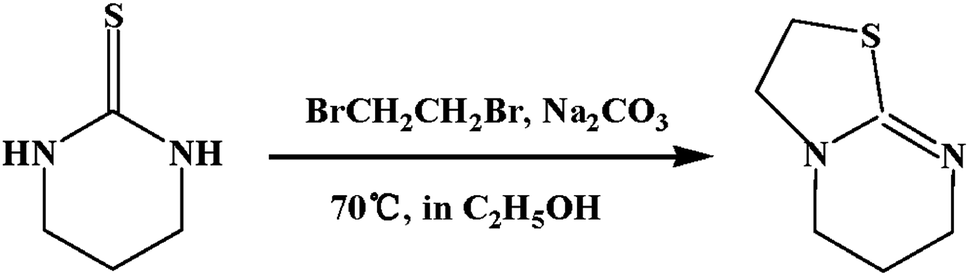DOI:
10.1039/D0RA04974C
(Paper)
RSC Adv., 2020,
10, 25979-25987
Binary catalytic system for homo- and block copolymerization of ε-caprolactone with δ-valerolactone†
Received
5th June 2020
, Accepted 30th June 2020
First published on 9th July 2020
Abstract
The combined interaction of 2,3,6,7-tetrahydro-5H-thiazolo[3,2-a] pyrimidine (ITU) as the organocatalytic nucleophile with YCl3 as Lewis acid cocatalyst, generating ITU/YCl3, was employed for homo- and copolymerization of ε-caprolactone (CL) with δ-valerolactone (VL). Poly(caprolactone) (PCL) and poly(caprolactone)–poly(ethylene glycol)–poly(caprolactone) (PCL–PEG–PCL) triblock copolymer and poly(valerolactone)–poly(caprolactone)–poly(ethylene glycol)–poly(caprolactone)–poly(valerolactone) (PVL–PCL–PEG–PCL–PVL) pentablock copolymer were successfully prepared by ring-opening polymerization (ROP) of CL employing ITU/YCl3 as catalyst in the presence of benzyl alcohol (BnOH) or poly(ethylene glycol) (PEG) as initiator, respectively. The reaction was systematically optimized, and the architecture, molecular weight and thermal properties of the polymers were characterized by NMR, FTIR, SEC and DSC analyses. Finally, a plausible polymerization mechanism was proposed.
Introduction
Biodegradable polymers have gained considerable attention over the past decades. Poly(caprolactone) (PCL) and their copolymers are widely used in the biomedical applications including drug delivery, cell therapy, and tissue engineering.1,2 PCL applicability as a biomaterial arises from the presence of polylactones generating high flexibility and biocompatibility. However, its high hydrophobicity and crystallinity can interfere with modulation of its mechanical properties and degradation rate, which can be efficiently adjusted via copolymerization.3,4 Poly(ethylene glycol) (PEG) is a useful material for segments in block copolymers due to its nontoxicity and high hydrophilicity. Additionally, poly(δ-valerolactone) (PVL) is commonly employed owing to its moderate mechanical properties. A–B–A block copolymer of PCL with PEG can provide materials with desirable degradation rates. Furthermore, copolymerization of CL with VL can potentially modify the mechanical, thermal properties.
At present, metal catalysts dominate the field of polymers due to their nearly endless combinations of metals and ligands.5–10 However, metal residues in polymers limited the application in biomedicine, food packaging, and microelectronics. In contrast, organocatalysts can be considered alternatives to promote ROP of various monomers owing to numerous advantages, including low toxicity, versatility and high activity.11–18 And organopolymerizations have been successfully employed in the preparation of functional and biodegradable materials.19–29
To the date, successful organocatalysts for living ROP of cyclic esters include guanidines and amidines, such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), N-methyl-TBD (MTBD), and 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), thiourea/amines, N-heterocyclic carbenes (NHCs), N-heterocyclic olefins (NHOs) and 4-dimethylaminopyridine (DMAP). Hedrick et al.30,31 reported that DBU and MTBD with similar basicities, can efficiently control ROP of CL and VL only when thiourea (TU) was used as the cocatalyst, producing high yields (80% CL; 95% VL) with predictable molecular weights at room temperature. In 2016, the ROP of CL and VL using MTBD and bis- (biTU) or trithiourea (triTU), was successfully reported by Kiesewetter group,32–36 and biTU displayed enhanced catalytic activity versus mono-TU catalyst in C6D6 solution at room temperature. It was found that triTU exhibited significantly reduced activity versus mono-TU or biTU. Moreover, bis- or trithiourea, along with an H-bond accepting base, exhibited the highly active and living characteristics. For all thiourea catalysts under solvent-free conditions, the more active catalysts are generally more controlled. Kiesewetter et al.37 developed TU/DBU dual catalytic system for the ROP of substituted cyclic carbonates to afford high functionalized oligocarbonates. Such cooperative catalytic systems are of interest, due to the combination of organic bases and Lewis acids for ROP with cyclic esters. For example, InCl3/NEt3 pair (Lewis acid/amine) promoted polymerization of CL and substituted derivatives, although slower polymerization rates and broader polydispersity index were observed.38 Buchmeiser et al.39 showed that free NHCs in combination with Lewis acids (Sn(II), Mg(II), or Zn(II) chlorides) promote efficient ROP of CL. Importantly, the polymerization activity depended both on the structure of NHCs and nature of Lewis acid. Naumann et al.40 also utilized four NHOs coupled with Lewis acids (MgCl2, MgI2, YCl3, or ZnI2) in order to control homopolymerization of CL and VL at room temperature, with an overall order of activity of MgI2 > YCl3 > ZnI2 > MgCl2. Chen groups41 reported that strong Lewis base NHO in combination with strong Lewis acid Al(C6F5)3, cooperatively promoted the living ring-opening (co)polymerization of VL and CL at 25 °C in toluene. Dove et al.42 employed DMAP and MgI2 or YCl3 in ROP of CL, VL and their random copolymerization in THF at 70 °C. Lately, Bai et al.43 investigated isothiourea-based Lewis pairs (such as MgX2, X = I, Br, Cl) dual catalyst system to homopolymerize ω-pentadecalactone (PDL) and random copolymerize PDL with CL.
Inspired by these pioneering works, novel 2,3,6,7-tetrahydro-5H-thiazolo [3,2-a]pyrimidine (ITU) were synthesized and further utilized in combination with YCl3 to achieve controlled homopolymerization of CL in the presence of BnOH. Additionally, PCL–PEG–PCL triblock copolymer and PVL–PCL–PEG–PCL–PVL pentablock copolymer were obtained successfully via ITU/YCl3 system in the presence of PEG. Moreover, the architectures of the prepared polymers were analyzed and characterized, and thermal properties were determined.
