Commun., 2015, 51, 8304–8306 RSC.
- S. Eissa and M. Zourob, Sci. Rep., 2017, 7, 1016 CrossRef.
- Z. Shekari, H. R. Zare and A. Falahati, Anal. Biochem., 2017, 518, 102–109 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05117a |
| This journal is © The Royal Society of Chemistry 2020 |