
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide quantum dots immobilized on mesoporous silica: preparation, characterization and electroanalytical application†
Albina Mikhraliieva a,
Vladimir Zaitsev*ab,
Oleg Tkachenkocd,
Michael Nazarkovskya,
Yutao Xinge and
Edilson V. Benvenuttic
a,
Vladimir Zaitsev*ab,
Oleg Tkachenkocd,
Michael Nazarkovskya,
Yutao Xinge and
Edilson V. Benvenuttic
aDepartment of Chemistry, Pontifical Catholic University of Rio de Janeiro, Marquês de São Vicente, 225, 22451-900, Rio de Janeiro, Brazil. E-mail: vnzaitsev@puc-rio.br
bNational University of Kyiv-Mohyla Academy, 2 Skovorody vul., Kyiv, 04070, Ukraine. E-mail: zaitsev@univ.kiev.ua
cMaterials Chemistry Department, V. N. Karazin Kharkiv National University, 4 Svoboda Square, Kharkiv, 61022, Ukraine
dInstitute of Chemistry, UFRGS, PO Box 15003, CEP, Porto Alegre, RS 91501-970, Brazil
eLaboratório de Microscopia Eletrônica de Alta Resolução, Centro de Caracterização Avançada para Indústria de Petróleo (LaMAR/CAIPE), Universidade Federal Fluminense, 24210-346, Niterói, RJ, Brazil
First published on 24th August 2020
Abstract
Because of its high surface area and combination of various functional groups, graphene oxide (GO) is currently one of the most actively studied materials for electroanalytical applications. It is not practical to utilize self-supported GO on its own and thus it is commonly integrated with different supporting carriers. Having a large lateral size, GO can only wrap the particles of the support and thus can significantly reduce the surface area of porous materials. To achieve synergy from the high surface area and polyfunctional nature of GO, and the rigid structure of a porous support, the lateral size of GO must essentially be decreased. Recently reported graphene oxide quantum dots (GOQDs) can fulfil this task. Here we report the successful preparation of an SiO2-GOQDs hybrid, where GOQDs have been incorporated into the mesoporous network of silica. The SiO2-GOQDs emit a strong luminescence with a band maximum at 404 nm. The Raman spectrum of SiO2-GOQDs shows two distinct peaks at 1585 cm−1 (G-peak) and 1372 cm−1 (D-peak), indicating the presence of a graphene ordered basal plane with aromatic sp2-domains and a disordered oxygen-containing structure. Covalent immobilization of GOQDs onto aminosilica via such randomly structured oxygen fragments was proven with the help of Fourier transform infrared spectroscopy, solid-state cross-polarization magic angle spinning 13C nuclear magnetic resonance, and X-ray photoelectron spectroscopy. SiO2-GOQDs were used as a modifier of a carbon paste electrode for differential pulse voltammetry determination of two antibiotics (sulfamethoxazole and trimethoprim) and two endocrine disruptors (diethylstilbestrol (DES) and estriol (EST)). The modified electrodes demonstrated a significant signal enhancement for EST (370%) and DES (760%), which was explained by a π–π stacking interaction between GOQDs and the aromatic system of the analytes.
1. Introduction
Graphene oxide (GO) is one of the most commonly used carbon modifiers for the preparation of various hybrid materials.1,2 Graphene oxide belongs to the class of 2D-nano objects (nanosheets) of up to several nanometres thickness with a lateral size >104 nm. A macromolecule of GO has a huge surface area (up to 2630 m2 g−1)3 and contains a basal plane of graphene with many oxygen-containing defects. Thus, it can interact with various molecules via non-covalent bonding, including electrostatic, hydrogen and dative bonds, π–π stacking and dispersion forces.4,5 Because of its high surface area and polyfunctional nature, GO has great potential for use in the preconcentration of organic compounds containing aromatic rings and electronegative functional groups.6–9It is not practical to utilize self-supported 2D materials on their own. Due to strong π–π stacking and a hydrophobic interaction between the graphene layers, GO nanosheets can easily agglomerate and thus drastically reduce their surface area. Therefore, GO is commonly integrated with different supporting carriers, such as mesoporous silica gels,10,11 magnetic nanoparticles,12 carbon nanotubes, and inorganic oxides, such as TiO2, Fe2O3, or MgO.13 Since the lateral size of GO particles is roughly 10–100 μm, which is 2–20 times bigger than common silica particles, GO can only wrap silica particles.10,14 If we take into account that at least 90% of the surface area of mesoporous silicas is found in the pores, it becomes obvious that immobilization of a flat non-porous nanosheet onto a porous support surface, instead of increasing the overall surface area of the hybrid material, can significantly reduce it due to pore blocking. As a result, the adsorption capacity of SiO2@GO hybrid materials towards analytes can be much lower than that of individual GO or even SiO2. For example, SiO2@GO demonstrates ten times lower total adsorption capacity for Cu and Pb than a silica-based adsorbent.14 Unfortunately, in many recent publications, this effect was ignored and, hence, hybrid SiO2@GO materials have not shown their full potential.1,15–17
To achieve synergy between the high surface area and polyfunctional nature of GO on the one hand, and the rigid structure of a porous support, on the other hand, the lateral size of GO must be considerably decreased. A lot of progress has been achieved in this direction, in order to tune the properties of GO by downsizing its particles to the several nanometre scale.18,19 New particles obtained by the downsizing of GO were signified as graphene oxide quantum dots (GOQDs).20 Immobilized GOQDs demonstrate improved properties as adsorbents for HPLC,21 in photonic/electronic devices,22 as carriers for the photo-thermal and redox-responsive release of medications,23 and as sensors in electrochemical analysis.20,24 In the latter case, smaller nanoparticles have better electrochemical properties.19
Because of the miniaturization of electronics in recent years, the interest in electroanalytical methods has been growing, particularly in the rapid and inexpensive determination of environmentally important contaminants with in-field portable instruments. The technical development associated with electrochemical sensors is an efficient, low-cost, fast-response and easy-to-operate alternative compared to spectroscopic or chromatographic sensors. Besides, electrochemical devices have the advantage of portability and miniaturization.25 Various materials have been used as working electrodes in such devices: conductive glasses,26 screen-printing,27 glassy carbon,28 ceramic carbon,29 and carbon paste.30,31 To increase the electroactive area and facilitate the kinetics of the electrode/solution interface, the electrodes have usually been modified with metal/metal oxide nanoparticles,32,33 carbon nanotubes,34 graphene or GO,18,35 or with hybrid silica-based materials.36 For example, it was found that the addition of mesoporous silica (SBA-15) to a carbon paste electrode (CPE) can considerably enhance its sensitivity toward diethylstilbestrol,37 by as much as immobilization of graphene on glassy carbon electrode.38 Application of GOQDs as an individual modifier, as well as part of a hybrid material is under intensive electrochemical study.27 For instance, the possibility of the simultaneous determination of dopamine and epinephrine using gold nanocrystals capped with graphene quantum dots in a silica network was demonstrated recently.39
Among biologically active compounds, which are inevitably discharged into the environment, antibiotics and hormones require special attention. The residue of antibiotics in the environment has resulted in bacterial resistance, which could seriously affect human health and the ecological balance.40 Environmental estrogens belong to a group of endocrine disruptors (ED) and can cause cancerous tumours, birth defects, and other developmental disorders of the endocrine system even at low concentrations.41–43 Hence, it is important to develop analytical methods for time- and cost-effective monitoring of ED in various media. Electrochemical sensors are proposed to answer this demand.44
In the current research, GOQDs have been incorporated into a silica network to obtain a mesoporous electrochemically active material with a high surface area. The porous structure and morphology of the new SiO2-GOQDs hybrid were investigated using an N2 adsorption/desorption experiment and high-resolution scanning electron microscopy (SEM) equipped with an energy-dispersive spectroscopy (EDS) detector. Covalent immobilization of GOQDs on aminosilica was confirmed by Cross-Polarisation Magic Angle Spinning Nuclear Magnetic Resonance (CP MAS NMR), X-ray photoelectron spectroscopy (XPS), and Raman and Fourier Transform Infrared (FTIR) spectroscopies. The electroanalytical properties of SiO2-GOQDs were studied as a modifier of a carbon paste electrode in differential pulse voltammetry determinations of four environmentally important endocrine disruptors: sulfamethoxazole (SMZ) and trimethoprim (TMP) (antibiotics), and diethylstilbestrol (DES) and estriol (EST) (hormones). The selected analytes have various polar as well as aromatic fragments, affecting their multiple-point interaction with the GOQDs surface (Fig. 1). The modified electrode demonstrated an essential increase in selectivity toward EST and DES, which was explained by a significant π–π stacking interaction between GOQDs and the aromatic system of the analytes.
