
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale†
Ahmed M. Sayeda,
Amira R. Khattabb,
Asmaa M. AboulMagd c,
Hossam M. Hassande,
Mostafa E. Ratebf,
Hala Zaidg and
Usama Ramadan Abdelmohsen*hi
c,
Hossam M. Hassande,
Mostafa E. Ratebf,
Hala Zaidg and
Usama Ramadan Abdelmohsen*hi
aDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, 62513 Beni-Suef, Egypt
bPharmacognosy Department, College of Pharmacy, Arab Academy for Science, Technology and Maritime Transport, 1029 Alexandria, Egypt
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University, 62513 Beni Suef, Egypt
dDepartment of Pharmacognosy, Faculty of Pharmacy, Beni-Suef University, 62514 Beni-Suef, Egypt
eDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, 62513 Beni-Suef, Egypt
fSchool of Computing, Engineering & Physical Sciences, University of the West of Scotland, Paisley PA1 2BE, UK
gMinistry of Health and Population, Cairo, Egypt
hDepartment of Pharmacognosy, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt. E-mail: usama.ramadan@mu.edu.eg; Tel: +2-86-2347759
iDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, Universities Zone, P. O. Box 61111 New Minia City, Minia, Egypt
First published on 27th May 2020
Abstract
The novel Coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 is a potential factor for fatal illness and a tremendous concern for global public health. The COVID-19 pandemic has entered a dangerous new phase. In the context of drug discovery, the structurally-unique and chemically-diverse natural products have been valuable sources for drug leads. In this review, we report for potential candidates derived from natural sources with well-reported in vitro efficacy against SARS-CoV during the last decade. Additionally, a library of 496 phenolic metabolites was subjected to a computer-aided virtual screening against the active site of the recently reported SARS-CoV Main protease (Mpro). Analysis of physicochemical properties of these natural products has been carried out and presented for all the tested phenolic metabolites. Only three of the top candidates, viz. acetylglucopetunidin (31), isoxanthohumol (32) and ellagic acid (33), which are widely available in many edible fruits, obey both Lipinski's and Veber's rules of drug-likeness and thus possess high degrees of predicted bioavailability. These natural products are suggested as potential drug candidates for the development of anti-SARS-CoV-2 therapeutics in the near future.
1. Introduction
Severe Acute Respiratory Syndrome Corona Virus (SARS-CoV) is rapidly re-evolving and causing a global outbreak named “COVID-19” that is threatening hundreds of thousands of people all over the world. Similar outbreaks with the first wave of SARS-CoV emerged in southern China in 2002. In 2012, another coronavirus outbreak emerged in Jeddah, Saudi Arabia and spread within and beyond the Middle East. A Middle East respiratory syndrome caused by coronavirus (MERS-CoV) was reported this time to be associated with severe pneumonia and multi-organ failure.1 A timeline depicting the reported SARS-CoV-2 outbreaks is shown in Fig. 1A. Etiologically, SARS-CoV belongs to the genus Coronaviridae, a positive strand family. The enveloped RNA viruses have a membrane comprising four viral proteins i.e. spike glycoprotein (S), membrane glycoprotein (M), nucleocapsid proteins (N) and an envelope protein (E) (Fig. 1B). These proteins function during host cell entry and viral morphogenesis and release S-glycoprotein on the surface of the virus, which is responsible for virus attachment to a host receptor named ACE-II.2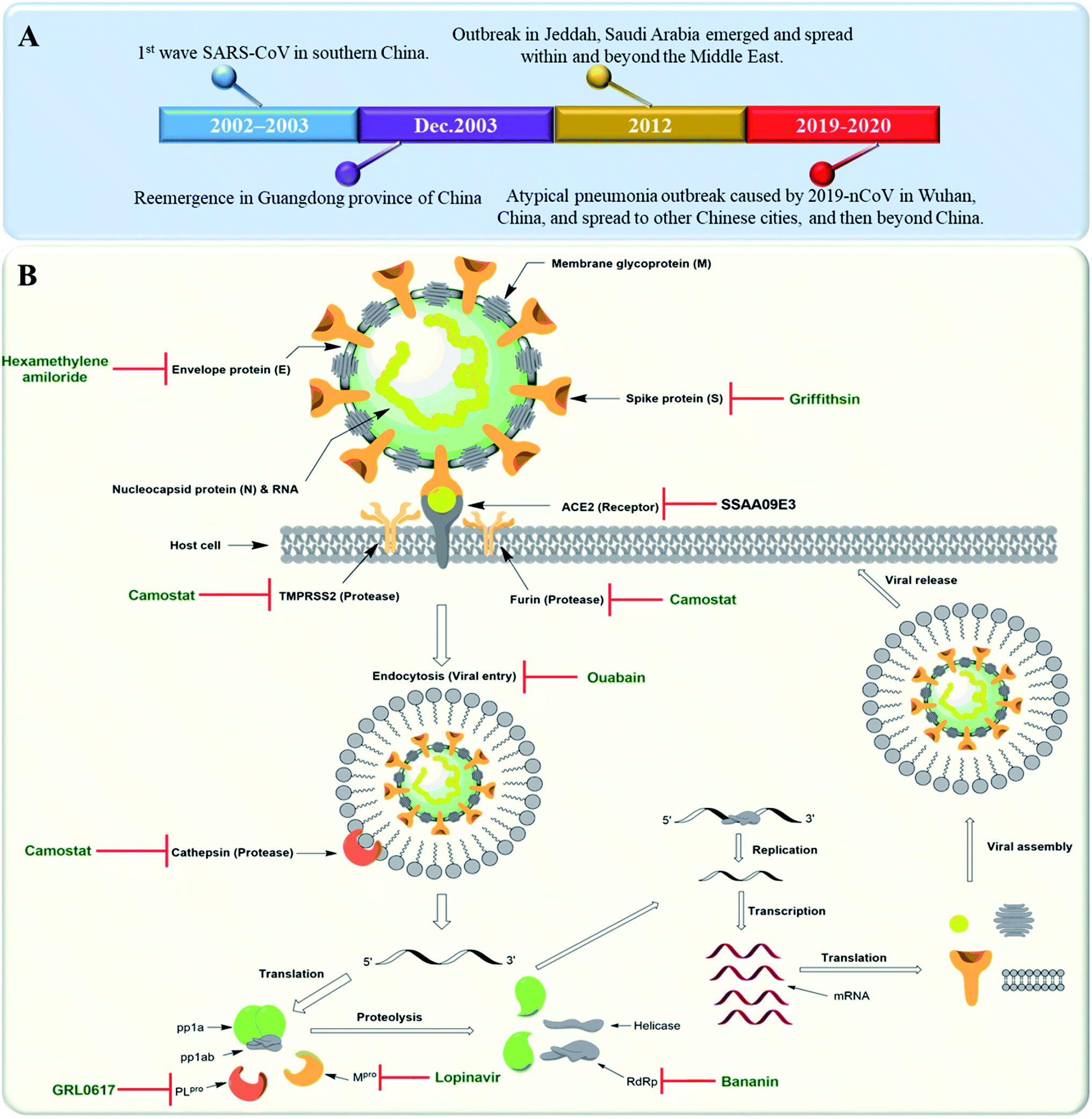 | ||
| Fig. 1 (A) Timeline of SARS-CoV outbreaks during the last two decades. (B) Coronavirus structure and its life cycle, illustrating the main molecular targets and their inhibitors. | ||
SARS-CoV genome is predominated by two open reading frames that are connected by a ribosomal frameshift site, and that encode two replicase proteins, viz. pp1a and pp1ab.3 pp1a and pp1b produce viral functional proteins via proteolytic process performed by the aid of two proteases i.e. papain-like cysteine protease (PLpro) and chemotrypsin-like cysteine protease (3CLpro). The latter is also called Main protease (Mpro), which is vital to the viral replication and virulence.4
We aimed in this review to compile an updated knowledge on the natural products with proven in vitro anti-SARS CoV reported during the period from 2005 till the beginning of 2020. We searched PubMed and Google Scholar for English language articles published without start date restrictions up to April 15th 2020 with the keywords: “anti-SARS CoV”, “natural products as Mpro, PLpro, ACE2-S-protein, helicase inhibitors”, “natural products against SARS-CoV”. Cross-referencing and “related articles” functions were used to expand the search criteria. Searches were further supplemented with publicly available information and reports from US Centers for Disease Control and Prevention and WHO websites. Furthermore, we carried out an in silico exploration of a library of phenolic natural products (496 compounds) via a computer-aided virtual screening against the active site of the SARS-CoV Main protease (Mpro) and a drug-likeness computation of the compounds reported in this review (75 compounds).
2. Anti-SARS CoV therapeutic drugs
2.1. Virus-based targets and their therapeutics
Many plant derived therapeutics exhibited an in vitro efficacy against multiple SARS viral targets viz. viral entry into host cells, viral processing (viral protease), viral replication (transcription and translation), and viral release from infected cells (Fig. 1B).SARS-CoV replicase gene has been revealed to encode a number of enzymatic functions.5 These include Mpro (nsp5), a papain-like protease (PLpro, nsp3), an RNA-dependent RNA polymerase (RdRp, nsp12), and a helicase (HEL1, nsp13). Such essential functional proteins have been selected as potential targets for the development of new anti-SARS CoV therapeutics during the last two decades. Both viral Mpro and PLpro cleave two large replicase polyproteins, viz. pp1a and pp1ab, that are encoded by the open reading frame 1a/b (ORF1a/b) to produce a number of non-structural proteins (nsp 1–15) such as RNA-dependent RNA polymerases (RdRp) and helicases that initiate the viral replication process. Such proteases can be considered key factors in the viral life cycle.6 Accordingly, their in vitro and in vivo inhibition has exhibited anti-SARS CoV potential (Fig. 1B).
