
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective in vitro delivery of paclitaxel by nanocargo of mesoporous polycaprolactone against triple negative breast cancer cells by minimalizing drug dose†
Himadri Medhi‡
a,
Saratchandra Singh Khumukcham‡b,
Bramanandam Manavathi*b and
Pradip Paik *ac
*ac
aSchool of Engineering Sciences and Technology, University of Hyderabad, Hyderabad 500046, India
bDepartment of Biochemistry, School of Life Sciences, University of Hyderabad, Hyderabad 500046, India. E-mail: manavbrahma@gmail.com
cSchool of Biomedical Engineering, Indian Institute of Technology, BHU, Varanasi 221 005, India. E-mail: paik.bme@iitbhu.ac.in; pradip.paik@gmail.com
First published on 24th June 2020
Abstract
Among the breast cancers, triple negative breast cancer (TNBC) has relatively poor outcomes with a lower survival rate and personalised chemotherapy is the only option available for treatment. Currently in the biomedical domain, nanomaterials with porous morphology have revealed their tremendous possibilities to be used as a nanocarrier in treating cancer by offering void space to encapsulate/entrap biological agents. However, the development of nanocarrier-based targeted therapy with high therapeutic efficacy and fewer side effects to normal cells is always a challenge. Here, we have developed nanocargos based on biodegradable mesoporous PCL (polycaprolactone) of approx. diameter of 75 nm by template removal synthesis techniques. Succeeding the comparative analysis of the nanocarriers, the efficiencies of core shell PCL-mZnO (PZ) and mesoporous PCL (HPZ) to deliver paclitaxel (Taxol/T) into breast cancer cells, is investigated. We found that HPZ nanocapsules have less cytotoxicity and drug loading efficiency of about 600 μg mg−1. The Taxol-loaded nanoparticles (T-HPZ) have exhibited more cytotoxicity than Taxol alone treated cancer cells. Furthermore, T-HPZ treated MDA-MB231 cells are accumulated at G2/M phase of the cell cycle and eventually undergo apoptosis. In support of this, anchorage independent growth of MDA-MB231 cells are significantly inhibited by T-HPZ treatment. Together, our findings suggest that T-HPZ-based paclitaxel (Taxol/T) loaded nanoparticles provide a novel therapeutic option in the treatment of TNBC.
Introduction
Breast cancer is one of the most frequently arising cancers among women with a transience rate of ∼15%. Among the breast cancer types, triple negative breast cancer (TNBC) is highly invasive in nature and has a lower survival rate.1 TNBC is characterized by the lack of expression of three main receptors such as estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2).1 Due to this, TNBC is resistant to the conventional therapies such as trastuzumab, tamoxifen and aromatase inhibitors, which are routinely used in breast cancer treatment. TNBC accounts for 15–20% of all breast cancers with a poor prognosis and high mortality of over 50% as compared to the other subtype of breast cancers.2,3 Chemotherapy is the only principle treatment and effective option for TNBC.4,5 However, treatment associated with this mode is accompanied with myriad side effects as the accessible anticancer drugs not only affect the malignant cells but also damage the healthy cells surrounding the malignant cells. Therefore, continuous efforts are being made in this field to improve the therapeutic efficiency of drugs carrying fewer side effects.6–8 The biodegradable ‘polymer based nano-sized capsules’ are becoming more promising as a drug delivery cargo9–13 because of their advantages over the other nanomaterials used for delivery applications.14 The polymer nanocapsules are engineered by encapsulating the drugs or other biomolecules on the surface or inside of the nanoparticle by chemical or physical approaches.15,16 The engineered nano-drugs formulation thus designed are delivered to the site of interest, and after delivery, these nanocarriers effortlessly degrade in the physiological environment.17Polycaprolactone (PCL) is one of the dexterous polymers that exhibits amalgamation of biodegradable and biocompatible properties, which makes it complimentary for all the biological applications.6,7 Moreover, it is approved by the FDA (Food and Drug Administration) for the delivery of the drugs.8 Paclitaxol (Taxol/T) is an efficient drug used in the treatment of various cancers that include breast, lung, ovarian, cervical, pancreatic cancer, etc.18 It is also widely utilized as a chemotherapeutic negotiator. Despite it exhibits several side effects, due to its wide range of application in cancer treatment, Taxol has been considered as one of the principle regimen in the chemotherapy.19 Due to its hydrophobic nature, dehydrated ethanol is supplemented in Taxol formulation to enhance the solubility.20,21 Considering all these limitations, quite a few straight forward approaches were adapted to improve the solubility of Taxol in aqueous medium and also the bioavailability of the drug in the physiological systems. Few strategies have been developed in the past which includes: (a) conjugation of Taxol with hydrophilic polymers/nucleic acids, (b) attached or encapsulated the drug molecules with polymeric nanomaterials or (c) attaching Taxol with albumin etc.22–26
Nanoscale aided polymer capsules have been established as Taxol-delivery carriers as they could increase the drug stability, permeability, solubility etc.27 Not only that, these nanocarriers could also facilitate the distribution of therapeutics in the biological fluids while maintaining drug efficiency and minimizing the amount of therapeutics, thereby diminishing the side effects.28,29 A recent study evaluated that Taxol is very effective for oral delivery against the drug-resistant ovarian cancer. Although PLGA (poly lacto-co-glycolic acid)-Taxol formulation could not show much efficacy as compared to the free drug, nonetheless showed significant activity towards the drug-resistant tumours supports exploring the concept of developing nanoparticles for the improved drug delivery.30
In this work, we have explored the biodegradable polymer, PCL to form nanostructure along with porous structures for the treatment of TNBC using Taxol as model anticancer drug. The porous structure of the PCL capsules have been created by templating mesoporous ZnO nanoparticles (mZnO) of size 12 nm in dia. The equivalent sizes of pores on the PCL nanoparticles have created via subsequent removal of the template mZnO. Comparative cytotoxic studies of PCL-mZnO (PZ), mZnO and hollow-mesoporous PCL (HPZ) have been carried out against MDA-MB231 cell lines. Subsequently, biocompatible HPZ is used as Taxol carrier to test various parameters in this cell line model system. Taxol drug is a cytotoxic drug that doesn't distinguish the cancerous from non-cancerous cells. Our approach is to minimise the side effects caused by Taxol with the help of PCL polymer NCs which can encapsulate Taxol drug inside its pores and release slowly to the cancer cells for a longer period. Therefore, we have checked the toxicity level associated with the NC along with few other sets of NCs like PZ, mZnO. Furthermore, T-HPZ treated MDA-MB231 cells are accumulated at G2/M phase of the cell cycle and eventually undergoes apoptosis. In support of this, anchorage independent growth of MDA-MB231 cells have significantly inhibited by T-HPZ treatment. It can be noted that, the Taxol-loaded nanoparticles (T-HPZ) have exhibited more cytotoxicity than Taxol alone treated cells which further has been studied herei systematically.
Material and method
Materials
For this work, precursor material for the template, zinc acetate dihydrate (Zn(COOCH3)2·2H2O) (98.5%) was purchased from Alfa Aesar and sodium hydroxide (NaOH) pellet (97%) were purchased from SDFCL. Polymeric shell precursor PCL (polycaprolactone) (99.9%), surfactant Igepal CO-50 (99.9%) and Taxol (99%) were purchased from Sigma Aldrich and etchant hydrofluoric acid (HF) (98.3%) obtained from Fisher Scientific.Methods
Synthesis methods of core shell PCL-mZnO NPs and mesoporous PCL NCs have been described as below.Characterizations
With the help of high resolution transmission electron microscopy (HRTEM) (Model: FEI Tecnai G2-TWIN 200 kV) the size and surface morphology of the synthesized nanomaterials was analysed. Surface chemical structure was studied by using Fourier transform infrared (FTIR) spectroscopy (Model: PerkinElmer FTIR/FIR Spectrometer). Phase, solid state crystal structure were characterized by X-ray diffractometer (Model: X Bruker D8 Advance). Pore size and BET surface area were calculated with the help of N2 adsorption–desorption isotherm experiment (Model: Tristar II 3020). UV-Vis-NIR spectrometer was used to record the absorption spectra of the drug concentrations. (Model: PerkinElmer LAMBDA 750).For MTT assay, the absorption intensity for the colour formation was recorded at 570 nm (background 665 nm) using Multimode Reader (iMark™ Microplate Absorbance Reader, Bio-Red). Apoptosis and change in the morphology of the cells were studied using a fluorescence microscope (Nikon, Tokyo, Japan) and an inverted light microscope (Olympus CKX41). For cell cycle analysis study we performed flow cytometry (FACS-Aria, Becton Dickinson, New Jersey, USA) and analysed the data using Flow Jo software.
Loading of anticancer drug Taxol in PZ and HPZ
Hollow mesoporous PCL NCs were loaded with the drug ‘Taxol’ (Taxol/T). For that purpose, PZ and HPZ were incubated with Taxol with a ratio of drug to NCs of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mg mL−1 each (PZ and HPZ) in PBS (pH 7.4). Taxol loaded NCs in PBS mixture were stirred (at 5000 rpm) for 1 h and kept in a dark place for one day without disturbing it at room temperature. The Taxol loaded NCs were separated by centrifugation (at 9000 rpm, 4 °C). Then drug loaded NCs were washed three times with PBS (pH 7.4) to remove the free drug molecules from the shell of the NCs. Then it was dried in a clean chamber by air at 25 °C and collected for the further experiments. The whole drug loading process was done at room temperature.
