
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and optical properties of langbeinite-related red-emitting K2Sc2(MoO4)(PO4)2:Eu phosphors†
Kateryna V. Terebilenko *a,
Serhii G. Nedilkoa,
Vitalii P. Chorniiab,
Vadym M. Prokopetsa,
Mykola S. Slobodyanika and
Volodymyr V. Boykob
*a,
Serhii G. Nedilkoa,
Vitalii P. Chorniiab,
Vadym M. Prokopetsa,
Mykola S. Slobodyanika and
Volodymyr V. Boykob
aTaras Shevchenko National University of Kyiv, Kyiv 01601, Ukraine. E-mail: kterebilenko@gmail.com
bNational University of Life and Environmental Sciences of Ukraine, Kyiv, 03041, Ukraine
First published on 7th July 2020
Abstract
The concentration series of langbeinite-related solid solutions K2Sc2(MoO4)(PO4)2:xEu (x = 0.1, 0.2, 0.6, 0.8, and 1.0 mol%) has been prepared via a solid state route and the effects of europium content on the phase composition, morphology, crystal structure and luminescence properties have been studied by scanning electron microscopy, X-ray powder diffraction, UV-vis, IR and luminescence spectroscopy. The band gap values have been estimated from UV-vis spectra and are in the range of 3.7–3.8 eV for all concentrations studied. The electronic band structure calculations have shown that Sc d, Mo d and Ophos p states dominate in the band edge region and determine the optical transitions in the K2Sc2(MoO4)(PO4)2 host. The photoluminescence (PL) spectra, intensity and decay time dependences on the Eu3+ concentration revealed complex behavior of europium-containing emitting centers. The PL characteristics indicated the presence of at least two types of luminescence centers. One of them (EuK) is shown to be formed by the Eu3+ ion located within K sites, while the other one is formed by the Eu3+ ions that reside in Sc sites (EuSc). The luminescence color coordinates calculated for K2Sc2(MoO4)(PO4)2:xEu indicated that these ceramics can be potential candidates for UV-based lighting applications as efficient red phosphors.
Introduction
Langbeinite-related complex oxides have been attracting lots of attention in recent decades owing to their prominent advantages such as high structural flexibility toward different anionic substitutions by phosphate,1 sulfate2 or molybdate3 anions showing high thermal and chemical stability.Among the diversity of these complex oxides a series of phosphate-containing ones has the general composition AxB2(PO4)3, where different combinations of mono- (A) and three/four-valent (B) metals can be found.4 These frameworks gained increasing attention due to their remarkable optical properties and promising application in light emitting diodes (LEDs), biomedical diagnostics, optical communication, high density optical data storage and sensors, etc.1,5–7
Mainly, these species containing PO43− groups have become one of the most widely investigated families of phosphates with three dimensional (3D) frameworks since Norberg8 has widened the group of langbeinite-related phosphates with a common formula K2BTi(PO4)3, where B – rare-earth metals. Incorporation of RE ions into langbeinite-related phosphate host allowed designing optical materials with remarkable luminescence properties. In particular, bright blue cooperative up-conversion emission of Yb3+ has been observed from K2Ti1.887Yb0.113(PO4)3 langbeinite single crystals grown from a phosphate flux.1 A possibility to get luminescence with tunable colors from blue to green and yellow has been reported for K2Tb1.5Ta0.5P3O12:Eu3+ and KBaSc2(PO4)3:Ce3+/Eu2+/Tb3+ compounds.5,9 Most of the reported RE-containing derivatives of langbeinite structure show outstanding luminescent properties due to the structural peculiarities and, consequently, they have possibility of application in the white LEDs. In this area, study of the langbeinite-related compounds have been mainly focused on alkaline and lanthanide-containing systems5,9–11 owing to the simple iso- and aliovalent substitution within the cationic sites of langbeinite structure. Meanwhile, the designed assembly of poly-oxoanionic frameworks is seen to be more difficult to obtain due to stability that inherent to langbeinite structure. Up to now there are only a several poly-anionic derivatives of langbeinite structure have been successfully obtained, for example, K2MgTi(SO4)(PO4)2,12 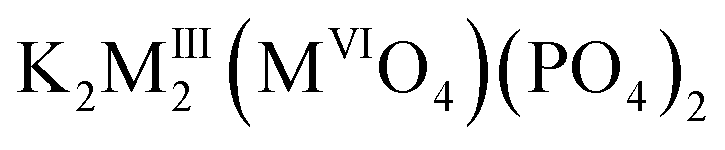 , (MIII = Fe, Sc; MVI = Mo, W)13 and KBaMIIIMIV(SiO4)(PO4)2 (MIV = Zr, Sn; MIII = Y, La, Nd, Sm, Eu, Gd, Dy).14 The key feature of these structures is presence of one unique position for a tetrahedrally coordinated atom, X, (XO4), where X = P, Si, Mo, S, Cr. Thus, in spite of different sizes and oxidation states phosphorus and other atoms (tungsten, molybdenum, silicon or sulfur) occupy the same position with a ratio P/X = 2
, (MIII = Fe, Sc; MVI = Mo, W)13 and KBaMIIIMIV(SiO4)(PO4)2 (MIV = Zr, Sn; MIII = Y, La, Nd, Sm, Eu, Gd, Dy).14 The key feature of these structures is presence of one unique position for a tetrahedrally coordinated atom, X, (XO4), where X = P, Si, Mo, S, Cr. Thus, in spite of different sizes and oxidation states phosphorus and other atoms (tungsten, molybdenum, silicon or sulfur) occupy the same position with a ratio P/X = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This feature leads to a significant change in the X–O distances in respect to the typical P–O distances in pure phosphate langbeinites. Elongation of X–O bonds results in changes of oxygen surrounding of metals in six-fold coordination and can affect luminescence properties of the compound.
:1. This feature leads to a significant change in the X–O distances in respect to the typical P–O distances in pure phosphate langbeinites. Elongation of X–O bonds results in changes of oxygen surrounding of metals in six-fold coordination and can affect luminescence properties of the compound.
In this paper we report the influence of Eu3+ doping concentration on morphology, structure and optical properties of K2Sc2(MoO4)(PO4)2:xEu (x = 0.1, 0.2, 0.6, 0.8, and 1.0 mol%) ceramics. The procedure of synthesis and infra-red (IR) spectroscopy for undoped K2Sc2(MoO4)(PO4)2 compound containing mixed (P/Mo)O4 tetrahedra have been reported so far.13
The crystal structure of the K2Sc2(MoO4)(PO4)2:xEu ceramics have been studied by powder X-ray diffraction. The grain size distribution and morphology have been revealed by scanning electron microscopy (SEM). Electronic band structure of the perfect K2Sc2(MoO4)(PO4)2 crystal, luminescent properties as well as chromaticity characteristics of the K2Sc2(MoO4)(PO4)2:xEu samples are reported in this work for the first time.
Experimental
Synthetic procedures
The powdered polycrystalline samples of K2Sc2(MoO4)(PO4)2:xEu (x = 0.1, 0.2, 0.6, 0.8, and 1.0 mol%) have been synthesized through a conventional solid-state reaction technique. Analytically pure MoO3, Eu2O3, Sc2O3, KH2PO4 and K2MoO4 have been taken as starting materials without further purification. Stoichiometric amounts of the reagents have been thoroughly mixed in an agate mortar with a pestle into fine powders. At the first stage, the mixtures have been preheated at 550 °C for 2 h to get rid of water. Then, the resulting powders have been mixed with a drop of ethanol, thoroughly reground and sintered at 600, 700, 800, 900 and 1000 °C, for 5 h at each temperature, and finally fired at 1050 °C for 20 h in a muffle furnace with intermediate grindings at each annealing temperature. The coated alumina crucibles have been used as a reaction vessel. For photoluminescence study powdered samples have been compacted into pellets with 15 mm diameter and 2 mm thickness.Calculation details
The electronic structure of K2Sc2(MoO4)(PO4)2 perfect crystal has been calculated with the use of WIEN2k program package,15 in which the full-potential linear-augmented-plane-wave (FP-LAPW) method is implemented within the framework of density-functional theory (DFT). The Perdew and Wang generalized gradient approximation has been employed for the exchange–correlation potential.16 The relativistic effects have been treated in the scalar relativistic approximation. The modified tetrahedron method has been used for the Brillouin zone (BZ) integration.17 The muffin-tin (MT) radii have been chosen from condition of ‘almost touching’ MT spheres. In all cases, the energy of separation between core and valence states has been chosen as 6.0 Ry. The potential and charge density in the MT spheres have been expanded in spherical harmonics with lmax = 10. The plane wave cutoff parameter RMT × Kmax was equal to 6.0. The magnitude of the largest vector Gmax in the charge density Fourier expansion was 14.0. The convergence criterion of total energy has been chosen as the 0.0001 Ry. Calculations of partial densities of electronic states (PDOSes) were carried out for 16 nonequivalent sampling k-points in the irreducible part of the Brillouin zone. The partial densities of electronic states (PDOS) were calculated using well-known relations implemented in the WIEN2k program code.15 Atomic coordinates and lattice parameters utilized in the electronic band structure calculations have been taken from literature.13 It is worth noting that the partial occupation of crystal sites by P and Mo with ratio 2:1 is inherent to K2Sc2(MoO4)(PO4)2. The phosphorus or molybdenum atoms were placed on each of 12 appropriate positions within unit cell with attempt to obtain uniform distribution of these elements in the crystal.
