
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituted adenine quartets: interplay between substituent effect, hydrogen bonding, and aromaticity†
Halina Szatylowicz *a,
Paulina H. Marekab,
Olga A. Stasyuk*c,
Tadeusz M. Krygowskib and
Miquel Solàc
*a,
Paulina H. Marekab,
Olga A. Stasyuk*c,
Tadeusz M. Krygowskib and
Miquel Solàc
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: halina@ch.pw.edu.pl
bUniversity of Warsaw, Faculty of Chemistry, Pasteura 1, 02-093 Warsaw, Poland
cDepartment of Chemistry, Institute of Computational Chemistry and Catalysis, University of Girona, C/ M. Aurèlia Capmany, 69, 17003 Girona, Spain. E-mail: o.a.stasuk@gmail.com
First published on 18th June 2020
Abstract
Adenine, one of the components of DNA/RNA helices, has the ability to form self-organizing structures with cyclic hydrogen bonds (A4), similar to guanine quartets. Here, we report a computational investigation of the effect of substituents (X = NO2, Cl, F, H, Me, and NH2) on the electronic structure of 9H-adenine and its quartets (A4-N1, A4-N3, and A4-N7). DFT calculations were used to show the relationships between the electronic nature of the substituents, strength of H-bonds in the quartets, and aromaticity of five- and six-membered rings of adenine. We demonstrated how the remote substituent X modifies the proton-donating properties of the NH2 group involved in the H-bonds within quartets and how the position of the substituent and its electronic nature affect the stability of the quartets. We also showed the possible changes in electronic properties of the substituent and aromaticity of adenine rings caused by tetramer formation. The results indicate that the observed relationships depend on the A4 type. Moreover, the same substituent can both strengthen and weaken intermolecular interactions, depending on the substitution position.
Introduction
Purine derivatives play a significant role in medicinal chemistry and they are also important intermediates for the synthesis of biologically-active nucleoside analogues, and frequently exhibit antiviral or antitumor properties.1 The nature and position of the substituents determine their reactivity and biological activity. Adenine (6-amino substituted purine derivative), one of the building blocks of DNA/RNA helices, can participate in intermolecular interactions which may lead to substantial changes in its electronic structure and, in consequence, to changes in its chemical/physicochemical/biochemical properties. In this perspective, the knowledge of the changes in the electronic structure of adenine caused by well-defined factors, such as introduction of substituents with specific electron donating/accepting properties, is of fundamental importance. It should be stressed that the effects of the intermolecular interactions (H-bonding, π-stacking, etc.) on the electronic structure and molecular properties of nucleic acid bases have long been a subject of wide and intensive studies.2 However, the effects of substituents on the properties of nucleic acid bases are much less represented in the literature. It was reported that in the synthesis of deoxyribonucleosides, substituents at the C8 position of purine can greatly affect the rate of deoxyribosyl transfer to the base and the nature of the nucleoside formation.3 An electron-donating substituent directs deoxyribosyl transfer to N9 of the base, while an electron-withdrawing substituent leads to the N3-product. Halogen substituents at the C8 position of adenine do not change the tautomeric preference in the gas phase but induce significant changes in vibrational frequencies and Raman intensities.4 Substituent effects on hydrogen bonding were most frequently studied in structurally-modified Watson–Crick base pairs (adenine–thymine and guanine–cytosine).5–11One of the interesting features of DNA/RNA bases is their ability to form self-assembled structures (clusters). Among them, the most commonly studied are cyclic hydrogen-bonded quartets (tetramers),12,13 which occur naturally in telomeres,14 immunoglobulin gene switch regions,15 and certain gene promoters.16 The guanine quartet is the most well-known for a long time, for reviews see ref. 17 and 18. Although adenine quartets (A4) have been studied experimentally19–22 and have also been the subject of computational investigations,23–26 the effect of remote substituents on hydrogen bonds in adenine tetramers has not yet been studied. Introduction of additional functionalities to the adenine with subsequent formation of quartet with intermolecular adenine⋯adenine hydrogen-bond interactions is one of the design strategies to enrich chemistry of purine derivatives with ability to bind many metal ions.27 Since the stabilization of such complexes depends on the functional groups, the simulation of adenine quartets with certain substituents of different electronic nature is a good step toward design of synthetic functional materials. Moreover, changes in aromaticity of the adenine rings in the tetramer could be used to fine-tune π–π stacking interactions.
In this work, we have investigated computationally the effect of substituents (X = NO2, Cl, F, H, Me, NH2) on electronic structure of 9H-adenine and its quartets (A4-N1, A4-N3, and A4-N7) with dispersion-corrected Density Functional Theory. We have demonstrated how the position of the substituent and its electronic nature affect the stability of the quartets. Additionally, we have analyzed the influence of the electronic nature of the substituents on the strength of H-bonds in the quartets and aromaticity of five- and six-membered rings of adenine.