:1 mg mL−1 each (PZ and HPZ) in PBS (pH 7.4). Taxol loaded NCs in PBS mixture were stirred (at 5000 rpm) for 1 h and kept in a dark place for one day without disturbing it at room temperature. The Taxol loaded NCs were separated by centrifugation (at 9000 rpm, 4 °C). Then drug loaded NCs were washed three times with PBS (pH 7.4) to remove the free drug molecules from the shell of the NCs. Then it was dried in a clean chamber by air at 25 °C and collected for the further experiments. The whole drug loading process was done at room temperature.
Taxol release behaviour from PZ and HPZ NCs
Separately, Taxol loaded 0.5 mg of each T-PZ and T-HPZ NCs were suspended in DMEM and the release behaviour of Taxol from the NCs was studied for 3 days in a regular time interval (e.g., 5 min, 1 h, 5 h, 7 h, 24 h, 36 h, 48 h, 60 h and 72 h) at 37 °C using UV-Vis spectrometer. The extent of drug molecules released in the medium was calculated from UV-Vis absorption spectra and the extent of release of Taxol was calculated by considering the absorbance band of the spectra at λmax = 278 nm and using a standard plot of Taxol.Cell culture
Breast cancer cell line MDA-MB231 and human embryonic kidney cell line (HEK-293T) were obtained from the National Centre for Cell Science (NCCS), Pune, India. Cells were maintained in Dulbecco's modification of Eagle medium (DMEM, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 100 units per mL penicillin and 100 μg mL−1 streptomycin under a humidified incubator maintaining 5% CO2 at 37 °C.Western blotting
Western blotting experiments were performed as described previously.32 Briefly, cells were lysed in RIPA lysis buffer (100 mM NaCl, 50 mM Tris–HCl pH 7.5, 1% Triton X-100, 1 mM EDTA, 10 mM β-glycerophosphate, 2 mM sodium vanadate and protease inhibitor) and cell extracts were estimated for protein concentration by BIO-RAD's RC-DC protein assay. Equal amount of protein lysates were resolved on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred to nitrocellulose blotting membrane (NCBM) (GE Healthcare Life Science, USA). The membrane was blocked with 5% skim milk powder (HIMEDIA) for 1 h and then incubated with primary antibody against cleaved-caspase 3 (1:3000; Cell Signalling technology; #9661) at 4 °C overnight. β-Actin (1:25000; Sigma, USA; #A3854) was used as internal control. Membranes were washed three times (10 min each) with TBST buffer and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature [Goat anti-Rabbit HRP-conjugated secondary antibody (1:4000; #A120-101P)]. After thorough washing with TBST buffer, blots were developed using chemiluminescence reagent (femtoLUCENTTM PLUS-HRP #786–003, G-Bioscience, USA) and chemiluminescence was captured by FUSION imaging system (FUSION FX VILBER LOURMAT, USA).
Cytotoxicity assay (MTT assay)
To test the cytotoxicity, HPZ, PZ, and mZnO or Taxol loaded HPZ (T-HPZ) were dispersed in DMEM medium with mild sonication for uniform dispersion and performed serial dilution to different test concentrations (of HPZ, PZ, and mZnO), such as 1000 μg mL−1, 712 μg mL−1, 356 μg mL−1, 128 μg mL−1, 64 μg mL−1, 32 μg mL−1, 16 μg mL−1, 8 μg mL−1 and 0 μg mL−1, for T-HPZ: 250 μg mL−1, 200 μg mL−1, 150 μg mL−1, 100 μg mL−1, 50 μg mL−1 and 0 μg mL−1. Then the samples with different test concentrations were treated with respective cell lines (5 × 103 cells per well) in 96-well plates followed by incubation for 24 h at 37 °C maintaining, 5% CO2. Similarly, Taxol (Sigma; #T7402-1MG) treatment with different test concentration with MDA-MB231 for 24 h was performed. The cell viability was assessed by adding 10 μL of MTT (Sigma; #M5655-1G) solution (stock solution 5 mg mL−1) per well and incubated at 37 °C for 3 h. The medium was discarded and cells were dispersed in DMSO (100 μL). Then the absorption intensity for the colour formation was recorded at 570 nm (background 665 nm) using Multimode Reader (iMark™ Microplate Absorbance Reader, Bio-Red). The percentage of cell viability was calculated and compared with the control sample (without test compound). With increase in the concentration of samples the morphologies of the both cancer and normal cells could change which was a sign of potential cytotoxicity and death of the cells. The morphology of the cells has been checked with the microscopy study before and after treatment with the samples.Fluorescence microscopy analysis of apoptotic cell
For each cell (MDA-MB231, and HEK293T), after following treatment with different concentration of HPZ, PZ, mZnO, and T-HPZ, the culture plates were captured using an inverted light microscope (Olympus CKX41) to see the morphological changes in the cells.The MDA-MB231 cells were seeded in 35 mm plates (triplicate) and kept for 24 h in 5% CO2 with humidified atmosphere allowing complete attachment to the surface of the plates. Cells were treated with the concentration (IC50 value) of Taxol, only particle (HPZ), and the same amount to T-HPZ, for 24 h in CO2 incubator. After 24 h of treatment, cells were stained with AO/EtBr (20 μg, EB; Sigma-Aldrich) following incubation and images were then captured using a fluorescence microscope (Nikon, Tokyo, Japan). We counted the number of apoptotic cells based on the fluorescence colour (live or health cell-green colour, apoptotic cell-red or yellow or orange colour).
Anchorage independent assay (in vitro tumour formation)
In vitro tumour formation was performed using MDA-MB231 cells.33 A layer of 0.5% base agarose (0.5% agarose in DMEM with 10% FBS) in 35 mm plates were prepared and after solidifying, a mixture of 1 mL 0.3% agarose (DMEM with 10% FBS) containing suspended 104 cells was poured above the bottom layer. Triplicate culture plates were used for the experiment. Cells in the agarose were fed by DMEM with 10% FBS along with IC50 value of Taxol or Taxol loaded HPZ (T-HPZ) or particle (HPZ) and incubated in 5% CO2 humidified incubator at 37 °C for 14 days. The fresh DMEM media with respective required treatment were added to the wells every 3 days and colonies were counted. The percentage of the colony formation was calculated with reference to the control values (without test compound).Cell cycle analysis
The MDA-MB231 cells were seeded in 60 mm dishes for one day. Cells were treated with the concentration (IC50 value) of Taxol, only particle (HPZ), and the same amount to T-HPZ, for 24 h in CO2 incubator. Cells were washed in cold PBS two times and then fixed with 1% paraformaldehyde for 10 min at room temperature. Fixed cells were permeabilized with 70% ethanol. After the following washing with PBS twice, cells were treated with RNAse A (250 μg mL−1) and nuclear DNA was stained with 50 μg mL−1 propidium iodide (PI) (Sigma-Aldrich, St Louis, MO, USA) for 20 min at room temperature. The samples were processed by flow cytometry (FACS-Aria, Becton Dickinson, New Jersey, USA) and analyzed the data using Flow Jo software.Statistical analysis
Statistical significance of the data was analysed by student's t-test. Data are presented as the mean ± standard deviation. Statistical analysis was performed using Graph Pad 5.0 (Graph Pad Software, USA). P < 0.05 was considered as statistically significant. The IC50 values for inhibition of cell viability were calculated using Graph Pad 5.0 software.Mechanism of formation of porous PCL NCs
During the synthesis of core shell PZ NPs, the oxygen of surfactant Igepal-50 provides its electron pairs to the electron deficient carbonyl of PCL. The electronic transfer has occurred with other ionic interaction for the completion of synthesis of PZ structure. Covalent interaction, van der Waal's forces of interactions, etc., may have involved in the whole synthesis procedure. While removing core mZnO from the PZ nanostructure, NaOH has added and thereby making some pores in the polymer shell. In the whole reaction process, water has acted as a solvent medium, resulting in a possibility to produce a compound Na2Zn(OH)4 which will be removed completely after repeated centrifugation at 10000 rpm for 10 min and finally leaving behind PCL shell with few porous structure. Proposed chemical reaction of ZnO with NaOH in aqueous medium is, ZnO + 2NaOH + 2NaOH = Na2Zn(OH)4. The whole reaction mechanism has been shown schematically in Fig. 1.
