
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of the site-unspecified peptide identification method for proteolytic peptide mapping
H. B. Wang†
,
F. Zeng†,
Y. Y. Wang,
X. Li,
S. H.,
Y. M. Li,
Y. F. Wang,
Y. H. Liu* and
F. P. Lu *
*
Key Laboratory of Industrial Fermentation Microbiology, Ministry of Education, Tianjin Key Laboratory of Industrial Microbiology, National Engineering Laboratory for Industrial Enzymes, The College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, P. R. China. E-mail: lfp@tust.edu.cn; lyh@tust.edu.cn
First published on 7th October 2020
Abstract
Proteases are widely used in the food industry to hydrolyze proteins and prepare bioactive peptides. Peptide mapping identification supports the application of proteases in the food industry. The site-specified peptide identification method, which was developed for site-specific proteases like trypsin, is relatively mature and reliable but cannot be applied using most industrial proteases with weak site specificity. To address this issue, the performance and reliability of the site-unspecified peptide identification method should be investigated and evaluated. In this study, tryptic hydrolysates of a single protein and a protein mixture were used to evaluate the site-unspecified identification method. The species origin of the hydrolyzed proteins was not specified in a database search, meaning that millions of protein sequences were included for calculating and matching. At least 98% of the tryptic peptides were successfully identified via the site-unspecified method, demonstrating that the site-unspecified method shows promising reliability. Moreover, the site-unspecified method identified more peptides than the site-specified method, including those from the low-frequency site-unspecific hydrolysis of trypsin, suggesting that the method has strong capabilities for peptide mapping. The results indicate the applicability of the site-unspecified peptide identification method in the study of site-unspecific industrial proteases.
Introduction
Proteases are widely used in the food, medicine, and chemical industries.1,2 The sale of proteases accounts for more than half of all enzymes sold in the world each year. In the food industry, proteases are mainly used for the hydrolysis of food proteins and the preparation of bioactive peptides.3,4 Proteases perform proteolysis through the hydrolysis of peptide bonds. Different kinds of proteases have different levels of site specificity in the selection of cleavage sites and therefore produce different peptide mappings. Some kinds of proteases have strong site specificity. For example, trypsin cleaves peptide chains mainly at the carboxyl side of the amino acid lysine or arginine, except when either is followed by proline.5,6 In contrast, other proteases such as the alkaline proteases from subtilisin have weak site specificity and choose many amino acids as cleavage sites.7,8 Obviously, the analysis of proteolytic peptide mapping is important for understanding the proteolytic process and the characteristics of industrial proteases.An approach based on mass spectrometry and database searching has been used to analyze peptide mapping in proteomics research.6,9 This method can identify proteolytic peptides by matching the experimental data with the theoretical data from the database.10 This method has the advantages of high sensitivity and throughput.11 However, the main existing method for peptide identification is suitable for proteases with high site specificity (e.g., trypsin). In this method, the specified cleavage sites need to be known beforehand. However, most proteases used in the food industry have weak site specificity and uncertain cleavage sites; thus, a different method is needed to identify their peptide mappings.
According to the site specificity of the protease, there are two methods for peptide identification: the site-specified identification method is suitable for highly site-specific proteases, while the site-unspecified identification method is suitable for proteases with weak or no site specificity or for the identification of endogenous peptides.12 The principle and process of the site-specified identification method are as follows.9,13,14 First, according to the site specificity of trypsin, all the sequences belonging to the selected species in the protein database are cleaved in theory, producing sets of theoretical peptide mappings. Second, according to the fragmentation rules of the collision-induced dissociation of peptide ions, a set of theoretical MS2 data corresponding to the set of theoretical peptide mappings are produced. Third, the MS2 data for each peptide are matched with the set of theoretical MS2 data, and the matches that meet the statistical requirements are accepted for positive identification. The site-unspecified method follows a similar process; however, the sequences in the protein database cannot be cleaved in theory based on the site specificity before matching them with the experimental data because the site specificity is not given, or the protease under consideration has no site specificity. Though some identification tools such as Mascot, Spectrum Mill, and INSPIRE15,16 include site-unspecified analysis algorithms, their reliability for the study of site-unspecific industry proteases has not been thoroughly evaluated, limiting peptide mapping analyses of industrial proteases in the food industry. In this study, the site-unspecified identification method was investigated and evaluated for application to the tryptic hydrolysates of proteins and then applied to analyze proteolytic peptides using a widely used industrial protease (2709 alkaline protease).