Several small molecules inhibitors for these two enzymes have been reported since 2002 (outlined in Fig. 1B), of which both lopinavir and ritonavir showed a broad spectrum in vitro Mpro inhibitory activity7 as well as clinical efficacy in patients infected by COVID-19.8 Similarly, a number of specific and selective inhibitors have been developed against PLpro, and two of them with a common naphthalene benzamide scaffold are under clinical trials now.9,10 Broad-spectrum RdRp and helicase inhibitors like ribavirin and bananin, respectively have shown some clinical efficacy against SARS-CoV and MERS-CoV.11,12 The CoV small hydrophobic envelope protein (E-protein) was found to contain cation-selective ion channels similar to the mouse hepatitis virus (MHV) and human immunodeficiency virus (HIV). Inhibitors for such ion channels like hexamethylene amiloride have a well-proven in vitro and in vivo anti-CoV efficacy.13
2.2. Host-based targets and their therapeutics
ACE-II enzyme was found to be a functional and specific receptor for the viral spike-glycoproteins (S-protein). Hence, its antibodies were able to interfere with the viral entry to the host cells, and thus blocking viral replication.14 The spike glycoproteins (S1 and S2 subunits) were also reported to be potential targets for neutralizing antibodies and oligosaccharide-specific binding proteins (e.g. Griffithsin, a natural product derived from the red algae Griffithsia), in addition to several small molecules fusion inhibitors (S2 subunit inhibitors).15 Other host proteases that could be targeted for drug development include the endosomal cysteine protease i.e. cathepsins that facilitate endosomal cell entry of CoV,16 and both the transmembrane protease serine 2 (TMPRSS2) and furin, that cleave and activate spike proteins into the S1 and S2 subunits to mediate the non-endosomal virus entry at the plasma membrane.17 The broad-spectrum serine protease inhibitor camostat possessed in vitro inhibitory activity toward cathepsins, furin and TMPRSS2 and significant in vivo efficacy. Moreover, viral entry via endocytosis was also found to be inhibited by cardiotonic steroids such as ouabain and bufalin at nanomolar concentrations18 (Fig. 1B).3. Anti-SARS CoV natural therapeutics
3.1. Viral entry inhibitors
Natural therapeutic agents that target viral entry are considered an important class of antiviral therapeutics because of their potential to block the first step of viral infection and hence, preventing its propagation and ability to evolve and acquire drug resistance. SARS-CoV virus entry is initiated when the spike glycoprotein (S) efficiently bind with a metallopeptidase, ACE-II on host cell.19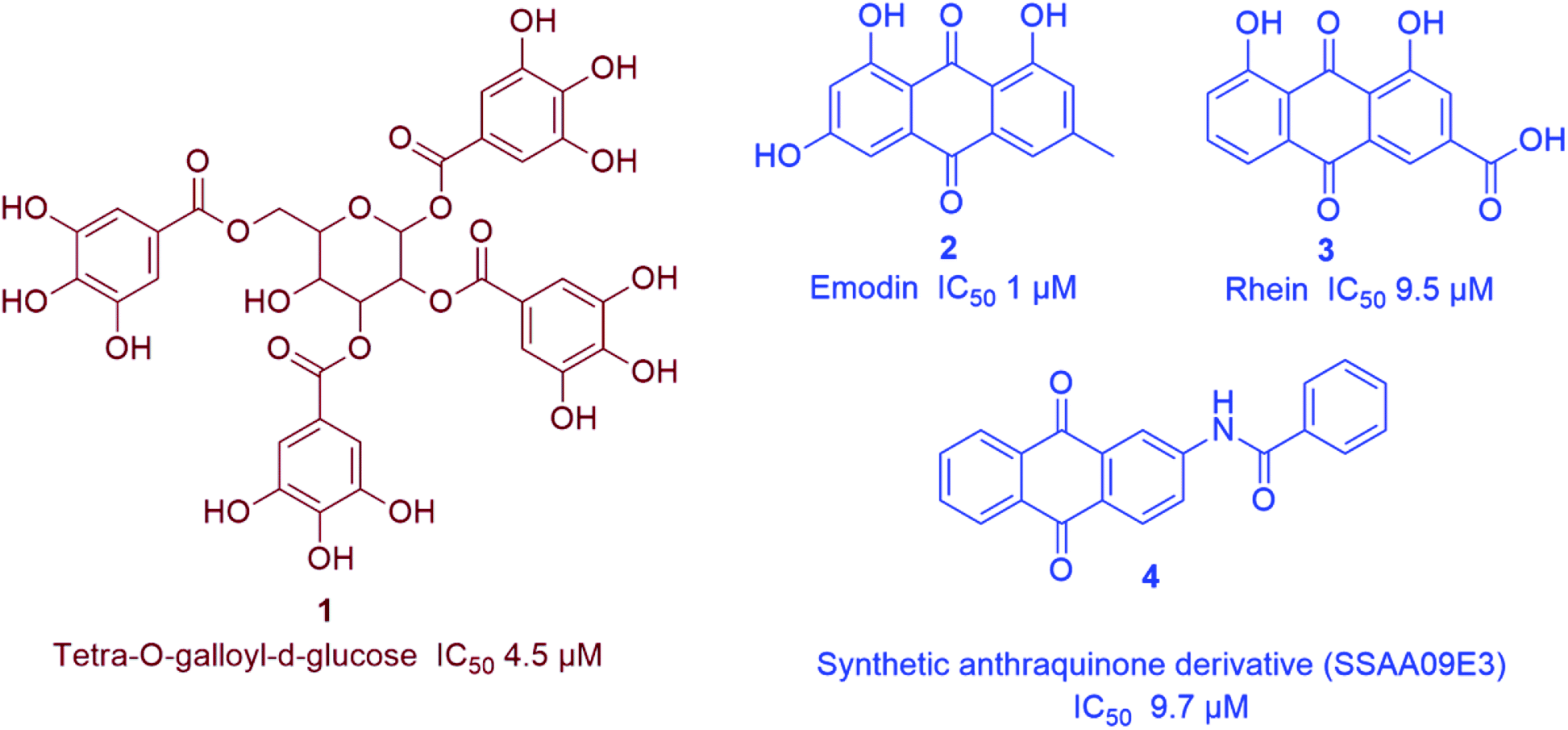 | ||
| Fig. 2 Representative natural products with SARS CoV S-protein-ACE-II binding inhibitory action (in vitro) viz. anthraquinones (blue) and tannins (brown). | ||
3.2. Inhibitors of viral replication
Being one of the most prominent molecular targets in COVID-19, Mpro and PLpro inhibitors were selected for the development of potential anti-SARS CoV therapeutics. Several small molecules have been developed during and after the first and second CoV spread waves. Among which, many edible plants-derived natural products and their related synthetic derivatives have offered a number of interesting potential inhibitors with well-proven safety and efficacy.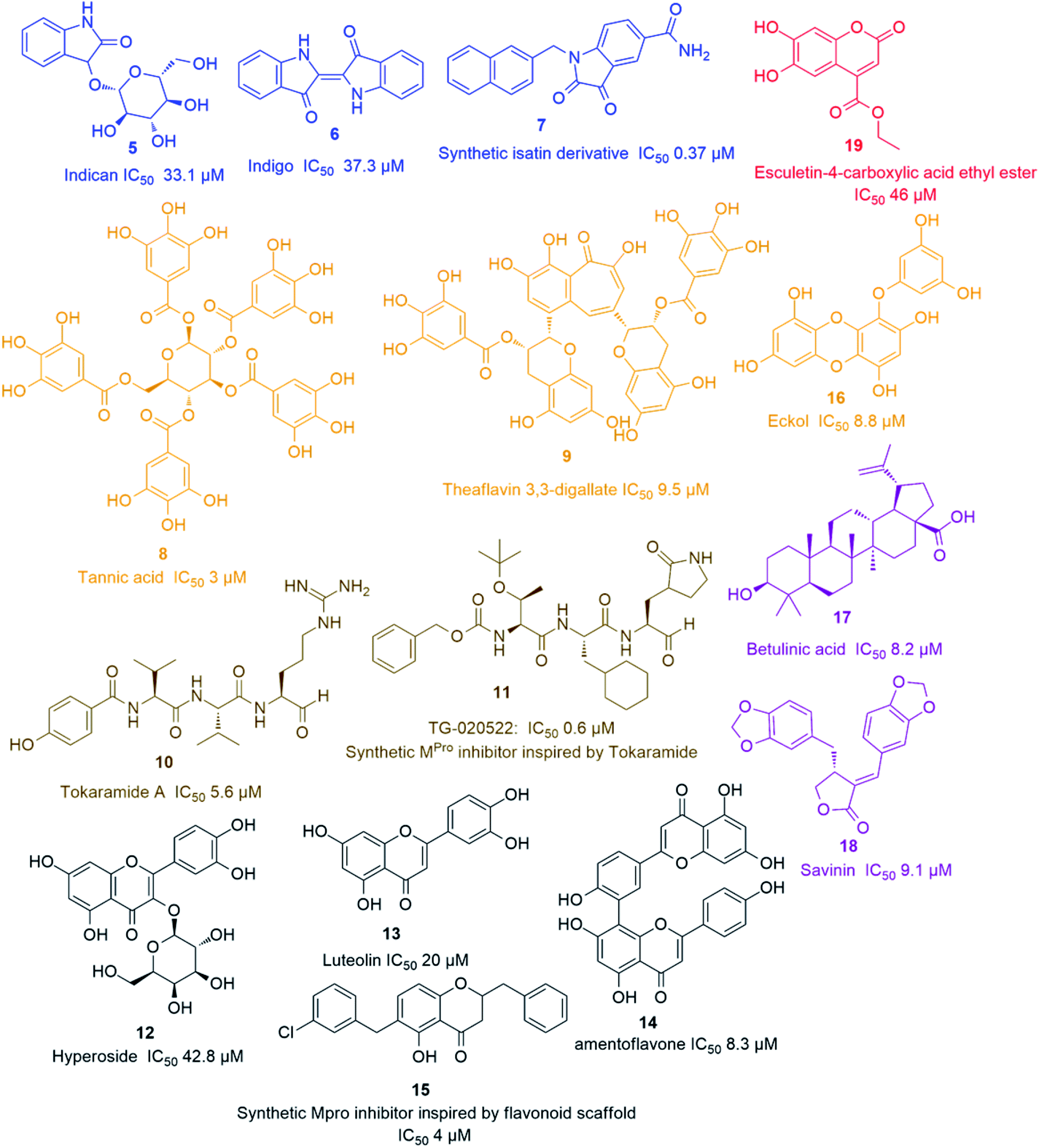 | ||
| Fig. 3 Representative natural products with SARS CoV Mpro in vitro inhibitory action viz. alkaloids (blue), coumarins (red), tannins (orange), peptides (brown), terpenes (purple) and flavonoids (black). | ||
The rapid development of computer-aided drug design (CADD) used for the in silico molecular modelling along with the growing small molecules libraries derived by the aid of combinatorial chemistry have dramatically improved the drug discovery and development process. During the last coronavirus waves, such computer-based approaches have contributed to finding a wide array of new and potent drug leads. Natural product libraries and databases have also been utilized to find potential anti-SARS CoV therapeutics. In 2004, shortly after the first SARS-CoV spread, a number of polyphenolic metabolites of which tannic acid (8), and theaflavin-3,3′-digallate (9) (Fig. 3) were reported as potent viral Mpro inhibitors (IC50 3 and 9.5 μM, respectively). These compounds were filtered out from a library of phenolic compounds consisted of 720 compounds using an in silico approach called structure-based virtual screening.25