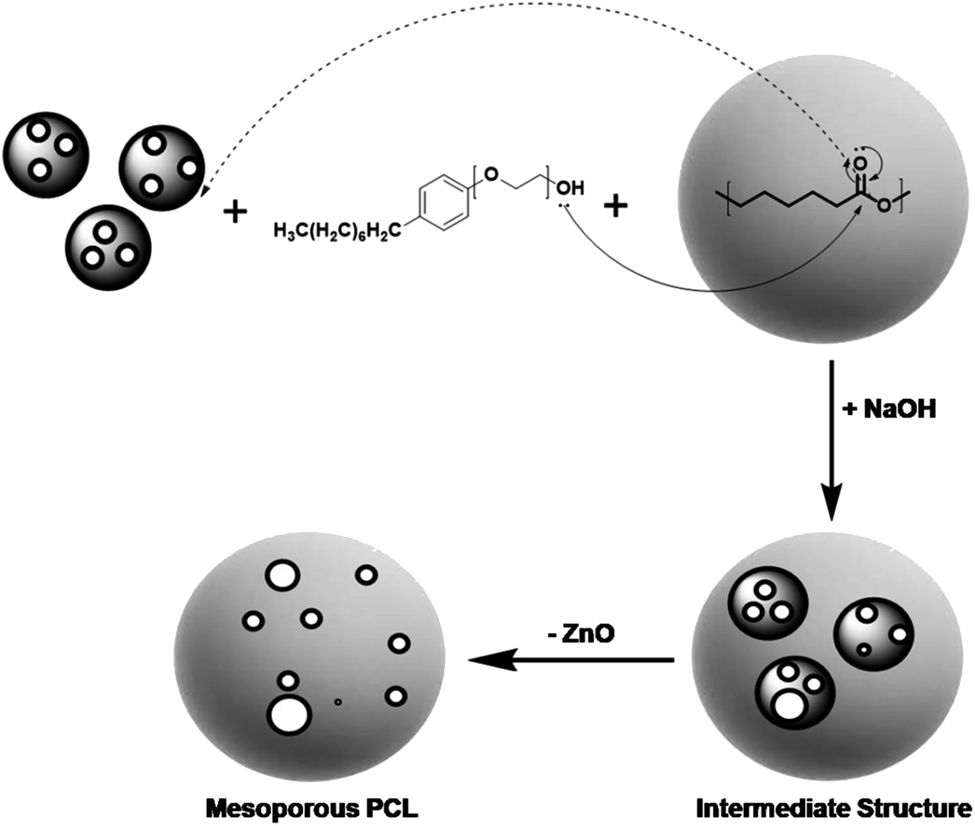 | ||
| Fig. 1 Schematically represented synthesis mechanism of core shells PCL-mZnO NPs and mesoporous PCL NCs after removal of template mZnO. In the first step, PCL polymer has attached to the mesoporous ZnO NPs with the help of surfactant Igepal-50, to develop the core shell structure. In the next step, NaOH has added to remove the template mZnO and leaving behind a porous polymeric nanostructure. | ||
Results and discussion
Designing mesoporous PCL NCs
PZ and HPZ were synthesized followed by the method as described in the Experimental section. HRTEM study was performed to acquire micrographs, which revealed the surface morphology, size, shape, etc. of the synthesized nanomaterials. TEM micrographs of Fig. 2A and C represent almost uniformly dispersed PZ and HPZ nanostructures. Although the synthesized nanostructures are not uninform in size but they appeared to be spherical in shape. In Fig. 2A, it is evident from the size of the core in PZ that, aggregation of 1 or 2 numbers of mZnO have occurred and resulted in the formation of the mZnO core while polymer chains of PCL have been deposited and making a shell structure. Fig. 2C shows the TEM micrographs of the porous PCL NCs (HPZ) after the removal of template mZnO from the core of PZ. Fig. 2B and D represents the average particle size and their distribution of individual samples e.g., PZ as well as HPZ, respectively. The average sizes (diameter) are calculated and found to be ∼32 nm (with maximum number of particles in between 20–50 nm, see Fig. 2B) and 75 nm (maximum number of particles in between 50 to 100 nm, see Fig. 2D) for PZ and HPZ, respectively (calculated from the TEM micrographs, Fig. 2A and C). Similar results have been obtained from the DLS experiments for both PZ and HPZ NCs (see ESI results Fig. S1†). The distribution of the particle size has obtained due to the distribution of molecular weight (MW) of polymers and from TEM it is visible that few cases the core containing multiple numbers of mZnO template. Detail characterization results for the template mZnO have been reported previously by the authors of this article31 and hence not provided here. However, we have considered here 1:1 ratio of mZnO to PCL for the formation of core shell nanoparticles (PZ). With the change of the concentration ratio of mZnO to PCL e.g., 1:2 and 1:3 or 2:1 and 3:1 we have achieved core shell nanoparticles of comparatively bigger in size or the particles are unable to form core shell structure, mesoporous and porous PCL (HPC) properly and which are not preferable for drug delivery applications and could not solve our purpose.
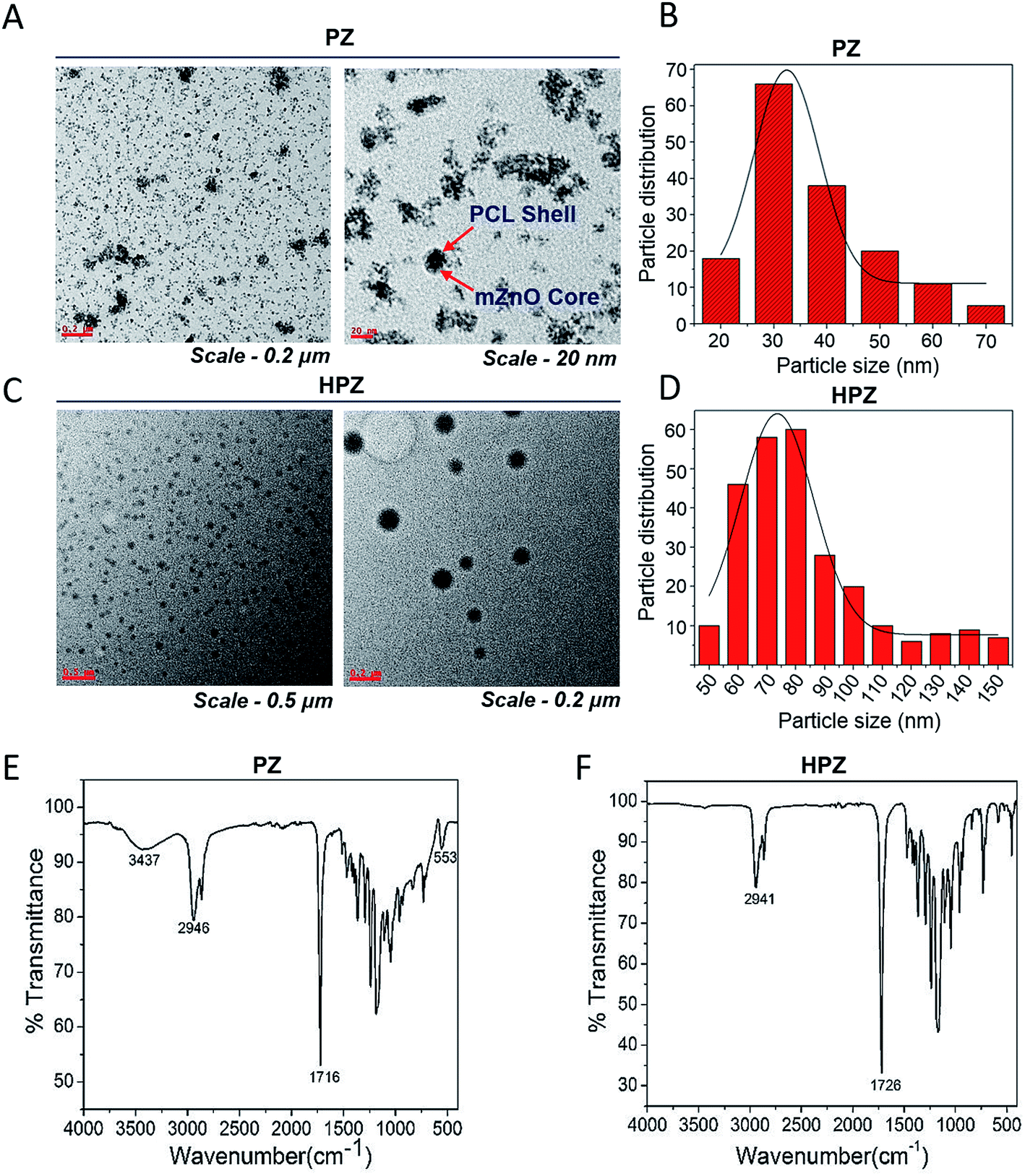 | ||
| Fig. 2 Morphology and surface chemical structure analysis of PZ and HPZ by HRTEM and FTIR respectively. TEM micrographs of (A) core shell (PZ) from lower to higher magnifications, (C) porous PCL NCs (HPZ) from lower to higher magnifications, histogram for the average particle size distribution of (B) PZ calculated from HRTEM image (A), (D) HPZ calculated from HRTEM image (B). (E) and (F) FTIR spectra of PCL-mZnO core shell (PZ) NPs and porous PCL (HPZ) NCs respectively. | ||
The surface chemical structure of the PZ and HPZ analysed by FTIR spectroscopy and results are shown in Fig. 2E and F. In Fig. 2E, the band at 3437 cm−1 arises for stretching vibration mode of a hydroxyl group (–OH) present on the surface of mZnO core. The band appears at 2946 cm−1 for C–H(–CH2) group of PCL polymer and the band arises at 1716 cm−1, which is due to the presence of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of polymer PCL.34 At 553 cm−1, the band arises due to the vibration mode of Zn and O bond in ZnO lattice.35,36 Bands are overlapping in the region from 1500 cm−1 to 750 cm−1 due to the polymer chains entangled over mZnO. Thus, FTIR spectrum (Fig. 2E) has suggested the presence of polymer PCL on the surface of mZnO NPs and thus confirmed the formation of PZ NPs. Non-appearance of any other additional bands in the FTIR spectrum confirms that PZ is free from impurities. Fig. 2F shows the FTIR spectrum of PCL NCs after etching the mZnO. The band at 2941 cm−1 arises for C–H(–CH2) group of PCL and the band arises at 1726 cm−1, which is due to the presence of –CO group of PCL.34 Absenteeism of any additional bands accountable for mZnO have evidence that the template has been removed from the core entirely and the non-appearance of additional bands confirm the absence of impurities or the other component of Zn(II) in the PCL nanostructure.
O group of polymer PCL.34 At 553 cm−1, the band arises due to the vibration mode of Zn and O bond in ZnO lattice.35,36 Bands are overlapping in the region from 1500 cm−1 to 750 cm−1 due to the polymer chains entangled over mZnO. Thus, FTIR spectrum (Fig. 2E) has suggested the presence of polymer PCL on the surface of mZnO NPs and thus confirmed the formation of PZ NPs. Non-appearance of any other additional bands in the FTIR spectrum confirms that PZ is free from impurities. Fig. 2F shows the FTIR spectrum of PCL NCs after etching the mZnO. The band at 2941 cm−1 arises for C–H(–CH2) group of PCL and the band arises at 1726 cm−1, which is due to the presence of –CO group of PCL.34 Absenteeism of any additional bands accountable for mZnO have evidence that the template has been removed from the core entirely and the non-appearance of additional bands confirm the absence of impurities or the other component of Zn(II) in the PCL nanostructure.