Methodology
Quartets of substituted 9H adenine tautomer were chosen as models for studying interrelationships between electronic nature of substituents, aromaticity of transmitting moiety, and intermolecular H-bonding between individual adenine molecules. Three types of adenine quartets were considered: A4-N1, A4-N3 and A4-N7. They are formed by one hydrogen bond between two neighbouring bases as shown in Fig. 1. These quartets have a non-planar S4-symmetric structure as the global minimum. However, they are forced into planarity by stacking interactions which compensate for the energy spent on planarization. The planarization energy of A4-N3 and A4-N7 was found to be 0.2 and 11 kcal mol−1, respectively, while for A4-N1 system it was 22.3 kcal mol−1.28 The low stability of planar A4-N1 quartet is probably a main reason why this type of quartet has not been characterized by X-ray crystallography in contrast to A4-N3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 and A4-N7.20
29 and A4-N7.20
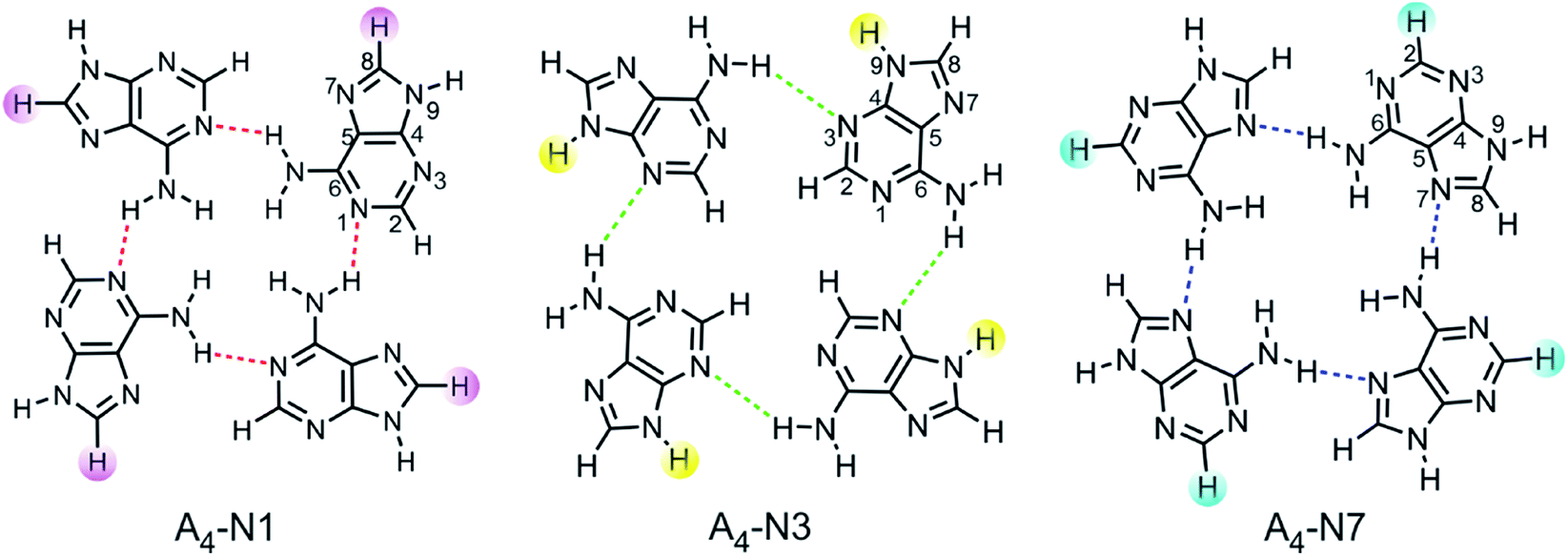 | ||
| Fig. 1 Analyzed tetramers of adenine with places of substitution marked, C8, N9, and C2, respectively. | ||
Selected substituents, varying in their electronic properties (X = NO2, Cl, F, H, Me, NH2), were inserted into each adenine molecule at the same position within a certain quartet. For A4-N1 and A4-N3 tetramers, it was done either at the C8 or N9 position, whereas in the case of A4-N7 tetramer they were introduced either at the C2 or N9 position, resulting with 33 systems to be analysed. Although N9 substitution is less common, some organic ligands with functional groups can be incorporated into the adenine molecule at the N9 position.27
Geometry optimization of each system was performed using ADF program30–32 at the BLYP/TZ2P level of theory33–35 with Grimme D3 dispersion correction.36,37 To ensure that the obtained geometries are in energy minima, the vibrational analysis was performed and no imaginary frequencies were found.
The bonding energy of the tetramers, or hydrogen bond energy, EHB, was calculated as sum of interaction and preparation energies.
| EHB = Eint + Eprep | (1) |
Interaction energy, Eint, was calculated as the difference between the energy of the tetramer and the sum of the energies of its components (adenines) in the tetramer geometry.
| Eint = Etetramer − 4Emonomer in tetramer geometry | (2) |
Preparation energy, Eprep, was calculated as the difference between energies of adenine monomers in the tetramer geometry and adenine monomers in equilibrium geometry.
| Eprep = 4Emonomer in tetramer geometry − 4Emonomer | (3) |
Analysis of intermolecular interactions within the framework of Bader's Quantum Theory of Atoms in Molecules (QTAIM) was performed using the AIMAll program package.38 Such parameters as the electron density in bond critical points (BCPs), ρBCP, its Laplacian, ∇2ρBCP, density of the total electron energy in BCP, HBCP, and its two components, potential and kinetic electron energy densities, VBCP and GBCP, respectively, were taken into account.
Substituent effect was described using cSAR (charge of the Substituent Active Region) concept.39,40 The cSAR(X) value was calculated as the sum of Voronoi charges41 of the atoms of substituent X and ipso C- or N-atom of adenine.
| cSAR(X) = q(X) + q(Cipso or Nipso) | (4) |
The choice of VDD charges is based on our previous research into the electronic structure of the nitro group in para-substituted nitrobenzene derivatives42 and studies of adenine systems substituted in C-X and N-X positions.43
Electron delocalization was characterized using geometry-based aromaticity index HOMA (Harmonic Oscillator Model of Aromaticity),44,45 which is defined as:
 | (5) |
The local HOMA index was calculated for both five- and six-membered adenine rings. This aromaticity descriptor was previously successfully used to describe the aromaticity changes in adenine rings due to H-bonding and complexation of metal ions.46 The obtained values of the bonding energies and all substituent effect descriptors are gathered in Tables S1–S4.† cSAR and HOMA parameters were calculated for each individual adenine molecule in the tetramer. Because their values are similar within a given tetramer for all molecules, the mean values are given in the Tables S2 and S4,† respectively.
Results and discussion
The optimized A4-N3 and A4-N7 structures with substituents resemble flat geometry in symmetry close to C4h, while the substituted A4-N1 quartets are more distorted and adopt a similar pattern to that described by the S4 point group. The unconstrained optimized geometries of systems with different substituents do not change significantly within one quartet type (Fig. S1†). The obtained H-bond lengths for unsubstituted A4-N3 (dN⋯N = 3.06 Å) and A4-N7 (dN⋯N = 2.93 Å) are close to the experimental values, 3.05 and 2.91 Å for A4-N3 and A4-N7, respectively.20,29 The three quartets have similar stabilities. Our calculations show that unsubstituted A4-N3 tetramer is the most stable followed by A4-N7 at only 0.4 kcal mol−1 and A4-N1 at 2.7 kcal mol−1 (Table S1†). However, substituents affect the electronic structure of the tetramers and change this stability order. In general, among N9-substituted tetramers the A4-N3 is the most stable quartet. For C2/C8-substituted quartets, the A4-N3 tetramer is more favourable only with H and Me substituents, while for the other studied substituents the A4-N7 tetramer is preferred. It should be noted that the most stable substituted A4-N7 tetramers (except X = H and Me) have the shortest hydrogen bonds (dN⋯H < 1.9 Å). According to the substitution position, C2/C8-substituted tetramers are considerably more stable than the N9-substituted ones.Intramolecular interactions, the so-called substituent effect, can be considered from three points of view, as classical and reverse substituent effects, as well as the influence of the substituent on the properties of transmitting moiety. The classical substituent effect, introduced and quantified by Hammett,47,48 characterizes how a substituent X affects properties of a fixed group Y (so-called “reaction site”) in a substituted system X–R–Y (R – transmitting moiety). In our case, properties of the reaction site – the amino group – can be described either by the cSAR(NH2) value or by the strength of H-bonds.