Experimental section
Materials
CL (Alfa Aesar) and VL were dried over CaH2, distilled under vacuum, and stored over molecular sieves under argon. 3,4,5,6-Tetrahydro-2-pyrimidinethiol was purchased from FLUORO CHEM Ltd (Shanghai, China). 1,2-Dibromoethane was purchased from Tianjin Guangfu Fine Chemicals Ltd (Tianjin, China). YCl3 (Alfa Aesar, “ultra dry”, 99.99%) was purchased from Alfa Aesar. Dihydroxyl-PEG2000 (Mn = 2000) was dried by azeotropic distillation with dry toluene prior to use. BnOH was distilled over fresh CaH2 for 48 h under argon atmosphere and stored over 4 Å molecular sieves. Toluene was dried with CaCl2 before distillation over sodium/benzophenone. Methanol was used without further purification.
Equipment
Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker spectrometer operating at 600 MHz with deuterated chloroform as solvent and tetramethylsilane as internal reference. The molecular weight (Mn) and molecular weight distribution (Đ) of polymers determined by gel permeation chromatography (SEC) (ACQUITY APC) were based on experiments conducted in THF at a flow rate of 1.0 mL min−1 at 40 °C. Polystyrene standards with narrow Đ were used to generate a calibration curve. Infrared (IR) spectra were recorded by a Varian 660-IR spectrometer using KBr pellet technique. The differential scanning calorimetry (DSC) analyses were carried out on a DSC (NETZSCH DSC 200F3) instrument. The thermograms were recorded according to the following cycles: samples were heated from room temperature to 200 °C at a rate of 10 °C min−1 under a nitrogen purge and held for 2 min to erase the thermal history, then cooled to −60 °C at a rate of 40 °C min−1 and held for another 2 min. The second heating scan from −60 °C to 200 °C was then recorded. 3 mg of the sample was used for each analysis, using aluminum capsules. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) of the obtained polymer was performed using a mass spectrometer (autoflexspeed; Bruker) equipped with a Smartbeam II modified Nd:YAG laser. Five hundred shots were accumulated for the spectra at a 25 kV acceleration voltage in the positive linear mode. The polymer sample was dissolved in THF at a concentration of 5 mg mL−1 and the matrix was trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propylidene]malonitrile (DCTB) dissolved in THF (20 mg mL−1). The sample for MALDI-TOF MS was prepared by mixing the matrix solution, polymer solution and sodium trifluoroacetate (0.1 M in THF) with a volume ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1. Then 0.5 μL of which was spotted onto the MALDI-TOF sample plate before being air-dried.
:5:1. Then 0.5 μL of which was spotted onto the MALDI-TOF sample plate before being air-dried.
Catalyst preparation
ITU was synthesized according to the literature (Scheme 1). 3,4,5,6-Tetrahydro-2-pyrimidinethiol (5.7 g, 0.05 mol), Na2CO3 (6.0 g, 0.06 mol), absolute ethanol (50 mL) and 1,2-dibromethane (4.5 mL, 0.05 mol) were added to a round bottomed flask, stirred, and refluxed under nitrogen atmosphere at 70 °C. The reaction was monitored by TLC. After 15 h the reaction mixture was evaporated, the resultant residue were diluted with water (100 mL), then adjusted to pH = 14 with 20% NaOH and immediately extracted with dichloromethane (3 × 100 mL). The organic phases were dried over Na2SO4 and filtered. The solvent was then removed under vacuum to give a pale yellow oil. The pure product was isolated by column chromatography (5% MeOH, 5% Et3N and 90% CH2Cl2) to yield the product as a clear oil. Yield: 70%; 1H NMR44 (600 MHz, CDCl3): δ 3.56 (t, 2H), 3.40 (t, 2H), 3.27 (t, 2H), 3.15 (t, 2H), 1.88 (app. 2H).
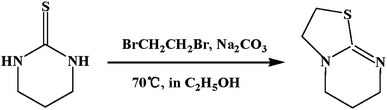 |
| | Scheme 1 Synthesis of ITU. | |
Synthesis of PCL homopolymer
All polymerizations were carried out in 20 mL glass ampoules that were previously flamed and purged with dry argon. CL (1.0 g, 8.77 mmol) was first syringed into an ampoule containing the exact amount of YCl3 (0.024 g, 8.77 mmol). In a typical procedure, BnOH (9.1 μL, 8.77 mmol) dissolved in toluene (2.0 mL) and ITU (0.0013 g, 0.0877 mmol) was then added into the ampoule to start the polymerization. After the desired reaction time, the product was terminated and precipitated with methanol, isolated by filtration, and dried under reduced pressure at 40 °C to constant mass. Conversion: 98.8%; Mn,SEC, 24.8 kg mol−1; Đ, 1.09. 1H NMR (CDCl3): δ (ppm) 1.32 (m, 2H, CH2CH2CH2CO–O), 1.57 (m, 4H, CH2CH2CH2CH2CO–O), 2.24 (t, 2H, CH2CO–O), 3.57 (t, 2H, CH2OH), 4.00 (t, 2H, CH2O–CO), 5.05 (s, 2H, ArCH2), 7.28 (s, 5H, aromatic);13C NMR (CDCl3): δ (ppm), 173.49 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 64.12 (CH2O), 34.11 (COCH2), 28.35 (CH2CH2–O), 25.53 (COCH2–CH2–CH2), 24.57 (COCH2CH2CH2CH2CH2O).
O), 64.12 (CH2O), 34.11 (COCH2), 28.35 (CH2CH2–O), 25.53 (COCH2–CH2–CH2), 24.57 (COCH2CH2CH2CH2CH2O).
Synthesis of PCL–PEG–PCL triblock copolymer
The triblock copolymerizations were performed under argon by using standard Schlenk techniques. YCl3 (0.0078 g, 0.0877 mmol) and CL (1.0 g, 8.77 mmol) were added to a glass ampoule, and then polymerization was initiated with the addition of dihydroxyl-PEG2000 (0.17 g, 0.0877 mmol) and ITU (0.013 g, 0.0877 mmol) in dry THF. After a specific reaction time, the reaction was terminated. The work-up procedure was similar to that described above, generating a white product. Conversion: 97.8%; Mn,SEC, 25.5 kg mol−1; Đ, 1.08. 1H NMR (CDCl3): δ (ppm), 1.38 (m, 2H, CH2CH2CH2CO–O), 1.65 (m, 4H, CH2CH2CH2CH2CO–O), 2.31 (t, 2H, CH2CO–O), 3.64 (s, OCH2CH2O), 4.06 (t, 2H, CH2O–CO); 13C NMR (CDCl3): δ (ppm), 173.49 (CO), 70.78 (OCH2CH2O), 64.12 (CH2O), 34.11 (COCH2), 28.35 (CH2CH2–O), 25.53 (COCH2CH2CH2), 24.57 (COCH2CH2CH2CH2CH2O).
