
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uncovering the origin of enhanced field emission properties of rGO–MnO2 heterostructures: a synergistic experimental and computational investigation†
Sachin R. Rondiya *a,
Indrapal Karbhalb,
Chandradip D. Jadhavc,
Mamta P. Nasaned,
Thomas E. Daviesa,
Manjusha V. Shelkeb,
Sandesh R. Jadkard,
Padmakar G. Chavane and
Nelson Y. Dzade*a
*a,
Indrapal Karbhalb,
Chandradip D. Jadhavc,
Mamta P. Nasaned,
Thomas E. Daviesa,
Manjusha V. Shelkeb,
Sandesh R. Jadkard,
Padmakar G. Chavane and
Nelson Y. Dzade*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Wales, UK. E-mail: RondiyaS@cardiff.ac.uk; DzadeNY@cardiff.ac.uk
bPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune 411008, MH, India
cThe State Key Laboratory of Refractories and Metallurgy, Institute of Advanced Materials and Nanotechnology, College of Materials and Metallurgy, Wuhan University of Science and Technology, Wuhan 430081, P. R. China
dDepartment of Physics, Savitribai Phule Pune University, Pune 411007, India
eDepartment of Physics, School of Physical Sciences, Kavayitri Bahinabai Chaudhari North Maharashtra University, Jalgaon 425001, India
First published on 10th July 2020
Abstract
The unique structural merits of heterostructured nanomaterials including the electronic interaction, interfacial bonding and synergistic effects make them attractive for fabricating highly efficient optoelectronic devices. Herein, we report the synthesis of MnO2 nanorods and a rGO/MnO2 nano-heterostructure using low-cost hydrothermal and modified Hummers' methods, respectively. Detailed characterization and confirmation of the structural and morphological properties are done via X-ray Diffraction (XRD), Field Emission Scanning Electron Microscopy (FESEM) and Transmission Electron Microscopy (TEM). Compared to the isolated MnO2 nanorods, the rGO/MnO2 nano-heterostructure exhibits impressive field emission (FE) performance in terms of the low turn-on field of 1.4 V μm−1 for an emission current density of 10 μA cm−2 and a high current density of 600 μA cm−2 at a relatively very low applied electric field of 3.1 V μm−1. The isolated MnO2 nanorods display a high turn-on field of 7.1 for an emission current density of 10 μA cm−2 and a low current density of 221 μA cm−2 at an applied field of 8.1 V μm−1. Besides the superior FE characteristics of the rGO/MnO2 nano-heterostructure, the emission current remains quite stable over the continuous 2 h period of measurement. The improvement of the FE characteristics of the rGO/MnO2 nano-heterostructure can be ascribed to the nanometric features and the lower work function (6.01 and 6.12 eV for the rGO with 8% and 16% oxygen content) compared to the isolated α-MnO2(100) surface (Φ = 7.22 eV) as predicted from complementary first-principles electronic structure calculations based on density functional theory (DFT) methods. These results suggest that an appropriate coupling of rGO with MnO2 nanorods would have a synergistic effect of lowering the electronic work function, resulting in a beneficial tuning of the FE characteristics.
1. Introduction
Nanoscale heterostructure design comprising different material compositions is emerging as an attractive strategy and essential building block for functional devices to achieve improved performance. The desired physicochemical properties of the participating nanomaterials in nanostructured hybrids/composites complement each other by tuning their electronic properties to meet the requirements for the fabrication of efficient electronic devices.1 Reduced Graphene Oxide (rGO) is an attractive and ideal nanomaterial to be paired with another suitable semiconductor for the development of multifunctional heterostructures because of its unique electronic properties, high electrical conductivity (5 × 10−3 S cm−1), flexible structure, high aspect ratio, and high specific surface area (2630 m2 g−1).2–5 Owing to its unique physicochemical properties, rGO is being recognized as a material of great interest for potential applications in nanoelectronics,6 nanoelectromechanical systems,7 sensors,8 catalysis,9 energy storage devices,10,11 optics,12 and field emission (FE).13–15 There exist several reports of the successful synthesis of rGO or modified graphene heterostructures with various semiconducting nanomaterials such as TiO2, SnO2, ZnO, Si, CdSe, etc. in the literature.16–18For field emission applications, where electrons are extracted from the surface of a metal/semiconductor by an electrostatic field through quantum mechanical tunneling, rGO-based nanocomposites such are rGO-Bi2S19 and WS2-RGO,20 have demonstrated superior field emission properties. Among transition metal oxides, manganese dioxide (MnO2) has attracted increasing interest for field emission applications, owing to their wide structural diversity combined with unique chemical and physical properties.21,22 The advantages of MnO2 as field emitter are the lower cost for raw materials and the fact that manganese is more environmentally friendly than other metal oxide.23,24 MnO2 has also attracted a lot of attention as an electrochemical pseudocapacitor material due to its high theoretical capacitance (1370 F g−1).25–27 Wu et al. reported inspiring results such as low turn-on field value of 8.4 V μm−1 at current density of 1 μA cm−2 and maximum emission current density of 160 μA cm−2 at an applied field 18 V μm−1.21 The field emission applications of MnO2 is, however, limited by its low specific surface area and poor electrical conductivity (10−5 to 10−6 S cm−1). Compared to its flat films, by fabricating rGO/MnO2 nanocomposite the interface area can be significantly enlarged, which is desirable for field emission application.28 Besides, rGO is solution-processable and thus can be deposited in large areas onto different kinds of substrates enabling simple and cost-effective fabrication of field electron emitters for display applications. The formation of rGO–MnO2 nanostructures and their electrochemical performance have been extensively investigated and it was demonstrated that compared to the single metal-oxide, rGO/MnO2 nanocomposites show superior electric conductivity, electric capacity and charge/discharge efficiency for supercapacitor performance.29–34 These characteristics make rGO/metal-oxide nanocomposites promising materials for energy applications. Considering that field emission is geometry (shape, size, aspect ratio, alignment, and areal density of the nanostructure) and work function dependent phenomenon, well-aligned rGO/MnO2 nanostructures is promising for enhancement of field emission characteristics.35
Herein, we report a simple and cost-effective solution-based method to prepared MnO2 nanorods and rGO/MnO2 nano-heterostructure. The structural and morphological verifications have been done by using X-ray diffraction (XRD), Field Emission Scanning Electron Microscope (FESEM), and Transmission Electron Microscopy (TEM). Finally, the field emission properties of the as-prepared MnO2 nanorods and rGO/MnO2 nano-heterostructure was systematically characterized and compared. The rGO/MnO2 nano-heterostructure exhibits superior field emission characteristics compared to the MnO2 nanorod. The rGO/MnO2 nano-heterostructure demonstrates a low turn-on field of 1.4 V μm−1 for an emission current density of 10 μA cm−2 compared to 7.1 V μm−1 for MnO2 nanorod. The combined contribution of the sharp edges of the thin rGO sheets and high aspect ratio of the MnO2 nanorods, coupled with synergetic effect in the rGO/MnO2 nano-heterostructure are responsible for the observed enhanced field emission behavior. Consistent with the experimental data, our complementary first-principles DFT calculations predict lower work function for the rGO/MnO2 nano-heterostructure compared to the isolated MnO2 as the primary origin for improved field emission.
