
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proteomics and metabolomics analysis reveal potential mechanism of extended-spectrum β-lactamase production in Escherichia coli†
He Ma ab,
Bingjie Laic,
Yufen Jind,
Chang Tiana,
Jiaying Liua and
Ke Wang*a
ab,
Bingjie Laic,
Yufen Jind,
Chang Tiana,
Jiaying Liua and
Ke Wang*a
aDepartment of Respiratory Medicine, The Second Hospital of Jilin University, Changchun, China. E-mail: kewangm1@hotmail.com
bDepartment of Anesthesiology, The Second Hospital of Jilin University, Changchun, China
cDepartment of Intensive Care Unit, The Second Hospital of Jilin University, Changchun, China
dClinical Laboratory, The Second Hospital of Jilin University, Changchun, China
First published on 17th July 2020
Abstract
In this study, ten clinical susceptible strains and ten clinical ESBL-EC (extended-spectrum β-lactamase-producing Escherichia coli) were screened and obtained by microbial identification using ITEK® 2 Compact. TMT (Tandem Mass Tag) proteomics analysis discovered 1553 DEPs (differentially expressed proteins) between ESBL-EC and non-ESBL-EC. In addition, an untargeted metabolomics assay by using UHPLC-MS (ultra-high-performance liquid chromatography-mass spectrometry) was applied to compare the differential profiles of metabolites between β-lactam antibiotic-sensitive E. coli and multidrug-resistant ESBL-producing E. coli strains. The PCA (principal component analysis) score plots and OPLS-DA (orthogonal projections to latent structures discriminant analysis) plots clearly discriminated ESBL-EC and non-ESBL-EC, and volcano analysis presented 606 and 459 altered metabolites between ESBL-EC vs. non-ESBL-EC in positive and negative ion modes, respectively. Interestingly, the bioinformatics analysis demonstrated that the purine metabolism pathway was enriched in ESBL-EC. These results suggest that the existence of extended-spectrum β-lactamase affects the metabolite and protein profiles of E. coli. The correlation analysis of metabolomics and proteomics data established a correlation between DEPs and differential metabolites in the purine metabolism pathway. Moreover, three metabolite candidates in the purine metabolism pathway were validated by the UPLC-MRM-MS (ultra-performance liquid chromatography multiple reaction monitoring mass spectrometry) method. Our data suggest that these DEPs and differential metabolites may play important roles in the antibiotic resistance of ESBL-EC. Our study can provide scientific data for the mechanism study of antibiotic resistance of ESBL-EC at the metabolite and protein levels and targeting modulators to these pathways may be effective for treatment of ESBL-EC strains.
1. Introduction
E. coli (Escherichia coli) has the characteristics of being both a widespread gut commensal of vertebrates and a versatile pathogen.1 E. coli is a common cause of urinary tract infection and intra-abdominal infection in humans of all ages,2 and it kills more than 2 million humans annually in the form of intra- and extra-intestinal diseases.3,4 ESBLs (Extended-spectrum β-lactamases) are plasmid-encoded β-lactamases resistant to penicillins, cephalosporins, β-lactams, and other antimicrobials, such as aminoglycosides and fluoroquinolones.5 The emergence of multidrug-resistant bacteria renders the treatment of bacterial infections an urgent global challenge.6 Thus, the mechanisms of E. coli resistance should be clearly understood in the future development of antibiotics, and new therapy methods should be sought.Detecting and identifying metabolome-scale changes in microorganisms is essential in understanding their roles in cellular processes. Metabolites may be used to realize the molecular basis for a biological phenomenon, including cell signaling.7 Metabolomics, one of the most powerful and promising tools for analyzing metabolism,8 is a high-throughput analytical technology used to identify and quantify small amounts of metabolites.9 This tool provides a broad range of information on integrated cellular response.10 Furthermore, metabolomics has been applied to a variety of pathophysiological process, including cancer and diabetes, with the goal of identifying the biomarkers predictive of a disease onset and ensuring treatment efficacy monitoring.11 Studies have been reported on metabolomics techniques that focus on the role of bacterial metabolism in constraining the evolution of antibiotic resistance.12 Wei et al. characterized the mechanisms of p-aminosalicylic acid resistance in Mycobacterium tuberculosis might be associated with the increased metabolites level in phenylalanine metabolism pathway using multi-omics (genome, proteome, and metabolome) analysis.13 Zhao et al. used multi-omics (LC-MS based metabolomic, and iTRAQ labeling proteomic) analysis to assess capreomycin resistance on tlyA deficient and point mutation (G695A) Mycobacterium tuberculosis strains, with the finding that the greater drug tolerance of CAPr1 strains may be associated with the weakening of S-adenosyl-L-methionine-dependent methyltransferase activity and abnormal membrane lipid metabolism.14
In addition to metabolomics, proteomics is also a potent tool for improving the characterization of complex biological systems or pathologies. Proteomics methods have been used to identify protein network alterations, thus helping researchers understand the responses to various diseases.15,16 Protein expression studies based on proteomics have unique advantages, such as large-scale, high-throughput, high-sensitivity, and dynamic formats.17 The proteomics methods for several types of drug resistance have attracted increasing attention in previous studies.18 Investigating metabolome-scale changes may provide insights into the widespread modes of acquired bacterial resistance in clinics.7
In this study, a conjoint analysis of TMT (Tandem Mass Tag) proteomics and metabolomics was applied to compare the differential profile of proteins and metabolites between β-lactam antibiotic-sensitive E. coli and multidrug-resistant ESBL-producing E. coli strains and reveal the mechanisms of antibiotic resistance of clinical E. coli strains. The novelty of this study lies in revealing the metabolic characteristics of enzyme-producing bacteria and finding potential important pathways and genes for regulating enzyme production.
2. Materials and methods
2.1. Chemicals and reagents
Ultrapure water was produced by a Ming Che D24 UV water purification system (Merck Millipore, Burlington, USA). MeOH, ACN, NH4OAc, and NH4OH (CNW Technologies, Düsseldorf, Germany) were all MS grade. 2-Chloro-L-phenylalanine (Sigma, St. Louis, MO, USA) was used as an internal standard. HEPES, SDS, TFA, triethylammonium bicarbonate (TEAB), ammonium formate, Columbia agar, MacConkey agar, and Chocolate agar were obtained from Sigma (St. Louis, MO, USA). Dithiothreitol (DTT), iodoacetamide (IAA), and trypsin (sequencing grade) were obtained from Promega Life Sciences (Madison, USA). Urea was provided by GibcoBRL (MD, USA). EDTA and PMSF were obtained from Amesco (USA). Ethanol, formic acid, acetone, BCA protein quantification kit, TMT 6 standard kit, Acclaim PepMap C18 Column (100 μm × 20 mm, 75 μm × 250 mm) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). TEAB and 25% ammonia were obtained from Santa Cruz (CA, USA). Sep Pak C18 cartridges (1 cc, 100 mg) and the Acquity UPLC®BEH C18 column (50 mm × 2.1 mm, 1.7 μm) were obtained from Waters (Milford, USA). The Luria–Bertani (LB) broth and LB agar were purchased from Becton Dickinson (Sparks, MD, USA). The lysis buffer for protein analysis was obtained from Roche Ltd (Basel, Switzerland). All other reagents were of ACS reagent grade.2.2. Clinical bacterial specimens collection and cultivation
Clinical specimens of human phlegm, urine, blood, secretions were randomly collected on January–June 2018 from patients of the Second Hospital of Jilin University, the specimens were divided as experimental group (n = 10) and control group (n = 10). Detailed sample information was list in Table 1. The patients signed informed consent forms for the experimental study. All experimental procedures were performed in accordance with the Guidelines for the Collection and Application of Human Related Specimens of the Second Hospital of Jilin University and approved by the Ethics Committee of the Second Hospital of Jilin University. The age and gender of all patients included in this research were recorded. The phlegm samples must comply with the requirements of more than 25 leukocytes and less than 10 epithelial cells in the low-power field. The phlegm and urine samples were planted with Columbia agar with blood solution, MacConkey agar, and Chocolate agar plates and kept in a 5% CO2 incubator at 35 °C. Blood samples were cultured in automated blood culture flask system. The blood samples with bacterial infection were used for further plate culture. A single colony was selected and cultured in the LB broth to 0.5 MCF. After microbial identification and sensitivity analysis with the aid of VITEK® 2 Compact, the bacterial suspension was coated on a plate, then ceftazidime, ceftazidime/clavulanic acid and cefotaxime, and cefotaxime/clavulanic acid were used as indicators to detect the ESBLs in the E. coli according to the CLSI (2014) Performance Standards for Antimicrobial Susceptibility Testing.| Sample number | Experimental group | Collection time | Gender | Control group | Collection time | Gender |
|---|---|---|---|---|---|---|
| 1 | Urine | 2018/1/25 | Male | Urine | 2018/2/15 | Male |
| 2 | Blood | 2018/3/1 | Male | Phlegm | 2018/3/9 | Female |
| 3 | Blood | 2018/3/12 | Female | Phlegm | 2018/3/21 | Female |
| 4 | Phlegm | 2018/3/29 | Male | Blood | 2018/3/27 | Male |
| 5 | Phlegm | 2018/4/20 | Female | Blood | 2018/4/1 | Female |
| 6 | Blood | 2018/5/3 | Female | Urine | 2018/4/27 | Male |
| 7 | Urine | 2018/5/12 | Male | Blood | 2018/5/6 | Male |
| 8 | Phlegm | 2018/5/20 | Female | Urine | 2018/5/24 | Female |
| 9 | Urine | 2018/5/21 | Female | Phlegm | 2018/6/1 | Male |
| 10 | Vaginal secretion | 2018/5/27 | Female | Phlegm | 2018/6/5 | Female |
2.3. Proteomics analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 30 min at 4 °C, the supernatant was collected and measured for protein concentration with BCA. Then, the 100 μg protein was selected and set to the constant volume of 100 μL per test with 100 mM of TEAB.
