
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhizovagine A, an unusual dibenzo-α-pyrone alkaloid from the endophytic fungus Rhizopycnis vagum Nitaf22†
Ali Wang,
Siji Zhao,
Gan Gu,
Dan Xu,
Xuping Zhang,
Daowan Lai * and
Ligang Zhou*
* and
Ligang Zhou*
Department of Plant Pathology, College of Plant Protection, China Agricultural University, Beijing 100193, China. E-mail: dwlai@cau.edu.cn; lgzhou@cau.edu.cn
First published on 27th July 2020
Abstract
Rhizovagine A (1), a novel dibenzo-α-pyrone alkaloid with an unprecedented 5/5/6/6/6 fused pentacyclic skeleton, was isolated from the endophytic fungus Rhizopycnis vagum Nitaf22. The structure was elucidated by comprehensive spectroscopic analysis, in combination with quantum chemical 13C NMR and electronic circular dichroism (ECD) calculations for configurational assignment. A plausible biosynthetic pathway for 1 was proposed. Compound 1 displayed acetylcholinesterase inhibitory activity.
Introduction
Dibenzo-α-pyrones, also known as 6H-dibenzo[b,d]pyran-6-ones, are a class of polyketides mainly produced by fungi.1 Structurally, the diversity of this type of metabolites mainly arises from ring A by having different substitutions and different degrees of unsaturation, while ring B is exclusively aromatic though it might have different substituents at C-7 and C-9 (i.e. OMe, OH) (Fig. 1). In some unusual cases, ring A is contracted to a five-membered ring, as found in cephalosol,2 or spiro-fused to a γ-lactone ring at C-4, as in rhizopycnolides A and B,3 and penicilliumolide A,4 or fused to a tetrahydrofuran, as in xinshengin,5 or attached to a C3 unit to form a diepoxy-cage structure, as in alternarilactone A.6 Ring C is six-membered in all reported structures except graphislactone D that contains a seven-membered ring.7 These metabolites display diverse biological activities, such as antimicrobial,2,3 cytotoxic,3,4 antioxidant,8 and larvicidal9 activities. The highly structural novelty as well as the interesting bioactivities made them attractive to the synthetic chemists. For example, the total synthesis of cephalosol, and graphislactones A–D has been completed.10–12 | ||
| Fig. 1 Structure of rhizovagine A (1), and the basic skeleton of dibenzo-α-pyrones (with possible substitutions and unsaturation pattern indicated). | ||
Rhizopycnis vagum Nitaf22 is a fungal endophyte that was isolated from Nicotiana tabacum, which was found to produce dibenzo-α-pyrones in our previous research,3 as well as anisic acid derivatives.13 In our course to searching for new bioactive fungal metabolites, this fungal strain was re-fermented in the rice medium in a large scale, which led to the isolation of a novel alkaloid, rhizovagine A (1) (Fig. 1). Herein, we report the isolation, structure elucidation, and the bioactivity of 1. The biosynthetic pathway of 1 was also discussed.
Results and discussion
The fungal EtOAc extract was successively subjected to repeated column chromatography over silica gel, and ODS as well as semi-preparative HPLC to afford compound 1 (Fig. 1).Rhizovagine A (1) was isolated as a minor, yellow amorphous solid. It exhibited a prominent pseudomolecular ion peak at m/z 386.1221 in the high-resolution electrospray ionization mass spectrometry (HRESIMS), indicating its molecular formula of C20H19NO7, with 12 degrees of unsaturation. The UV spectrum displayed maximum absorptions at 203, 249, 340, and 400 nm, which were similar to the reported dibenzo-α-pyrones.3 The 13C NMR spectrum (Table 1) displayed a total of 20 resonances, which were assigned to twelve sp2 carbons (including two sp2 CH at δC 101.7, 104.7), one oxygenated quaternary sp3 carbon (δC 86.2), one methine (δC 97.3), three methylene (δC 30.7, 25.4, 48.8), two methoxyl (δC 60.9, 55.9), and one methyl group (δC 26.2) by the aid of HSQC spectrum. Two meta-coupled aromatic protons (δH 7.20, 6.62, each d, J = 2.4 Hz) were seen in the downfield region of the 1H NMR spectrum (Table 1), which were characteristic of the 7,9-dioxygenated substitutions in B ring, commonly found in dibenzo-α-pyrones.3 One oxymethine proton (δH 5.65, t), and six methylene protons (δH 1.74–3.70) were observed in the upfield region, together with two methoxyl groups (δH 3.91, 3.83) and one singlet methyl group (δH 1.86). In addition, one chelated hydroxyl group (δH 11.41) was present.