2. Experimental, characterization, and computational methods
2.1 Synthesis of MnO2 and rGO/MnO2
The rGO has been synthesized by modified Hummer's method36 whereas the MnO2 nanorods has been synthesized by the hydrothermal method.37 For the preparation of the rGO/MnO2 composite, 1 mg ml−1 rGO was dispersed in the 100 ml of DI water in a beaker. Later, 10 mM of KMnO4 and 10 mM of MnSO4 were added into the rGO solution and stirred for 30 min form a homogenous solution. The prepared solution was transferred into a stainless steel autoclave and kept at 160 °C for 24 h. After cooling to room temperature, the material was filter washed with DI water and ethanol to obtian rGO/MnO2 composite, which was dried in an oven at 80 °C for 12 hours and used for various characterizations presented next. Scheme 1 represents the synthesis steps which were followed for synthesis of rGO, MnO2 and rGO/MnO2 heterostructure.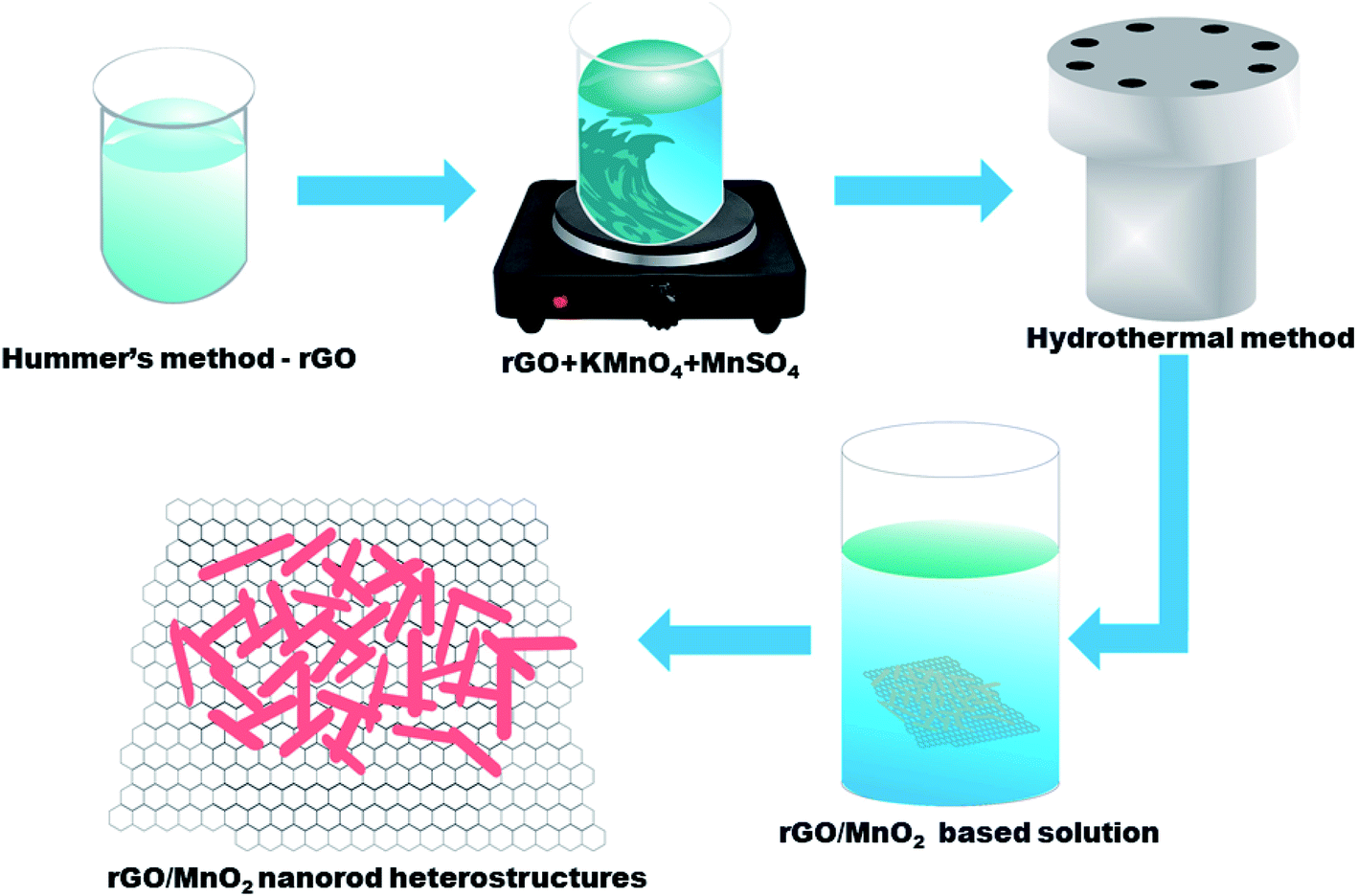 | ||
| Scheme 1 The facile synthesis of MnO2 nanorods and rGO/MnO2 heterostructure. | ||
2.2 Materials characterization
The MnO2 nanorods and rGO/MnO2 nano-heterostructure were characterized by various complementary experimental methods. The XRD patterns were obtained with a Bruker D8 Advance X-ray diffractometer using the Cu Kα line (λ = 1.54 Å) at 1° grazing angle. The HR-TEM micrographs and selected area electron diffraction (SAED) patterns were obtained with JEOL-JEM 2100 microscope operating at 200 kV. The samples were dry dispersed over 300 mesh copper grids coated with holey carbon film. A Field Emission Scanning Electron Microscope (FEG-SEM Model – Tescan MAIA3) was used to examine the morphology and surface topography of the MnO2 nanorods and rGOs/MnO2 composite. The accelerating voltage was 15 kV. X-ray spectroscopy (EDS) measurements were done using Oxford Instruments X-MaxN 80 detector and analyzed using Aztec software. X-ray Photoelectron Spectroscopy (XPS) was carried on the samples using a Kratos Axis Ultra DLD photoelectron spectrometer utilizing monochromatic AlKα radiation operating at an energy of 120 W (10 × 12 kV). Data were analyzed using Casa XPS and modified Wagner sensitivity factors as supplied by the instrument manufacturer after subtraction of a Shirley background. All spectra were calibrated to the C(1s) line taken to be 284.8 eV.2.3 Field emission
The field emission studies of the MnO2 nanorods and rGO/MnO2 heterostructure were carried out in the Ultra-High Vacuum (UHV) chamber at a base pressure of ∼1 × 10−8 mbar (Excel Instruments model: I-100). Detail experimental procedure may found in our earlier paper.38 The configuration of the field emission experiment steps up is shown in ESI (scheme S1).† The distance between inter-electrode was maintained at 1 mm. The area of both specimens (MnO2 nanorods and rGOs/MnO2heterostructure) was 0.25 cm2.2.4 Computational methods
The first-principles spin polarized density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP),39–41 a periodic plane wave DFT code which includes the interactions between the core and valence elections using the Project Augmented Wave (PAW) method.42 An energy cut-off of 600 eV, and Monkhorst–Pack43 k-point mesh of 7 × 7 × 3 was used to sample the sample the Brillouin zone of bulk α-MnO2. Geometry optimizations were performed based on the conjugate-gradient algorithm until the residual Hellmann–Feynman forces on all relaxed atoms reached 10−3 eV Å−1. The electronic exchange–correlation potential was calculated using the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) functional.44 To accurately reproduce the experimentally known band gaps and density of states features of α-MnO2 and rGO, the screened hybrid functional HSE06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 was used with the exchange value of 25%. The projected density of states (PDOS) was calculated using tetrahedron method with Bloch correction.46
45 was used with the exchange value of 25%. The projected density of states (PDOS) was calculated using tetrahedron method with Bloch correction.46