000×g for 30 min at 4 °C, the supernatant was collected and measured for protein concentration with BCA. Then, the 100 μg protein was selected and set to the constant volume of 100 μL per test with 100 mM of TEAB.2.4. Metabolomics analysis
2.5. Statistical analysis
The data were analyzed with student's T test by using Excel 2013. A p-value of <0.05 was considered significant.3. Results
3.1. TMT quantitative proteomics analysis
The 1553 DEPs (differentially expressed proteins) (fold change < 0.83 or fold change > 1.2; p-value < 0.05) were identified by TMT MS; among the DEPs, 18 were upregulated and 1535 were downregulated in the ESBL-EC (Fig. 1A, B and S1†). The detailed results of the DEPs are shown in Table S1.†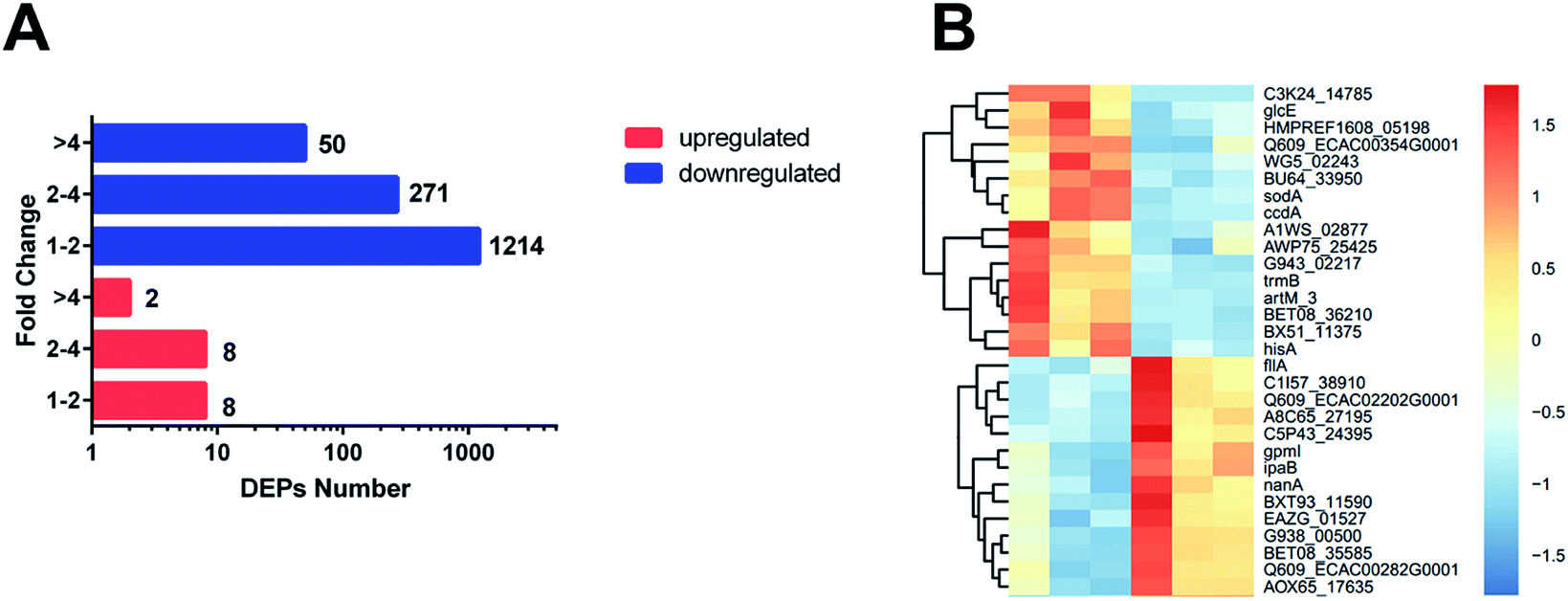 | ||
| Fig. 1 (A) Number of proteins identified as upregulated and downregulated between ESBL-EC and non-ESBL-EC. The red bars represent the upregulated proteins. The blue bars represent the downregulated proteins. The x-axis represents the number of DEPs. The y-axis represents the fold change value. (B) Heatmap based on representative hierarchical clustering analysis with 30 DEPs between ESBL-EC and non-ESBL-EC (the detailed heatmap is shown in Fig. S1†). The relative protein level is depicted in color scale, in which red indicates upregulation, and blue indicates downregulation. | ||
3.2. Function analysis results
Systematic and integrative analysis was performed using DAVID to gain a comprehensive understanding of the DEPs.22 The properties of the genes and gene products of the DEPs were classified into three categories: GO_BP (GO Term Biological Process), GO_CC (Cellular Component), and GO_MF (Molecular Function). The classifications were used to identify the potential affected pathways. The DEPs were significantly enriched in 53 GO terms (p-value < 0.05). Among them, 29 were BP terms, 7 were CC terms, and 17 were MF terms (Table S2†). The 30 most representative and markedly enriched GO terms within the three main functional modules are shown in Fig. 1C. The leading seven BP terms were “translation”, “transcription antitermination”, “glycolytic process”, “isopentenyl diphosphate biosynthetic process, methylerythritol 4-phosphate pathway,” “negative regulation of translational initiation”, “nucleobase-containing small molecule interconversion” and “Ubiquinone biosynthetic process” (Fig. 2).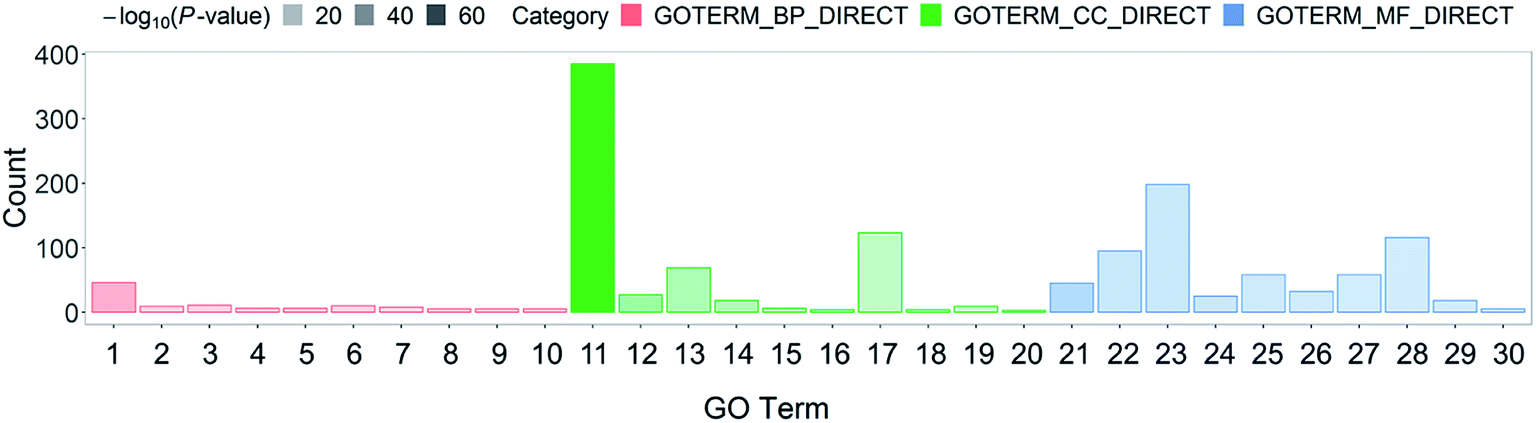 | ||
| Fig. 2 Top 10 GO classification histograms of DEPs for ESBL-EC vs. non-ESBL-EC. The x-axis represents GO_BP (GO Term Biological Process), GO_CC (Cellular Component), and GO_MF (Molecular Function). (1) Translation, (2) transcription antitermination, (3) glycolytic process, (4) isopentenyl diphosphate biosynthetic process, methylerythritol 4-phosphate pathway, (5) negative regulation of translational initiation, (6) nucleobase-containing small molecule interconversion, (7) ubiquinone biosynthetic process, (8) nucleotide phosphorylation, (9) terpenoid biosynthetic process, (10) DNA-templated transcription, termination, (11) cytosol, (12) cytosolic large ribosomal subunit, (13) membrane, (14) cytosolic small ribosomal subunit, (15) chromosome, (16) DNA topoisomerase complex (ATP-hydrolyzing), (17) cytoplasm, (18) glycerol-3-phosphate dehydrogenase complex, (19) cell division site, (20) cell surface, (21) structural constituent of ribosome, (22) identical protein binding, (23) protein binding, (24) rRNA binding, (25) zinc ion binding, (26) RNA binding, (27) magnesium ion binding, (28) ATP binding, (29) tRNA binding, (30) mRNA 5′-UTR binding. The y-axis represents the number of involved DEPs between ESBL-EC and non-ESBL-EC. | ||
In addition, significantly enriched metabolic pathways of DEPs were identified using the KEGG database to determine the systemic difference between ESBL-EC and non-ESBL-EC.23 A total of 82 significant systemically changed KEGG pathways were detected (Table S3†). PPI network analyses are important in the discovery of the variation tendency of DEPs in proteomics analysis, and they are helpful in targeting the key node between DEPs.