Later on, a number of natural peptides i.e. tokaramide A (10) (Fig. 3) derived from the marine organism Theonella mirabilis26,27 inspired another research group to develop a semisynthetic peptide derivative i.e. TG-0205221 (11) with a nanomolar Mpro inhibition activity (IC50 0.6 μM), where they exhibited a non-competitive inhibition through the formation of covalent bonds with their binding sites (Fig. 3).28 Following the screening of natural product databases, another research group reported for hyperoside (quercetin-3-β-galactoside) (12) as being a promising Mpro inhibitor with IC50 of 42.79 μM. They also studied the structure–activity relationship of this flavonoid in comparison with a number of related derivatives and found that a hydroxyl group at C-3′ is an essential structural requirement for hyperoside competitive inhibitory action (Fig. 3).29
As part of the ongoing investigation of flavonoids-based antiviral candidates, Ryu and co-workers in 2010, reported the Mpro inhibitory activity (IC50 20–200 μM) of some common flavonoids i.e. quercetin, apigenin and luteolin (13) and bioflavonoids such as amentoflavone (14), with the latter possessing better in vitro inhibitory activity (IC50 8.3 μM). These findings revealed that larger polyhydroxylated compounds are more preferred to develop potent Mpro noncompetitive inhibitors (Fig. 3).30 Such interesting activity together with the simplicity of flavonoids-based inhibitors encouraged another researcher group to develop potent inhibitors analogs such as compound (15) based on the flavonoids scaffold which were subjected for a preclinical trial (Fig. 3).31 In 2013, eight phlorotannins, isolated from an edible brown algae Ecklonia cava, namely eckol (16), dieckol, triphloretol A, dioxinodehydroeckol, 2-phloroeckol, 7-phloroeckol, fucodiphloroethol G and phlorofucofuroeckol A, with the first two compounds (Fig. 3) possessing the most potent SARS-CoV Mpro inhibitory activities (IC50 8.8 and 2.7 μM, respectively).32 Epigallocatechin gallate and gallocatechin gallate showed good Mpro inhibition activity (IC50 73 and 47 μM, respectively) with their galloyl moiety at 3-OH position required for the antiviral action.33
Herbacetin, rhoifolin and pectolinarin showed anti-SARS-CoV Mpro action (IC50 33.17, 27.45 and 37.78 μM, respectively) using a tryptophan-based fluorescence method.34
Scutellarein, myricetin, quercetagetin, and robinetin were reported in a Chinese patent to inhibit SARS Mpro activity in a fluorescent assay.2 The former two compounds also were proved to inhibit the SARS-CoV helicase protein in vitro by affecting the ATPase activity.35 High throughput screening of natural product libraries led to the discovery of two interesting Mpro competitive inhibitors i.e. betulinic acid (17) and savinin (18) (IC50 8.2 and 9.1 μM, respectively). Both compounds were able to bind the enzyme's active site constituting S1 and S2 subsites via multiple hydrogen bonds with Asn 142, Gln 189 and Cys 145 amino acid residues. Thus, they could be promising starting points for developing more potent derivatives, particularly, betulinic acid (17) (Fig. 3) which is abundant in many edible and medicinal plants.36
Four quinone-methide triterpenes were reported to exhibit Mpro inhibitory actions namely celastrol, pristimerin, tingenone and iguesterin (IC50 10.3, 5.5, 9.9, and 2.6 μM, respectively), which were isolated from the bark of Tripterygium regelii (F. Celastraceae), a woody vine used in traditional Chinese medicine in the treatment of inflammatory and autoimmune diseases.37
A coumarin compound named esculetin-4-carboxylic acid ethyl ester (19) (Fig. 3), isolated from tropical marine sponge Axinella corrugata, was reported to possess Mpro inhibitory action with IC50 value of 46 μM.38 Isolinoleic acid (at a concentration of 50 μM) possessed Mpro inhibitory potential as reported in another Chinese patency.2
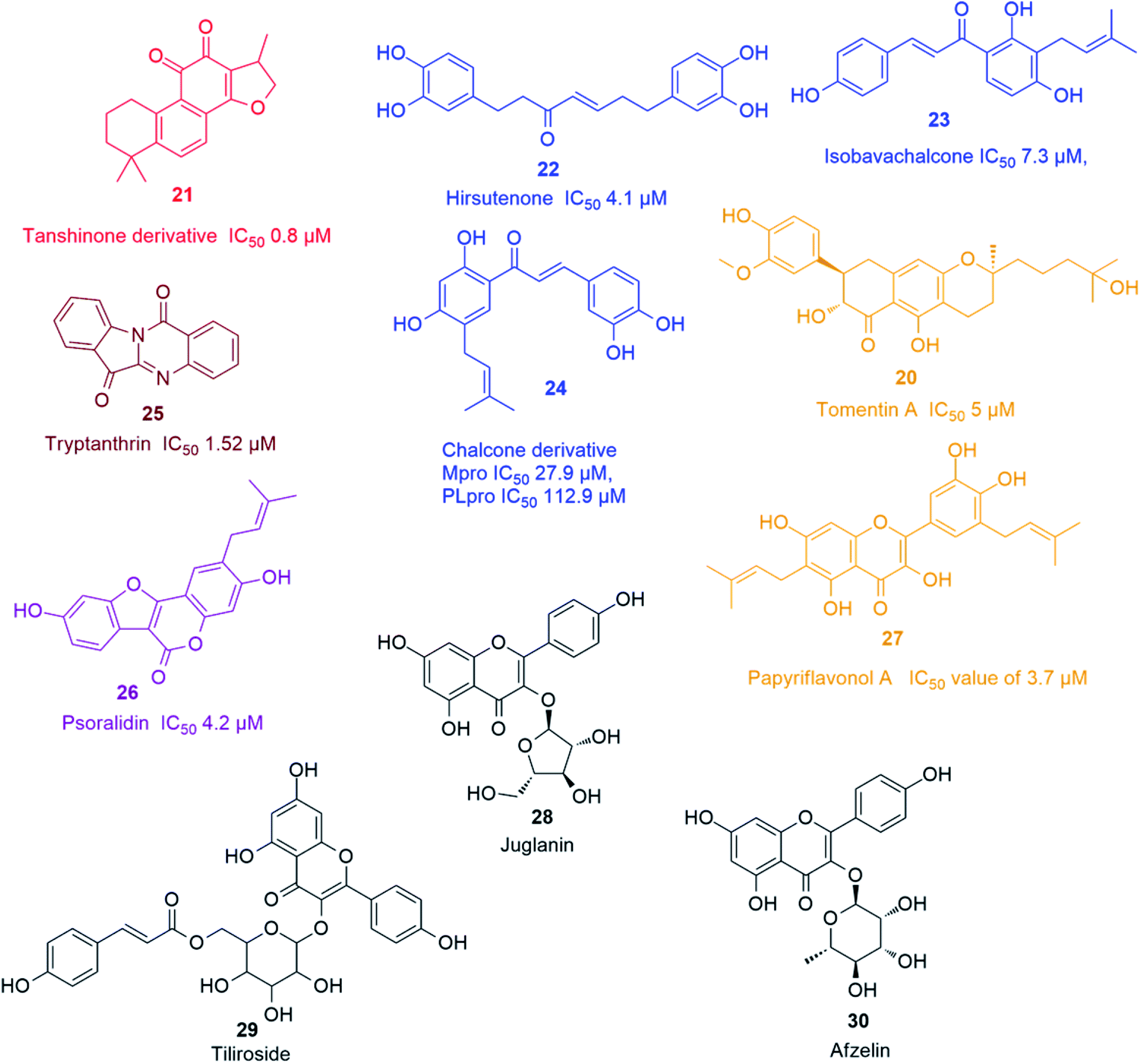 | ||
| Fig. 4 Representative natural products with SARS CoV PLpro in vitro inhibitory action viz. chalcones (blue), terpenes (Red), flavonoids (orange), alkaloids (brown), coumarins (purple), and viral release inhibitors viz. flavonoid glycosides (black). | ||
