
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensible graphene oxide differentiates macrophages and Leishmania: a bio-nano interplay in attenuating intracellular parasite†
Aakriti Singh‡
a,
Sandeep Sharma‡b,
Ganesh Yadagiria,
Shabi Parveza,
Ritika Guptac,
Nitin Kumar Singhal c,
Nikhil Koratkard,
Om Prakash Singhe,
Shyam Sundarf,
Vijayakumar Shanmugam*b and
Shyam Lal Mudavath*a
c,
Nikhil Koratkard,
Om Prakash Singhe,
Shyam Sundarf,
Vijayakumar Shanmugam*b and
Shyam Lal Mudavath*a
aInfectious Disease Biology Laboratory, Chemical Biology Unit, Institute of Nano Science and Technology, Habitat Centre, Phase-10, Sector-64, Mohali, Punjab 160062, India. E-mail: shyamlal@inst.ac.in; shavs0502@gmail.com
bInstitute of Nano Science and Technology, Habitat Centre, Phase-10, Sector-64, Mohali, Punjab 160062, India. E-mail: psvijayakumar@inst.ac.in; vijayakumarshanmugham@gmail.com
cFood and Nutritional Biotechnology, National Agri-Food Biotechnology Institute, Mohali, Punjab, India
dMechanical, Aerospace and Nuclear Engineering, Rensselaer Polytechnic Institute, 110, 8th Street, Troy, New York 12180, USA
eDepartment of Biochemistry, Institute of Science, Banaras Hindu University, Varanasi, Uttar Pradesh 221005, India
fInfectious Disease Research Laboratory, Department of Medicine, Institute of Medical Sciences, Banaras Hindu University, Varanasi, Uttar Pradesh 221005, India
First published on 22nd July 2020
Abstract
Leishmania is an obligate intracellular protozoan parasite, which resides in human macrophage vacuoles that are referred to as parasitophorus vacuoles. Amphotericin B (AmB) is the first-line drug with 99% cure rates; however, overdose-induced toxic side effects are a major limitation. To improve the efficacy at lower dose and subsequently to avoid toxicity and to further investigate the role of charge dynamics on the efficacy, a graphene oxide (GO)-based composite of AmB was developed with native negatively charged GO and amine-conjugated positively charged AGO. The AGO composite resulted in enhanced uptake as confirmed by confocal and FACS analysis. Thus, AGO caused a strong inhibition of amastigotes, with IC50 values 5-fold lower than free AmB. The parasitophorus vacuoles harbour a hydrolytic and acidic environment, which is favourable for the parasites, as they don't attenuate this condition. AGO–AmB was able to modify the intracellular pH of the Leishmania donovani-infected macrophages, generating unfavourable conditions for the amastigote, and thus improving its efficacy.
1. Introduction
Visceral leishmaniasis (VL) is a neglected infectious disease caused by Leishmania donovani, an obligate intra-macrophage protozoan parasite, which resides and multiplies inside the phagolysosomes of the macrophages.1 Clinically, VL is the most severe form of leishmaniasis, with an estimated global annual occurrence of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–90000 cases.2 Due to the unavailability of effective vaccine(s) against VL, the treatment mainly relies on chemotherapy. The current anti-leishmanial chemotherapy (pentavalent antimonials, miltefosine, liposomal amphotericin B and pentamidine) has several limitations, such as toxic adverse effects, cost and long duration of treatment.1,3 Amphotericin B (AmB) is the approved first-line drug for VL-infected patients, but requires close observation due to its toxic effects, including hypokalaemia, nephrotoxicity and myocarditis.4 Hence, a nanoformulation-assisted enhanced uptake at lower drug concentration could improve the therapeutic index and decrease the toxic effects.5 Liposomal formulation of AmB approved by the U.S. FDA is available under the brand name “AmBisome” and have shown improved efficacy and safety.4,6 Nanosized formulations are easily phagocytosed by the macrophages to release chemotherapeutic agents to clear intracellular parasites.7 For treating fungal infections, various nanoformulations of AmB have been developed using polysaccharides, dendrimers, silver nanoparticles, iron nanoparticles and carbon nanoparticles as vectors.7–9 These AmB formulations with different functional moieties showed specific binding to fungal ergosterol with enhanced efficacy and reduced toxicity in comparison to free AmB at lower concentration.10 Nanoformulations of AmB,11 functionalized carbon nanotubes (f-CNTs)–AmB,12 mannose-anchored thiolated chitosan (MTC)-coated AmB13 and AmB–SLNs have been used to effectively treat experimental VL infections with enhanced efficacy and safety compared to free AmB.
000–90000 cases.2 Due to the unavailability of effective vaccine(s) against VL, the treatment mainly relies on chemotherapy. The current anti-leishmanial chemotherapy (pentavalent antimonials, miltefosine, liposomal amphotericin B and pentamidine) has several limitations, such as toxic adverse effects, cost and long duration of treatment.1,3 Amphotericin B (AmB) is the approved first-line drug for VL-infected patients, but requires close observation due to its toxic effects, including hypokalaemia, nephrotoxicity and myocarditis.4 Hence, a nanoformulation-assisted enhanced uptake at lower drug concentration could improve the therapeutic index and decrease the toxic effects.5 Liposomal formulation of AmB approved by the U.S. FDA is available under the brand name “AmBisome” and have shown improved efficacy and safety.4,6 Nanosized formulations are easily phagocytosed by the macrophages to release chemotherapeutic agents to clear intracellular parasites.7 For treating fungal infections, various nanoformulations of AmB have been developed using polysaccharides, dendrimers, silver nanoparticles, iron nanoparticles and carbon nanoparticles as vectors.7–9 These AmB formulations with different functional moieties showed specific binding to fungal ergosterol with enhanced efficacy and reduced toxicity in comparison to free AmB at lower concentration.10 Nanoformulations of AmB,11 functionalized carbon nanotubes (f-CNTs)–AmB,12 mannose-anchored thiolated chitosan (MTC)-coated AmB13 and AmB–SLNs have been used to effectively treat experimental VL infections with enhanced efficacy and safety compared to free AmB.
Poly(o-phenylenediamine)-based nanocomposites have been developed for selective leishmaniasis treatment in the macrophage, but here the dose was above 50 μg mL−1.14 The drug-delivery efficiency of the nanocarriers has demonstrated a large dependence on the surface charge, and it is predominantly believed that a positive surface charge could increase the uptake and toxicity.15–18 Hence to avoid such nonspecific binding, a smart system having a negative charge but which could attain a positive charge in the targeted cell environment is desired. To achieve this, a nanogel with 2-diisopropylamino, which can turn negatively to positively charged in the acidic environment at the extracellular tumour cell space has been previously demonstrated.19 Further, amastigotes (the infective form of L. donovani in humans) dwell in the parasitophorus vacuoles of macrophages. This is crucial for the amastigotes, since an acidic environment is essential for the multiplication of the parasite.20 We envisaged attenuating this environment with a nanocarrier, which could complement the uptake through charge dynamics to improve drug efficacy.
Graphene oxide (GO) has been used for targeted drug delivery thanks to its ability to bind and release in response to different stimuli.21 In GO, the double polymer chitosan (CS) and a dimethyl maleic anhydride-modified chitosan (DMMC)-coated composite was found to have such dynamics.22 Often the bottleneck for the translation of GO to biology applications is the residual effects.23 Recently, the biodegradation of GO was confirmed with different enzymes, which facilitates their use as a residue-free drug-delivery platform.24,25 To the best of our knowledge, the use of a GO-based composite for leishmaniasis drug delivery has been limited to our previous study, where an amino acid-coated composite was shown to have less toxicity to the host cell, but the effect of the charge and its dynamics was ignored.4 Here, epoxy-targeted selective amine-functionalized GO has been developed by ethylenediamine conjugation.26,27 Thus, the carboxyl groups present in the GO offers a negative charge at neutral pH and the acidic pH in the protozoa vicinity may neutralize this charge to enhance the uptake. Our earlier amino acid-modified GO conjugated with AmB composite had neutral charge, which challenged its stability. Hence in the present study, we developed GO–AmB and amine modified GO (AGO)-AmB nanocomposite (AGO–AmB) and investigated in detail the charge dynamics mechanism, which, to the best of our knowledge, is the first time this has been reported. Further, the in vitro release behaviour is documented along with uptake via confocal microscopy and FACS analysis and finally the anti-leishmanial effect is quantified.