| Position | δC, type | δH, mult. (J in Hz) |
|---|---|---|
| 1 | 86.2, C | |
| 2 | 160.3, C | |
| 3 | 128.7, C | |
| 4 | 171.9, C | |
| 4a | 143.7, C | |
| 6 | 164.4, C | |
| 6a | 100.9, C | |
| 7 | 164.7, C | |
| 8 | 101.7, CH | 6.62, d (2.4) |
| 9 | 166.6, C | |
| 10 | 104.7, CH | 7.20, d (2.4) |
| 10a | 134.2, C | |
| 10b | 121.7, C | |
| 1-CH3 | 26.2, CH3 | 1.86, s |
| 3-OCH3 | 60.9, CH3 | 3.83, s |
| 7-OH | 11.41, s | |
| 9-OCH3 | 55.9, CH3 | 3.91, s |
| 1′ | 97.3, CH | 5.65, t (5.6) |
| 2′ | 30.7, CH2 | 2.23, m |
| 1.81, m | ||
| 3′ | 25.4, CH2 | 2.11, m |
| 4′ | 48.8, CH2 | 3.61, m |
The gross structure of 1 was established by extensive analysis of the 2D NMR spectra (Fig. 2). The B ring was easily constructed by analysis of the HMBC spectra. The correlations from the chelated OH (δH 11.41) to C-7 (δC 164.7), C-6a (δC 100.9), and C-8 (δC 101.7), and from the methoxyl group (δH 3.91) to C-9 (δC 166.6), allowed the assignment of a 7-hydroxyl-9-methoxyl substituted B ring. The correlations from the methyl group (δH 1.86, s) to C-1 (δC 86.2), C-2 (δC 160.3), and C-10b (δC 121.7), together with the correlation from H-10 (δH 7.20, d) to C-10b, clearly positioned the methyl group to C-1. The second methoxyl group (δH 3.83) was attached to C-3 (δC 128.7) as inferred from the cross-peak between them in the HMBC spectrum. Two sp2 carbons (δC 171.9, 143.7) remained to be assigned, among which the former was a carbonyl group, while the latter had to be part of a double bond. By comparing the chemical shifts of these carbons with those of the co-occurring rhizopycnin B,3 suggested that this carbonyl was located at C-4, while the other positioned at C-4a (δC 143.7) to complete the A and C rings.