Recently, Park and his co-workers identified several polyhydroxylated chalcone derivatives, i.e. isobavachalcone (23), 4-hydroxyderricin, xanthoangelol, xanthoangelol F, xanthoangelol D, xanthoangelol E, xanthoangelol B, xanthoangelol G and xanthokeistal A, and coumarins from Angelica keiskei, i.e. psoralen, bergapten, xanthotoxin and isopimpinellin (Fig. 4), as being potent competitive inhibitors of both Mpro and PLpro with IC50 values ranging from 5.8 to 11.9 μM. Xanthoangelol E, containing the perhydroxyl group, exhibited the most potent Mpro and PLpro inhibitory activity with IC50 values of 11.4 and 1.2 μM, respectively.43 Another group of polyphenolic compounds, were isolated from the medicinal plant Broussonetia papyrifera, of which a chalconoid derivative (24) showed the best inhibitory potential against both Mpro and PLpro (IC50 27.9 and 112.9 μM, respectively).44 Tryptanthrin (25) isolated from Strobilanthes cusia leaf inhibited the cleavage activity of PLpro and the post-entry stage of viral replication with IC50 of 1.52 μM.45 Six cinnamic amides i.e. N-trans-caffeoyltyramine, N-trans-coumaroyltyramine, N-trans-feruloyltyramine, terrestriamide and N-trans-feruloyloctopamine, isolated from Tribulus terrestris fruits, showed significant PLpro inhibition (IC50 values ranging from 15.8 to 70.1 μM). The latter compound possessed the most potent inhibition with IC50 of 15.8 μM.46 Six phenolic compounds with good PLpro inhibitory actions (IC50 ranging between 4.2 and 38.4 μM), namely bavachinin, neobavaisoflavone, 4′-O-methylbavachalcone, corylifol A, isobavachalcone and psoralidin (26) were isolated from Psoralea corylifolia seeds. The latter two compounds showed the most potent actions with IC50 values of 7.3 and 4.2 μM, respectively.47 The inhibitory potential of ten polyphenols derived from Broussonetia papyrifera roots, i.e. broussochalcone A, broussochalcone B, 4-hydroxyisolonchocarpin, papyriflavonol A (27), 3'-(3-methylbut-2-enyl)-3′,4,7-trihydroxyflavane, kazinol A, kazinol B, broussoflavan A, kazinol F and kazinol J, were tested against the two SARS CoV proteases with a more potent inhibition recorded against PLpro than that of 3CLpro. The most potent PLpro inhibition was exhibited by the prenylated flavone derivative viz. papyriflavonol A (27) with IC50 value of 3.7 μM, exceeding the inhibitory potential of non-prenylated flavone derivatives viz. quercetin and kaempferol (IC50 8.6 and 16.3 μM, respectively). This signified the crucial role of the prenyl group in forming stronger hydrophobic interactions with the enzyme as well as the increase in the hydroxylation in the flavone backbone.48
3.3. Viral release inhibitors
Several kaempferol derivatives (Fig. 4) were reported to block the 3a channel of Coronavirus, which are formed by the ORF 3a-coded proteins, thus counteracting the viral production and release from the host cells. This gives the body a chance to adjust its immune system to counteract the viral attack. Kaempferol glycosides were proved to possess more potent inhibitory effect than kaempferol and this pointed out to the significance of sugar residues for the antiviral activity. Juglanin (kaempferol-3-O-α-L-arabinofuranoside) (28) was the most effective glycoside with an IC50 value of 2.3 μM. Other kaempferol glycosides i.e. tiliroside (kaempferol-3-O-(6-p-coumaroyl)-glucoside) (29) and afzelin (kaempferol-3-O-α-L-rhamnoside) (30) were less potent than juglanin (28) but showed similar activity to that of emodin (2).494. Analysis of drug-likeness properties
The druggability of any molecule is determined depending on its pharmacokinetic properties as well as its excellent pharmacodynamics. Calculating the physicochemical properties (e.g. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and molecular weight) of a bioactive compound can predict its possible pharmacokinetics (e.g. absorption and bioavailability). Lipinski's rule of five considers a given bioactive compound as a drug candidate if it possess the following physicochemical parameters i.e. logP ≤ 5, molecular weight ≤ 500, hydrogen bond donor [HBD] ≤ 5, and hydrogen bond acceptor [HBA] ≤ 10. Despite, 90% of orally active drugs that have reached phase 2 clinical status50 follow these parameters range, however, meeting the rule of five does not guarantee that a molecule is a drug-like.51 Drug's cellular permeability and in turn its distribution and excretion are associated with its topological polar surface area (tPSA) and molecular flexibility (i.e. the number of rotatable bonds). Hence, bioactive molecules with tPSA of 140 Å or less and rotatable bonds of ten or fewer (i.e. Veber's oral bioavailability) can be considered as potential drug candidates.51
P and molecular weight) of a bioactive compound can predict its possible pharmacokinetics (e.g. absorption and bioavailability). Lipinski's rule of five considers a given bioactive compound as a drug candidate if it possess the following physicochemical parameters i.e. logP ≤ 5, molecular weight ≤ 500, hydrogen bond donor [HBD] ≤ 5, and hydrogen bond acceptor [HBA] ≤ 10. Despite, 90% of orally active drugs that have reached phase 2 clinical status50 follow these parameters range, however, meeting the rule of five does not guarantee that a molecule is a drug-like.51 Drug's cellular permeability and in turn its distribution and excretion are associated with its topological polar surface area (tPSA) and molecular flexibility (i.e. the number of rotatable bonds). Hence, bioactive molecules with tPSA of 140 Å or less and rotatable bonds of ten or fewer (i.e. Veber's oral bioavailability) can be considered as potential drug candidates.51