We constructed the PPI of E. coli by using the STRING database23 to uncover the potential protein interaction of DEPs and the biological functions in the bacterial cellular processes for ESBL-EC (Fig. 3). Moreover, the PPI networks analysis was used to obtain different central nodes, including hisA, glcE, soda, and GroL.
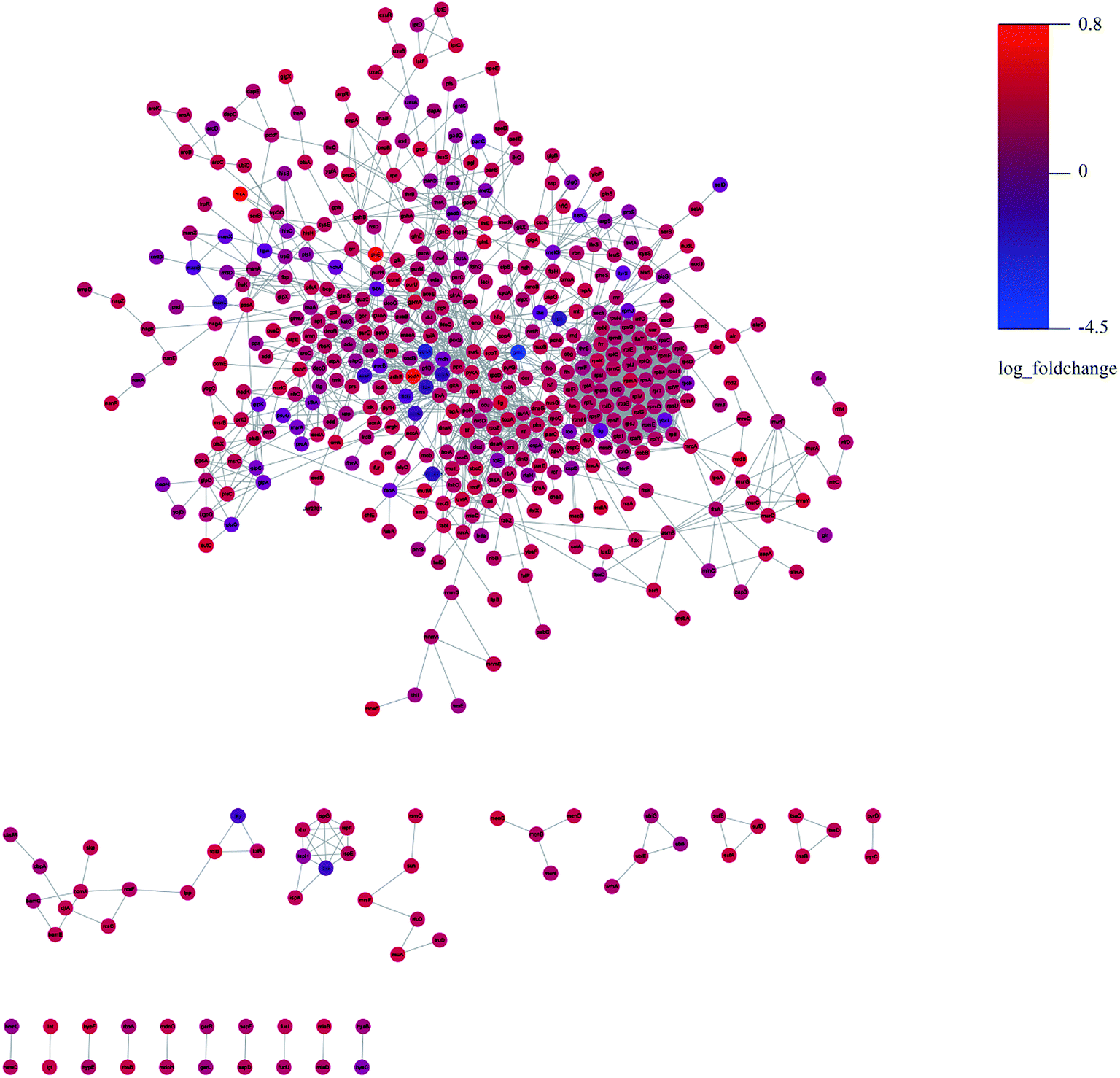 | ||
| Fig. 3 PPI (protein–protein interaction) network of DEPs between ESBL-EC and non-ESBL-EC. Large sizes and dark colors represent the high impact and level of DEPs. | ||
3.3. Multivariate analysis of UPLC-QTof/MS data for the metabolomics study
Unsupervised pattern recognition PCA (principal component analysis) was conducted to visualize clearly the separation of the metabolites between ESBL-EC and non-SBL-EC (Fig. 4). QC injections were closely clustered in the PCA score plots in both positive mode (R2X = 0.525) and negative mode (R2X = 0.531). This study has verified the stability of the UPLC-QTof/MS system.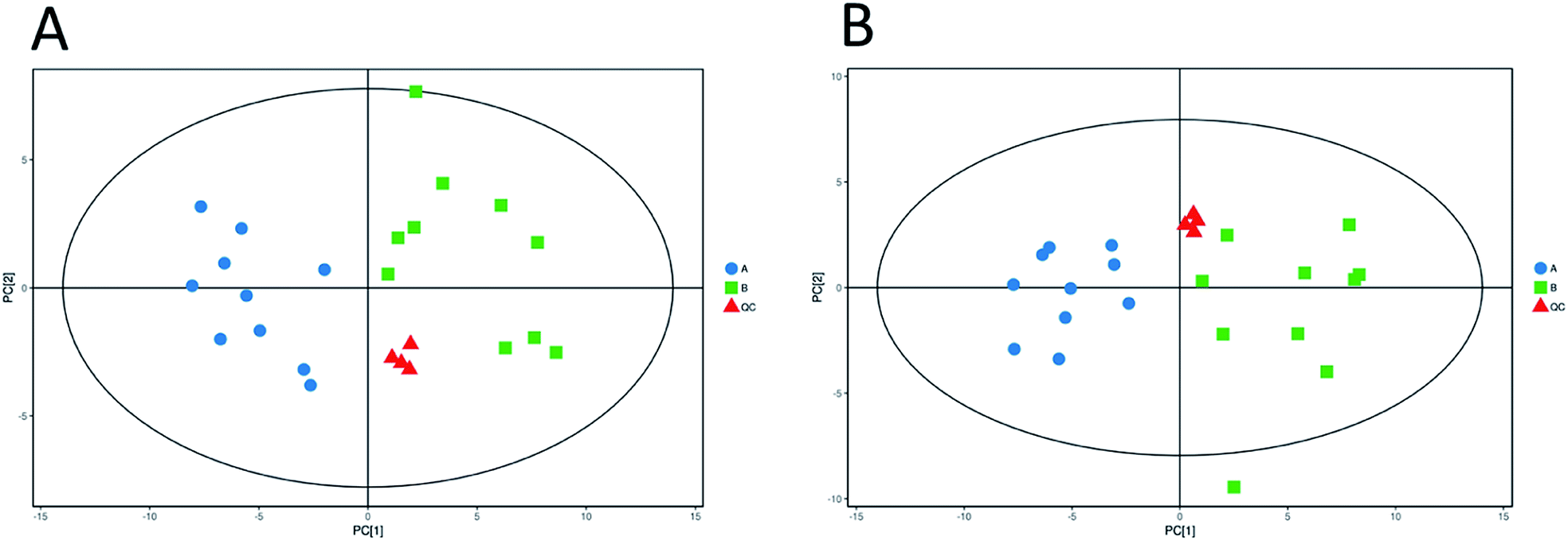 | ||
| Fig. 4 Multivariate analysis of untargeted metabolomics data. PCA score plots of metabolic profile from different groups in positive ESI mode (A) and negative ESI mode (B). | ||
In addition, OPLS-DA (orthogonal projections to latent structures discriminant analysis) was performed to remove the descriptor variables that were not correlated to the property variables and determine the differential metabolite profiles that was responsible for the classification between the two groups (Fig. 5A and 6A). The R2X and R2Y of the OPLS-DA model were 0.364 and 0.982 for the ESI+ mode and 0.352 and 0.976 for the ESI− mode, respectively. Subsequently, seven-fold cross validation was implemented and applied with 200 random permutations to validate the OPLS-DA models. All blue Q2 values at the left part were lower than the original points on the right part (Fig. 5B and 6B). This finding indicates that the original models are robust and have no overfitting.