Substituent effect on H-bonding – classical substituent effect
An illustrative example of the classical substituent effect is the dependence of cSAR(NH2) on cSAR(X) shown in Fig. 2. The dependence was considered not only for quartets but also for substituted adenine monomers. Slopes of linear equations and determination coefficients are presented in Table 1. Note that correlations are acceptable or even good with R2 > 0.84, except for C2-substituted A4-N7 series, where very narrow range of cSAR(NH2) values was found. In all cases the slope a is negative meaning that electron-donating properties of the amino group decrease with increasing electron-donating ability (more positive cSAR(X) value) of substituent X. According to the slope a value, the C8 position in monomer is the most sensitive to the electronic nature of substituent X. This is consistent with results for adenine based on other substituent effect descriptors.49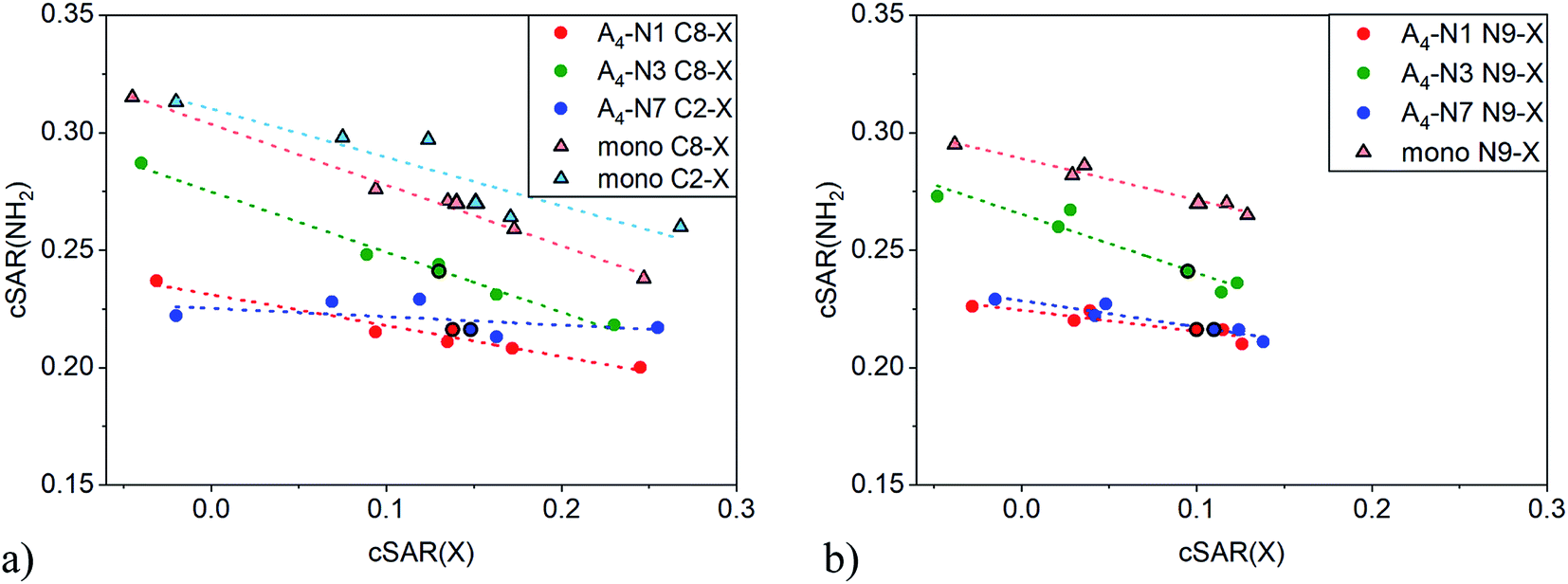 | ||
| Fig. 2 Dependences of cSAR(NH2) on cSAR(X) for C2-/C8- (a) and N9- (b) substituted adenine monomers and tetramers. More positive cSAR(X) values correspond to more electron-donating substituents. | ||
| Position | C2/C8 | N9 | ||
|---|---|---|---|---|
| a | R2 | a | R2 | |
| Monomer C2 | −0.206 | 0.835 | — | — |
| Monomer C8 | −0.259 | 0.992 | — | — |
| Monomer N9 | — | — | −0.176 | 0.969 |
| A4-N1 | −0.133 | 0.957 | −0.090 | 0.845 |
| A4-N3 | −0.257 | 0.987 | −0.250 | 0.921 |
| A4-N7 | −0.036 | 0.248 | −0.112 | 0.896 |
As expected, cSAR(NH2) values are higher in monomers (triangles) than in quartets (circles) where amino group is involved in H-bond. The reason is accumulation of negative charge on the N atom of proton-donating –NH part of the amino group during formation of hydrogen bond. As can be seen in Fig. 2, electron-donating power of the NH2 group is less affected by the tetramer formation in A4-N3 quartets than in the others, because the formed H-bonds in A4-N3 are the longest among all quartets, dN⋯H > 2.0 Å. This observation was also confirmed by direct comparison of cSAR(NH2) of NH2 attached to C6 in monomers and tetramers, see Fig. S2 and Table S5.† In A4-N3, cSAR(NH2) values of monomers and tetramers are comparable, while for A4-N1 and A4-N7, cSAR(NH2) in tetramers are significantly smaller than in monomers. Moreover, A4-N3 quartet is the most responsive to the electronic nature of substituent X either at C8 or N9 position.
As a consequence of the substituent effect on the properties of the amino group, H-bond energy in quartets also changes. Fig. 3 shows the dependence of bonding energy, EHB, in A4-N1, A4-N3, and A4-N7 tetramers on the electronic properties of remote substituents, where substituent X is described by cSAR(X) value. More positive cSAR values indicate better electron-donating properties of substituents. According to Fig. 2, substituent X changes properties of the amino group in the same direction regardless to the quartet type. However, the substituent effect on H-bond energy is different. This means that in addition to the substituent effect on the amino group, the substituent X also affects the proton-accepting abilities of N atoms of both rings. Positive and negative slopes of linear equations (Table 2) show the direction of the substituent effect, whereas their magnitude displays a sensitivity of intermolecular interactions to electronic properties of the substituents X.