Sample characterization
Phase composition has been investigated by powder XRD using of Shimadzu XRD-6000 diffractometer (curved pyrolytic graphite counter monochromator, CuKα radiation with λ = 1.54184 Å) operating in Bragg–Brentano (θ/2θ) geometry (2θ = 5 ÷ 90°).IR spectra have been measured on a PerkinElmer Spectrum BX FTIR spectrometer in the frequency range 400–4400 cm−1 using KBr technique.
Particles size and morphology have been studied by scanning electron microscope (SEM) Tescan Mira 3 LMU with a 20 nm electronic beam diameter during the measurements. Detector of the secondary electrons (InBeam) enhances spatial resolution up to 1 nm. Besides the SEM imaging, microelement analysis of various areas of the samples was also performed using the same SEM.
Diffuse reflectance spectra have been collected at room temperature using 80 Thermo Scientific Evolution 600 UV-vis spectrometer equipped with R-928 red sensitive photomultiplier tube. The spectra were recorded in the range of 190–600 nm with 0.2 nm resolution with BaSO4 as a reference.
The PL emission and excitation spectra have been recorded at room temperature of the samples using the DFS-12 spectrometer equipped with FEU-100 photomultiplier. The powerful xenon arc lamp (DXeL-1000) combined with DMR4 prism monochromator was used as source of excitation light. All the spectra have been corrected on system response.
The PL kinetics have been measured with use of the MSA-300 multiscaler photon counter and LEDs (λrad = 365 and 400 nm) operating at pulse regimes as sources of the PL excitation.
Results and discussion
Crystal structure and morphology
The X-ray diffraction patterns for solid solutions K2Sc2(MoO4)(PO4)2:xEu (x = 0.1–1.0 mol%) ceramics sintered at 1050 °C are presented in Fig. 1 in comparison to a standard card of K2Sc2(MoO4)(PO4)2 (ICSD #262501). A langbeinite-like structure for a series of solid solutions is obtained across the narrow range x = 0.1–0.6 mol%. When the x value increases to 0.8 and 1.0 mol%, a trace of the admixture phase peaks could be revealed, which indicates that the solid solubility toward rare-earth metal for the single-phase langbeinite materials reached a maximum here. Increasing of the annealing time and temperature has not influenced the XRD pattern for x = 0.8–1.0 mol%. The impurity composition seems to be a highly stable ScPO4 that is also known for the KPO3–K2MoO4–MoO3–Sc2O3 molten system.13 Some admixture peaks (namely those at 2Θ = 27.1, 36.6, and 41.5°) correlate well with standard pattern for ScPO4, while these ones at 2Θ = 21.6 and 30.7° cannot belong to ScPO4 are also observed for the two samples with the highest Eu content.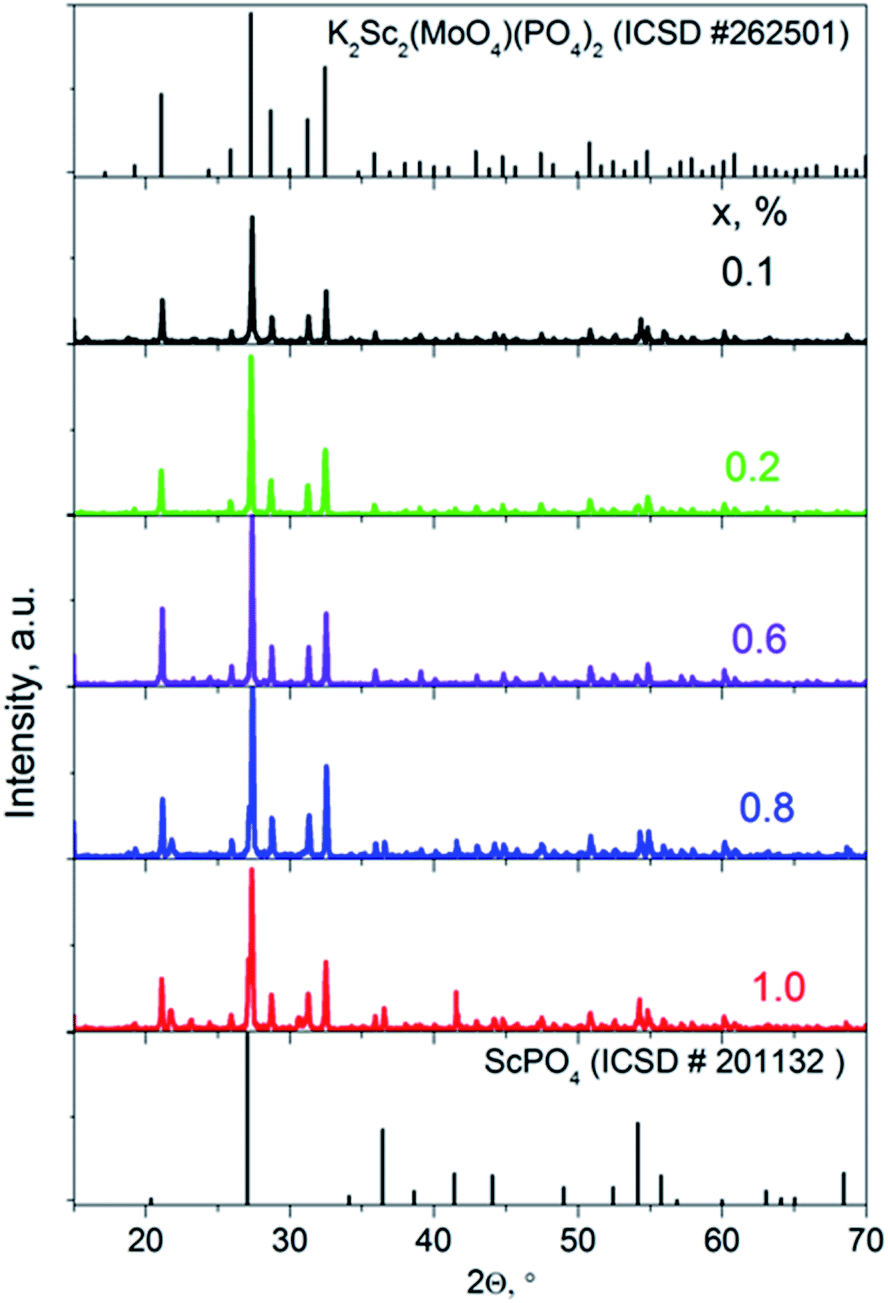 | ||
| Fig. 1 XRD patterns of K2Sc2(MoO4)(PO4)2:xEu, where x = 0.1–1.0 mol%. | ||