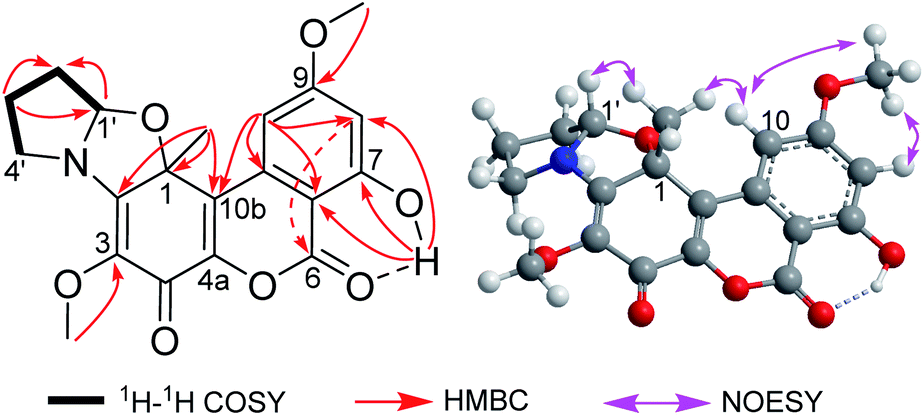 | ||
| Fig. 2 Key 2D NMR correlations of 1. | ||
In addition, a 2-oxygenated pyrrolidine ring was recognized by analysis of the 1H–1H COSY spectrum, in which correlations from H-1′ (δH 5.65, t) to H2-2′ (δH 2.23, 1.81, m), H2-2′ to H2-3′ (δH 2.11, m), and in turn H2-3′ to H2-4′ (δH 3.61, m) were seen (Fig. 2), as well as by taken into consideration of the chemical shifts of these hydrocarbon groups, especially CH2-4′ (δH 3.61/δC 48.8) and CH-1′ (δH 5.65/δC 97.3).14 The above functionalities only accounted for 11 degrees of unsaturation, thus hinting the presence of one additional ring to satisfy the required unsaturation index. Then, C-1 and C-1′ had to be connected via an epoxy bridge, while C-2 was linked to the nitrogen atom to form a five-membered ring. This can explain the unusual downfield shift for C-1 (δC 86.2), comparing to that of rhizopycnin B (δC 69.5).3 Moreover, the NOESY correlation between H-1′ and 1-CH3 not only confirmed the above fusion pattern, but also defined the relative configuration of 1 (Fig. 2).
Meanwhile, in order to confirm the deduced structure, a GIAO 13C NMR calculation was performed on mPW1PW91/6-311+G(2d,p)//B3LYP/6-31+G(d,p) level of theory using the reported procedure and scaling parameters.15 As shown in Table 2, the calculated 13C NMR data of the proposed structure fitted well with the experimental data with MAE 1.59 ppm and RMSD 1.82 ppm. Therefore, the structure of 1 was elucidated to be a hexahydropyrrolo[2,1-b]oxazole fused dibenzo-α-pyrone, hitherto not reported in nature.
| Position | Exp. δC | Cal. δCa | Δδ (cal. − exp.) |
|---|---|---|---|
| a Cal. δC was scaling corrected from calculated shielding tensor (σ) using the formula δC = (186.5242 − σ)/1.0533. | |||
| 1 | 86.2 | 88.0 | 1.8 |
| 2 | 160.3 | 161.0 | 0.7 |
| 3 | 128.7 | 128.4 | −0.3 |
| 4 | 171.9 | 169.4 | −2.5 |
| 4a | 143.7 | 144.8 | 1.1 |
| 6 | 164.4 | 162.6 | −1.8 |
| 6a | 100.9 | 100.0 | −0.9 |
| 7 | 164.7 | 163.0 | −1.7 |
| 8 | 101.7 | 99.2 | −2.5 |
| 9 | 166.6 | 164.4 | −2.2 |
| 10 | 104.7 | 102.9 | −1.8 |
| 10a | 134.2 | 133.6 | −0.6 |
| 10b | 121.7 | 122.7 | 1.0 |
| 1-CH3 | 26.2 | 24.8 | −1.4 |
| 3-OCH3 | 60.9 | 56.7 | −4.2 |
| 9-OCH3 | 55.9 | 53.2 | −2.7 |
| 1′ | 97.3 | 96.4 | −0.9 |
| 2′ | 30.7 | 31.8 | 1.1 |
| 3′ | 25.4 | 26.8 | 1.4 |
| 4′ | 48.8 | 49.9 | 1.1 |
| MAE | 1.59 | ||
| RMSD | 1.82 | ||
To determine the absolute configuration of 1, the TDDFT ECD computations were carried out on different level of theory (pbe0/TZVP, B3LYP/TZVP, BH&HLYP/TZVP) for the B3LYP/6-31G(d)-optimized conformers of 1. The calculated spectrum of (1R,1′S)-1 matched the experimental data (Fig. S1, ESI†), and the best agreement was found for the BH&HLYP/TZVP-calculated one (Fig. 3), thus the absolute configuration of 1 was unambiguously assigned.