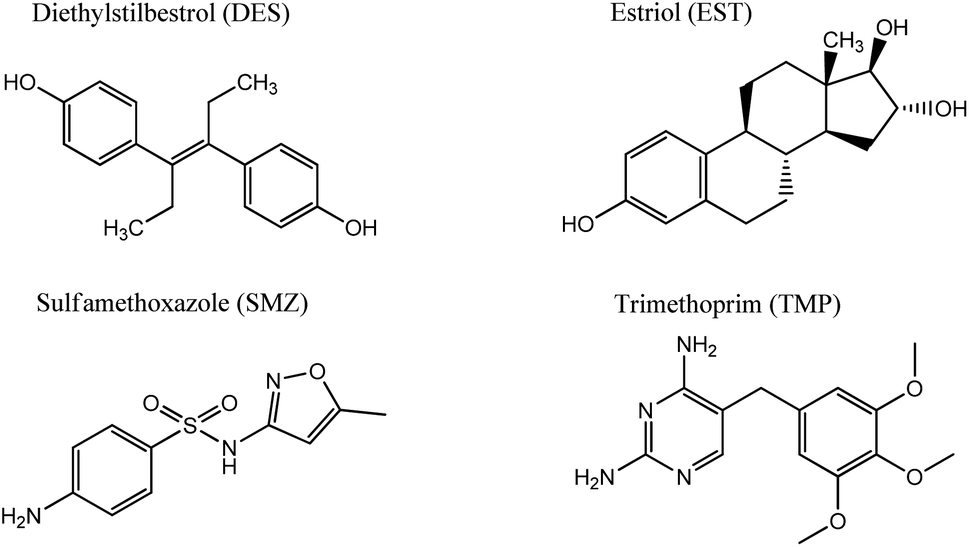 | ||
| Fig. 1 Chemical formulas of medications studied in this research. | ||
2. Materials and methods
2.1 Chemicals and reagents
Silica gel with a 4 nm average pore size and 63–200 μm particle size distribution was purchased from Merck, (3-aminopropyl) triethoxysilane (APTES, ≥98%), anhydrous toluene, N,N′-dicyclohexylcarbodiimide (DCC, 99%), potassium permanganate (KMnO4, ≥99.0%), hydrogen peroxide solution (H2O2, 30% (w/w)) were purchased from Sigma-Aldrich. Ninhydrin (99%) and sulfuric acid (95–98%) were provided by QHEMIS (Brazil), graphite and dimethylformamide (99.8%) came from Synth (Brazil). Diethylstilbestrol (DES), estriol (EST) (both 97%), sulfamethoxazole (SMZ, 98%) and trimethoprim (TMP, 98%) were provided by Sigma-Aldrich. Britton–Robinson buffer solution (BRbs) (0.04 mol L−1) was prepared from the following reagents: boric acid (Neon, 99.5%), acetic acid (Dinamica, 99.7%), phosphoric acid (Vetec, 85%), hydrochloric acid (Merck, 37%), sodium hydroxide (Vetec, 97%), sodium nitrate (Quimica Moderna, >99%). After distillation with calcium hydride (Sigma-Aldrich), toluene was kept in a dark bottle with 3 Å molecular sieves (4–8 mesh, Sigma-Aldrich) as a readily available solvent for the synthesis. The aqueous solutions were prepared using ultra-pure water from PURELAB Classic, (Elga, UK).2.2 Characterization techniques
The solution pH was measured using a PHS-3E pH-meter with a BioTrode (Hamilton, USA) ion-selective electrode. The electrical conductivities of the suspensions were measured using an HI 8633 conductivity meter (Hanna instruments, UK). The concentrations of manganese ions in solution were determined by an Optima 7300 DV inductively coupled plasma optical emission spectrometer (PerkinElmer, USA). The specific surface area was calculated with the standard BET method and the pore size distribution was determined using the modified Nguyen–Do approach45–47 from the nitrogen adsorption/desorption isotherms on a Tristar II 3020 Kr instrument (Micromeritics, USA). Elemental analysis (CHN) of the samples was made on a PE-2400 elemental analyser (PerkinElmer, USA). Photoluminescence measurements were performed using an LS 55 luminescence spectrometer (PerkinElmer, USA) with powder holders. Fourier transform infrared (FTIR) spectra were recorded in the region 4000 to 400 cm−1 on an FTLA-2000 spectrometer (Thermo Scientific Nicolet). The solid-state Cross-Polarization Magic Angle Spinning Carbon-13 Nuclear Magnetic Resonance (CP/MAS 13C NMR) spectrum of the sample was obtained on an Agilent Technologies DD2 500/54A (Agilent, USA) – 100.6 MHz (13C) instrument. UV-Vis absorption spectra were measured on a Cary 100 machine (Agilent, USA). The surface composition of the hybrid materials was determined from X-ray photoelectron spectroscopy (XPS), using a Kα X-ray photoelectron spectrometer (Thermo Fisher Scientific, UK) equipped with a hemispherical electron analyser and an aluminium anode X-ray source (Kα = 1486.6 eV) at an energy resolution of 1 eV. The morphology of the samples was studied on a JEOL JSM 7100F field-emission scanning electron microscope (JEOL, Japan) with a silicon-drift EDS detector from Oxford. Differential pulse voltammograms (DPV) were recorded on an IviumStat potentiostat/galvanostat (Ivium Technologies, The Netherlands) with a conventional three-electrode cell.The concentration of immobilized aminopropyl groups (CNH2) was calculated from elemental and XPS analyses of modified silicas, using eqn (1),48,49 and eqn (2),50 respectively.
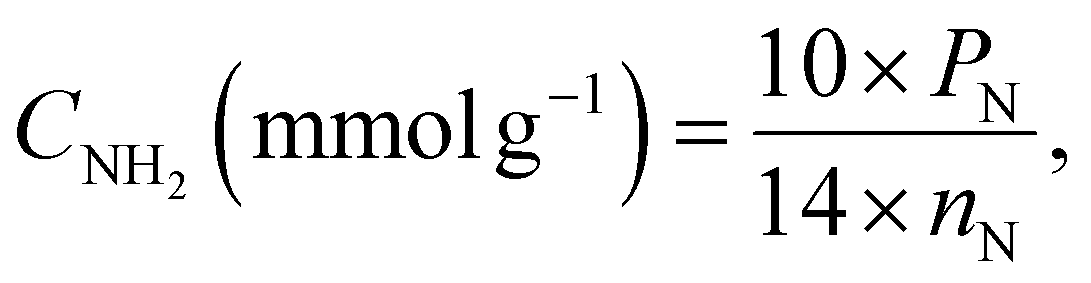 | (1) |
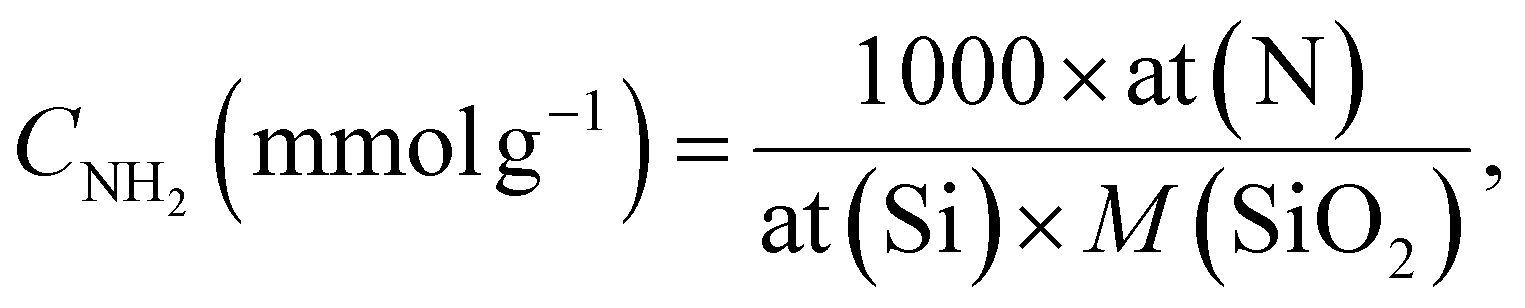 | (2) |
A modified carbon paste electrode (CPE, working electrode), platinum wire (the auxiliary electrode) and Ag/AgCl (reference electrode) were mounted in the cell. For DPV the following parameters were set: pulse amplitude of 50 mV, a pulse time of 50 ms, a step potential of 1 mV and a scan rate of 10 mV s−1. The background was subtracted from each voltammogram, as recommended by convention.51 The limit of detection (LOD) and limit of quantification (LOQ) were calculated as follows: LOD = 3Sb/b, and LOQ = 10Sb/b, where Sb is the standard deviation of the blank (n = 10) and b is the slope of the calibration curve. For organic analytes, the octane–water distribution coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) were found from the PubChem database.52
P) were found from the PubChem database.52
2.3 Synthesis of graphene oxide quantum dots
GOQDs were prepared in a one-step ultrasonic synthesis.53 In brief, 200 mL of a mixture of concentrated H2SO4 and H3PO4 (9:1 v/v) was added to graphite powder (1.5 g) in a 500 mL round-bottom flask equipped with a mechanical stir bar. Then KMnO4 (9 g) was added slowly at room temperature under stirring and after the flask was heated to 50 °C and kept at this temperature for 12 h. The obtained light-pink mixture was cooled down, poured slowly onto ice (400 mL) with 30% H2O2 (20 mL) giving an orange suspension. The solid was separated by centrifugation and washed with water. To remove manganese impurities, the precipitate was immersed in HCl (2%) then separated from the solution by centrifugation. The procedure was repeated until negative results on Mn impurity in solution with ICP-OES were confirmed. Finally, 40 mg of the freeze-dried film (Fig. S1a†) was sonicated in 50 mL of DMF for about 2 h. Emitting green-blue light in UV irradiation, the dispersion was subjected to further immobilization.