Synthesis of PVL–PCL–PEG–PCL–PVL pentablock copolymer
As a typical pentablock copolymerization, a glass ampoule was charged with YCl3 (0.0078 g, 0.0877 mmol) and CL (1.0 g, 8.77 mmol), dried toluene was added to maintain the initial concentration of CL at 2.0 mol L−1. To this solution, dihydroxyl-PEG2000 (0.17 g, 0.0877 mmol) and ITU (0.013 g, 0.0877 mmol) were added to start the polymerization. After the first polymerization mixture was proceed for 60 min at 60 °C, the block copolymerization was started by the second monomer VL (0.8 mL, 8.77 mmol). After 2 h, the residual mixture was poured into methanol to precipitate the polymer. Then the resulting copolymer was dried under a vacuum to a constant weight. Conversion: 91.4%; Mn,SEC, 36.9 kg mol−1; Đ, 1.16. 1H NMR (CDCl3): δ (ppm), 1.37 (m, 2H, CH2CH2CH2COO), 1.67–1.70 (m, 4H, COCH2CH2CH2 and CH2CH2CH2O), 2.34 (t, 4H, OCOCH2CH2), 3.64 (s, 2H, OCH2CH2O), 4.07 (t, 4H, CH2OCO); 13C NMR (CDCl3): δ (ppm), 173.55 (CO), 71.00 (OCH2CH2O), 64.06 (CH2O), 34.01 (COCH2), 28.21 (CH2CH2–O), 25.53 (COCH2CH2CH2), 24.57 (COCH2CH2CH2CH2CH2O).
Results and discussion
Synthesis of PCL homopolymers using BnOH
ROP of CL with ITU/YCl3 as the catalyst in the presence of BnOH as the initiator was investigated under different reaction conditions. The first polymerization reactions were performed at 25 °C in toluene, in CL/catalyst ratio of 100:1. The results are summarized in Table 1, where the effect of monomer/catalyst ([CL]/[ITU]) on polymerization is observed. At [CL]/[ITU] molar ratio of 100, PCL yield is 98.8% and molecular weight is 24.8 kg mol−1 (Table 1, entry 3). Both the monomer conversion and molecular weight of the product increase with increasing [CL]/[ITU] molar ratio from 50 to 100. However, at higher molar ratio ([CL]/[ITU] > 100), the monomer conversion and molecular weight of PCL decrease. Thus, for higher molecular weight polymers, high monomer/catalyst ratios are necessary in order to prevent undesired transesterification and cyclic oligomers formation.
Table 1 Effects of [CL]/[ITU] feed ratios on the synthesis of PCLa
| Entry |
[CL]/[ITU] |
Conv.b (%) |
Mnc (kg mol−1) |
Đc |
| Polymerization conditions: [CL] = 2.0 mol L−1, [CL]/[I] = 100:1, [YCl3]/[ITU] = 1:1, 25 °C, 60 min, in toluene. Monomer conversion determined by 1H NMR spectroscopy. Mn and Đ determined by SEC. |
| 1 |
50 |
98.7 |
24.5 |
1.58 |
| 2 |
80 |
98.7 |
24.7 |
1.22 |
| 3 |
100 |
98.8 |
24.8 |
1.09 |
| 4 |
150 |
57.3 |
10.2 |
1.05 |
| 5 |
200 |
21.1 |
7.8 |
1.01 |
The influence of polymerization temperature and time on polymerization behavior was examined (Table 2). Enhanced polymerization rate for ring-opening polymerization of CL was obtained at higher temperature. As expected, the conversion increases significantly compared to experiments achieved at 25 °C, but the molecular weight distribution broadens to 1.43 at 40 °C (Table 2, entry 6). In terms of polymerization time, after 80 min at 25 °C, 94.7% of monomer was consumed (Table 2, entry 10). Prolongation of the reaction time, generates polymers with lower molecular weight and broader molecular weight distribution, which may be due to the transesterification reaction occurring (Table 2, entry 11). The data show that 25 °C and 60 min are optimal reaction conditions for the ring-opening polymerization of CL in toluene (Table 2, entry 9).
Table 2 Effects of reaction temperature and time on PCL synthesisa
| Entry |
Tb (°C) |
Timec (min) |
Conv.d (%) |
Mne (kg mol−1) |
Đe |
| Polymerization conditions: [CL] = 2.0 mol L−1, [CL]/[ITU]/[YCl3]/[BnOH] = 100:1:1:1, in toluene. Polymerization temperature. Polymerization time. Monomer conversion determined by 1H NMR spectroscopy. Mn and Đ determined by SEC. |
| 1 |
10 |
60 |
20.2 |
— |
— |
| 2 |
15 |
60 |
45.0 |
0.80 |
1.04 |
| 3 |
20 |
60 |
75.1 |
18.6 |
1.06 |
| 4 |
30 |
60 |
98.5 |
24.7 |
1.25 |
| 5 |
35 |
60 |
98.8 |
24.9 |
1.28 |
| 6 |
40 |
60 |
98.8 |
25.2 |
1.43 |
| 7 |
25 |
40 |
86.4 |
21.2 |
1.06 |
| 8 |
25 |
50 |
95.0 |
22.3 |
1.07 |
| 9 |
25 |
60 |
98.8 |
24.8 |
1.09 |
| 10 |
25 |
80 |
94.7 |
22.2 |
1.07 |
| 11 |
25 |
110 |
87.4 |
21.0 |
1.21 |
The chemical structure of the resulting PCL was analyzed using 1H and 13C NMR spectra (Fig. S1 and S2†). As expected, the corresponding peak at δ = 7.28 and 5.05 ppm can be respectively recognized as benzyl protons present in the initiator BnOH. δ = 3.57 ppm is assigned to the protons of CH2OH end group. Above results revealed the ROP of CL was initiated by BnOH. In addition, obtained PCL was also analyzed by MALDI-TOF mass spectroscopy (Fig. 1). In Fig. 1, the peaks are separated by m/z 114, corresponding to linear PCL with BnO/H chain ends [Mn = 114.14 + 108.14 (BnOH) + 23.0 (Na+) (g mol−1)].