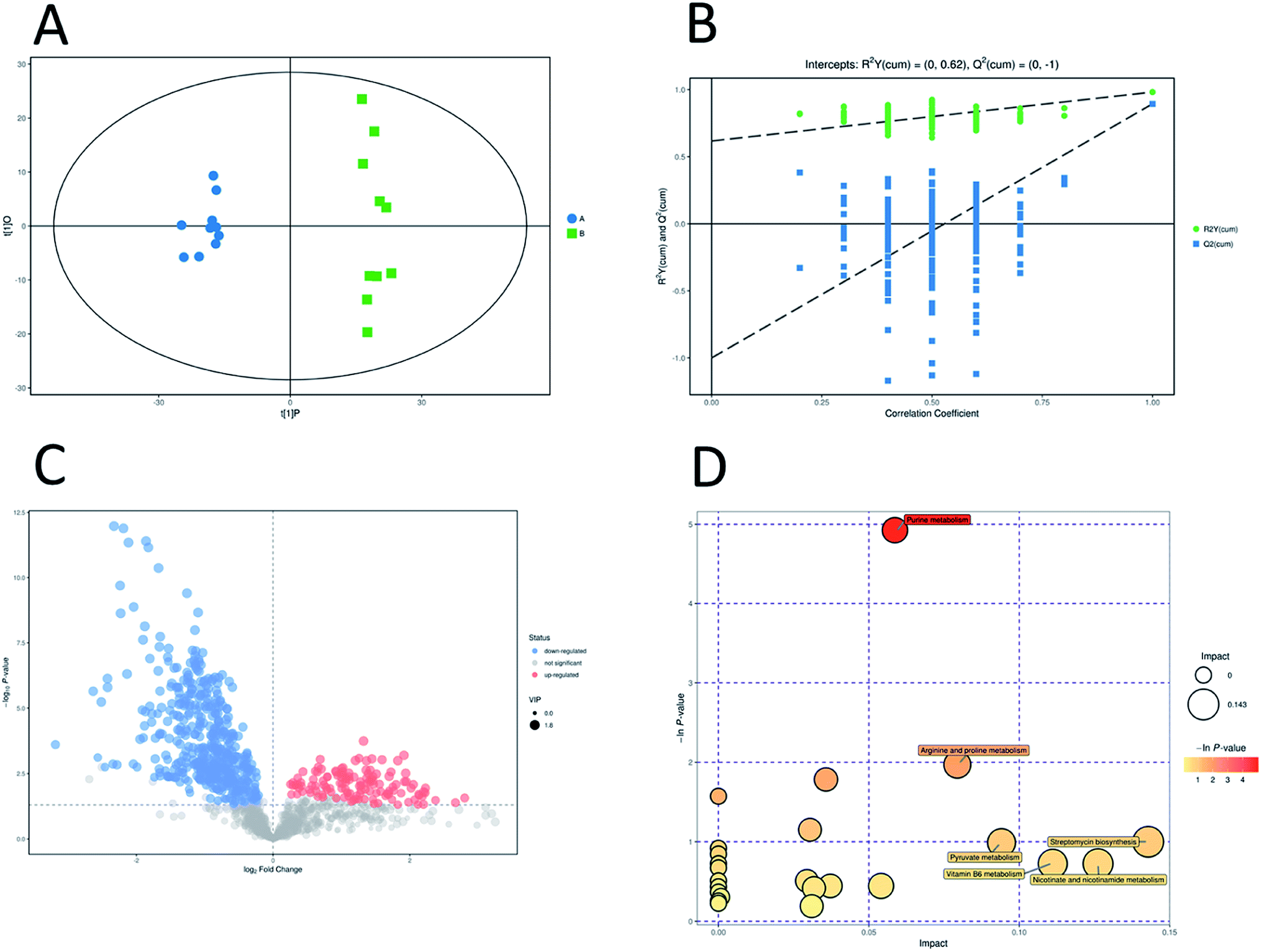 | ||
| Fig. 5 Multivariate analysis of untargeted metabolomics data and pathway analysis in positive mode. OPLS-DA score plots of metabolic profiling of different groups (A). The corresponding 200 random permutations plots of the OPLS-DA models (B). Volcano plots for ESBL-EC vs. non-ESBL-EC (C). The relative metabolite level is depicted according to color scale. Red represents the upregulation and blue represents the downregulation of the metabolites in ESBL-EC vs. non-ESBL-EC. Large sizes represent high VIP values of metabolites. Overview of significantly changed pathway analysis for ESBL-EC vs. non-ESBL-EC (D). The x-axis represents pathway enrichment. The y-axis represents the pathway impact. Large sizes and dark colors represent the major pathway enrichment and high pathway impact values, respectively. | ||
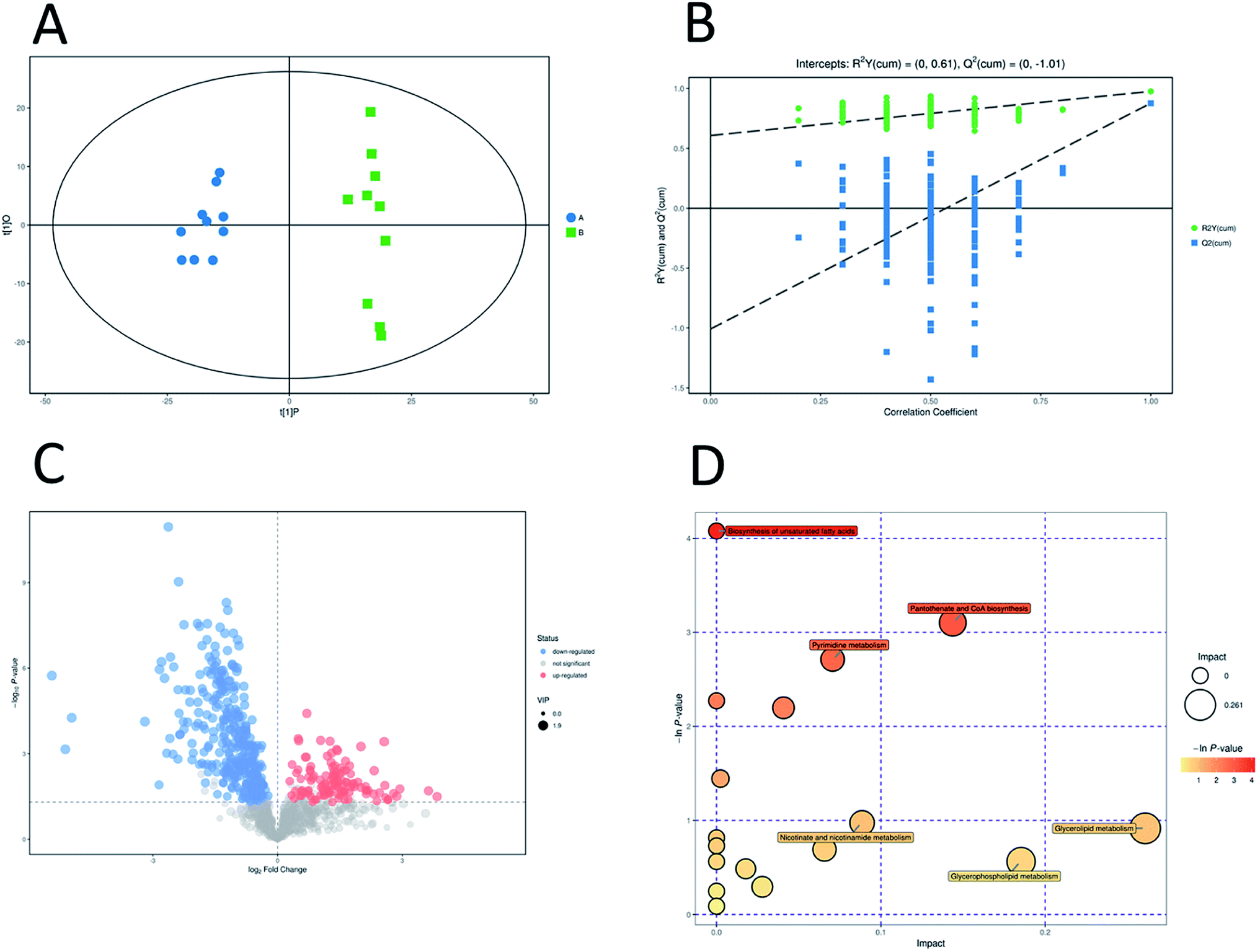 | ||
| Fig. 6 Multivariate analysis of untargeted metabolomics data and pathway analysis in negative mode. OPLS-DA score plots of metabolic profiling of different groups (A). The corresponding 200 random permutations plots of the OPLS-DA models (B). Volcano plots for ESBL-EC vs. non-ESBL-EC (C). The relative metabolite level is depicted according to color scale. Red represents the upregulation and blue represents the downregulation of the metabolites in ESBL-EC vs. non-ESBL-EC. Large sizes represent high VIP values of metabolites. Overview of significantly changed pathway analysis for ESBL-EC vs. non-ESBL-EC (D). The x-axis represents pathway enrichment. The y-axis represents the pathway impact. Large sizes and dark colors represent the major pathway enrichment and high pathway impact values, respectively. | ||
3.4. Potential marker identification and the involved metabolic pathway attribution
The volcano plots, which were produced on the basis of OPLS-DA, depict the contribution of the variables between the two groups (Fig. 5C and 6C). MetaboAnalyst was used to search for the pathways of the changed metabolites. The pathway analysis based on the potential biomarkers showed 18 different metabolic pathways between the two groups (Fig. 5D, 6D; Tables 2 and 3).| Pathway | Total | Hits | Raw p | −ln(p) | FDR | Impact |
|---|---|---|---|---|---|---|
| Purine metabolism | 73 | 9 | 0.007253 | 4.9264 | 0.63099 | 0.05866 |
| Arginine and proline metabolism | 41 | 4 | 0.13937 | 1.9706 | 1 | 0.0794 |
| Pyrimidine metabolism | 44 | 4 | 0.1679 | 1.7844 | 1 | 0.03574 |
| Glycerophospholipid metabolism | 23 | 2 | 0.31626 | 1.1512 | 1 | 0.03043 |
| Streptomycin biosynthesis | 9 | 1 | 0.36767 | 1.0006 | 1 | 0.14286 |
| Pyruvate metabolism | 26 | 2 | 0.37182 | 0.98934 | 1 | 0.09409 |
| Vitamin B6 metabolism | 13 | 1 | 0.48491 | 0.72379 | 1 | 0.11111 |
| Nicotinate and nicotinamide metabolism | 13 | 1 | 0.48491 | 0.72379 | 1 | 0.12617 |
| Butanoate metabolism | 18 | 1 | 0.60186 | 0.50774 | 1 | 0.02941 |
| Citrate cycle (TCA cycle) | 20 | 1 | 0.64096 | 0.44479 | 1 | 0.0372 |
| Propanoate metabolism | 20 | 1 | 0.64096 | 0.44479 | 1 | 0.05405 |
| Glutathione metabolism | 21 | 1 | 0.65908 | 0.41692 | 1 | 0.03175 |
| Valine, leucine and isoleucine biosynthesis | 26 | 1 | 0.73704 | 0.30511 | 1 | 0.00085 |
| Cysteine and methionine metabolism | 34 | 1 | 0.82693 | 0.19004 | 1 | 0.03099 |