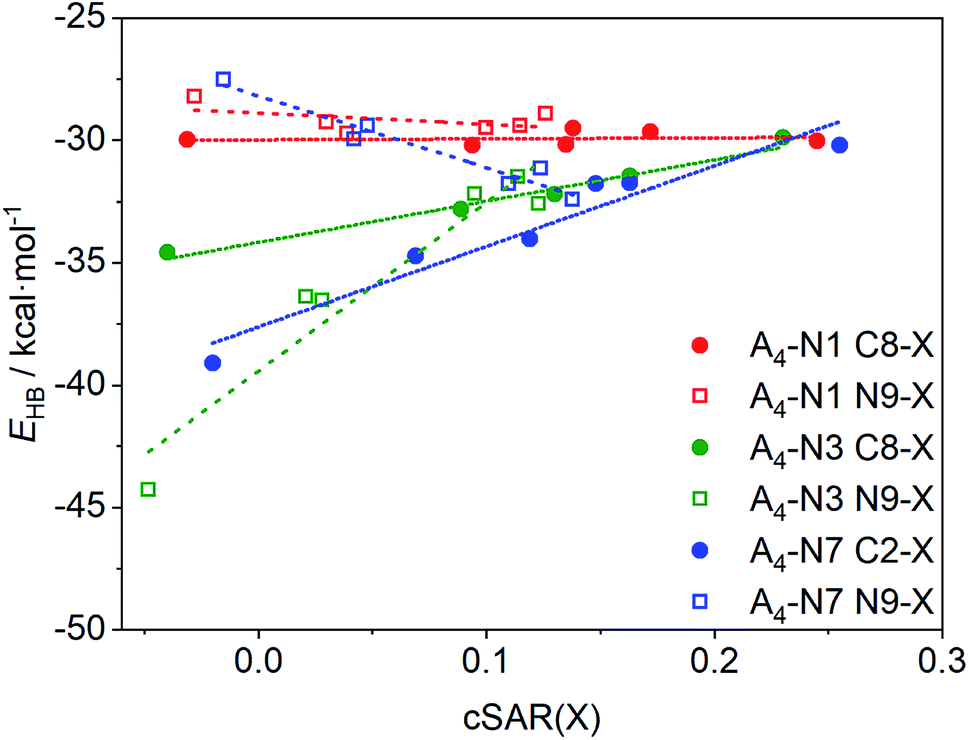 | ||
| Fig. 3 Relationships between the bonding energies, EHB, and cSAR(X) of substituent for the studied adenine quartets. | ||
| Substitution position | C2/C8 | N9 | ||
|---|---|---|---|---|
| Quartet type | a | R2 | a | R2 |
| A4-N1 | 0.413 | 0.018 | −4.468 | 0.244 |
| A4-N3 | 16.86 | 0.970 | 68.91 | 0.921 |
| A4-N7 | 32.91 | 0.934 | −29.23 | 0.936 |
Two groups can be distinguished in the studied systems: (i) with positive slope, where H-bond weakens with increase in electron-donating properties of substituent X, and (ii) with negative slope, where H-bond strengthens with the same substituent change. The first group consists of all adenine quartets with the substituents at the carbon atom (either C2 or C8) and N9-substituted A4-N3 quartet. The second group is represented by A4-N1 and A4-N7 quartets with the N9-substituents. In the case of A4-N3 and A4-N7 tetramers (R2 > 0.92), the substituents notably change the energy of the intermolecular interactions. The largest changes were found in the N9-substituted A4-N3 system. This is consistent with observation of substituent effect X on properties of the NH2 group (Fig. 2). On the other hand, for substituted A4-N1 quartets the changes in the H-bonds strength caused by the substituent X at any position are very small (slopes a are only 0.413 and −4.468, respectively) and R2 of these regressions are less than 0.25. Such low coefficients of determination (Table 2) can be explained by very narrow energy range. This is due to the fact that both C8 and N9 substitution positions in A4-N1 quartets are far from the atoms participated in the H-bond formation and the substituent effect is not transmitted so far. Different slopes for the A4-N7 substituted tetramers can be explained as follows: when substituent X is attached to the N9 position it changes the electron density on the N7 atom and modifies its proton-accepting power to a greater extent, increasing EHB for electron-donating X groups, whereas the substitution at the C2 position affects mostly the proton-donating ability of the amino group decreasing EHB for electron-donating X groups. Thus, depending on the place of substitution the same substituent X can either weaken or enhance the intermolecular H-bonds in A4-N7 tetramers.
Summarizing, the strong electron-accepting substituents contribute to strengthening of H-bonds in A4-N3 (at the C8 and N9 position) and A4-N7 (at the C2 position) quartets. The electron-donating substituents make H-bonds stronger only in A4-N7 quartet if they are located at the N9 position. Introducing different substituents into A4-N1 quartet has relatively small effect.