In this review, the 74 anti-CoV natural products (Table S1, ESI†) were found to belong to four classes of compounds viz. phenolics, terpenoids, alkaloids and peptides, where phenolics were the most prominent compounds of which flavonoids were the major class (72%) (Fig. 5). The majority of compounds were found targeting Mpro and PLpro (55% and 26%, respectively, Fig. 5) indicating that they are the most susceptible targets towards natural products, particularly the phenolics. The antiviral activity of these compounds possessed activity ranging from potent to moderate (IC50 ranging from 0.0025 to 90 μM, average IC50 20.13 μM), where phenolics were also the most active ones (Fig. 5). The physicochemical characters, viz. molecular weight, logP, HBD, HBA, tPSA and rotatable bonds, of the 74 anti-SARS CoV were calculated using LigandScout 4.3 (Vienna, Austria) and projected onto a drug-likeness cut-off.52
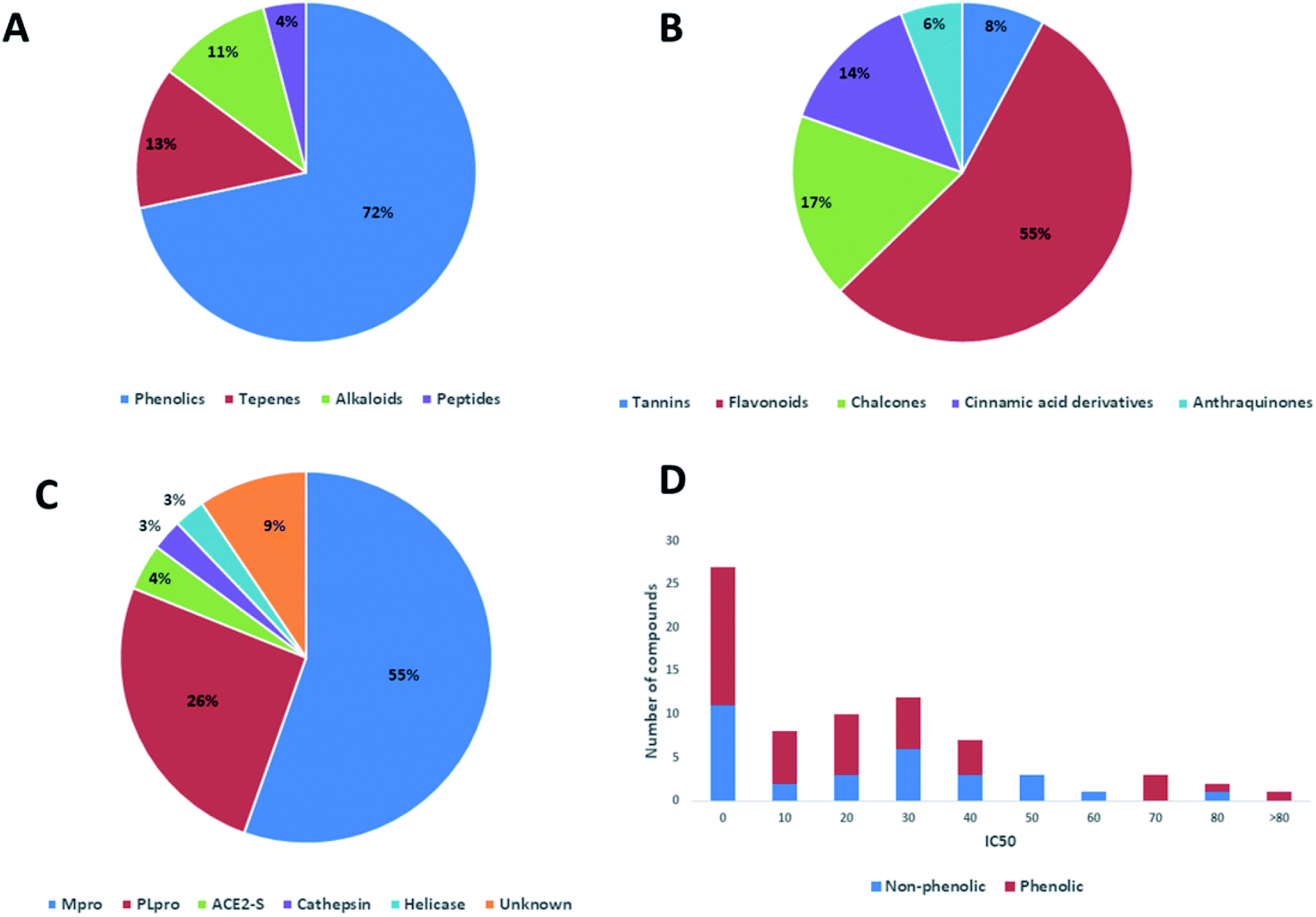 | ||
| Fig. 5 Pie charts indicating the reported anti-SARS CoV natural therapeutic classes (A & B), and their common viral targets (C). A bar chart depicting activity range of the reported natural products illustrating phenolic compounds as being the most prominent class of compounds (D). | ||
Regarding the drug-likeness properties, 76% of the reviewed molecules followed Lipinski's and Veber's rules (Fig. 7), indicating the promising potential of natural products to serve as drug candidates against the newly emerged anti-SARS CoV. The molecular weights of the 74 compounds distributed between 248.24 and 1701.2 Da (average of 138.248 Da), and most of them were between 200 and 400 Da (Fig. 6). Both tannic acid and β-aescin were the largest molecules. However, they showed potent anti-SARS CoV (IC50 3 and 6 μM, respectively). The logP of the compounds under study followed a normal distribution with the major was around 2, and 79% of the compounds were of favourable logP values (≤5) (Fig. 6). Regarding HBD and HBA parameters (average of 3 and 4, respectively, Fig. 6), they showed a distribution pattern similar to that of logP (Fig. 6) with more than 80% of compounds were of acceptable numbers. Additionally, 74% of compounds can demonstrate good oral bioavailability (Fig. 6). Being the most predominant and active compounds, phenolic's tPSA and to some extent of flexibility (number of rotatable bonds) which are interestingly linked to their activity. The compounds with higher tPSA and number of rotatable bonds (i.e. flexible) were found to exhibit higher IC50 values (Fig. 7). Such observation was also found in the peptides class, as they were among the most active compounds (IC50 0.0025 to 6.8 μM), and also have high tPSA (>140 Å) and high flexibility (rotatable bonds 12 to 27). Natural products are not always available to supply the drug industry due to their limited natural occurrence for some ecological considerations. Accordingly, combinatorial chemistry can aid in providing an alternative source for the natural product itself or its analogs. In this context, we examined the complexity and the synthetic accessibility of our reviewed natural products. As illustrated in Fig. 7, most of the compounds (>85%) got a score ranging from 2 to 5, indicating that they or their analogs could be synthesized if the natural product itself cannot be readily accessible.
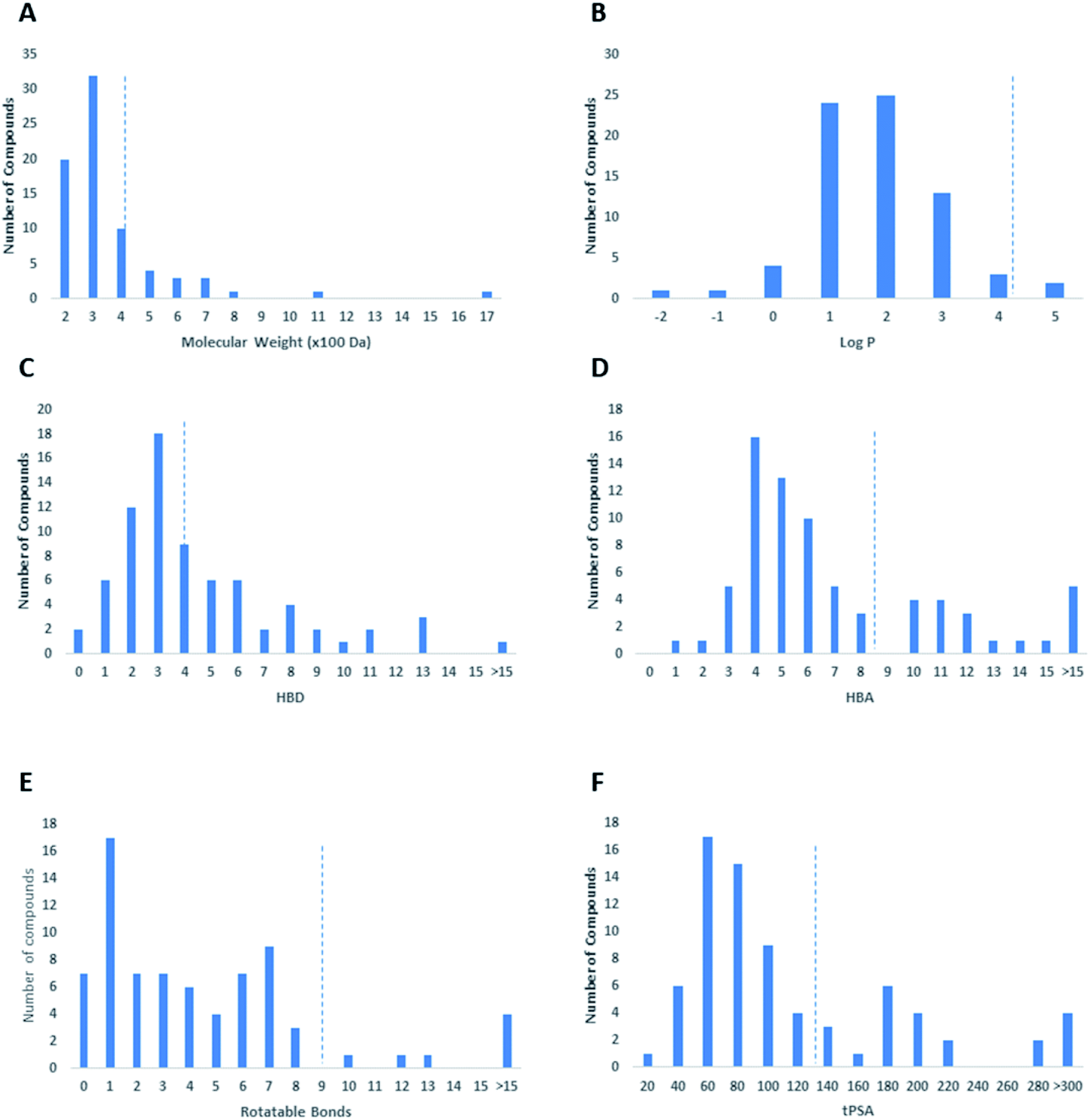 | ||
| Fig. 6 Analysis of the physicochemical properties of anti-SARS CoV natural products displayed in terms of molecular weight (A), calculated logP (B), HBD (C), HBA (D), number of rotatable bonds (E), and tPSA (F). | ||
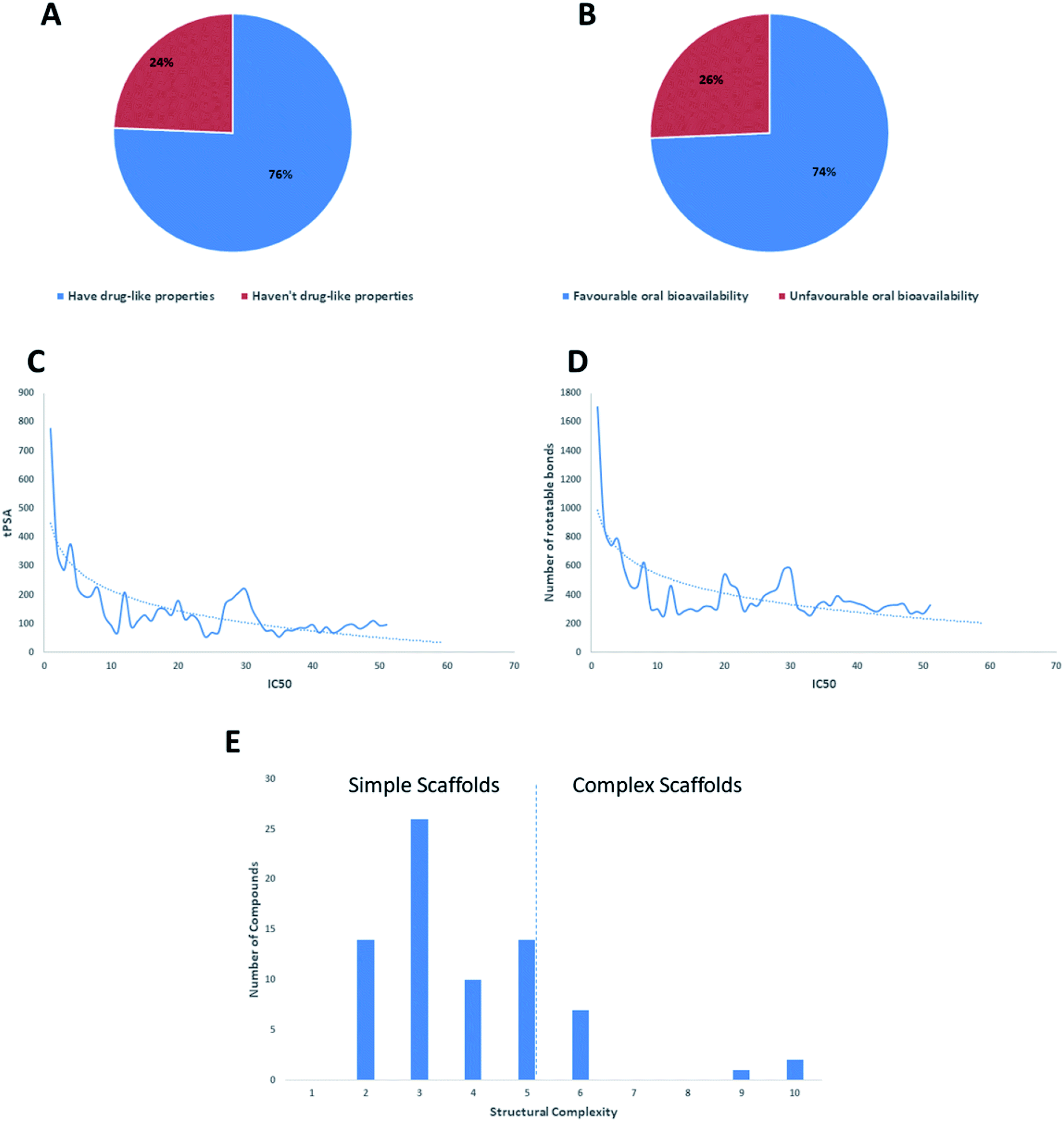 | ||
| Fig. 7 The compliance of the tested natural products with drug-likeness rules Lipinski's and Veber's rules (A & B, respectively). The relationship between the IC50 and both tPSA and the number of rotatable bond (C & D, respectively). Distribution of the reported anti-SARS CoV natural products depending on their scaffold's complexity (E). | ||
5. In silico analysis of potential anti SARS-CoV 2 agents