BET experiments were performed on PZ and HPZ further to analyse their specific surface areas available to load the drug molecules. Fig. 3 demonstrates the N2-absorption–desorption isotherm as well as the pore size distribution of both PZ and HPZ NCs. In case of HPZ, the clear hysteresis loop (from relative pressure P/P0 0.7 to 0.99) is observed, which confirms the porous nature of the nanomaterial with a type-IV adsorption isotherm. BET surface area for porous HPZ NCs has been calculated as 68.67 m2 g−1 at relative pressure P/P0 = 0.283 and adsorption average pore size has been achieved as 22.1 nm from the Fig. 3 inset image. In case of PZ, due to the lack of pores in the structure, the BET surface has been decreased to 36.6 m2 g−1 at relative pressure P/P0 = 0.284 and adsorption average pore size attained as ∼12 nm. Thus the BET confirms the formation of hollow and porous structure of polymer nanocapsules synthesized in this work.
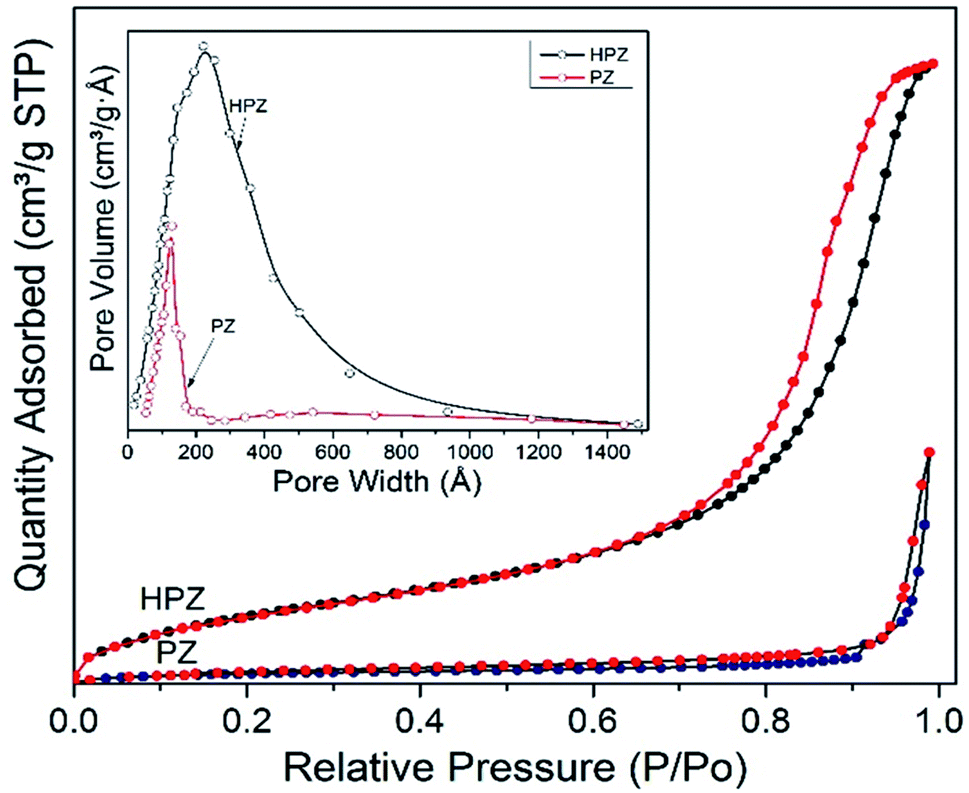 | ||
| Fig. 3 BET results. N2 absorption–desorption isotherm of PZ and HPZ with BJH pore size distribution of PZ and HPZ from adsorption isotherm (inset figure). | ||
The solid-state crystal structure of the synthesized samples was characterized through the powder XRD and the diffraction pattern is represented in Fig. S2A.† Template mZnO is found to have hexagonal crystal structure with characteristic diffraction peaks that appeared at 2θ = 32.2°, 34.6°, 36.6°, 47.9°, 57.1°, 63.1°, 66.8°, 68.4°, 69.6°, 72.8°, 77.3° and 81.8° for the corresponding to the (100), (002), (101), (102), (110), (103), (200), (112), (201), (004) and (202) planes, respectively. The crystal structure corresponds to the phase pure wurtzite-type ZnO (hexagonal phase with space group P63mc, JCPDS card no. 36-1451, a polar axis parallel to c-axis). XRD pattern of PZ is found matching well with all diffraction peaks of mZnO, along with some additional peaks those arise at 2θ = 21.4° and 23.0°, corresponds to the orthorhombic crystal structure [(110) and (200) plans] of PCL polymer, respectively. Accordingly, the XRD pattern verifies the presence of both mZnO (as core) and PCL (as shell) in PZ structure. XRD pattern of HPZ sample showed the diffraction peaks only at 2θ = 21.4° and 23.0°, corresponding to the orthorhombic crystal structure with characteristic (110) and (200) crystalline planes of PCL, respectively.37,38 Absence of any other additional diffraction peaks of mZnO in HPZ is the evidence for the absence of template in PCL nanostructure.
To check the colloidal stability and surface charge of the synthesized samples the zeta potential values of bare PZ and mesoporous PCL (HPZ) was calculated. To find out zeta potential 500 μg of each sample was dispersed in 1 mL of DI water. The values were recorded at room temperature (25 °C) by using a He–Ne LASER with a wavelength of 633 nm. Fig. S2B and C,† represent the zeta potential values of PZ and HPZ which have been found to be −28.3 mV and −29.9 mV, respectively and thus it can be concluded that both the samples are colloidal stable. It can be noted that due to the surface potential of nanoscale materials, they have the natural tendency to agglomerate and thereby reduce energy spread and it causes thermodynamic instability. Consequently, the synthesised NCs are showing relatively high zeta potential values, which can results: (i) monodispersed and lesser degree of agglomeration due to high electrostatic repulsive force between the particles in a medium, (ii) they acquire high colloidal stability in the liquid medium, (iii) possesses high kinetic stability of NCs' and slower sedimentation and (iv) additionally high zeta potential leads to lower cytotoxicity of the nanoparticles and improved cellular uptake.
Analysis of drug loading efficiency and time dependent drug release kinetics of Taxol from PZ and HPZ NCs
The extent of encapsulation of Taxol in the formulations T-PZ and T-HPZ have been calculated with the help of the formulae entrapment efficiency, % = (weight of drug in NPs/weight of drug fed initially) × 100. 0.5 mg of anticancer drug loaded T-PZ and T-HPZ NCs were suspended in DMEM medium in two different cuvettes and behaviour of the drug release from NCs was studied separately for a period of 3 days with a time interval at room temperature (37 °C). The calculated entrapment efficiency values for both PZ and HPZ are found to be 38.6%, and 60.8%, respectively.The extent of Taxol molecules released from T-PZ and T-HPZ in the culture medium was calculated with the help of UV-Vis absorption spectrometer for various time points e.g., 5 min, 1 h, 5 h, 7 h, 24 h, 36 h, 48 h, 60 h, 72 h. Fig. 4 shows the nature of time dependent release kinetic profile of Taxol for PZ and HPZ NCs. The percentage of Taxol release from the NCs is gradually increased with increased in time. The release amounts of Taxol molecules from the NCs have been calculated as 12.6 ± 1.3%, 31.2 ± 1.2%, 34.8 ± 1.4%, 41.8 ± 3.9%, 43.2 ± 2.0%, 48.5 ± 2.1%, 55.4 ± 3.7%, 66.2 ± 2.0%, 70.1 ± 1.1% and 8.2 ± 2.3%, 32.2 ± 4.3%, 35.7 ± 1.4%, 49.1 ± 2.9%, 50.4 ± 2.0%, 52.3 ± 1.0%, 68.8 ± 1.7%, 75.6 ± 2.0%, 78.2 ± 3.1% on the time periods of 5 min, 1 h, 5 h, 7 h, 24 h, 36 h, 48 h, 60 h, 72 h for PZ and HPZ, respectively. Thus, the release of Taxol from the T-PZ and T-HPZ are found slow, controlled and it is a sustained release (Fig. 4). The reason for the slow release is that the Taxol molecules are entrapped inside the pores and hollow cores due to the electrostatic interaction of forces. Further it is noted that the initial release (within 5 min) of Taxol from T-PZ is faster than the rate of release from T-HPZ. Because PZ morphology doesn't have any pores on it. Therefore the drug molecules are poorly loaded on the surface of the nanoparticles and whenever it is coming in contact with the favourable medium the attachment breaks and it is started to diffuse to the medium very fast. However, presence of pores in HPZ acts as reservoirs for the drug molecules and therefore their loading efficiency is also more and release of these drugs from the pores in the media occurs via diffusion mechanism and is slow.