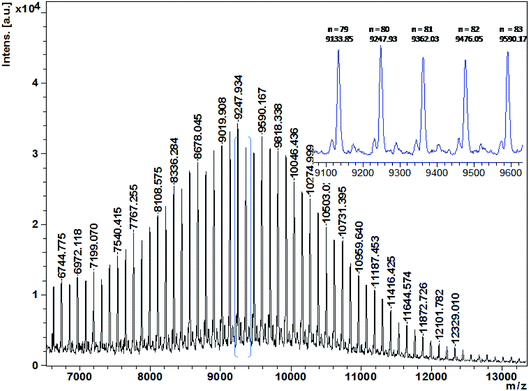 |
| | Fig. 1 MALDI-TOF mass spectrum of PCL produced with [CL]/[ITU]/[YCl3]/[BnOH] = 100:1:1:1 (Table 1, entry 3). | |
Living polymerization nature of ITU/YCl3-catalyzed ROP of CL
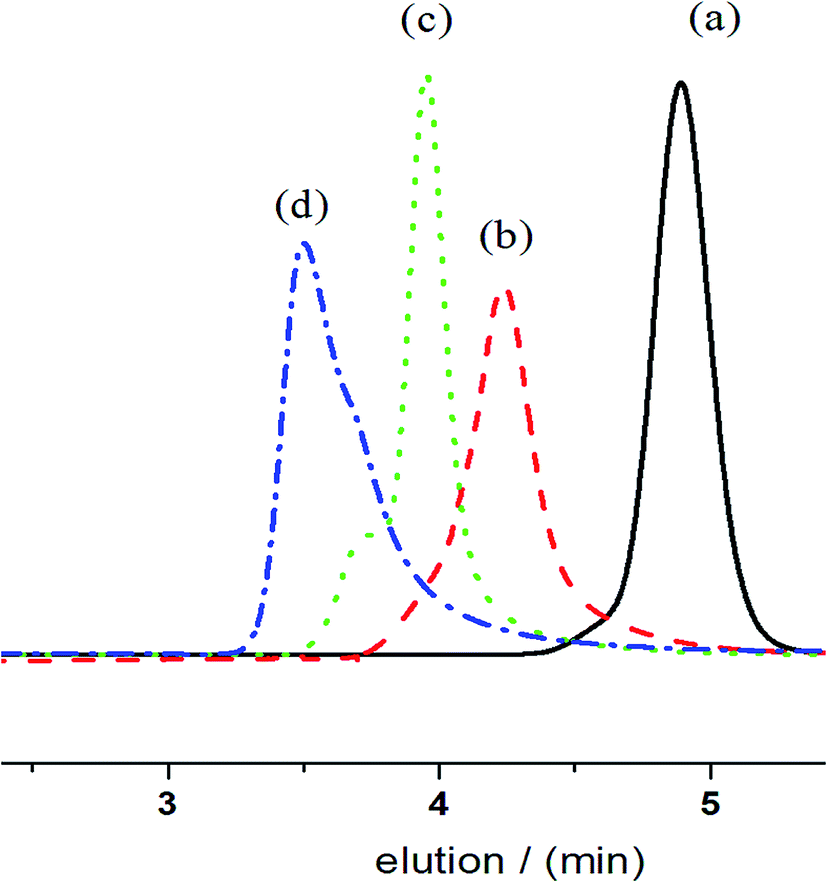
The ratios of [CL]/[BnOH] were changed to evaluate the living nature of the reaction under catalytic conditions. As shown in Fig. 2, SEC traces of PCLs shift shorter elution times, which reflects the average molecular weights of the corresponding polymers that increase linearly with increasing molar ratio of [CL]/[BnOH] from 50 to 200 to 400 while maintaining the equivalent of [ITU]/[YCl3]. The observed distributions are narrow even at high molecular weights, which strongly suggest the controlling nature of ROP catalyzed by ITU/YCl3.
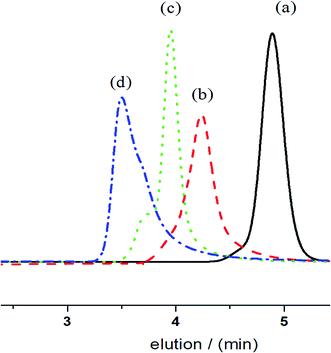 |
| | Fig. 2 SEC curves of obtained PCLs with [CL]/[BnOH] molar ratio of (a) 50, (b) 100, (c) 200, (d) 400. | |
To evaluate the living nature of ITU/YCl3-catalyzed polymerization, the kinetics of ITU/YCl3-catalyzed ROP of CL performed at 25 °C in toluene was examined (Fig. S3†). The linear dependence of monomer consumption on reaction time for different catalyst concentrations reveals a distinct first-order reaction rate. Moreover, the average molecular weight of obtained PCLs increase linearly as a function of conversion (Fig. 3), highlighting the controlling character of the polymerization process. Interestingly, Đ values of PCLs remain relatively narrow.
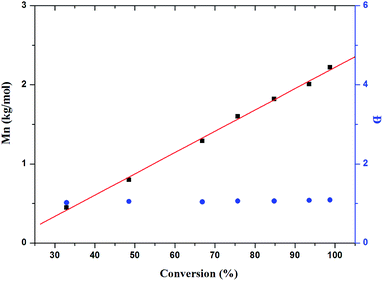 |
| | Fig. 3 The relationship of Mn and Đ versus conversion. | |
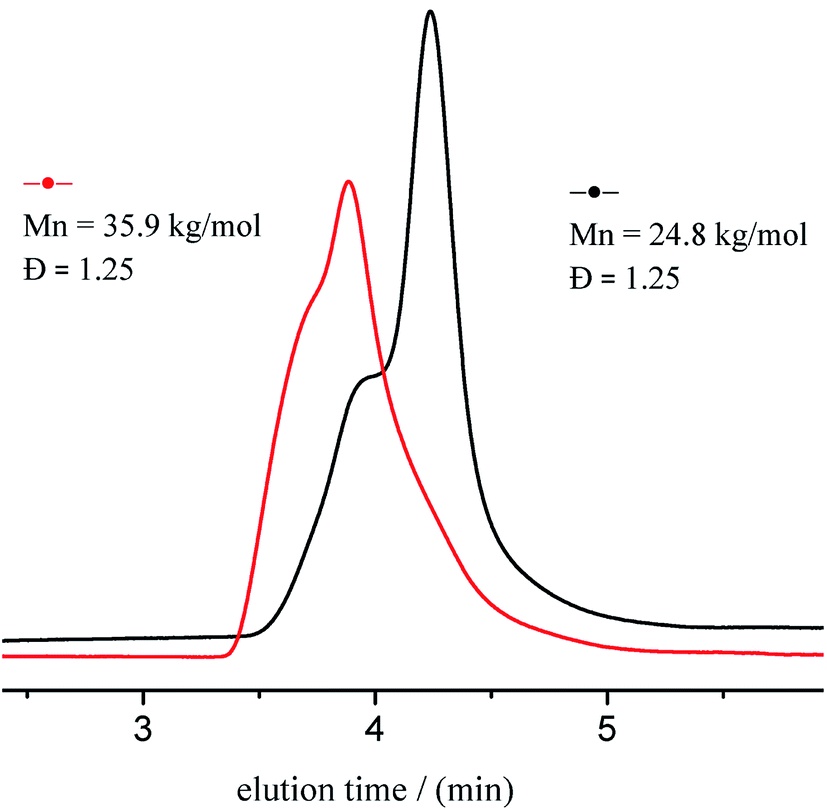
Chain extension experiments were conducted to study the living character of ITU/YCl3-catalyzed ROP of CL. SEC traces of obtained polymers are shown in Fig. 4. PCL with Mn of 24.8 kg mol−1 and Đ of 1.25, produced 98.5% conversion using 80 equiv. of CL. The reaction proceeded further with the addition of another 25 equiv. of CL, yielding PCL with Mn of 35.9 kg mol−1 and Đ of 1.25. Hence, the chain end group of PCL retained its living character.