2. Materials and methods
4′,6-Diamidino-2-phenylindole (DAPI), fluorescein isothiocyanate isomer I (FITC), amphotericin B (AmB), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), graphite powder and ethylenediamine (EDA) (ReagentPlus) were procured from Sigma Aldrich, USA. Phosphotungstic acid (PTA), dimethylsulfoxide (DMSO), phosphate buffer saline (PBS), methanol, 4% paraformaldehyde and a cellulose dialysis tube were obtained from HiMedia Laboratories, Mumbai, India. Glass-bottomed dishes were purchased from Thermo Scientific (USA). Carbon-coated TEM grids were procured from Beta Tech Equipment Pvt. Ltd. (India). Hydrogen peroxide (ISO grade), orthophosphoric acid (BP grade), potassium permanganate, ethanol and sulphuric acid were purchased from Merck. All chemicals used were of analytical grade.2.1 Synthesis of GO
GO was synthesized by the well-known improved Hummer's method.28 Briefly, 500 mg of graphite flakes were added to 66.6 mL of a concentrated H2SO4/H3PO4 (9:1 ratio) mixture in a 250 mL conical flask. The reaction mixture was stirred for 15 minutes followed by the slow addition of 3 g KMnO4. After that, the mixture was incubated overnight at 50 °C under vigorous stirring. Then, the mixture was allowed to cool to room temperature (R.T) followed by the addition of 67 mL of chilled distilled water and 0.5 mL of 30% H2O2. Again, the reaction mixture was allowed to cool to R.T and centrifuged at 12000 × g for 20 minutes. The supernatant was decanted and the obtained pellet was washed three times each with HCl, ethanol and distilled water. Finally, the obtained material was neutralized by dialysis in distilled water and dried overnight at 50 °C.
2.2 Amine functionalization of GO (AGO)
The GO obtained in the previous step was amine functionalized as previously reported by us and others.29,30 Briefly, 50 mg of GO was dispersed in 50 mL of 95% ethanol by probe sonication and 325 μL of EDA was added. Then, the reaction mixture was allowed to react with EDA for 24 h at R.T under continuous stirring. After completion of the reaction, the product was pelleted down by centrifugation at 12000 × g for 10 min. Then, the obtained product was washed three times each with ethanol, methanol and acetone and finally, the obtained product was dried in a vacuum oven.
2.3 Cell culture and the parasite
The murine J774A.1 macrophage cell line was purchased from the National Centre for Cell Science, Pune, India. The cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (HIFBS; Gibco), 1% penicillin/streptomycin (Pen/Strep, 10000 IU mL−1; Gibco), sodium bicarbonate (Sodium Bicarbonate Solution; Gibco) and HEPES (Gibco; HEPES). The cells were maintained in a humidified incubator at 37 °C with a 5% CO2 environment.
In vitro anti-leishmanial activity was evaluated using L. donovani (LEM 138) promastigotes. L. donovani promastigotes were cultured in M199 medium supplemented with 10% heat-inactivated fetal bovine serum (HIFBS), 100 IU per mL penicillin, 100 μg per mL streptomycin and 50 μg per mL gentamycin and maintained at 26 °C in a BOD incubator. Cultures were passaged twice a week and maintained for not more than 1 month.
2.4 Conjugation of GO and AGO with AmB
First, AmB was conjugated onto the surface of GO and AGO by dispersing 20 mg each of GO and AGO in 10 mL of milli Q water, followed by sonication for 10 min to obtain a homogeneous suspension. AmB (20 mg) in 5 mL methanol was subsequently added drop-wise into individual homogeneous suspensions to facilitate the electrostatic interactions between AmB and GO/AGO. The suspension was then continuously stirred at room temperature for 24 h. The resulting mixture was then centrifuged at 14000 × g for 30 min and washed three times with milli Q water to remove any unbound AmB. The obtained pellet was freeze-dried and thus obtained GO–AmB and AGO–AmB nanocomposites were used for further studies.
2.5 Characterization
Absorption spectra were measured using a UV-visible spectrophotometer (UV-2600 Shimadzu). Powder X-ray diffraction (XRD) pattern analysis was carried out using a Cu Kα1 radiation source (λ = 1.5406 Å) at 25 mA and 40 kV with a Bruker D8 ADVANCE diffractometer. ATR-FTIR spectroscopy was employed to measure the FT-IR spectra (Bruker Vertex 70v spectrophotometer). The scanning range was 400–4000 cm−1 for each spectrum. The morphology of the GO and AGO nanostructures was analyzed by transmission electron microscopy (JEOL JEM-2100 operating at 200 kV). The samples were prepared by placing a drop of homogeneous solution of GO and AGO on a carbon-coated copper grid followed by vacuum drying in a desiccator overnight. Energy dispersive X-ray (EDX) spectra were recorded by a Bruker Nano GmbH instrument equipped with Esprit 1.9 software. A Malvern Zetasizer Nano ZSP instrument was employed to determine the zeta potential and hydrodynamic diameter at 25 °C. Well-dispersed samples were added into a clear glass DLS cuvette for hydrodynamic diameter measurements. Raman spectra were recorded at 532 nm laser excitation with a confocal Raman system (WITec Focus Innovations).2.6 Encapsulation efficiency (% EE) and drug loading (% DL)
The percentage of AmB conjugated with GO–AmB and AGO–AmB nanostructures was determined by quantifying the amount of free drug in the supernatant. Briefly, the dispersion of conjugated GO–AmB and AGO–AmB nanocomposites was centrifuged by using an ultracentrifuge (Beckman Coulter) at 14000 × g for 30 min at room temperature to pellet down the suspended nanostructures to settle. The supernatant was collected and free AmB in the supernatent was analyzed using a UV-visible spectrophotometer at 408 nm. The encapsulation efficiency and drug loading capacity were determined by using the following formulae.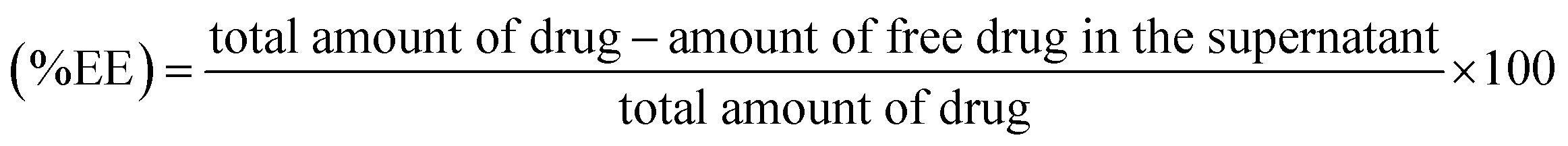