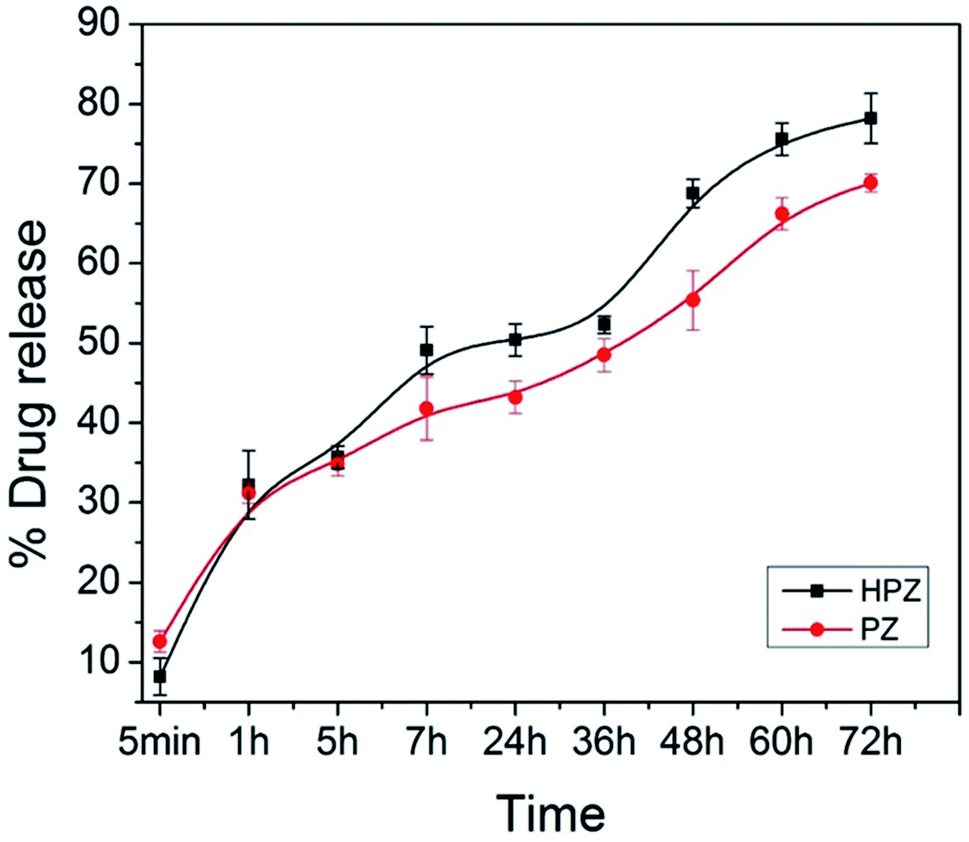 | ||
| Fig. 4 The drug release profile. Taxol from PZ (T-PZ) and Taxol from HPZ (T-HPZ) at 25 °C temperatures for 72 h in DMEM. Data are presented as mean ± SD (n = 3). | ||
Effect of different concentrations of HPZ, PZ, and mZnO on the viability of breast cancer cells
Both MDA-MB231, a triple negative breast cancer cell line and HEK293T, human embryonic kidney (considered as normal cell line) were used to check the cytotoxic effect of the nanoparticles. MDA-MB231 cells were treated with various concentrations of HPZ, PZ, and mZnO ranging from 0 to 1000 μg mL−1, for 24 h and MTT assay was performed to evaluate the cytotoxicity. As shown in Fig. 5A, PZ and mZnO treatment resulted in a dose-dependent reduction in the cell viability with estimated IC50 values of 54.9 μg mL−1 and 37.71 μg mL−1, respectively. Next we analysed the cellular morphology of MDA-MB231 cells post treatment of these nanoparticles. The optical microscopic image of the cells for each respective particle showed dynamic changes from spindle shape to round shape morphology with the increasing concentration of particles, a sign of potential cytotoxicity and death of the cells (Fig. 5C and D). On the other hand, HPZ didn't show any significant cytotoxicity even at the higher concentration i.e., 1000 μg mL−1 and their morphology remain unchanged (Fig. 5A and B).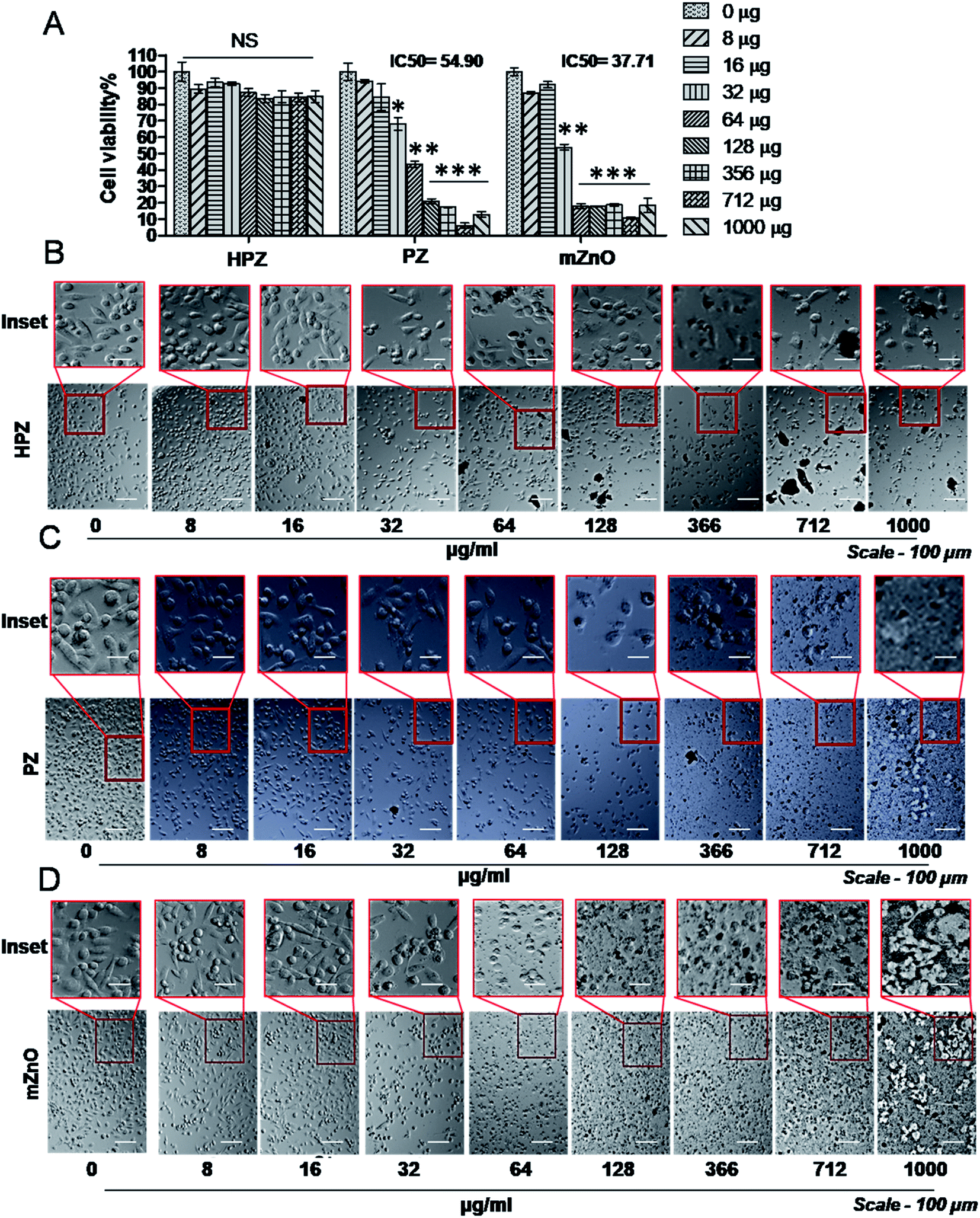 | ||
| Fig. 5 Effect of various concentrations of HPZ, PZ, and mZnO on the viability of breast cancer cells. (A) MTT assay demonstrates the cytotoxic effect of free HPZ, PZ, or mZnO on MDA-MB231 cells 24 h post treatment at specified concentrations. Each bar represents mean ± SD (n = 3). *P < 0.01, **P < 0.001, ***P < 0.0001 were considered as significant. Microscopic images showing the morphology of the MDA-MB231 cells at various mention concentrations of free HPZ (B), PZ (C), and mZnO (D). Scale bar-100 μm, magnification-10× mentioned. | ||
To verify whether our nanocapsules have a similar effect on the normal cells, we used HEK293T cells and evaluated the cell viability by MTT assay. As expected, except HPZ (Fig. 6B), both PZ and mZnO exhibited significant cytotoxicity in a dose-dependent fashion (Fig. 6A, C and D). The IC50 values of PZ and HPZ were found to be 15.36 μg mL−1 and 15.90 μg mL−1, respectively (Fig. 6A). Together this data indicated that HPZ could not show the cytotoxicity neither on the cancer cells (MDA-MB231) nor on the normal cells (HEK293T). Thus, it prompted us to use HPZ as a Taxol carrier for further study for therapeutic applications as well as for the treatment of cancer.