Materials and methods
Materials
Sequencing-grade modified trypsin was purchased from Promega (USA). The purified 2709 alkaline protease was supplied by Tianjin Nuoao Technology (China). High-performance liquid chromatography (HPLC)-grade acetonitrile and formic acid were purchased from Fisher Scientific (USA). Ammonium bicarbonate was purchased from Beijing Chemical Company (China). Soy protein was purchased from Shanghai Jianglai Biotechnology Company (China). Bovine serum albumin (BSA) and all other chemicals were purchased from Sigma (USA). Water was prepared by a Milli-Q system (Millipore, USA).Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (total protein:trypsin, w/w) at 37 °C for 16 h.
:1 (total protein:trypsin, w/w) at 37 °C for 16 h.Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analysis
Peptide samples were analyzed using an 1100 series HPLC system (Agilent, USA) coupled to an ESI-Q/TOF mass spectrometer (Agilent, USA). A Vydac C18 column (300 Å, 2.1 × 150 mm, Grace Vydac, USA) was used at a flow rate of 0.2 mL min−1. HPLC was performed with buffer A (0.1% FA in H2O) and buffer B (0.1% FA in acetonitrile) using the following gradient: 3% buffer B from 0–5 min; 3–50% buffer B from 5–75 min; 50–95% buffer B from 75–80 min; 95% buffer B from 80–85 min; and 95–3% buffer B from 85–90 min. The post time was 10 min. ESI-Q/TOF was performed under the following conditions: drying gas flow rate and temperature, 12 L min−1 and 300 °C, respectively; nebulizer pressure, 45 psi; capillary voltage, 3500 V; fragmentor, 175 V; collision energy slope and offset, 3.7 and 2.5 V, respectively; MS scan range and rate, 300–1500 and 3 Hz, respectively; MS/MS scan range and rate, 100–3000 and 3 Hz, respectively; and auto MS/MS, 5 precursors with active exclusion on and 2 repeat and release after 0.5 min. The collected data were used for the identification of peptide mappings.The site-specified peptide identification method
The raw LC-MS/MS data were first processed using MassHunter software (Agilent), and mgf files were exported for identification. The site-specified identification of peptide mapping was performed with Mascot software (Matrix Science) under the following conditions: protein database, Swissprot; species, bovine for BSA sample and soybean for soy protein sample; protease, trypsin; missed cleavages, no more than 1; fixed modification, cysteine carbamidomethylation; variable modifications, methionine oxidation; precursor mass tolerance, 50 ppm; and fragment ion tolerance, 50 ppm.The site-unspecified peptide identification method
The raw LC-MS/MS data were first processed by MassHunter software for identification. Site-unspecified peptide identification was also performed using Mascot software (Matrix Science). The site-unspecified identification conditions were the same as those for the site-specified method except that “none” and “all entries” were selected for the protease and species options, respectively.Results and discussion
Trypsin is widely used in proteomics research because it has strong site specificity for the lysine or arginine site on the carboxyl side. The identification of peptides hydrolyzed by trypsin uses the site-specified identification method, which has been shown to have high reliability.17,18 For proteases with weak site specificity, only the site-unspecified method can be used. However, for proteases with strong site specificity (e.g., trypsin), both the site-specified and site-unspecified methods can be used. The site-specified method is considered to have higher reliability for peptide identification, while the site-unspecified method has higher peptide coverage.17 In this study, the tryptic hydrolysates of a single protein (BSA) and a mixture of soybean proteins were used to evaluate the site-unspecified identification method.Identification of tryptic peptides using the site-specified method
For the site-specified identification method, bovine and soybean were selected in the species option for the BSA sample and the soybean protein mixture, respectively. In the Swissprot protein database, bovine has 31872 protein sequences, and soybean has 74440 protein sequences. Therefore, the method needs to identify BSA peptides from 31872 sequences and soy protein peptides from 74440 sequences; thus, a lot of calculations and matching are required. Finally, the method identified 52 peptides from BSA hydrolysate with 75% coverage of the entire BSA sequence. For soy protein hydrolysate, 14 proteins and 137 peptides were identified.
Peptide identification of tryptic hydrolysates using the site-unspecified method
Site-unspecified identification via Mascot searching combines the mass-based matching of precursor ions and the MS2 data of fragment ions. Because the cleavage sites are not specified beforehand, this method involves more calculations than the site-specified method. Moreover, in the species option for database searching, “all entries” was selected rather than “bovine” or “soybean”, meaning that millions of protein sequences were included for calculation and matching. Bovine and soybean have 31872 and 74440 protein sequences, respectively, while all entries in the Swissprot database include 180740843 protein sequences (https://www.uniprot.org/). The aim was to increase the difficulty of correct searching and matching to make the evaluation of the site-unspecified method stricter. Although the sequences of BSA and soy protein are known and collected in many protein databases, we selected the Swissprot protein database for identification because it is a comprehensive and non-redundant database that contains most of the publicly available protein sequences in the world.19 The site-unspecified method with Mascot software identified 64 peptides from the BSA sample with 83% coverage of the entire BSA sequence. For the soy protein sample, the site-unspecified method identified 14 proteins and 203 peptides. In the actual proteolysis process, some peptide bonds without arginine and lysine at the carboxyl side may also be cleaved to a lesser extent.5,20 Therefore, in the tryptic hydrolysates, the majority of peptides are site-specifically cleaved, while only a very small number of peptides are site-unspecifically cleaved. Obviously, the peptides produced by unspecific cleavage cannot be identified by the site-specified identification method. Both the quantity and coverage of peptides identified by the site-unspecified method were greater than those for the site-specified method.
Evaluation of the site-unspecified peptide identification method using tryptic hydrolysates
Cleaved sites were not specified beforehand in the site-unspecified method; thus, the probability of correct identification was the same for the peptides produced by site-specific and site-unspecific cleavage. Therefore, the reliability of the site-unspecified method can be assessed by the identification quality of the peptides from specific cleavage. Due to the high reliability of the site-specified method, the repetition of its identified peptides in the results of the site-unspecified method can be used to evaluate the reliability of the site-unspecified method. Thus, a higher degree of repeatability indicates a higher reliability of the site-unspecified method.Fig. 1 shows the repetition of peptides identified by the site-specified method in those identified by the site-unspecified method. The site-specified method identified 52 peptides from BSA hydrolysate, all of which were also identified by the site-unspecified method. The site-specified method identified 137 peptides from soy protein hydrolysate, 98.5% of which were also identified by the site-unspecified method. Moreover, it should be noted that the site-unspecified method identified peptides under the condition that “all entries” was selected for the species option in the database search. The high repeatability indicates that the site-unspecified method had high reliability for the identification of peptide mapping. Soy protein is a mixture containing glycinin, conglycinin, seed lipoxygenase, trypsin inhibitor, and so on.21 Table 1 compares the peptide mapping results obtained for soy protein hydrolysate using the two identification methods. As shown in the table, the repetition of peptides identified by the site-specified method in those identified by the site-unspecified method reached 100% for most protein components, and the lowest repetition percentage was 94%, reflecting the high reliability of the site-unspecified method. In addition, for most of the soy protein components, more peptides were identified by the site-unspecified method than by the site-specified method. The percentage increase in identified peptides reached 113% and was 40% on average, indicating that the site-unspecified method provided more peptide mapping information than the site-specified method.
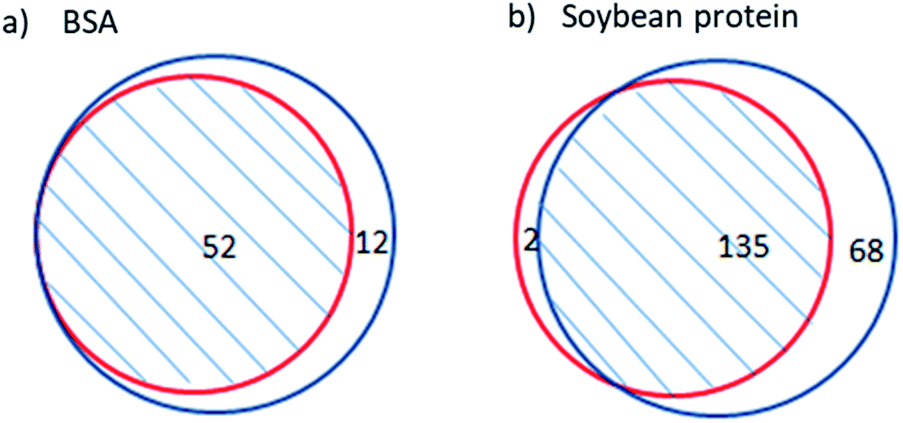 | ||
Fig. 1 Repeatability of the identified peptides between the site-specified and site-unspecified methods for the peptide mapping analysis of BSA protein (a) and soy protein (b) hydrolysis ( : site-specified identification method of peptide mapping; : site-specified identification method of peptide mapping;  : site-unspecified identification method of peptide mapping). : site-unspecified identification method of peptide mapping). | ||
| Protein name | Number of peptides identified using the site-specified method (A) | Number of peptides identified using the site-unspecified method (B) | Number of peptides in common (C) | Repetition rate (percentage of C in A) | Percentage increase of B compared to A |
|---|---|---|---|---|---|
| Glycinin G1 | 22 | 37 | 21 | 95% | 68% |
| Glycinin G2 | 16 | 34 | 15 | 94% | 113% |
| Glycinin G3 | 11 | 16 | 11 | 100% | 45% |
| Beta-conglycinin, α-chain | 17 | 23 | 17 | 100% | 35% |
| Beta-conglycinin, α′-chain | 8 | 14 | 8 | 100% | 75% |
| Beta-conglycinin, β-chain | 11 | 13 | 11 | 100% | 18% |
| Glycinin G4 | 10 | 17 | 10 | 100% | 70% |
| Glycinin | 5 | 8 | 5 | 100% | 60% |
| Sucrose-binding protein | 11 | 12 | 11 | 100% | 8% |
| Seed lipoxygenase-1 | 9 | 10 | 9 | 100% | 11% |
| Seed lipoxygenase-2 | 3 | 3 | 3 | 100% | 0 |
| Seed lipoxygenase-3 | 8 | 8 | 8 | 100% | 0 |
| Trypsin inhibitor A | 3 | 5 | 3 | 100% | 67% |
| Kunitz-type trypsin inhibitor | 3 | 3 | 3 | 100% | 0 |
| Total | 137 | 203 | 135 | 98.5% | 48% |
Distribution analysis of the tryptic cleavage sites on BSA and soy protein
The site-unspecified identification method of peptide mapping can reveal the proteolysis process of any kind of protease. Based on the identified peptides and their relative abundances, the frequency distribution of trypsin cleavage sites on BSA protein and soy protein were revealed (Fig. 2). As shown in the figure, trypsin had strong site specificity for lysine or arginine, and the relative frequency of other unspecified cleavage sites was less than 5%.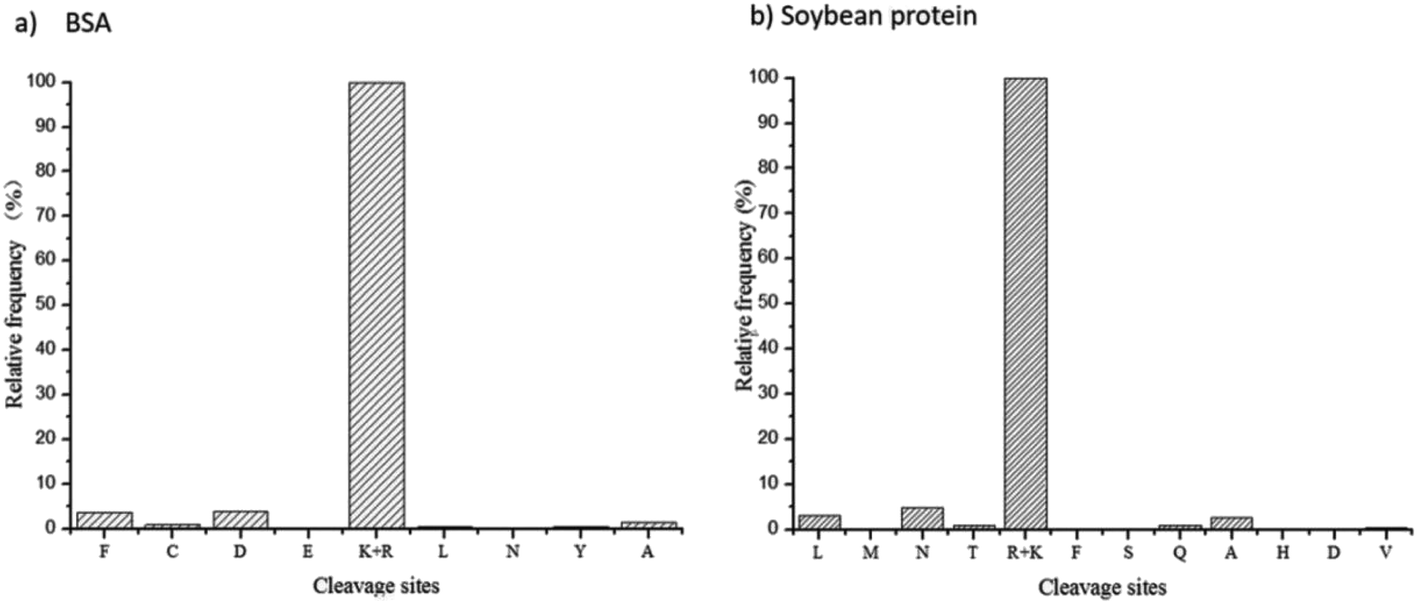 | ||
| Fig. 2 Frequency distributions of tryptic cleavage sites on BSA protein (a) and soy protein (b). | ||
Peptide mapping analysis of the hydrolysates of 2709 alkaline protease using the site-unspecified identification method
The site-unspecified peptide identification method makes it possible to analyze the peptide mapping of the hydrolysates of industrial proteases with weak site specificity. 2709 alkaline protease is an industrial protease produced by the fermentation of Bacillus licheniformis no. 2709.22 It is widely used in the food industry to hydrolyze proteins and has very weak site specificity. Therefore, the hydrolysates of BSA and soy protein obtained using 2709 alkaline protease were analyzed only by the site-unspecified identification method. Fig. 3 shows the structural distribution of the main hydrolysis products of 2709 alkaline protease for BSA protein and glycinin G1. As shown in the figure, the main peptide products were mainly distributed on the outside of the protein molecular structure, indicating a strong influence of the spatial structure on the proteolysis process.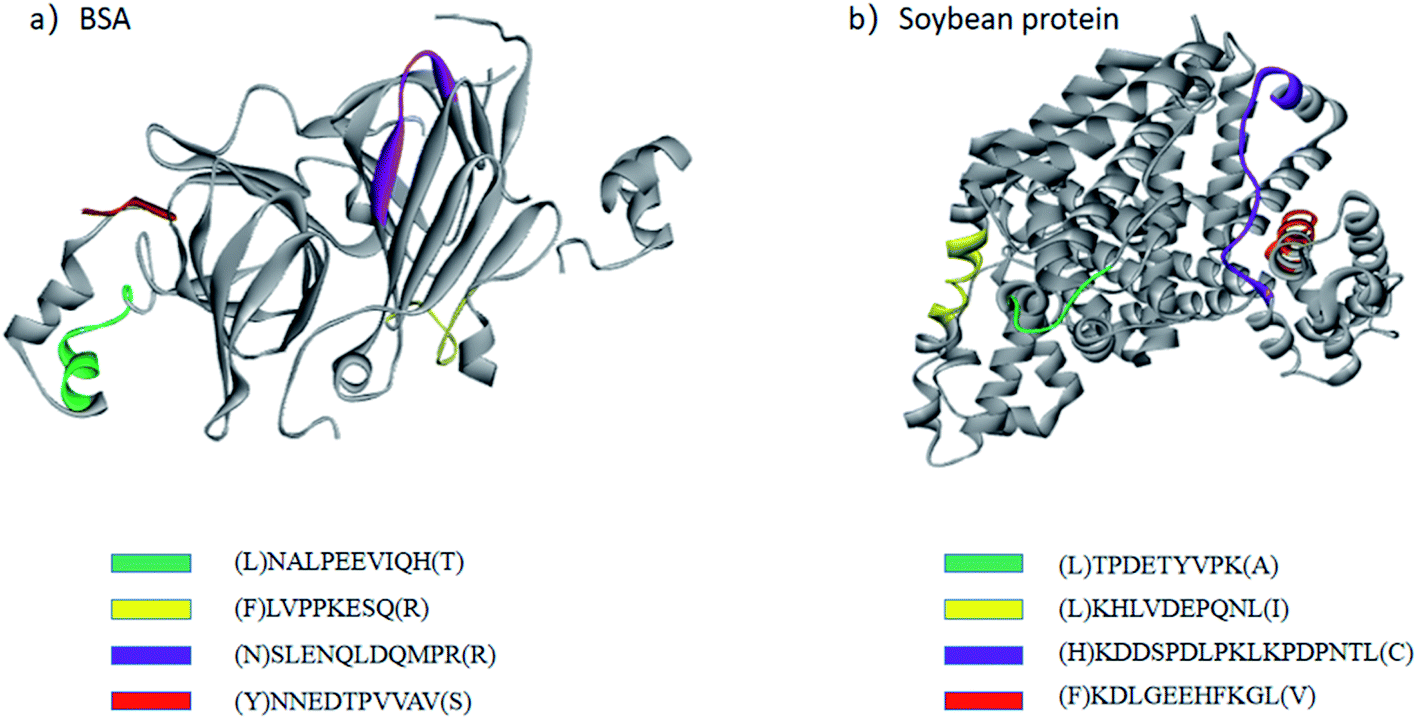 | ||
| Fig. 3 Structural distributions of the main peptides in BSA protein (a) and glycinin G1 (b) after one hour of hydrolysis by 2709 alkaline protease. BSA used PDB 3V03, and glycinin G1 used PDB 1FXZ. | ||
In conclusion, this study evaluated and demonstrated the site-unspecified identification method for peptide mapping using tryptic hydrolysates of the single protein BSA and a soybean protein mixture. Because the site-unspecified method is not dependent on site specificity, it is suitable for most industrial proteases with weak site specificity in theory. Even the species origin of the hydrolyzed proteins was not specified in the database search. At least 98% of the tryptic site-specific peptides were successfully identified via the site-unspecified method, demonstrating that the site-unspecified method also has high reliability in the identification of peptide mapping. Compared to the site-specified method, the site-unspecified method identified 23% and 54% more peptides from BSA and soy protein hydrolysates, respectively, suggesting that the site-unspecified method has greater sequence coverage and can provide more peptide mapping information. The results provide an important methodological basis for the analysis of proteolysis and help promote the rational choice and application of proteases in industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation (31671806 and 31871740).References
- A. Razzaq, S. Shamsi, A. Ali, Q. Ali, M. Sajjad, A. Malik and M. Ashraf, Front. Bioeng. Biotechnol., 2019, 7, 1–20 CrossRef.
- Q. Li, L. Yi, P. Marek and B. L. Iverson, FEBS Lett., 2013, 587, 1155–1163 CrossRef CAS.
- O. L. Tavano, A. Berenguer-Murcia, F. Secundo and R. Fernandez-Lafuente, Compr. Rev. Food Sci. Food Saf., 2018, 17, 412–436 CrossRef.
- H. B. Wang, J. Wang, Z. J. Lv, Y. H. Liu and F. P. Lu, Eur. Food Res. Technol., 2013, 236, 419–424 CrossRef CAS.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4524 CrossRef CAS.
- M. Enoksson, J. Li, M. M. Ivancic, J. C. Timmer, E. Wildfang, A. Eroshkin, G. S. Salvesen and W. A. Tao, J. Proteome Res., 2007, 6, 2850–2858 CrossRef CAS.
- P. Ellaiah, B. Srinivasulu and K. Adinarayana, J. Sci. Ind. Res., 2002, 61, 690–704 CAS.
- R. C. Kasana, R. Salwan and S. K. Yadav, Crit. Rev. Microbiol., 2011, 37, 262–276 CrossRef CAS.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS.
- S. Gessulat, T. Schmidt, D. P. Zolg, P. Samaras, K. Schnatbaum, J. Zerweck, T. Knaute, J. Rechenberger, B. Delanghe, A. Huhmer, U. Reimer, H. C. Ehrlich, S. Aiche, B. Kuster and M. Wilhelm, Nat. Methods, 2019, 16, 509–518 CrossRef CAS.
- B. H. J. van den Berg and A. Tholey, Proteomics, 2012, 12, 516–529 CrossRef CAS.
- Y. L. Chen, W. H. Chang, C. Y. Lee and Y. R. Chen, Analyst, 2019, 144, 3045–3055 RSC.
- M. A. Baldwin, Mol. Cell. Proteomics, 2004, 3, 1–9 CrossRef CAS.
- J. M. Chick, D. Kolippakkam, D. P. Nusinow, B. Zhai, R. Rad, E. L. Huttlin and S. P. Gygi, Nat. Biotechnol., 2015, 33, 743–749 CrossRef CAS.
- I. Losito, F. Mavelli, A. D. Loiotile and F. Palmisano, Anal. Chim. Acta, 2012, 718, 70–77 CrossRef CAS.
- L. Bozzacco, H. Q. Yu, H. A. Zebroski, J. Dengjel, H. T. Deng, S. Mojsov and R. M. Steinman, J. Proteome Res., 2011, 10, 5016–5030 CrossRef CAS.
- W. Yu, J. A. Taylor, M. T. Davis, L. E. Bonilla, K. A. Lee, P. L. Auger, C. C. Farnsworth, A. A. Welcher and S. D. Patterson, Proteomics, 2010, 10, 1172–1189 CrossRef CAS.
- M. The and L. Käll, Nat. Commun., 2020, 11, 3234 CrossRef CAS.
- E. Boutet, D. Lieberherr, M. Tognolli, M. Schneider, P. Bansal, A. J. Bridge, S. Poux, L. Bougueleret and I. Xenarios, Methods Mol. Biol., 2016, 1374, 23–54 CrossRef CAS.
- O. Schilling, M. L. Biniossek, B. Mayer, B. Elsasser, H. Brandstetter, P. Goettig, U. H. Stenman and H. Koistinen, Biol. Chem., 2018, 399, 997–1007 CAS.
- K. Nishinari, Y. Fang, S. Guo and G. O. Phillips, Food Hydrocolloids, 2014, 39, 301–318 CrossRef CAS.
- C. Zhou, H. Zhou, D. Li, H. Zhang, H. Wang and F. Lu, Microb. Cell Fact., 2020, 19, 45–57 CrossRef CAS.
Footnote |
| † Co-first author. |
| This journal is © The Royal Society of Chemistry 2020 |