The most stable α-MnO2 (100) surface47 was employed to form the nano-heterostructure with rGO (rGO/α-MnO2). The α-MnO2 (100) surface was created from the optimized bulk material using the METADISE code, which ensures the creation of surfaces with zero dipole moment perpendicular to the surface plane. The rGO/α-MnO2nano-heterostructure was constructed with (2 × 4)-α-MnO2(100) and (5 × 5)-rGO supercells. We used k-point meshes of 9 × 9 × 1 for the rGO monolayer, 5 × 5 × 1 for the α-MnO2 (100) surface, and 5 × 5 × 1 for the rGO/α-MnO2 composite. In each simulation cell, a vacuum region of length 20 Å was added perpendicular to the surface to avoid interactions between periodic slabs. The electrostatic potential of each surface was averaged along the c-direction, using the Macro Density package.48–50 The work function (Φ) was calculated as Φ = Vvacuum − EF, where Vvacuum and EF are the vacuum and Fermi level, respectively. Dipole correction perpendicular to all surfaces was accounted for, which ensured that there is no net dipole perpendicular to the surfaces that may affect the potential in the vacuum level.51–53
3. Results and discussions
3.1 Characterization of MnO2 and rGO/MnO2
The crystalline structures of the MnO2 nanorods and rGO/MnO2 nano-heterostructure were confirmed by XRD and the corresponding results are presented in Fig. S1.† All the diffraction peaks in Fig. S1† can be indexed to the tetragonal crystal structure of MnO2 (ICDD card no. 72-1982) with lattice constant a = b = 9.815 Å and c = 2.847 Å. We have observed the highest growth of α-MnO2 in the (211) plane.54 The XRD diffraction pattern of the rGO–MnO2 nanostructure is shown in inset of Fig. S1.† The broad peak at 2θ around 26° corresponds to the (002) plane of the reduced graphene oxide.55 The field emission scanning electron microscope (FESEM) images in Fig. 1, reveal the morphological properties of the MnO2 nanorods and rGO/MnO2 nano-heterostructure. The FESEM images recorded at different magnifications (panels a–c of Fig. 1) show the formation of randomly distributed MnO2 nanorods. The low magnification image shown in Fig. 1a depicts large coverage of the MnO2 nanorods. The diameter of the formed ultra-long nanorods is estimated in the range of 140–150 nm as revealed by the high magnification FESEM images analyses (Fig. 1b and c). It is evident from the FESEM image of the rGO/MnO2 nano-heterostructure that the MnO2 nanorods are embedded in the rGO network (Fig. 1b–d). The high magnification image in Fig. 1f reveals the enormous coverage of the rGO/MnO2 nano-heterostructure. | ||
| Fig. 1 FESEM images of MnO2 nanorods (a–c) and rGO–MnO2 heterostructures (d–f) recorded at different magnification. | ||
Energy dispersive X-ray spectroscopy (EDX) composition analysis in the 0–10 keV energy range (ESI, Fig. S2†) confirmed that the MnO2 nanorods and rGO/MnO2 nano-heterostructure have the optimal stoichiometric atomic Mn:O and Mn:O:C ratios, respectively. Moreover, the FESEM-EDS elemental mapping (ESI, Fig. S2†) confirms an even distribution of the chemical constituents (Mn, O, and C). For detail structural analysis of the as-synthesized MnO2 nanorods and rGO/MnO2 nano-heterostructure, TEM studies were carried out. The TEM micrograph (Fig. 2a) reveals the morphology of the MnO2 nanorods, with sizes ranging between 50 and 70 nm in width and the average length of 1 μm. The selected area electron diffraction (SAED) pattern, depicted in Fig. 2b, confirms the polycrystalline nature of the MnO2 nanorods. The lattice-resolved high-resolution transmission electron microscopy (HRTEM) image of the MnO2 nanorod (Fig. 2c) clearly reveals its crystalline nature. The lattice fringes are clearly observed in the HR-TEM image (Fig. 2c and d) and a 0.69 nm interplanar distance indicates these planes to be of the (110) character.56 The inverse FFT HR-TEM image and the corresponding profile plot of the MnO2 nanorod are shown in Fig. S3(a and b).† A number of firmly attached α-MnO2 nanorods onto the rGO sheets can be clearly seen from Fig. 2e and f. X-ray photoelectron spectroscopy (XPS) analysis was applied to determine the oxidation state and elemental composition of the prepared MnO2 nanorods and rGO/MnO2 nano-heterostructure. The XPS results for the MnO2 nanorods are shown in Fig. S4(a and b)† and the rGO/MnO2 nano-heterostructure in Fig. S4(c–e).† The peaks centered at 642.63 and 654.53 eV in the high-resolution spectrum of Mn 2p (Fig. S4a†) can be assigned to the Mn 2p3/2 and Mn 2p1/2 peaks, respectively, confirming the presence of MnO2.57–60 The deconvolution peaks of the O 1s spectrum (Fig. S4b†) can be divided into three peaks which correspond to O–Mn bonding. The high-resolution spectrum of Mn 2p and O 1s shown in Fig. S4c and S4d,† respectively, for rGO/MnO2 nano-heterostructure, confirm the presence of MnO2, whereas the C (1s) spectra (Fig. S4e†) exhibit peaks that originates from the rGO sheets.
 | ||
| Fig. 2 (a) TEM images of MnO2 nanorods (b) selected area electron diffraction (SAED) pattern of MnO2 nanorods (c) HR-TEM image of MnO2 nanorods with clear lattice resolution (d) Inverse Fourier transform of area shown in (c) with interplanar spacing of MnO2 (e and f) TEM images of rGO–MnO2 heterostructures. | ||
3.2 Field emission investigations
Fig. 3(a and b) shows the emission current density as a function of the applied electrical field (J–E curves) for the MnO2 nanorods and rGO/MnO2 nano-heterostructure. In this work, the turn-on field defined as the field required to draw an emission current density (J) of 10 μA cm−2 is found to be 7.1 and 1.4 V μm−1 for the MnO2 nanorods and the rGO/MnO2 nano-heterostructure, respectively. The rGO/MnO2 nano-heterostructure also attains an impressive current density of 600 μA cm−2 at an applied field of 3.1 V μm−1 compared to the isolated MnO2 which displayed a lower current density of 221 μA cm−2 at a relatively higher applied field of 8.1 V μm−1. A comparison between the turn-on field values obtained in the present study and previously reported MnO2 nanostructures and the MnO2/rGO nano-heterostructures is provided in Table 1.21,22,61–66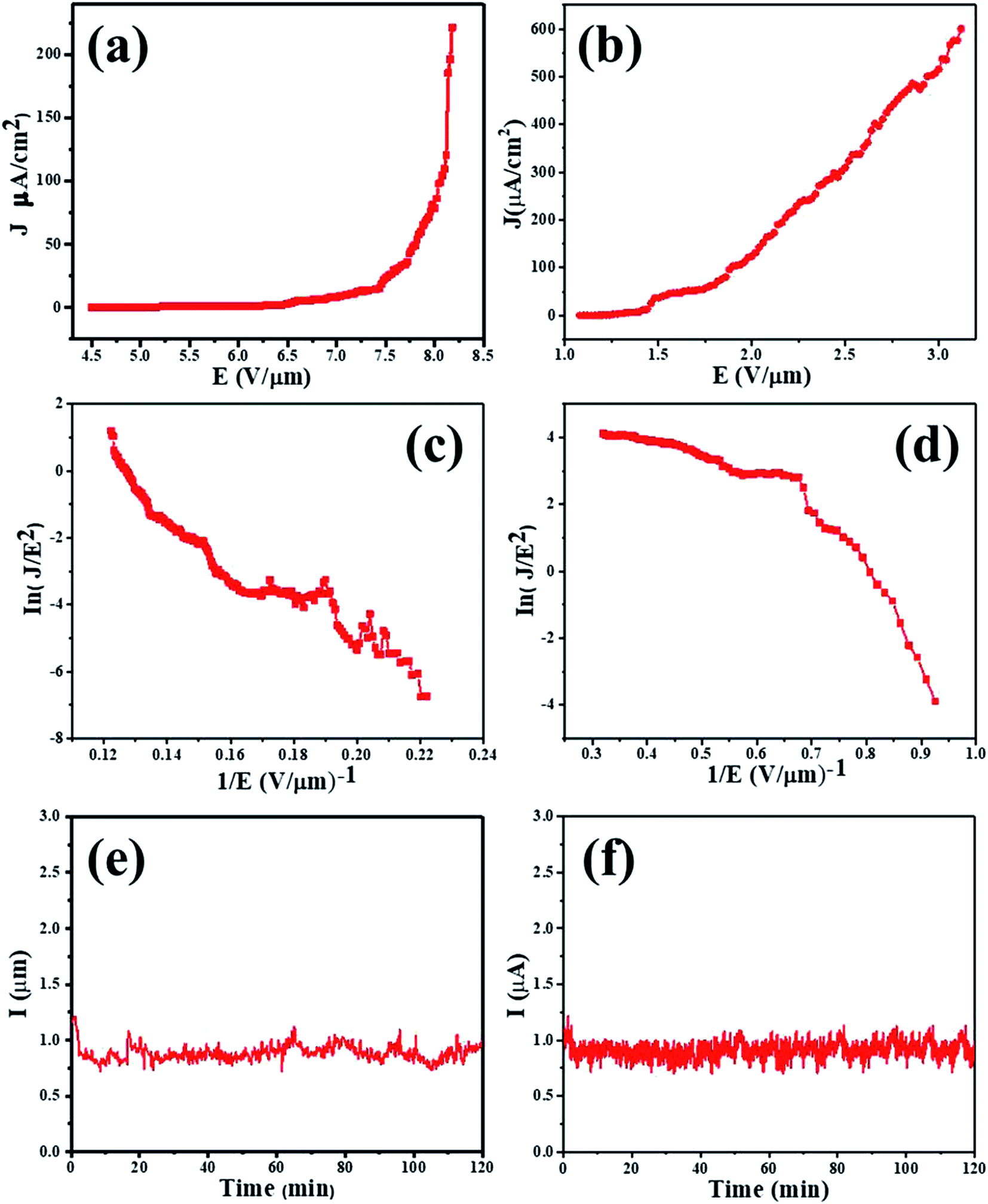 | ||
| Fig. 3 FE characteristics of the MnO2 nanorods and rGO–MnO2 heterostructures: (a and b) emission current density versus applied electric field (J–E) curve, (c and d) F–N plot, and (e and f) emission current versus time (I–t) plot. | ||
| Sr. no. | Specimen | Turn-on field (V μm−1) (for J = 10 μA cm−2) | Reference |
|---|---|---|---|
| 1 | MnO2 nanotubes | 8.4 (1 μA cm−2) | 21 |
| 2 | MnO2 nanorods | 5.90 (1 μA cm−2) | 22 |
| 3 | MnO2/rGO nanocomposite | 3.6 (1 μA cm−2) | |
| 4 | rGO/TiO2 nanocomposite | 2.6 | 61 |
| 5 | T-ZnO/rGO nanocomposite | 1.54 (1 μA cm−2) | 62 |
| 6 | SnO2/rGO nanocomposite | 1.8 (1 μA cm−2) | 63 |
| 7 | ZnO/graphene nanocomposite | 1.3 (1 μA cm−2) | 64 |
| 8 | ZnO/graphene nanocomposite | 2.1 | 65 |
| 9 | SnO2/graphene nanocomposite | 3.85 (1 μA cm−2) | 66 |
| 10 | MnO2 nanorods | 7.1 | Present work |
| 11 | MnO2/rGO nano-heterostructure | 1.4 |
The Fowler–Nordheim (F–N) plot for MnO2 nanorods and rGO/MnO2 nano-heterostructure obtained from ln(J/E2) verses (1/E) is shown in Fig. 3(c and d). Consistent previous reports,21,22 the FN plots of the MnO2 nanorods and rGO/MnO2 nano-heterostructure exhibit linear behavior in good agreement with their semiconducting nature. The nanometric features of the rGO/MnO2 nano-heterostructure coupled with the high electrical conductivity of rGO are suggested as the key factors behind the observed superior field emission properties.2–5 Apart from the improved field emission performance, the electron emission current stability is an important parameter for device fabrication considerations. We have therefore measured the emission current as a function of time in order to ascertain the robustness of the rGO/MnO2 nano-heterostructure. The emission current versus time (I–t) plot of the MnO2 nanorods and rGO/MnO2 nano-heterostructure were recorded continuously for 2 h at a preset value of emission current of ∼1 μA as shown in Fig. 3(e and f). Generally, the results show that the emission current remains quite stable without showing any sign of diminishing over the 2 h period of continuous testing. Instabilities in the form of spikes can be attributed to the presence of residual gas molecules across the emitter surface.