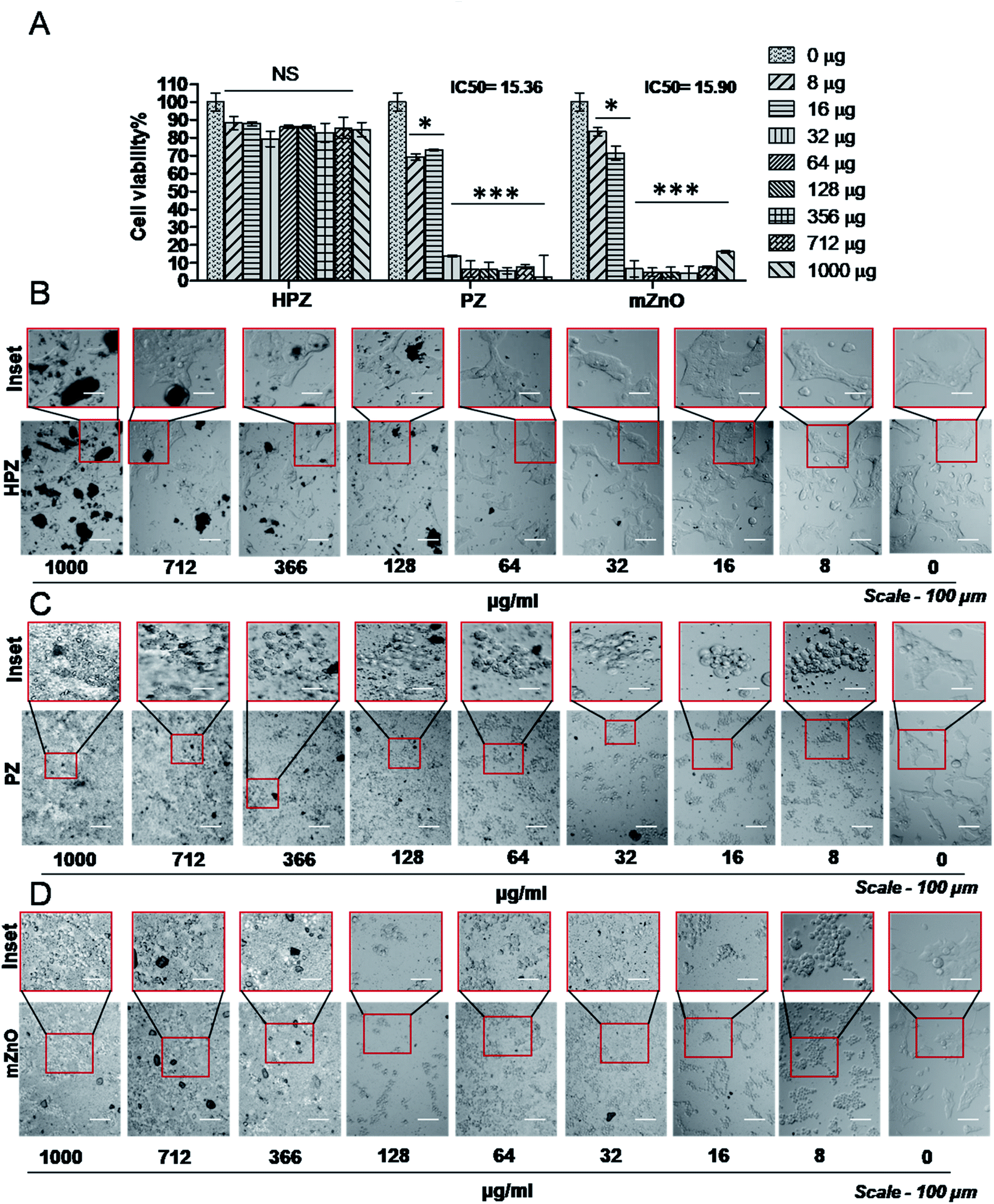 | ||
| Fig. 6 Dosage effect of HPZ, PZ or mZnO on the viability of normal cells. (A) MTT assay demonstrates the cytotoxic effect of free HPZ, PZ, or mZnO on HEK293T cells 24 h post treatment at specified concentrations. Each bar represents mean ± SD (n = 3). *P < 0.01, ***P < 0.0001 were considered as significant. Microscopic images showing the morphology of the HEK293T cells at various mention concentrations of free HPZ (B), PZ (C), and mZnO (D). Scale bar-100 μm, magnification-10× mentioned. | ||
Taxol induced breast cancer cell death or apoptosis is sensitized by HPZ
It is known that Taxol induces apoptosis by arresting the cells at the G2/M phase of the cell cycle.39,40 We therefore next investigated the cytotoxicity effect of Taxol-loaded nanoparticles (T-HPZ) on MDA-MB231 cells by MTT assay. As shown in Fig. 7A, the IC50 value for T-HPZ on MDA-MB231 cells was found to be 43.30 μg mL−1. Whereas the IC50 value of Taxol was found to be 31.9 μg mL−1 (Fig. S2A and B†) which is significantly very less as compared to the Taxol-loaded particles used in this work. Next we studied the morphology of the cells post treatment with these drugs. The prominent spherical shape of cells with increasing concentration of loaded drug showed the induction of apoptosis (Fig. 7B). Meanwhile, HPZ can carry an amount of 58.8 μg of Taxol per 100 μg of particle i.e., 43 μg (IC50 value) of particles can carry 25 μg of Taxol. The Taxol-loaded nanoparticles (T-HPZ) exhibited more cytotoxicity than Taxol alone following 24 h treatment implying that HPZ could neither sensitize nor enhance the cytotoxicity of the Taxol. Further, in the earlier section we have demonstrated that HPZ is viable to the MDA-MB231 cells. Therefore, Taxol loaded with HPZ (T-HPZ) is very much potential in monitoring the growth and killing of MDA-MB231 cells.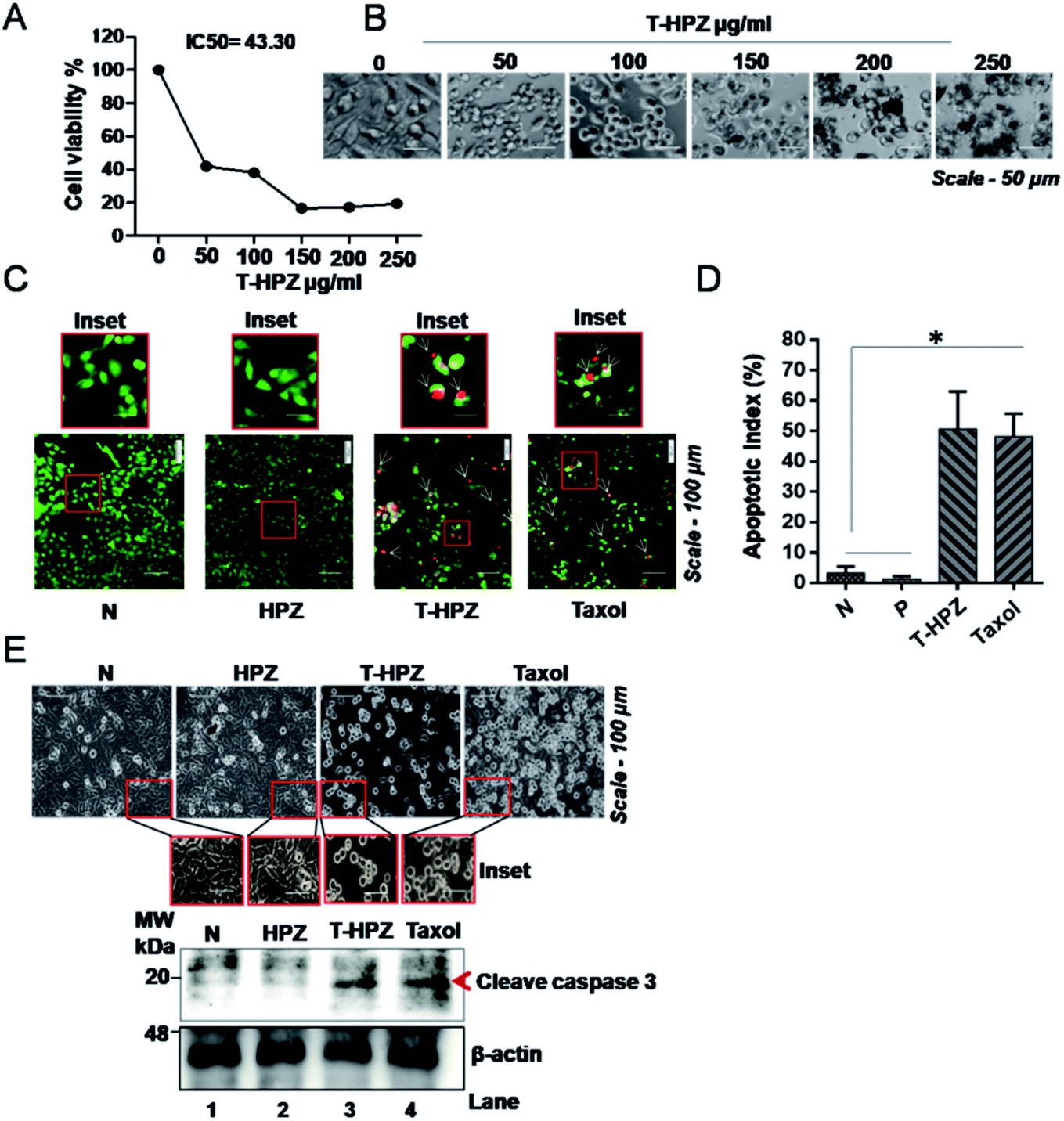 | ||
| Fig. 7 Effect of Taxol loaded HPZ nanoparticles on Taxol-induced apoptosis in MDA-MB231 cells. (A) In vitro cytotoxic effect of Taxol loaded HPZ (T-HPZ) on MDA-MB231 cells determined by MTT assay 24 h post treatment at specified concentration. Data are presented as mean ± SD (n = 3). (B) Microscopic images showing the morphology of MDA-MB231 cells at various concentrations of T-HPZ. Scale bar-50 μm, magnification-10×. (C) Apoptosis assay-fluorescence microscopy images of normal cells (untreated, N) or post treated cells with free HPZ or Taxol loaded T-HPZ (IC50 value) or Taxol (IC50) after AO/EtBr staining. (Live or health cell – green colour, apoptotic cell – red or yellow or orange colour). (D) Quantitative graph showing the percentage of apoptotic cells from (C). Each bar represents mean ± SD (n = 3). *P < 0.01 was considered significant. (E) MDA-MB231 cells were treated with free HPZ or Taxol loaded T-HPZ (IC50 value) or Taxol (IC50) for 24 h and cell morphology was captured (upper panel). Western blotting demonstrates the effect of free HPZ or Taxol loaded T-HPZ (IC50 value) or Taxol (IC50) on cleaved-caspase 3. β-Actin is used as internal control. Scale bar-100 μm, magnification-10× were blotted. | ||
T-HPZ induces apoptosis in TNBC cells
Resistance to apoptosis is an important hallmark of cancer.41 In view of this, the apoptotic activity of the Taxol loaded HPZ particles on breast cancer cell line was analysed by acridine orange staining method. Cells were treated with particle alone, loaded particle or T-HPZ on MDA-MB231 cells and stained the cells with acridine orange (AO) and ethidium bromide (EtBr) followed by fluorescence microscopy imaging. We measured the number of apoptotic cells based on the fluorescence colour (green colour represents live or healthy cell; red, yellow or orange colour represents apoptotic cell). The cells treated with loaded particle (T-HPZ) or Taxol showed significantly higher percentage of apoptosis as compared to untreated or only particle (HPZ) treated cells (Fig. 7C and D). Activation of caspases is a feature of cell undergoing apoptosis and caspase-3 is cleaved by apoptosome.42 Therefore we next analysed the cleaved caspase 3 levels post-treatment of MDA-MB231 cells with particles or loaded particles. In line with the results of cytotoxicity, the results revealed that cells treated with loaded particle (T-HPZ) or Taxol showed an elevated cleaved-caspase 3 expression as compared to unloaded or only particle (HPZ) treated cells which is accompanied by altered morphology (Fig. 7E). Together, these results indicate the potential apoptotic activity of T-HPZ particles on MDA-MB231 cells.T-HPZ block G2/M phase of the cell cycle in MDA-MB231 cells
Normal cells undergo a series of strictly monitoring stages, G1, S, G2 and M in a cyclic manner (Fig. 8E).43 Abnormality or altered cell-cycle progression can trigger apoptosis in the normal cells. Taxol blocks the cells in the G2/M phase of the cell cycle and induces apoptosis (Fig. 8E).39 To investigate the anti-proliferative activity of the Taxol-loaded particles (T-HPZ), MDA-MB231 cells were stained with PI for 24 h post treatment with the loaded particles (T-HPZ) and subjected to flow cytometer analysis. As shown in Fig. 8F, treatment of MDA-MB231 cells with Taxol (Fig. 8D) or Taxol loaded particles (T-HPZ) (Fig. 8D) could arrest large percentage of the cells at G2/M stage of the cell cycle as compared to either untreated cells (Fig. 8A) or particle treated (HPZ) cells (Fig. 8B). Taken together, our data indicate that the Taxol-loaded particle (T-HPZ) possibly induces apoptosis in MDA-MB231 cells by inhibiting the G2/M phase of the cell cycle. | ||