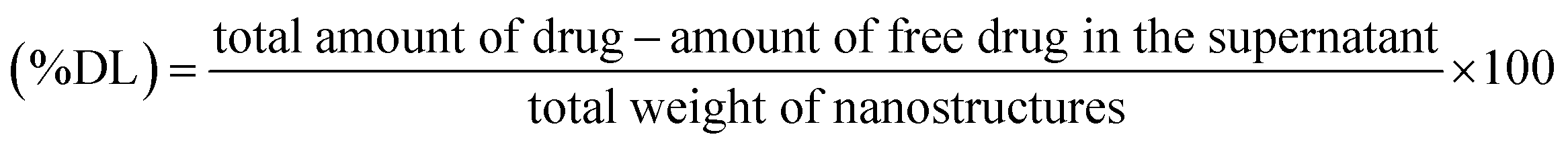
2.7 In vitro release study
The GO–AmB and AGO–AmB nanocomposites (1 mg each) were dispersed in 2 mL aqueous solution and placed into a pre-soaked dialysis bag with a 12 kDa cut-off size and dialyzed in 20 mL of buffer solution of pH 7.4 under continuous shaking at 100 rpm and maintained at 37 ± 2 °C. At regular time intervals, aliquots were withdrawn and replaced with fresh buffer to maintain the sink condition. The samples withdrawn at different time intervals were analyzed by UV-visible spectroscopy (Shimadzu UV-2600) at 408 nm. A standard curve was plotted and used to calculate the corresponding concentration of released AmB and hence the concentration obtained was plotted against various time points to obtain the drug-release profile.2.8 In vitro cytotoxicity assay
The cytotoxicities of AmB, GO, AGO, GO–AmB and AGO–AmB were assessed by MTT assay on the J774A.1 macrophage cell line. Briefly, J774A.1 cells were cultured on 96 well plates at an initial cell density of 5 × 104 cells per well and incubated for 24 h at 37 °C in a 5% CO2 environment allowing the cells to adhere, followed by treatment with 1 μg mL−1 of AmB, GO, AGO, GO–AmB and AGO–AmB for 24 h at 37 °C in 5% CO2; untreated cells served as the control. After treatment, a volume of 20 μL MTT reagent (5 mg mL−1) was added to each well and then incubated for 4 h to form reduced MTT formazan crystals. The precipitated crystals were dissolved in 100 μL of DMSO. Finally, the reduced MTT was spectrophotometrically analyzed at 595 nm, using a microplate reader (Infinite 200 PRO microplate plate reader). The percentage viability was expressed as the percentage of [1 − (ODcontrol − ODparticle)/(ODcontrol − ODblank)] × 100.2.9 Cellular uptake study
000 × g for 30 min and washed three times with milli Q water to remove any unbound FITC. Briefly, 5 × 104 J774A.1 cells were seeded onto glass-bottomed dishes and allowed to adhere for 24 h at 37 °C in a 5% CO2 environment. The cells were washed with incomplete RPMI-1640 media and then treated with 50 μM FITC@GO–AmB and FITC@AGO–AmB for 24 h. After treatment, the cells were fixed with 4% paraformaldehyde for 20 min followed by washing (3 times) with 1× PBS. The cells were then stained with 4′,6-diamidino-2-phenylindole dye (DAPI) for 5 min followed by washing (3 times) with 1× PBS. Lastly, the images were acquired at 63× using a confocal laser scanning microscope (CLSM, LSM 880 NLO, Carl Zeiss, Germany).2.10 In vitro anti-leishmanial activity against intra-macrophage amastigotes of L. donovani
In vitro anti-leishmanial activity of GO–AmB and AGO–AmB was performed against intra-cellular amastigotes of L. donovani. Briefly, J774A.1 macrophages (2.5 × 105 cells per mL) were cultured in complete RPMI-1640 and seeded (200 μL per well) in eight chamber Lab Teck tissue culture slides (Nunc, USA) followed by incubation at 37 °C in a 5% CO2 environment for 2 h to allow the cells to adhere. The adherent macrophages were washed (3 times) with pre-warmed incomplete RPMI-1640 and infected with metacyclic promastigotes in a 1:10 ratio, followed by incubation at 37 °C in a 5% CO2 environment for 12 h. Non-phagocytosed promastigotes were removed by washing (3 times) with warm incomplete RPMI-1640 and the infected macrophages were incubated in the presence and absence of the test compounds (GO–AmB and AGO–AmB) in different concentrations in complete RPMI-1640 at 37 °C in a 5% CO2–air atmosphere for 72 h. The treated wells were washed with PBS and stained with Wright's stain to assess the intra-cellular amastigote growth by counting at least 100 cells per slide under an oil immersion lens microscope (100×). The percentage inhibition of amastigote multiplication was calculated by the following formula: | PI = 100 − (AT/AC) × 100 |
3. Results and discussion
In this study, the anti-leishmanial potential of GO and AGO were evaluated along with details of drug loading, slow and sustained drug delivery, cellular uptake and FACS analysis. GO was synthesized by an improved Hummer's method, with epoxide and other groups. GO was modified with amine groups through the attack of nucleophilic EDA on the electrophilic epoxide ring in GO as previously reported.30 After synthesis, GO and AGO were characterized through various techniques, including XRD, TEM, UV-vis, Raman and EDX.3.1 Characterization of GO and AGO