Additional intermolecular interactions
Some interesting observation was done when considering the relationships between strength of intermolecular interactions and length of the hydrogen bonds, dN⋯H, shown in Fig. 4. Despite the fact that H-bond lengths in A4-N3 quartets are longer (dN⋯H > 2.0 Å) than in the A4-N7 ones (dN⋯H < 1.9 Å), the bonding energies in A4-N3 tetramers are comparable in most cases with the energies in A4-N7 systems or even stronger. It is worth mentioning that the two systems with the strongest and the weakest H-bonds have a common element, namely the nitro group as a substituent. These terminal cases were found in the A4-N3 and A4-N7 tetramers with the nitro substituent at the N9 position (Fig. 5). Their energies are −44.3 and −27.5 kcal mol−1 for A4-N3 and A4-N7, although H-bond lengths are 2.01 and 1.91 Å, respectively. The reduced EHB in A4-N7 N9-NO2 substituted tetramer is expected because of the electron-accepting character of the nitro group as discussed above, whereas the large EHB in A4-N3 N9-NO2 substituted tetramer is understood not only by the electron-accepting character of the substituent (vide supra), but also by the presence of additional intermolecular interactions.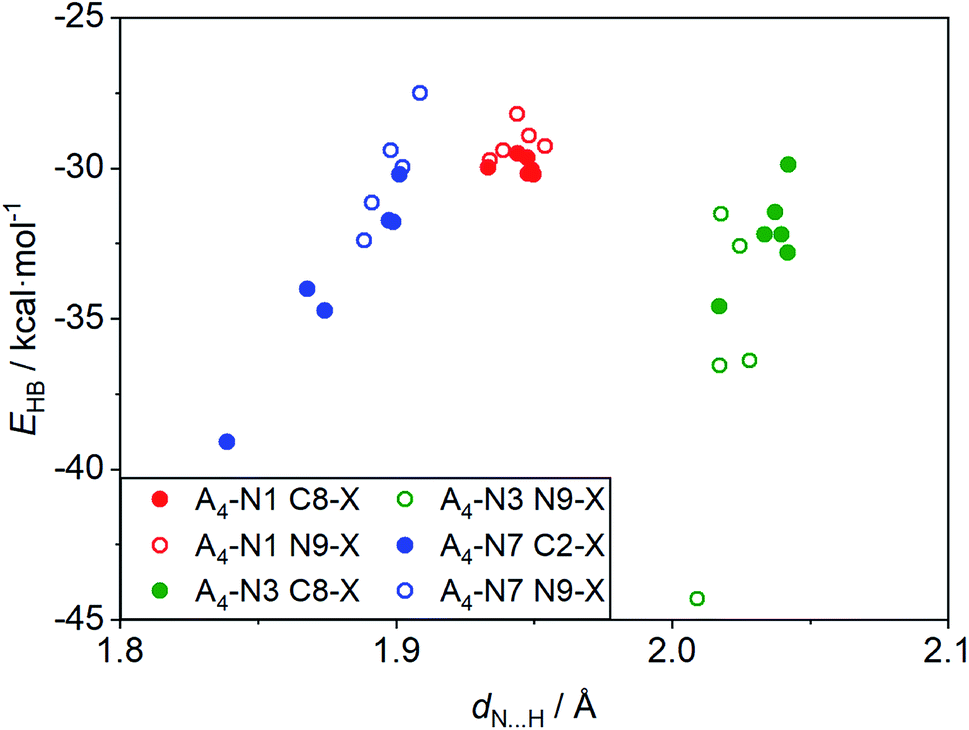 | ||
| Fig. 4 Relationships between bonding energy and N⋯H length, dN⋯H, for the studied substituted tetramers. | ||
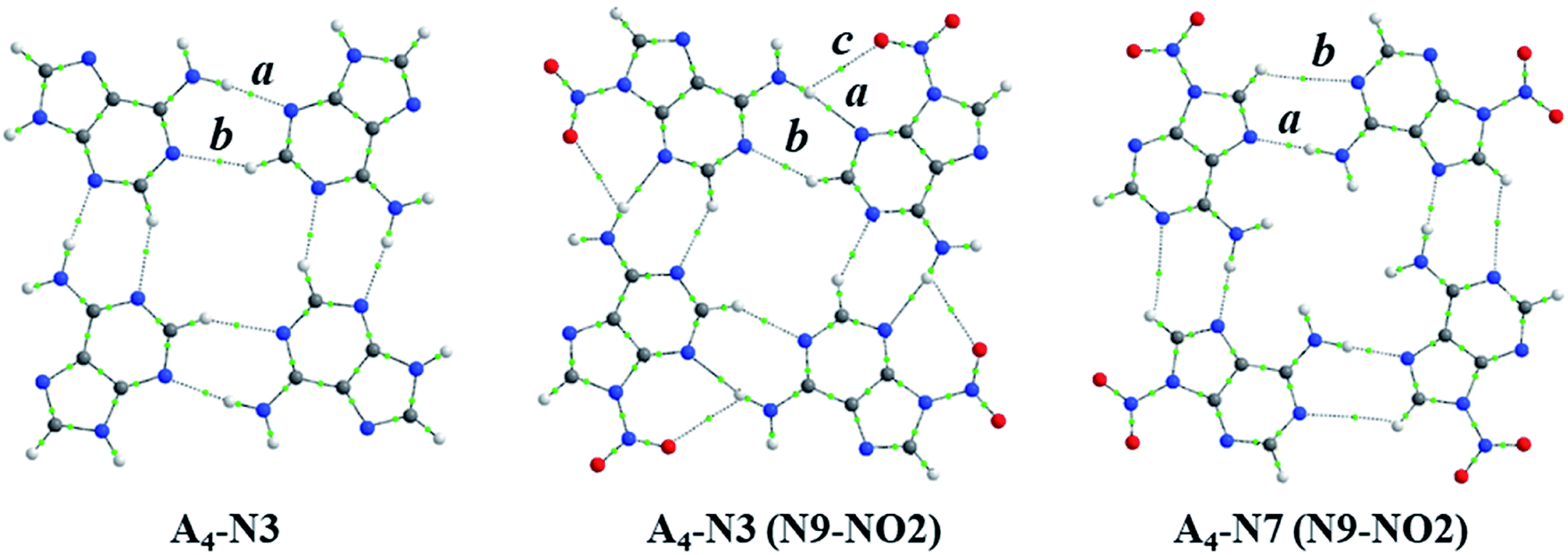 | ||
| Fig. 5 Structures of A4-N3, A4-N3(N9-NO2) and A4-N7(N9-NO2) systems with bond critical points: a is a typical H-bond in quartet, b – additional CH⋯N contact, c – additional O⋯HN contact. | ||
Upon a detailed examination of their structures we found two additional close contacts in A4-N3 quartets (Fig. 5). One of them is formed between C–H unit of one adenine molecule and N1 atom of the other adenine (CH⋯N), whereas the second contact is made between NO2 group and NH2 group (O⋯HN). To characterize these extra contacts quantitatively we performed QTAIM analysis for 3 systems: unsubstituted A4-N3, and N9-NO2-substituted A4-N3 and A4-N7. Particular attention was paid to CH⋯N interactions in A4-N3 quartets. The distance dH⋯N is 2.33 Å in A4-N3, and 2.27 Å in its N9-NO2-substituted analogue, whereas such dH⋯N in adenine–thymine pair is 2.75 Å. The parameters of the corresponding BCPs are collected in Table 3. Their values confirm that CH⋯N and O⋯HN contacts are weak closed-shell interactions. Although the interactions between NO2 and NH2 groups are very week, the CH⋯N contacts in A4-N3 quartets can be considered as weak H-bonds,50 and therefore additionally contribute to their stability.