| Pathway | Total | Hits | Raw p | −ln(p) | FDR | Impact |
|---|---|---|---|---|---|---|
| Pantothenate and CoA biosynthesis | 23 | 3 | 0.045 | 3.1011 | 1 | 0.14377 |
| Pyrimidine metabolism | 44 | 4 | 0.066497 | 2.7106 | 1 | 0.07066 |
| Purine metabolism | 73 | 5 | 0.11116 | 2.1967 | 1 | 0.04076 |
| Pyruvate metabolism | 26 | 2 | 0.23588 | 1.4444 | 1 | 0.00239 |
| Nicotinate and nicotinamide metabolism | 13 | 1 | 0.37763 | 0.97384 | 1 | 0.08859 |
| Glycerolipid metabolism | 14 | 1 | 0.40008 | 0.91609 | 1 | 0.26087 |
| Peptidoglycan biosynthesis | 17 | 1 | 0.50104 | 0.69108 | 1 | 0.06571 |
| Glycerophospholipid metabolism | 23 | 1 | 0.56972 | 0.56262 | 1 | 0.18536 |
| Valine, leucine and isoleucine biosynthesis | 26 | 1 | 0.6151 | 0.48597 | 1 | 0.0178 |
| Galactose metabolism | 37 | 1 | 0.745 | 0.29438 | 1 | 0.02786 |
3.5. Correlation analysis of metabolomics and proteomics
The omic-domain data integration for ESBL-EC was obtained by subjecting the content information of the DEPs and differential metabolites to relevant calculations by using the Spearman algorithm. The p-value of the correlation matrix was used for the follow-up analysis. A correlation network graph (Fig. 7 and 8) was constructed on the basis of the data that could meet both the correlation coefficient of >0.9 and the p-value of <0.05. In the figure, the green squares represent differential metabolites, the yellow circles represent the differential protein genes, the red line represents a positive correlation between the differential metabolites and the differential protein genes, and the blue line represents a negative correlation. Tables S8–S11† show all of the binary relations of the subnetworks.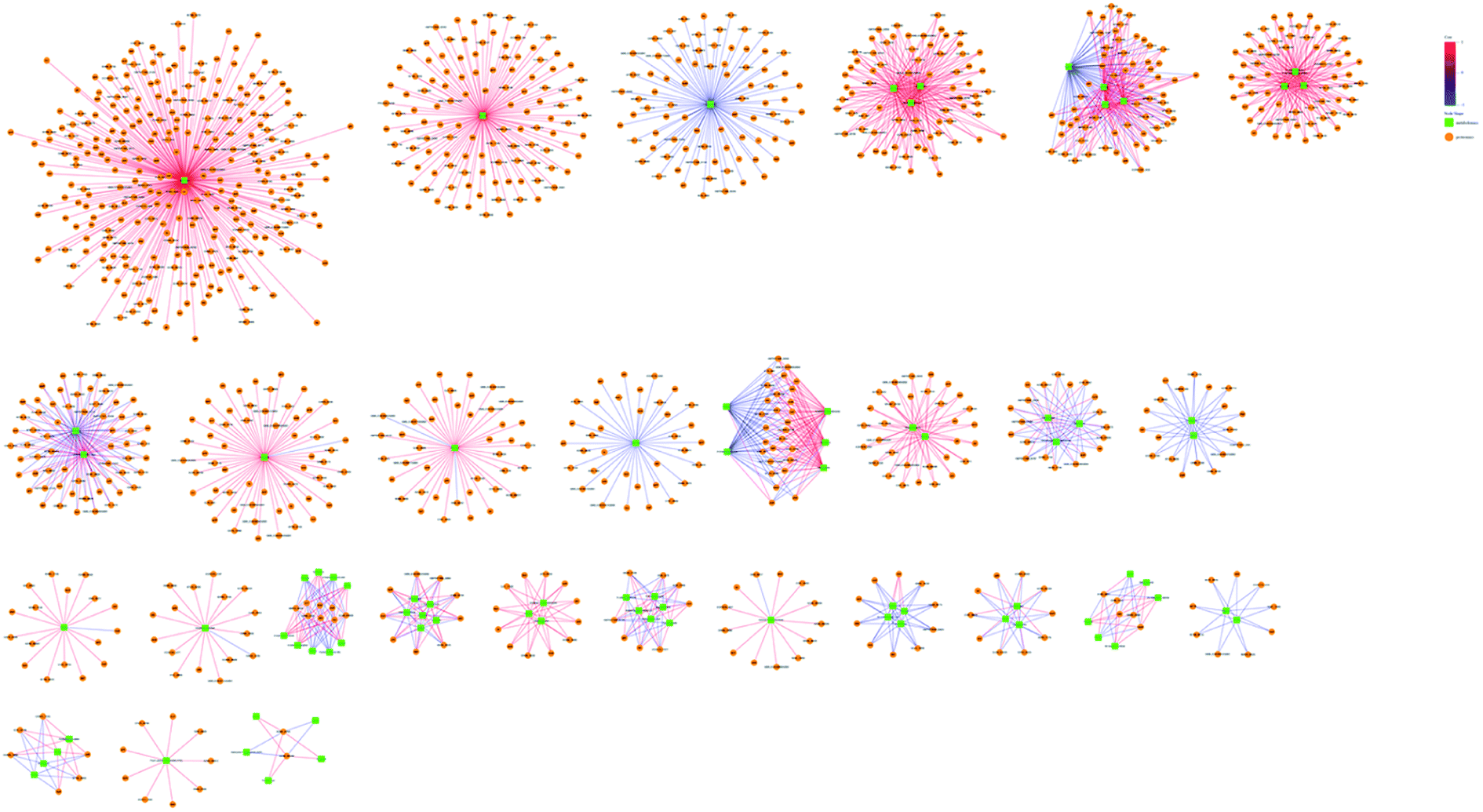 | ||
| Fig. 7 Metabolite–protein interaction network based on the differential metabolites and DEPs for ESBL-EC vs. non-ESBL-EC in positive mode. The green squares represent differential metabolites. The yellow circles represent differential protein genes. The red line represents a positive correlation between the differential metabolites and the differential protein genes. The blue line represents a negative correlation. | ||
 | ||
| Fig. 8 Metabolite–protein interaction network based on the differential metabolites and DEPs for ESBL-EC vs. non-ESBL-EC in negative mode. The green squares represent differential metabolites. The yellow circles represent differential protein genes. The red line represents a positive correlation between the differential metabolites and the differential protein genes. The blue line represents a negative correlation. | ||
3.6. Target metabolomics analysis
The drug resistance mechanism of ESBL-EC was further investigated, then the non-target metabolomics results were verified in this study. The sequential research was focused on the differential metabolites in the purine metabolism pathway (eco00230-E. coli K-12 MG1655). Three differential metabolites (2-deoxyadenosine monohydrate, 2,6-dihydroxypurine, and xanthosine) in the purine metabolism pathway were selected and subjected to target metabolomics analysis assay development by using UHPLC-QQQ-MS (MRM). As shown in Fig. S2,† the baseline separation of the metabolites can be successfully obtained, and the retention time and peak shapes of all of