In the XRD pattern of GO, the (0 0 2) crystal plane of GO was evident at 2θ = 11.4° (Fig. S1†).32,33 The TEM image of GO showed flakes with an irregular shape and size (Fig. 1A). In the case of AGO, the flake size was found to be reduced; this may be due to the undesired fragmentation of the GO sheet through reduction (Fig. 1B). The UV-vis spectra of the colloidal solution of GO showed the absorbance maxima at 230 nm and a hump at 300 nm, which could be attributed to π–π* and n–π* transitions of carbonyl groups, respectively (Fig. 1C; black curve).28 Whereas in the case of AGO, the peak was found to be red-shifted to 265 nm as reported before (Fig. 1C; red curve).30,34 Raman analysis showed the characteristics GO peaks at 1604 cm−1, corresponding to the G band, which arose due to the E2g phonon of sp2 carbon atoms, while the peak at 1352 cm−1 corresponded to D bands, assigned to the zone boundary phonons with an A1g symmetry (Fig. 1D; black curve).32,35–38 In reference to GO, the G band in AGO showed a marginal shift from 1604 to 1597 cm−1, which may be due to the electron-donating nature of EDA in AGO or restoration of the sp2 domain (Fig. 1D; red curve).39–41 Further, the restoration of the sp2 domain or reduction of GO led to the increase in the ID/IG ratio in the case of AGO.42 The other reason for the increase in the ID/IG ratio may be due to the C–N bond formation and distortion. The distribution of functional groups in GO and AGO was confirmed by EDX. In the EDX spectra of GO, only carbon (64%) and oxygen (35%) peaks appeared; whereas in the case of AGO, an intense peak corresponding to nitrogen (17%) appeared and the oxygen peak was reduced by 19%, which confirmed the successful amine functionalization (Fig. S2†). The distribution of amines on GO sheets in AGO was confirmed by EDX mapping (Fig. S3†). AGO showed a homogeneous distribution of carbon, oxygen and nitrogen groups (which appeared due to the amine) on the whole surface, thus confirming the successful functionalization. Similar, EDX mapping confirmation of the amine conjugation to GO has been reported before.43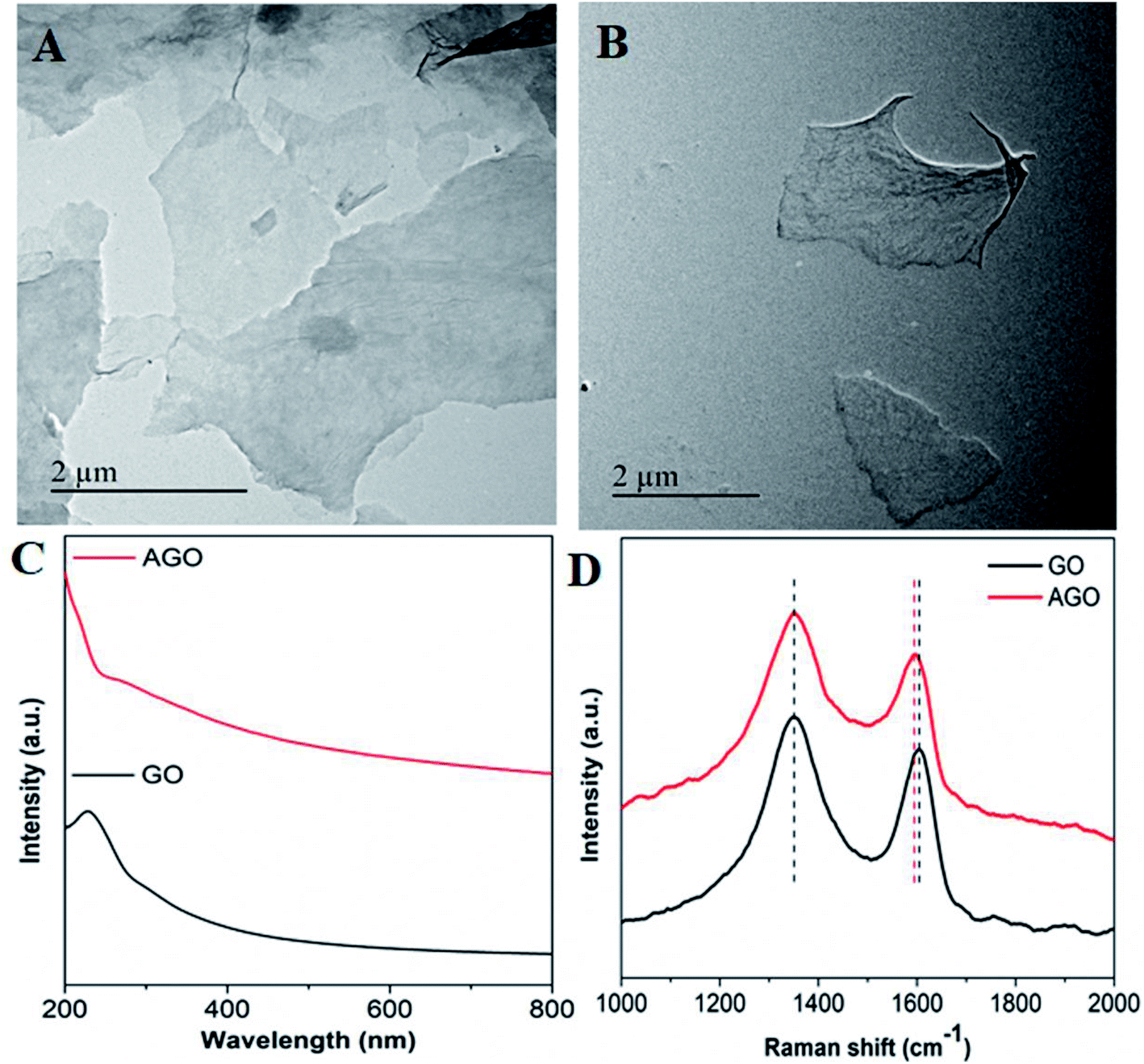 | ||
| Fig. 1 TEM images of (A) GO, (B) AGO, (C) UV-vis spectra of GO and AGO, and (D) Raman spectra of GO and AGO. | ||
Further, the presence of primary amines on GO in AGO was confirmed by ninhydrin assay. Fig. S4A† shows the appearance of a purple colour upon the reaction of ninhydrin with different concentrations of AGO. The absorbance at 510 nm was found to increase with increasing the AGO concentration, which confirmed the presence of primary amines in AGO (Fig. S4B†); whereas in the case of GO, it was absent.
After successful characterization, GO and AGO were loaded with AmB, which is currently the most commonly used first line drug for the treatment of VL, by a lyophilization technique to form a nanodrug composite viz., GO–AmB and AGO–AmB respectively. Following this the unbound drug was washed in deionized water (3 times) to eliminate any unbound drug. The loading and entrapment efficiencies of GO and AGO were calibrated by subtracting the drug amount present in the supernatant, which was quantified with UV-vis spectroscopy. The drug loading reflects the mass ratio of the drug to the nanostructures, while the entrapment efficiency indicates the amount of drug conjugated with the nanostructures. GO and AGO showed loading efficiencies of 43% and 55%, respectively. Collectively, this loading could be attributed to the hydrogen bonding, π–π interaction and hydrophobic interaction.44,45 Thus, the loading efficiency confirmed that as compared to GO, AGO served as a better carrier, which can be attributed to the increased hydrogen bonding with the amine functionalization. Since the epoxy rings, which cannot participate in hydrogen bonding in the GO, is opened up into hydroxyls and amines in AGO, which, has the capacity to form hydrogen bonds. All the possible hydrogen bonding options in the GO and AGO drug composite are given below. There is possibility for six types of hydrogen bond formation between GO and AmB nanostructures: –COOH of GO with –OH, –NH2 and –COOH of AmB, –OH of GO –OH, –NH2 and –COOH of AmB.46 The amine functionalization in AGO adds three more hydrogen bond formation possibilities: –NH2 of AGO with –OH, –NH2 and –COOH of AmB. The entrapment efficiencies of AmB in GO and AGO was found to be approximately the same at 99% and 99.5%, respectively (Table 1).
| GO–AmB | AGO–AmB | |
|---|---|---|
| Drug loading (% DL) | 43.09 | 55.29 |
| Encapsulation efficiency (% EE) | 99.11 | 99.53 |
The loading of AmB was further confirmed by FTIR, zeta potential and the corresponding change in hydrodynamic size. The FTIR spectra of GO, AGO, AmB drug and the nanodrug composite (GO–AmB and AGO–AmB) are shown in Fig. 2A. The FTIR spectrum of GO showed signature peaks at 3000–3500, 1725, 1630, 1170 and 1053 cm−1, which corresponded to –OH stretching, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, CC bending, epoxy C–O–C stretching and alkoxy C–O stretch vibrations, respectively (Fig. 2A; black curve).28 After amine conjugation, a few of the GO peaks (1725 and 1170 cm−1) disappeared in AGO, due to EDA binding. A new intense peak corresponding to the in-plane N–H stretching vibrations appeared at 1600 cm−1 (Fig. 2A; red curve).30 After AmB loading, GO–AmB and AGO–AmB showed the co-appearance of 1630 and 1590 cm−1 peaks corresponding to GO and AGO, respectively, with additional peaks at 1070 (C–O stretch) and 870 cm−1 (C–H stretch) corresponding to AmB (Fig. 2A; blue and pink curve), confirms successful loading of AmB.
O stretching, CC bending, epoxy C–O–C stretching and alkoxy C–O stretch vibrations, respectively (Fig. 2A; black curve).28 After amine conjugation, a few of the GO peaks (1725 and 1170 cm−1) disappeared in AGO, due to EDA binding. A new intense peak corresponding to the in-plane N–H stretching vibrations appeared at 1600 cm−1 (Fig. 2A; red curve).30 After AmB loading, GO–AmB and AGO–AmB showed the co-appearance of 1630 and 1590 cm−1 peaks corresponding to GO and AGO, respectively, with additional peaks at 1070 (C–O stretch) and 870 cm−1 (C–H stretch) corresponding to AmB (Fig. 2A; blue and pink curve), confirms successful loading of AmB.