| BCP | Atoms | dH⋯N Å−1 | ρBCP | ∇2ρBCP | V | G | H | |V|/G | DI |
|---|---|---|---|---|---|---|---|---|---|
| A4-N3(N9-H) | |||||||||
| a | N–H | 2.03 | 0.024 | 0.077 | −0.016 | 0.018 | 0.001 | 0.921 | 0.080 |
| b | N–H | 2.33 | 0.012 | 0.056 | −0.008 | 0.011 | 0.003 | 0.754 | 0.041 |
|
|||||||||
| A4-N3(N9-NO2) | |||||||||
| a | N–H | 2.01 | 0.025 | 0.079 | −0.017 | 0.019 | 0.001 | 0.936 | 0.082 |
| b | N–H | 2.27 | 0.014 | 0.063 | −0.010 | 0.013 | 0.003 | 0.782 | 0.046 |
| c | O–H | 2.67 | 0.003 | 0.024 | −0.002 | 0.004 | 0.002 | 0.565 | 0.004 |
|
|||||||||
| A4-N7(N9-NO2) | |||||||||
| a | N–H | 1.91 | 0.033 | 0.095 | −0.023 | 0.023 | 0.000 | 0.989 | 0.104 |
| b | N–H | 3.07 | 0.002 | 0.011 | −0.001 | 0.002 | 0.001 | 0.559 | 0.005 |
Change in substituent electronic properties – reverse substituent effect
The cSAR concept allows us to study the reverse substituent effect,42 which describes how the electronic properties of substituents X depend on the properties of the moiety R–Y to which they are attached. Various substituent constant scales51–53 “represent” a fixed electronic structure of substituents or, in other words, its fixed electron-donating/attracting power. In turn, cSAR(X) value characterizes not the fixed electronic properties of substituent X but shows its properties depending on the substituted moiety. The reverse substituent effect can be illustrated by changes in cSAR(X) values for a particular substituent X when it is considered in different adenine quartets (Fig. S3†). The range of cSAR(X) values (Δ in Table S4†) documents that the properties of X depend on the moiety to which it is attached. For C-substituted systems, the largest change is observed for the NH2 substituent, while for the N-substituted ones the cSAR(NO2) changes the most; these changes correspond to approximately 13 and 20% of the total variation of the cSAR(X) values in the series, respectively.The effect of tetramer formation on the electronic properties of X can be demonstrated by comparing cSAR(X) in the quartet with the values in the adenine monomer (Fig. S4 and Table S6†). Undoubtedly, very high determination coefficients, R2 > 0.99 and slopes close to 1.0 indicate a large similarity of interactions between X and substituted moieties in both cases. For N9-substitution in A4-N3 systems, the slope a is slightly larger than in other series. This means that in this case the substituent X is more responsive to tetramer formation most probably due to additional spatial interactions with the amino group of neighboring adenine molecule (Fig. 5). In adenine monomer, cSAR(X) values of all substituents are slightly greater than their values in tetramers (Table S4†), where these values depend on the position of substituent X relative to the proton-acceptor place (N1, N3 or N7) and proton-donor group (NH2). Only in N9-substituted A4-N7 tetramer, cSAR(X) values become more positive than in monomer due to stronger withdrawal of electronic density to the H-bonding area. Importantly, the H-bond formation may decrease the electron-attracting ability of the nitro group by ∼60%.
Substituent effect on aromaticity
The substituent effect on aromaticity of the transmitting moiety can be demonstrated by changes in HOMA index for five- and six-membered rings in the substituted quartets, as presented in Fig. S5 and Table S7.† Correlations between HOMA indices and cSAR(X) values indicate that electron-donating substituents at the C2 and C8 positions decrease the aromaticity of the five-membered rings, while at the N9 position cause the opposite effect; the latter is in line with previous studies54 showing that N-substitution in imidazole rings increases the aromaticity for electron-donating groups. The six-membered rings are highly aromatic (HOMA > 0.9) and is slightly responsive only to the substituents at the C8 position, whereas C2- and N9-substitution does not significantly affect them. Increase in the aromaticity of the five-membered ring by substitution, in general, goes with a reduction of the aromaticity of the six-membered ring. Aromaticity changes by N9-substitution in the five-membered ring translate into small variation in the six-membered ring. Moreover, minor aromaticity changes by C2-substitution are in line with previous studies indicating a high resistance of the π-electron structure to the substituent effect.55 According to the slopes a, the highest HOMA sensitivity to the substituent effect was observed in A4-N3 quartets, most likely due to multiple noncovalent interactions between adenine molecules. Different slopes observed for five- and six-membered rings may depend on the number of bonds between the substituent X and the “reaction site” (NH2). A similar observation is well known for meta and para substituted benzene derivatives, where the substituent effect depends on the number of bonds between X and Y.56Comparison of HOMA values for tetramers with values for monomers of substituted adenine (Table S3†) clearly shows that aromaticity of both rings slightly decreases upon tetramer formation. The five-membered rings of A4-N7 quartet become more aromatic than in monomer because their N7 atom participates in the H-bond as a proton-acceptor. Great correlations between HOMA values of five-membered rings for substituted monomers and tetramers (R2 > 0.94; Fig. S6 and Table S8†) with slopes a close to 1.0 point out that aromaticity of five-membered rings does not change much upon tetramer formation. Aromaticity of six-membered rings in tetramer becomes either more sensitive (a > 1.0) or less sensitive (a < 1.0) to the nature of substituent X in comparison with monomer.
Conclusions
The substituent X, depending on the position, modifies the proton-donating properties of the NH2 group and thus the strength of hydrogen bonds in adenine quartets. According to the findings, to increase the stability of the A4-N3 quartet, it is recommended to introduce a strong electron-accepting substituent into each adenine molecule at the C8 or N9 positions. On the other hand, to stabilize A4-N7 quartet it is necessary to substitute C2 position with electron-accepting substituent or N9 position with electron-donating substituent. Substituent effect in the A4-N1 quartet is not pronounced and cannot be considered as a tool for tuning stability of the quartet.The obtained results show that the electronic properties of the substituent X and aromaticity of five- and six-membered rings are affected by the tetramer formation and are slightly different depending on the A4 type. For C-substituted systems, the largest change in cSAR(X) values is observed for the NH2 group, while for the N-substituted ones the cSAR(NO2) changes the most. Aromaticity of both adenine rings in A4-N3 tetramer is more sensitive to electronic nature of the substituents than in other tetramers. Its “special properties” are most likely due to the fact that additional weak hydrogen bonds occur here as documented by the results of QTAIM analysis. In general, to increase aromaticity of the five-membered rings the electron-accepting substituents should be introduced at the C2 or C8 positions, or the electron-donating substituent at the N9 position. In turn, six-membered rings are highly aromatic (HOMA > 0.9) and slightly responsive to the substituents at the C8 position, whereas C2- and N9-substitution does not affect them.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. S. and T. M. K. thank the National Science Centre of Poland for supporting this work under the grant no. UMO-2016/23/B/ST4/00082. P. H. M. would like to acknowledge Operational Project Knowledge Education Development 2014–2020 co-financed by European Social Fund. The work has been performed under the Project HPC-EUROPA3 (INFRAIA-2016-1-730897), with the support of the EC Research Innovation Action under the H2020 Programme; in particular, P. H. M. gratefully acknowledges the support of the Institute of Computational Chemistry and Catalysis of the University of Girona and the computer resources and technical support provided by Barcelona Supercomputing Center. M. S. and O. A. S. are grateful to the Ministerio de Economía y Competitividad (MINECO) of Spain (project CTQ2017-85341-P and Juan de la Cierva formación contract FJCI-2017-32757 to O. A. S.) and the Generalitat de Catalunya (project 2017SGR39).References
- M. Legraverend and D. S. Grierson, The Purines: Potent and Versatile Small Molecule Inhibitors and Modulators of Key Biological Targets, Bioorg. Med. Chem., 2006, 14, 3987–4006 CrossRef CAS.