Fig. 2 shows the SEM images of the samples K2Sc2(MoO4)(PO4)2:xEu. It can be seen that the samples exhibit dense microstructures with almost no pores with relatively uniform particles (Fig. 2a–d). The product is crystallized in a compact form with an average size of the particles of about 3–7 μm, which are well separated. A grain boundary between the microcrystals is clearly seen for all range of x value studied. The octahedrally shaped particles develop loose agglomerates as it has been shown for single crystals prepared from a melt.13 Importantly, the morphology of the microcrystals is maintained to be highly symmetrical octahedral when the synthetic approach changes from high temperature growth to solid state route. When the x value increases up to 1.0 mol% the particles become slightly more agglomerated and some different particles with irregular shape can be found over the sample studied (Fig. 2d). The average particles' size increase can be related with the melting-point depression when the Eu content increases. It had been shown for mixed potassium-europium molybdate-phosphate powder to melt at 1050 °C.18 On the contrary, K2Sc2(MoO4)(PO4)2 is thermally stable at more than 1200 °C.13 The SEM-EDS analysis performed for the samples with x = 0.1 (Fig. 2e) and 1.0 (Fig. 2f) has showed that in the case of x = 1.0 the bigger particles of irregular shape possess composition of the admixture ScPO4 phase. The details of EDS analysis are given in Fig. S1 and Table S1, ESI.†
 | ||
| Fig. 2 The SEM images of the K2Sc2(MoO4)(PO4)2:xEu powders; x = 0.1 (a); 0.6 (b); 0.8 (c); 1.0 mol% (d) and areas where EDS analysis was performed for x = 0.1 (e) and 1.0 (f) indicating K2Sc2(MoO4)(PO4)2 as Phase 1 and ScPO4 as Phase 2. | ||
IR and UV-vis spectroscopy
The Fig. 3 shows the IR spectra of solid solutions K2Sc2(MoO4)(PO4)2:xEu. The broad bands centred at 1045 and 847 cm−1 have to be assigned to the stretching asymmetric and symmetric vibrations ν3 and ν1 of PO43− and MoO42− tetrahedral, respectively as it has been admitted for a structure of K2Bi(PO4)(MoO4).19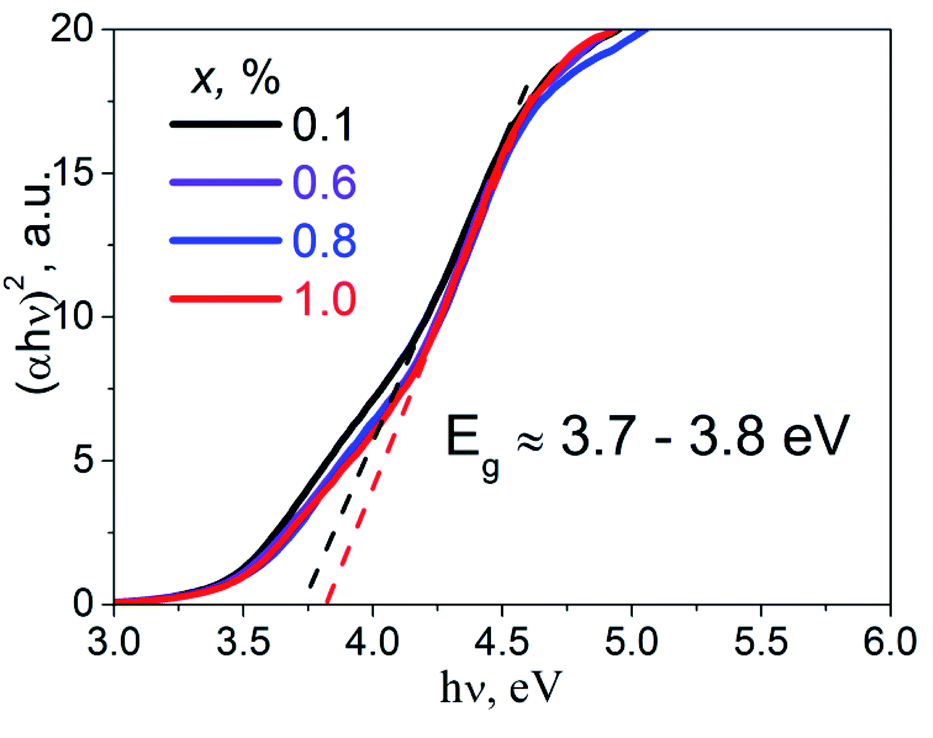 | ||
| Fig. 3 The FTIR spectra of K2Sc2(MoO4)(PO4)2:xEu samples, where x = 0.1–1.0 mol%. | ||
These bands are significantly widened, if compare to the same bands for noted bismuth-containing compound, where they are quite narrow and multiple. The broadening of the bands indicates simultaneous presence of both P and Mo in the same crystallographic position,20 in spite of the K2Bi(PO4)(MoO4) case, where P and Mo occupy two distinct positions. Three bands at 557, 593 and 630 cm−1 have been attributed to a PO4 bending vibration δ4, while the band at 450 cm−1 corresponds to the symmetric δ2 vibration. Consequently, IR spectra confirm the co-existence of both P and Mo in the same positions with tetrahedral surrounding.
Diffuse reflectance spectra of the studied samples were transformed into absorbance spectra, α(ν), using the well-known Kubelka–Munk equation.21 The Tauc plot, (αhν)2 = f(hν), was performed in order to estimate direct band gap values, Eg, of the studied K2Sc2(MoO4)(PO4)2:xEu samples (Fig. 4). Experimental band gaps were in the range from 3.7 to 3.8 eV that corresponds to the data for the samples of 0.1 and 1.0 mol% of Eu concentration, respectively. The Eg values obtained have been used for analysis of the calculation and luminescence spectroscopy results.
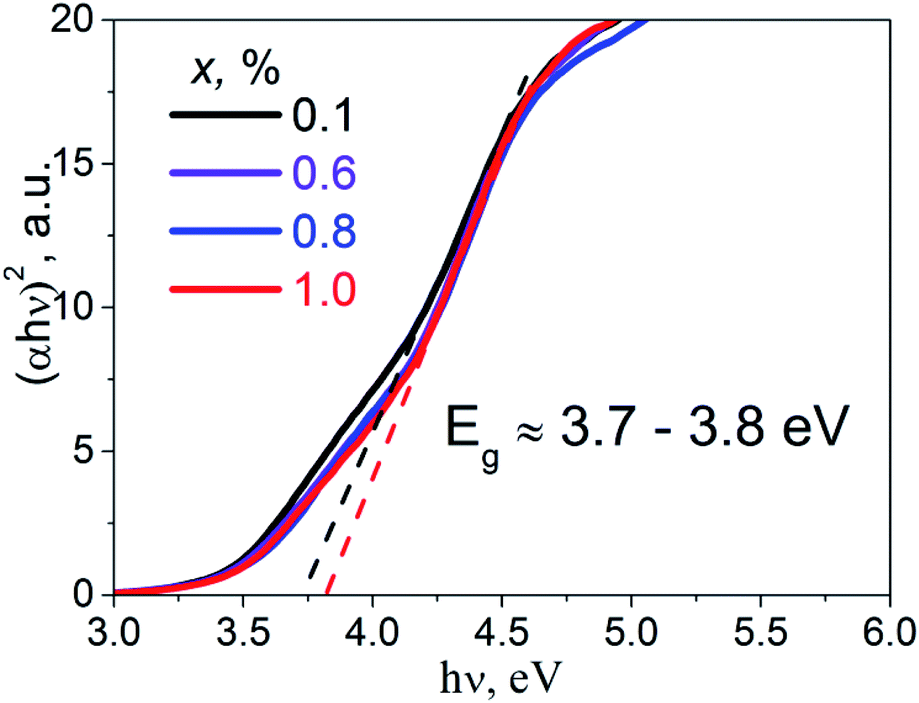 | ||
| Fig. 4 The Tauc plots for the K2Sc2(MoO4)(PO4)2:xEu samples; x = 0.1, 0.6, 0.8, and 1.0 mol%. | ||
Partial densities of states
The used FP-LAPW method is based on the DFT formalism and therefore the calculated values of dielectric band-gaps, Ecalcg, are usually underestimated.22 That is why, calculated by us Ecalcg of perfect K2Sc2(MoO4)(PO4)2 (3.3 eV) has been corrected to experimental value (Eexperg = 3.7 eV). This corrected value was used for calculated partial densities of states shown in the Fig. 5. The partial densities of electronic states of the K and Sc cations are shown in the Fig. 5a. It is seen that only Sc d states and K p states have notable densities in the energy range −8 to 8 eV. The Sc d states are located at the bottom part of conduction band (CB) of crystal, while potassium states appear at upper part of CB. The partial densities of elements, those belong to the PO43− and MoO42− molecular anions are shown separately on Fig. 5b and c, respectively.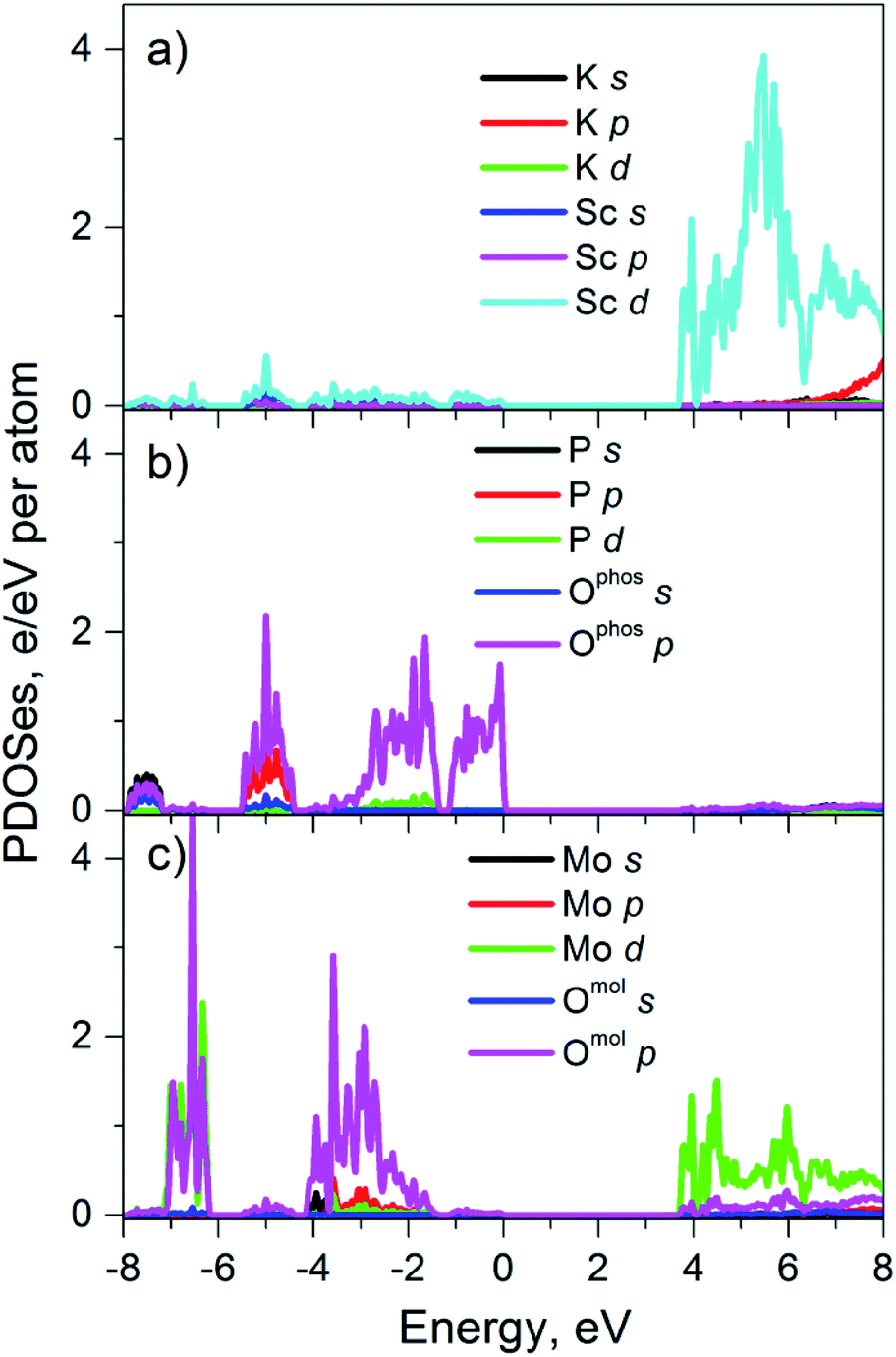 | ||
| Fig. 5 Calculated PDOS'es for perfect K2Sc2(MoO4)(PO4)2 crystal. | ||
Phosphate group is represented mainly by P p and O p states which are in the valence band (VB) of crystal. Moreover, the oxygen p states, Ophos p, dominate at the top of the VB. As for the conduction band, there are no significant electronic densities of elements of phosphate groups, while the molybdate anions reveal states at both VB and CB. The O p states of molybdates groups are located in the deep part of VB and the Mo d states are located near the bottom of CB. As it can be seen from Fig. 5 the top of the VB of perfect K2Sc2(MoO4)(PO4)2 is formed mainly by p states of oxygen that belong to phosphate groups and the bottom of conduction band is formed by Sc d states and Mo d states. Thus, only Ophos p, Sc d and Mo d states should be considered when analyzing optical properties of K2Sc2(MoO4)(PO4)2 crystal at the photon energy (hν) range near Eg (hν ≥ Eg).
Luminescence spectroscopy
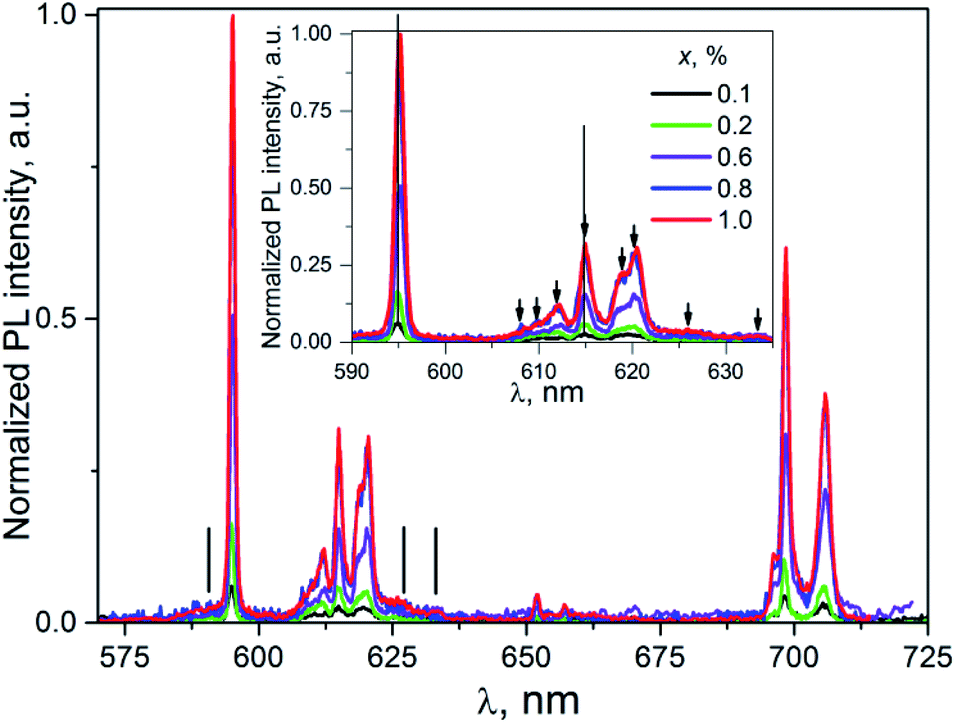 | ||
| Fig. 6 The PL emission spectra of K2Sc2(MoO4)(PO4)2:xEu, λex = 398 nm, T = 300 K. Inset: enlarged part within 590–640 nm. | ||
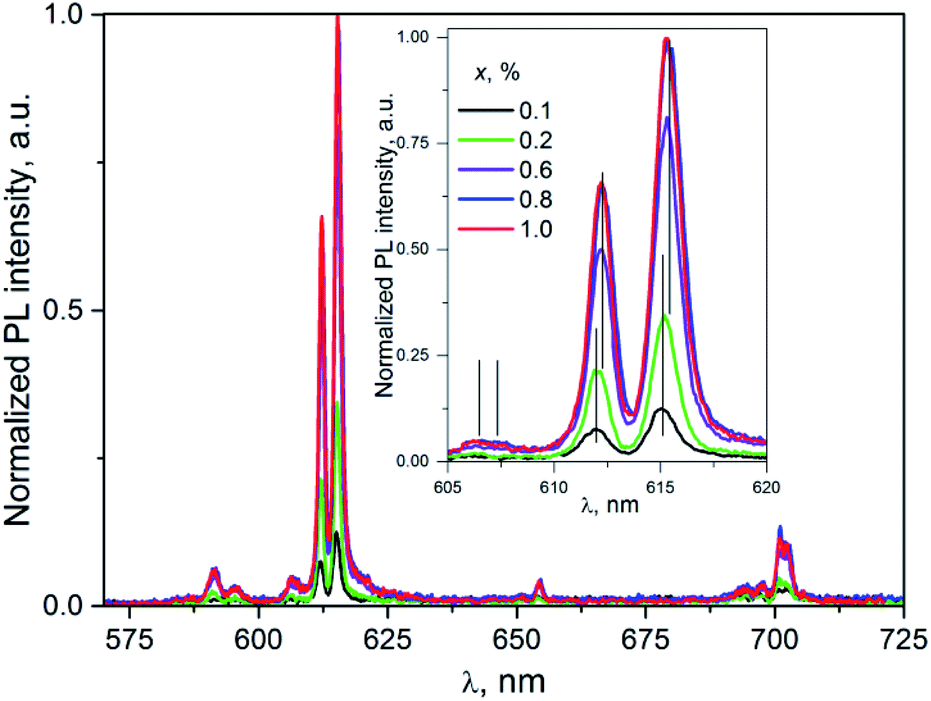 | ||
| Fig. 7 The PL spectra of K2Sc2(MoO4)(PO4)2:xEu; λex = 331 nm; T = 300 K. Inset: enlarged part of the spectra corresponding to the 5D0 → 7F2 transitions. | ||
Taking into consideration that the number of the PL lines for the 5D0 → 7F2 transition exceeds their theoretical maximal number (5 lines), one can conclude at least of two PL centers formed by the Eu3+ ions present in the host described. It should be pointed one more that being discussed centers are revealed under direct f–f excitation Eu3+ ions in the K2Sc2(MoO4)(PO4)2:xEu compounds.
Importantly, in case of PL excitation with λex = 331 nm which correspond to absorbed photon energy to be near Eg value, there is noticeable change in PL spectra for ceramics studied (Fig. 7). Thus, the group of lines attributed to 5D0 → 7FJ transitions are blue-shifted in comparison to their position at λex = 398 nm. The value of the shift for the 5D0 → 7F1 and 5D0 → 7F2 transitions can be evaluated as 150 and 100 cm−1, respectively. The most intensive emission lines for λex = 331 nm are related with ED radiation transitions 5D0 → 7F2 with maxima at 606, 612, and 615.5 nm. The lines ascribed to MD transitions 5D0 → 7F1 with maxima at 591 and 595.5 nm have about of 20 times lower peak intensity in comparison to ED ones.
The increase of Eu3+ content to x = 0.6, 0.8, and 1.0 mol% leads to the red-shift of peak positions, for example in case of lines attributed 5D0 → 7F2 (vertical lines in the inset to Fig. 7) this shift is ≈0.2 nm. This observation clearly indicates on complex nature of these lines. This conclusion is also supported by the non-symmetrical shape of a PL band located at 600–608 nm. Thus, in case of luminescence excitation by band to band transitions there are two types of the PL centers in the host studied as it has been also observed in the case of f–f inner excitation.
The values of PL intensity (calculated as area under the PL lines) for both ED and MD transitions as well as the total the PL intensity integrated over the range 550–720 nm are collected in the Table 1. All of the PL intensity have been normalized on value of total the PL intensity for K2Sc2(MoO4)(PO4)2:0.8% Eu sample excited at λex = 331 nm. The total PL intensity as well as intensity of the ED or MD transitions increases proportionally with Eu3+ content increase. However, the increase rate is twice higher for the 331 nm excitation in comparison to the 398 nm excitation. One can propose at least two possible explanations of the difference in the PL intensity change. In particular, it may be observed due to a difference in number of the emitting centers under different excitation. On the other hand, there may be different pathways for absorbed energy transfer to luminescent Eu3+ ions.
| x, mol% | 0.1 | 0.2 | 0.6 | 0.8 | 1.0 | |
|---|---|---|---|---|---|---|
| λex = 398 nm | I(5D0 → 7F1) | 0.007 | 0.021 | 0.058 | 0.093 | 0.112 |
| I(5D0 → 7F2) | 0.014 | 0.064 | 0.082 | 0.156 | 0.168 | |
| I(total) | 0.050 | 0.095 | 0.359 | 0.464 | 0.507 | |
| RI | 2.00 | 3.05 | 1.41 | 1.68 | 1.50 | |
| λex = 331 nm | I(5D0 → 7F1) | 0.021 | 0.021 | 0.040 | 0.057 | 0.039 |
| I(5D0 → 7F2) | 0.066 | 0.148 | 0.330 | 0.441 | 0.414 | |
| I(total) | 0.194 | 0.289 | 0.612 | 1.000 | 0.85 | |
| RII | 3.14 | 7.05 | 8.25 | 7.74 | 10.62 | |
Slight decrease in the PL intensity for x = 1.0 mol% is more likely to be related with an impurity influence on morphology and structure of the samples.
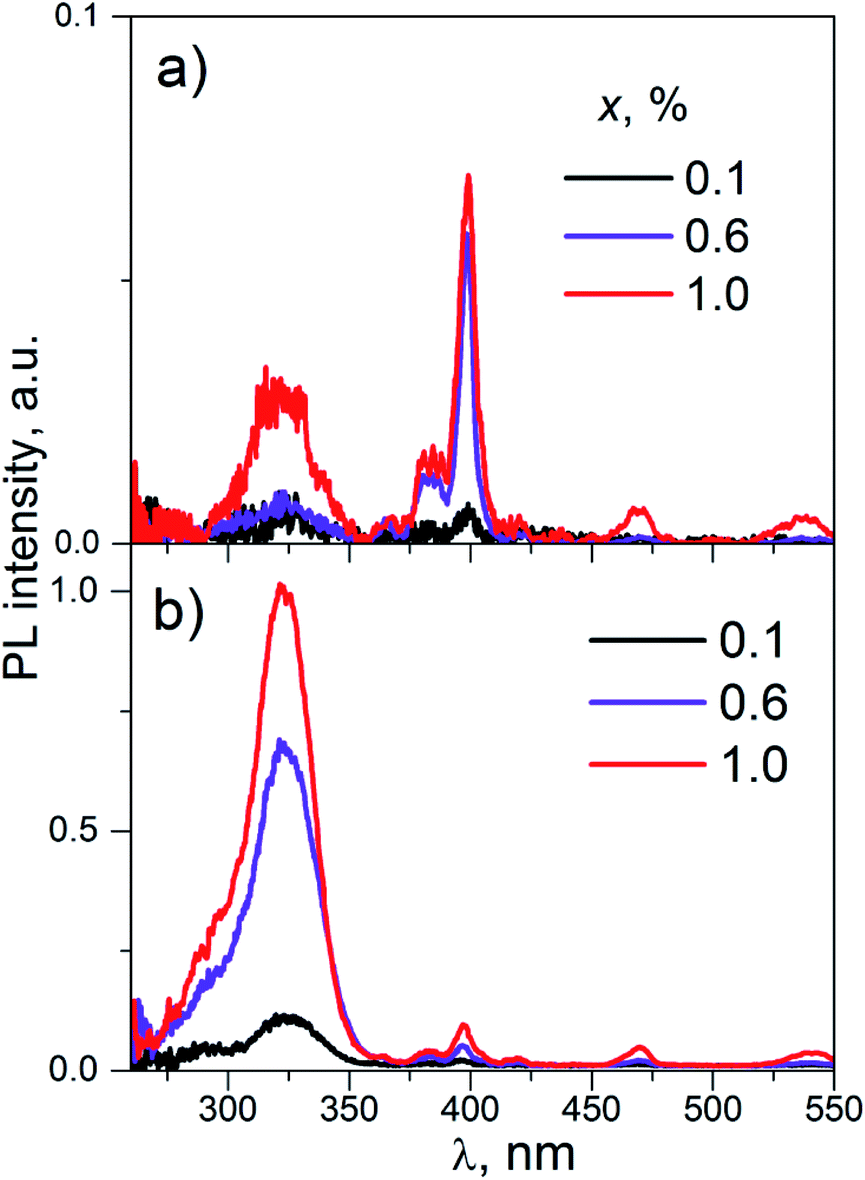 | ||
| Fig. 8 The PL excitation spectra of K2Sc2(MoO4)(PO4)2:xEu (concentrations, x, are shown on the figure); λem = 595 (a) and 615.5 nm (b), T = 300 K. | ||
The wide band with maxima near 325 nm is ascribed to the band-to-band absorption transitions in the K2Sc2(MoO4)(PO4)2 host. The set of narrow lines is attributed to inner f–f transitions in the Eu3+ ions. These lines are located at λmax ≈ 398, 470, 535 nm and are related to 7F0 → 5L6, 7F0 → 5D2, and 7F0–2 → 5D1 transitions, respectively. The PL excitation spectra on Fig. 8a differ from Fig. 8b by intensity ratio of band-to-band and inner transitions. In fact, the intensity of narrow PL excitation lines are alike for the PL registrations both at ED and MD transitions. Particularly, the line caused by 7F0 → 5L6 inner absorption transition (λmax ≈ 398 nm) is the most intensive in the PL excitation spectra monitored at emission caused by 5D0 → 7F1 MD radiation transition (λmax ≈ 595 nm). At the same time, peak intensity of wide band at 325 nm is about of 20 times higher for the case of the PL registration at 615.5 nm (ED radiation transition).
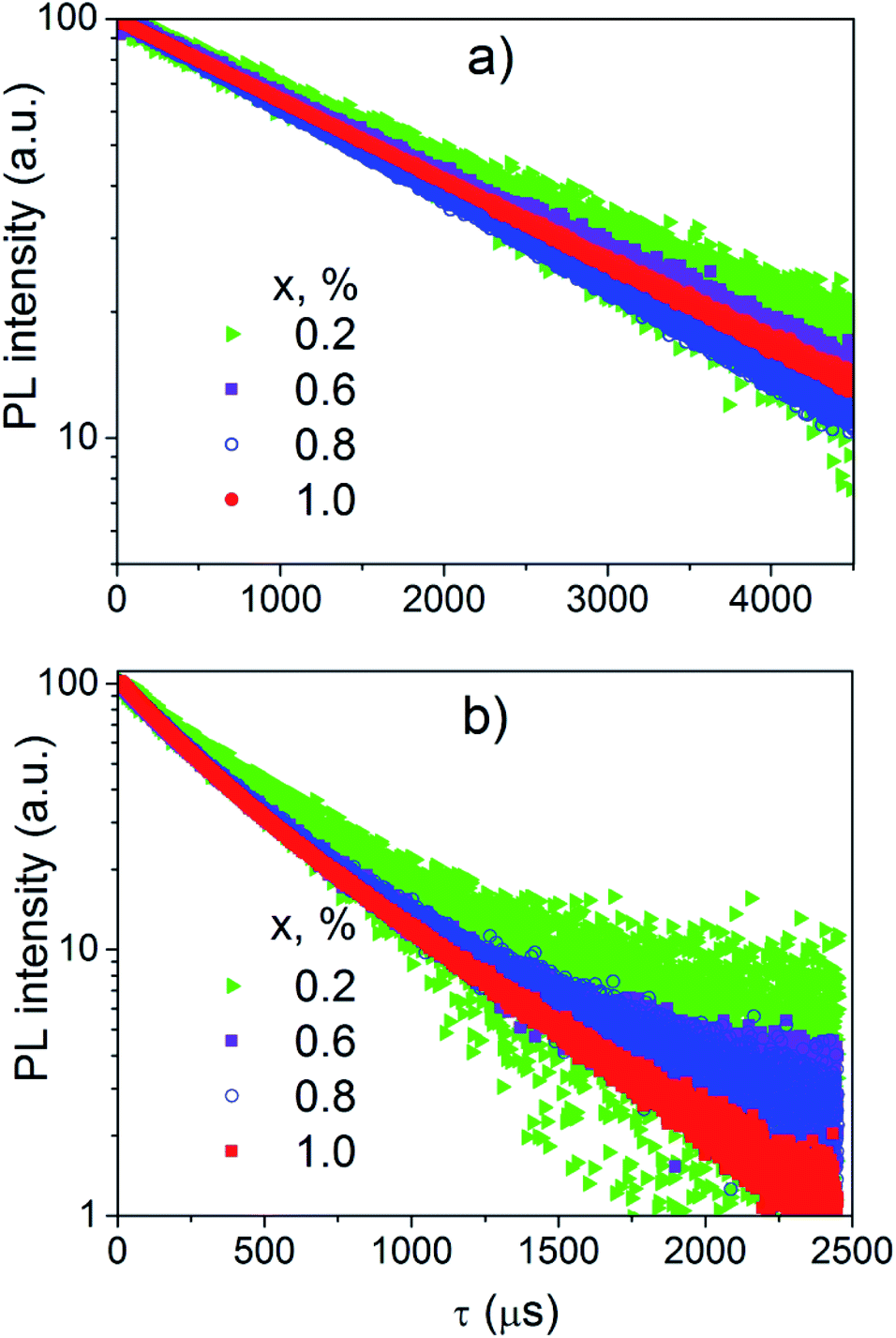 | ||
| Fig. 9 The PL decay curves of K2Sc2(MoO4)(PO4)2:xEu samples; λem = 595 (a) and 615.5 nm (b). | ||
The decay curves of the PL excited directly by inner f–f transitions in the Eu3+ ions (λex = 400 nm) and registered in the range of the 5D0 → 7F1 electronic transitions (λem = 595 nm) are linear in a semi-logarithmic scale (Fig. 9a). Consequently, the decay of this PL is described by single exponential dependence
| I(t) = A + I1e−t/τ. | (1) |
The decay time (τ) has tendency to decrease when the Eu3+ concentration increases (Table 2).
| x | λem = 595 | λem = 615.5 | |||
|---|---|---|---|---|---|
| τ | τ1 | τ2 | I1 | I2 | |
| 0.1 | 2.5 ± 0.3 | 0.12 ± 0.03 | 0.4 ± 0.3 | 5 | 95 |
| 0.2 | 2.5 ± 0.2 | 0.12 ± 0.02 | 0.5 ± 0.2 | 7 | 93 |
| 0.6 | 2.3 ± 0.1 | 0.16 ± 0.02 | 0.5 ± 0.2 | 19 | 81 |
| 0.8 | 2.1 ± 0.1 | 0.29 ± 0.02 | 0.7 ± 0.2 | 59 | 41 |
| 1.0 | 2.2 ± 0.1 | 0.24 ± 0.02 | 0.6 ± 0.2 | 43 | 57 |
Decay curves of the PL excited near edge of the band-to-band transitions (λex = 365 nm) and registered in the range of the 5D0 → 7F2 electronic transitions (λem = 615.5 nm) are not linear in semi-logarithmic scale (Fig. 9b). These decay curves have been successfully fitted by double exponential function:
| I(t) = A + I1e−t/τ1 + I2e−t/τ2, | (2) |
It should be admitted strong difference in the PL kinetic for two described cases regarding the luminescence kinetics data obtained.
Discussion
Based on the PL properties experimental data for ceramics studied the presence of two fundamentally different types of luminescence centers (denoted as type I and II, respectively) formed by Eu3+ ions in the K2Sc2(MoO4)(PO4)2 host should be taken for further consideration. Moreover, the detailed analysis of PL emission and excitation spectra revealed identification at least two PL sub-centers within each type of the centers I and II (denoted as Ia, Ib and IIa, IIb). These sub-centers are responsible for shifts of PL emission lines maxima on Fig. 6 and 7.The crystal structure K2Sc2(MoO4)(PO4)2 is characterised by four unique cationic positions within the framework. Two of them are occupied by K atoms, while other ones – by Sc atoms.13 Thus, there are four different positions for Eu3+ ions placement in the crystal structure. Importantly, there are 12 oxygen atoms in the nearest K surrounding, while Sc atoms are six-fold oxygen coordinated.13 All of the cationic sites possess C3v symmetry providing highly symmetrical environment for all cations found for the un-doped K2Sc2(MoO4)(PO4)2 host. The incorporation of Eu3+ ions into the host described leads to formation of two main emitting centers namely, I and II, that corresponds to impurity localization within K and Sc sites, respectively. In a light of structural peculiarities, the sub-centers a and b represent the possibility of Eu3+ ions to occupy two crystallographically distinct positions for both K and Sc ions.
Type I placement of Eu3+ ions
The photoluminescence of type Ia and Ib centers is excited mainly by internal f–f transitions in the electron shell of the Eu3+ ions, in particular, at λex = 398 nm. The most intensive PL bands of these centers located near 595 nm are MD transitions in Eu3+ ions. This peculiarity clearly indicates quite high symmetry nearest oxygen surrounding for the position where Eu3+ ions reside. The level of distortion of the Eu3+ ions surrounding in solid hosts can be estimated using the asymmetry ratio R = I(5D0 → 7F2)/I(5D0 → 7F1). It is well-known, that the 5D0 → 7F2 transition is so-called as a hypersensitive one and its intensity, in contrast to 5D0 → 7F1 transition, strongly depends both on local symmetry of the site occupied by Eu3+ ion and on the covalence of the Eu3+ ions bonding with nearest environment. Table 1 shows that the calculated RI values being in a range of 1.41–3.05 do not essentially change when the concentration of Eu3+ increases and are close to these ones reported for Eu3+ ions with C3 local symmetry.23 The PL kinetics of the type I centers is described by single exponent law with decay time constant τ in the range of 2.1–2.5 ms for x = 0.1–1.0 mol%. This dependency in PL kinetic is typical for Eu3+ in symmetrical surroundings (e.g. C3 or C3v site symmetry in EuBaB9O16 compound24).Based on the peculiarities of crystal structure change by an impurity doping the PL centers of type I can be ascribed to Eu3+ ions on K+ sites. From a simple comparison of the cationic sizes and polyhedral available within the K2Sc2(MoO4)(PO4) structure, when Eu3+ replaces K+ the charge compensation is required. Consequently, quite a few different possibilities can be anticipated. Below, using Kröger–Vink notation shows the schematic representation of the most probable:
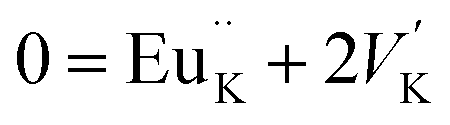
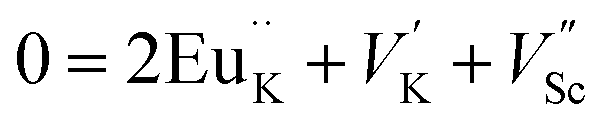
The formation of cationic vacancies followed by Eu3+ aliovalent entering K position are more likely to influence the second coordination sphere of the Eu3+ surrounding. Moreover, the oxygen polyhedra after K+ by Eu3+ replacement should be shrinked due to a difference in Eu–O and Sc–O bond distances. From crystallo-chemical point of view, the polyhedral distortion should be slight due to a fact that d(K–O) ≥ 2.94 Å in K2Sc2(MoO4)(PO4), while Eu–O distances in similar compounds are somewhat shorter (e.g., average d(Eu–O) is 2.48 Å for the EuPO4 compound25). However, taking into account that there are 8 oxygen atoms from PO4 moiety and other 4 from MoO4 ones in the initial KO12 polyhedra being discussed, the Eu3+ entrance should lead to bond lengths redistribution. The main reason for this phenomenon is connected with higher affinity of Eu3+ toward phosphate groups than to molybdate ones,26 as a result the overall symmetry of EuK oxygen environment should lower from C3v to C3.
Type II placement of Eu3+ ions
The photoluminescence of type IIa and IIb centers is excited mainly in the range of the band-to-band transitions, in particular, at λex = 331 nm. The most intensive PL bands of this type centers are located near 615 nm and are attributed to ED transitions in Eu3+ ions. The RII values for these centers increase from ∼3 up to ∼11 when concentration of Eu3+ increases (Table 1) clearly indicating on significant lowering of site symmetry. Similar R values have been reported for the Ca5(PO4)3F apatite host containing the same doping agent. The asymmetry ratio has been found to be 1.8 for the Eu3+ ions in the site with symmetry C3, while R value equal to 10.0 has been found for site symmetry Cs.23,27The PL decay kinetics has been considered as containing two components. The slower one is characterized by the time constant τ2 with values of 0.4–0.7 ms that are similar to these ones reported for binary oxide compounds and Sc2O3 with τ = 0.55 ms as well.28–30 The faster decay component is characterized by constant τ1 = 0.12–0.29 ms and is similar to those ones reported for scandium-containing complex oxides. In particular, decay time is 0.29 ms for the PL of the K0.5Mg0.5Sc(Lu)1.5(MoO4)3:Eu3+ solid solutions.31 Importantly, that both mentioned above and studied structures possess Sc3+ cation in similar octahedral oxygen environment. On the contrary, K+ cation is surrounded by 6 oxygen atoms in K0.5Mg0.5Sc(Lu)1.5(MoO4)3, while there are KO12 polyhedra found in the structure studied. Similarities in the PL decay times indicate that the Eu3+ ions which are responsible for the red emission at 615.5 nm for both K0.5Mg0.5Sc(Lu)1.5(MoO4)3 and K2Sc2(MoO4)(PO4)2 compounds are located more likely at scandium sites (EuSc) than at potassium ones.
Relatively low symmetry of Eu3+ oxygen environment in comparison to expected ones can be explained by distortions of oxide polyhedra provoked by the second coordination sphere change (Fig. 10). This assumption was done taking to consideration available cationic sites within the framework studied.
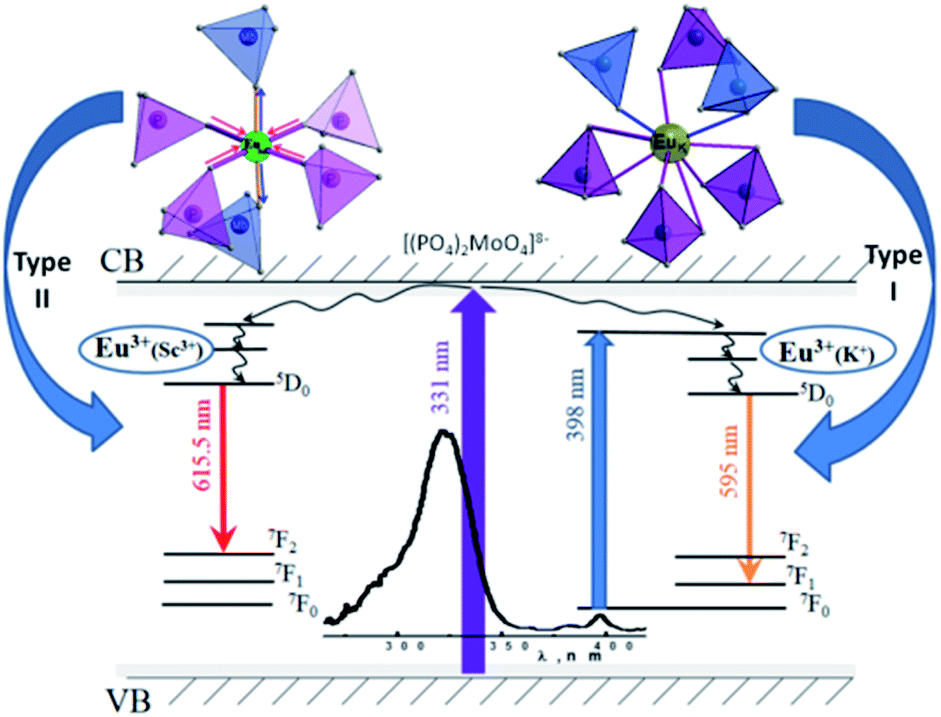 | ||
| Fig. 10 The scheme of the oxide polyhedra distortion for emitting centers and schematic view of the energy level diagram and electron transitions process. | ||
Mainly, when Eu3+ ions enter Sc sites the corresponding oxygen polyhedra should expand due to significantly longer distances of Eu–O in comparison to these ones for d(Sc–O) = 2.04–2.09 Å. On average, within the first coordination sphere there are 4 oxygen atoms from phosphate group and 2 – from molybdate ones to fulfil the distribution of the P/Mo atoms in the structure studied. Taking into account the values of the ionic radii of molybdenum (0.73 Å) and phosphorus (0.52 Å)32 and high affinity of Eu3+ to phosphate groups one can assume that length of these P/Mo–O bonds should be redistributed when Eu3+ enter Sc3+ position. It is expected, that the influence of the second coordination sphere may lead to Eu–O–Mo bonding elongation and Eu–O–P ones shortening consequently lowering the local symmetry of the emitting Eu3+ polyhedra from C3v much more than it can be observed for Eu3+ ion within potassium site.
The peculiarities of the possible localization of Eu ions within the framework studied agree well with bi-exponential character of PL kinetics.
The random distribution of P and Mo atoms over one crystallographically unique position in the host studied causes the deviation in the second coordination sphere environment. Thus, any Sc polyhedra may have from zero to six molybdate groups in the nearest surrounding. Incorporation of the Eu3+ ions into Sc site should change this distribution. As it has been discussed above the Sc3+ substitution by Eu3+ ion leads to elongation of the EuSc–O bonds that should be accompanied with shortening of the P/Mo–O bonds in the nearest to EuSc oxygen tetrahedra (Fig. 8). The lengths of the P/Mo–O bonds for K2Sc2(MoO4)(PO4)2 compounds are in the 1.57–1.61 Å range, that is somewhat longer than for phosphate-based langbeinites (1.53–1.55 Å for K2YZr(PO4)3 (ref. 33) and 1.49–1.51 Å for K2BiZr(PO4)3 (ref. 34)), while pure Mo–O bonds are much longer (d(Mo–O) = 1.74–1.79 Å for K2Bi(PO4)(MoO4)19). Consequently, the reorganization of EuSc–O–Mo bonds can be seen as forced overlapping of electronic clouds of europium, oxygen and molybdenum ions providing a complex structural defect acting as complex luminescence center of type II. Moreover, the time constant τ1 reveals the values which are typical for Eu3+ ions luminescence in some coordination complexes.35 This fact indicates not only a decrease in symmetry, but also the increase in the covalent bonding contribution for EuSc polyhedra within K2Sc2(MoO4)(PO4)2 host. The increase in Eu3+ dopant concentration should lead to general redistribution of the P/Mo ratio in the second coordination environment providing change in the PL characteristics of type II centers.
The assumptions concerning two types of Eu3+ ions placements in lattice are also supported by PL excitation and computation results. In particular, origin of the broad band in PL excitation spectra for the centers of both types can be specified accounting the results of our calculations. The electronic structure calculations have revealed that only Ophos p states of phosphate groups as well as Mo d states and Sc d states can be involved to the absorption processes near band edge. This peculiarity of calculated PDOSs indicates that wide PL excitation band at 325 nm is not connected with intramolecular transitions neither in PO43− nor in MoO42− anions. The light absorption mechanisms in the studied ceramics can be similar to ones in some Sc-containing oxides compounds, such as Sc2O3 and ScPO4. These compounds are wide gap insulators having top of the VB formed by O p states and the bottom of CB – by Sc d states. The formation of spatially correlated electrons and holes has been reported for Sc2O3 where Sc atoms are surrounded by 6 oxygen atoms like in the structure studied.36,37 Formation of electron–hole pairs under PL excitation in the ultraviolet and vacuum ultraviolet spectral ranges (hν > 7 eV) has also been reported for ScPO4.38 Thus, it is assumed that band-to-band light absorption in the K2Sc2(MoO4)(PO4)2 is accompanied with formation of the electron–hole pairs. These pairs consist of electron on scandium site and a hole located on the oxygen atom that belongs to phosphate group. Due to the fact that scandium has doubtful stability of oxidation state +2, the O2− → Sc3+ electron transfer can lead to formation of weakly-bonded electron–hole pair that should be resulted in (1) fast radiation-less annihilation or (2) transfer of electron–hole energy to the nearest surrounding. The way #2 can be realized by two manners: energy absorbed by nearest (2.1) molybdate group or (2.2) Eu3+ ion. Any traces of the molybdate groups luminescence have not been detected in spite of the fact that it is well known.39,40 Consequently, it has been assumed that way 2.1 is less probable or energy of the excited MoO42− anions is released without radiation. Thus, the way 2.2 is more likely to be realized with higher efficiency for EuSc-related centers luminescence in respect to EuK ones.
The further studies of the both RE-doped and un-doped K2Sc2(MoO4)(PO4)2 materials like low-temperature and time-resolved measurements should be performed in order to establish the precise mechanisms of luminescence and structure of luminescence centers in the ceramics studied.
The ceramics studied possess useful for solid state lighting applications luminescence properties. In particular, the plasma display phosphors should have luminescence decay time below 2.0 ms (ref. 41) and this requirement is satisfied by K2Sc2(MoO4)(PO4)2:xEu ceramics in case of PL excitation in the region of the host absorption. Another important characteristic of these types of phosphors is their CIE colour coordinates. These coordinates have been calculated for both type of PL excitations (inner f–f transitions in Eu3+ ions and band-to-band transitions in host) and have been collected in the Table 3 illustration the good fitting to the standard requirements for the pure red colour which are X = 0.67; Y = 0.33.
| x, mol% | 0.1 | 0.2 | 0.6 | 0.8 | 1.0 | |
|---|---|---|---|---|---|---|
| λex = 398 nm | X | 0.471 | 0.624 | 0.605 | 0.636 | 0.636 |
| Y | 0.529 | 0.376 | 0.395 | 0.364 | 0.364 | |
| λex = 331 nm | X | 0.569 | 0.633 | 0.634 | 0.627 | 0.658 |
| Y | 0.431 | 0.367 | 0.366 | 0.373 | 0.342 | |
Conclusions
The set of K2Sc2(MoO4)(PO4)2:xEu (x = 0.1–1.0 mol%) ceramics has been prepared by high temperature solid-state reaction technique. The optical band gap values have been estimated in a range of 3.7–3.8 eV. Calculations of the electronic band structure of K2Sc2(MoO4)(PO4)2 crystal has showed that top of its valence band is formed mainly by p states of oxygen ions belonging to PO4 groups. The bottom of the conduction band is formed simultaneously by d states of Sc and d states of Mo.The ceramics reveal intense red photoluminescence (PL) under excitation in the range of the band-to-band absorption and via inner f–f electronic transitions in the Eu3+ ions.
Experimental data have shown that the admixture Eu3+ ions are more likely to be distributed over potassium and scandium sites within K2Sc2(MoO4)(PO4)2 framework.
Two main types of luminescence centers correspond to noted two types of Eu3+ions placement. The first one reveals intense 5D0 → 7F1 transition and has been ascribed to Eu3+ ions located at K sites. The second one is characterized by higher intensity of the 5D0 → 7F2 transition and has been attributed to Eu3+ ions located at Sc sites.
Their nature of two additive Eu3+ luminescence centers has been discussed in a light of the Eu3+ ions second coordination sphere changes.
Luminescent and chromaticity characteristics indicates that K2Sc2(MoO4)(PO4)2:xEu ceramics are perspective red phosphors. The possibility to influence the polyhedral symmetry of emitting centers by the bond length redistribution in a second coordination sphere can be successfully applied for tunable luminescent materials design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the State Budget of Ukraine via Ministry of Education and Science of Ukraine (the projects #19BF037-01, #18BF051-01, and #110/14-pr-2019).References
- S. Sadhasivam, P. Manivel, K. Jeganathan, C. K. Jayasankar and N. P. Rajesh, Mater. Lett., 2017, 188, 399–402 CrossRef CAS.
- A. Souamti, I. R. Martín, L. Zayani, M. A. Hernández-Rodríguez, K. Soler-Carracedo, A. D. Lozano-Gorrín and D. Ben Hassen Chehimi, Opt. Mater., 2016, 53, 190–194 CrossRef CAS.
- B. G. Bazarov, R. F. Klevtsova, T. T. Bazarova, S. I. Arkhincheeva, L. A. Glinskaya, K. N. Fedorov and Z. G. Bazarova, Russ. J. Inorg. Chem., 2006, 51, 1577–1580 CrossRef.
- I. Yamai and T. Ota, J. Am. Ceram. Soc., 1992, 75, 2276–2282 CrossRef CAS.
- Y. L. Xue, D. Zhao, S. R. Zhang, Y. N. Li and Y. P. Fan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2019, 75, 213–220 CrossRef CAS PubMed.
- A. E. Kanunov and A. I. Orlova, Rev. J. Chem., 2018, 8, 1–33 CrossRef CAS.
- A. Bohre and O. P. Shrivastava, Int. J. Appl. Ceram. Technol., 2013, 10, 552–563 CrossRef CAS.
- S. T. Norberg, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 743–749 CrossRef PubMed.
- M. Jiao, W. Lü, B. Shao, L. Zhao and H. You, ChemPhysChem, 2015, 16, 2663–2669 CrossRef CAS PubMed.
- Z. J. Zhang, J. L. Yuan, H. H. Chen, X. X. Yang, J. T. Zhao, G. B. Zhang and C. S. Shi, Solid State Sci., 2009, 11, 549–555 CrossRef CAS.
- Z. J. Zhang, X. Lin, J. T. Zhao and G. B. Zhang, Mater. Res. Bull., 2013, 48, 224–231 CrossRef CAS.
- A. E. Kanunov, E. A. Asabina and A. I. Orlova, Russ. J. Gen. Chem., 2016, 86, 18–25 CrossRef CAS.
- N. S. Slobodyanik, K. V. Terebilenko, I. V. Ogorodnyk, I. V. Zatovsky, M. Seredyuk, V. N. Baumer and P. Gütlich, Inorg. Chem., 2012, 51, 1380–1385 CrossRef CAS PubMed.
- S. P. Kumar and B. Gopal, J. Alloys Compd., 2016, 657, 422–429 CrossRef CAS.
- P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka and J. Luitz, WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties, Karlheinz Schwarz Technische Universität, Wien, Austria, 2001 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- P. E. Blochl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef CAS PubMed.
- M. A. Ryumin, V. V. Pukhkaya and L. N. Komissarova, Russ. J. Inorg. Chem., 2010, 55(7), 1010–1013 CrossRef CAS.
- I. V. Zatovsky, K. V. Terebilenko, N. S. Slobodyanik, V. N. Baumer and O. V. Shishkin, J. Solid State Chem., 2006, 179, 3550–3555 CrossRef CAS.
- S. P. Kumar and B. Gopal, J. Am. Ceram. Soc., 2011, 94, 1008–1013 CrossRef CAS.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38, 448–457 CrossRef CAS PubMed.
- J. P. Perdew, Int. J. Quantum Chem., 1985, 28, 497–523 CrossRef.
- M. Gaft, R. Reisfeld, G. Panczer, S. Shoval, B. Champagnon and G. Boulon, J. Lumin., 1997, 72, 572–574 CrossRef.
- Y. Huang, H. Lin and H. J. Seo, J. Electrochem. Soc., 2010, 157, J405–J409 CrossRef CAS.
- D. F. Mullica, D. A. Grossie and L. A. Boatner, Inorg. Chim. Acta, 1985, 109, 105–110 CrossRef CAS.
- K. V. Terebilenko, D. V. Kyselov, V. N. Baumer, M. S. Slobodyanik, O. V. Petrenko, O. V. Khomenko and V. P. Dotsenko, Cryst. Res. Technol., 2018, 53, 1800158 CrossRef.
- M. Gaft, G. Panczer, R. Reisfeld and E. Uspensky, Phys. Chem. Miner., 2001, 28, 347–363 CrossRef CAS.
- G. H. Lee and S. Kang, J. Lumin., 2011, 131, 2582–2588 CrossRef CAS.
- Y. Pan, X. Xie, Q. Huang, C. Gao, Y. Wang, L. Wang, B. Yang, H. Su, L. Huang and W. Huang, Adv. Mater., 2018, 30, 1705256 CrossRef PubMed.
- G. A. West and N. S. Clements, J. Lumin., 1992, 54, 245–248 CrossRef CAS.
- N. M. Kozhevnikova, S. Y. Batueva and R. M. Gadirov, Inorg. Mater., 2018, 54, 460–465 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- U. Guth, B. Löscher, P. Schmidt, H. Wulff and H. H. Möbius, Solid State Ionics, 1992, 51, 183–185 CrossRef CAS.
- V. Chornii, Y. Hizhnyi, S. G. Nedilko, K. Terebilenko, I. Zatovsky, I. Ogorodnyk and V. Boyko, Solid State Phenom., 2015, 230, 55–61 Search PubMed.
- T. Yamase, T. Kobayashi, M. Sugeta and H. Naruke, J. Phys. Chem. A, 1997, 101, 5046–5053 CrossRef CAS.
- A. Lushchik, M. Kirm, C. Lushchik, I. Martinson and G. Zimmerer, J. Lumin., 2000, 87, 232–234 CrossRef.
- O. M. Bordun, I. M. Bordun and A. B. Lyskovich, J. Appl. Spectrosc., 1999, 66, 738–741 CrossRef CAS.
- A. Trukhin and L. A. Boatner, Mater. Sci. Forum, 1997, 239, 573–576 Search PubMed.
- Y. Hizhnyi, I. Zatovsky, S. Nedilko, R. Boiko, J. Li, W. Han and I. Klyui, J. Lumin., 2019, 211, 127–137 CrossRef CAS.
- D. Spassky, A. Vasil'ev, A. Belsky, N. Fedorov, P. Martin, S. Markov, O. Buzanov, N. Kozlova and V. Shlegel, Opt. Mater., 2019, 90, 7–13 CrossRef CAS.
- D. S. Zang, J. H. Song, D. H. Park, Y. C. Kim and D. H. Yoon, J. Lumin., 2009, 129, 1088–1093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04975a |
| This journal is © The Royal Society of Chemistry 2020 |