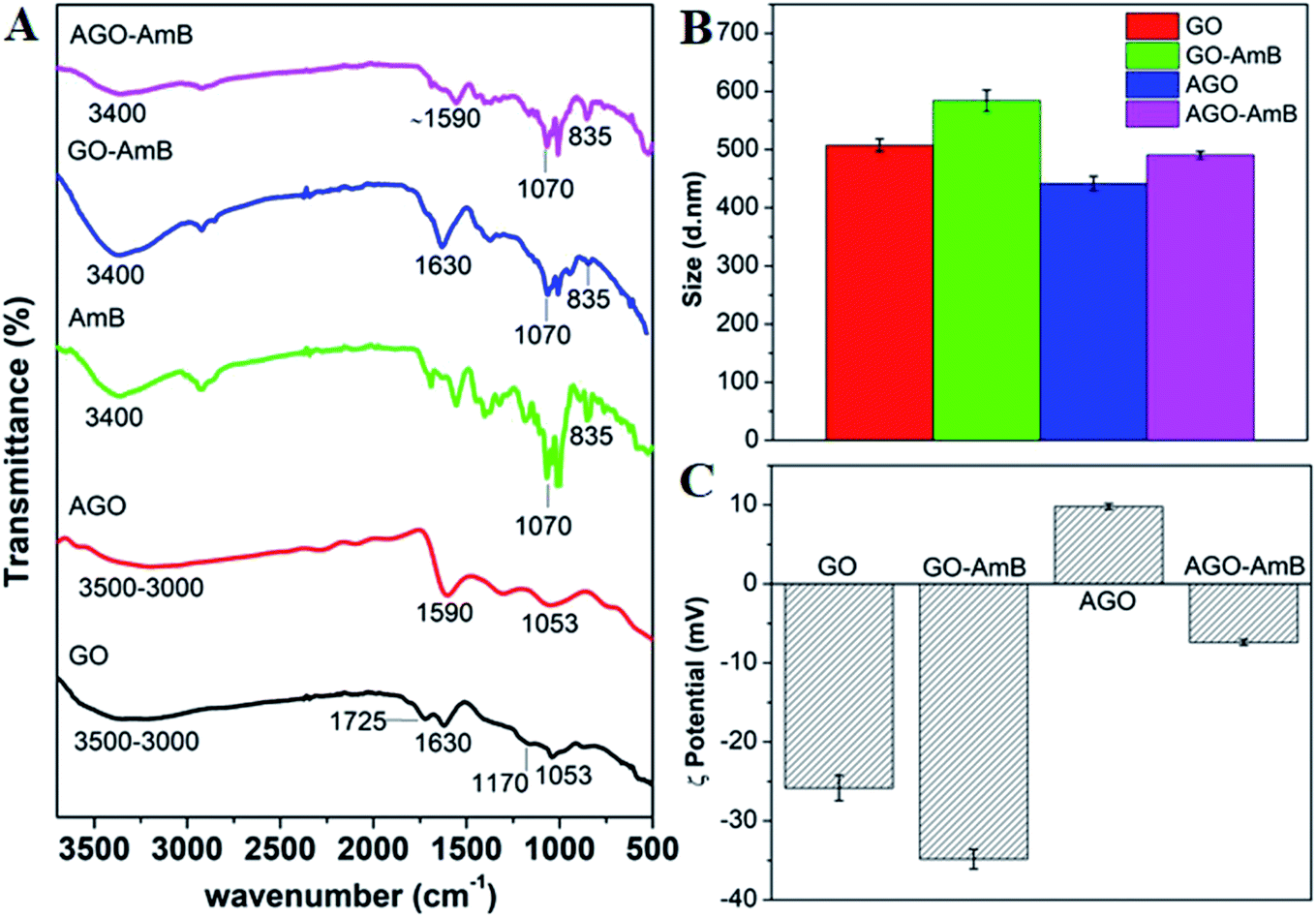 | ||
| Fig. 2 (A) FTIR spectra of GO, AGO, AmB, GO–AmB and AGO–AmB, (B) DLS, and (C) zeta potential. | ||
The hydrodynamic sizes of GO and AGO before and after AmB loading were checked by dynamic light scattering (Fig. 2B). The average hydrodynamic sizes of GO and AGO were found to be ∼510 and ∼440 nm, respectively. Here, the small reduction in size of AGO may be due to the fragmentation of GO sheets through reduction, which further corroborated the results in the TEM images. After AmB loading, the sizes of GO and AGO were found to increase to ∼585 and ∼490 nm, respectively. A similar increment in the size after drug loading in nanoparticles has been reported.47 The surface charges of particles before and after drug loading are presented in Fig. 2C. The neutralized GO showed an average negative charge of −26 mV, which was due to the presence of negatively charged functional groups on the surface of GO. In the case of AGO, due to amine conjugation the negative charge of GO was shifted to +9 mV.30 After the loading of AmB in GO and AGO, the average value of charge was shifted to the negative side with −35 mV in GO–AmB and −7 mV in AGO–AmB. This may be due to the carboxyl groups in the AmB drug molecules.47
3.2 In vitro release study
As the Leishmania parasite is an intra-macrophage protozoan parasite, to minimize toxicity to the host cell, AmB needs to be released slowly.48 Therefore, the release profile of AmB from the nanostructures at biological pH (7.4) was studied. The release profiles of AmB from GO–AmB and AGO–AmB nanostructures were assessed by using the dialysis bag diffusion method in a release medium of pH 7.4, respectively, maintained at 37 °C under continuous shaking. Both the nanostructures displayed biphasic release kinetics. At pH 7.4, the AGO–AmB nanostructures exhibited an initial burst release of 31.02%, while the GO–AmB nanostructures exhibited a slightly lower burst release, with 28.5% drug release over the first 6 h followed by a sustained release. The initial burst release of a minor amount was due to the rapid desorption and diffusion of AmB molecules located close to the surface of the nanostructures. After the burst release phase, the rate of release stabilised as the dominant release mechanism was changed to drug diffusion. It can be seen in Fig. 3A that the drug-modified nanostructures provided a sustained release. Moreover, the release of AmB was not affected by the conjugation.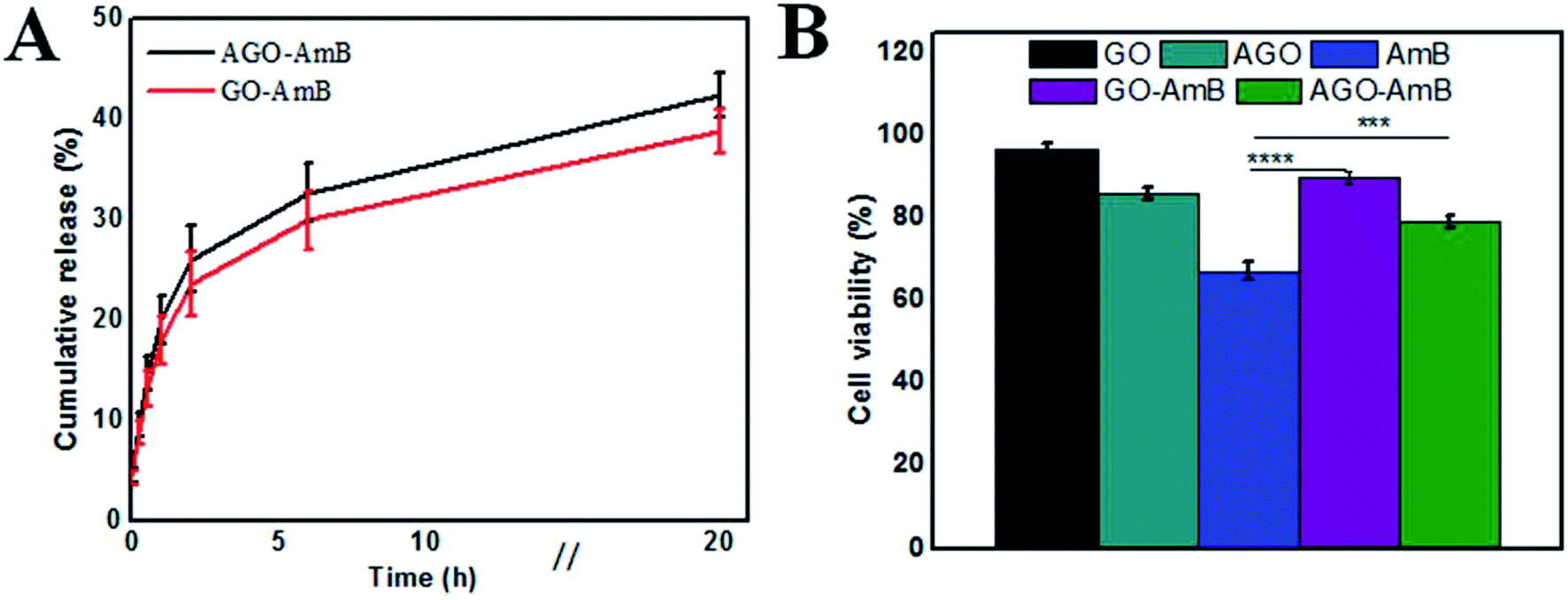 | ||
| Fig. 3 (A) In vitro drug-release profile from AGO–AmB and GO–AmB composites in buffer of pH 7.4, and (B) Cell viability percentage of J774A.1 cells treated with 1 μg mL−1 of AmB in the form of free AmB, AGO–AmB and GO–AmB along with an equivalent amount of AGO and GO in the treatment with AGO–AmB and GO–AmB, respectively (***p < 0.001; ****p < 0.0001). | ||
3.3 In vitro cytotoxicity assay
Before examining the cellular uptake and activity, the in vitro cytotoxicity of the composite (GO–AmB and AGO–AmB) and the free drug along with GO and AGO was assessed by performing the MTT assay on J774A.1 cells after 24 h of treatment. The cell viability data shown in Fig. 3B revealed that GO–AmB and AGO–AmB were less toxic compared to free AmB. Considering the widely accepted 80% viability as the threshold to consider a material as toxic,49 the composites GO–AmB and AGO–AmB at 1 μg mL−1 did not show any significant toxicity. Whereas the treatment with equal amount of free drug causes the cell viability to drop down to less than 80% (67%), which highlights the ability of GO and AGO to ameliorate the drug toxicity to the host.3.4 Cellular uptake