- J. Sponer, J. Leszczynski and P. Hobza, Electronic Properties, Hydrogen Bonding, Stacking, and Cation Binding of DNA and RNA Bases, Biopolym, 2001, 61, 3–31 CrossRef CAS PubMed.
- M. C. Huang, J. A. Montgomery, M. C. Thorpe, E. L. Stewart, J. A. III Secrist and R. L. Blakley, Formation of 3-(2′-Deoxyribofuranosyl) and 9-(2′-Deoxyribofuranosyl) Nucleosides of 8-Substituted Purines by Nucleoside Deoxyribosyltransferase, Arch. Biochem. Biophys., 1983, 222, 133–144 CrossRef CAS PubMed.
- Y.-L. Chen, D.-Y. Wu and Z.-Q. Tian, Theoretical Investigation on the Substituent Effect of Halogen Atoms at the C8 Position of Adenine: Relative Stability, Vibrational Frequencies, and Raman Spectra of Tautomers, J. Phys. Chem. A, 2016, 120, 4049–4058 CrossRef CAS PubMed.
- C. Fonseca Guerra, T. van der Wijst and F. M. Bickelhaupt, Substituent Effects on Hydrogen Bonding in Watson–Crick Base Pairs. A Theoretical Study, Struct. Chem., 2005, 16, 211–221 CrossRef.
- C. Fonseca Guerra, T. van der Wijst and F. M. Bickelhaupt, Supramolecular Switches Based on the Guanine–Cytosine (GC) Watson–Crick Pair: Effect of Neutral and Ionic Substituents, Chem.–Eur. J., 2006, 12, 3032–3042 CrossRef PubMed.
- C. Fonseca Guerra, T. van der Wijst and F. M. Bickelhaupt, Nanoswitches Based on DNA Base Pairs: Why Adenine–Thymine is Less Suitable than Guanine–Cytosine, ChemPhysChem, 2006, 7, 1971–1979 CrossRef PubMed.
- A. Ebrahimi, S. M. Habibi Khorassani, H. Delarami and H. Esmaeeli, The Effect of CH3, F and NO2 Substituents on the Individual Hydrogen Bond Energies in the Adenine–Thymine and Guanine–Cytosine Base Pairs, J. Comput.-Aided Mol. Des., 2010, 24, 409–416 CrossRef CAS PubMed.
- V. Nikolova and B. Galabov, Effects of Structural Variations on the Hydrogen Bond Pairing between Adenine Derivatives and Thymine, Maced. J. Chem. Chem. Eng., 2015, 34, 159–167 CrossRef CAS.
- K. Jana and B. Ganguly, In Silico Studies with Substituted Adenines to Achieve a Remarkable Stability of Mispairs with Thymine Nucleobase, New J. Chem., 2016, 40, 1807–1816 RSC.
- A. C. Castro, M. Swart and C. Fonseca Guerra, The Influence of Substituents and the Environment on the NMR Shielding Constants of Supramolecular Complexes Based on A–T and A–U Base Pairs, Phys. Chem. Chem. Phys., 2017, 19, 13496–13502 RSC.
- C. Roberts, J. C. Chaput and C. Switzer, Beyond Guanine Quartets: Cation-Induced Formation of Homogenous and Chimeric DNA Tetraplexes Incorporating iso-Guanine and Guanine, Chem. Biol., 1997, 4, 899–908 CrossRef CAS PubMed.
- C.-D. Xiao, T. Ishizuka, X.-Q. Zhu, Y. Li, H. Sugiyama and Y. Xu, Unusual Topological RNA Architecture with an Eight-Stranded Helical Fragment Containing A-, G-, and U-Tetrads, J. Am. Chem. Soc., 2017, 139, 2565–2568 CrossRef CAS PubMed.
- D. Rhodes and R. Giraldo, Telomere Structure and Function, Curr. Opin. Struct. Biol., 1995, 5, 311–322 CrossRef CAS PubMed.
- D. Sen and W. Gilbert, Formation of Parallel Four-Stranded Complexes by Guanine-Rich Motifs in DNA and its Implications for Meiosis, Nature, 1988, 334, 364–366 CrossRef CAS PubMed.
- A. I. Murchie and D. M. Lilley, Retinoblastoma Susceptibility Genes Contain 5′ Sequences with a High Propensity to Form Guanine-Tetrad Structures, Nucleic Acids Res., 1992, 20, 49–53 CrossRef CAS.
- J. T. Davis, G-Quartets 40 Years Later: From 5′-GMP to Molecular Biology and Supramolecular Chemistry, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Interaction of Metal Complexes with G-Quadruplex DNA, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- P. K. Patel, A. S. Koti and R. V. Hosur, NMR Studies on Truncated Sequences of Human Telomeric DNA: Observation of a Novel A-Tetrad, Nucleic Acids Res., 1999, 27, 3836–3843 CrossRef CAS PubMed.
- B. Pan, Y. Xiong, K. Shi, J. Deng and M. Sundaralingam, Crystal Structure of an RNA Purine-Rich Tetraplex Containing Adenine Tetrads: Implications for Specific Binding in RNA Tetraplex, Structure, 2003, 11, 815–823 CrossRef CAS PubMed.
- B. Pan, Y. Xiong, K. Shi and M. Sundaralingam, An Eight-Stranded Helical Fragment in RNA Crystal Structure: Implications for Tetraplex Interaction, Structure, 2003, 11, 825–831 CrossRef CAS PubMed.
- M. S. Searle, H. E. L. Williams, C. T. Gallagher, R. J. Grant and M. F. G. Stevens, Structure and K+ Ion-Dependent Stability of a Parallel-Stranded DNA Quadruplex Containing a Core A-Tetrad, Org. Biomol. Chem., 2004, 2, 810–812 RSC.
- J. Gu and J. Leszczynski, The Structure, Stability, H-Bonding Pattern, and Electrostatic Potential of Adenine Tetrads, Chem. Phys. Lett., 2001, 335, 465–474 CrossRef CAS.
- M. Meyer and J. Suhnel, Density Functional Study of Adenine Tetrads with N6–H6⋯N3 Hydrogen Bonds, J. Phys. Chem. A, 2008, 112, 4336–4341 CrossRef CAS PubMed.
- C. Fonseca Guerra, T. van der Wijst, J. Poater, M. Swart and F. M. Bickelhaupt, Adenine versus Guanine Quartets in Aqueous Solution: Dispersion-Corrected DFT Study on the Differences in π-Stacking and Hydrogen-Bonding Behavior, Theor. Chem. Acc., 2010, 125, 245–252 Search PubMed.