| Fig. 8 Effect of Taxol loaded HPZ nanoparticles on cell cycle progression in MDA-MB231 cells. Analysis of distribution of cells at different stages of the cell cycle (duplicated untreated cells, N1 and N2) (A) or post treatment of the cells with free HPZ (duplicated treated cells, HPZ1 and HPZ2) (B) or Taxol loaded T-HPZ (IC50 value) (duplicated treated cells, T-HPZ1 and T-HPZ2) (C) or Taxol (IC50) (duplicated treated cells, Taxol-1 and Taxol-2) (D) by flow cytometer. (E) Representative pictorial image showing the action of Taxol on cell cycle stages. It arrests the cells at G2/M stage of the cell cycle leading to apoptosis. (F) Quantitative graph showing the percentage of distribution of cells at different stages of cell cycle derived from (C). Each bar represents mean ± SD (n = 2). | ||
T-HPZ inhibits anchorage independent growth of MDA-MB231 cells
Normal cells require a basement membrane (substratum) to attach and grow, otherwise cells undergo anoikis, a type of apoptotic cell death.33 However, the cancer cells have the ability to grow independently of the solid surface, a phenomenon called as anchorage-independent growth or anoikis resistance, which is a hallmark of cancer (Fig. 9A). To evaluate the tumour suppressive role of Taxol loaded nanoparticle (T-HPZ) in vitro, we performed a well-established soft agar formation assay (Fig. 9B).44 MDA-MB231 cells were grown in suspension on semi-solid agar mixed with culture medium, which prevents cells from attaching to the basement. Three days post seeding of the cells on semi-solid agar, cells were treated with Taxol alone, HPZ or T-HPZ and incubated for 7 days. Following 7 days of treatment with drugs, the number of colonies formed ware counted (Fig. 9B). As shown in (Fig. 9C and D), the ability of cancer cells to grow in the absence of substratum (anchorage independent growth) was significantly decreased as compared to the cells treated with either Taxol loaded particles (T-HPZ) or Taxol alone. This finding suggests that less amount of Taxol can be utilized using HPZ particles as a carrier to target the cancer cells with fewer side effects.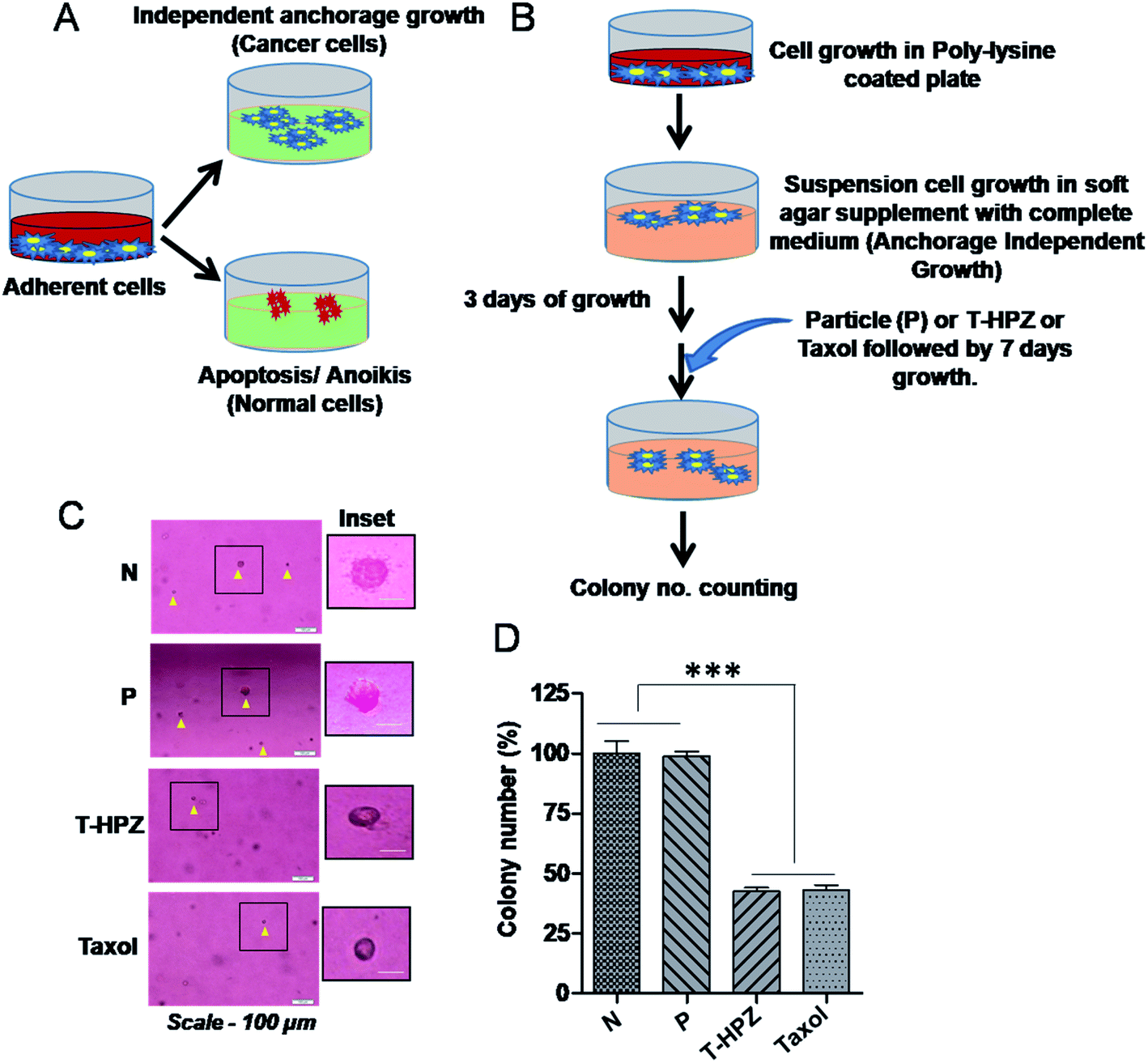 | ||
| Fig. 9 Effect of Taxol loaded nanoparticle (T-HPZ) on the anchorage independent growth ability of MDA-MB231 cells. (A) Representative pictorial image showing the fate of growing cells with or without substratum (basement membrane). (B) Pictorial diagram showing the brief protocol of anchorage independent assay. (C) Representative image of colonies form after the completion of anchorage independent assay, untreated cells (N) or post treatment of the cells with free HPZ or Taxol loaded (T-HPZ) (IC50 value) or Taxol (IC50). Scale bar-100 μm, magnification-10×. (D) Quantitative graph showing the percentage of colony number derives from (C). Each bar represents mean ± SD (n = 3). ***P < 0.0001 was considered significant. | ||
Conclusions
There is a paradigm shift in application of drugs using biodegradable polymer based nanocarriers as they are more competent as well as efficient in delivering the drug.45 In the current study we have designed mesoporous PCL nanocapsules (HPZ) to encapsulate anticancer drug Taxol and to deliver it in the physiological environment for a longer period of time and to kill TNBC. Novel PCL nanocapsules (HPZ) are quite stable in a biological fluid and possessing a slow rate of degradation. The encapsulation efficiency of Taxol in the nanocarrier is excellent. Time dependent diffusion controlled release of Taxol has played a crucial role in killing the TNBC cells. The porous PCL nanocarrier has minimized the amount of drug obligatory for TNBC treatment imply that the side effect instigated by the large amount of administered Taxol can be minimized. However, the in vivo experiments are warranted to further understand the efficacies of the nanoencapsulated Taxol and to extend the drug for future clinical trials.Author contributions
PP, BM, HM and SSK designed the experiments and wrote the paper. HM and SSK carried out the experiments and analysed data under the supervision of PP and BM. All authors analysed the results and approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest with the contents of this article.Acknowledgements
Authors acknowledge the Research support grants awarded by DST-Nanomission, India (Ref: SR/NM/NS-1005/2015) and Science and Engineering Research Board, India (Ref: EEQ/2016/000040), India to P. Paik. This work is also supported by the Department of Science and Technology (DST), Grant No. SB/SO/BB/013/2013, India (to BM).References
- C. Yam, S. A. Mani and S. L. Moulder, Oncologist, 2017, 22, 1086–1093 CrossRef PubMed.