As Leishmania amastigotes reside inside the host's macrophages, the cellular uptake of the composite is an important factor to understand the therapeutic efficiency. Hence, we investigated the cellular uptake of GO–AmB and AGO–AmB in J774A.1 cells using confocal microscopy with the respective equivalent amount of FITC-labelled composites (FITC@GO–AmB and FITC@AGO–AmB) after 24 h of incubation (Fig. 4A–H). FITC fluorescence appeared localized intracellularly inside the cell, suggesting that the nanostructures were taken up by the cell. The image showed an enhanced uptake of the amine-modified composite FITC@AGO–AmB compared to the FITC@GO–AmB composite. To understand this enhanced uptake, we tested the pH-dependent charge dynamics in the AGO–AmB composite (Fig. S5†). The weak negative charge on the surface of AGO–AmB got neutralized at acidic pH, whereas in the GO composite, the strong negative surface did not allow for the complete neutralization. Hence the enhanced uptake of FITC@AGO–AmB could be explained as being due to the neutralisation of the negative charge on the AGO–AmB composite in the endocytosis-mediated pH environment. This charge dynamics allowed the AGO–AmB composite to cross the negatively charged cell membrane more easily than GO–AmB.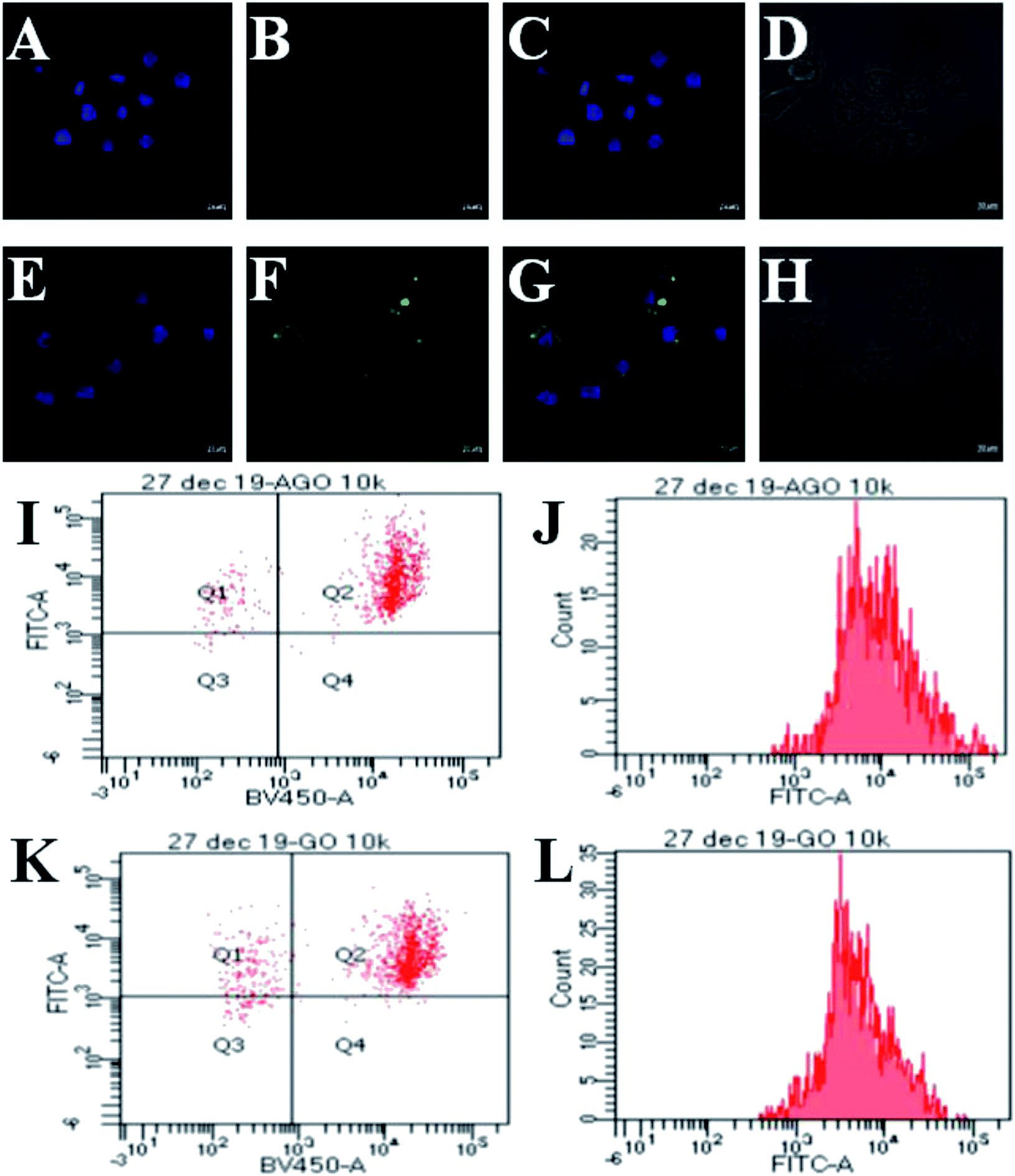 | ||
| Fig. 4 CLSM images (A and E) DAPI (λex: 405 nm)-stained nucleus of J774A.1 cells, colocalization of FITC (λex: 488 nm)@GO–AmB (B) FITC@AGO–AmB (F) in J774A.1 cells after 24 h, (C and G) merged images and (D and H) bright field images. Scale bar in 63× are denoted as 10 μm. Dot plot FACS analysis of internalized (I) FITC@AGO–AmB and (K) FITC@GO–AmB by J774A.1 cells. The fluorescent intensity of (J) FITC@AGO–AmB and fluorescent intensity of (L) FITC@GO–AmB. | ||
3.5 FACS
The dot plot analysis shown in Fig. 4I shows a population of DAPI-stained nucleus of J774A.1 cells after 24 h of treatment with FITC@AGO–AmB localized in quadrant Q2. Fig. 4K represents a population of DAPI-stained J774A.1 cells in quadrant Q2 localized with FITC@GO–AmB after 24 h of treatment. It could be observed that the majority of J774A.1 cells were internalized with FITC@AGO–AmB, when compared to FITC@GO–AmB. The fluorescent intensity of FITC@AGO–AmB was ∼90% (Fig. 4J), while the fluorescent intensity of FITC@GO–AmB was ∼80% (Fig. 4L). This difference in intensities indicates that more FITC@AGO–AmB was internalized per cell when compared to FITC@GO–AmB. The results obtained by the FACS study provide a more accurate quantitative analysis for the cellular uptake study.3.6 In vitro anti-leishmanial activity of AGO–AmB, GO–AmB, AmBisome and AmB against L. donovani-infected J774A.1 macrophages
The anti-leishmanial activity of AGO–AmB and GO–AmB were compared with AmBisome and AmB under in vitro conditions with L. donovani-infected macrophages. All the samples were stained and the intra-cellular amastigotes were enumerated. AGO–AmB (1 μg mL−1) had significantly (p < 0.001) diminished intra-cellular amastigotes compared to free AmB. The IC50 values of AGO–AmB and GO–AmB were observed to be 0.057 ± 0.017 and 0.141 ± 0.034 μg mL−1, respectively, which were significantly (p < 0.05) less than the IC50 value of free AmB (0.288 ± 0.037 μg mL−1). The IC50 values of AGO–AmB and GO–AmB were ∼5-fold and ∼2-fold lower than the IC50 value of free AmB, respectively. L. donovani amastigotes reside in the parasitophorus vacuoles (strongly hydrophilic and acidic environment; pH 5.0) of macrophages. The acidic environment of parasitophorus vacuoles supports the growth, cellular respiration, nucleic acid synthesis and replication of amastigotes.50 AGO–AmB has been found to be able to modify the intracellular pH of parasitophorus vacuoles, which cause unfavourable conditions for amastigote survival. After internalisation, AGO–AmB creates a basic pH environment in parasitophorus vacuoles, which results in the inhibition of the metabolic activities of amastigotes and ultimately kills the parasites.51 AGO–AmB showed a maximum percentage of inhibition (92.4%) on intra-cellular amastigote growth of L. donovani. Interestingly, an equivalent amount of the GO and AGO without the drug has not shown any inhibition of amastigote growth, thus served as an inert drug carrier (Fig. 5).