2.4 Preparation of SiO2-GOQDs hybrid material
GOQDs were covalently immobilized on the silica gel surface via the silica-immobilized aminosilane and carboxylic groups of GOQDs, as generally recommended for carboxylic compounds.7 Typically, activated in HNO3 and dried at 500 °C, silica gel (10 g) was suspended in 100 mL of dry toluene, and 3 mL of APTES was added under constant stirring. The reaction mixture was refluxed for 10 h, filtrated, washed in a Soxhlet apparatus with toluene for 24 h and finally dried under vacuum at 120 °C for 7 h. The resulting aminosilica (SiO2-NH2), (3 g) was added to the suspension of GOQDs in 50 mL of DMF and 40 mg of DCC were added under stirring. The suspension was heated at 85 °C for 60 h with periodic sonification for 30 min. The solid phase was separated by decantation, and washed with DMF, methanol and water under ultrasonic treatment (5 min). Finally, the precipitate was dried at 120 °C for 8 h to obtain approximately 3 g of SiO2-GOQDs. Chemical analysis of SiO2-NH2 and SiO2-GOQDs revealed augmented carbon content in the samples – from 2.89 to 3.22% (Table S1†), while the percentage of nitrogen did not change. This indicated that about 3.3 mg of GOQDs had been immobilized per gram of SiO2-GOQDs, which constitutes 25% of that initially loaded for synthesis (13 mg g−1).2.5 Preparation of CPE modified electrode
Typically, 8 mg of SiO2-GOQDs and 12 mg of graphite powder were carefully mixed in an agate mortar with the addition of mineral oil (5 mg). The prepared homogeneous paste was placed in a Teflon cavity (1 mm in depth and 2 mm in diameter), covered with a platinum disk fused to a glass tube with copper wire as an electrical conductor. The fabricated electrode was denoted CPE/SiO2-GOQD. An unmodified CPE electrode prepared in the same way without SiO2-GOQDs was used for comparative analyses.3. Results and discussion
3.1 Synthesis approach
Hybrid SiO2-GOQDs materials can be obtained by adsorption and further chemical anchoring of the preliminarily prepared GOQDs on the support surface.4,23,54 An alternative way is incomplete pyrolysis of small organic compounds trapped inside the adsorbed pores.50 In the latter case, a porous matrix can better confine the size and shape of the resulting GOQDs.55,56 However, preparation of SiO2-GOQDs inside the host porous system can drastically decrease the specific surface area of the resulting hybrid. For example, an SBA-15-GQDs nanocomposite prepared by incomplete pyrolysis of pyrene adsorbed in SBA-15 pores has only 26 m2 g−1 while the specific surface area of the pristine host was 719 m2 g−1.55 Therefore, the first scheme was selected and GOQDs were prepared from graphite in a one-step ultrasonic synthesis with further immobilization of ready nanoparticles in the porous network of mesoporous silica. The ability of GOQDs to penetrate silica pores has been confirmed recently.24,57 Samples of GO were prepared from graphite powder by oxidation with KMnO4 in a mixture of concentrated H2SO4 and H3PO4.It was of key importance to obtain a metal-free nanocomposite,55 since even traces of electrocatalytically active metal ions such as Mn2+ can essentially alter the properties of SiO2-GOQDs electrodes.58–60 To ensure the Mn-free composition of GO samples, the concentration of Mn ions in solution in the course of stepwise washing was monitored. The results presented in Fig. S2† demonstrate that, for removal of Mn2+ ions impurities from GO, washing in water is less efficient than washing with 2% HCl. This fact can be explained by the good adsorption properties of GO towards metal ions.1,61
Covalent immobilization of GOQDs on the silica surface was performed via an earlier-established procedure for the covalent attachment of carboxyl-containing organic compounds on SiO2-NH2, by the acylation of immobilised aminosilane in anhydrous solvent (DMF) in the presence of DCC.10,14 An excess of GOQDs was separated from the final product by multiple decantations of the precipitate alternating with ultrasonic treatment of SiO2-GOQDs in DMF. The finally obtained brown-grey product demonstrates greenish luminescence under irradiation with UV light at 365 nm (Fig. S1†).
3.2 Morphology of SiO2-GOQDs
As can be seen from Fig. 2a, SiO2-GOQDs have an identical shape of the N2 adsorption/desorption isotherm to pristine SiO2-NH2. This suggests that immobilization of GOQDs does not change the porous structure of the silica support with a type IV isotherm with a distinct hysteresis H1 loop within the p/p0 range of 0.4–1.0, accounting for mesoporosity. At high relative pressure, saturation of the isotherms is observed, and this feature indicates the complete filling of the mesopores and the absence of macropores (Fig. 2b). Very similar isotherm profiles for pristine SiO2-NH2 and SiO2-GOQDs are evidence that immobilisation of GOQDs does not produce significant textural changes. The similarity in morphology and average pore sizes, together with the simultaneous enlargement in the SiO2-GOQDs surface area (Table 1) could indicate the incorporation of GOQDs into the porous structure of the hybrid material.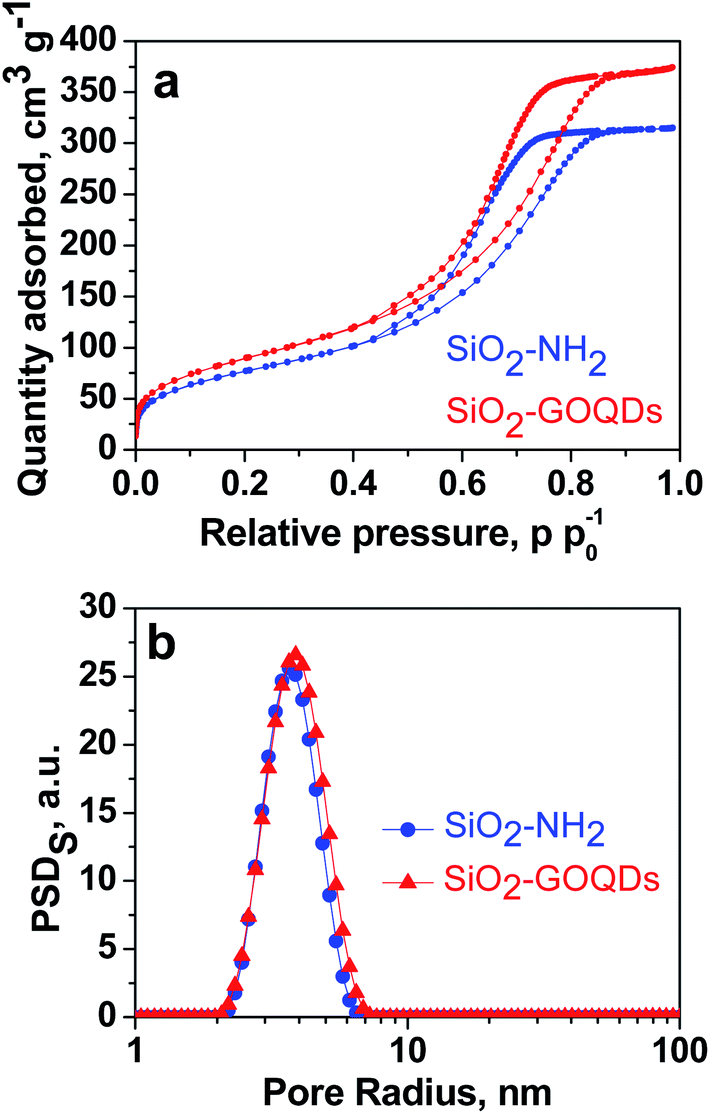 | ||
| Fig. 2 N2 adsorption–desorption isotherms of SiO2-NH2 and SiO2-GOQDs (a); incremental pore size distribution by surface area (PSD) (b). | ||
| Material | SBET, m2 g−1 | Average pore size, nm |
|---|---|---|
| SiO2-NH2 | 278 | 5.4 |
| SiO2-GOQDs | 324 | 5.2 |
The morphology of GO and SiO2-GOQDs was investigated by the SEM technique. Freeze-dried GO demonstrates a closely packed lamellar texture, reflecting its multi-layered microstructure (Fig. 3a inset). With the exfoliation of graphite oxide into GO, the edges of the GO sheets become crumpled and folded (Fig. 3a). Low-magnification SEM images of SiO2-GOQDs demonstrate that the silica particles are well separated with none of the agglomeration commonly observed for silica-decorated GO62,63 (Fig. 3b). Also, from the high-magnification SEM images of SiO2-GOQDs it can be seen that the silica gel particles are not wrapped by GO (Fig. 3c and d). In some cases, the porous structure of SiO2-GOQDs can be distinguished (Fig. 3d). This result agrees with the conclusion made from the N2-adsorption experiment, suggesting the incorporation of GOQDs into the SiO2 porous network.