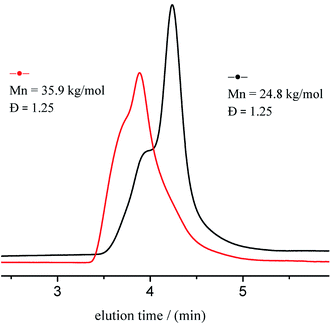 |
| | Fig. 4 SEC traces of first (black line) sequence and post-polymerization (red line). | |
Synthesis of triblock PCL–PEG–PCL copolymers with PEG 2000
The influences of monomer concentration on CL copolymerization with PEG are systematically presented in Fig. 5. The polymerization conversion is quite high, with higher monomer concentration generating broadly dispersed products, however, lower monomer concentration gave narrow distributions. We found that PCL–PEG–PCL with 97.8% conversion and 25.5 kg mol−1 molecular weight was obtained at 2.0 mol L−1 monomer concentration.
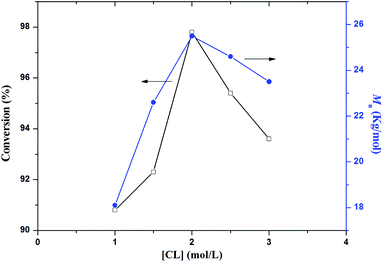 |
| | Fig. 5 Effect of monomer concentration on PCL–PEG–PCL synthesis. | |
Selected results for the polymerization reactions are listed in Table 3 and show the effect of monomer/catalyst ([CL]/[ITU]) and monomer/initiator molar ratio ([CL]/[PEG]) on copolymerization. Both the conversion of CL and molecular weight of the copolymer increase with increasing [CL]/[ITU] molar ratio, but decrease rapidly at [CL]/[ITU] molar ratio above 100. This may be due to the lack of active species in the reaction medium, which decreases the rate of polymerization. Additionally, Table 3 shows that the molecular weight of copolymer was increasingly realized by adjusting [CL]/[PEG] molar ratio from 50 to 100. However, at higher molar ratio ([CL]/[PEG] > 100), the monomer conversion of PCL–PEG–PCL decreases and molecular weight distribution broadens. Therefore, copolymer with 97.8% conversion and molecular weight of 25.5 kg mol−1 was obtained at [CL]/[ITU] molar ratio of 100 and [CL]/[PEG] molar ratio of 100 (Table 3, entry 3).
Table 3 Effects of [CL]/[ITU] and [CL]/[PEG] feed ratios on PCL–PEG–PCL synthesisa
| Entry |
[CL]/[ITU] |
[CL]/[PEG] |
Timeb (min) |
Conv.c (%) |
Mnd (kg mol−1) |
Đd |
| Copolymerization conditions: [CL] = 2.0 mol L−1, 60 °C, in toluene. Polymerization time. Total monomer conversion determined by 1H NMR spectroscopy. Mn and Đ determined by SEC. |
| 1 |
50 |
100 |
30 |
97.7 |
23.8 |
1.18 |
| 2 |
80 |
100 |
50 |
98.0 |
24.0 |
1.14 |
| 3 |
100 |
100 |
60 |
97.8 |
25.5 |
1.08 |
| 4 |
150 |
100 |
150 |
97.7 |
25.1 |
1.07 |
| 5 |
200 |
100 |
1440 |
34.6 |
7.0 |
1.05 |
| 6 |
100 |
50 |
60 |
97.7 |
7.6 |
1.06 |
| 8 |
100 |
200 |
90 |
97.8 |
41.2 |
1.32 |
| 9 |
100 |
400 |
120 |
96.8 |
62.0 |
1.37 |
As shown in Table 4, ITU/YCl3 binary catalyst exhibits excellent activities toward CL polymerization in the presence of PEG at 60 °C for 60 min in toluene (Table 4, entry 8). When the polymerization temperature is over 60 °C, Đ gradually broadens (Table 4, entries 3–5). According to the results, higher temperatures promote weak control over copolymerization. Similarly, prolongation of the reaction time from 60 min to 80 min leads to undesirable side-reactions such as inter- and intra-transesterifications, which results in relatively low molecular weights and wide Đ of PCL–PEG–PCL copolymer (Table 4, entry 9). However, when continuing prolongating time from 80 min to 110 min, both molecular weight and Đ are almost unchanged (Table 4, entry 10).
Table 4 Effects of reaction temperature and time on PCL–PEG–PCL synthesisa
| Entry |
Tb (°C) |
Timec (min) |
Conv.d (%) |
Mne (kg mol−1) |
Đe |
| Copolymerization conditions: [CL] = 2.0 mol L−1, [CL]/[ITU]/[YCl3]/[PEG] = 100:1:1:1, in toluene. Polymerization temperature. Polymerization time. Monomer conversion determined by 1H NMR spectroscopy. Mn and Đ determined by SEC. |
| 1 |
40 |
960 |
97.1 |
21.4 |
1.17 |
| 2 |
50 |
150 |
97.4 |
21.9 |
1.15 |
| 3 |
70 |
40 |
97.7 |
23.5 |
1.18 |
| 4 |
90 |
18 |
97.9 |
24.7 |
1.19 |
| 5 |
110 |
10 |
98.3 |
24.9 |
1.20 |
| 6 |
60 |
40 |
59.7 |
17.0 |
1.07 |
| 7 |
60 |
50 |
85.9 |
18.7 |
1.08 |
| 8 |
60 |
60 |
97.8 |
25.5 |
1.08 |
| 9 |
60 |
80 |
97.4 |
24.4 |
1.19 |
| 10 |
60 |
100 |
97.2 |
24.3 |
1.19 |
The purified polymer was analyzed by SEC, DSC, NMR and FTIR analysis. In the SEC profile (Fig. 6) peak ‘‘a’’ corresponds to PCL–PEG–PCL using PEG as the macroinitiator, whereas peak ‘‘b’’ corresponds to homopolymer PCL using BnOH. The SEC curves of the purified polymer show unimodal traces, which indicates that only one active species is presented in the polymerization system. Hence, monomer CL has been completely converted to PCL–PEG–PCL successfully using ITU/YCl3 cocatalyst. Moreover, SEC trace also confirmed the formation of the triblock structure and absence of any detectable amount of unreacted PEG in triblock copolymers.
