DOI:
10.1039/D0RA05086E
(Paper)
RSC Adv., 2020,
10, 27884-27893
A theoretical study of the hydrolysis mechanism of A-234; the suspected novichok agent in the Skripal attack†
Received
9th June 2020
, Accepted 17th July 2020
First published on 27th July 2020
Abstract
A-234, [EtO–P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(F)–NC(Me)–N(Et)2], is the suspected A-type nerve agent used in the Skripal attack on the 4th of March 2018. Studies related to the structure and reactivity of this compound are limited. We, therefore, aimed at understanding the underlying hydrolysis mechanism of A-234 within the DFT framework. The attack of the water molecule can occur at the phosphinate and acetoamidine reactive centres. Our theoretical findings indicate that the hydrolysis at the acetoamidine centre is thermodynamically favoured compared to the hydrolysis at the phosphinate centre. The hydrolysis at the acetoamidine moiety may proceed via two pathways, depending on the nitrogen atom participating in the hydrolysis. The main pathway consists of four distinct channels to reach the final product, with the concerted 1,3-proton shift favoured kinetically and thermodynamically in the gas phase and water as solvent. The results are in good agreement with the literature, although some differences in the reaction mechanism were observed.
O)(F)–NC(Me)–N(Et)2], is the suspected A-type nerve agent used in the Skripal attack on the 4th of March 2018. Studies related to the structure and reactivity of this compound are limited. We, therefore, aimed at understanding the underlying hydrolysis mechanism of A-234 within the DFT framework. The attack of the water molecule can occur at the phosphinate and acetoamidine reactive centres. Our theoretical findings indicate that the hydrolysis at the acetoamidine centre is thermodynamically favoured compared to the hydrolysis at the phosphinate centre. The hydrolysis at the acetoamidine moiety may proceed via two pathways, depending on the nitrogen atom participating in the hydrolysis. The main pathway consists of four distinct channels to reach the final product, with the concerted 1,3-proton shift favoured kinetically and thermodynamically in the gas phase and water as solvent. The results are in good agreement with the literature, although some differences in the reaction mechanism were observed.
1 Introduction
The chemical attack in the UK on the 4th of March 2018 caused the world to be more alert to the detection and destruction of novichoks. Sergei Skripal and his daughter, Yulia, were found unconscious after being exposed to a novichok nerve agent, A-234.1–4 The scientific literature on novichoks is incomplete and sometimes contradictory.5,6 There are several discrepancies around novichoks mostly due to the secrecy surrounding the investigations, which were conducted as a part of the Soviet military program.7 Most information available in the literature was provided by Mirzayanov, who worked on the development of novichoks.1 The latter claimed that these A-series nerve agents are 7–8 times more toxic than VX,8,9 although scientific information to support this argument is elusive. Mirzayanov also published chemical structures for novichoks, including A-234 (Scheme 1, structure A).1,8 At the same time, Hoenig10 and Ellison11 proposed alternative structures for novichoks, including A-234 (Scheme 1, structure B). The main difference between the two structures is related to their functional groups: while Mirzayanov's structure (A) is a phosphoramidate, Hoenig's structure (B) is a phosphorylated oxime. Further to the work of Chai et al.12 and Harvey et al.,13 as well as the amendment to the Chemical Weapons Convention (CWC),14 there is some consensus on the phosphoramidate nature of the A-series nerve agents. Therefore, this research work is based on the structure proposed by Mirzayanov (structure A, N′-[ethoxy(fluoro)phosphoryl]-N,N-diethylethanimidamide).
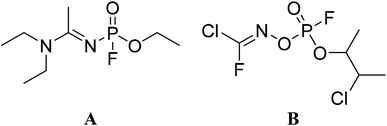 |
| | Scheme 1 Proposed chemical structures of A-234. | |
Scientific research on novichoks has only recently started to emerge. Hosseini and co-workers15 are among the first research groups who reported the synthesis and spectroscopic data of some novichok derivatives, which are included in the Schedule 2.B.04 of the CWC. Chai et al.12 detailed the development of novichoks and their known toxicity. Another historical overview mentioned that novichoks were designed to be undetectable by the standard North Atlantic Treaty Organisation (NATO) chemical detection equipment.1 Nonetheless, much progress is being made and the ultrasensitive detection of the novichok nerve agent A-232 using vibrational spectroscopy was recently reported.16 Vale et al.2 reviewed the features and clinical treatment of well-studied nerve agents and extrapolated them to novichoks. They suggested that the administration of atropine, pralidoxime, diazepam, and breathing support could help during detoxification from novichoks. Carlsen3 studied the toxicology and environmental fate of novichok nerve agents, including A-234, by using quantitative structure–activity relationship (QSAR) models. Interestingly, their results contradicted Mirzayanov's claim about the relative toxicity of novichoks and the hydrolysis half-life of A-234, at a pH of 6.5–7.4, was estimated to be moderate (10–30 days).3 Enzymatic hydrolysis of novichoks was also studied both theoretically and experimentally.13,17 The hydrolysis rate of novichok agents A-230, A-232, and A-234 was further probed and compared with common G- and V-series nerve agents at 25 °C and a pH of 7.2 in mM bis-tris-propane.13 The measured hydrolysis rate for A-234 (0.0032 μM min−1) is comparatively low with respect to GB (6.68 μM min−1) and VX (0.246 μM min−1). An activation energy of 37.98 kJ mol−1 was determined via the Arrhenius plot for the hydrolysis of A-234.13
Bhakhoa et al.4 carried out a theoretical study on A-234 (Scheme 1, structure A), highlighting: (i) its molecular, electronic, and spectroscopic properties; (ii) its toxicity; (iii) its poisoning action and antidotes based on model reactions, and (iv) its hydrolysis and thermal degradation. Natural bond orbital (NBO) analysis showed that the acetoamidine carbon and the phosphorus atoms of A-234 are positively charged and are susceptible to nucleophilic attack. The enthalpy and Gibbs free energy changes for the hydrolysis at both electropositive centres were calculated. Based on the energetics, it was deduced that hydrolysis will take place at the acetoamidine carbon centre preferentially to yield N,N-diethylacetamide and ethyl phosphoramidofluoridate. However, this was a preliminary study and a detailed mechanistic study is required to substantiate the conclusion made on the hydrolysis of A-234. Moreover, it is known that Skripal and his daughter were discharged from the hospital a few weeks after being exposed.2 However, novichok A-234 may still be present in the environment.18 In an attempt to study its persistence in the environment, we herein present a detailed mechanistic study on the neutral hydrolysis reaction of A-234.