 | ||
| Fig. 3 Calculated and experimental ECD spectra of 1. | ||
Structurally, compound 1 was characterized as one pyrrolidine fused to a dibenzo-α-pyrone uniquely via an oxazolidine ring. The plausible biosynthetic pathway of 1 was proposed (Scheme 1). This should involve the formation of a key metabolite, TMC-264 (3), which was co-isolated from the titled fungus, and was synthesized via the polyketide pathway.16 This compound should be derived from alternariol (2) that was the first product of PKS,17 by several tailoring steps. The nucleophilic attack from Δ1-pyrroline that derived from L-ornithine, to the substrate 3, would yield the iminium cation intermediate (1a), which then give rise to the derivative 1b by ejecting the chloride ion (as a leaving group). The followed-up attack from 1-OH to C-1′ in 1b should give the oxazolidine (1). Iminium cations were reported to be widely involved in the biosynthesis of a range of alkaloids in nature.18,19
 | ||
| Scheme 1 Plausible biosynthetic pathway of 1. | ||
Hexahydropyrrolo[2,1-b]oxazole or pyrrolidinooxazolidine alkaloids were not commonly found in nature. So far only 15 natural ones that incorporated in eight different skeletons have been reported, to our knowledge, which included two derivatives from the ladybird beetles, three from marine invertebrates, and ten from plants (Fig. S2, ESI†), however, none were reported from fungi. Some of these metabolites exhibited antibacterial, cytotoxic, antiplasmodial, antiviral, and phytotoxic activities,20–23 or playing a defense role against predators.24
Compound 1 was thus screened for the antibacterial activity, however, it was inactive against the tested bacteria (MIC > 128 μg mL−1). This compound was also tested for the antioxidant activity due to the presence of a phenolic group, whereas it did not exhibit strong activity in the DPPH radical scavenging assay (IC50 > 200 μg mL−1). Meanwhile, in view of alkaloids being a major class of naturally-occurring acetylcholinesterase (AchE) inhibitors,25 which could be of use as insecticides or a treatment for Alzheimer's disease, compound 1 was hence evaluated for its inhibitory activity against AchE. And a moderate inhibition (IC50 43.1 μM) was found comparing to the positive control (tacrine hydrochloride, IC50 6.1 μM).
Conclusions
In this study, rhizovagine A (1), the first fungal pyrrolidinooxazolidine alkaloid, bearing an unprecedented pentacyclic skeleton was isolated. The structure was elucidated by comprehensive spectroscopic analysis, together with quantum-chemical 13C NMR and ECD calculations. Compound 1 showed AchE-inhibitory activity, which could serve as a scaffold for developing new AchE inhibitors. Moreover, due to the structural novelty, 1 could be an intriguing target for realizing a total synthesis or biosynthesis.Experimental section
General experimental procedures
The optical rotations were recorded on a Rudolph Autopol III automatic polarimeter (Rudolph Research Analytical, Hackettstown, New Jersey). Ultraviolet (UV) spectra were recorded on a TU-1810 UV/Vis spectrophotometer (Beijing Persee General Instrument Co., Ltd., Beijing, China). Circular dichroism (CD) spectra were recorded on a JASCO J-815 CD spectrometer (JASCO Corp., Tokyo, Japan). High-resolution electrospray ionization mass spectrometry (HRESIMS) spectra were recorded on a LC 1260/Q-TOF-MS 6520 machine (Agilent Technologies, Santa Clara, CA). 1H, 13C, and 2D NMR spectra were measured on Bruker Avance 400 NMR spectrometers (Bruker BioSpin, Zürich, Switzerland). 1H and 13C NMR chemical shifts were expressed in δ (ppm) referring to the inner standard tetramethylsilane (TMS), and coupling constants in Hertz. HPLC-DAD analysis was performed on a Shimadzu LC-20A instrument equipping with a SPD-M20A photodiode array detector (Shimadzu Corp., Tokyo, Japan) using an analytic C18 column (250 mm × 4.6 mm i.d., 5 μm; Phenomenex Inc., Torrance, California). Semi-preparative HPLC separation was carried out on a Lumtech instrument (Lumiere Tech. Ltd., Beijing, China) equipped with a K-501 pump (flow rate: 3 mL min−1) and a K-2501 UV detector (detection was set at 370 nm) using a Luna-C18 column (250 mm × 10 mm i.d., 5 μm, Phenomenex Inc., Torrance, California) eluting with a mixture of MeOH and water.Fungal material and fermentation
The endophytic fungus Rhizopycnis vagum Nitaf22 was isolated from Nicotiana tabacum previously in our lab.3The fungus was cultured on potato dextrose agar for 5 days at 25 °C, and the resulting hyphae were used to inoculate 150 mL potato dextrose broth in a 250 mL flask. The culture was shaking for 7 days at 150 rpm and 25 °C, which was then used as inoculum to inoculate the autoclaved rice media in 1 L Erlenmeyer flasks each containing 100 g of rice and 110 mL of distilled water. The fermentation was carried out on a total of 10 kg of rice under static conditions at room temperature (RT) in the dark for 50 days.
Extraction and isolation
The culture was combined, dried and ground. The dry materials were extracted with EtOAc three times at RT, each for 5 days, which resulted in 163 g of extract after removal of the solvent.The extract was subjected to vacuum liquid chromatography over silica gel (i.d. 8 cm × 30 cm) by eluting with a different mixture of petroleum ether and acetone, then CH2Cl2–MeOH. Fractions were pooled according to TLC, and five fractions were obtained (Frs. A–E). Fr. B was found to contain dibenzo-α-pyrones, which was then chromatographed by medium pressure liquid chromatography over silica gel using a gradient of CH2Cl2–MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–1:1, v/v) as the eluent to yield seven subfractions (Frs. B1–B7). Fr. B1 was further processed by chromatography over ODS using a gradient of MeOH/H2O (35–100%) as the mobile phase, to afford nine further fractions (Frs. B1-1–B1-9). Among which, compound 1 (2.9 mg) was isolated from Fr. B1-5 by semi-preparative HPLC (63% MeOH/H2O).
:0–1:1, v/v) as the eluent to yield seven subfractions (Frs. B1–B7). Fr. B1 was further processed by chromatography over ODS using a gradient of MeOH/H2O (35–100%) as the mobile phase, to afford nine further fractions (Frs. B1-1–B1-9). Among which, compound 1 (2.9 mg) was isolated from Fr. B1-5 by semi-preparative HPLC (63% MeOH/H2O).
ε) 203 (4.57), 249 (4.66), 340 (3.86), 400 (3.84) nm; ECD (c = 4.33 × 10−4 M, MeOH) λ (Δε) 218 (+11.33), 247 (−27.33), 289 (+10.46), 352 (−0.97) nm; 1H NMR (CDCl3, 400 MHz), 13C NMR (CDCl3, 100 MHz) see Table 1; HRESIMS m/z 386.1221 [M + H]+ (calcd for C20H20NO7, 386.1234).ECD calculation
Conformational searches using Merck Molecular Force Field (MMFF) were done by the Macromodel software. Geometry optimization and frequency calculations of the MMFF-conformers using the DFT method were performed by the Gaussian 09 package.26 TDDFT ECD calculations of the low-energized conformers (≥1%) without imaginary frequencies were carried out by Gaussian 09 using different functions (PBE0, B3LYP, BH&HLYP) and TZVP basis set with the polarizable continuum model (PCM) for MeOH. The ECD spectrum of each conformer was simulated by the program SpecDis27 using a Gaussian band shape with an exponential half-width σ of 0.25–0.3 eV, using the dipole-length computed rotational strengths. The equilibrium population of each conformer at 298.15 K was calculated from its relative Gibbs free energies using Boltzmann statistics. The Boltzmann-averaged ECD spectrum was then generated accordingly. The generated ECD spectra were compared with the experimental one to determine the absolute configuration. The calculated ECD spectra were UV-shifted (+7, +3, and +28 nm for PBE0, B3LYP, and BH&HLYP, respectively), for comparison with the measured spectrum.13C NMR calculation
For the calculation of 13C NMR chemical shifts, B3LYP/6-31+G(d,p) method was used to optimize the selected conformers. NMR calculations were performed at the level of mPW1PW91/6-311+G(2d,p) with the gauge-independent atomic orbital (GIAO) method using the SMD model for CHCl3. The 13C NMR chemical shifts were considered as the average value of the same atom in different conformers according to their Boltzmann distributions, using the relative Gibbs free energies as the weighting factors. The empirical scaling method was used to decrease the systematic error. The chemical shift (cal. δC) of each atom was scaling corrected from the calculated shielding tensor (σ) by using the formula δC = (186.5242 − σ)/1.0533, using the scaling factors reported by Lodewyk, et al.15 The differences Δδ were determined by subtracting the experimental chemical shifts (δexp.) from the calculated chemical shifts (δcal.). The results were evaluated in terms of the mean absolute deviation (MAE), and root mean square deviation (RMSD).Antibacterial assay
The antibacterial activity of compound 1 was evaluated against six pathogenic bacteria, Agrobacterium tumefaciens, Bacillus subtilis, Pseudomonas lachrymans, Ralstonia solanacearum, Staphylococcus hemolyticus, and Xanthomonas vesicatoria, as described previously.3DPPH radical scavenging assay
The DPPH radical scavenging activity of compound 1 was determined based on the reduction of 1,1-diphenyl-2-picrylhydrazyl (DPPH) as reported previously.8 Butylated hydroxytoluene (BHT) was used as the positive control.AchE inhibitory assay
The AchE inhibitory bioassay was performed using the method described by Meng et al.28 Briefly, the assay was performed in 96-well plates, and in each well phosphate buffered saline (PBS, pH 7.7) solution (160 μL), AChE solution (1.0 U mL−1, 10 μL), 5,5′-dithio-bis-(2-nitrobenzoic acid) solution (7.5 mM in PBS, 10 μL), and the tested compound of various concentrations (dissolved in 30% of DMSO–PBS, 10 μL) were added. After pre-incubation at 30 °C for 15 min, 10 μL of acetylthiocholine iodide (10 mM in PBS) was added. The incubation was maintained for 30 min, then the absorbance was measured at 405 nm using a microplate spectrophotometer. Tacrine hydrochloride was used as the positive control, while PBS as the negative control. The percentage (%) of inhibition was determined using the following expression: [(Ac − At)/Ac] × 100, where Ac is the absorbance of the negative control (n = 3), while At is that of the tested compounds (n = 3). The median inhibitory concentration (IC50) was calculated using the linear regression between the inhibitory probability and concentration logarithm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided by the National Key R&D Program of China (No. 2017YFD0201105 and 2017YFC1600905).Notes and references
- Z. Mao, W. Sun, L. Fu, H. Luo, D. Lai and L. Zhou, Molecules, 2014, 19, 5088–5108 CrossRef.
- H. W. Zhang, W. Y. Huang, J. R. Chen, W. Z. Yan, D. Q. Xie and R. X. Tan, Chem.–Eur. J., 2008, 14, 10670–10674 CrossRef CAS.
- D. Lai, A. Wang, Y. Cao, K. Zhou, Z. Mao, X. Dong, J. Tian, D. Xu, J. Dai, Y. Peng, L. Zhou and Y. Liu, J. Nat. Prod., 2016, 79, 2022–2031 CrossRef CAS.
- C. Darsih, V. Prachyawarakorn, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, RSC Adv., 2015, 5, 70595–70603 RSC.
- F. Ye, G.-D. Chen, J.-W. He, X.-X. Li, X. Sun, L.-D. Guo, Y. Li and H. Gao, Tetrahedron Lett., 2013, 54, 4551–4554 CrossRef CAS.
- J.-W. Tang, H.-C. Xu, W.-G. Wang, K. Hu, Y.-F. Zhou, R. Chen, X.-N. Li, X. Du, H.-D. Sun and P.-T. Puno, J. Nat. Prod., 2019, 82, 735–740 CrossRef CAS PubMed.
- T. Tanahashi, M. Kuroishi, A. Kuwahara, N. Nagakura and N. Hamada, Chem. Pharm. Bull., 1997, 45, 1183–1185 CrossRef CAS.
- J. Tian, L. Fu, Z. Zhang, X. Dong, D. Xu, Z. Mao, Y. Liu, D. Lai and L. Zhou, Nat. Prod. Res., 2017, 31, 387–396 CrossRef CAS.
- Z. Mao, D. Lai, X. Liu, X. Fu, J. Meng, A. Wang, X. Wang, W. Sun, Z. L. Liu, L. Zhou and Y. Liu, Pest Manage. Sci., 2017, 73, 1478–1485 CrossRef CAS PubMed.
- Y. Xie, N. Wang, B. Cheng and H. Zhai, Org. Lett., 2012, 14, 3–5 CrossRef CAS PubMed.
- A. Arlt and U. Koert, Synthesis, 2010, 2010, 917–922 CrossRef.
- H. Abe, K. Nishioka, S. Takeda, M. Arai, Y. Takeuchi and T. Harayama, Tetrahedron Lett., 2005, 46, 3197–3200 CrossRef CAS.
- A. Wang, P. Li, X. Zhang, P. Han, D. Lai and L. Zhou, Molecules, 2018, 23, 591 CrossRef.
- W.-L. Chen, L.-Y. Wang and Y.-J. Li, Eur. J. Org. Chem., 2020, 2020, 103–107 CrossRef CAS.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS.
- M. Sakurai, M. Nishio, K. Yamamoto, T. Okuda, K. Kawano and T. Ohnuki, Org. Lett., 2003, 5, 1083–1085 CrossRef CAS PubMed.
- Y.-H. Chooi, M. J. Muria-Gonzalez, O. L. Mead and P. S. Solomon, Appl. Environ. Microbiol., 2015, 81, 5309 CrossRef CAS PubMed.
- T. Hemscheidt, in Biosynthesis: aromatic polyketides, isoprenoids, alkaloids, ed. F. J. Leeper and J. C. Vederas, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000, pp. 175–206, DOI:10.1007/3-540-48146-x_4.
- T. Hartmann and D. Ober, in Biosynthesis: aromatic polyketides, isoprenoids, alkaloids, ed. F. J. Leeper and J. C. Vederas, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000, pp. 207–243, DOI:10.1007/3-540-48146-x_5.
- M. Tadesse, J. Svenson, M. Jaspars, M. B. Strom, M. H. Abdelrahman, J. H. Andersen, E. Hansen, P. E. Kristiansen, K. Stensvag and T. Haug, Tetrahedron Lett., 2011, 52, 1804–1806 CrossRef CAS.
- J. H. Cardellina, R. L. Hendrickson, A. A. Stierle and G. E. Martin, Tetrahedron Lett., 1991, 32, 2347–2350 CrossRef CAS.
- Z.-X. Hu, H.-Y. Tang, J. Guo, H. A. Aisa, Y. Zhang and X.-J. Hao, Tetrahedron, 2019, 75, 1711–1716 CrossRef CAS.
- G. Komlaga, G. Genta-Jouve, S. Cojean, R. A. Dickson, M. L. K. Mensah, P. M. Loiseau, P. Champy and M. A. Beniddir, Tetrahedron Lett., 2017, 58, 3754–3756 CrossRef CAS.
- P. Radford, A. B. Attygalle, J. Meinwald, S. R. Smedley and T. Eisner, J. Nat. Prod., 1997, 60, 755–759 CrossRef CAS PubMed.
- P. J. Houghton, Y. Ren and M.-J. Howes, Nat. Prod. Rep., 2006, 23, 181–199 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- T. Bruhn, A. Schaumloeffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- X. Meng, Z. Mao, J. Lou, L. Xu, L. Zhong, Y. Peng, L. Zhou and M. Wang, Molecules, 2012, 17, 11303–11314 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ECD and 13C NMR calculation data, 1D and 2D NMR, and MS spectra of 1. See DOI: 10.1039/d0ra05022a |
| This journal is © The Royal Society of Chemistry 2020 |