3.3 Density functional theory analyses
Considering that field emission is a geometry and work function (Φ) dependent phenomenon, with lower Φ enhancing the field emission characteristics, we have carried out first-principles density functional theory calculations to gain atomic-level insight into the electronic structure and work function of the isolated rGO, α-MnO2 (100) surface, and the rGO/α-MnO2 (100) nanocomposite. As the electronic band gap of rGO vary depending on the degree of reduction, the rGO monolayer with two concentrations of epoxide functional groups with 8% and 16% oxygen contents were modelled as shown in Fig. 4(a and b). The band gap of the rGO with 8% and 16% oxygen contents are predicted at 0.48 eV (Fig. 4d) and 0.81 eV (Fig. 4e). This is consistent with experimental data that showed that the band gap of rGO can be tuned from 0.264–0.786 eV by controlling the surface concentration of epoxide groups.67 The bulk α-MnO2 was modelled tetragonal crystal structure (space group – I4/m, no. 87) in the antiferromagnetic AFM-C2 configuration68 as shown in Fig. 4c. A full unit cell relaxation yielded a strain-free α-MnO2 with lattice parameters a = b = 9.763 Å, c = 2.872 Å, which compares closely with known experimental data (a = b = 9.71 Å and c = 2.88 Å).69,70 The electronic band gap of α-MnO2 is predicted at 2.42 eV (Fig. 4f), in close agreement with an experimental estimate of 2.23 eV71 and previous theoretical prediction of 2.7 eV.72 The valence band edge of α-MnO2 is demonstrated to consist mainly of O-p states whereas the conduction band edge is dominated by Mn-d states. The smaller band gap of the rGO compared to the MnO2 suggests that the rGO has better electrical conductivity than the metal oxide.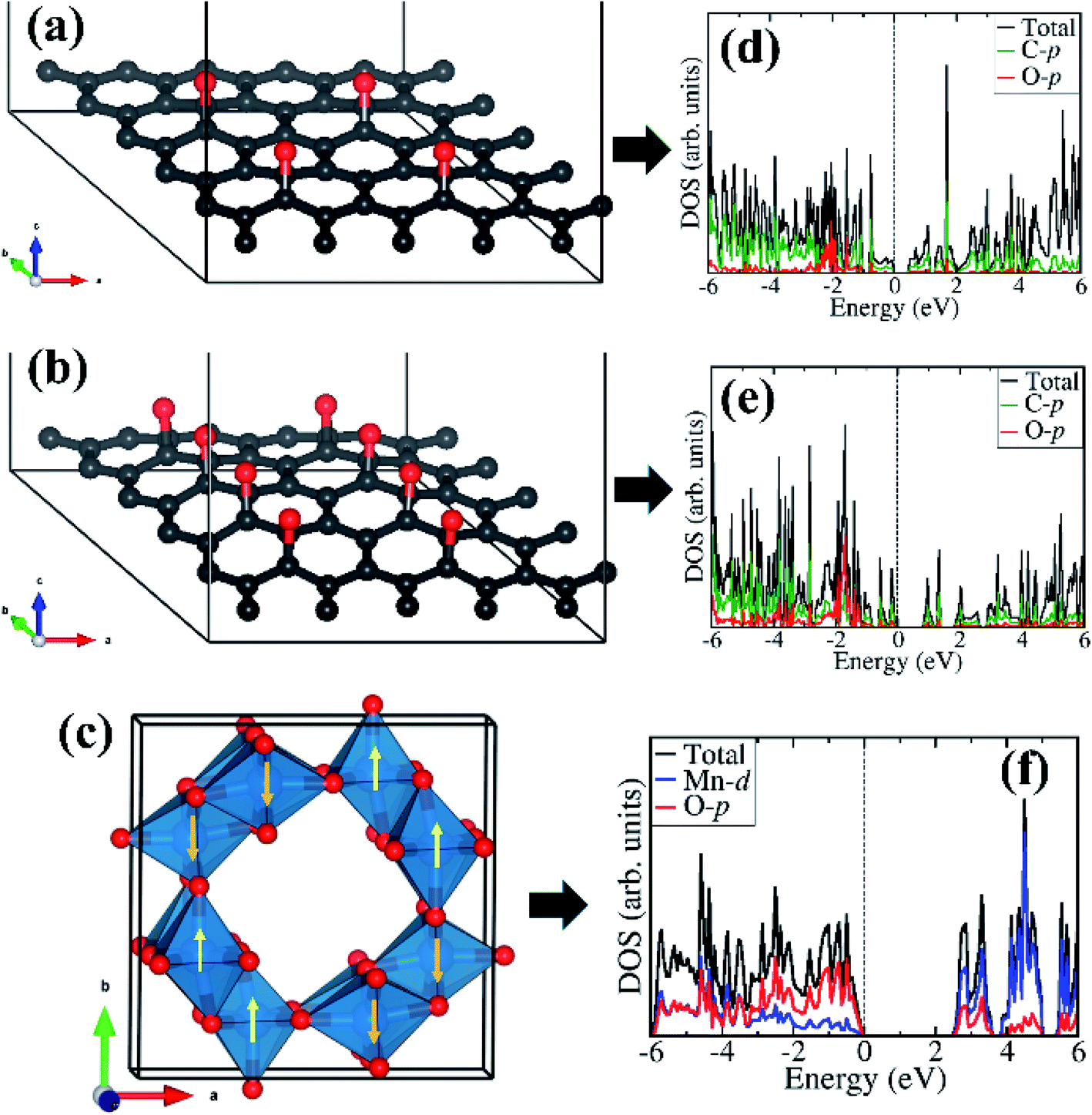 | ||
| Fig. 4 Optimized structure of rGO with epoxide functional group with (a) 8% and (b) 16% oxygen content. The tetragonal structure of α-MnO2 showing the AFM spin ordering in (c). The corresponding partial density of states are shown in the right column (d–f). Atomic color: C = grey, O = oxygen, Mn = blue. | ||
The optimized structures of the rGO/α-MnO2(100) nanocomposite formed by rGO with epoxide functional group with 8% and 16% oxygen contents are shown in Fig. 5a and d, respectively. The rGO is stabilized on the MnO2 surface via C–O and C–Mn chemical bonds through the terminal C atoms. The interactions of the rGO with the MnO2 surface gave rise to electron density redistribution within the rGO/α-MnO2(100) nanocomposite, which was analyzed by determining the three-dimensional-charge density difference iso surface contours as shown in Fig. 5b for the rGO with 8% oxygen content and Fig. 5e for rGO with 16% oxygen content. The yellow and cyan regions represent charge depletion and accumulation in the space, respectively. We observe electron density accumulations mainly in the interfacial bonding regions in the rGO/α-MnO2(100) nanocomposite, suggesting strong interactions between rGO and MnO2(100) surface. Consistent with the strong interaction, we observe strong hybridization between the C-p orbitals of the rGO with the Mn-d and O-p of the MnO2 surface as shown in Fig. 5c and f, resulting in metallic conductivity of the composite systems.
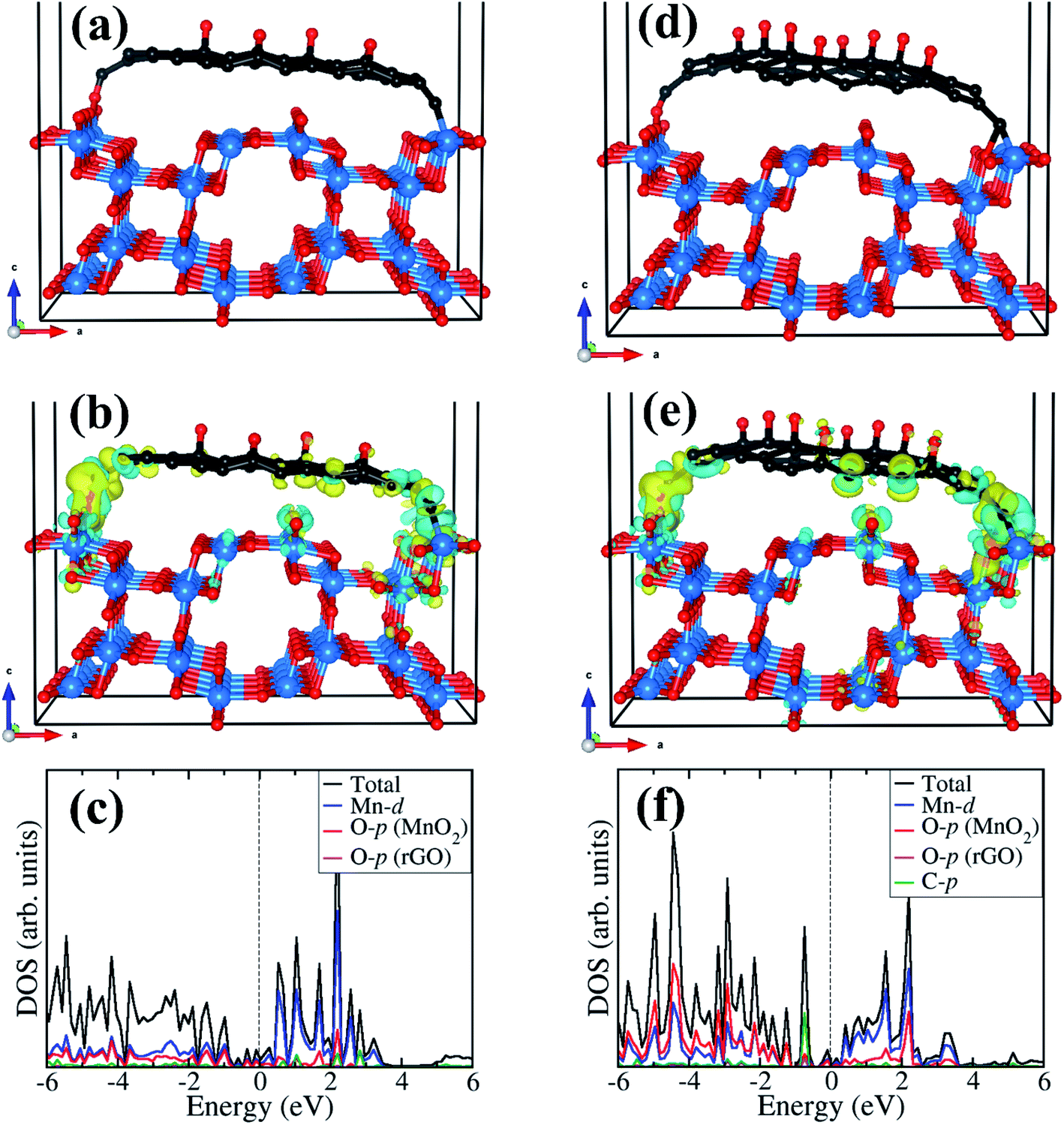 | ||
| Fig. 5 Optimized structures of the rGO/α-MnO2(100) nanocomposite formed by rGO with epoxide functional group (a) 8% and (d) 16% oxygen content. The corresponding differential charge density iso surface contours and partial density of states are shown in (b) and (c) for 8% oxygen content, and in (e) and (f) for 16% oxygen content. The yellow and cyan regions indicate electron density depletion and accumulation by 0.003 e Å−3, respectively. Atomic color: C = grey, O = oxygen, Mn = blue. | ||
The analysis of the work function (Φ) for the rGO monolayer, α-MnO2(100) and the rGO/α-MnO2(100) nanocomposite can help us to understand the origin/direction of charge transfer at the rGO/α-MnO2(100) interface. The Φ for the isolated rGO is predicted at 5.21 and 5.85 eV for the 8% and 16% oxygen contents, respectively (Fig. 6a and b). This is consistent with a previous theoretical investigation which predicted a work function of rGO with epoxy groups to be 4.35 eV for 1.5% oxygen content and 5.6 eV for 20% oxygen content.73 The Φ for the α-MnO2(100) surface is predicted at 7.22 eV (Fig. 6c), also in good agreement with earlier theoretical pH-corrected work function of 7.7 eV for the α-MnO2 (110) surface.72 The work function of the rGO/α-MnO2(100) nanocomposite is predicted at 6.01 and 6.12 eV for the rGO with 8% and 16% oxygen contents (Fig. 6d and e), both of which are lower than that of the isolated α-MnO2(100) surface (Fig. 6c). The reduction in the work function of rGO/α-MnO2(100) nanocomposite relative to the isolated α-MnO2(100) surface can be ascribed to the interfacial bonding, electronic interaction and synergistic effects. Considering that the electron emission capability of a material is dictated by its work function, the observed superior field emission characteristics of the rGO/α-MnO2 nanocomposite compared to the isolated α-MnO2 material can be attributed to the predicted lower work function for the rGO/α-MnO2(100) nanocomposite. Reduction in the work function has been observed in other composite materials compared to the isolated materials.52,53 For instance, Susaki et al., have shown that the deposition of a single unit cell of MgO on an Nb:SrTiO3 substrate reduces the work function by about 0.8 eV.74 Similarly, by decorating SnSe nanosheets with Au nanoparticles (Au/SnSe) and porous ZnO nanosheets with CuSCN nanocoins (CuSCN/ZnO) resulted in significant improvements in the FE characteristics owing to predicted lower functions.52,53 The higher work function predicted for the α-MnO2(100) surface compared to rGO monolayer (Fig. 6) suggests that spontaneous electrons transfer will flow from the rGO monolayer to the α-MnO2(100) after the two are coupled together. Besides the reduction of the work function, the formation of nano-protrusions (denoted by red circles) in the rGO/MnO2 nano-heterostructure (Fig. S5†) can act as effective emission sites. In addition, the better electrical conductivity (5 × 10−3 S cm−1) of rGO is expected to plays an important role in electron transportation. The rGO as backbone may results in easy and high percolation of electrons from the rGO to MnO2 nanorods giving rise to the observed superior field emission behavior of the rGO/MnO2 nano-heterostructure.
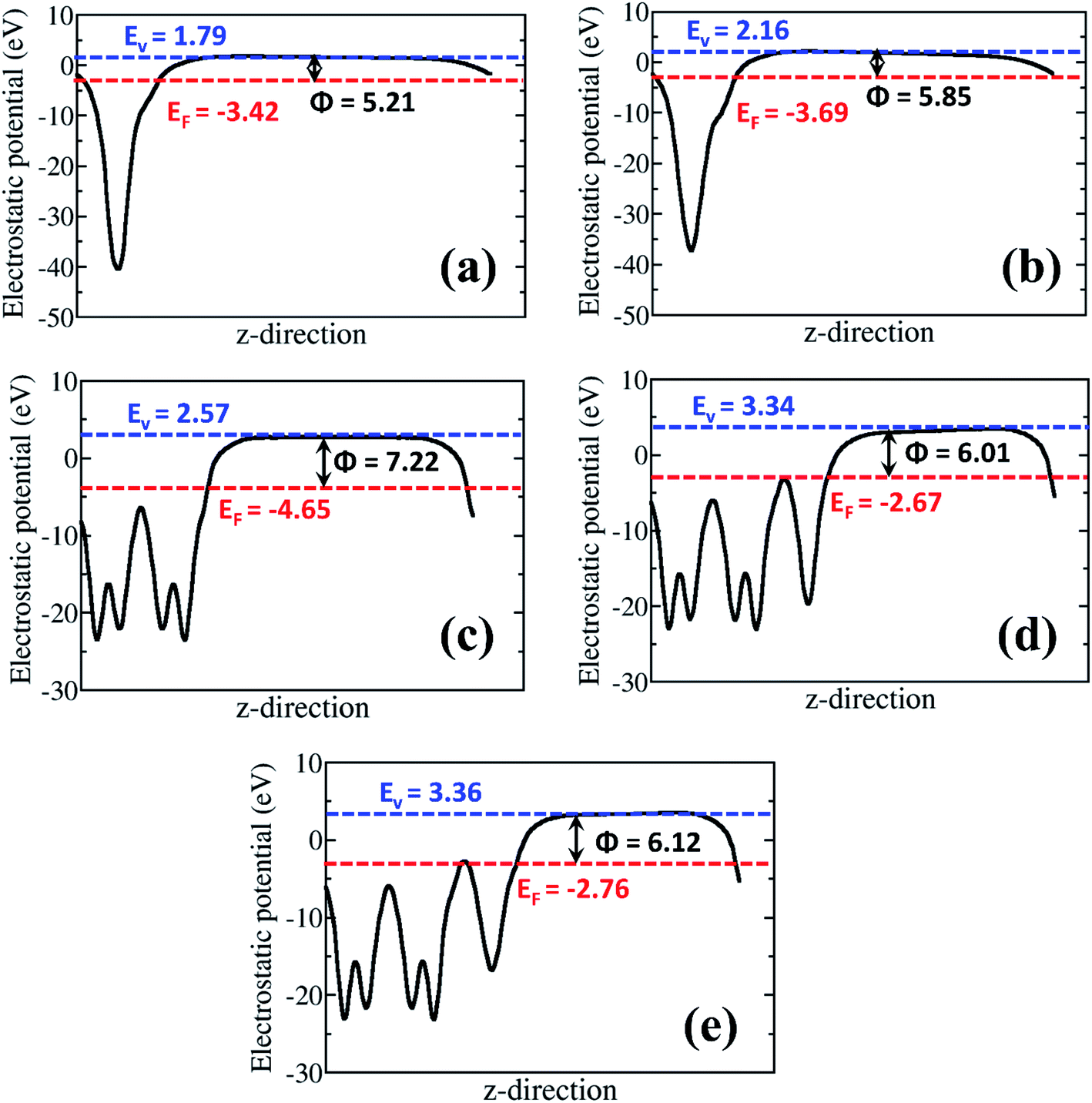 | ||
| Fig. 6 The electrostatic potentials for isolated epoxide-rGO with (a) 8% and (b) 16% oxygen content; (c) isolated α-MnO2(100) surface, and rGO/α-MnO2(100) nanocomposite formed rGO with (d) 8% and (e) 16% oxygen content. The blue and red dashed lines represent the vacuum level (Evac) and the Fermi level (EF), respectively. Φ denotes the work function. | ||
4. Conclusion
In summary, we report the successful synthesis of MnO2 nanorods and rGO/MnO2 nano-heterostructure using cost effective hydrothermal and modified Hummer's methods, respectively. The coupling of rGO sheets with MnO2 nanorods is demonstrated to have a synergistic effect in improving the FE characteristics of formed rGO/MnO2 nano-heterostructure. The dramatic reduction of the turn-on field by 5.7 V μm−1 for an emission current density of 10 μA cm−2 and the achieved high current density of 600 μA cm−2 with an applied field of 3.1 V μm−1 demonstrate the superior FE characteristics of the rGO/MnO2 nano-heterostructure compared to the isolated porous MnO2 nanorods. The results are corroborated by first principles DFT calculations, which predict lower work function for the rGO/MnO2 nano-heterostructure (6.01 and 6.12 eV for the rGO with 8% and 16% oxygen contents, respectively) compared to the isolated MnO2 (7.22 eV) as the primary origin for the improved field emission of the rGO/MnO2 nanocomposite. The controlled nanofabrication of rGO/MnO2 heterostructure reported here provides a promising approach for designing highly efficient MnO2-based next generation FE electron sources and extend their practical applications in micro/nano electronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. R. R. and N. Y. D. acknowledge the UK Engineering and Physical Sciences Research Council (EPSRC) for funding (Grant No. EP/S001395/1). This work has also used the computational facilities of the Advanced Research Computing at Cardiff (ARCCA) Division, Cardiff University, and HPC Wales. This work also made use of the facilities of ARCHER (http://www.archer.ac.uk), the UK's national supercomputing service via the membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202). P. G. C. acknowledges the Consortium for Scientific Research (CSR) Indore, India for financial support (CSR project-CSR-IC-MSRSR-19/CRS-227/2017-2018/1308). Information on the data that underpins the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2020.0111099343.References
- D. J. Milliron, S. M. Hughes, Y. Cui, L. Manna, J. Li, L. W. Wang and A. P. Alivisatos, Nature, 2004, 430, 190–195 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8(10), 3498–3502 CrossRef CAS PubMed.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- G. A. M. Ali, S. A. Makhlouf, M. M. Yusoff and K. F. Chong, Rev. Adv. Mater. Sci., 2015, 41, 35–43 CAS.
- G. A. M. Ali, M. M. Yusoff and K. F. Chong, ARPN J. Eng. Appl. Sci., 2016, 16, 9712–9717 Search PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- J. T. Robinson, M. Zalalutdinov, J. W. Baldwin, E. S. Snow, Z. Wei, P. Sheehan and B. H. Houston, Nano Lett., 2008, 8(10), 3441–3445 CrossRef CAS PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Q. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS PubMed.
- D. R. Dreyer, H. Jia and C. W. Bielawski, Angew. Chem. Int. Ed., 2010, 49, 6813–6816 CAS.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS PubMed.
- W. Gao, N. Singh, L. L. Song, A. L. Reddy, L. Ci, R. Vajtai, Q. Zhang, B. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6, 496–500 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- H. Yamaguchi, K. Murakami, G. Eda, T. Fujita, P. Guan, W. Wang, C. Gong, J. Boisse, S. Miller, M. Acik, K. Cho, Y. Chabal, M. Chen, F. Wakaya, M. Takai and M. Chhowalla, ACS Nano, 2011, 5(6), 4945–4952 CrossRef CAS PubMed.
- R. Kumar, R. Singh, A. Vaz, R. Yadav, C. Rout and S. Moshkalev, New J. Chem., 2017, 41, 8431–8436 RSC.
- R. Roy, A. Jha, D. Sen, D. Banerjee and K. K. Chattopadhyay, J. Mater. Chem. C, 2014, 2, 7608–7613 RSC.
- C. Chen, W. Cai, M. Long, B. Zhou, Y. Wu, D. Wu and Y. Feng, ACS Nano, 2010, 4(11), 6425–6432 CrossRef CAS PubMed.
- C. Zhu, S. Guo, P. Wang, L. Xing, X. Y. Fang, Y. M. Zhai and S. J. Dong, Chem. Commun., 2010, 46, 7148–7150 RSC.
- Z. Rujia, Z. Zhang, L. Jiang, K. Xu, Q. Tian, S. Xue, J. Hu, Y. Bando and D. Golberg, Mater. Chem., 2012, 22, 19196–19201 RSC.
- G. H. Gote, S. R. Bhopale, M. A. More and D. J. Late, Phys. Status Solidi A, 2019, 216, 1900121 CrossRef.
- C. S. Rout, P. D. Joshi, R. V. Kashid, D. S. Joag, M. A. More, A. J. Simbeck, M. Washington, S. K. Nayak and D. J. Late, Sci. Rep., 2013, 3, 3282 CrossRef PubMed.
- M. Wu, J. Lee, Y. Wang and C. Wan, J. Phys. Chem. B, 2004, 108(42), 16331–16333 CrossRef CAS.
- K. Hareesh, S. R. Suryawanshi, A. B. Phatangare, M. A. More, V. N. Bhoraskar and S. D. Dhole, Mater. Lett., 2016, 185, 472–475 CrossRef CAS.
- J. Wu, H. Huang, L. Yu and J. Hu, Adv. Mater. Phys. Chem., 2013, 3, 201–205 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- T. Bordjiba and D. Belanger, Electrochim. Acta, 2010, 55, 3428–3433 CrossRef CAS.
- K. Sun, H. Wang, H. Peng, Y. Wu, G. Ma and Z. Lei, Int. J. Electrochem. Sci., 2015, 10, 2000–2013 CAS.
- S. R. Majid Rusi, Solid State Ionics, 2014, 262, 220–225 CrossRef CAS.
- G. A. M. Alia, M. M. Yusoff, H. Algarnic and K. F. Chonga, Ceram. Int., 2018, 44, 7799–7807 CrossRef.
- S. Ghasemi, S. R. Hosseini and O. Boore-talari, Ultrason. Sonochem., 2018, 40, 675–685 CrossRef CAS PubMed.
- R. Rajagopal and K. Ryu, ChemElectroChem, 2018, 5, 2218 CrossRef CAS.
- S. Ghosal and P. Bhattacharyya, IEEE Trans. Electron. Dev., 2019, 66, 3882 Search PubMed.
- J. Hong, T. T. Mengesha, S. Hong, H. Kim and Y. Hwang, J. Korean Phys. Soc., 2020, 76, 264–272 CrossRef CAS.
- O. Sadak, W. Wang, J. Guan, A. K. Sundramoorthy and S. Gunasekaran, ACS Appl. Nano Mater., 2019, 2, 4386–4394 CrossRef CAS.
- J. G. Radich and P. V. Kamat, ACS Catal., 2012, 2, 807–816 CrossRef CAS.
- B. Q. Zeng, G. Y. Xiong, S. Chen, H. Jo, W. Z. Wang, D. Z. Wang and Z. F. Ren, Appl. Phys. Lett., 2006, 88, 213108 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4(8), 4806–4814 CrossRef CAS PubMed.
- S. Santhanagopalan, A. Balram and D. D. Meng, ACS Nano, 2013, 7(3), 2114–2125 CrossRef CAS PubMed.
- G. P. Patil, P. K. Baviskar, V. S. Bagal, R. D. Ladhe, A. B. Deore, M. A. More, B. R. Sankapal and P. G. Chavan, RSC Adv., 2016, 6, 71958 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and G. J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11189 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- I. Biaggio, R. W. Hellwarth and J. P. Partanen, Phys. Rev. Lett., 1997, 78, 891 CrossRef CAS.
- V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106-1–224106-5 CrossRef PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed.
- D. A. Tompsett, S. C. Parker and M. S. Islam, J. Mater. Chem. A, 2014, 2, 15509–15518 RSC.
- A. Walsh and K. T. Butler, Acc. Chem. Res., 2013, 47, 364–372 CrossRef PubMed.
- M. Ganose, K. T. Butler, A. Walsh and D. O. Scanlon, J. Mater. Chem. A, 2016, 4, 2060–2068 RSC.
- L. Wu, N. Y. Dzade, L. Gao, D. O. Scanlon, Z. Öztürk, N. Hollingsworth, B. M. Weckhuysen, E. J. M. Hensen, N. H. L. Leeuw and J. P. Hofmann, J. Adv. Mater., 2017, 28, 9602–9607 CrossRef PubMed.
- N. Y. Dzade and N. H. Leeuw, Phys. Chem. Chem. Phys., 2017, 19, 27478–27488 RSC.
- P. K. Baviskar, S. R. Rondiya, G. P. Patil, B. R. Sankapal, H. M. Pathan, P. G. Chavan and N. Y. Dzade, ACS Omega, 2020, 5(12), 6715–6724 CrossRef CAS PubMed.
- S. R. Rondiya, C. D. Jadhav, P. G. Chavan and N. Y. Dzade, Sci. Rep., 2020, 10(1), 1–10 CrossRef PubMed.
- B. He, G. Cheng, S. Zhao, X. Zeng, Y. Li, R. Yang, M. Sun and L. Yu, J. Solid State Chem., 2019, 269, 305–311 CrossRef CAS.
- P. K. S. Mural, M. Sharma, A. Shukla, S. Bhadra, B. Padmanabhan, G. Madras and S. Bose, RSC Adv., 2015, 5, 32441–32451 RSC.
- H. Wang, Z. Lu, D. Qian, Y. Li and W. Zhang, Nanotechnology, 2007, 18, 115616 CrossRef.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404–6410 CrossRef CAS PubMed.
- H. Huang and X. Wang, Nanoscale, 2011, 3, 3185–3191 RSC.
- G. P. Patil, V. S. Bagal, C. R. Mahajan, V. R. Chaudhari, S. R. Suryawanshi, M. A. More and P. G. Chavan, Vacuum, 2016, 123, 167–174 CrossRef CAS.
- C. Wu, T. W. Kim and T. Guo F. Li, Sci. Rep., 2016, 6, 38613 CrossRef CAS PubMed.
- S. R. Bansode, R. T. Khare, K. K. Jagtap, M. A. More and P. Koinkar, Mater. Sci. Semicond. Process., 2017, 63, 90–96 CrossRef CAS.
- W. T. Zheng, Y. M. Ho, H. W. Tian, M. Wen, J. L. Qi and Y. A. Li, J. Phys. Chem. C, 2009, 113, 9164 CrossRef CAS.
- R. Zou, G. He, K. Xu, Q. Liu, Z. Zhang and J. Hu, J. Mater. Chem. A, 2013, 1, 8445 RSC.
- J. Ding, X. Yan, J. Li, B. Shen, J. Yang, J. Chen and Q. Xue, ACS Appl. Mater. Interfaces, 2011, 3, 4299 CrossRef CAS PubMed.
- Y. Jin, Y. Zheng, S. G. Podkolzin and W. Lee, J. Mater. Chem. C, 2020, 8, 4885–4894 RSC.
- G. Kruthika, J. Karthikeyan and P. Murugan, Phys. Chem. Chem. Phys., 2017, 19, 3770–3776 RSC.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem. Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- C. S. Johnson, D. W. Dees, M. F. Mansuetto, M. M. Thackeray, D. R. Vissers, D. Argyriou, C. K. Loong and L. Christensen, J. Power Sources, 1997, 68, 570–577 CrossRef CAS.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Phys. Chem. B, 2005, 109, 9651–9655 CrossRef CAS PubMed.
- M. J. Young, A. M. Holder, S. M. George and C. B. Musgrave, Chem. Mater., 2015, 27, 1172–1180 CrossRef CAS.
- P. V. Kumar, M. Bernardi and J. C. Grossman, ACS Nano, 2013, 7, 1638–1645 CrossRef CAS PubMed.
- T. Susaki, A. Makishima and H. Hosono, Phys. Rev., 2011, 83, 115435 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03360j |
| This journal is © The Royal Society of Chemistry 2020 |