- L. A. Carey, C. M. Perou, C. A. Livasy, L. G. Dressler, D. Cowan, K. Conway, G. Karaca, M. A. Troester, C. K. Tse, S. Edmiston, S. L. Deming, J. Geradts, M. C. Cheang, T. O. Nielsen, P. G. Moorman, H. S. Earp and R. C. Millikan, J. Am. Med. Assoc., 2006, 295, 2492–2502 CrossRef CAS PubMed.
- K. R. Bauer, M. Brown, R. D. Cress, C. A. Parise and V. Caggiano, Cancer, 2007, 109, 1721–1728 CrossRef PubMed.
- G. Early Breast Cancer Trialists' Collaborative, Lancet, 2005, 365, 1687–1717 CrossRef.
- D. P. Silver, A. L. Richardson, A. C. Eklund, Z. C. Wang, Z. Szallasi, Q. Li, N. Juul, C. O. Leong, D. Calogrias, A. Buraimoh, A. Fatima, R. S. Gelman, P. D. Ryan, N. M. Tung, A. De Nicolo, S. Ganesan, A. Miron, C. Colin, D. C. Sgroi, L. W. Ellisen, E. P. Winer and J. E. Garber, J. Clin. Oncol., 2010, 28, 1145–1153 CrossRef CAS PubMed.
- C. X. Lam, D. W. Hutmacher, J. T. Schantz, M. A. Woodruff and S. H. Teoh, J. Biomed. Mater. Res., Part A, 2009, 90, 906–919 CrossRef PubMed.
- B. Azimi, P. Nourpanah, M. Rabiee and S. Arbab, J. Eng. Fibers Fabr., 2014, 9 Search PubMed.
- A. S. A. Cipitria, T. R. Dargaville, P. D. Dalton and D. W. Hutmacher, J. Mater. Chem., 2011, 21, 9419–9453 RSC.
- P. Paik and Y. Zhang, Nanoscale, 2011, 5, 2215–2219 RSC.
- V. B. Kumar, H. Medhi, Z. Yong and P. Paik, Nanomed. Nanotechnol. Biol. Med., 2016, 12(3), 579–588 CrossRef CAS PubMed.
- C. Amgoth, G. Dharmapuri, A. M. Kalle and P. Paik, Nanotechnology, 2016, 27(12), 125101 CrossRef PubMed.
- A. K. Yamala, V. Nadella, Y. Mastai, H. Prakash and P. Paik, Nanoscale, 2017, 9, 14006–14014 RSC.
- R. Ferrari, M. Sponchioni, M. Morbidelli and D. Moscatelli, Nanoscale, 2018, 10, 22701–22719 RSC.
- W. Paul and C. P. Sharma, Inorganic nanoparticles for targeted drug delivery, Biointegration of Medical Implant Materials, Elsevier Science, 2019 Search PubMed.
- G. H. Son, B. J. Lee and C. W. Cho, J. Pharm. Invest., 2017, 47(4), 287–296 CrossRef CAS.
- A. P. Singh, A. Biswas, A. Shukla and P. Maiti, Signal Transduction Targeted Ther., 2019, 4, 33 CrossRef PubMed.
- C. X. Lam, M. M. Savalani, S. H. Teoh and D. W. Hutmacher, Biomed. Mater., 2008, 3(3), 34108 CrossRef PubMed.
- C. M. Spencer and D. Faulds, Drugs, 1994, 48, 794–847 CrossRef CAS PubMed.
- R. C. Donehower, E. K. Rowinsky, L. B. Grochow, S. M. Longnecker and D. S. Ettinger, Cancer Treat. Rep., 1987, 71, 1171–1177 CAS.
- R. C. Alves, R. P. Fernandes, J. O. Eloy, H. R. N. Salgado and M. Chorilli, Crit. Rev. Anal. Chem., 2018, 48, 110–118 CrossRef CAS PubMed.
- G. Deepa, N. Ashwanikumar, J. J. Pillai and G. S. Kumar, Curr. Med. Chem., 2012, 19, 6207–6213 CrossRef CAS PubMed.
- G. S. Gomes, T. R. Maciel, E. M. Piegas, L. R. Michels, L. M. Colome, R. J. Freddo, D. S. Avila, A. Gundel and S. E. Haas, AAPS PharmSciTech, 2018, 19, 551–564 CrossRef CAS PubMed.
- M. R. Green, G. M. Manikhas, S. Orlov, B. Afanasyev, A. M. Makhson, P. Bhar and M. J. Hawkins, Ann. Oncol., 2006, 17, 1263–1268 CrossRef CAS PubMed.
- J. Zou, Y. Yu, Y. Li, W. Ji, C. K. Chen, W. C. Law, P. N. Prasad and C. Cheng, Biomater. Sci., 2015, 3, 1078–1084 RSC.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavia-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 6972 CrossRef CAS PubMed.
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138, 10834–10837 CrossRef CAS PubMed.
- S. Yao, L. Li, X. T. Su, K. Wang, Z. J. Lu, C. Z. Yuan, J. B. Feng, S. Yan, B. H. Kong and K. Song, J. Exp. Clin. Cancer Res., 2018, 37, 29 CrossRef PubMed.
- S. Lv, Z. Tang, M. Li, J. Lin, W. Song, H. Liu, Y. Huang, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 6118–6129 CrossRef CAS PubMed.
- X. Duan, J. Xiao, Q. Yin, Z. Zhang, H. Yu, S. Mao and Y. Li, ACS Nano, 2013, 7, 5858–5869 CrossRef CAS PubMed.
- V. Bhardwaj, J. A. Plumb, J. Cassidy and M. N. Ravi Kumar, Cancer Nanotechnol., 2010, 1, 29–34 CrossRef CAS PubMed.
- S. Afroz, H. Medhi, S. Maity, G. Minhas, S. Battu, J. Giddaluru, K. Kumar, P. Paik and N. Khan, Nanoscale, 2017, 9, 14641–14653 RSC.
- S. S. Khumukcham, V. S. K. Samanthapudi, V. Penugurti, A. Kumari, P. S. Kesavan, L. R. Velatooru, S. R. Kotla, A. Mazumder and B. Manavathi, J. Biol. Chem., 2019, 294, 10236–10252 CrossRef CAS PubMed.
- S. M. Frisch and R. A. Screaton, Curr. Opin. Cell Biol., 2001, 13, 555–562 CrossRef CAS PubMed.
- A. K. Bassi, J. E. Gough, M. Zakikhani and S. Downes, J. Tissue Eng., 2011, 2011, 615328 CAS.
- S. B. Kalpana Handore, A. Horne, P. Chhattise, K. Mohite, J. Ambekar, N. Pande and V. Chabukswar, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51 DOI:10.1080/10601325.2014.967078.
- M. M. S. Mallakpoure, Bull. Mater. Sci., 2012, 35, 333–339 CrossRef.
- S. Agarwal and C. Speyerer, Polymer, 2010, 51, 1024–1032 CrossRef CAS.
- H. S. X. Liu, J. Mater. Sci., 2007, 42, 8113–8119 CrossRef.
- S. B. Horwitz, Trends Pharmacol. Sci., 1992, 13, 134–136 CrossRef CAS PubMed.
- D. R. Kohler and B. R. Goldspiel, Pharmacotherapy, 1994, 14(1), 3–34 CrossRef CAS PubMed.
- R. S. Wong, J. Exp. Clin. Cancer Res., 2011, 30, 87 CrossRef CAS PubMed.
- V. Kumar, A. K. Abbas, N. Fausto, S. L. Robbins and S. R. Cotran, Robbins and Cotran pathologic basis of disease, Elsevier Saunders, Philadelphia, PA, 2010, pp. 25–32 Search PubMed.
- D. O. Morgan, Nature, 1995, 374, 131–134 CrossRef CAS PubMed.
- S. Borowicz, M. Van Scoyk, S. Avasarala, M. K. Karuppusamy Rathinam, J. Tauler, R. K. Bikkavilli and R. A. Winn, J. Visualized Exp., 2014, 92, 51998 Search PubMed.
- S. Zhang, J. Zou, M. Elsabahy, A. Karwa, A. Li, D. A. Moore, R. B. Dorshow and K. L. Wooley, Chem. Sci., 2013, 4, 2122–2126 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S3. See DOI: 10.1039/d0ra04505e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |