
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence enhancement of quinolines by protonation†
Essi Tervola a,
Khai-Nghi Truonga,
Jas S. Warda,
Arri Priimagib and
Kari Rissanen*a
a,
Khai-Nghi Truonga,
Jas S. Warda,
Arri Priimagib and
Kari Rissanen*a
aDepartment of Chemistry, University of Jyvaskyla, P.O. Box 35, Survontie 9B, 40014 Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi
bSmart Photonic Materials, Faculty of Engineering and Natural Sciences, Tampere University, P.O. Box 541, FI-33101 Tampere, Finland
First published on 12th August 2020
Abstract
A study of the fluorescence enhancement of isoquinoline, acridine (benzo[b]quinoline) and benzo[h]quinoline is reported with six organic acids of different pKa values. Protonation was found to be an effective tool in the fluorescence enhancement of quinolines. A significant increase in the fluorescence intensity is observed only when strong acids are used, resulting in an over 50-fold increase in fluorescence with trifluoroacetic or benzenesulfonic acid and isoquinoline in a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The benzenesulfonic acid was found to be the most effective in the protonation of the bases despite its higher pKa value compared to trifluoro- and trichloroacetic acid. The X-ray crystal structures of 14 salts reveal the charge-assisted hydrogen bond O⋯N distances to vary very little, from 2.560(2)–2.714(3) Å, with the exception of the isoquinolinium benzenesulfonate where the O⋯N distance of 2.862(7) Å is caused by additional intermolecular interactions in the solid-state.
:1 ratio. The benzenesulfonic acid was found to be the most effective in the protonation of the bases despite its higher pKa value compared to trifluoro- and trichloroacetic acid. The X-ray crystal structures of 14 salts reveal the charge-assisted hydrogen bond O⋯N distances to vary very little, from 2.560(2)–2.714(3) Å, with the exception of the isoquinolinium benzenesulfonate where the O⋯N distance of 2.862(7) Å is caused by additional intermolecular interactions in the solid-state.
Introduction
Supramolecular chemistry has been defined as “the chemistry of the intermolecular bond, covering the structures and functions of the entities formed by association of two or more chemical species”.1 The field of supramolecular chemistry has gained notable interest in the past decades due to its great potential in many subfields of molecular self-assembly, molecular recognition, and molecular machinery, all gaining advantage from non-covalent bonds.2 Non-covalent interactions cover electrostatic, hydrogen-bonding, halogen-bonding, aromatic donor–acceptor, cation– and anion–π interactions, and solvophobic effects. Understanding the chemistry and intermolecular interactions of molecular entities and their associations provides significant advantages in the synthetic design of new systems, and are essential for understanding the world around us.The importance of luminescence control is apparent not only in biological applications, e.g., as a biosensing tool,3 but also as an emission control in optical applications, such as lighting or organic light-emitting diodes.4 A pH-dependent fluorescence has been reported in various occasions: de Silva et al. reported pH-dependent fluorescence of naphthalenic derivatives for molecular sensing and switching,5,6 Ma et al. have reported pH-dependent fluorescent probes for cancer cell imaging,7 and already decades ago Mataga et al. studied the hydrogen bonding effect on the fluorescence of some N-heterocycles.8
N-Heterocycles, such as quinolines, are known to be weakly fluorescent in comparison to their isoelectronic hydrocarbons. These heterocycles possess non-bonding electrons which give rise to (n–π*) excited states resulting in an increased spin–orbit coupling.9–11 This coupling leads to an enhanced intersystem crossing, effectively decreasing the fluorescence quantum yield. It is also commonly observed that the protonation of the “aromatic” nitrogen results in a loss of emission fine structure and a notable red-shift in the emission wavelength.12 Thus, studies of structurally simple N-heterocycles are vital for understanding further the effects of protonation on the emission of molecules.
The first report on the fluorescence of isoquinoline was recorded in frozen ethanol by Zimmermann and Joop nearly 50 years ago,13 and later Ziegler and El-Sayed reported the phosphorescence of isoquinoline.14 Additionally, the luminescence properties of nitrogen-containing heterocycles have been known to be strongly affected by the choice of solvent.15 The excited-state proton transfer in N-heterocycles, including isoquinoline and benzo[h]quinoline, has been previously studied in acidic media.9,16–18 Benzo[h]quinoline and acridine (benzo[b]quinoline), belonging to a group of polycyclic aromatic N-heterocycles, have been discovered to be particularly interesting since they possess different pKa values in the ground and excited state.19–22 However, there has been no studies that are based on comparing different acids and the effects of the resulting counter-anion. The previous research in this area has been scarce and focused on studying the emission of these compounds with a single base and, to the best of our knowledge, there are no reports of studies using single crystal X-ray diffraction to study the structural effects of protonation on these N-heterocycles.
Herein, we have compiled a comprehensive study on the effects of protonation on the fluorescence of three different quinolines using six different organic acids in solution and the structures of the resulting salts in the solid-state using single crystal X-ray diffraction.
Results and discussion
The protonation of isoquinoline, acridine, and benzo[h]quinoline (see Scheme 1) was studied using absorption and fluorescence spectroscopy in dichloromethane (DCM, 0.5–1.0 × 10−4 M). The compounds were chosen due to their aromatic nature, commercial availability and the single site for protonation. The protonation was studied using six organic acids with different pKa values (see Scheme 2). Dichloromethane as an aprotic solvent was chosen to reduce the effects of solvation. | ||
| Scheme 1 The structures of isoquinoline, acridine, and benzo[h]quinoline used as bases and their pKb values in water.23 | ||
 | ||
| Scheme 2 The structures and pKa values of the acids used for the protonation of the bases. | ||
The full protonation of the base leads to an ion pair (a salt). An example of the protonation is depicted in Scheme 3 and investigated further in the following sections.
 | ||
| Scheme 3 The protonation of isoquinoline with trifluoroacetic acid resulting in the isoquinolinium trifluoroacetate salt. | ||
Spectroscopic studies in solution
:1 acid–base ratio are presented in Fig. 1 (for other acids, see ESI†). Based on the pKa value of 0.52,24 TFA is the strongest acid used in the experiments. The natural absorbance of isoquinoline in DCM is shown in red. Isoquinoline has three slightly overlapping absorption bands at 268 nm, 305 nm, and 318 nm. As the protonation of the isoquinoline increases, the band at 268 nm decreases and experiences a bathochromic shift of 5 nm. During the protonation process, the band at 305 nm remains roughly the same and a new band arises at 328 nm. At 2.0 equivalents of the acid the spectrum shows saturation indicating a complete protonation of the base, as higher amounts of the acid do not induce any more spectral changes. Interestingly enough, based on the fluorescence spectra isoquinoline seems to be only 68% protonated with 1.0 equivalent of TFA.