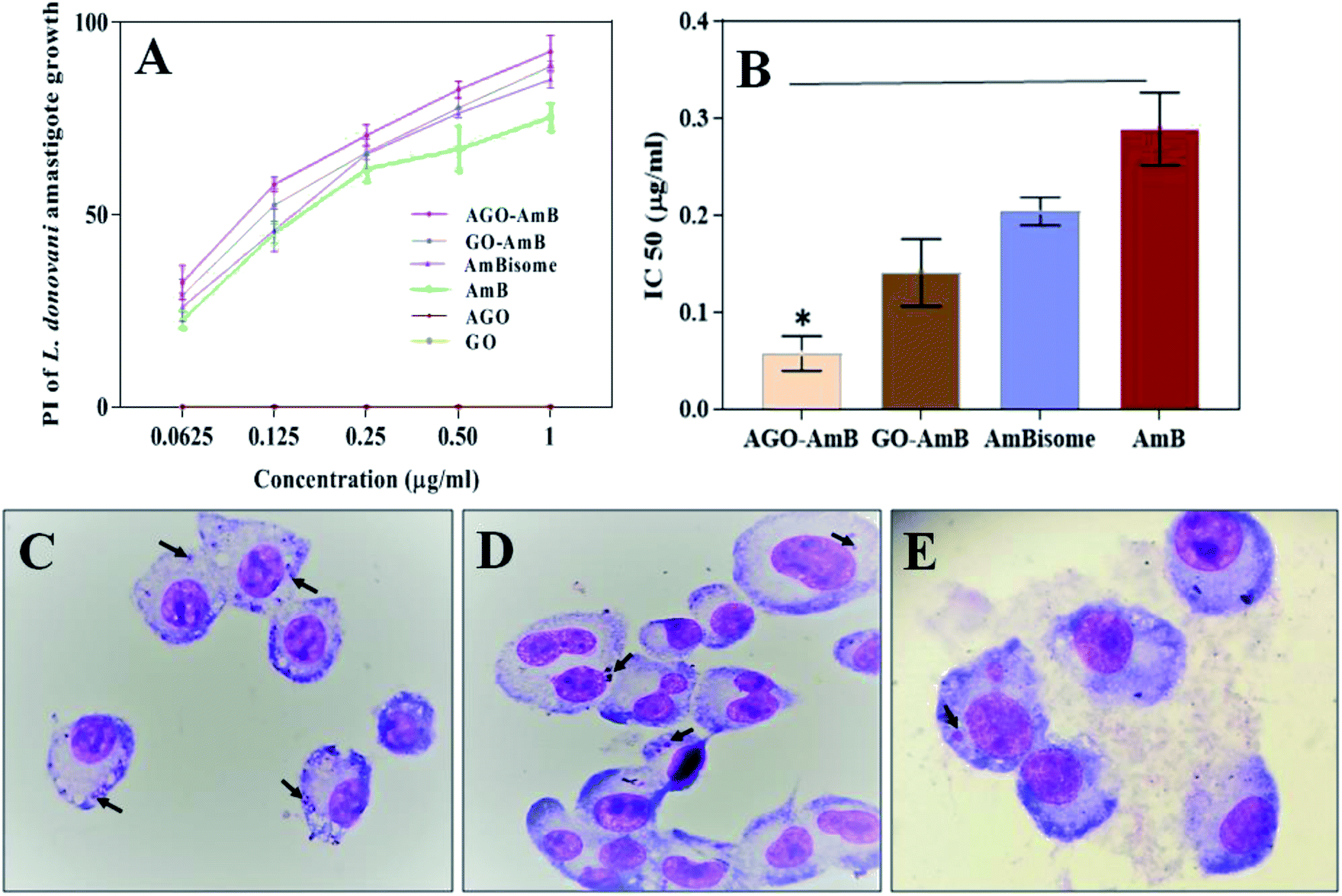 | ||
| Fig. 5 (A) Percentage inhibition of AGO–AmB, GO–AmB, AmBisome and AmB against L. donovani-infected J774A.1 macrophages, (B) IC50 values of AGO–AmB, GO–AmB, AmBisome and AmB against L. donovani-infected macrophages. Results are presented as the mean ± standard deviation and were analyzed by Graph Pad Prism 5 software (AGO–AmB, GO–AmB and AmBisome) vs. AmB, *(p < 0.05). Microscopic images of (C) control, (D) GO–AmB-treated and (E) AGO–AmB-treated infected J774A.1 macrophages. Black arrows indicate L. donovani amastigotes. | ||
4. Conclusions
GO and AGO nanostructures were successfully prepared and characterized using FTIR, TEM, UV-vis, Raman, EDX and XRD. The nanostructures were then loaded with the anti-leishmanial drug AmB and the loading efficiency was calculated using UV-vis spectroscopy. Consequently, the nanocomposites showed a sustained biphasic release pattern, which curtailed the dose-dependent toxic side effects. Confocal microscopy revealed that amine conjugation of the negatively charged GO imparted a slight positive charge, which favour increased permeation through the cell membrane as compared to GO. FACS analysis provided more accurate quantitative insight into the results from the cellular uptake studies. In vitro cytotoxicity studies on J774A.1 cells revealed that the nanocomposites were potentially non-toxic at lower concentrations, while in vitro anti-leishmanial studies revealed that the AGO–AmB nanocomposites showed a higher (92%) inhibition of amastigote growth, with IC50 values 5-fold less than for free AmB. The nanocomposites could, therefore, be used as potential therapeutic anti-leishmanial agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Aakriti Singh, Sandeep Sharma and Shabi Parvez thanks Institute of Nano Science and Technology for the Doctoral Fellowship. Ganesh Yadagiri is thankful to Department of Science & Technology (DST) for providing financial support. This work is supported by Government of India for funding under DST-SERB Early Career Research Award (ECR/2016/000977), DST-SERB Core Research Grant (CRG/2019/006317) and in part by DST Nano Mission (SR/NM/NS-57/2016).References
- G. Yadagiri and P. P. Singh, in Infectious Diseases and Your Health, Springer Singapore, 2018, pp. 63–97 Search PubMed.
- Leishmaniasis|European Vaccine Initiative, http://www.euvaccine.eu/vaccines-diseases/diseases-of-poverty/leishmaniasis, accessed 4 June 2020.
- L. Monzote, Current Treatment of Leishmaniasis: A Review, 2009 Search PubMed.
- S. L. Mudavath, M. Talat, M. Rai, O. N. Srivastava and S. Sundar, Drug Des., Dev. Ther., 2014, 8, 1235–1247 CAS.
- E. Palma, A. Pasqua, A. Gagliardi, D. Britti, M. Fresta and D. Cosco, Materials, 2018, 11, 1–16 Search PubMed.
- A. Meyerhoff, Clin. Infect. Dis., 1999, 28, 42–48 CAS.
- M. Y. Want, M. Islammudin, G. Chouhan, H. A. Ozbak, H. A. Hemeg, A. P. Chattopadhyay and F. Afrin, Int. J. Nanomed., 2017, 12, 2189–2204 CrossRef CAS PubMed.
- V. Kesarwani, H. G. Kelly, M. Shankar, K. J. Robinson, S. J. Kent, A. Traven and S. R. Corrie, ACS Appl. Mater. Interfaces, 2019, 11, 34676–34687 CrossRef CAS PubMed.