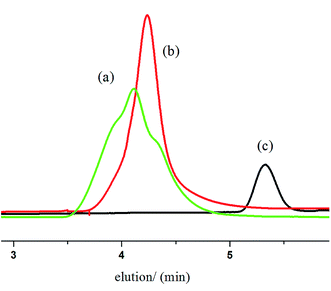 |
| | Fig. 6 SEC curves of PCL–PEG–PCL (a) and PCL (b) and PEG (c). | |
In order to elaborate on the molecular structure of the copolymer, it was analyzed by 1H and 13C NMR spectroscopy. The 1H NMR spectrum of PCL–PEG–PCL and its assigns are shown in Fig. 7. Peaks at 4.06, 2.31, 1.64 and 1.38 ppm correspond to methylene protons of –OCH2–, –(CH2)2–, –CH2– and –CH2O of PCL units, respectively. The peak at 3.64 ppm (–CH2CH2–) relates to PEG units. In the case of 13C NMR spectrum of PCL–PEG–PCL (Fig. 8), the peak at 173.49 ppm corresponds to the PCL blocks, whereas the peak at 70.78 ppm is attributed to the PEG block.
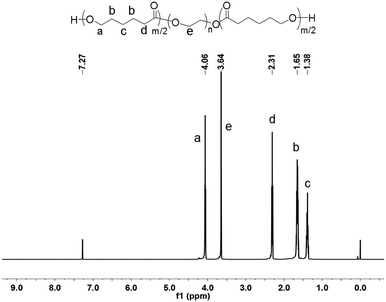 |
| | Fig. 7 1H NMR spectrum of PCL–PEG–PCL. | |
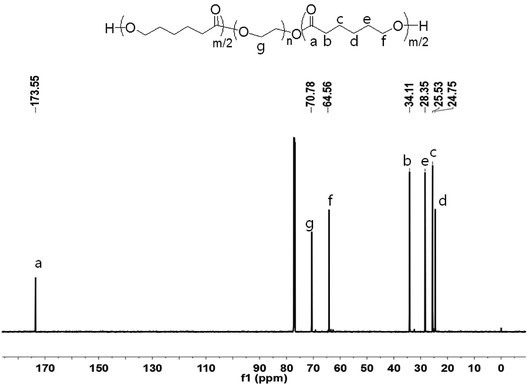 |
| | Fig. 8 13C NMR spectrum of PCL–PEG–PCL. | |
FTIR measurements further confirmed the molecular structure of the copolymers. As shown in Fig. 9, the peak at approx. 1730 cm−1 corresponds to the carbonyl group stretching of the ester groups, and the peaks at 1105 cm−1 are attributed to the characteristic –CH2–O–CH2– stretching vibrations of –OCH2CH2– units from the PEG segment. From these observations, we verified that triblock copolymer was produced without any impurities.
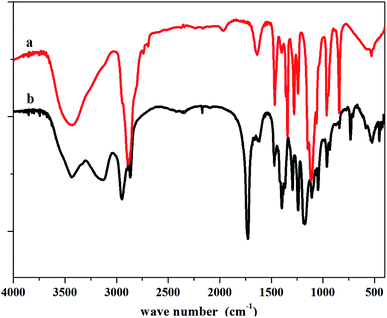 |
| | Fig. 9 IR spectrum of (a) PEG2000, (b) PCL–PEG–PCL. | |
The thermal analyses are depicted in Fig. 10. During the second heating cycle, DSC curves revealed polymers with Tm values at 49.9 °C (PEG), 63.4 °C (PCL) and 61.7 °C (PCL–PEG–PCL). Tm of PCL–PEG–PCL copolymer is located between the homopolymers PEG and PCL. This may be due to reduced mobility of PEG chain in the presence of PCL chains (PCL–PEG–PCL, Mn = 25.5 kg mol−1, Đ = 1.08).
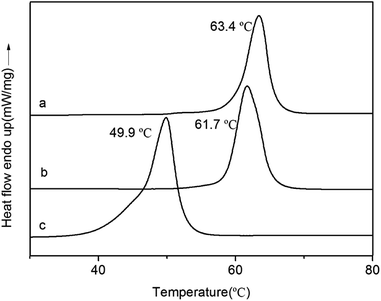 |
| | Fig. 10 DSC curves of PCL (a), PCL–PEG–PCL (b), PEG (c). | |
Synthesis of pentablock PVL–PCL–PEG–PCL–PVL copolymers
The highly efficient and controlled ROP of CL and VL by ITU/YCl3 catalytic system allowed the preparation of pentablock copolymers via sequential copolymerization, as shown in Scheme S1.† ROP of CL was first conducted using PEG as the initiator without quenching at 60 °C after 60 min in toluene. Subsequently, VL was added to polymerize as the second monomer to afford PVL–PCL–PEG–PCL–PVL pentablock copolymer at 60 °C in toluene. Time and temperature dependence of the conversion and molecular weight of PVL–PCL–PEG–PCL–PVL are listed in Table 5. From the data, the total conversion of 91.4% and high Mn = 36.9 kg mol−1 were obtained within 120 min at 60 °C, retaining the optimized ratio of [CL]/[ITU]/[YCl3]/[PEG] = 100:1:1:1. Improving reaction temperature led to a rapid copolymerization and the copolymers were easily degraded (Table 5, entries 4 and 5). Prolong polymerization time after monomer consumption usually broaden the polydispersity, which indicated some transesterification of the copolymers backbone occurring (Table 5, entries 8 and 9).