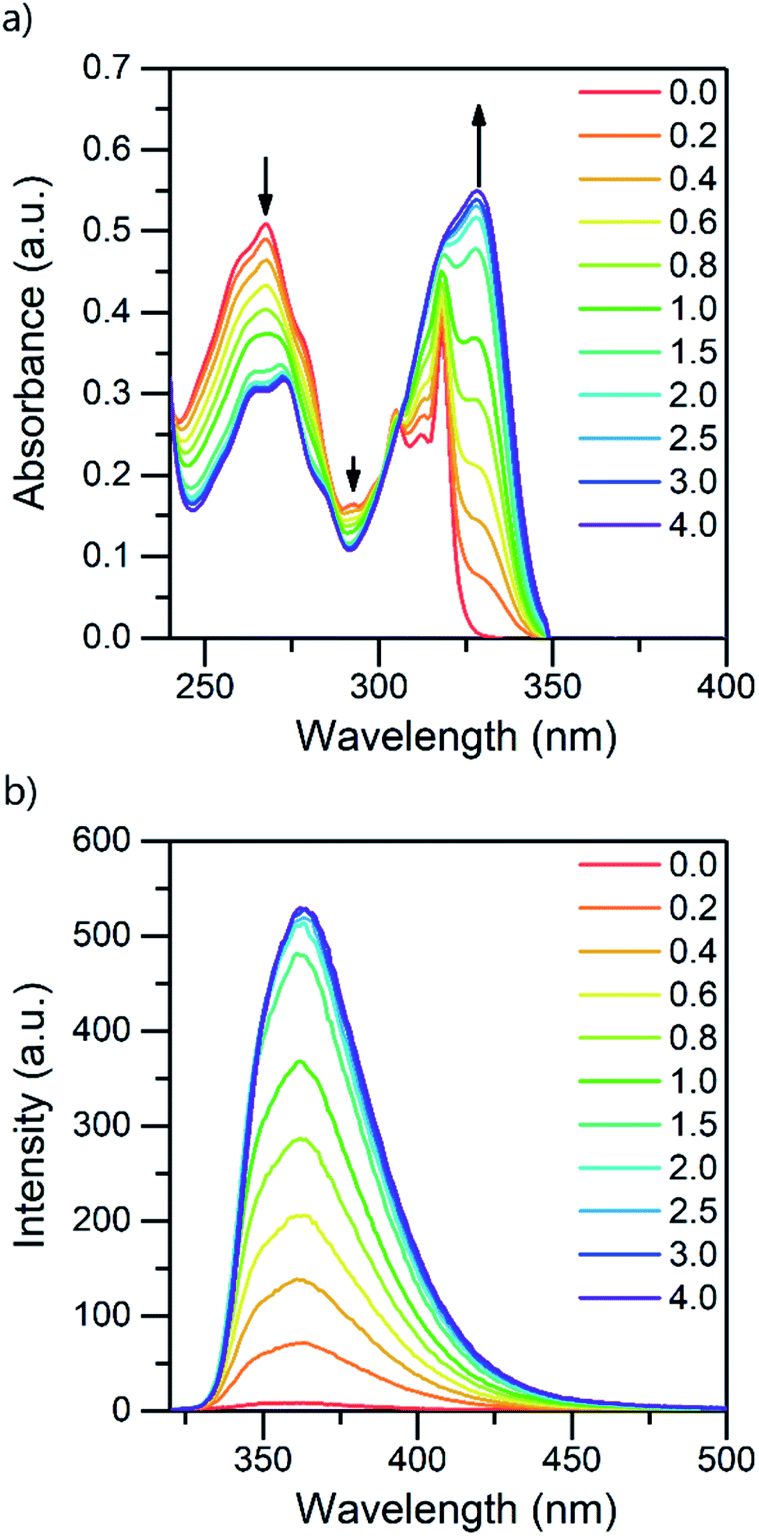 | ||
| Fig. 1 Absorption (a) and fluorescence (b) spectra of isoquinoline while titrating with TFA from 0.0 to 4.0 equivalents of acid in DCM (10−4 M, λex = 310 nm). | ||
The protonation of isoquinoline occurs in the ground state as deduced from the characteristics observed in the absorption spectrum. From the observed changes during the titration, the absorption at 268 nm can be assigned to the neutral species, whereas the band arising at 328 nm is assigned to the protonated species due to the (π,π*) transitions at longer wavelengths. Joshi et al. observed similar structureless broadband absorption in acidic medium at 330 nm for isoquinoline.9 The absorption spectra of acridine and benzo[h]quinoline (see ESI†) show similar spectral changes at longer wavelengths due to protonation, but from these spectra, the absorption bands of the neutral and protonated species are difficult to distinguish. The smaller changes in the spectra could indicate that in those compounds, protonation occurs more easily in the excited state than in the ground state.20
The fluorescence intensity of isoquinoline increases significantly upon titration with acid. Similar to the absorption spectra, protonation induces an increase in the fluorescence intensity until the base is fully protonated. Isoquinoline and acridine (Fig. S4†) show only an increase in the intensity while the structure of the spectra remains the same. However, for benzo[h]quinoline a bathochromic shift of 50 nm of the fluorescence maxima of the neutral and the cationic species is observed. The absorption and fluorescence spectra of all the bases with different acids are presented in the ESI (see Fig. S1–S6).†
The absorption and fluorescence properties of aromatic N-heterocyclic compounds are strongly dependent on the properties of the solvent environment showing stronger fluorescence in polar solvents and weaker fluorescence in nonpolar solvents.25a In dichloromethane, isoquinoline itself is only weakly fluorescent due to its non-bonded electrons giving rise to an (n,π*) excited state.26 The lone-pair of the nitrogen atom in the heterocyclic system interacts with the π-electrons restricting the freedom of the π-electrons and making the system π-deficient, and therefore restricting the fluorescence of the whole system.27 Once the nitrogen gets protonated, the lone pair is shared with the hydrogen atom and the S1 excited state is converted to a (π,π*) excited state resulting in a state more likely to undergo fluorescence which is observed as an increase in the fluorescence intensity. The fluorescence intensity increases until full protonation after which the intensity remains unchanged. Titration with TFA causes an over 50-fold increase in the fluorescence intensity of isoquinoline. Similar titrations were performed with all the acids shown in Scheme 2. Once the pKa value of the acid is higher than 1, the changes in fluorescence decrease significantly to an extent where chloroacetic acid (pKa = 2.87) induces virtually no changes to the fluorescence properties of isoquinoline due to insufficient donating power of the acid for protonation to occur. The fluorescence spectra of all studied quinolines treated with 1.0 equivalent of acid are presented in Fig. 2.
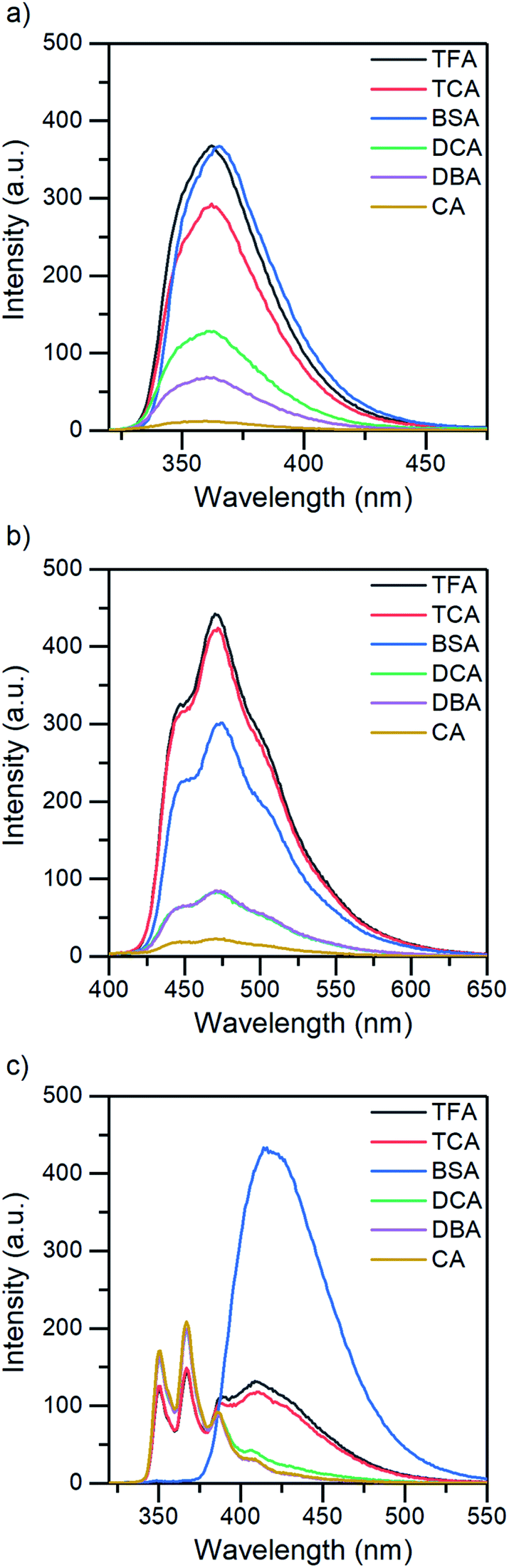 | ||
| Fig. 2 The fluorescence spectra of (a) isoquinoline, (b) acridine, and (c) benzo[h]quinoline treated with 1.0 equivalent of acid in DCM (5 × 10−5 M, λex = 355 nm for acridine; and 10−4 M, λex = 310 nm for isoquinoline and benzo[h]quinoline). | ||
 | ||
| Fig. 3 (a) The integrated relative intensity change, and (b) the changes in the fluorescence quantum yield of isoquinoline while titrating with various acids (DCM, 10−4 M, λex = 310 nm, 0.0 to 4.0 acid equivalents). | ||
The fluorescence quantum yield (QY) was studied using eqn (1)25b and 9,10-diphenylanthracene as reference (ΦFL = 0.90, cyclohexane28).
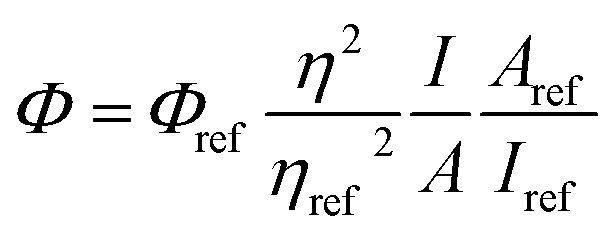 | (1) |
Quantum yields for the quinolines in their native state were determined to be less than 1% for isoquinoline and acridine and 15% for benzo[h]quinoline. Again, by excluding BSA from the analysis, the QY evolution follows the trend of the pKa values. The greatest increase in QY can be observed for isoquinoline, changing from virtually zero to up to 27% (see Fig. 3b). The maximum fluorescence quantum yields obtained for all the compounds with each acid are listed in Table 1.
| Isoquinoline | Acridine | Benzo[h]quinoline | |
|---|---|---|---|
| TFA | 0.241 | 0.089 | 0.157 |
| TCA | 0.201 | 0.082 | 0.156 |
| BSA | 0.274 | 0.101 | 0.148 |
| DCA | 0.102 | 0.035 | 0.157 |
| DBA | 0.057 | 0.031 | 0.156 |
| CA | 0.010 | 0.007 | 0.158 |
| Free base | 0.004 | 0.008 | 0.147 |
The change from the (n,π*) to (π,π*) excited state increases the fluorescence quantum yield. This is due to the (n,π*) states having longer radiative lifetimes than the (π,π*) states, and shorter lifetimes are more likely to undergo fluorescence before intersystem crossing.26 However, the fluorescence quantum yield is defined as ΦFL = 1 − ΦISC − ΦIC, where ΦISC is the phosphorescence quantum yield and ΦIC is the quantum yield of internal conversion.
Isoquinoline experiences significant changes in its absorption spectra upon protonation. Distinctive absorption bands can be assigned to the neutral and cationic species differentiating isoquinoline from the other two N-heterocycles. Similar changes can be observed for acridine as well, merely to a smaller extent. Acridine has a different electronic structure leading to very narrow absorption bands and rendering the observation of the change and isosbestic points more challenging. As found for isoquinoline and acridine, the absorption of the cationic species can also be observed for benzo[h]quinoline at higher wavelengths, but contrary to isoquinoline and acridine, the absorption increases throughout the spectrum and no isosbestic points are observed.
In addition to that, benzo[h]quinoline also differs in terms of fluorescence: isoquinoline and acridine only experience an increase in their fluorescence intensity until eventually reaching a saturation point, whereas the spectra of benzo[h]quinoline changes significantly. Neutral benzo[h]quinoline has a fluorescence maximum at 367 nm, whereas after protonation, the cationic species arises at 416 nm, shifting the fluorescence maximum 50 nm and changing the spectra from three distinctive bands to one broadband fluorescence. These drastic changes affect the overall fluorescence intensity change making it minuscule in comparison to the isoquinoline and acridine 50-fold and 25-fold increases, respectively.
All three compounds also share similarities in their fluorescence intensity. The intensity changes vary between the compounds, but in each case, BSA has the largest effects, while the acetic acids follow the trend of their respective pKa values. Considering the chemical structure of benzo[h]quinoline, it is apparent that it is more prone to steric hindrance than isoquinoline and acridine due to the hydrogen at C10 (see ESI, Fig. S35†) which causes a severe clash with the hydrogen at the nitrogen atom during the protonation.
Due to the unsuitability of the NMR spectroscopy (see ESI, Fig. S11–S14†) for binding constant determination, the absorption and fluorescence spectra of the bases were used instead. Due to the difficulty in determining pH values in DCM, the binding constants were determined only for isoquinoline, for which the absorbance of the neutral and cationic species could be easily determined from the absorption spectrum. For isoquinoline, the absorption band at 268 nm signifies the absorbance of the neutral species, which decreases upon addition of the acids, whereas the band at 328 nm is contributed by the cationic species (see Fig. 4). These changes can be used to determine the ratio of c(bound) and c(unbound). Binding constants for the ground state are determined using eqn (2)29
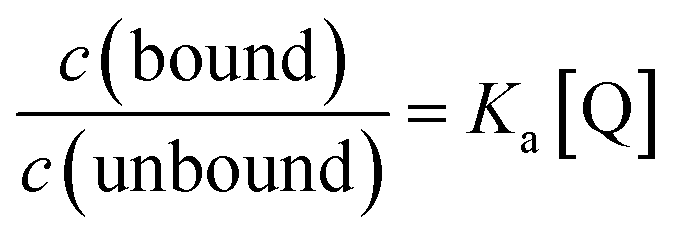 | (2) |
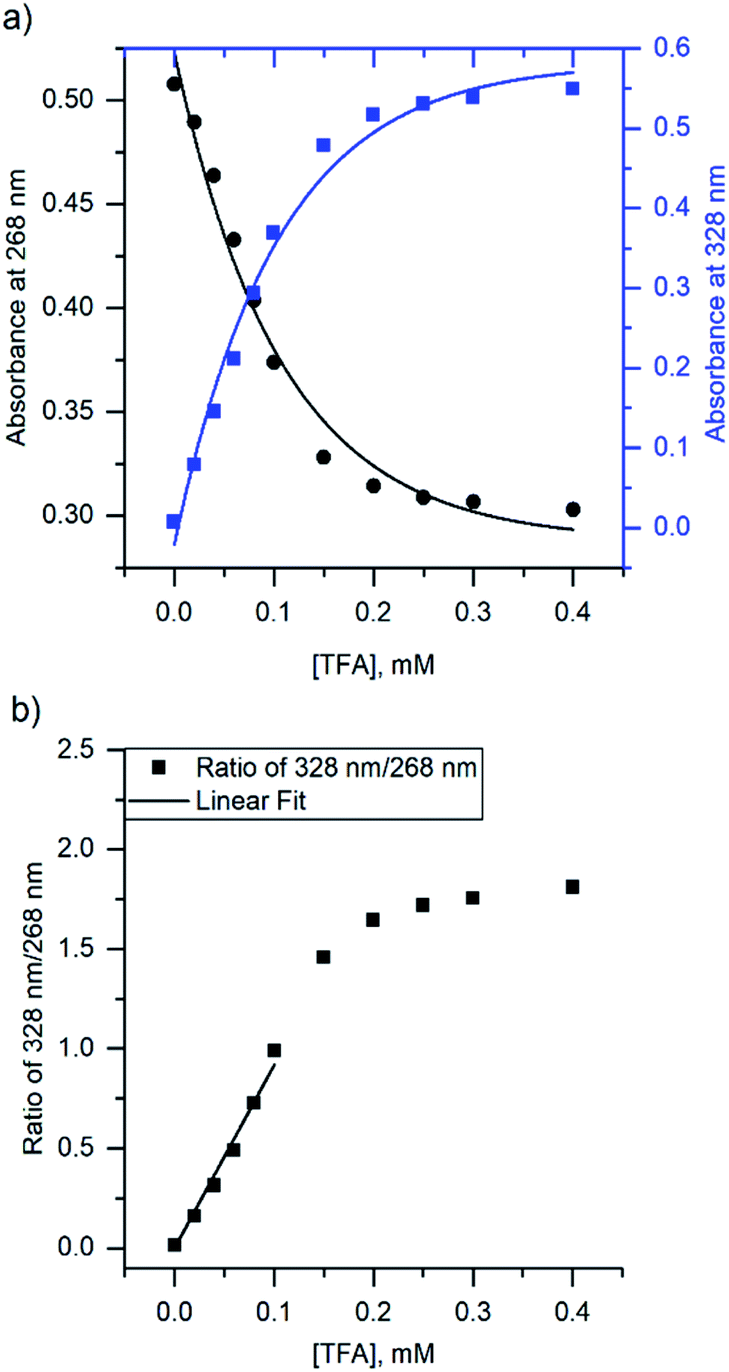 | ||
| Fig. 4 (a) The change in absorbance of isoquinoline at 268 nm and 328 nm upon addition of TFA, and (b) ratio of the intensities at 268 nm and 328 nm with linear fit. | ||
| Ka (M−1) | |
|---|---|
| TFA | 9200 ± 330 |
| TCA | 8300 ± 190 |
| BSA | 32700 ± 1590 |
| DCA | 4000 ± 100 |
| DBA | 3100 ± 120 |
| CA | N/A |
The binding constant for the protonation of isoquinoline with CA was not determined simply because the changes in the spectra are too small and within experimental error. The determined binding constants (see ESI, S9 and S10†) obey the trend of the pKa values of the acids yet again excluding BSA, which has a significantly higher binding constant for isoquinoline than the other acids. Although the binding constants for acridine and benzo[h]quinoline were not determined, similar results are expected based on the fluorescence spectra.
Single crystal X-ray crystallography
The crystallizations of the acid–base pairs were carried out with 1:1 acid–base ratio in chloroform and the single crystals of the corresponding salts were obtained either by slow evaporation of the chloroform solution or by vapor diffusion using chloroform/hexane or dichloromethane/diisopropyl ether as the solvent/antisolvent system. Unfortunately, not all of the crystallizations were successful and some of the salts from weaker acids were obtained as a gel or amorphous powder. The suitable single crystals were subjected to single crystal X-ray study. To simplify the crystallographic nomenclature, the salts are referred to as numbers, e.g. isoquinolinium trifluoroacetate is denoted as 1. The numbering of all the salts is shown in Table 3.
| TFA | TCA | BSA | DCA | DBA | CA | |
|---|---|---|---|---|---|---|
| Isoquinoline | 1 | 2 | 3 | — | — | — |
| Acridine | 4 | 5 | 6 | 7 | 8 | 9 |
| Benzo[h]quinoline | 10 | 11 | 12 | 13 | 14 | — |
The salt formation, viz. full protonation of isoquinoline, acridine, and benzo[h]quinoline, was observed in the solid-state in all cases, regardless of the pKa values of the used acid. All solid-state structures, except 7, 8, 13 and 14, have the expected 1:1 stoichiometry (see Fig. 5 and 6). The deprotonated acid dimer (R–COOH⋯OOC–R) found in 7, 8, 13 and 14 is H-bonded to the H–N+ moiety from the deprotonated acid, unlike in the other studied structures, and has formally a 2:1 stoichiometry (see Fig. 7).
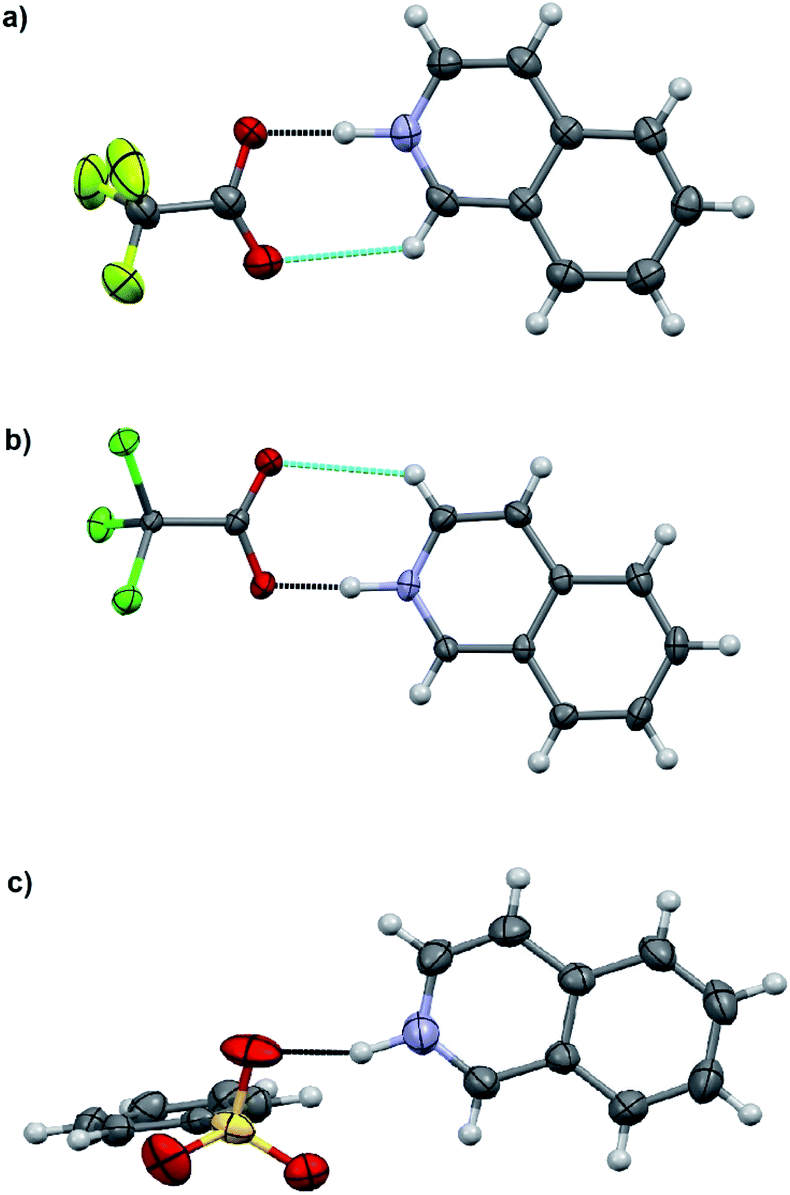 | ||
| Fig. 5 X-ray structures of (a) 1, (b) 2, and (c) 3. Displacement ellipsoids are drawn at the 50% probability level. The black-dotted lines are the N–H⋯O hydrogen bonds and the blue-dotted lines represent weak C–H⋯O interactions. | ||
 | ||
| Fig. 6 X-ray structures of (a) 4, (b) 5, (c) 6, and (d) 9. Displacement ellipsoids are drawn at the 50% probability level. The black-dotted lines are the N–H⋯O hydrogen bonds and the blue-dotted lines represent C–H⋯O interactions. | ||
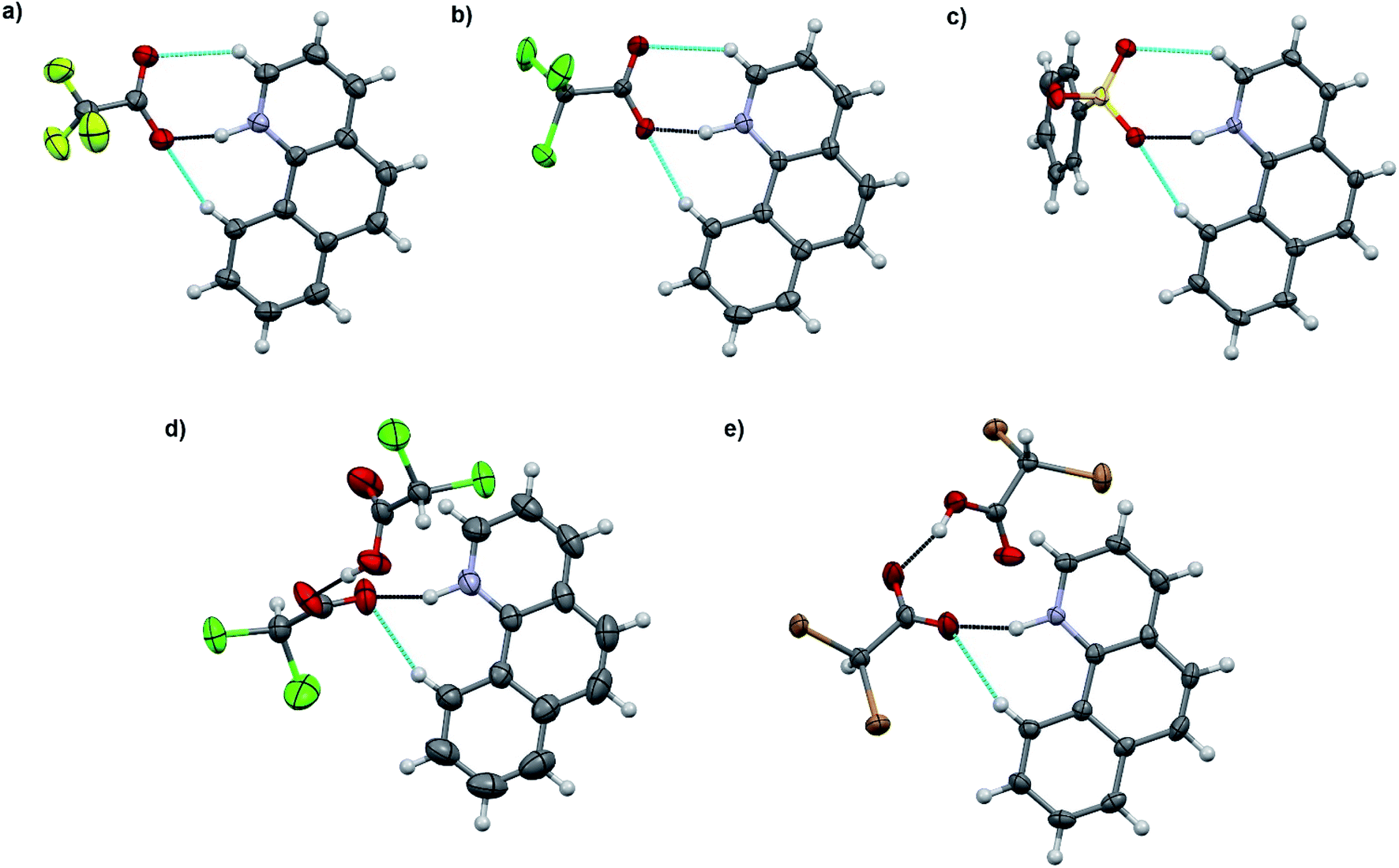 | ||
| Fig. 7 X-ray structures of (a) 10, (b) 11, (c) 12, (d) 13, and (e) 14. Displacement ellipsoids are drawn at the 50% probability level. The black-dotted lines are the N–H⋯O hydrogen bonds and the blue-dotted lines represent weak C–H⋯O interactions. | ||
The charge-assisted hydrogen bond distances between the corresponding acetate or sulfonate oxygen and the nitrogen of the protonated base (O⋯N) are listed in Table 4. Due to the unreliable positioning of the hydrogen atom from X-ray data, only the distances between the heavier atoms (O and N) are discussed. However during the refinements, a realistic H–N bond distance of 1.04 Å, based on an average of neutron diffraction values,30 was used.
| Isoquinoline | Acridine | Benzo[h]quinoline | |
|---|---|---|---|
| a Two independent 1:1 acid–base pairs in the asymmetric unit.b Acridine moiety is disordered over a symmetry element resulting in unreliable O⋯N distances. |
|||
| TFA | 1: 2.645(4) | 4: 2.636(2) | 10: 2.644(3) |
| TCA | 2: 2.617(4) | 5: 2.617(3) | 11: 2.633(4)a, 11: 2.634(4)a |
| BSA | 3: 2.862(7) | 6: 2.666(3) | 12: 2.714(3) |
| DCA | Gel | 7: 3.031(4)b | 13: 2.657(3) |
| DBA | Gel | 8: 3.102(6)b | 14: 2.709(6) |
| CA | Gel | 9: 2.560(2) | Powder |
The charge-assisted H-bond distances for TFA and TCA (1, 2, 4, 5, 10, and 11) salts are very close to each other (Δδ = 0.02–0.05 Å). From the solution studies (see above), it was concluded that the protonation of isoquinoline by BSA was the most effective as it had the greatest effect on the fluorescence intensity and induced the largest chemical shift change in the 1H NMR spectra (see ESI,† NMR studies). Yet in the solid-state, the O⋯N distance for 3 is found to be the longest, 2.862(7) Å, being 0.245–0.148 Å longer than for 1, 2, 6 or 12. The plausible explanation for the longer O⋯N distance in the BSA–isoquinoline salt are the slightly acidic H-atoms of the phenyl ring of the benzenesulfonate anion, resulting in a multitude of additional H-bonds to the adjacent anions, and thus elongating the charge-assisted N–H⋯O–S hydrogen bond. The same phenomenon does not occur in the corresponding acridine (6) and benzo[h]quinoline (12). This is evidenced by the Hirshfeld surface analysis of 3, 6 and 12 (see ESI, Fig. S45†) which clearly indicates a multitude of H⋯O interactions to adjacent benzenesulfonate anions only in the case of 3. The DFT-level (SPARTAN18,31 M062X, def2-TZVP, nonpolar solvent model) calculations for 3, 6 and 12 also support this reasoning, as all optimized structures give the nonpolar solvent phase O⋯N distances between 2.6–2.7 Å (see ESI, Fig. S46†).
The acridinium cation in the DCA and DBA salts (7 and 8, see Fig. S27–S32,† respectively) reside on a symmetry element resulting in disorder of the +N–H and C–H parts of the acridine skeleton. This is manifested by the abnormally long O⋯N distances, as they actually represent the average of the 2.7 Å (+N–H⋯O−) and 3.5 Å (C(arom)–H⋯O) distances, thus these XRD based distances for 7 and 8 are excluded from the discussion. The same DFT-level (SPARTAN18,31 M062X, def2-TZVP, nonpolar solvent model) calculations for 7 and 8 confirm the normal solution phase O⋯N distances to be 2.6 Å (see ESI, Fig. S47†). In contrast to the 1:1 salts, the structures of 7 and 8 are formed in a 2:1 ratio in the solid-state, consisting of one (disordered) acridinium cation and a H-bonded DCA or DBA dimer (see ESI, Fig. S27 and S28†). Similar acid dimers in a 2:1 acid–base ratio were observed for benzo[h]quinoline with DCA (13) and DBA (14), respectively. Yet, due to the non-disordered structures of 13 and 14, the O⋯N distances are very similar to the 1:1 salts (see Table 4). Surprisingly, the shortest O⋯N interaction distance is observed in 9 with 2.560(2) Å which is even 0.03 Å shorter than in the reported quinolinium trifluoroacetate structure,32 and very close to the DFT-level (SPARTAN18,31 M062X, def2-TZVP, nonpolar solvent model) calculated O⋯N distance of 2.61 Å for 9 (see ESI, Fig. S47†).
A search in the Cambridge Structural Database (CSD)33 for protonated isoquinoline, acridine and benzo[h]quinoline resulted in 22 hits, respectively. The average charge-assisted hydrogen bond O⋯N distance calculated from the CSD structures34–47 is 2.658 Å, precisely matching the average of 2.644 Å calculated from 1, 2, 4, 5, 6, 9, 10, 11, 12, 13 and 14 (excluding the abnormally long O⋯N distances in 3, 7 and 8).
Conclusions
The protonation of simple N-heterocycles was successfully used as a preliminary study to gain further information on the use of acid–base interactions for supramolecular emission control. The protonation was found to significantly boost fluorescence emission, hence being an effective tool to be used in fluorescence enhancement and avoiding time-consuming synthetic procedures. In the solution studies, the protonation was observed to increase the fluorescence intensity with an over 50-fold increase observed for isoquinoline. Surprisingly, the protonation was found to be incomplete in a 1:1 acid–base ratio, despite the single protonation site of the bases. No structural changes were observed in the fluorescence spectra of isoquinoline or acridine, but interestingly, benzo[h]quinoline experiences a bathochromic shift of 50 nm upon protonation. Benzo[h]quinoline and acridine were also observed to be more easily protonated in the excited state than in the ground state, whereas the isoquinoline absorption spectra display effective protonation in the ground state.
The relative fluorescence intensities of the N-heterocycles become larger as the donating powers of the proton donors increase. The increase in the fluorescence intensity followed the same trend with the pKa values of the acids with the exception of BSA, which was the most effective in protonating all the bases. Acids with pKa values higher than 1 did not have significant effects on the fluorescence of the compounds due to insufficient donating power of the acid for protonation to occur. Binding constants for isoquinoline in the ground state were determined based on the absorption spectra and similar results were observed in these values as well, with BSA having a three times larger binding constant compared to the other, stronger acids (TFA and TCA).
In the solid-state, the protonation of isoquinoline, acridine and benzo[h]quinoline was observed in each case regardless of the pKa value of the acid. In solution, BSA was determined to be the most effective in the protonation of the bases, yet, in single-crystal X-ray structures, the O⋯N distance was found to be the longest in the BSA–isoquinoline salt due to additional intermolecular interactions in the solid-state. The solid-state studies suggested that the O⋯N distances were mainly affected by the lattice interactions and do not directly reflect the acid–base or the anion–cation interactions in solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Academy of Finland (KR: grant no. 317259; AP: grant no. 311142) and the University of Jyväskylä, Finland.Notes and references
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- J. Steed and J. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd., Chippenham, United Kingdom, 2009 Search PubMed.
- S. Vigneshvar, C. C. Sudhakumari, B. Senthilkumaran and H. Prakash, Front. Bioeng. Biotechnol., 2016, 4, 11 CAS.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-K. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- A. P. de Silva, A. Goligher, N. Gunaratne and T. E. Rice, ARKIVOC, 2003, 7, 229–243 Search PubMed.
- S. Zheng, P. L. M. Lynch, T. E. Rice, T. S. Moody, H. Q. N. Gunaratne and A. P. de Silva, Photochem. Photobiol. Sci., 2012, 11, 1675–1681 RSC.
- J. Ma, W. Li, J. Li, R. Shi, G. Yin and R. Wang, Talanta, 2018, 182, 464–469 CrossRef CAS PubMed.
- N. Mataga and S. Tsuno, Bull. Chem. Soc. Jpn., 1957, 30, 368–374 CrossRef CAS.
- N. K. Joshi, H. C. Joshi, R. Gahlaut, N. Tewarj, R. Rautela and S. Pant, J. Phys. Chem. A, 2012, 116, 7272–7278 CrossRef CAS PubMed.
- N. Mataga, Bull. Chem. Soc. Jpn., 1958, 31, 459–462 CrossRef CAS.
- M. F. Anton and W. R. Moomaw, J. Chem. Phys., 1977, 66, 1808–1818 CrossRef CAS.
- S. A. Tucker, W. E. Acree Jr and C. Upton, Polycyclic Aromat. Compd., 1993, 3, 221–229 CrossRef CAS.
- H. Zimmermann and N. Joop, Z. Elektrochem., 1961, 65, 61–66 CAS.
- S. M. Ziegler and M. A. El-Sayed, J. Chem. Phys., 1970, 52, 3257–3268 CrossRef CAS.
- I. Janic and A. Kawski, Adv. Mol. Relax. Processes, 1973, 5, 185–191 CrossRef CAS.
- K. Kumari, N. Tewari, M. S. Mehta, N. Pandey, K. Tiwari, R. K. Ratnesh, H. C. Joshi and S. Pant, J. Mol. Struct., 2019, 1180, 855–860 CrossRef CAS.
- M. Norek, J. Dresner and J. Prochorow, Acta Phys. Pol., A, 2003, 104, 425–439 CrossRef CAS.
- M. Norek, B. Kozankiewicz and J. Prochorow, Acta Phys. Pol., A, 2004, 106, 77–94 CrossRef CAS.
- E. T. Ryan, T. Xiang, K. P. Johnston and M. A. Fox, J. Phys. Chem. A, 1997, 101, 1827–1835 CrossRef CAS.
- M. Nakamizo, Spectrochim. Acta, 1966, 22, 2039–2053 CrossRef CAS.
- A. Grabowska, B. Pakula and J. Pancir, Photochem. Photobiol., 1969, 10, 415–425 CrossRef CAS PubMed.
- E. Vander Donckt, R. Dramaix, J. Nasielski and C. Vogels, Trans. Faraday Soc., 1969, 65, 3258–3262 RSC.
- H. C. Brown, et al., in Determination of Organic Structures by Physical Methods, ed. E. A. Braude, and F. C. Nachod, Academic Press, New York, 1955 Search PubMed.
- Acid Dissociation Constants, CRC Handbook of Chemistry and Physics, retrieved on 20.12.2018 Search PubMed.
- (a) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH, New York, 2001, p. 59 CrossRef; (b) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH, New York, 2001, p. 161 CrossRef.
- B. Wardle, Principles and Applications of Photochemistry, John Wiley & Sons, Ltd., United Kingdom, 2009 Search PubMed.
- R. T. Williams and J. W. Bridges, J. Clin. Pathol., 1964, 17, 371–394 CrossRef CAS PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CAS.
- G. Grynkiewicz, M. Poenie and R. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CAS.
- F. H. Allen and I. J. Bruno, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 380–386 CrossRef CAS PubMed.
- SPARTAN18, Wavefunction Inc., Irvine, CA, USA, 2018 Search PubMed.
- Effendy, P. C. Junk, C. J. Kepert, L. M. Louis, T. C. Morien, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2006, 632, 1312–1325 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- X.-H. Chang, Z. Kristallogr. – New Cryst. Struct., 2019, 234(6), 1253–1254 CAS.
- N. Kobayashi, T. Naito and T. Inabe, Bull. Chem. Soc. Jpn., 2003, 76, 1351–1362 CrossRef CAS.
- H. Eshtiagh-Hosseini, A. Hassanpoor, M. Mirzaei and A. R. Salimi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2996 CrossRef CAS PubMed.
- P. Bora, B. Saikia and B. Sarma, Cryst. Growth Des., 2018, 18(3), 1448–1458 CrossRef CAS.
- L. Plasseraud and H. Cattey, C. R. Chim., 2013, 16, 613–620 CrossRef CAS.
- H. Aghabozorg, J. A. Gharamaleki, M. Parvizi and Z. Derikvand, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m83–m84 CrossRef CAS PubMed.
- N. Bedekovic, V. Stilinovi and T. Piteša, Cryst. Growth Des., 2017, 17, 5732–5743 CrossRef CAS.
- X. Mei and C. Wolf, Eur. J. Org. Chem., 2004, 4340–4347 CrossRef CAS.
- Z. Derikvand, M. M. Olmstead and J. A. Gharamaleki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o416 CrossRef CAS PubMed.
- K. Fennig and A. Sikorski, Z. Kristallogr. – New Cryst. Struct., 2018, 233(4), 675–676 CAS.
- P. P. Devi and D. Kalaivani, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 570–574 CrossRef PubMed.
- D. E. Lynch, A. N. Kirkham, M. Z. J. Chowdhury, E. S. Wane and J. Heptinstall, Dyes Pigm., 2012, 94, 393–402 CrossRef CAS.
- A. Garai, S. Mukherjee, S. K. Ray and K. Biradha, Cryst. Growth Des., 2018, 18, 581–586 CrossRef CAS.
- E.-Y. Xia, J. Sun, R. Yao and C.-G. Yan, Tetrahedron, 2010, 66, 3569–3574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2000976–2000989. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04691d |
| This journal is © The Royal Society of Chemistry 2020 |