Virtual screening (VS) proved to be a helpful tool for drug hit/lead identification. Many studies have recently employed in silico exploration of large sets of natural products with the aid of online databases in attempt to discover new hits against potential molecular targets. Among which, a study by ul Qamar M. T. reported for structural insights into anti-viral phytochemicals/traditional Chinese medicinal compounds with in silico screening of 32297 natural products for their SARS-CoV-2 3CLpro inhibitory action.53 As shown in our in silico analysis, phenolic derivatives were the most prominent and active compounds against previous CoVs (Fig. 5). Thus, we subjected a library of phenolic metabolites,54 to a computer-aided virtual screening against the active site of the recently reported COVID-19 Mpro (PDB code: 5R7Z) based on high-throughput flexible docking technique for the sake of finding possible candidate acting as inhibitors for the newly emerged SARS-CoV2. Thirty four compounds were ranked by highest energy score and the poses that maintained the essential interactions. Twelve compounds were retrieved as the top-hits that fit into the Mpro target pocket (Table S2, ESI†). Only three of these top candidates obey both Lipinski's and Veber's rules of drug-likeness (Fig. 8 and 9). The structure–activity relationship analyses of the final top-hits presented in Fig. 9(A) revealed that they shared convergent interactions with the active site's amino acid residues that are comparable with the previous CoV-main proteases inhibitors. All top hits exhibited hydrogen bonding interaction with Thr 190 via the hydroxyl phenolic group and the carbonyl oxygen of Thr 190 residue. Additional hydrogen bonding interactions were found to exist between ellagic acid (33) and Met-165 or isoxanthohumol (32) and Cys-145. Regarding van der Waal interactions, all the hits showed interaction with Gln-189, which is assumed to be essential for the activity (Fig. 9). Additionally, the presence of Gln-191, Glu-166 and Cys-145 with some top-hits to form the hydrophobic subsite in other CoV Mpro which may create an extra van der Waal interactions. These essential interactions were found to be missing in low-scoring compounds. The selected hits with drug-like properties are well reported to be abundant in many edible fruits such as raspberry and pomegranate. Hence, they are readily available for the in vitro investigation to validate them as potential anti-SARS CoV2 drug leads (Fig. 9). Regarding the possibility of the metabolic unstability of acetylglucopetunidin (31) and (2S)-isoxanthohumol (32), both compounds show the presence of acetylester or methoxy groups that could be easily hydrolyzed inside the human body. Accordingly, we redocked their respective metabolites (34 & 35) to explore their inhibitory potential. Both metabolites were found to reserve the same hydrophobic and hydrogen bond interactions with His-41 and Gln-189 amino acid residues, respectively however, they gained an additional hydrogen bond interaction with Gln-189. Moreover, both metabolites bind with Cys-44 and Cys-145 via two hydrogen bond donors with binding affinity of −7.147 kcal mol−1. The metabolite of (2S)-isoxanthohumol (35) was found to bind with the same three amino acid residues i.e. Gln-189, Thr-190, and Glu-166, with a binding affinity of −6.021 kcal mol−1, which was comparable to its parent compound (32), however, an extra hydrogen bond donor with Leu 141 was recorded (Fig. 9(D)).
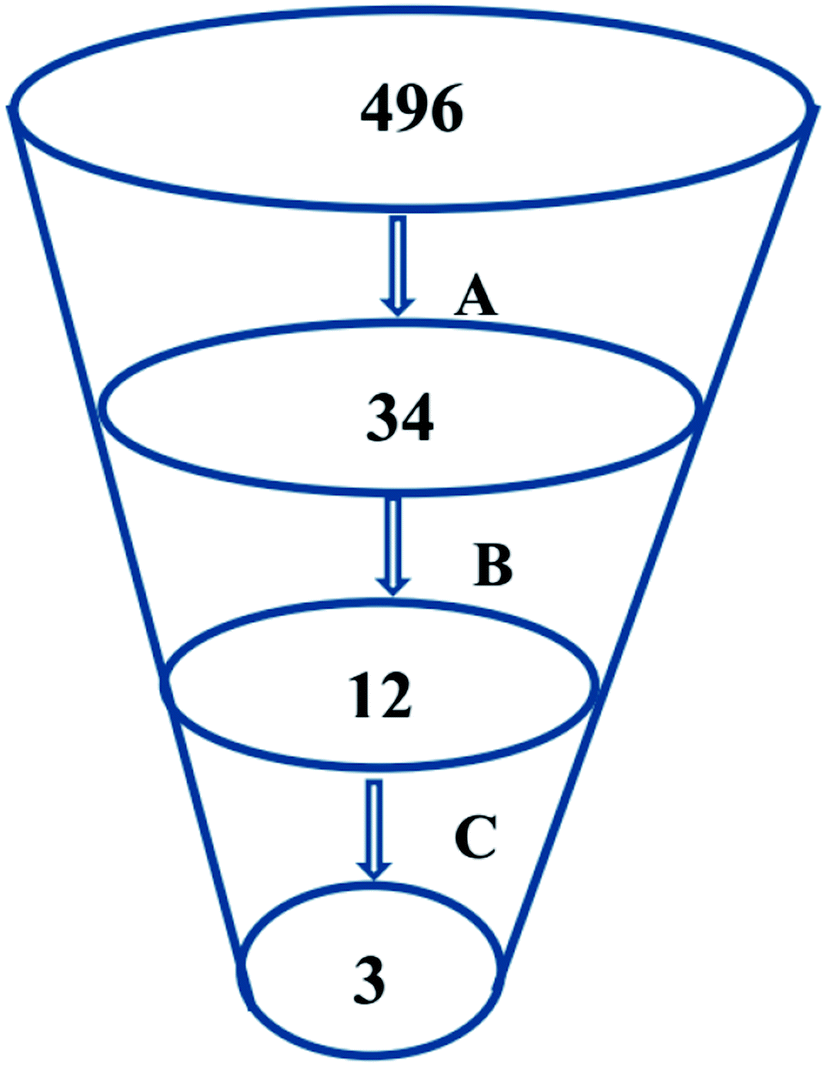 | ||
| Fig. 8 An outline of the employed virtual screening methodology that comprised of (A) phenolic compounds library virtual screening via molecular docking, (B) selection of the docking poses maintaining the essential binding interactions and (C) selection of top phenolic hits with the most acceptable drug-likeness properties. | ||
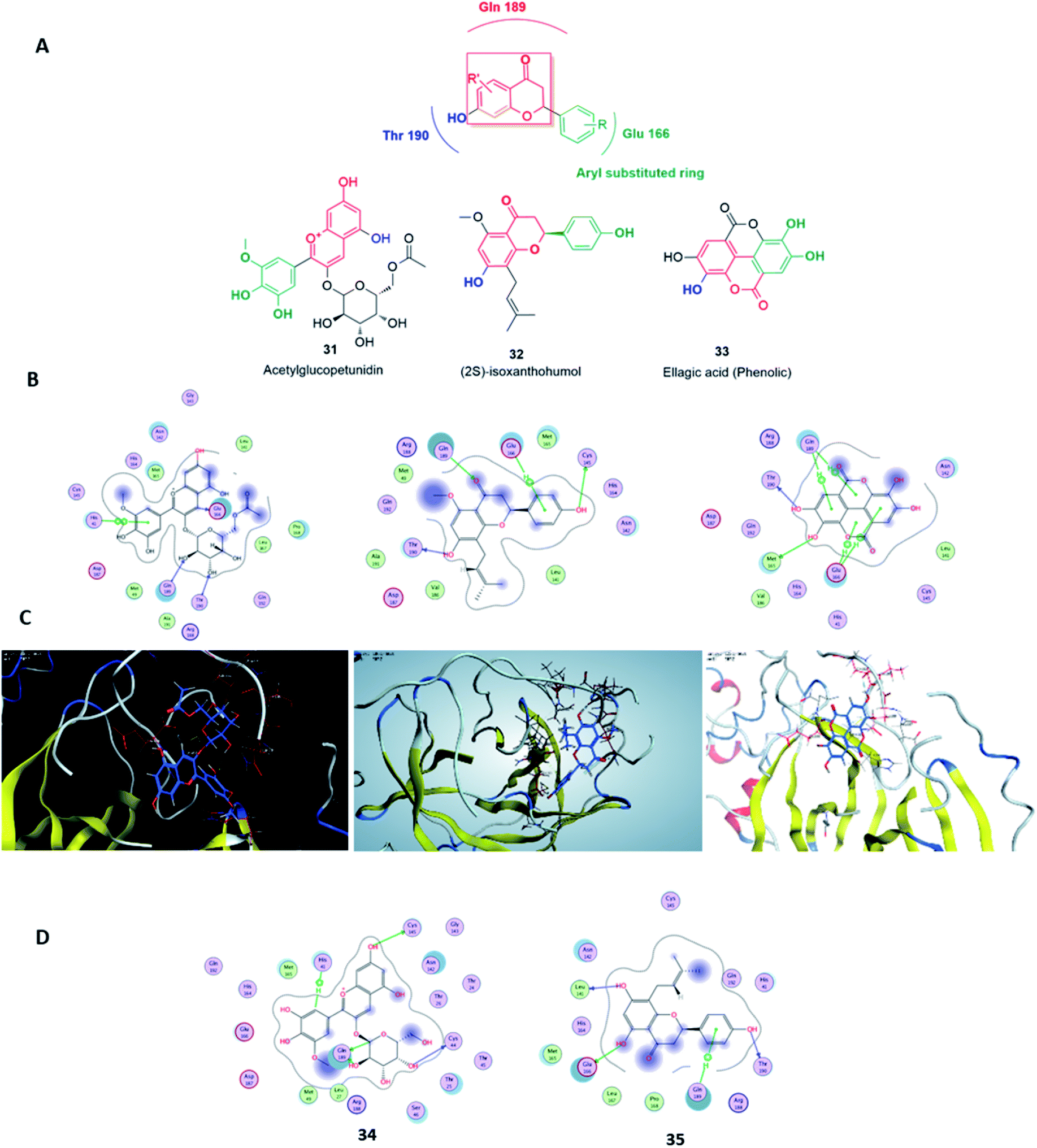 | ||
| Fig. 9 The main pharmacophore of the top three hits (A), along with their binding modes inside SARS CoV Mpro active sites (B & C). The binding modes of the hydrolysis metabolites derived from compounds 31 and 32 (D). | ||
6. Concluding remarks
The rapid identification of effective therapeutic interventions against COVID-19 is a major challenging issue nowadays. Taking into consideration that the development of new synthetic drugs is a time-consuming process, so repurposing the already known medications is the most convenient choice under the current situation. Hence, natural products can provide a valuable source for the rapid drug discovey of effective as well as safe anti-SARS CoV therapeutics. As has been illustrated in the current review, and other recently published reviews, a wide array of chemically-diverse natural products have proven to be excellent candidates as anti-SARS CoV, among which phenolic derivatives, viz. flavonoids were the most reported active agents, due to their promising pharmacokinetic profiles.55Being the most susceptible targets in CoV, both Mpro and PLpro of SARS-CoV in addition to a number of host-based proteases viz. cathepsins and furin, can be utilized for the sake of developing broad-spectrum anti-SARS CoV therapeutic agents that target multiple viral and non-viral proteins. Some phenolics such as tannins and flavonoids have exhibited a broad-spectrum activity not only against SARS-CoV but also other viruses56 Based on our findings, tPSA and molecular flexibility are the most essential descriptors that should be taken into consideration for selecting active and bioavailable phenolic compounds. The reported IC50 values of SARS natural products-inhibitors were mostly recorded in a micromolar range. However, more potent synthetic or semi-synthetic derivatives can be developed from the current scaffolds as they showed low to moderate molecular complexity. With the tremendous ongoing efforts to develop broad-spectrum antiviral therapeutics to combat coronaviruses and stop the rapid spread of COVID-19 worldwide, we hope that the current outbreak to settle down in a few months, as has been witnessed previously with SARS and MERS pandemics.
Contributors
AMS, AMA and ARK initiated the review. URA, HMH and MER developed the scope of the manuscript. AMS, AMA and ARK did the literature search and prepared the first draft. AMS designed the figures and interpreted the findings. URA, HZ, HMH and MER critically reviewed the data and draft. All authors subsequently modified the manuscript jointly. URA is the guarantor of the final version, which was read and approved by all the authors.Conflicts of interests
We declare that we have no conflicts of interest.Acknowledgements
The authors thank Nahda University in Beni Suef (NUB), Minia, Beni Suef Universities and Arab Academy for Science, Technology and Maritime Transport for supporting this work. We thank Jörn Piel (Institute of Microbiology, ETH Zürich, Zürich, Switzerland), Prof. Hossam Eldin Elmalahi (Nahda University, Beni Suef, Egypt) and Prof. Emad Hamdi Ghoz (Deraya University, New Minia, Egypt) for their support and fruitful discussion.References
- E. De Wit, N. Van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS PubMed.
- V. Kumar, Y. S. Jung and P. H. Liang, Expert Opin. Ther. Pat., 2013, 23, 1337–1348 CrossRef CAS PubMed.
- V. Thiel, K. A. Ivanov, Á. Putics, T. Hertzig, B. Schelle, S. Bayer, B. Weißbrich, E. J. Snijder, H. Rabenau, H. W. Doerr, A. E. Gorbalenya and J. Ziebuhr, J. Gen. Virol., 2003, 84, 2305–2315 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- E. J. Snijder, P. J. Bredenbeek, J. C. Dobbe, V. Thiel, J. Ziebuhr, L. L. M. Poon, Y. Guan, M. Rozanov, W. J. M. Spaan and A. E. Gorbalenya, J. Mol. Biol., 2003, 331, 991–1004 CrossRef CAS PubMed.
- S. van Boheemen, M. de Graaf, C. Lauber, T. M. Bestebroer, V. S. Raj, A. M. Zaki, A. D. M. E. Osterhaus, B. L. Haagmans, A. E. Gorbalenya, E. J. Snijder and R. A. M. Fouchier, mBio, 2012, 3, 1–9 CrossRef PubMed.
- K. S. Chan, S. T. Lai, C. M. Chu, E. Tsui, C. Y. Tam, M. M. L. Wong, M. W. Tse, T. L. Que, J. S. M. Peiris, J. Sung, V. C. W. Wong and K. Y. Yuen, Hong Kong Med. J., 2003, 9, 399–406 CAS.
- B. Cao, Y. Wang, D. Wen, W. Liu, J. Wang, G. Fan, L. Ruan, B. Song, Y. Cai, M. Wei, X. Li, J. Xia, N. Chen, J. Xiang, T. Yu, T. Bai, X. Xie, L. Zhang, C. Li, Y. Yuan, H. Chen, H. Li, H. Huang, S. Tu, F. Gong, Y. Liu, Y. Wei, C. Dong, F. Zhou, X. Gu, J. Xu, Z. Liu, Y. Zhang, H. Li, L. Shang, K. Wang, K. Li, X. Zhou, X. Dong, Z. Qu, S. Lu, X. Hu, S. Ruan, S. Luo, J. Wu, L. Peng, F. Cheng, L. Pan, J. Zou, C. Jia, J. Wang, X. Liu, S. Wang, X. Wu, Q. Ge, J. He, H. Zhan, F. Qiu, L. Guo, C. Huang, T. Jaki, F. G. Hayden, P. W. Horby, D. Zhang and C. Wang, N. Engl. J. Med., 2020, 382(19), 1787–1799 CrossRef PubMed.
- Y. M. Báez-Santos, S. J. Barraza, M. W. Wilson, M. P. Agius, A. M. Mielech, N. M. Davis, S. C. Baker, S. D. Larsen and A. D. Mesecar, J. Med. Chem., 2014, 57, 2393–2412 CrossRef PubMed.
- K. Ratia, S. Pegan, J. Takayama, K. Sleeman, M. Coughlin, S. Baliji, R. Chaudhuri, W. Fu, B. S. Prabhakar, M. E. Johnson, S. C. Baker, A. K. Ghosh and A. D. Mesecar, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16119–16124 CrossRef CAS PubMed.
- C. M. Booth, L. M. Matukas, G. A. Tomlinson, A. R. Rachlis, D. B. Rose, H. A. Dwosh, S. L. Walmsley, T. Mazzulli, M. Avendano, P. Derkach and I. E. Ephtimios, J. Am. Med. Assoc., 2003, 289, 2801–2809 CrossRef CAS PubMed.
- J. A. Tanner, B. J. Zheng, J. Zhou, R. M. Watt, J. Q. Jiang, K. L. Wong, Y. P. Lin, L. Y. Lu, M. L. He, H. F. Kung, A. J. Kesel and J. D. Huang, Chem. Biol., 2005, 12, 303–311 CrossRef CAS PubMed.
- L. Wilson, P. Gage and G. Ewart, Virology, 2006, 353, 294–306 CrossRef CAS PubMed.
- W. Li, M. J. Moore, N. Vasllieva, J. Sui, S. K. Wong, M. A. Berne, M. Somasundaran, J. L. Sullivan, K. Luzuriaga, T. C. Greeneugh, H. Choe and M. Farzan, Nature, 2003, 426, 450–454 CrossRef CAS PubMed.
- J. Gao, G. Lu, J. Qi, Y. Li, Y. Wu, Y. Deng, H. Geng, H. Li, Q. Wang, H. Xiao, W. Tan, J. Yan and G. F. Gao, J. Virol., 2013, 87, 13134–13140 CrossRef CAS PubMed.
- Z. Qian, S. R. Dominguez and K. V. Holmes, PLoS One, 2013, 8, e76469 CrossRef CAS PubMed.
- K. Shirato, M. Kawase and S. Matsuyama, J. Virol., 2013, 87, 12552–12561 CrossRef CAS PubMed.
- C. Burkard, M. H. Verheije, B. L. Haagmans, F. J. van Kuppeveld, P. J. M. Rottier, B.-J. Bosch and C. A. M. de Haan, J. Virol., 2015, 89, 4434–4448 CrossRef CAS PubMed.
- A. K. Ghosh, K. Xi, M. E. Johnson, S. C. Baker and A. D. Mesecar, Annu. Rep. Med. Chem., 2006, 41, 183–196 CAS.
- L. Yi, Z. Li, K. Yuan, X. Qu, J. Chen, G. Wang, H. Zhang, H. Luo, L. Zhu, P. Jiang, L. Chen, Y. Shen, M. Luo, G. Zuo, J. Hu, D. Duan, Y. Nie, X. Shi, W. Wang, Y. Han, T. Li, Y. Liu, M. Ding, H. Deng and X. Xu, J. Virol., 2004, 78, 11334–11339 CrossRef CAS PubMed.
- T. Y. Ho, S. L. Wu, J. C. Chen, C. C. Li and C. Y. Hsiang, Antiviral Res., 2007, 74, 92–101 CrossRef CAS PubMed.
- A. O. Adedeji, W. Severson, C. Jonsson, K. Singh, S. R. Weiss and S. G. Sarafianos, J. Virol., 2013, 87, 8017–8028 CrossRef CAS PubMed.
- C. W. Lin, F. J. Tsai, C. H. Tsai, C. C. Lai, L. Wan, T. Y. Ho, C. C. Hsieh and P. D. L. Chao, Antiviral Res., 2005, 68, 36–42 CrossRef CAS PubMed.
- L. Zhou, Y. Liu, W. Zhang, P. Wei, C. Huang, J. Pei, Y. Yuan and L. Lai, J. Med. Chem., 2006, 49, 3440–3443 CrossRef CAS PubMed.
- C. N. Chen, C. P. C. Lin, K. K. Huang, W. C. Chen, H. P. Hsieh, P. H. Liang and J. T. A. Hsu, Evid. base Compl. Alternative Med., 2005, 2, 209–215 CrossRef PubMed.
- N. Fusetani, M. Fujita, Y. Nakao, S. Matsunaga and R. W. Van Soest, Bioorg. Med. Chem. Lett., 1999, 9, 3397–3402 CrossRef CAS PubMed.
- Y. akao, M. Fujita, K. Warabi, S. Matsunaga, N. Fusetani and A. Miraziridine, J. Am. Chem. Soc., 2000, 122, 10462–10463 CrossRef.
- S. Yang, S. J. Chen, M. F. Hsu, J. D. Wu, C. T. K. Tseng, Y. F. Liu and W. C. Chen, J. Med. Chem., 2006, 49, 4971–4980 CrossRef CAS PubMed.
- L. Chen, J. Li, C. Luo, H. Liu, W. Xu, G. Chen and H. Jiang, Bioorg. Med. Chem., 2006, 14, 8295–8306 CrossRef CAS PubMed.
- Y. B. Ryu, H. J. Jeong, J. H. Kim, Y. M. Kim, J. Y. Park, D. Kim and M. C. Rho, Bioorg. Med. Chem., 2010, 18, 7940–7947 CrossRef CAS PubMed.
- A. O. Adedeji and S. G. Sarafianos, Curr. Opin. Virol., 2014, 8, 45–53 CrossRef CAS PubMed.
- J. Y. Park, J. H. Kim, J. M. Kwon, H. J. Kwon, H. J. Jeong, Y. M. Kim and Y. B. Ryu, Bioorg. Med. Chem., 2013, 21, 3730–3737 CrossRef CAS PubMed.
- T. T. H. Nguyen, H. J. Woo, H. K. Kang, V. D. Nguyen, Y. M. Kim, D. W. Kim and D. Kim, Biotechnol. Lett., 2012, 34, 831–838 CrossRef CAS PubMed.
- Y. Yang, M. S. Islam, J. Wang, Y. Li and X. Chen, Int. J. Biol. Sci., 2020, 16, 1708 CrossRef PubMed.
- M. S. Yu, J. Lee, J. M. Lee, Y. Kim, Y. W. Chin, J. G. Jee and Y. J. Jeong, Bioorg. Med. Chem. Lett., 2012, 22, 4049–4054 CrossRef CAS PubMed.
- C. C. Wen, Y. H. Kuo, J. T. Jan, P. H. Liang, S. Y. Wang, H. G. Liu and C. C. Hou, J. Med. Chem., 2007, 50, 4087–4095 CrossRef CAS PubMed.
- Y. B. Ryu, S. J. Park, Y. M. Kim, J. Y. Lee, W. D. Seo, J. S. Chang and W. S. Lee, Bioorg. Med. Chem. Lett., 2010, 20, 1873–1876 CrossRef CAS PubMed.
- P. Hamill, D. Hudson, R. Y. Kao, P. Chow, M. Raj, H. Xu and F. Jean, Biol. Chem., 2006, 387, 1063–1074 CAS.
- J. K. Cho, M. J. Curtis-Long, K. H. Lee, D. W. Kim, H. W. Ryu, H. J. Yuk and K. H. Park, Bioorg. Med. Chem., 2013, 21, 3051–3057 CrossRef CAS PubMed.
- J. Y. Park, J. H. Kim, Y. M. Kim, H. J. Jeong, D. W. Kim, H. H. Park and Y. B. Ryu, Bioorg. Med. Chem., 2012a, 20, 5928–5935 Search PubMed.
- J. Y. Park, H. J. Jeong, J. H. Kim, Y. M. Kim, S. J. Park, D. Kim and Y. B. Ryu, Biol. Pharm. Bull., 2012, 35, 2036–2042 CrossRef CAS PubMed.
- H. Wang, S. Xue, H. Yang and C. Chen, Virol. Sin., 2016, 31, 24–30 CrossRef CAS PubMed.
- J. Y. Park, J. A. Ko, D. W. Kim, Y. M. Kim, H. J. Kwon, H. J. Jeong and Y. B. Ryu, J. Enzyme Inhib. Med. Chem., 2016, 31, 23–30 CrossRef CAS PubMed.
- J. Y. Park, H. J. Yuk, H. W. Ryu, S. H. Lim, K. S. Kim, K. H. Park, Y. B. Ryu and W. S. Lee, J. Enzyme Inhib. Med. Chem., 2017, 32, 504–512 CrossRef CAS PubMed.
- Y. C. Tsai, C. L. Lee, H. R. Yen, Y. S. Chang, Y. P. Lin, S. H. Huang and C. W. Lin, Biomolecules, 2020, 10, 366 CrossRef CAS PubMed.
- Y. H. Song, D. W. Kim, M. J. Curtis-Long, H. J. Yuk, Y. Wang, N. Zhuang and K. H. Park, Biol. Pharm. Bull., 2014, 37, 1021–1028 CrossRef CAS PubMed.
- D. W. Kim, K. H. Seo, M. J. Curtis-Long, K. Y. Oh, J. W. Oh, J. K. Cho and K. H. Park, J. Enzyme Inhib. Med. Chem., 2014, 29, 59–63 CrossRef CAS PubMed.
- J. y. Park, H. J. Yuk, H. W. Ryu, S. H. Lim, K. S. Kim, K. H. Park and W. S. Lee, J. Enzyme Inhib. Med. Chem., 2017, 32, 504–512 CrossRef CAS.
- S. Schwarz, D. Sauter, K. Wang, R. Zhang, B. Sun, A. Karioti and W. Schwarz, Planta Med., 2014, 80, 177–182 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discov. Today Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- G. Wolber and T. Langer, J. Chem. Inf. Model, 2005, 45, 160–169 CrossRef CAS PubMed.
- M. T. ul Qamar, S. M. Alqahtani, M. A. Alamri and L. L. Chen, J. Pharm. Anal., 2020 DOI:10.1016/j.jpha.2020.03.009.
- V. Neveu, J. Perez-Jiménez, F. Vos, V. Crespy, L. du Chaffaut, L. Mennen and A. Scalbert, Database, 2010, 2010, bap024 CrossRef CAS PubMed.
- M. T. Islam, C. Sarkar, D. M. El-Kersh, S. Jamaddar, S. J. Uddin, J. A. Shilpi and M. S. Mubarak, Phytother Res., 2020, 1–22 CAS.
- O. S. Martins, I. A. Santos, D. M. Oliveira, R. V. Grosche and A. C. G. Jardim, Viruses, 2020, 12, 272 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04199h |
| This journal is © The Royal Society of Chemistry 2020 |