Table 5 Effects of reaction temperature and time on PVL–PCL–PEG–PCL–PVL synthesisa
| Entry |
Timeb (min) |
Tc (°C) |
Conv.d (%) |
Mne (kg mol−1) |
Đe |
| Copolymerization conditions: first stage of CL polymerization: [CL] = 2.0 mol L−1, [CL]/[ITU]/[YCl3]/[PEG] = 100:1:1:1, [CL]/[VL] = 50:50, 60 °C, 60 min, in toluene. Polymerization time. Polymerization temperature. Total monomer conversion determined by 1H NMR spectroscopy. Mn and Đ determined by SEC. |
| 1 |
120 |
40 |
66.8 |
30.7 |
1.11 |
| 2 |
120 |
50 |
78.4 |
31.5 |
1.13 |
| 3 |
120 |
60 |
91.4 |
36.9 |
1.16 |
| 4 |
120 |
70 |
91.3 |
36.9 |
1.22 |
| 5 |
120 |
80 |
89.9 |
35.2 |
1.26 |
| 6 |
60 |
60 |
79.2 |
31.5 |
1.12 |
| 7 |
90 |
60 |
83.7 |
33.1 |
1.15 |
| 8 |
150 |
60 |
91.3 |
34.9 |
1.17 |
| 9 |
180 |
60 |
90.1 |
34.8 |
1.19 |
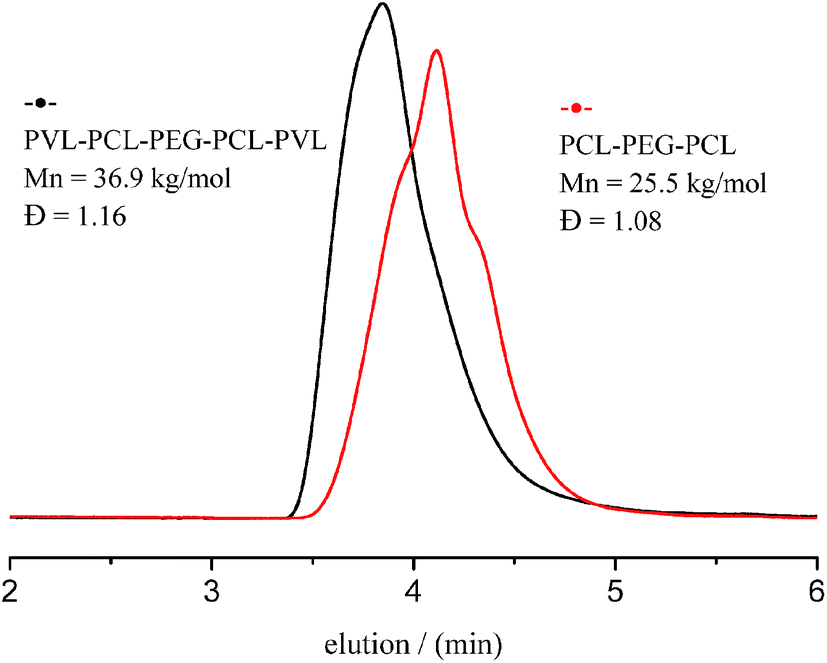
Fig. 11 gives typical SEC trace of triblock PCL–PEG–PCL precursor with Mn of 25.5 kg mol−1 and Đ of 1.08 (Fig. 11, black curve) before the addition of VL. After the addition of VL, a shift of SEC curves of resulting pentablock copolymer toward higher Mn of 36.9 kg mol−1 (Fig. 11, red curve) together with monomodal and narrow distribution (Đ = 1.16) suggests excellent control of this “one-pot” ROP synthesis. The 1H and 13C NMR spectra show that PVL–PCL–PEG–PCL–PVL copolymer was synthesized successfully. The well-designed structure comparatively analyzed by stacking 1H NMR spectra of PVL, PCL and PVL–PCL–PEG–PCL–PVL (Fig. S4 and S5†). These results show that ITU/YCl3 can be considered a good binary catalytic system for the preparation of block copolymer from PCL and PVL with different block sequences and topologies.
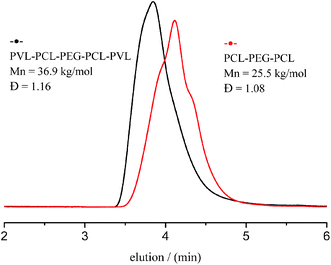 |
| | Fig. 11 SEC traces of PCL–PEG–PCL and PVL–PCL–PEG–PCL–PVL. | |
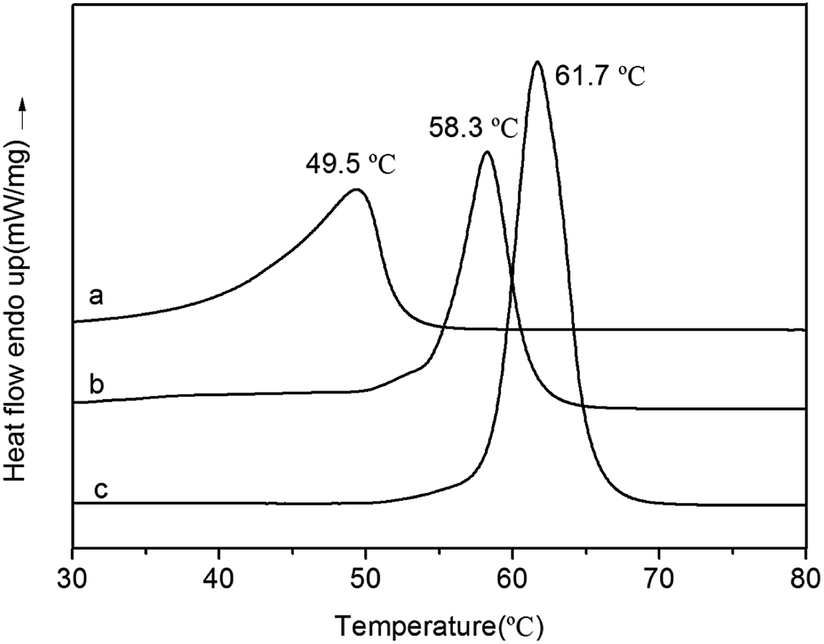
DSC curves of PVL, PCL–PEG–PCL and PVL–PCL–PEG–PCL–PVL are shown in Fig. 12. Only a Tm value was detected at 49.5 °C (PVL), 58.3 °C (PVL–PCL–PEG–PCL–PVL) and 61.7 °C (PCL–PEG–PCL) at the second heating, respectively. It noted that Tm of PVL–PCL–PEG–PCL–PVL was lower than PCL–PEG–PCL at 61.7 °C. This decreased melting point can be attributed to the presence of PVL blocks attached to PCL blocks, probably disrupting orderly fold pattern of the crystal.