2 Computational details
All geometries were optimised without symmetry constraint within the density functional theory (DFT) framework using the B3LYP19,20 functional in conjunction with the 6-31+G(d,p)21 basis set. The method used was based on the findings of Flinn et al.22 and Wu et al.,23 which showed that the B3LYP/6-31+G(d,p) method provides consistent results. All computations were performed using Gaussian 1624 available on SEAGrid.25–29 The stationary points were located by means of Berny algorithm30 using redundant internal coordinates. Analytical Hessians were computed to determine the nature of stationary points (one and zero imaginary frequencies for transition states (TSs) and minima, respectively)31 as well as the unscaled zero-point energies (ZPEs), thermal corrections and entropy effects using the standard statistical–mechanics relationships for an ideal gas.32 TS structures show only one negative eigenvalue in their diagonalised force constant matrices and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration using the intrinsic reaction coordinate (IRC) method.33 Unless otherwise stated, the Gibbs free energies were computed at 298.15 K and 1 atm. Solvent effects were taken into account using the self-consistent reaction field method, based on the conductor-like polarisable continuum model (CPCM),34,35 and using the universal force field to account for cavity radii. Water was used as the solvent. It should be noted that A-234 exists in several conformations. In this study, the most stable conformer, reported by Bhakhoa et al.,4 was used.
3 Results and discussion
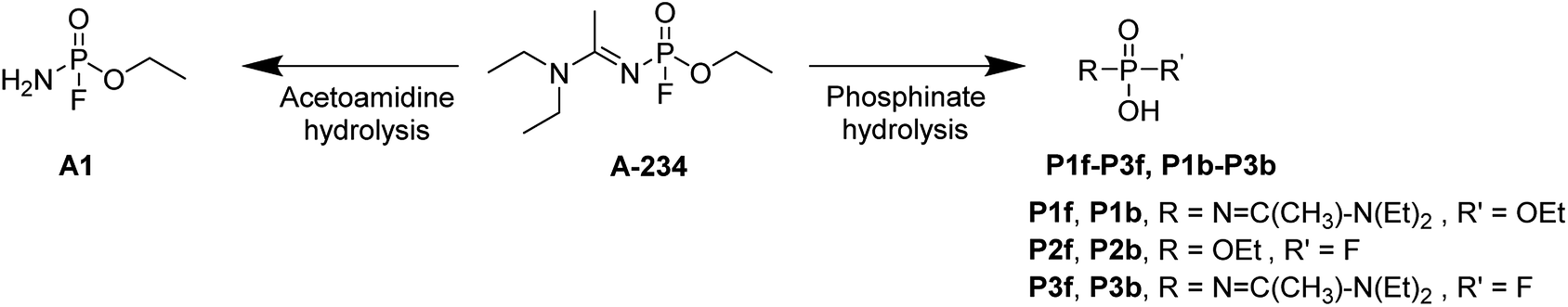
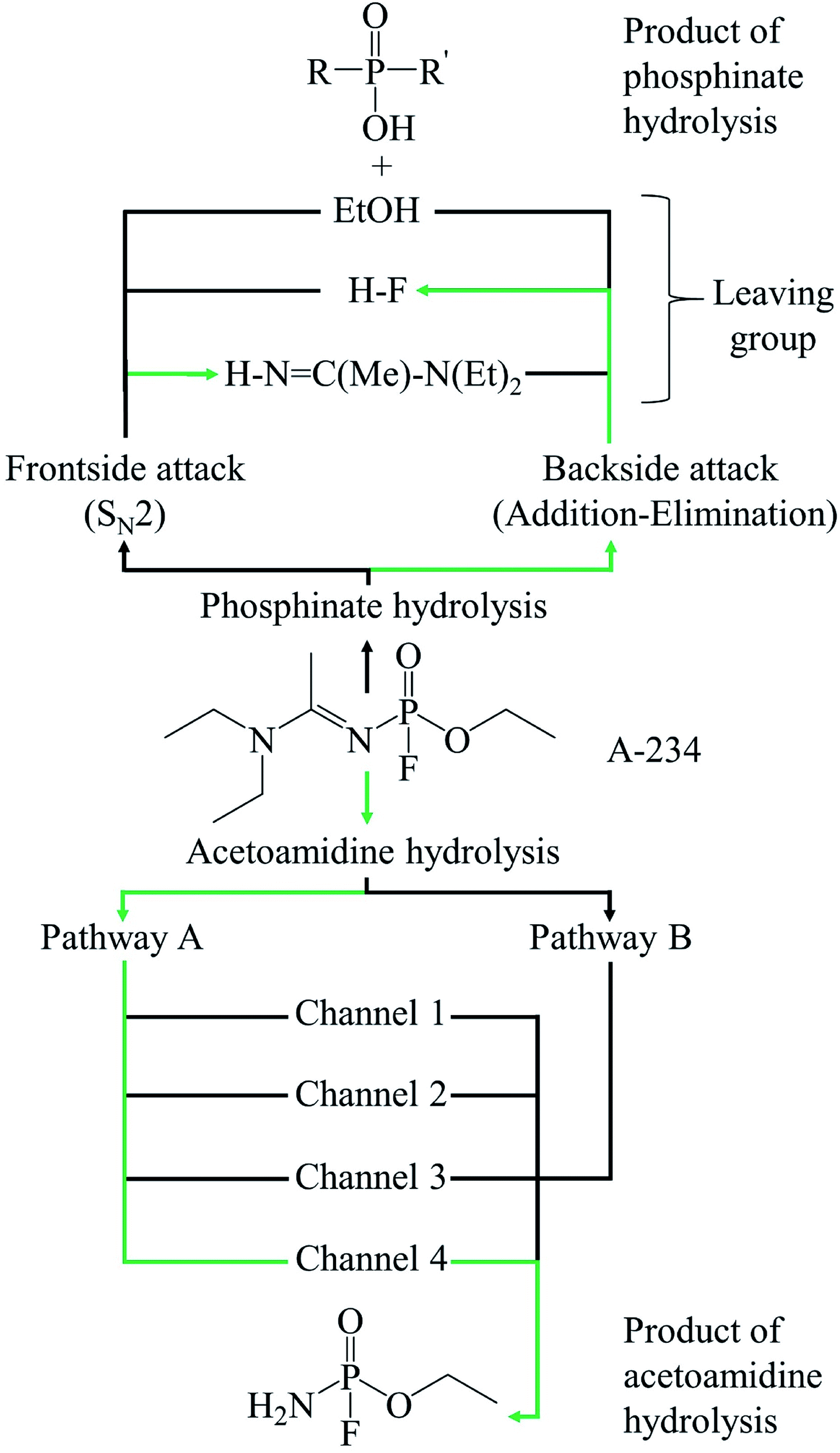
The hydrolysis of A-234 potentially occurs either at the acetoamidine moiety, leading to product A1, or at the phosphinate moiety, leading to products P1f–P3f and P1b–P3b (Scheme 2).4,36,37
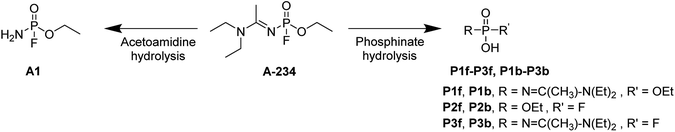 |
| | Scheme 2 The hydrolysis products of A-234. | |
3.1 Acetoamidine hydrolysis
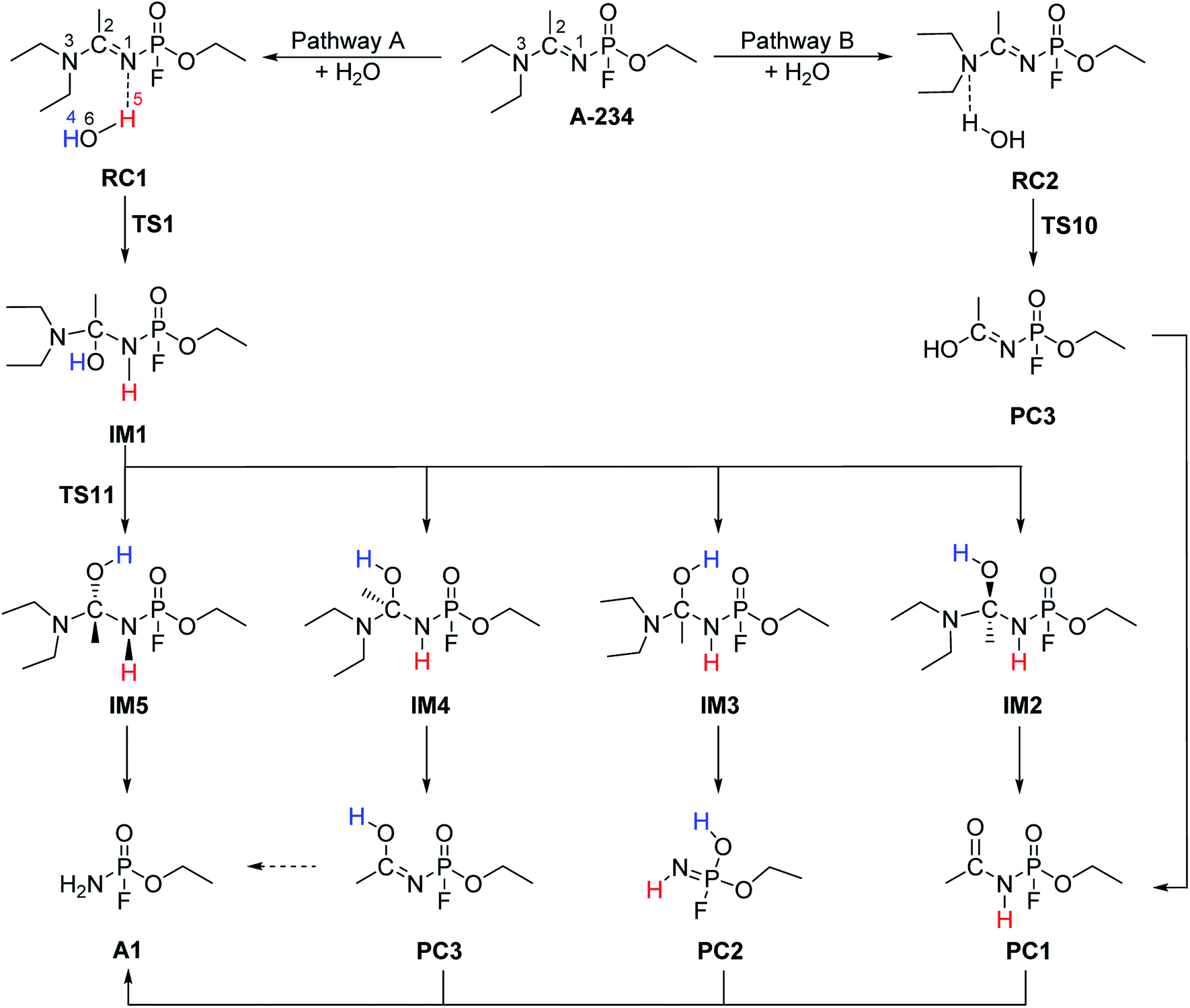
The hydrolysis at the acetoamidine moiety was studied using the same approach as that of Flinn et al.22 and Wu et al.23 Different possibilities for the acetoamidine hydrolysis are shown in Scheme 3. The hydrolysis at the amidine moiety starts with N–H hydrogen bond formation either at the sp2 nitrogen (N1) or at the sp3 nitrogen (N3), followed by the nucleophilic attack of the water molecule on the electropositive carbon (C2). The first case is denoted as Pathway A, while the second case is denoted as Pathway B. Pathway A results in the formation of a tetrahedral intermediate, IM1, which has several conformers, IM2–IM5, each leading to a distinct proton shift. Pathway B results in the formation of an imidic acid, PC3, which tautomerises to an amide, PC1, and is further hydrolysed to A1. In Scheme 3, PC1, PC2, PC3, and A1 consist of an interaction with X, where X = diethyl amine, acetic acid, or diethyl acetamide; X is shown in Schemes 4–7.
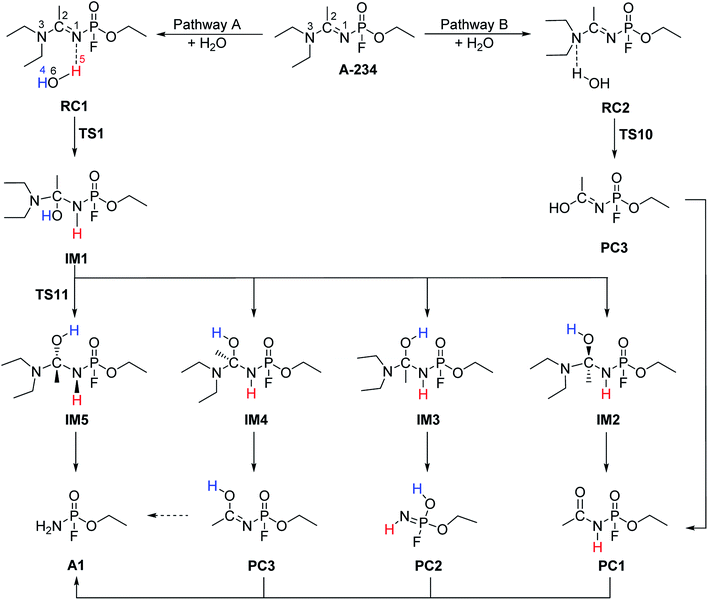 |
| | Scheme 3 Hydrolysis at the acetoamidine centre of A-234. PC1, PC2, PC3, and A1 consist of an interaction with X, where X = diethyl amine, acetic acid, or diethyl acetamide. | |
3.1.1 Pathway A. A reactant complex, RC1, is formed between A-234 and a water molecule, through a hydrogen bond between the hydrogen atom of water (H5) and the lone pair of sp2 N1 atom in A-234. The binding energy is found to be −3.59 kcal mol−1 in the gas phase. The transition state, TS1, which leads to the formation of IM1, has bond distances of 1.061 Å (N1⋯H5) and 2.274 Å (C2⋯O6). The imaginary frequency indicates a mode of vibration corresponding to a bond formation between N1 and a proton from water (H5), and a bond formation between the oxygen atom of water (O6) and C2. The activation barriers of this step are 41.53, 43.85, and 42.90 kcal mol−1 for Ea(g), ΔG‡(g), and ΔG‡(aq), respectively. A slight decrease in the Gibbs free activation barrier is observed in solution. This part of the mechanism has reaction energies (ΔGreac) of 19.78 and 23.87 kcal mol−1 in the gas phase and water, respectively, implying an endergonic reaction.IM1, which is formed during the first step of Pathway A, has several conformations due to the change of the C2 hybridisation from sp2 to sp3. Each of these conformers is associated to distinct proton shifts denoted as Channels 1–4. Hence, the second step of Pathway A is a proton shift in the conformers of IM1, which leads to the final product, A1.
3.1.2 Channel 1. Channel 1 is associated with the conformer IM2. Compared to IM1, the structure has an inversed N(C2H5)2 moiety and a rotated N–C bond. This change brings H4 into a favourable position, directly in front of the lone pair of electrons of N3 for the reaction to proceed (Scheme 4). A 1,3-proton shift from O6 to N3 has a ΔG‡(g) of 25.63 kcal mol−1. The solvent medium lowers the barrier by 0.90 kcal mol−1. The transition state, TS2, includes a four-membered ring structure with distances of 1.302 Å between H4 and O6 and 1.255 Å between H4 and N3 for the gas-phase structure. The IRC computation shows that the proton shift to N3 occurs before the cleavage of the C2–N3 bond. The resulting product complex, PC1, involves diethyl amine and ethyl acetylphosphoramido fluoridate, a secondary amide. PC1 can interact with another water molecule to form RC3, which undergoes further hydrolysis to form A1. This reaction occurs either via a concerted or a stepwise mechanism.38 To complete the pathway, the concerted mechanism was considered herein. The concerted mechanism proceeds through TS3 with a ΔG‡(g) of 49.70 kcal mol−1 and a ΔG‡(aq) of 47.29 kcal mol−1. The last step of Channel 1 has a ΔGreac of −8.46 kcal mol−1 in the gas phase and a ΔGreac of −9.81 kcal mol−1 in water, indicating an exergonic process.
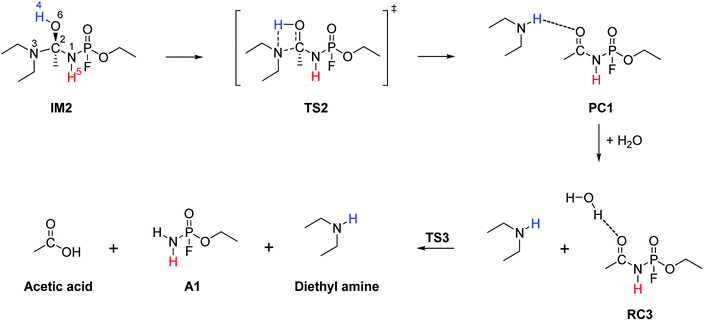 |
| | Scheme 4 The reaction mechanism for Channel 1. | |
3.1.3 Channel 2. The pre-reactive complex for Channel 2 is IM3, where the hydroxyl proton H4 is on the same plane as the oxygen atom (O7) of the phosphinate group. The migration of H4 to the phosphinate moiety (1,5-proton shift) forms an O7–H4 bond (Scheme 5). The corresponding transition state, TS4, has a six-membered cyclic structure and its IRC leads to an unexpected four-membered cyclic intermediate, IMA. The IRC shows that further to the proton shift, the resulting negatively-charged O6 atom is only 2.208 Å away from the electropositive phosphorus and consequently, a covalent P–O6 bond is formed. This step has a ΔG‡(g) of 27.79 kcal mol−1 and bulk solvation decreases the activation energy by 0.90 kcal mol−1. Due to the strain of its four-membered ring structure, IMA is relatively unstable and hence, dissociates into the product complex, PC2, via TS5. PC2 then undergoes another 1,3-proton shift via TS6 to form A1.
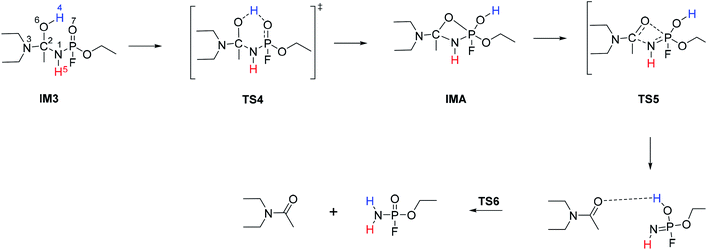 |
| | Scheme 5 The reaction mechanism for Channel 2. | |
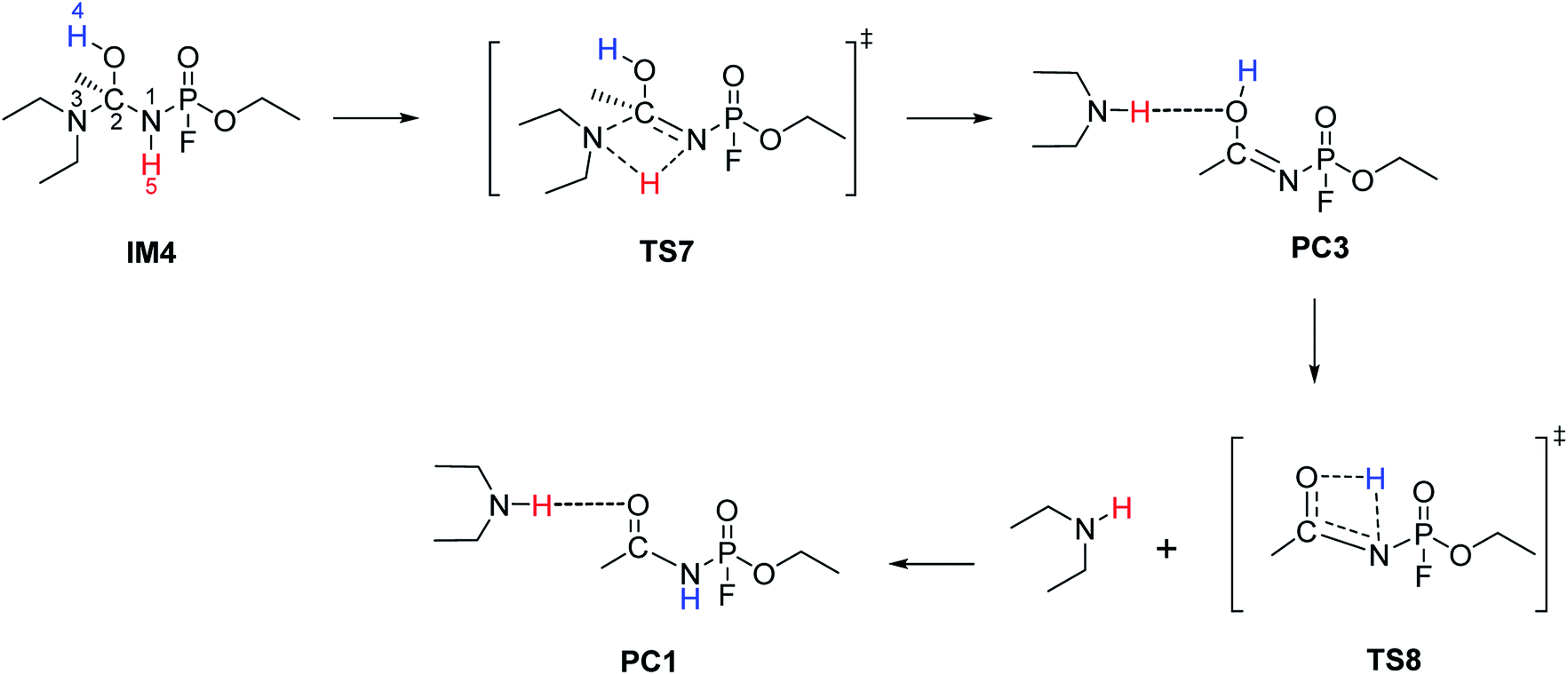
3.1.4 Channel 3. For Channel 3 (Scheme 6), the IRC computation of TS7 leads to the conformer IM4 as a minimum. Compared to IM1, the P–N1–C2 bond angle changes from 125.4° to 120.9° and a rotation around the N1–C2 bond occurs in the gas-phase structure. Channel 3 consists of a 1,3-proton shift from N1 to N3. The transition state, TS7, consists of a four-membered cyclic structure, with the H5 atom being almost equidistant from the two nitrogen atoms with an average distance of 1.30 Å. This channel has a ΔG‡(g) of 27.04 kcal mol−1 and ΔG‡(aq) of 24.60 kcal mol−1. Bulk solvation decreases the activation energy by 2.44 kcal mol−1. TS7 leads to the formation of product complex, PC3, which is a tautomer of PC1 and their interconversion occurs via TS8. ΔG‡(g) of the tautomerisation step is 26.59 kcal mol−1 while the corresponding ΔG‡(aq) is 7.62 kcal mol−1. This step of the reaction is highly influenced by bulk solvation, with a decrease of 18.97 kcal mol−1 in the Gibbs free activation energy. PC1 will ultimately convert to A1, as described in Section 3.1.2 (Channel 1).
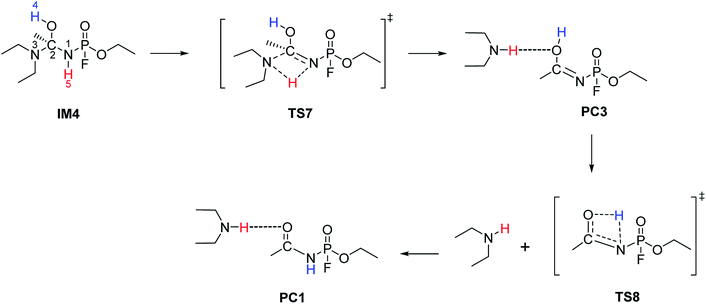 |
| | Scheme 6 The reaction mechanism for Channel 3. | |
3.1.5 Channel 4. Channel 4 consists of a 1,3-proton shift from O6 to N1 (Scheme 7). The transition state, TS9, consists of a four-membered cyclic structure with H4 being 2.078 Å away from N1 in the gas-phase structure. The IRC computation of TS9 leads to the conformer IM5 as a pre-reactive complex. This structure is different from IM1 with respect to the O7–P–N1–H5 and N1–C2–O6–H4 dihedral angles, which change from −136.0° to −112.6° and from 59.9° to 54.2°, respectively. This channel leads to the product complex, A1⋯X, where X = dimethyl acetamide, without the formation of any intermediates. The transition state, TS11, which connects conformers IM1 and IM5, was also located. This conformational change has Gibbs free energy barriers of 4.81 kcal mol−1 in the gas phase and 5.19 kcal mol−1 in water. Therefore, the small barrier and reaction reversibility lead to the formation of the thermodynamically more stable intermediate IM5.
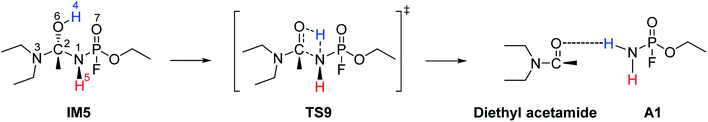 |
| | Scheme 7 The reaction mechanism for Channel 4. | |
The Gibbs free energy barriers and reaction energies associated with the Channels 1–4 were compared (Table 1). Channel 4 has the lowest Gibbs free energy barrier in both the gas phase and in water and hence, it is kinetically favoured. Bulk solvation has a significant effect on the Gibbs free energy barrier of Channel 4. This occurs due to the electrostatic nature of the nitrogen and oxygen atoms in TS9; the atoms are strongly charged. The step from IM5 to A1⋯X, where X = diethyl acetamide, is also the most exergonic, with a ΔGreac of −32.91 kcal mol−1 in the gas phase and −35.48 kcal mol−1 in water. The preference for Channel 4 is explained by the short distance between H4 and N1 in IM5, making the proton shift feasible. The by-product diethyl acetamide (X) is a tertiary amide and its resonance stabilisation explains the reaction exergonicity. Similar trends are observed in terms of the Gibbs free energies and the electronic energies. The product complex (A1⋯X, where X = diethyl acetamide) of both Channel 4 and Channel 2 are comparable. However, the product complex of Channel 4 is more stable than that of Channel 2 by 4.32 (ΔE) and 4.08 kcal mol−1 (ΔG) in the gas phase and are of comparable stability in water. This is because the stabilising interaction (H4⋯O6) is not present in the gas-phase structure of the product complex of Channel 2. Although, the same product complex is obtained via Channel 4 and Channel 2, the high energy barrier of the latter channel makes the reaction less likely to occur.
Table 1 Gibbs free energy barriers and reaction energies of Channels 1–4 from IM2–IM5 to the respective product complexes (PC1–PC3 and A1⋯Xa)
| Channel |
ΔG‡(g) (kcal mol−1) |
ΔG‡(aq) (kcal mol−1) |
ΔGreac,(g) (kcal mol−1) |
ΔGreac,(aq) (kcal mol−1) |
| X = diethyl acetamide. |
| 1 (IM2 → PC1) |
25.63 |
24.71 |
−20.60 |
−24.17 |
| 2 (IM3 → PC2) |
27.80 |
28.69 |
−13.91 |
−18.40 |
| 3 (IM4 → PC3) |
27.04 |
24.60 |
−13.78 |
4.93 |
| 4 (IM5 → A1⋯X) |
13.69 |
5.36 |
−32.91 |
−35.48 |
3.1.6 Pathway B. The hydrolysis of the acetoamidine moiety can also occur at the N3 atom and it is initiated by the formation of the pre-reactive complex, RC2 (Scheme 3, Pathway B). It proceeds via the transition state, TS10, to yield PC3. This pathway has higher energy barriers than Pathway A, with a ΔG‡(g) of 54.32 kcal mol−1 and a ΔG‡(aq) of 53.81 kcal mol−1. Pathway B is also thermodynamically less feasible, since it yields the product of Channel 3. Pathway B is unfavourable and this is attributed to the resonance effect in A-234, where the lone pair on N3 becomes less available for bonding with the proton from the water molecule. This is in line with previous reports22,23 and it can be concluded that this reaction will not occur under the reaction condition (298.15 K) considered in this work.
3.1.7 Summary of the hydrolysis mechanism at the acetoamidine moiety. The mechanism of the hydrolysis at the acetoamidine moiety of A-234 may proceed via two multi-step routes, namely, Pathway A and Pathway B. Both pathways start with the formation of a hydrogen bond at the sp2 N1 and sp3 N3 atoms, respectively. This step is followed by a nucleophilic attack at the C2 atom in both pathways. Pathway A is the more favoured route, leading to the tetrahedral intermediate, IM1, which exists as conformers IM2–IM5. From IM1, Pathway A is sub-divided into four channels, depending on the conformer participating in the reaction. Channels 1, 3, and 4 correspond to 1,3-proton shifts, while Channel 2 involves a 1,5-proton shift. Channel 4 is favoured both thermodynamically and kinetically, with Gibbs free energy barriers of 13.69 kcal mol−1 in the gas phase and 5.36 kcal mol−1 in water. Therefore, the hydrolysis mechanism at the amidine moiety proceeds via Pathway A, through Channel 4, with the formation of the intermediate, IM1, being the rate-determining step. The overall energy profile for the favoured pathway-channel combination, associated with the acetoamidine hydrolysis, is illustrated in Fig. 1.
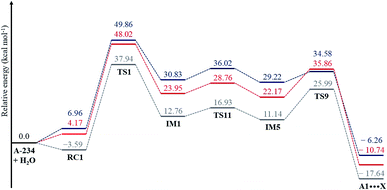 |
| | Fig. 1 The energy profile for the acetoamidine hydrolysis (Pathway A-Channel 4). ΔG(aq) (in blue) was obtained using the CPCM(water)-B3LYP/6-31+G(d,p) method, and ΔE(g) (in grey) and ΔG(g) (in red) were obtained using the B3LYP/6-31+G(d,p) method. | |
3.2 Hydrolysis at the phosphinate moiety
The hydrolysis mechanism at the phosphinate moiety depends on the position of the water molecule with respect to the leaving group (HF, EtOH, or HNC(Me)–N(Et)2). When the water molecule is on the same side of the leaving group, a frontside SN2 mechanism occurs; when the water molecule is on the opposite side of the leaving group, an addition–elimination reaction occurs.4,39–42
3.2.1 Frontside attack. In the frontside SN2 mechanism, the water molecule approaches the electropositive phosphorus from the same side as the leaving group. Three possible leaving groups were distinguished and labelled as P1f, P2f, and P3f (Scheme 8). IRC computations show that in the initial reaction stage, a bond is formed between the electronegative leaving group and a hydrogen atom from water, resulting in the temporary formation of OH−. This bond formation is simultaneous with the bond cleavage between the leaving group and phosphorus atom. OH− then attacks the electropositive phosphorus atom to form the respective product. The conclusions, made from the mechanism obtained, are similar for all three leaving groups considered. In all three cases, there is a retention of configuration with the frontside SN2 mechanism.
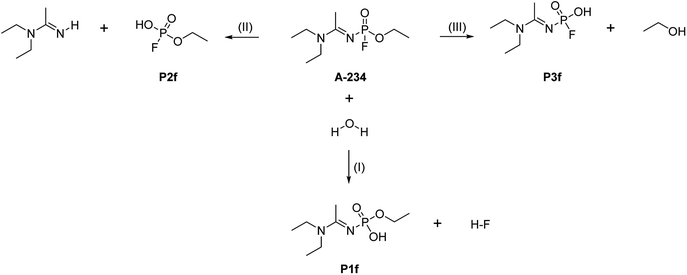 |
| | Scheme 8 Possible products in the frontside SN2 reaction. | |
3.2.2 Backside attack. The backside attack of the water molecule on A-234 occurs in an addition–elimination manner, similar to that reported by Šečkute et al.43 The mechanism is stepwise, where the first step is the addition of a water molecule to the phosphorus atom while one hydrogen atom is transferred to the oxygen atom of the PO functional group (Scheme 9). As with the frontside reaction, the backside reaction has three possible outcomes. The mechanism for each generates a trigonal bipyramidal intermediate (IMB1, IMB3, or IMB5). Substitutions at the apical positions of these intermediates are usually more favourable than substitutions at the equatorial ones.44 Since the water molecule approaches the phosphorus centre from the opposite side of the leaving group, the hydrogen atom in the P–O–H moiety is directed away from the leaving group. A rotation around the P–O bond occurs via TSP5, TSP8, or TSP11, which results in the reorientation of the hydrogen atom and thus, allowing the elimination step to take place. The leaving group is thus able to interact with the hydrogen atom, which causes the P–LG and the O–H bonds to break (Scheme 9). The backside mechanism leads to an inversion of configuration compared to the frontside mechanism and gives the products P1b, P2b, and P3b (Scheme 10).
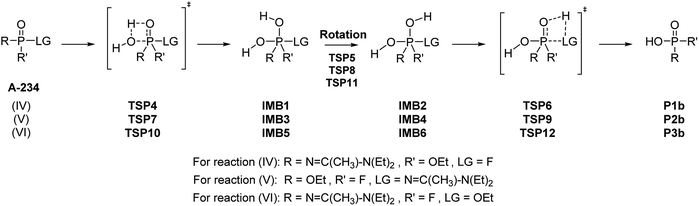 |
| | Scheme 9 The backside mechanism for the addition–elimination reaction. | |
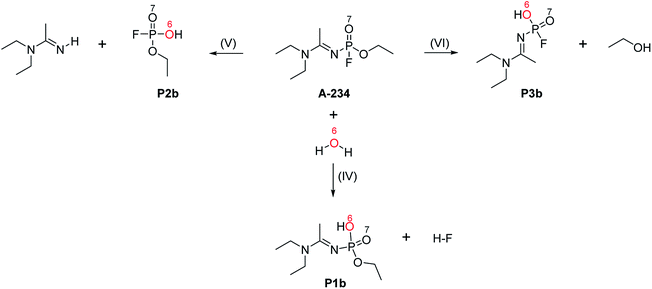 |
| | Scheme 10 Possible products in the backside addition–elimination reaction. | |
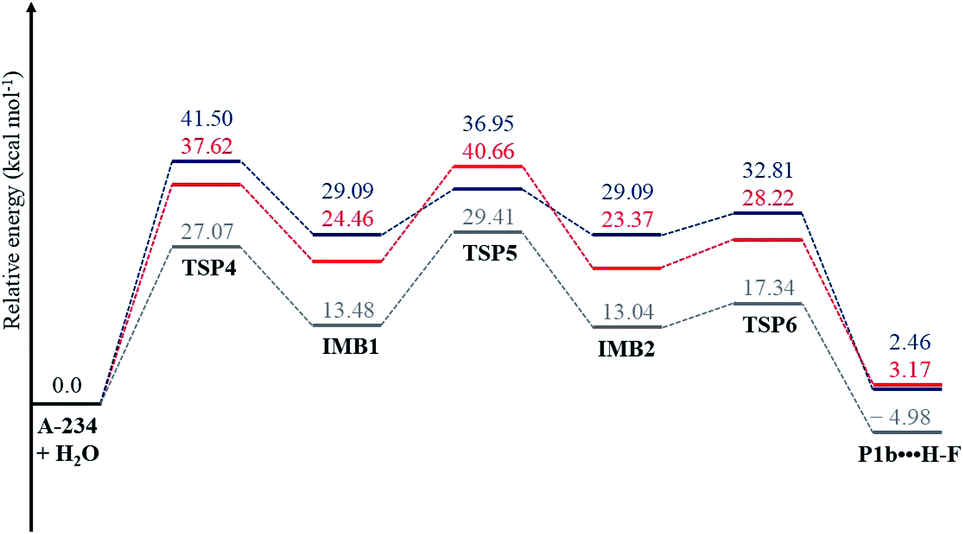
In the reaction (IV), the pentacoordinated intermediate has a fluorine atom at the apical position, which makes this channel favourable both kinetically and thermodynamically and this is in accordance with literature.44 As for the other two backside reactions, the addition of the water molecule causes the fluorine atom to shift to the equatorial position and the bulkier groups [NC(Me)–N(Et)2 in reaction (V) and OEt in reaction (VI)] to shift to the apical positions in the trigonal bipyramidal intermediates. This explains why these two reactions are less likely to occur. Bulk solvation increases the Gibbs free energy barriers due to differential solvation of the reactant and transition state; this phenomenon occurs due to the higher charge-delocalised transition state. Hydrolysis at the phosphinate moiety is also compared with that at the acetoamidine moiety. With H–F as the leaving group, the addition–elimination mechanism is slightly more favoured kinetically (Fig. 2) than the rate-determining step in Pathway A-Channel 4 of the acetoamidine hydrolysis (Fig. 1). However, the addition–elimination mechanism is not favoured thermodynamically (Fig. 1 and 2).
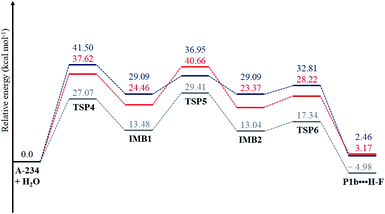 |
| | Fig. 2 The energy profile of the phosphinate hydrolysis for reaction (IV); see Scheme 9. ΔG(aq) (in blue) was obtained using the CPCM(water)-B3LYP/6-31+G(d,p) method, and ΔE(g) (in grey) and ΔG(g) (in red) were obtained using the B3LYP/6-31+G(d,p) method. | |
3.3 Comparison with other chemical warfare agents
The novichok nerve agents differ significantly from other nerve agents due to the presence of the amidine moiety. In this research work, we found that the acetoamidine moiety of A-234 is thermodynamically more susceptible to hydrolysis than the phosphinate moiety, while for other nerve agents, like GB and VX, the hydrolysis takes place at the phosphoryl moiety. The phosphor-amidine group increases the hydrophilicity of the novichoks when compared to VX,3 which is in agreement with our findings discussed herein (Table 2).
Table 2 Kinetic and thermodynamic data for hydrolysis at the phosphorus centre
| Reaction |
ΔG‡(g) (kcal mol−1) |
ΔG‡(aq) (kcal mol−1) |
ΔGreac,(g) (kcal mol−1) |
ΔGreac,(aq) (kcal mol−1) |
| Values within brackets are from the B3LYP/6-31++G(d,p) method as this method was used to study the hydrolysis at a phosphinate moiety.45 |
| (I) |
56.90 (56.87)a |
56.62 (56.60) |
7.30 (7.29) |
8.91 (8.95) |
| (II) |
53.87 (53.86) |
52.83 (52.76) |
7.84 (7.93) |
12.79 (11.12) |
| (III) |
55.29 (55.29) |
56.06 (56.03) |
5.84 (5.84) |
4.01 (4.02) |
| (IV) |
37.62 (37.57) |
41.50 (41.39) |
3.17 (3.17) |
2.46 (3.69) |
| (V) |
44.39 (44.36) |
51.86 (51.76) |
6.33 (6.44) |
11.25 (12.10) |
| (VI) |
48.53 (48.44) |
51.95 (47.70) |
9.38 (9.37) |
11.56 (12.42) |
4 Conclusions
The neutral hydrolysis mechanism of the novichok nerve agent, A-234, was studied using one water molecule (Scheme 11). This chemical warfare agent has two electropositive centres, which can undergo nucleophilic attack, namely the phosphorus atom and the sp2 carbon atom of the acetoamidine moiety. Our theoretical results indicate that the reaction barriers are comparable for the hydrolysis reaction at both the amidine and phosphinate moieties. However, the reaction at the acetoamidine moiety is thermodynamically preferred. The preference for the amidine hydrolysis is explained by the formation of the resonance-stabilised diethyl acetamide as the by-product of the reaction. It should also be noted that at room temperature, the phosphinate hydrolysis is expected to be slow and can be acid- or base-catalysed. In the acetoamidine hydrolysis, Pathway A, followed by the subsequent proton shift denoted as Channel 4, was found to be preferred. This corresponds to a nucleophilic attack by the water molecule at the sp2 carbon atom to form an intermediate, IM1, which then undergoes a conformational change ending with a 1,3-proton shift, resulting in the final product, A1. The formation of IM1 is the rate-determining step as it has the highest Gibbs free energy barrier. These theoretical outcomes provide an insight toward the efficient destruction of A-234.
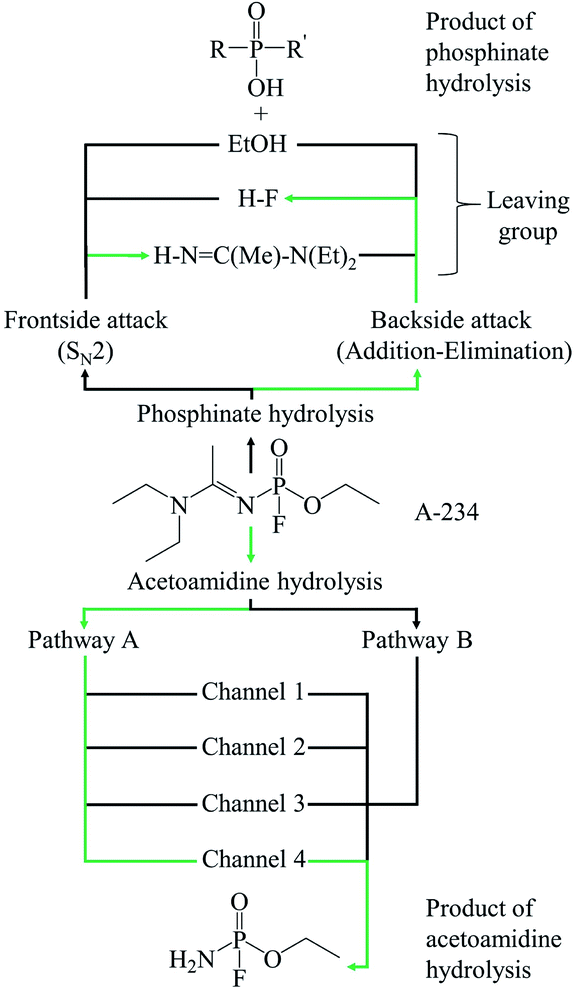 |
| | Scheme 11 Possible paths in the hydrolysis, starting from A-234 (Green arrows represent the preferred pathways according to the results). | |
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors acknowledge the facilities offered from their respective Universities. SEAGrid (https://seagrid.org/) is also acknowledged for the computational resources and services provided. NS acknowledges the funding provided by the Higher Education Commission (HEC) of Mauritius.
References
- E. Nepovimova and K. Kuca, Food Chem. Toxicol., 2018, 121, 343–350 CrossRef CAS.
- J. A. Vale, T. C. Marrs and R. L. Maynard, Clin. Toxicol., 2018, 56, 1093–1097 CrossRef CAS PubMed.
- L. Carlsen, Mol. Inf., 2018, 37, 1800106 Search PubMed.
- H. Bhakhoa, L. Rhyman and P. Ramasami, R. Soc. Open Sci., 2019, 6, 181831 CrossRef.
- S. Costanzi and G. D. Koblentz, Nonproliferation Rev., 2019, 26, 599–612 CrossRef.
- M. Kloske and Z. Witkiewicz, Chemosphere, 2019, 221, 672–682 CrossRef CAS.
- T. C. C. Franca, D. A. S. Kitagawa, S. F. d. A. Cavalcante, J. A. V. da Silva, E. Nepovimova and K. Kuca, Int. J. Mol. Sci., 2019, 20, 1222 CrossRef CAS.
- V. S. Mirzayanov, State Secrets: An Insider's Chronicle of the Russian Chemical Program, Outskirts Press, Inc., 2008 Search PubMed.
- M. Peplow, Chem. Eng. News, 2018, 96, 3–10 Search PubMed.
- S. L. Hoenig, Compendium of Chemical Warfare Agents, Springer, New York, 2008 Search PubMed.
- D. H. Ellison, Handbook of Chemical and Biological Warfare Agents, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- P. R. Chai, B. D. Hayes, T. B. Erickson and E. W. Boyer, Toxicol. Commun., 2018, 2, 45–48 CrossRef.
- S. P. Harvey, L. R. McMahon and F. J. Berg, Heliyon, 2020, 6, e03153 CrossRef.
- L. K. Sydnes, Chem. Int., 2020, 42, 26–27 Search PubMed.
- S. E. Hosseini, H. Saeidian, A. Amozadeh, M. T. Naseri and M. Babri, Rapid Commun. Mass Spectrom., 2016, 30, 2585–2593 CrossRef CAS.
- Y. B. Tan, I. R. Tay, L. Y. Loy, K. F. Aw, Z. L. Ong and S. Manzhos, Molecules, 2019, 24, 776 CrossRef.
- I. Lyagin and E. Efremenko, Catal. Commun., 2018, 120, 91–94 CrossRef.
- BBC 2018, Novichok: Why Nerve Agent Stays Deadly for So Long, 2018, https://www.bbc.com/news/health-44778697, accessed on 27 Sep 2019 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- C. G. Flinn, R. A. Poirier and W. A. Sokalski, J. Phys. Chem. A, 2003, 107, 11174–11181 CrossRef CAS.
- Y. Wu, L. Jin, Y. Xue, D. Q. Xie, C. K. Kim, Y. Guo and G. Sen Yan, J. Comput. Chem., 2008, 29, 1222–1232 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Pamidighantam, S. Nakandala, E. Abeysinghe, C. Wimalasena, S. R. Yogade, S. Marru and M. Pierce, Procedia Comput. Sci., 2016, 80, 1927–1939 CrossRef.
- N. Shen, Y. Fan and S. Pamidighantam, J. Comput. Sci., 2014, 5, 576–589 CrossRef.
- R. Dooley, K. Milfeld, C. Guiang, S. Pamidighantam and G. Allen, J. Grid Comput., 2006, 4, 195–208 CrossRef.
- K. Milfeld, C. Guiang, S. Pamidighantam and J. Giuliani, Cluster Computing through an Application-Oriented Computational Chemistry Grid, in Proc. 2005 Linux Clusters, The HPC Revolution, 2005 Search PubMed.
- This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- J. W. McIver and A. Komornicki, J. Am. Chem. Soc., 1972, 94, 2625–2633 CrossRef CAS.
- P. Atkins and J. de Paula, Physical Chemistry, Oxford University Press, New York, 8th edn, 2006 Search PubMed.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- J. H. Downing and M. B. Smith, Phosphorus Ligands, in Comprehensive Coordination Chemistry II, Elsevier, 2004, pp. 253–296 Search PubMed.
- W. E. White and J. B. Wright, Phosphorus, Sulfur, Silicon Relat. Elem., 1999, 144, 785–788 CrossRef.
- L. Gorb, A. Asensio, I. Tuñón and M. F. Ruiz-López, Chem.–Eur. J., 2005, 11, 6743–6753 CrossRef CAS PubMed.
- R. D. Cook, C. E. Diebert, W. Schwarz, P. C. Turley and P. Haake, J. Am. Chem. Soc., 1973, 95, 8088–8096 CrossRef CAS.
- M. K. Kesharwani, M. A. S. Khan, T. Bandyopadhyay and B. Ganguly, Theor. Chem. Acc., 2010, 127, 39–47 Search PubMed.
- D. Marciano, I. Columbus, S. Elias, M. Goldvaser, O. Shoshanim, N. Ashkenazi and Y. Zafrani, J. Org. Chem., 2012, 77, 10042–10049 CrossRef CAS.
- E. Dyguda-Kazimierowicz, S. Roszak and W. A. Sokalski, J. Phys. Chem. B, 2014, 118, 7277–7289 CrossRef CAS.
- J. Šečkute, J. L. Menke, R. J. Emnett, E. V Patterson and C. J. Cramer, J. Org. Chem., 2005, 70, 8649–8660 CrossRef.
- R. J. G. Thatcher and R. Kluger, Adv. Phys. Org. Chem., 1989, 25, 99–265 CrossRef.
- H. Chen, T. J. Giese, M. Huang, K.-Y. Wong, M. E. Harris and D. M. York, Chem.–Eur. J., 2014, 20, 14336–14343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05086e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tetiana Sergeievab,
Sergi Danésb,
Nandini Savoo
a,
Tetiana Sergeievab,
Sergi Danésb,
Nandini Savoo