- J. Yang, X. Zhang, Y. H. Ma, G. Gao, X. Chen, H. R. Jia, Y. H. Li, Z. Chen and F. G. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 32170–32181 CrossRef CAS PubMed.
- P. Kumar, P. Shivam, S. Mandal, P. Prasanna, S. Kumar, S. R. Prasad, A. Kumar, P. Das, V. Ali, S. K. Singh and D. Mandal, Int. J. Nanomed., 2019, 14, 6073–6101 CrossRef CAS PubMed.
- V. K. Prajapati, K. Awasthi, S. Gautam, T. P. Yadav, M. Rai, O. N. Srivastava and S. Sundar, J. Antimicrob. Chemother., 2011, 66, 874–879 CrossRef CAS PubMed.
- V. K. Prajapati, K. Awasthi, T. P. Yadav, M. Rai, O. N. Srivastava and S. Sundar, J. Infect. Dis., 2012, 205, 333–336 CrossRef CAS PubMed.
- H. S. Sarwar, M. F. Sohail, N. Saljoughian, A. U. Rehman, S. Akhtar, A. Nadhman, M. Yasinzai, H. E. Gendelman, A. R. Satoskar and G. Shahnaz, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 521–531 CrossRef CAS PubMed.
- U. Riaz, S. Jadoun, P. Kumar, M. Arish, A. Rub and S. M. Ashraf, ACS Appl. Mater. Interfaces, 2017, 9, 33159–33168 CrossRef CAS PubMed.
- M. M. Falinski, M. A. Garland, S. M. Hashmi, R. L. Tanguay and J. B. Zimmerman, Carbon, 2019, 155, 587–600 CrossRef CAS.
- S. R. S. Dev and G. S. Vijaya Raghavan, in Physical Properties of Foods: Novel Measurement Techniques and Applications, CRC Press, 2012, pp. 119–130 Search PubMed.
- Q. Zhang, P. Chen, L. Chen, M. Wu, X. Dai, P. Xing, H. Lin, L. Zhao and Y. He, J. Colloid Interface Sci., 2020, 568, 117–129 CrossRef CAS PubMed.
- T. Malina, K. Poláková, J. Skopalík, V. Milotová, K. Holá, M. Havrdová, K. B. Tománková, V. Čmiel, L. Šefc and R. Zbořil, Carbon, 2019, 152, 434–443 CrossRef CAS.
- L. Li, K. Raghupathi, C. Yuan and S. Thayumanavan, Chem. Sci., 2013, 4, 3654–3660 RSC.
- T. A. Glaser, J. E. Baatz, G. P. Kreishman and A. J. Mukkada, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7602–7606 CrossRef CAS PubMed.
- C. Hu, Y. Liu, J. Qin, G. Nie, B. Lei, Y. Xiao, M. Zheng and J. Rong, ACS Appl. Mater. Interfaces, 2013, 5, 4760–4768 CrossRef CAS PubMed.
- C. Hu, N. Ma, F. Li, Y. Fang, Y. Liu, L. Zhao, S. Qiao, X. Li, X. Jiang, T. Li, F. Shen, Y. Huang, Q. Luo and J. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 4603–4613 CrossRef CAS PubMed.
- R. Kurapati, S. P. Mukherjee, C. Martín, G. Bepete, E. Vázquez, A. Pénicaud, B. Fadeel and A. Bianco, Angew. Chem., Int. Ed., 2018, 57, 11722–11727 CrossRef CAS PubMed.
- R. Kurapati and A. Bianco, Nanoscale, 2018, 10, 19316–19321 RSC.
- R. Kurapati, F. Bonachera, J. Russier, A. R. Sureshbabu, C. Ménard-Moyon, K. Kostarelos and A. Bianco, 2D Mater., 2018, 5, 015020 CrossRef.
- A. Yang, J. Li, C. Zhang, W. Zhang and N. Ma, Appl. Surf. Sci., 2015, 346, 443–450 CrossRef CAS.
- T. Ramanathan, F. T. Fisher, R. S. Ruoff and L. C. Brinson, Chem. Mater., 2005, 17, 1290–1295 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Wang, P.-J. Chia, L.-L. Chua, L.-H. Zhao, R.-Q. Png, S. Sivaramakrishnan, M. Zhou, R. G.-S. Goh, R. H. Friend, A. T.-S. Wee and P. K.-H. Ho, Adv. Mater., 2008, 20, 3440–3446 CrossRef CAS.
- S. Sharma, B. K. Biswal, D. Kumari, P. Bindra, S. Kumar, T. Stobdan and V. Shanmugam, ACS Appl. Mater. Interfaces, 2018, 10, 18478–18488 CrossRef CAS PubMed.
- M. M. Sari, Mater. Chem. Phys., 2013, 138, 843–849 CrossRef CAS.
- Y. Cai, Y. Fadil, F. Jasinski, S. C. Thickett, V. Agarwal and P. B. Zetterlund, Carbon, 2019, 149, 445–451 CrossRef CAS.
- S. Sharma, S. Singh, A. K. Ganguli and V. Shanmugam, Carbon, 2017, 115, 781–790 CrossRef CAS.
- T. Kuila, P. Khanra, S. Bose, N. H. Kim, B. C. Ku, B. Moon and J. H. Lee, Nanotechnology, 2011, 22, 305710 CrossRef PubMed.
- B. Yang, Z. Liu, Z. Guo, W. Zhang, M. Wan, X. Qin and H. Zhong, Appl. Surf. Sci., 2014, 316, 22–27 CrossRef CAS.
- A. C. De Leon, L. Alonso, J. D. Mangadlao, R. C. Advincula and E. Pentzer, ACS Appl. Mater. Interfaces, 2017, 9, 14265–14272 CrossRef CAS PubMed.
- Y. Fadil, L. N. M. Dinh, M. O. Y. Yap, R. P. Kuchel, Y. Yao, T. Omura, U. A. Aregueta-Robles, N. Song, S. Huang, F. Jasinski, S. C. Thickett, H. Minami, V. Agarwal and P. B. Zetterlund, ACS Appl. Mater. Interfaces, 2019, 11, 48450–48458 CrossRef CAS PubMed.
- Y. Sui, A. Hess-Dunning, P. Wei, E. Pentzer, R. M. Sankaran and C. A. Zorman, Adv. Mater. Technol., 2019, 4, 1900834 CrossRef CAS.
- S. Sharma, B. Sahu, S. Srinivasan, M. Singh, J. Govindasamy and V. Shanmugam, Carbon, 2020, 162, 147–156 CrossRef CAS.
- W. Li, X. Z. Tang, H. Bin Zhang, Z. G. Jiang, Z. Z. Yu, X. S. Du and Y. W. Mai, Carbon, 2011, 49, 4724–4730 CrossRef CAS.
- Z. Li, C. He, Z. Wang, Y. Gao, Y. Dong, C. Zhao, Z. Chen, Y. Wu and W. Song, Photochem. Photobiol. Sci., 2016, 15, 910–919 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- A. Navaee and A. Salimi, RSC Adv., 2015, 5, 59874–59880 RSC.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- B. Li, G. Pan, N. D. Avent, R. B. Lowry, T. E. Madgett and P. L. Waines, Biosens. Bioelectron., 2015, 72, 313–319 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
- C. M. B. Santos, S. W. da Silva, L. R. Guilherme and P. C. Morais, J. Phys. Chem. C, 2011, 115, 20442–20448 CrossRef CAS.
- M. Ghorbani and R. Farhoudi, Drug Des., Dev. Ther., 2018, 12, 25–40 CrossRef CAS PubMed.
- ISO – ISO 10993-5:2009 – Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity, https://www.iso.org/standard/36406.html, accessed 19 February 2020.
- D. C. Miguel, J. K. U. Yokoyama-Yasunaka, W. K. Andreoli, R. A. Mortara and S. R. B. Uliana, J. Antimicrob. Chemother., 2007, 60, 526–534 CrossRef CAS PubMed.
- T. A. Glaser, J. E. Baatz, G. P. Kreishman and A. J. Mukkada, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7602–7606 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04266h |
| ‡ Equally contributed authors. |
| This journal is © The Royal Society of Chemistry 2020 |