 | ||
| Fig. 3 Low (a and b) and high (c and d) magnification SEM images of GO (a) and SiO2-GOQDs (b–d) with EDS element mapping of O, Si, C and N on SiO2-GOQDs (e). | ||
3.3 Composition of SiO2-GOQDs
Immobilisation of GOQDs on silica gel was also confirmed by electron microscope energy-dispersive spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), FTIR, CP/MAS 13C NMR and Raman spectroscopy measurements. For example, it is apparent from Fig. 3e, which illustrates the EDS mapping for the O, Si, C and N of SiO2-GOQDs, that the C and N elements were uniformly distributed in the silica matrix. This serves as confirmatory evidence for the immobilisation of GOQDs.Photoluminescent spectra of SiO2-GOQDs also strengthen the suggestion of GOQD immobilization on the SiO2 surface. It is known that neither aminosilica nor GO exhibit photoluminescence, but samples of SiO2-GOQDs exhibit a strong luminescent band with the maximum at 404 nm (Fig. 4) – likewise GOQDs in water suspension22 and other hybrid silica-based materials with loaded GOQDs.24,64 Additionally, the photoluminescence of SiO2-GOQDs can even be seen by the naked eye (Fig. S1†).
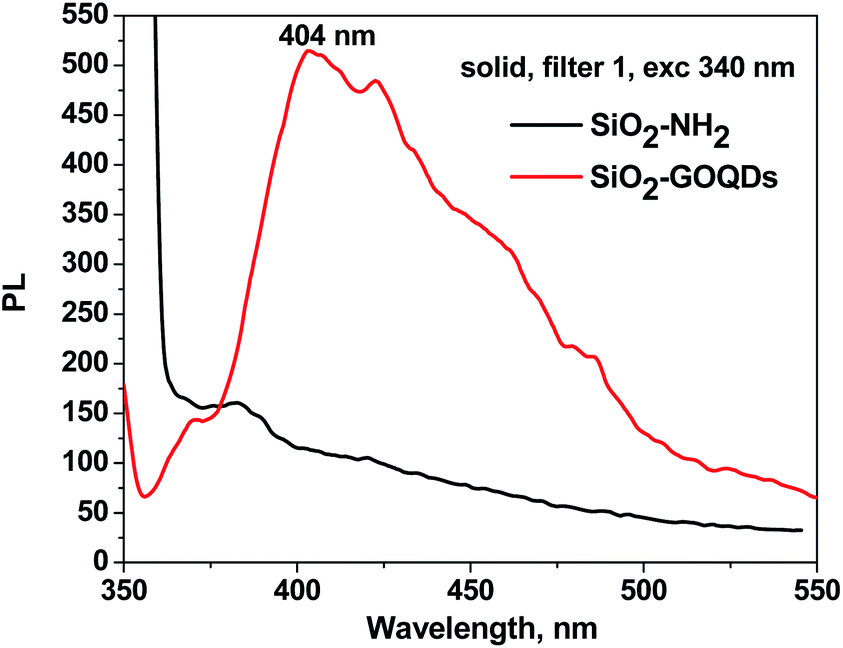 | ||
| Fig. 4 Photoluminescent spectra of solid SiO2-NH2 (black line) and SiO2-GOQDs (red line) under excitation at 340 nm. | ||
For the investigation of the GOQD chemisorption process, the Raman spectra of GOQDs and SiO2-GOQDs were recorded (Fig. 5). From Fig. 5 it is evident that the spectral profile of GOQDs shows two distinct peaks at 1585 cm−1 (G-peak) and 1372 cm−1 (D-peak). The D peak is considered an indication of the disordered structure of graphene in GOQDs due to oxidation. For the experimental samples ID/IG < 1, presumably indicating a decrease in the fraction of aromatic sp2 domains in GOQDs with an increase in the number of detected oxygen-containing sites.65
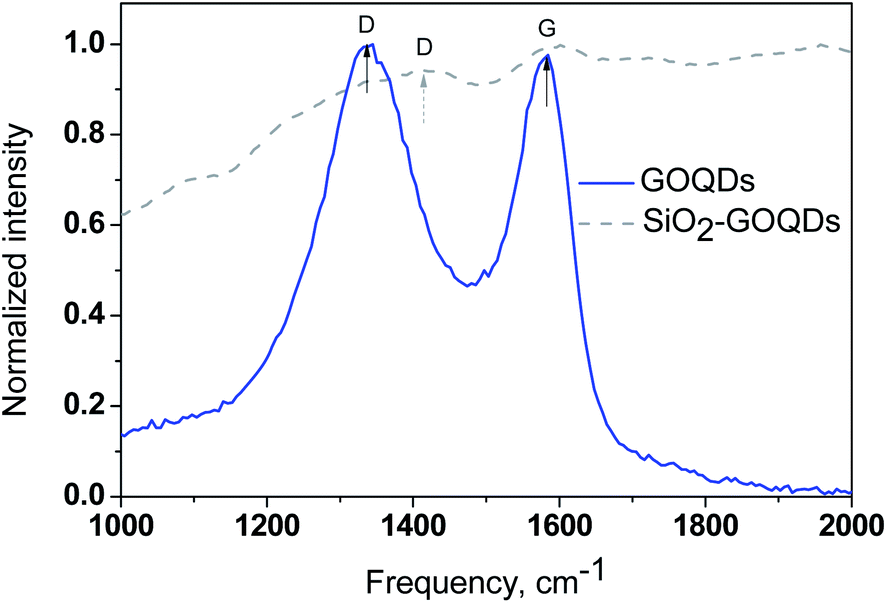 | ||
| Fig. 5 Raman spectra of GOQDs (—) and SiO2-GOQDs (- -). | ||
Despite the strong fluorescence background and low carbon content, we were able to record a Raman spectrum from SiO2-GOQDs and it confirms the immobilization of GOQDs (Fig. 5). An ordered G band was detected at about 1585 cm−1 that matches the position of the G band of individual GOQDs, indicating negligible interaction between the silica scaffold and the basal plane of the immobilized GOQDs. In contrast, the position of the D-band in the hybrid material is shifted to 1412 cm−1, suggesting the anchoring of GOQDs via oxygen-containing sites. There is little doubt that not all these sites related to GOQDs will react with immobilized aminosilane fragments in SiO2-NH2. Therefore, the D-band in the Raman spectra of SiO2-GOQDs is augmented (Fig. 5).
Further evidence for oxygen-containing groups in GOQDs was received from FTIR, CP 13C MAS NMR and XPS spectroscopy of the SiO2-GOQDs. The FTIR spectrum of the GOQDs as well as the Raman spectrum suggests the presence of oxygen-containing sites in GOQD nanoparticles, including C(O)O–H (νO–H at 3390 cm−1), CO–H (νO–H at 3250 cm−1) and carboxyl (νOC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1730 cm−1)6 (Fig. 6). Apart from the silica gel matrix, the pristine SiO2-NH2 shows several bands at around 2950 cm−1 (νCH3 and νCH2), and at peaks at 1560, 1475, 1450, 1420 and 1390 cm−1 that correspond to the stretching vibrations of the propylamine chain.
O at 1730 cm−1)6 (Fig. 6). Apart from the silica gel matrix, the pristine SiO2-NH2 shows several bands at around 2950 cm−1 (νCH3 and νCH2), and at peaks at 1560, 1475, 1450, 1420 and 1390 cm−1 that correspond to the stretching vibrations of the propylamine chain.
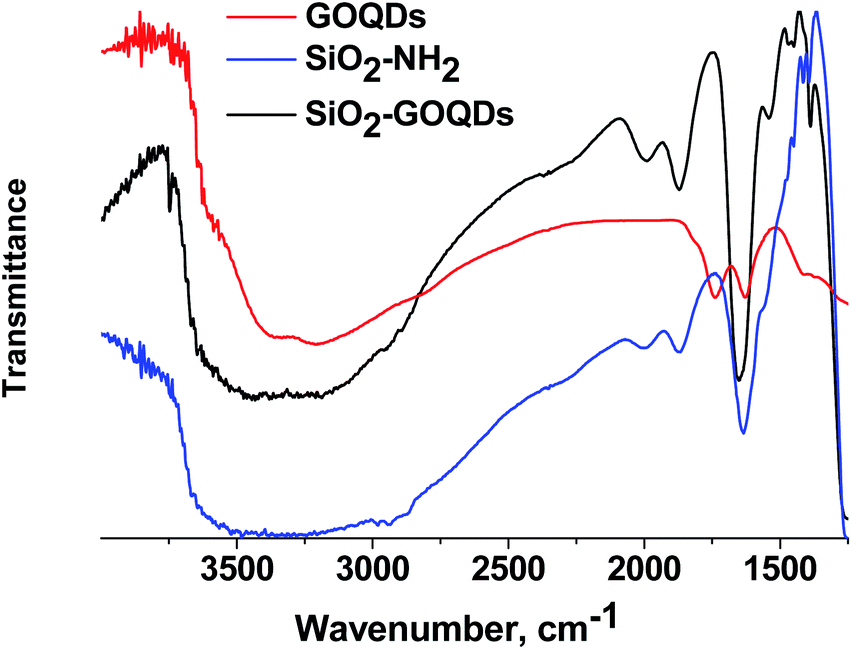 | ||
| Fig. 6 FTIR spectra of GOQDs, SiO2-NH2, SiO2-GOQDs. | ||
The FTIR spectrum of the GOQDs essentially changed after immobilization. In particular, the stretching vibration of the carboxylic group at 1730 cm−1 disappeared, and bands at 1650 cm−1 and 1574 cm−1 corresponding to the stretching vibration of CO and the bending vibrations of N–H in NHC(O) fragments of immobilized moieties emerged instead (Fig. 6).
From these observations, the covalent immobilization of GOQDs via carboxyl fragments of the nanoparticle and amino group of the silane can be assumed.6,21
The SiO2-GOQDs have low total carbon content; therefore, the 13C NMR spectra are onerous to obtain. Nevertheless, we were able to record the spectra and identify the signals. The most intense peaks at 10, 25, and 40 ppm were correspondingly assigned to Si–CαH2, CH2–CβH2 and CγH2–NH– in the immobilized moiety (Fig. 7). A signal at 165 ppm was attributed to the carbonyl fragment of the GOQDs,66,67 while a series of signals at 100–115 ppm were assigned to sp2 carbons in the basal plane of graphene.68 Consequently, the 13C NMR spectrum of SiO2-GOQDs as well as the FTIR and Raman spectra demonstrates an essential fraction of oxygen-containing fragments in GOQDs and proves the immobilization of the GOQDs in the porous network of silica.
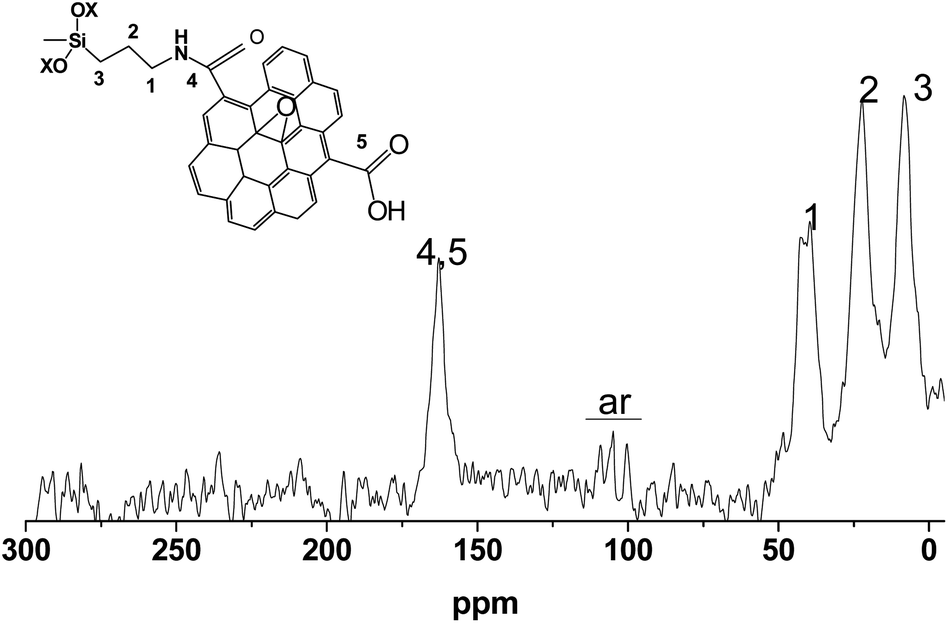 | ||
| Fig. 7 CP/MAS 13C NMR spectrum of SiO2-GOQDs. | ||
The survey XPS spectrum of SiO2-GOQDs demonstrates that SiO2-GOQDs is an Mn-free material since its XPS does not contain Mn 2p peaks at 641.3 and 653.2 eV (ref. 69) (Fig. 8a). The spectrum data also confirmed the immobilization of GOQD nanoparticles. As follows from Fig. 8a, the nitrogen to silicon atomic ratio for SiO2-NH2 and SiO2-GOQDs remains the same (0.060 ± 0.008), while the carbon-to-silicon ratio is increased for the latter, indicating a higher loading of carbon-containing moieties in SiO2-GOQDs. These results are in good agreement with the CHN analysis of the materials, which also demonstrates 3.22% of carbon loading in SiO2-GOQDs (Table S1†).
 | ||
| Fig. 8 Survey (a) and fitted XPS spectra of the C1s (b) and N1s (c) of SiO2-GOQDs and pristine SiO2-NH2 with deconvoluted data. | ||
The high-resolution XPS spectra demonstrate an essential difference between the C1s bands for SiO2-NH2 and SiO2-GOQDs (Fig. 8b). The C1s signal from SiO2-NH2 can be deconvoluted into three components attributed to C–C, C–N and C–O bonds in SiO2-immobilized aminopropyl fragments (Table 2). The relative intensity of the C–C and C–N peaks in the spectrum is about 3:1, which correlates with the composition of the immobilized fragment. Also, about 6% of carbon atoms in SiO2-NH2 are bonded with oxygen, which lets us assume the occurrence of incomplete hydrolysis of ethoxy groups of aminosilane in the immobilization process, as demonstrated in Fig. 7 (inset).
| C1s | ||||||||
|---|---|---|---|---|---|---|---|---|
| SiO2-NH2 | SiO2-GOQDs | |||||||
| C–H, C–C | C–N | C–O | CC |
CH, C–C | C–N | C–O | OCO | |
| eV | 285.1 | 286.3 | 287.7 | 284.7 | 285.1 | 286.2 | 287.7 | 288.9 |
| % | 69 | 25 | 6 | 5 | 47 | 31 | 8 | 9 |
| N1s | ||||
|---|---|---|---|---|
| SiO2-NH2 | SiO2-GOQDs | |||
| NH2 | NH3+ | NH2 | NH3+ | |
| eV | 399.7 | 401.6 | 400.2 | 401.6 |
| % | 86 | 14 | 79 | 21 |
The high-resolution XPS C1s spectrum of SiO2-GOQDs shows additional peaks among the signals from SiO2-NH2, attributed to CC and CO bonds in O–CO or N–CO fragments (Table 2), indicating the successful immobilization of GOQDs.14
Fig. 8c illustrates the high-resolution N1s XPS spectra of the synthesized hybrids. A band of pristine SiO2-NH2 consists of components attributed to neutral and protonated primary amines. On GOQD immobilization, the fraction of H-bonded amines noticeably increased, while another fraction of amine fragments was transformed to amide (Table 2). This effect reflects a peculiarity of the surface reaction of the immobilized amine with nanoparticles containing several carboxyl fragments. It should be readily apparent that only a few carboxyl groups of GOQDs can acylate immobilized amines due to steric restrictions. Others will be ionized and will protonate the remaining amines, as illustrated in Fig. 9.
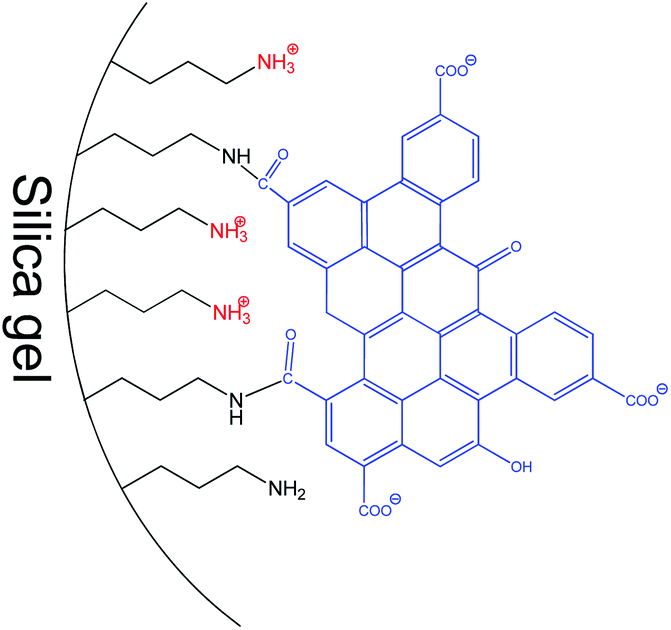 | ||
| Fig. 9 Schematic structure of surface layer in SiO2-GOQDs. | ||
3.4 Electrochemical properties of the carbon paste electrode modified with SiO2-GOQDs
A carbon paste electrode (CPE) was selected for modification because it is cheap, can be reproducibly fabricated in any laboratory and SiO2-GOQDs can be easily integrated into the electrode.31 Two antibiotics and two hormones were selected for investigation, namely: sulfamethoxazole and trimethoprim, diethylstilbestrol and estriol (Fig. 1). DES is the first synthetic estrogen that has been extensively used in the treatment of estrogen-deficiency disorders. Although it has been prohibited as a growth promoter for years,70 these estrogens are still found in rivers,71 fish,72 milk,73 and meat.8All selected analytes have aromatic rings that can form π–π stacking complexes with GOQDs, but various polar fragments are also present in their structure. The latter can weaken or enhance such interactions and, hence affect the sensitivity of the electrochemical analysis. First, the CPE electrode modified with SiO2-GOQDs was tested in differential pulse voltammetry (DPV) oxidations of a mixture containing sulfamethoxazole and trimethoprim (5:1). The results were compared with those obtained on the CPE electrode without any additives.
From the data presented in Fig. 10, it can be seen that being only slightly higher than TMP, the response of CPE/SiO2-GOQDs to SMZ is reasonably superior compared with bulk CPE. Also, the oxidation peaks are shifted to lower potentials for both components but differently (−155 mV for SMZ and −95 mV for TMP). This is impacted by the difference in anodic peaks of the analytes of 280 mV, making simultaneous determination of the presence of SMZ and TMP more reliable.
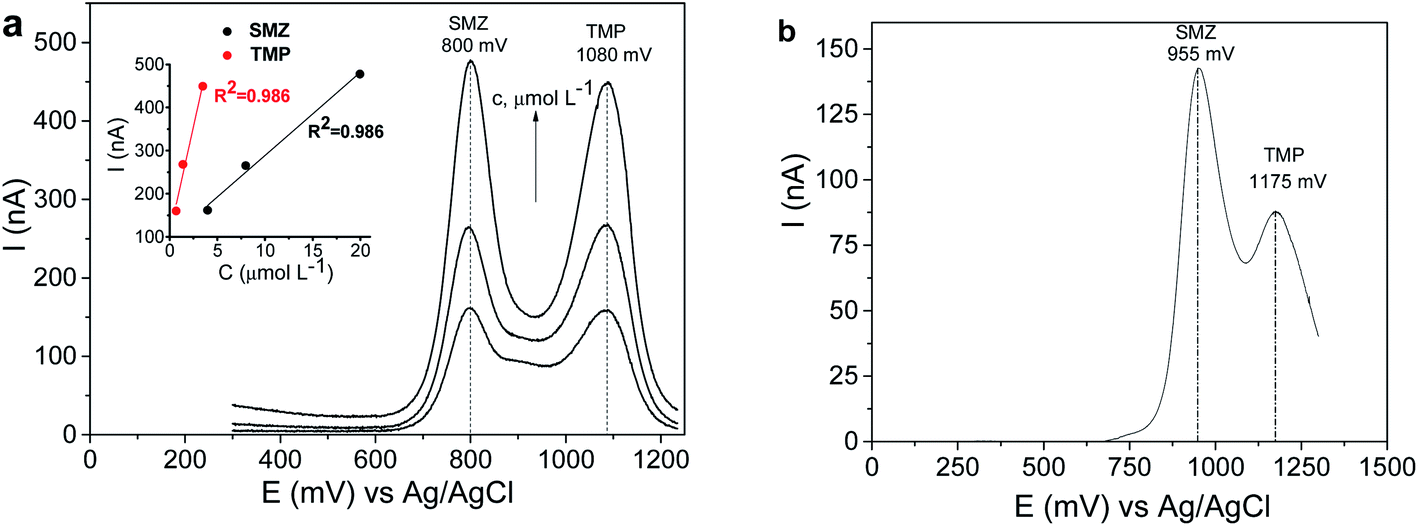 | ||
| Fig. 10 DPV curves of SMZ and TMP mixture on CPE/SiO2-GOQDs (a) and CPE (b), and linear relationship between peak currents and the concentrations of the analytes (inset). The analytes were present in a mixture (5:1) with the following concentrations of SMZ: (a) 4.0, 8.0 and 20 μmol L−1; (b) 4.0 μmol L−1. Supporting electrolyte: 0.04 mol L−1 of BRbs (pH 5.8), 0.5 mol L−1 of NaNO3. | ||
The significant improvement in the electrocatalytic performance of the fabricated CPE/SiO2-GOQDs electrode was observed in the DPV analysis of the selected hormones (Fig. 11). Similar to the results of the antibiotics analysis, the oxidation peaks of both hormones are shifted to lower potentials for CPE/SiO2-GOQDs in comparison with CPE, attesting to a better interaction of the analytes with the active centres of the electrode. The peak current of EST on the modified electrode was 11.4 nA versus 3.1 nA on the CPE, and correspondingly 70.9 nA versus 9.3 nA for DES (Fig. 11).
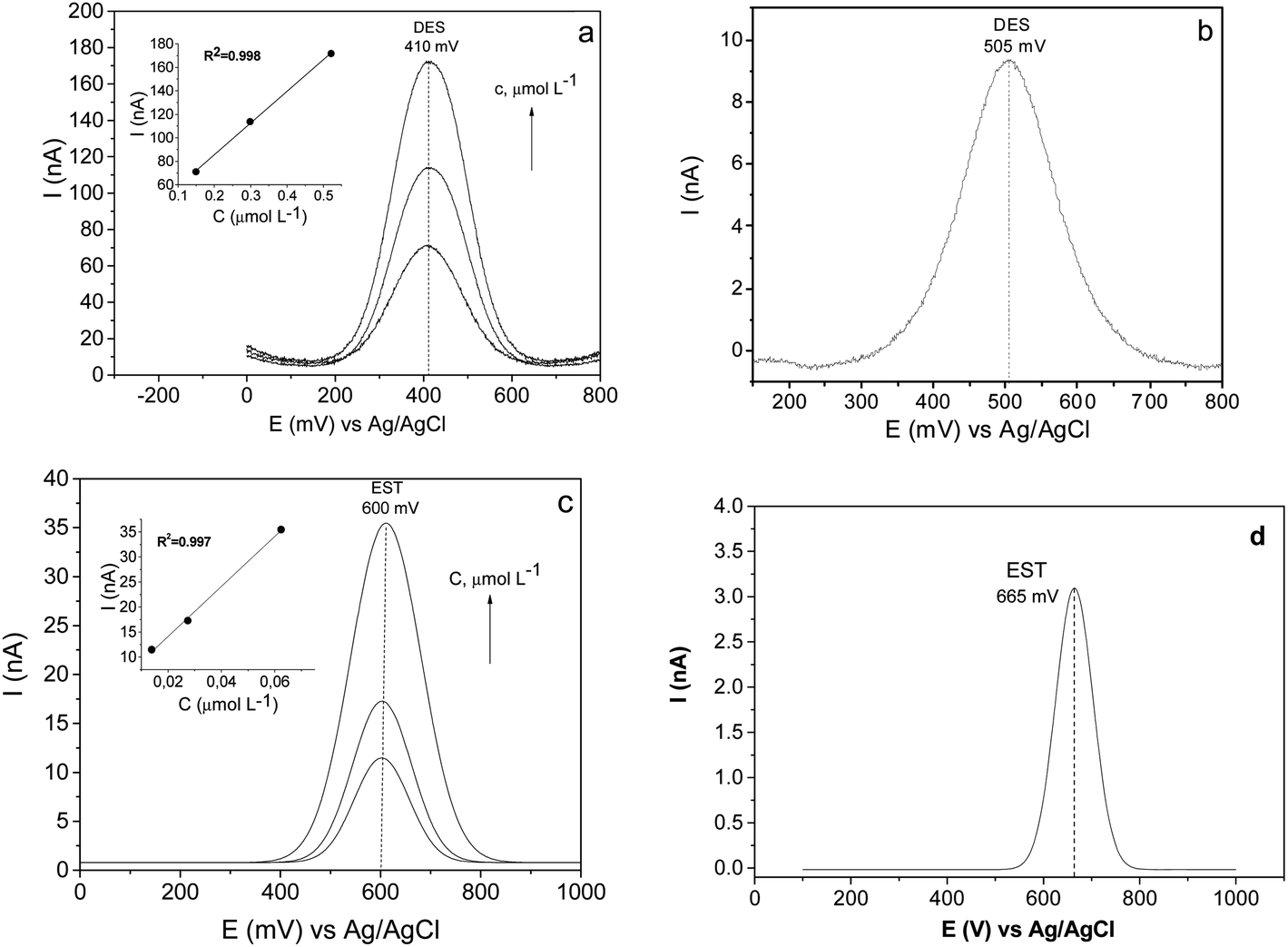 | ||
| Fig. 11 DPV curves of DES (a and b) and EST (c and d) on CPE/SiO2-GOQDs (a and c) and bulk CPE (b and d), and linear relationship between peak currents and the concentrations of the analytes (insets). Concentration of the analytes: DES – 0.15, 0.30 and 0.52 μmol L−1, EST – 0.014, 0.027, 0.062 μmol L−1. Supporting electrolyte: 0.04 mol L−1 of BRbs (pH 5.8), 0.5 mol L−1 of NaNO3. | ||
The modified electrodes demonstrated a linear signal response vs. concentration of the analytes, according to the equations presented in Table S2† along with other analytical characteristics of the subject modified electrode and other electrodes reported earlier. As is evident from the data presented, despite less impressive results in the determination of SMZ and TMP on the CPE/SiO2-GOQDs, the fabricated electrode can compete in terms of LOD and sensitivity to EST and DES with the most advanced modern glassy carbon electrodes modified with metal nanoparticles (Table S2†).
The overall sensitivity enhancement of CPE/SiO2-GOQDs toward selected analytes can be explained by considering the textural characteristics of SiO2-GOQDs. As discussed earlier, the integration of mesoporous silica into a graphite paste electrode can enhance its electrochemical response due to areal enlargement of the electroactive sites over the electrode surface.36 It seems that immobilisation of GOQDs on the silica surface can further enhance such a response thanks to the amplified affinity of the resulting modified electrode to the analytes. The higher affinity of CPE/SiO2-GOQDs towards the analytes reveals itself in a decrease in the oxidation potential of the analytes on 65–155 mV, compared with CPE.
The SiO2-GOQDs demonstrate an essential difference in analytical characteristics toward the selected analytes. In particular, the sensitivity of the modified electrode to EST is 500 nA L μmol−1, whereas to SMZ it is only 19 nA L μmol−1 (Table S2†). To understand the reasons, electroanalytical properties of the electrodes were analysed against molecular descriptors of the analytes, such as logP, the topological polar surface area (tPSA) and Hückel aromaticity.52 From Fig. S4,† which demonstrates the relationship between logP and normalised peak currents (I (nA)/C (μmol L−1)) on CPE and CPE/SiO2-GOQDs, it can be seen that the sensitivity of the modified electrode to analytes with higher logP is generally increased. A similar tendency was found for tPSA. But it is also clear that the electrode sensitivity to EST with logP = 2.4 is essentially higher than to DES with logP = 5.2. Apparently, logP is not the only factor determining the selectivity of the modified electrodes.
A clearer picture and a better explanation of the electroanalytical properties of CPE/SiO2-GOQDs can be obtained if the properties of modified and bulk PCE are compared. Fig. S3b† demonstrates that the most essential enhancement in the current on the modified electrode was observed for DES (760%), then EST (370%), TMP (180%) and finally SMZ (110%). According to the Hückel model, DES has two times higher aromaticity than EST, which has a similar molecular geometry (Fig. S5†). The higher aromaticity of DES can explain the reason for the most essential enhancement in CPE/SiO2-GOQDs sensitivity toward this analyte, which occurs due to stronger π–π stacking interactions between the aromatic system of DES and the graphene basal plane of the immobilized GOQDs. The EST has only one aromatic ring and it demonstrates smaller enhancement in the sensitivity to CPE/SiO2-GOQDs. Like EST, TMP and SMZ have only one aromatic ring but they are much less hydrophobic than EST (Fig. S4†). Also, SMZ has a negatively charged sulfamide fragment (Fig. S5†), which can repel the analyte from the negatively charged GOQD surface (Fig. 9). This effect can decrease the interaction between immobilised GOQD particles and SMZ and thus makes SMZ the least sensitive among the four analytes.
4. Conclusions
In summary, GOQDs can be successfully incorporated into the porous structure of silica gel, resulting in a SiO2-GOQDs hybrid that maintains its high surface area without a change in the pore size distribution profile. CPE modified with SiO2-GOQDs demonstrated enhanced sensitivity towards DES and EST and less effectiveness towards TMP and SMZ. This fact was explained by the π–π stacking interaction between the immobilized GOQDs and the selected hormones. This view is favoured by theoretical parameters describing the molecular polar/non-polar balance (logP and tPSA), whose values correlate linearly with the experimental output. Oxidation peaks for all analytes were shifted to lower potentials for ca. 100–150 mV, demonstrating better interaction between the analytes and the active centres of the electrode.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
Authors are grateful for the financial support received from Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (grants E-26/010.000978/2019, E-26/210.547/2019). Tkachenko expresses his gratitude to the Ministry of Education and Science of Ukraine for the grant number 0119U002532. Mikhraliieva is grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (154820/2015-6) and (FAPERJ) (E-26/200.612/2018) for the conceded fellowships. Nazarkovsky thanks to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for receiving funds (grant no. 2013037-31005012005P5 – PNPD-PUC Rio) to carry out the research. We also appreciate the technical support received from Brazilian Nanotechnology National Laboratory (LNNano) in XPS and Institute of Chemistry at UFRGS in CP/MAS 13C NMR. The authors thank LabNano (Brazilian Center for Research in Physics, CBPF, Brazil) for continued assistance in the microscopy studies. Also, we thank Dr Omar Pandoli (PUC-Rio) for Raman measurements.Notes and references
- X. Wu, J. Hu, J. Qi, Y. Hou and X. Wei, Sep. Purif. Technol., 2020, 239, 116511 CrossRef CAS.
- S. Z. N. Ahmad, W. N. Wan Salleh, A. F. Ismail, N. Yusof, M. Z. Mohd Yusop and F. Aziz, Chemosphere, 2020, 248, 126008 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- Q. Wu, L. Chen, J. Gao, S. Dong, H. Li, D. Di and L. Zhao, Talanta, 2019, 194, 105–113 CrossRef CAS PubMed.
- R. Sitko, B. Zawisza and E. Malicka, TrAC, Trends Anal. Chem., 2013, 51, 33–43 CrossRef CAS.
- H. R. Nodeh, W. A. Wan Ibrahim, M. A. Kamboh and M. M. Sanagi, RSC Adv., 2015, 5, 76424–76434 RSC.
- R. Shi, L. Yan, T. Xu, D. Liu, Y. Zhu and J. Zhou, J. Chromatogr. A, 2015, 1375, 1–7 CrossRef CAS PubMed.
- J. Wang, Z. Chen, Z. Li and Y. Yang, Food Chem., 2016, 204, 135–140 CrossRef CAS PubMed.
- M. Mehrzad-Samarin, F. Faridbod and M. R. Ganjali, Spectrochim. Acta, Part A, 2019, 206, 430–436 CrossRef CAS PubMed.
- Q. Liu, J. Shi, J. Sun, T. Wang, L. Zeng and G. Jiang, Angew. Chem., Int. Ed., 2011, 50, 5913–5917 CrossRef CAS PubMed.
- K.-J. Huang, Y.-J. Liu, J. Li, T. Gan and Y.-M. Liu, Anal. Methods, 2014, 6, 194–201 RSC.
- S. Mahpishanian, H. Sereshti and M. Ahmadvand, J. Environ. Sci., 2017, 55, 164–173 CrossRef PubMed.
- E. Santoso, R. Ediati, Y. Kusumawati, H. Bahruji, D. O. Sulistiono and D. Prasetyoko, Mater. Today Chem., 2020, 16, 100233 CrossRef CAS.
- R. Sitko, B. Zawisza, E. Talik, P. Janik, G. Osoba, B. Feist and E. Malicka, Anal. Chim. Acta, 2014, 834, 22–29 CrossRef CAS PubMed.
- M. Li, S. Tang, Z. Zhao, X. Meng, F. Gao, S. Jiang, Y. Chen, J. Feng and C. Feng, Chem. Eng. J., 2020, 386, 123947 CrossRef CAS.
- W. Czepa, D. Pakulski, S. Witomska, V. Patroniak, A. Ciesielski and P. Samorì, Carbon, 2020, 158, 193–201 CrossRef CAS.
- L. Chen and J. Liang, Mater. Sci. Eng., C, 2020, 112, 110924 CrossRef CAS PubMed.
- S. Li, K. Huang, Q. Fan, S. Yang, T. Shen, T. Mei, J. Wang, X. Wang, G. Chang and J. Li, Biosens. Bioelectron., 2019, 136, 91–96 CrossRef CAS PubMed.
- Y. Liu, R. Wang, J. Lang and X. Yan, Phys. Chem. Chem. Phys., 2015, 17, 14028–14035 RSC.
- S. Tajik, Z. Dourandish, K. Zhang, H. Beitollahi, Q. Van Le, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 15406–15429 RSC.
- Q. Wu, Y. Sun, J. Gao, L. Chen, S. Dong, G. Luo, H. Li, L. Wang and L. Zhao, New J. Chem., 2018, 42, 8672–8680 RSC.
- S.-H. Choi, J. Phys. D: Appl. Phys., 2017, 50, 103002 CrossRef.
- Y. Gao, S. Zhong, L. Xu, S. He, Y. Dou, S. Zhao, P. Chen and X. Cui, Microporous Mesoporous Mater., 2019, 278, 130–137 CrossRef CAS.
- L. Lu, L. Zhou, J. Chen, F. Yan, J. Liu, X. Dong, F. Xi and P. Chen, ACS Nano, 2018, 12, 12673–12681 CrossRef CAS PubMed.
- S. Shrivastava, N. Jadon and R. Jain, TrAC, Trends Anal. Chem., 2016, 82, 55–67 CrossRef CAS.
- M. Mierzwa, E. Lamouroux, A. Walcarius and M. Etienne, Electroanalysis, 2018, 30, 1241–1258 CrossRef CAS.
- G. Ozcelikay, L. Karadurmus, S. I. Kaya, N. K. Bakirhan and S. A. Ozkan, Crit. Rev. Anal. Chem., 2020, 50, 212–225 CrossRef CAS PubMed.
- A.-M. Chiorcea-Paquim, T. A. Enache and A. M. Oliveira-Brett, Curr. Med. Chem., 2018, 25, 4066–4083 CrossRef CAS PubMed.
- A. Tkachenko, M. Onizhuk, O. Tkachenko, L. T. Arenas, E. V. Benvenutt, Y. Gushikem and A. Panteleimonov, Methods Objects Chem. Anal., 2019, 14, 5–14 CAS.
- A. Rana, N. Baig and T. A. Saleh, J. Electroanal. Chem., 2019, 833, 313–332 CrossRef CAS.
- S. Tajik, H. Beitollahi, F. G. Nejad, M. Safaei, K. Zhang, Q. Van Le, R. S. Varma, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 21561–21581 RSC.
- S. Bibi, M. I. Zaman, A. Niaz, A. Rahim, M. Nawaz and M. Bilal Arian, Microchim. Acta, 2019, 186, 595 CrossRef PubMed.
- A. R. Younus, J. Iqbal, N. Muhammad, F. Rehman, M. Tariq, A. Niaz, S. Badshah, T. A. Saleh and A. Rahim, Microchim. Acta, 2019, 186, 471 CrossRef PubMed.
- X. Xuan and J. Y. Park, Sens. Actuators, B, 2018, 255, 1220–1227 CrossRef CAS.
- N. M. El-Shafai, M. M. Abdelfatah, M. E. El-Khouly, I. M. El-Mehasseb, A. El-Shaer, M. S. Ramadan, M. S. Masoud and M. A. El-Kemary, Appl. Surf. Sci., 2020, 506, 144896 CrossRef.
- L. V. de Souza, D. S. da Rosa, O. S. Tkachenko, A. de A. Gomes, T. M. H. Costa, L. T. Arenas and E. V. Benvenutti, Ionics, 2019, 25, 3259–3268 CrossRef CAS.
- X. Xie, D. Sun, G. Liu and Q. Zeng, Anal. Methods, 2014, 6, 1640 RSC.
- C. Yu, W. Ji, Y. Wang, N. Bao and H. Gu, Nanotechnology, 2013, 24, 115502 CrossRef PubMed.
- V. Vinoth, L. N. Natarajan, R. V. Mangalaraja, H. Valdés and S. Anandan, Microchim. Acta, 2019, 186, 681 CrossRef CAS PubMed.
- Y. Zhao, F. Yuan, X. Quan, H. Yu, S. Chen, H. Zhao, Z. Liu and N. Hilal, Anal. Methods, 2015, 7, 2693–2698 RSC.
- E. Corsini, F. Ruffo and M. Racchi, Curr. Opin. Toxicol., 2018, 10, 69–73 CrossRef.
- T. J. Houston and R. Ghosh, Biochem. Pharmacol., 2020, 172, 113743 CrossRef PubMed.
- T. L. Gonzalez, J. M. Rae and J. A. Colacino, Toxicology, 2019, 421, 41–48 CrossRef CAS PubMed.
- A. Azzouz, S. K. Kailasa, P. Kumar, E. Ballesteros and K.-H. Kim, TrAC, Trends Anal. Chem., 2019, 113, 256–279 CrossRef CAS.
- V. M. Gun’ko, Appl. Surf. Sci., 2014, 307, 444–454 CrossRef.
- C. Nguyen and D. D. Do, Langmuir, 1999, 15, 3608–3615 CrossRef CAS.
- V. M. Gun’ko and D. D. Do, Colloids Surf., A, 2001, 193, 71–83 CrossRef.
- A. Y. Fadeev, R. Y. Novotortsev and G. V. Lisichkin, Kinet. Catal., 2000, 41, 99–107 CrossRef CAS.
- V. N. Zaitsev, L. S. Vasilik, J. Evans and A. Brough, Russ. Chem. Bull., 1999, 48, 2315–2320 CrossRef CAS.
- A. Mikhraliieva, V. Zaitsev, Y. Xing, H. Coelho-Júnior and R. L. Sommer, ACS Appl. Nano Mater., 2020, 3, 3652–3664 CrossRef CAS.
- R. Porada, K. Fendrych and B. Baś, Electroanalysis, 2020, 32, 1208–1219 CrossRef CAS.
- National Library of Medicine, PubChem, https://pubchem.ncbi.nlm.nih.gov, accessed 3 July 2020 Search PubMed.
- Y. Zhu, G. Wang, H. Jiang, L. Chen and X. Zhang, Chem. Commun., 2015, 51, 948–951 RSC.
- Q. Gu, T. C. A. Ng, I. Zain, X. Liu, L. Zhang, Z. Zhang, Z. Lyu, Z. He, H. Y. Ng and J. Wang, Appl. Surf. Sci., 2020, 502, 144128 CrossRef CAS.
- M. A. Wahab, J. Joseph, L. Atanda, U. K. Sultana, J. N. Beltramini, K. Ostrikov, G. Will, A. P. O'Mullane and A. Abdala, ACS Appl. Energy Mater., 2020, 3, 1439–1447 CrossRef CAS.
- A. Mikhraliieva, V. Zaitsev, R. Q. Aucélio, H. B. da Motta and M. Nazarkovsky, Nano Express, 2020, 1, 010011 CrossRef.
- L. He, Z. Jia and J. Zhou, Chin. Opt. Lett., 2016, 14, 041601–041604 CrossRef.
- F. Risplendi, M. Re Fiorentin and G. Cicero, 2D Mater., 2020, 7, 025017 CrossRef.
- D. M. Fernandes, P. Mathumba, A. J. S. Fernandes, E. I. Iwuoha and C. Freire, Electrochim. Acta, 2019, 319, 72–81 CrossRef CAS.
- Z. Lin, H. Huang, L. Cheng, Y. Yang, R. Zhang and Q. Chen, ACS Sustainable Chem. Eng., 2020, 8, 427–434 CrossRef CAS.
- L. Chaabane, E. Beyou, A. El Ghali and M. H. V. Baouab, J. Hazard. Mater., 2020, 389, 121839 CrossRef CAS PubMed.
- B. Ramezanzadeh, Z. Haeri and M. Ramezanzadeh, Chem. Eng. J., 2016, 303, 511–528 CrossRef CAS.
- Y. Du, L. Huang, Y. Wang, K. Yang, Z. Zhang, Y. Wang, M. J. Kipper, L. A. Belfiore and J. Tang, J. Mater. Sci., 2020, 55, 11188–11202 CrossRef CAS.
- M. Alvand and F. Shemirani, Microchim. Acta, 2017, 184, 1621–1629 CrossRef CAS.
- L. Wang, Y. Wang, T. Xu, H. Liao, C. Yao, Y. Liu, Z. Li, Z. Chen, D. Pan, L. Sun and M. Wu, Nat. Commun., 2014, 5, 5357 CrossRef CAS PubMed.
- J. L. Gómez-Urbano, J. L. Gómez-Cámer, C. Botas, N. Díez, J. M. López del Amo, L. M. Rodríguez-Martinez, D. Carriazo and T. Rojo, Carbon, 2018, 139, 226–233 CrossRef.
- T. K. Das, S. Banerjee, M. Pandey, B. Vishwanadh, R. J. Kshirsagar and V. Sudarsan, Int. J. Hydrogen Energy, 2017, 42, 8032–8041 CrossRef CAS.
- Y. Li, H. Chen, L. Y. Voo, J. Ji, G. Zhang, G. Zhang, F. Zhang and X. Fan, J. Mater. Chem., 2012, 22, 15021 RSC.
- Q. Jiangying, G. Feng, Z. Quan, W. Zhiyu, H. Han, L. Beibei, W. Wubo, W. Xuzhen and Q. Jieshan, Nanoscale, 2013, 5, 2999 RSC.
- P. Harremoes, in The Precautionary Principle in the 20th Century: Late Lessons from Early Warnings, ed. P. Harremoes, Routledge, 2013, pp. 161–169 Search PubMed.
- J. Tang, L. Xiang, F. Zhao, F. Pan, S. Wang and X. Zhan, Anal. Lett., 2015, 48, 796–808 CrossRef CAS.
- K. Yin, F. Yu, D. Liu, Z. Xie and L. Chen, Sens. Actuators, B, 2016, 223, 799–805 CrossRef CAS.
- Y. Yang, J. Chen and Y.-P. Shi, Talanta, 2012, 97, 222–228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04605a |
| This journal is © The Royal Society of Chemistry 2020 |