- A. Karatosun, M. Cankaya and A. Tekin, Symmetry-Adapted Perturbation Theory Potential for the Adenine Dimer, Phys. Chem. Chem. Phys., 2018, 20, 26303–26314 RSC.
- B. Mohapatra, Pratibha and S. Verma, Directed Adenine Functionalization for Creating Complex Architectures for Material and Biological Applications, Chem. Commun., 2017, 53, 4748–4758 RSC.
- T. van der Wijst, B. Lippert, M. Swart, C. Fonseca Guerra and F. M. Bickelhaupt, Differential Stabilization of Adenine Quartets by Anions and Cations, J. Biol. Inorg Chem., 2010, 15, 387–397 CrossRef CAS PubMed.
- H. Liu, R. Wang, X. Yu, F. Shen, W. Lan, P. Haruehanroengra, Q. Yao, J. Zhang, Y. Chen, S. Li, B. Wu, L. Zheng, J. Ma, J. Lin, C. Cao, J. Li, J. Sheng and J. Gan, High-Resolution DNA Quadruplex Structure Containing All the A-, G-, C-, T-Tetrads, Nucleic Acids Res., 2018, 46, 11627–11638 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Towards an Order-N DFT Method, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- ADF2019, S. C. M., Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.comsoft Search PubMed.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Development of the Colle–Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- J. G. Snijders, P. Vernooijs and E. J. Baerends, Roothaan–Hartree–Fock–Slater Atomic Wave Functions. Single-zeta, Double-zeta, and Extended Slater-Type Basis Sets for 87Fr–103Lr, At. Data Nucl. Data Tables, 1981, 26, 483–509 CrossRef CAS.
- S. Grimme, Accurate Description of van der Waals Complexes by Density Functional Theory Including Empirical Corrections, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 12.06.03), TK Gristmill Software, 2013, Overland Park KS, USA, http://aim.tkgristmill.com Search PubMed.
- N. Sadlej-Sosnowska, On the Way to Physical Interpretation of Hammett Constants: How Substituent Active Space Impacts on Acidity and Electron Distribution in p-Substituted Benzoic Acid Molecules, Pol. J. Chem., 2007, 81, 1123–1134 CAS.
- N. Sadlej-Sosnowska, Substituent Active Region – A Gate for Communication of Substituent Charge with the Rest of a Molecule: Monosubstituted Benzenes, Chem. Phys. Lett., 2007, 447, 192–196 CrossRef CAS.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, Voronoi Deformation Density (VDD) Charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD Methods for Charge Analysis, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- O. A. Stasyuk, H. Szatylowicz, C. Fonseca Guerra and T. M. Krygowski, Theoretical Study of Electron-Attracting Ability of the Nitro Group: Classical and Reverse Substituent Effects, Struct. Chem., 2015, 26, 905–913 CrossRef CAS.
- Unpublished results, article in preparation.
- J. Kruszewski and T. M. Krygowski, Definition of Aromaticity Basing on the Harmonic Oscillator Model, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- T. M. Krygowski, Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- O. A. Stasyuk, H. Szatyłowicz and T. M. Krygowski, Effect of H-bonding and complexation with metal ions on the π-electron structure of adenine tautomers, Org. Biomol. Chem., 2014, 12, 456–466 RSC.
- L. P. Hammett, The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- L. P. Hammett, Physical Organic Chemistry, McGraw-Hill, New York, 1940 Search PubMed.
- H. Szatylowicz, A. Jezuita, P. H. Marek and T. M. Krygowski, Substituent Effects on the Stability of the Four Most Stable Tautomers of Adenine and Purine, RSC Adv., 2019, 9, 31343–31356 RSC.
- U. Koch and P. L. A. Popelier, Characterization of C–H–O Hydrogen Bonds on the Basis of the Charge Density, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- T. M. Krygowski and B. T. Stępień, Sigma- and Pi- Electron Delocalization: Focus on Substituent Effects, Chem. Rev., 2005, 105, 3482–3512 CrossRef CAS PubMed.
- O. Exner and S. Bohm, Theory of Substituent Effects: Recent Advances, Curr. Org. Chem., 2006, 10, 763–778 CrossRef CAS.
- C. Curutchet, J. Poater, M. Solà and J. Elguero, Analysis of the Effects of N-Substituents on Some Aspect of the Aromaticity of Imidazoles and Pyrazoles, J. Phys. Chem. A, 2011, 115, 8571–8577 CrossRef CAS PubMed.
- T. M. Krygowski, K. Ejsmont, B. T. Stepień, M. K. Cyrański, J. Poater and M. Solà, Relation between the Substituent Effect and Aromaticity, J. Org. Chem., 2004, 69, 6634–6640 CrossRef CAS PubMed.
- M. Shahamirian, M. K. Cyrański and M. T. Krygowski, Conjugation Paths in Monosubstituted 1,2- and 2,3-Naphthoquinones, J. Phys. Chem. A, 2011, 115, 12688–12694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables: hydrogen bonds engaged in stabilizing freely optimized structures with total energy values (Table S1); interaction energy and cSAR values of analyzed systems (Table S2); HOMA aromaticity index observed in the molecules of substituted adenine (Table S3); cSAR values of X-substituted monomer and A4-N1, A4-N3, A4-N7 systems (Table S4); slope values and determination coefficients of cSARtetramer(NH2) and cSARmonomer(NH2) correlations (Table S5); slope values and determination coefficients of cSARtetramer(X) vs. cSARmonomer(X) correlations (Table S6); slope values and determination coefficients of HOMAtetramer(X) and cSAR(X) correlations for 5- and 6-membered rings (Table S7); slope values and determination coefficients of HOMAtetramer(X) and HOMAmonomer(X) correlations for 5- and 6-membered rings (Table S8). Figures: superposition of optimized structures of the substituted adenine tetramer depending on its type (Fig. S1); comparison of cSAR(NH2) values calculated for tetramers and monomers substituted at positions C-X and N-X (Fig. S2); changes in cSAR(X) values depending on the substitution position and type of adenine tetramer (Fig. S3); comparison of cSAR(X) values calculated for tetramers and monomers substituted at C2/C8 and N9 positions (Fig. S4); comparison of HOMA indices for both 5- and 6-membered rings calculated for tetramers substituted at the C2/C8 and N9 positions (Fig. S5); comparison of HOMA values for five- and six-membered rings calculated for substituted monomers and tetramers (Fig. S6). Cartesian coordinates of equilibrium geometries of substituted tetramer and monomer derivatives. See DOI: 10.1039/d0ra04585c |
| This journal is © The Royal Society of Chemistry 2020 |