the analytes present a good match between the standard solution and the real sample, i.e., 2-deoxyadenosine monohydrate (RT, 2.36 min), 2,6-dihydroxypurine (RT, 2.28 min), and xanthosine (RT, 3.30 min). Appropriate linear range with high linearity and low LOD and LOQ facilitate sensitive and selective analysis for the metabolites from biological samples. The LODs of the three metabolites were 1.22–19.53 nM, while the LOQs were 2.44–39.06 nM, with high linearity (R2 > 0.9997). The details of the calibration curves are shown in Table S4.† The five repeated injections presented a recovery from 93.5% to 104.2%, and the RSD of the targeted matabolites was less than 3.1% (Table S5†), which accords with the quantification of the targeted metabolomics analysis.Then the rapid MRM based target metabolomics analysis was successfully applied to absolutely quantify the changed metabolites in the clinical bacterial samples. The MRM result (Fig. 9) showed that the contents of the 2-deoxyadenosine monohydrate and 2,6-dihydroxypurine were downregulated (p-value < 0.01). This finding is consistent with the non-target metabolomics analysis. However, the xanthosine was relatively overexpressed in ESBL-EC (p-value > 0.05), a finding inconsistent with the non-target metabolomics analysis.
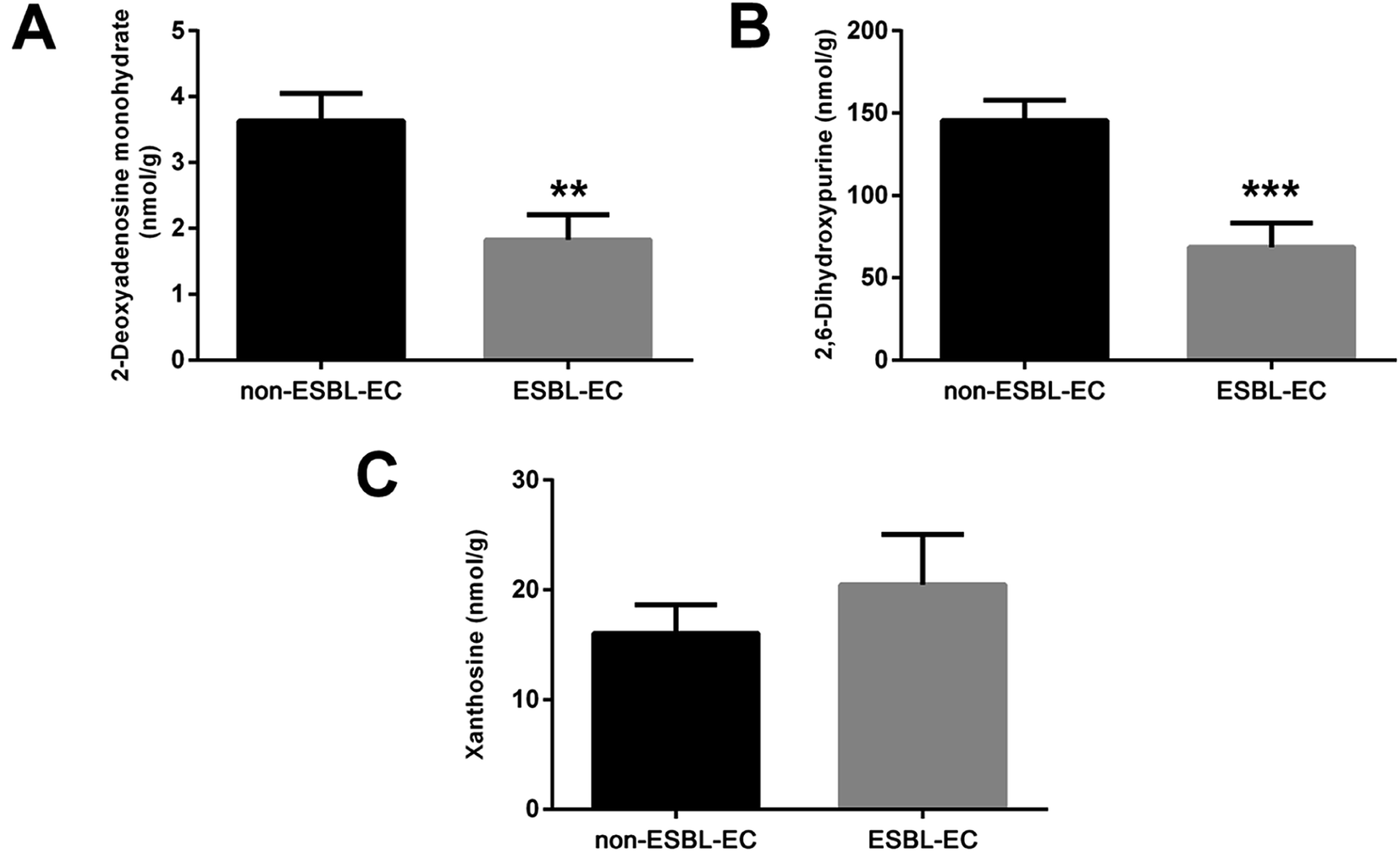 | ||
| Fig. 9 Target metabolomics analysis by using MRM for three differential metabolites for ESBL-EC vs. non-ESBL-EC: (A) 2-deoxyadenosine monohydrate, (B) 2,6-dihydroxypurine, and (C) xanthosine. The values are presented as mean ± SD, n = 10. **p-value < 0.01, and ***p-value < 0.001 and compared with the non-ESBL-EC group. | ||
4. Discussion
Antibiotics are commonly used as the first line of action in the treatment or prevention of certain bacterial infections.24 However, antibiotics do not work for patients infected with the ESBL-producing E. coli and thus a worldwide important public health concern.25 As discussed earlier, the antibiotic resistance and ESBL production capacity of E. coli remains poorly understood.18 Here, we examined for the first time the proteomics and metabolomics differences between ESBL-EC and non-ESBL-EC to clarify the potential mechanism of the ESBL-EC's antibiotic resistance and provide a scientific evidence for the discovery of novel targets for groundbreaking therapies.Among the 18 different metabolic pathways classified between ESBL-EC and non-ESBL-EC, the purine metabolism pathway showed a high enriched degree, as characterized by the lowest lnp-value and dark bubble in the bubble plots. Purine metabolism was found to be correlated to the drug resistance of parasites and the drug resistance and tumor relapse of childhood ALL in previous work.26,27 More recently, a comparative metabolomics study based on GC-MS showed that the metabolic profiles of purine metabolism fluctuates in multi-drug-resistant E. coli strains; this result from previous work18 agrees with our current findings. In addition, in using metabolomics and proteomics correlation analysis, we found drug resistance affects the metabolite and protein profiles of E. coli, as many central metabolite nodes were obtained, including 2-deoxyadenosine monohydrate, 2,6-dihydroxypurine, and xanthosine, which have significant correlations with DEPs.
Considering the potential significance of purine metabolism for the drug resistance of ESBL-EC, target metabolomics analysis was performed using MRM. We selected three different metabolites (2-deoxyadenosine monohydrate, 2,6-dihydroxypurine, and xanthosine) in the purine metabolism pathway for further quantitative verification analysis, with comprehensive consideration of the MS2 score, p-value, VIP, and metabolite–protein interactions in the correlation analysis. The MRM analytical results coincided with the non-targeted metabolomics data, suggesting the reliability of our analytical method. Moreover, the above metabolites may be involved in the drug resistance of the bacteria in ESBL-EC.
DEPs may play important roles in the antibiotic resistance and ESBL production capacity of ESBL-EC. The present work has provided scientific data for the metabolic and protein level characteristics and involved important pathways of clinical β-lactamase enzyme-producing bacteria, which revealed the potential mechanism for the antibiotic resistance of ESBL-EC.
The main purpose of the experiment is using the proteomic and metabolomics to explore the underline mechanism of extended-spectrum β-lactamase production in E. coli. On the basic of above study, our future work is going on, such as performing site-by-point editing of the critical node protein-related genes or the purine metabolism pathway in ESBL-EC by using gene editing tools, such as the CRISPR/Cas9 system, based on the above research results. We also plan to analyze the bacterial virulence, extended-spectrum β-lactamase activity, and antibiotic resistance of genetically engineered E. coli. Another plan is to construct a gRNA recombination vector-based vaccine carrier bacteria with the aim of eliminating or weakening the activity of β-lactam drug resistance genes. We also intend to test the efficacy of the vaccine carrier bacteria in vivo. Briefly, mice will be pre-stimulated with the vaccine carrier bacteria, then we shall establish the mouse infection model by using clinical ESBL-EC, and finally, we will observe the therapeutic effect of β-lactam drugs on the infection of mice.
5. Conclusion
This basic research has shown how metabolic and proteomics alterations can manifest in ESBL-EC. The untargeted metabolomics result was also confirmed by the LC-MRM-MS analysis. With the aid of metabolomics and proteomics correlation bioinformatics analysis, we obtained many central metabolite nodes and their corresponding DEPs. The findings can help in our future study of the antibiotic resistance mechanism in ESBL-EC and the corresponding research and development of biomedicine-based target therapy (Fig. 10).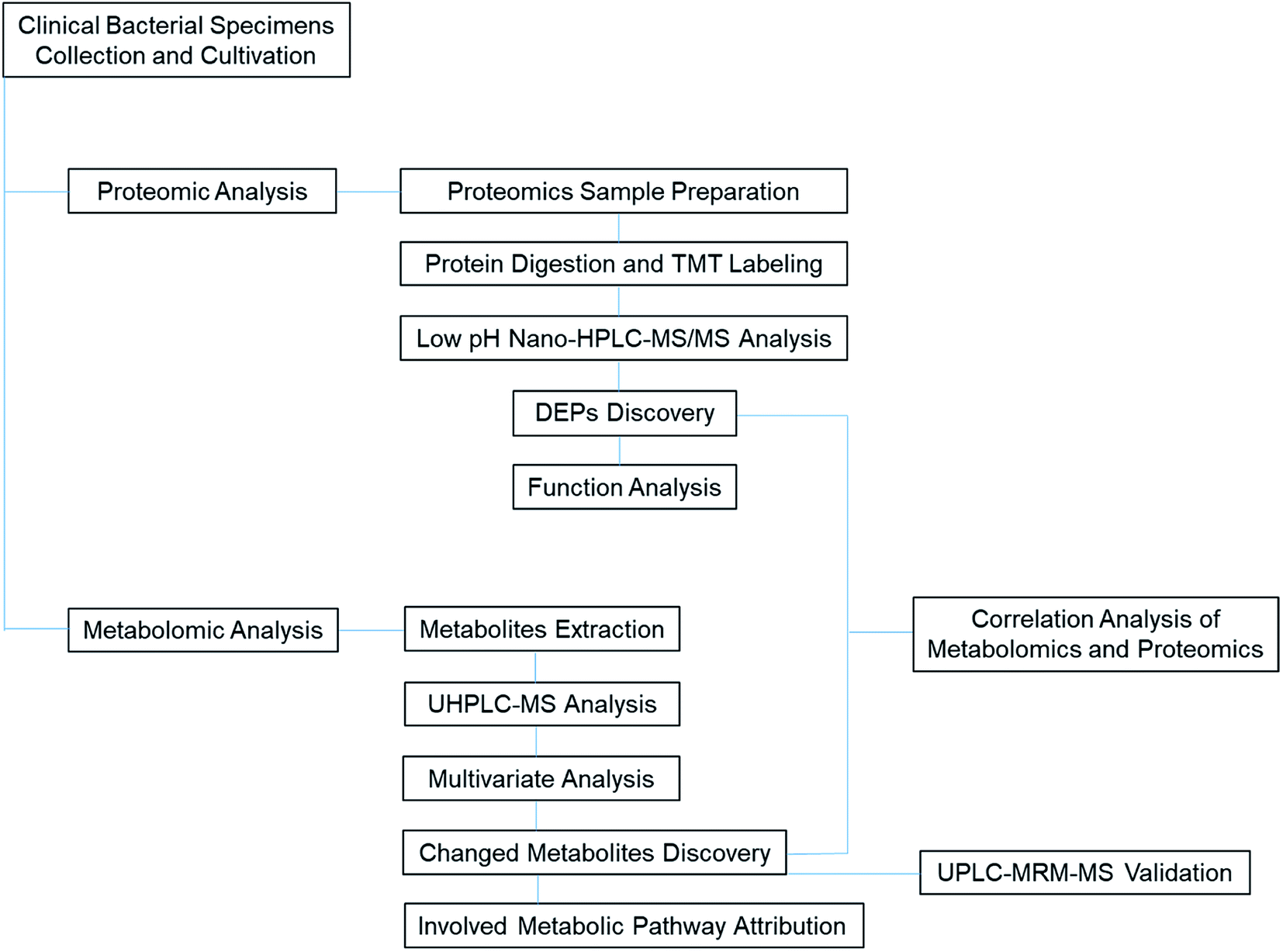 | ||
| Fig. 10 TOC graph for illustration of the proteomic and metabolomic analytical process of clinical ESBL specimens. | ||
Author contributions
Data curation: Bingjie Lai, Chang Tian; investigation: Jiaying Liu; project adminstration: He Ma, Yufen Jin; resources: Ke Wang; software: Bingjie Lai; supervision: Ke Wang; writing-original draft: He Ma; writing-review: Ke Wang.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work has been supported by Social Development Project of Jilin Science and Technology Department (20190303162SF), Advanced Program of Jilin Science and Technology Department (20191102012YY), and Natural Science Foundation of Jilin Province (20180101103JC).References
- O. Tenaillon, D. Skurnik, B. Picard and E. Denamur, The population genetics of commensal Escherichia coli, Nat. Rev. Microbiol., 2010, 8(3), 207–217 CrossRef CAS PubMed.
- C. I. Kang, M. K. Cha, S. H. Kim, K. S. Ko, Y. M. Wi, D. R. Chung, K. R. Peck, N. Y. Lee and J. H. Song, Clinical and molecular epidemiology of community-onset bacteremia caused by extended-spectrum beta-lactamase-producing Escherichia coli over a 6-year period, J. Kor. Med. Sci., 2013, 28(7), 998–1004 CrossRef PubMed.
- U. Klümper, M. Recker, L. Zhang, X. Yin, T. Zhang, A. Buckling and W. H. Gaze, Selection for antimicrobial resistance is reduced when embedded in a natural microbial community, ISME J., 2019, 2927–2937 CrossRef PubMed.
- X. Liu, J. Wang, M. Chen, R. Che, W. Ding, F. Yu, Y. Zhou, W. Cui, X. Xiaoxu, B. O. God'spower and Y. Li, Comparative proteomic analysis reveals drug resistance of Staphylococcus xylosus ATCC700404 under tylosin stress, BMC Vet. Res., 2019, 15(1), 224 CrossRef PubMed.
- J. Rodriguez-Bano, M. D. Navarro, L. Romero, M. A. Muniain, M. de Cueto, M. J. Rios, J. R. Hernandez and A. Pascual, Bacteremia due to extended-spectrum beta -lactamase-producing Escherichia coli in the CTX-M era: a new clinical challenge, Clin. Infect. Dis., 2006, 43(11), 1407–1414 CrossRef PubMed.
- M. Zampieri, T. Enke, V. Chubukov, V. Ricci, L. Piddock and U. Sauer, Metabolic constraints on the evolution of antibiotic resistance, Mol. Syst. Biol., 2017, 13(3), 917 CrossRef PubMed.
- D. K. Derewacz, C. R. Goodwin, C. R. McNees, J. A. McLean and B. O. Bachmann, Antimicrobial drug resistance affects broad changes in metabolomic phenotype in addition to secondary metabolism, Proc. Natl. Acad. Sci. U.S.A., 2013, 110(6), 2336–2341 CrossRef CAS PubMed.
- G. Monni, F. Murgia, V. Corda, C. Peddes, A. Iuculano, L. Tronci, A. Balsamo and L. Atzori, Metabolomic Investigation of beta-Thalassemia in Chorionic Villi Samples, J. Clin. Med., 2019, 8(6), 798 CrossRef CAS PubMed.
- H. T. Lin, M. L. Cheng, C. J. Lo, G. Lin, S. F. Lin, J. T. Yeh, H. Y. Ho, J. R. Lin and F. C. Liu, 1H Nuclear Magnetic Resonance (NMR)-Based Cerebrospinal Fluid and Plasma Metabolomic Analysis in Type 2 Diabetic Patients and Risk Prediction for Diabetic Microangiopathy, J. Clin. Med., 2019, 8(6), 874 CrossRef CAS PubMed.
- C. J. Lo, H. Y. Tang, C. Y. Huang, C. M. Lin, H. Y. Ho, M. S. Shiao and M. L. Cheng, Metabolic Signature Differentiated Diabetes Mellitus from Lipid Disorder in Elderly Taiwanese, J. Clin. Med., 2018, 8(1), 13 CrossRef PubMed.
- K. Wanichthanarak, J. F. Fahrmann and D. Grapov, Genomic, Proteomic, and Metabolomic Data Integration Strategies, Biomark. Insights, 2015, 10(Suppl 4), 1–6 CAS.
- D. Du, X. Wang-Kan, A. Neuberger, H. W. van Veen, K. M. Pos, L. J. V. Piddock and B. F. Luisi, Multidrug efflux pumps: structure, function and regulation, Nat. Rev. Microbiol., 2018, 16(9), 523–539 CrossRef CAS PubMed.
- W. Wei, H. Yan, J. Zhao, H. Li, L. Zhou, H. Guo, X. Wang, Y. Zhou, X. Zhang, J. Zeng, T. Chen and L. Zhou, Multi-omics comparisons of p -aminosalicylic acid (PAS) resistance in folC mutated and un-mutated Mycobacterium tuberculosis strains, Emerging Microbes Infect., 2019, 8(1), 248–261 CrossRef CAS PubMed.
- J. Zhao, W. Wei, H. Yan, Y. Zhou, Z. Li, Y. Chen, C. Zhang, J. Zeng, T. Chen and L. Zhou, Assessing Capreomycin Resistance on tlyA Deficient and Point Mutation (G695A) Mycobacterium Tuberculosis Strains Using Multi-Omics Analysis, Int. J. Med. Microbiol. Suppl., 2019, 309(7), 151323 CrossRef CAS PubMed.
- A. Marrugal, I. Ferrer, M. D. Pastor, L. Ojeda, A. Quintanal-Villalonga, A. Carnero, S. Molina-Pinelo and L. Paz-Ares, Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry, Cells, 2019, 8(8), 806 CrossRef CAS PubMed.
- L. Yuan, J. Wang, S. Xie, M. Zhao, L. Nie, Y. Zheng, S. Zhu, J. Hou, G. Chen and C. Wang, Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype, Int. J. Mol. Sci., 2019, 20(15), 3760 CrossRef CAS PubMed.
- M. Mann and N. L. Kelleher, Precision proteomics: The case for high resolution and high mass accuracy, Proc. Natl. Acad. Sci. U.S.A., 2008, 105(47), 18132–18138 CrossRef CAS PubMed.
- Y. Lin, W. Li, L. Sun, Z. Lin, Y. Jiang, Y. Ling and X. Lin, Comparative metabolomics shows the metabolic profiles fluctuate in multi-drug resistant Escherichia coli strains, J. Proteomics, 2019, 207, 103468 CrossRef CAS PubMed.
- L. Li, Q. Wu, Y. Wang, S. Morteza, Z. Ban, X. Zhang, H. Lu, D. Li, J. Yan, J. Limwachiranon and Z. Luo, Systematically quantitative proteomics and metabolite profiles offer insight into fruit ripening behavior in fragaria × ananassa, RSC Adv., 2019, 9, 14093–14108 RSC.
- H. Yao, P. Yu and C. Jiang, Metabolomics-driven identification of perturbations in amino acid and sphingolipid metabolism as therapeutic targets in a rat model of anorexia nervosa disease using chemometric analysis and a multivariate analysis platform, RSC Adv., 2020, 10, 4928 RSC.
- N. Yang, H. Wang, H. Lin, J. Liu, B. Zhou, X. Chen, C. Wang, J. Liu and P. Li, Comprehensive metabolomics analysis based on uplc-q/tof-mse and the anti-copd effect of different parts of celastrus orbiculatus thunb, RSC Adv., 2020, 10, 8396 RSC.
- B. Anindya, M. Biswajit and S. J. Susanna, Global transcriptome analysis reveals distinct bacterial response towards soluble and surface-immobilized antimicrobial peptide (lasioglossin- III), RSC Adv., 2015, 5(96), 78712–78718 RSC.
- A. Bilal, B. Madiha, N. M. Atif, K. Mohsin and R. M. Hidayat, Proteomics: Technologies and Their Applications, J. Chromatogr. Sci., 2016, 1–15 CAS.
- R. J. Fair and Y. Tor, Antibiotics and bacterial resistance in the 21st century, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- M. Melzer and I. Petersen, Mortality following bacteraemic infection caused by extended spectrum beta-lactamase (ESBL) producing E. coli compared to non-ESBL producing E. coli, J. Infect., 2007, 55(3), 254–259 CrossRef PubMed.
- W. B. Parker and M. C. Long, Purine metabolism in Mycobacterium tuberculosis as a target for drug development, Curr. Pharmaceut. Des., 2007, 13(6), 599–608 CrossRef CAS PubMed.
- H. Li, B. Li, F. Yang, C. Duan, Y. Bai, J. J. Yang, J. Chen, A. von Stackelberg, H. Chen, J. Tang, A. A. Ferrando, J. Zhang, S. Wang, R. Kirschner-Schwabe and B.-B. S. Zhou, De Novo Purine Biosynthesis in Drug Resistance and Tumor Relapse of Childhood ALL, Blood, 2015, 126(23), 2627 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04250a |
| This journal is © The Royal Society of Chemistry 2020 |