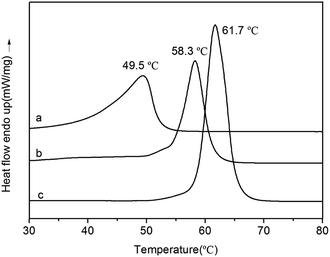 |
| | Fig. 12 DSC curves of PVL (a), PVL–PCL–PEG–PCL–PVL (b), PCL–PEG–PCL (c). | |
Proposed mechanism
The proposed reaction mechanism for ITU/YCl3 catalyzed ROP of CL is presented in Scheme 2. The process of ROP is assumed to occur via coordination of CL to yttrium-center, thereby increasing the electron deficiency on the carbonyl carbon, which facilitates ring opening via nucleophilic attack of activated alcohol. The mode of alcohol activation is often described as the deprotonation/activation of the initiating alcohol species by ITU in the presence of YCl3. Furthermore, ITU activates of the polymeric precursor chain ends generating active intermediates, similar to the mechanism proposed by Naumann.45–48 These intermediates attack the YCl3-activated monomers to propagate chains. The chain-growths enter the next chain of cyclic propagation to form linear PCLs. Further attempts to characterize the polymers and to gain insight into the mechanism of polymerization are in progress.
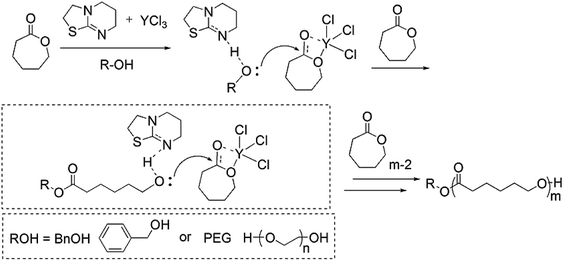 |
| | Scheme 2 Proposed mechanism for ROP of CL catalyzed by ITU/YCl3 binary system. | |
Conclusions
In summary, ITU showed excellent catalytic performances toward ROP of CL. The well-defined PCL homopolymers and PCL–PEG–PCL triblock copolymers were directly synthesized via ROP of CL in the presence of hydroxyl-terminated BnOH or α,ω-dihydroxyl PEG2000 as initiators, and ITU/YCl3 as an organocatalyst, respectively. Subsequently, PVL–PCL–PEG–PCL–PVL pentablock copolymers were successfully obtained using ITU/YCl3 catalyst. The architecture, molecular weight and thermal properties of PCL, PCL–PEG–PCL, and PVL–PCL–PEG–PCL–PVL were characterized using NMR, MALDI-TOF mass, FTIR, SEC and DSC analyses. A possible polymerization mechanism was speculated.
Conflicts of interest
There are no conflicts to declare.
References
- T. Ahmed, M. Shahid, F. Azeem, I. Rasul, A. Shah, M. Noman, A. Hameed, N. Manzoor, I. Manzoor and S. Muhammad, Environ. Sci. Pollut. Res., 2018, 25, 7287–7298 CrossRef CAS PubMed.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed.
- E. Stirling, Y. Champouret and M. Visseaux, Polym. Chem., 2018, 9, 2517–2531 RSC.
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. - Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS.
- S. Dagorne and C. Fliedel, Top. Organomet. Chem., 2013, 41, 125–171 CrossRef CAS.
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 39, 1800485 CrossRef PubMed.
- Q. L. Song, S. Y. Hu, J. P. Zhao and G. Z. Zhang, Chin. J. Polym. Sci., 2017, 35, 581–601 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- X. Wang, K. Liao, D. Quan and Q. Wu, Gaofenzi Xuebao, 2006, 229–235 Search PubMed.
- T. Shi, W. Luo, S. Liu and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 611–617 CrossRef CAS.
- S Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 1800485 CrossRef PubMed.
- X. Zhang, M. Fevre, G. O Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- S. Y. Hu, J. P. Zhao, G. Z. Zhang and H. Schlaad, Prog. Polym. Sci., 2017, 74, 34–77 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- Q.-L. Song, S.-Y. Hu, J.-P. Zhao and G.-Z. Zhang, Chin. J. Polym. Sci., 2017, 35, 581–601 CrossRef CAS.
- L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144–1164 CrossRef CAS.
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- D. Xie, B. Jiang and C. Yang, Gaofenzi Xuebao, 2000, 532–537 CAS.
- R. C. Prratt, B. G. G. Lohmeijer, D. A. Long, P. N. Pontus Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef.
- G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef.
- K. V. Fastnacht, S. S. Spink, N. U. Dharmaratne, J. U. Pothupitiya, P. P. Datta, E. T. Kiesewetter and M. K. Kiesewetter, ACS Macro Lett., 2016, 5, 982–986 CrossRef CAS.
- N. U. Dharmaratne, J. U. Pothupitiya, T. J. Bannin, O. I. Kazakov and M. K. Kiesewetter, ACS Macro Lett., 2017, 6, 421–425 CrossRef CAS.
- J. U. Pothupitiya, N. U. Dharmaratne, T. M. M. Jouaneh, K. V. Fastnacht, D. N. Coderre and M. K. Kiesewetter, Macromolecules, 2017, 50, 8948–8954 CrossRef CAS.
- J. U. Pothupitiya, R. S. Hewawasam and M. K. Kiesewetter, Macromolecules, 2018, 51, 3203–3211 CrossRef CAS.
- N. U. Dharmaratne, J. U. Pothupitiya and M. K. Kiesewetter, Org. Biomol. Chem., 2019, 17, 3305–3313 RSC.
- J. A. Edward, M. K. Kiesewetter, H. Kim, J. C. A. Flanagan and J. L. Hedrick, Biomacromolecules, 2012, 13, 2483–2489 CrossRef CAS PubMed.
- M. K. S. Bisht, L. A. Henderson, R. A. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 2705 CrossRef.
- S. Naumann, F. G. Schmidt, W. Frey and M. R. Buchmeiser, Polym. Chem., 2013, 4, 4172–4181 RSC.
- S. Naumann and D. R. Wang, Macromolecules, 2016, 49, 8869–8878 CrossRef CAS.
- Q. Wang, W. Zhao, J. He, Y. Zhang and E. Y. X. Chen, Macromolecules, 2017, 50, 123–136 CrossRef CAS.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
- J. Bai, X. Wang, J. Wang and L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1189–1196 CrossRef CAS.
- V. B. Birman, X. M. Li and Z. F. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed.
- E. Piedra-Arroni, C. Ladaviere, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13306–13309 CrossRef CAS PubMed.
- S. Naumann and D. R. Wang, Macromolecules, 2016, 49, 8869–8878 CrossRef CAS.
- P. Walther and S. Naumann, Macromolecules, 2017, 50, 8406–8416 CrossRef CAS.
- J. Meisner, J. Karwounopoulos, P. Walther, J. Kastner and S. Naumann, Molecules, 2018, 23, 432 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04974c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc