
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisulfide based prodrugs for cancer therapy
Qiang Wang
a,
Jiankun Guana,
Jiangling Wana and
Zifu Li
 *ab
*ab
aNational Engineering Research Center for Nanomedicine, College of Life Science and Technology, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan, 430074, China. E-mail: zifuli@hust.edu.cn
bHubei Key Laboratory of Bioinorganic Chemistry and Materia Medical, Huazhong University of Science and Technology, Wuhan, 430074, China
First published on 25th June 2020
Abstract
Advances in the tumor microenvironment have facilitated the development of novel anticancer drugs and delivery vehicles for improved therapeutic efficacy and decreased side effects. Disulfide bonds with unique chemical and biophysical properties can be used as cleavable linkers for the delivery of chemotherapeutic drugs. Accordingly, small molecule-, peptide-, polymer- and protein-based multifunctional prodrugs bearing cleavable disulfide bonds are well accepted in clinical settings. Herein, we first briefly introduce a number of prodrugs and divide them into three categories, namely, disulfide-containing small molecule conjugates, disulfide-containing cytotoxic agent–targeted fluorescent agent conjugates, and disulfide-containing cytotoxic agent–macromolecule conjugates. Then, we discuss the complex redox environment and the underlying mechanism of free drug release from disulfide based prodrugs in in vivo settings. Based on these insights, we analyze the impact of electronics, steric hindrance and substituent position of the disulfide linker on the extracellular stability and intracellular cleavage rate of disulfide containing prodrugs. Current challenges and future opportunities for the disulfide linker are provided at the end.
 Qiang Wang | Qiang Wang is currently a doctoral candidate under the supervision of Prof. Zifu Li at Huazhong University of Science and Technology. He received his MS degree in Medicinal Chemistry at Hubei University of Chinese Medicine in 2019. His current research interests focus on the hydroxyethyl starch based smart nanomedicines. |
 Zifu Li | Professor Zifu Li received B.S. degree at Huazhong University of Science and Technology in 2008 and PhD degree at the Chinese University of Hong Kong in 2012. From 2013 to 2015, he worked as a postdoctoral fellow at University of Alberta. He then joined in Georgia Institute of Technology as a research scientist. Since 2016, he has been a full professor at Huazhong University of Science and Technology. His group studies mechano-nanooncology and smart nanomedicine. |
1. Introduction
Prodrugs are molecules with little or no biological activity that can be metabolized into biologically active parent drugs in the body through enzymatic or chemical reactions or a combination of both.1–3 Over the past decade, prodrugs have accounted for more than 10% of the approved new chemical entities per year, making an amazing contribution to the arsenal of fighting disease.3 An attractive prodrug design strategy is to combine two or more different functional motifs with cleavable linkers. The rationale for using such a prodrug is to take advantage of the potential synergistic or targeted effects of multi-component prodrugs, thereby improving pharmacokinetics and reducing toxicity.4–9 There are several distinctive strategies to selectively cleave the linker and release the parent drugs. Some take advantage of unique aspects of disease pathophysiology, while others are based on disease-specific delivery technologies. A typical example of prodrug is the antibacterial agent Sultamicillin®, which consists of an irreversible β-lactam antibiotic ampicillin, the β-lactamase inhibitor penicillanic acid and a diester bond, and is simultaneously hydrolysed in vivo to release two drugs that effectively address the issue of bacterial resistance.9 Inspired by the multifunctionality of prodrugs, such as improved targeting properties and reversed multidrug resistance, the prodrug strategy that combine chemical units with different functions into a new chemical entity has become a robust solution to improving disease treatment.Cancer is regarded as an abnormal tissue with complex biology and a specific microenvironment, and displays complicated but unique characteristics, including mild acidity, high reductive potential and hypoxia.10–12 Advances in understanding the unique pathophysiological microenvironment of cancer have enabled the progress from conventional chemotherapy to smart multifunctional prodrugs, resulting in better therapeutic efficacy and alleviated side effects.
Disulfide bonds are the most important redox-reactive covalent bonds, formed by two cysteine residues in proteins. Disulfide bonds have already been widely found in proteins and play an important role in several important biological processes. Their key function is to accurately guide protein folding and enhance the stability of its tertiary and quaternary structures.13 Disulfide bonds can be used as cellular redox switches, involved in signal transmission through cascade reaction of thiol–disulfide conversion. With regard to the redox processes in vivo, the thiol pools in different biological compartments determine the redox-biological fate. It mainly includes glutathione/glutathione disulfide (GSH/GSSG), cysteine/cystine (Cys/CySS), thioredoxin-1 (Trx1), glutaredoxin (Grx) and protein disulfide isomerase (PDI). Furthermore, both the components and concentrations of thiol pools are largely different from the blood vessels to the intracellular environment.14 In plasma, the main thiol species is human serum albumin (HSA, 66.5 kDa) (∼422 μM). HSA's 585 amino acids residues possess 17 disulfide bridges and only one free thiol at Cys-34, which provides more than 80% of the free thiols in plasma.14,15 However, Cys-34 is located in a crevice with limited solvent exposure, severely hindering thiol–disulfide conversion.16 In contrast to the low free thiol concentration in plasma, GSH, an cysteine-containing tripeptide, is on average 1–10 mM in the cytoplasm. Moreover, tumor cells with active metabolism typically exhibit an elevated production of GSH in the cytoplasm.17 Therefore, the different thiol pools and the large differences in redox potential from blood vessels to the intracellular environment provide prerequisites for the specific drug release of disulfide-containing prodrug systems. Inspired by its chemical properties and functional roles, disulfide bonds have been used as candidate cleavable linkers in antitumor prodrug design. Connecting chemical units of different functions with disulfide bonds can form multifunctional anticancer prodrugs and achieve tumor-specific release.
In this review, we will briefly summarize few promising disulfide-containing prodrugs, highlighting several key issues in the research and development of disulfide-containing prodrugs: (a) is the release of the active ingredients tumor specific? (b) Which factors affect the self-immolation of disulfide bonds in vivo? (c) What are the current challenges of disulfide-containing prodrugs in clinical translation and their future development?
2. Disulfide-containing prodrugs
Chemotherapy is the most common cancer treatment. However, it suffers from a number of limitations, including low delivery efficiency and serious side effects.18,19 Therefore, targeted drug delivery systems have been extensively pursued. To this end, disulfide-containing prodrugs will be discussed in details. Based on the functional differences of the units on both sides of the disulfide bridge, we will divide them into disulfide-containing small molecule self-assembled nanomedicines, which are prepared from drug–drug conjugates, disulfide-containing targeting prodrugs, and disulfide-containing cytotoxic agent–macromolecule conjugates.2.1 Disulfide-containing small molecule self-assembled nanomedicines
Compared with small organic cytotoxic drugs, such as doxorubicin (DOX), paclitaxel (PTX) and camptothecin (CPT) or their derivatives, self-assembled nanoparticles prepared from drug–drug conjugates have significantly addressed the issues associated with parent drugs, such as low solubility and off-target toxicity. It was reported that disulfide bonds had a distinct preference for dihedral angles close to 90°. Such three-dimensional structure could better balance the intermolecular forces and promote the self-assembly of prodrugs into small-molecule nanomedicines rather than crystal formation caused by co-precipitation.20 As a result, various self-assembled nanoparticles have been prepared from disulfide-containing small molecule conjugates.DOX, an anthraquinone derivative, has multiple modifiable sites on the structure, but the three sites that are commonly used for derivatization are the amino group on the glycoside, the carbonyl group, and the C-14 hydroxyl group. To clarify the effect of linkers on the self-assembly and antitumor efficacy of DOX dimers, Zhang et al.21 designed and synthesized six DOX dimers with different linker types, lengths and linkage sites. The authors found that disulfide-containing prodrug modified with C-14 hydroxyl group exhibited higher antitumor efficacy than the modification of the amino group. Interestingly, among the six different DOX dimers, nanoparticles prepared from prodrug 1 (Fig. 1A) showed the highest release ratio in MCF-7 cells and the most effective antitumor efficacy in MCF-7 xenograft tumors. These results highlight that the antitumor efficacy of redox-selective DOX dimers are highly dependent on the modification site of DOX.
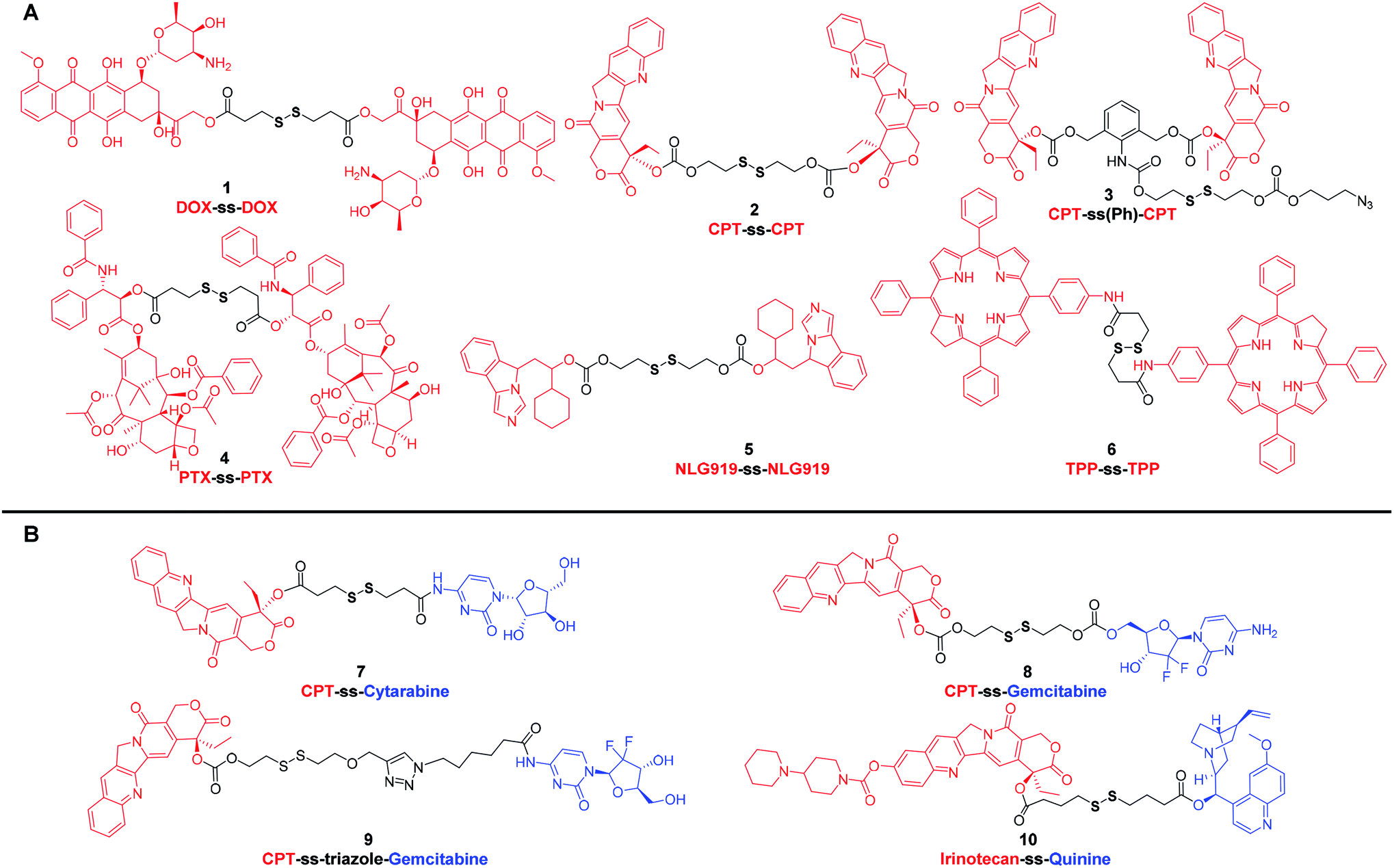 | ||
| Fig. 1 Chemical structures of disulfide-containing drug–drug conjugates. (A) Homodimers. (B) Heterodimers. | ||
CPT, an effective broad-spectrum DNA topoisomerase I inhibitor, exhibits potent toxicity against various tumor cells. However, CPT has a very low aqueous solubility. Many marketed CPT derivatives, such as topotecan and irinotecan, have thus been developed for improved pharmacokinetics. In addition to modification via introducing hydrophilic groups, nano drug delivery systems have also been extensively pursued for the delivery of CPT. Nonetheless, the conventional nanocarriers, such as liposomes,22 micelles,23 dendrimers,24,25 hyperbranched polymers,26 inorganic nanoparticle27 and so on, suffer from several drawbacks, including complicated synthesis, uncontrollable structure, low drug loading capacity, high reticuloendothelial system (RES) accumulation and potential immunogenic response.28 These deficiencies mirror the current state of the limited number of marketed nanomedicines. Carrier-free nanomedicines prepared with small molecule prodrugs have a defined structure and high drug-loading capacity and can be developed as next generation of drug delivery system. In 2015, Xie et al.29 reported a carrier-free nanomedicine prepared from CPT dimer. The disulfide bond bridged prodrug, CPT-ss-CPT (2) (Fig. 1A), was demonstrated to have reduction-responsive cytotoxicity toward HeLa cells, and the half-inhibitory concentration (IC50) was around 2 μM. In another instance, Cheng et al.30 employed a new disulfide-containing linker bearing σ bonds that could rotate freely to avoid the formation of large drug dimer particles. The resultant prodrug, CPT-ss(Ph)-CPT (3) (Fig. 1A), can also be easily cleaved and subsequently release the authentic form of CPT in the reducible environment. Compared with 2, 3 showed higher antiproliferative activity. The IC50 of the nanoparticles assembled from prodrug 3 on HeLa cells was 114 nM, much lower than 2. From these results, it is apparent that even if prodrug systems have the same cytotoxic payload, disulfide-containing linkers can affect their antitumor activity.
PTX is another widely used anticancer drug in clinic due to its potent antitumor activity against a broad range of solid tumors. Considering that the strong hydrophobicity reduces the amount of PTX reaching the tumor site, various types of nano-formulations have been developed. Abraxane®, an FDA-approved PTX nanoscale formulation, has been proposed to decrease systemic toxicity and enhance antitumor efficacy through enrichment at the tumor site based on the enhanced permeability and retention (EPR) effect.31 In addition to developing new nano drug delivery systems, efforts have also been devoted to obtaining PTX dimers self-assembling carrier-free nanomedicines.32–34 For instance, Xie et al.32 reported a series of nanomedicines prepared from PTX dimers with different linkers for the treatment of cervix carcinoma. Compared with the nanoparticles assembled from PTX dimer with non-cleavable linker, the nanoparticles assembled by PTX dimer 4 (Fig. 1A) of reduction-responsive disulfide linker showed superior antitumor effect in various cell lines. These results highlight that cleavable disulfide-containing linkers could be a preferred choice in the designing of drug–drug conjugates.
In addition to the design of drug–drug conjugates based on classic chemotherapeutic drugs, immunomodulator–immunomodulator conjugates and photosensitizer–photosensitizer conjugates are also frequently reported. For example, Li et al.35 reported a method of delivering binary synergistic prodrug nanoparticles (BCPN) that can be activated by tumor microenvironment. Based on the effect that IDO inhibitors can enhance the efficacy of common chemotherapeutics, BCPN was composed of DiPt-ASlink-PEG2k, an oxaliplatin (OXA) prodrug with acid instability, and NLG919-ss-NLG919 5 (Fig. 1A), a NLG919 prodrug with reduction response. OXA enhanced tumor immunogenicity by inducing immunogenic cell death (ICD), while NLG919 inhibited immunosuppressive tumor microenvironment by inactivating IDO-1. The disulfide bond in 5 not only enhanced the assembly stability of BCPN to ensure the circulation stability, but also realized the stimuli-responsive drug release in vivo. In another work, Xie et al.36 developed a multifunctional compound 6 (Fig. 1A), a disulfide-containing chlorin dimer, which can effectively convert the absorbed light into singlet oxygen (1O2) and thermal energy for photodynamic therapy (PDT) and photothermal therapy (PTT), respectively. The nanoparticles prepared from these disulfide-containing chlorin dimers exhibited higher PDT and PTT activity than their porphyrin counterparts upon laser irradiation. Furthermore, the nanoparticles also possessed the ability of photoacoustic imaging. These results indicate that the disulfide bonds can be used as effective redox-sensitive groups to fabricate multifunctional drug–drug conjugates for realizing therapeutic effects and imaging capability into one molecule.
Multidrug resistance (MDR) has become one of the major obstacles for effective cancer therapy.37 In most clinical cases, combination therapy is therefore used to overcome or delay the development of MDR. The synergistic effect of drugs with different mechanisms can prevent a single drug from triggering drug resistance. Therefore, small molecular-derived nano drug delivery systems that co-encapsulate multiple anticancer drugs or MDR inhibitors have been extensively pursued.38 To that end, prodrug self-assemblies composed of cytotoxic molecular units with different mechanisms have received widespread attention due to their high drug loading capacity and precise structures.39–41 For instance, in the context of redox-reactive conjugates, a variety of CPT-disulfide-cytotoxic prodrugs have been investigated. These heterodimers (7–10) (Fig. 1B) consisting of different toxic units were designed as a class of mutual prodrugs with dual modes of action. After bioreduction in tumor cells with high GSH content, these mutual prodrugs were demonstrated to release CPT and another active ingredient for overcoming MDR.42–45 Taking conjugate 10 as an example, it consisted of marketed anticancer drug irinotecan and P-glycoprotein (P-gp) inhibitor quinine. Compared with irinotecan and quinine, the self-assembled irinotecan-ss-quinine nanoparticles could be concentrated in tumor site and displayed higher cytotoxicity against irinotecan-resistant MCF-7/ADR cells. In vitro and in vivo results corroborated that 10 underwent GSH-triggered drug release. The released free drug quinine could inhibit the P-gp mediated drugs efflux from MCF-7/ADR cells, resulting in increased intracellular accumulation of irinotecan. Therefore, the co-delivery strategy of multiple drugs presents an attractive approach that can markedly improve anticancer activity by overcoming MDR.
Based on the above-mentioned small molecule conjugates and their self-assembled nanomedicines, it is apparent that the disulfide-containing dimer design strategy can be universally applied to various organic molecules, including not only cytotoxic drugs, but also immunomodulators and photosensitizers. Compared with conventional chemotherapy, these prodrugs assembled nanoparticles, which are composed of multiple drugs, exhibit enhanced antitumor efficacy and have the potential to overcome MDR. However, these carrier-free nanomedicines also suffer from several key challenges: (a) it is a dilemma to balance the hydrophilicity–hydrophobicity of each conjugate. Compared with conventional nanocarriers, these conjugates assembled nanomedicines might be less stable. Thus, they need to incorporate a small amount of amphiphilic macromolecular excipients, such as methoxypoly (ethylene glycol)-block-polylactide (mPEG-PLA), to solve this problem. Or it may work, by rationally designing the disulfide containing linker with increased hydrophilicity to improve the stability of the prodrugs assembled nanomedicines, but there has been no such report thus far. And (b) compared with carrier-assisted nano drug delivery systems, these carrier-free nanomedicines, consisting of drug conjugates solely, may have difficulties in modulating the charge, size and other key parameters.
2.2 Disulfide-containing targeting prodrugs
Although prodrug self-assembled nanoparticles containing disulfide bridges can increase the passive accumulation of drugs in tumor tissues through the EPR effect, they still suffer from insufficient delivery and cellular uptake. Multifunctional conjugates with active targeting ligands, such as biotin, folic acid, galactose, RGD peptide and antibodies, make it possible to have superior therapeutic efficacy. In this section, we will focus on a few cases rather than all the reported disulfide-containing targeting prodrugs.Folic acid (FA or Fol), due to its high affinity for folate receptor, which is ubiquitous in various tumors (such as breast, lung, kidney, and brain cancers), has become an ideal targeting ligand for selective delivery of therapeutic agents.46,47 Targeting agents consisting of Fol or its analog methotrexate (MTX) have been leveraged for the delivery of low molecular weight chemotherapeutic agents, immunotherapeutic agents, polymers, biomacromolecules (such as protein, enzyme and small interfering RNA) and so on.46,48 As an example, Epofolate (11) (Fig. 2A), also known as BMS-753493, is a novel epothilone–Fol conjugate linked through the self-immolating disulfide linker. Animal studies confirmed that 11 exhibited excellent selectivity for targeting FR over-expressing tumors. The safety, efficacy and pharmacokinetics of 11 had also been evaluated in patients with advanced solid tumors. Unfortunately, no progressing results have been posted after the phase I/II clinical trial initiated in 2007.49,50 Another example of this type of disulfide-containing targeting agent, namely Vintafolide (EC145, MK8109, 12) (Fig. 2A), exploited vinca alkaloid (desacetylvinblastine hydrazide; DAVBLH) as the cytotoxic agent and Fol as the targeting unit. In phase I clinical trial, 12 was found to possess an acceptable safety profile with rapid clearance through the kidneys and liver. Phase II investigations provided support for the notion that patients with platinum-resistant ovarian cancer were likely to benefit from 12. Unfortunately, in phase III, 12 failed to demonstrate a significant improvement on Progression-Free Survival (PFS) in patients with platinum-resistant ovarian cancer.51,52 With the selectivity and bioreductive cytotoxicity are realized by Fol and disulfide bond, respectively, many similar prodrugs have also been developed in preclinical studies. In 2011, Perez et al.53 reported a DOX-ss-Fol (13) (Fig. 2A) conjugate to reduce the systemic toxicity associated with DOX. The disulfide linker technology not only linked Fol to DOX, which made the specific internalization of DOX in folate receptor positive cells, but also ensured the subsequent redox-response DOX release in GSH-high cells. This targeted drug delivery strategy reduced the toxicity to normal tissues. In addition, Fol can also act as a fluorescent quencher for DOX. After incubating in GSH, the fluorescence of 13 increased 5-fold. These results corroborated that 13 showed significant cytotoxicity in Fol receptor positive cells and allowed the simultaneous tracking of drug release. Another example is podophyllotoxin (PPT), a microtubules disruptor, which has been widely used as a lead compound for further development of anticancer drugs, such as etoposide, teniposide and etoposide phosphate. Recently, one study involved PPT wherein PPT was conjugated to MTX via a disulfide bond. The obtained PPT-ss-MTX (14) (Fig. 2A) displayed significant cytotoxicity in folate receptor-positive KB cells while the self-assembled nanoparticles could suppress 4T1 xenograft tumors in BALB/c mice.54
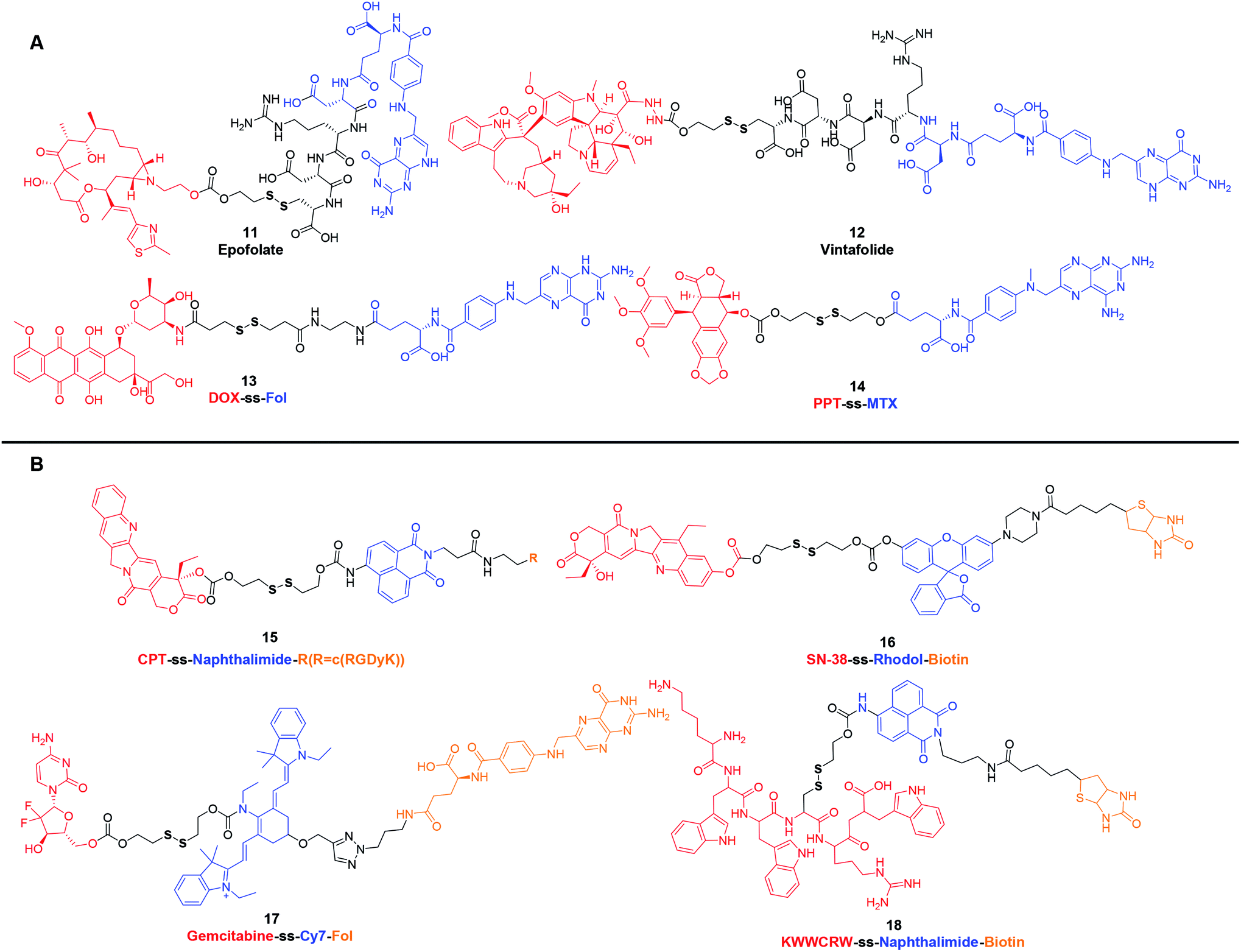 | ||
| Fig. 2 Structures of disulfide-containing multifunctional conjugates. (A) Fol or its analogue based targeting agents. (B) Theranostic agents. | ||
Another class of disulfide-containing targeting prodrugs are theranostic agents, which combine therapeutic effects and diagnostic functions within one defined structure. The general structural form of these multicomponent complexes is cytotoxic agent–disulfide–fluorescent reporter-tumor targeting ligand. Disulfide linkers enable these prodrugs to initiate theranostics in reducible-responsive way. These complexes can selectively deliver chemotherapeutic agents to tumors and generate special signals that can be easily monitored both in vitro and in vivo. To date, a variety of disulfide-containing targeting theranostic agents have been developed. Most of them employed traditional chemotherapeutic agents as cytotoxic drugs, such as DOX, CPT, PTX and gemcitabine (GEM), fluorophores as reporters, including naphthalimide, coumarin, BODIPY, rhodol, and Cy7, and site-specific entities as tumor targeting ligands, such as Fol, biotin, galactose and RGD peptide.55–58 Kim et al. has pioneered in designing and synthesizing new chemical entities to improve traditional chemotherapy. In 2015 and 2018, they systematically reviewed the disulfide-containing theranostic systems that were developed by their group.55,59 Here, we showcase several representative examples of these reductively driven theranostic agents. For instance, CPT-ss-naphthalimide-cRGDyK (15) (Fig. 2B) was composed of a CPT, a naphthalimide moiety, an RGD cyclic peptide, and disulfide-containing linker. These motifs were expected to act as the anticancer drug, an imaging reporter, a tumor targeting ligand and a GSH triggering cleavable linker, respectively. In this system, reactions between 15 and free thiols led to the disulfide cleavage. This dissociation released the parent drug CPT, and the fluorescence emission band was red-shifted from 473 to 535 nm, which enabled conjugate 15 to track CPT release at the subcellular level.60 In another study, SN-38-ss-rhodol-biotin (16) (Fig. 2B) included a fluorescent rhodol as a reporter unit that was tethered via a disulfide linker. Prodrug 16 exhibited a significantly increased fluorescence intensity, approximately 32-fold, upon GSH-mediated disulfide bond cleavage. The strong enhancement in the fluorescence intensity provided an opportunity to detect when, where, and how the CPT was delivered and released inside cells.61 For in vivo applications, the development of near-infrared (NIR) reporting system which is capable of deep tissue penetration is potentially useful. To that end, Kim et al. made efforts to develop a theranostic prodrug, namely Gem-ss-Cy7-Fol (17) (Fig. 2B), with an appended Cy7 as a NIR fluorescence reporter. The fluorescence intensity of the emission band at 735 nm increased with increasing concentration of GSH. The change in the fluorescence intensity of prodrug 17 provided an opportunity for real-time self-monitoring of drug release in vivo.62 Another investigation involved peptide-based prodrug KWWCRW-ss-naphthalimide-biotin (18) (Fig. 2B) wherein a Holliday junction (HJ) inhibitor peptide2 (KWWCRW) was conjugated to the naphthalimide via a disulfide linker. Similar to 15, prodrug 18 exhibited enhanced intracellular fluorescence upon GSH-mediated disulfide bond cleavage in HepG2 cells, and it also possessed the targeting effect of selective uptake by biotin-receptor-positive cells.63
It is evident that the above-mentioned disulfide-containing active targeted prodrugs hold great potential in future cancer therapy. Because of their well-defined structure, low cost, and easy to combine with versatile cytotoxic agents and fluorescent reporters, we anticipate that many more prodrugs as such will be reported. However, successful development of these targeted prodrug systems requires further consideration of more complex factors that affect the pharmacokinetics in vivo. Taking theranostic agents 15–18 as an example, they suffer from several grand challenges in clinical translation: (a) compared with conventional chemotherapeutic drugs, these new series of theranostic prodrugs generally exhibit lower cytotoxicity towards cancer cells, resulting in insufficient therapeutic efficacy. (b) The molecular weight of resultant targeted prodrugs is too large, which may lead to unsatisfactory pharmacokinetics. (c) Due to the lack of optimization of the disulfide-containing linker, difficulties exist in non-specific biotransformation of theranostic prodrugs to release parent drugs. Steric hindrance of the carbon atom around the disulfide bond greatly affects the self-immolation rate of the disulfide bond.64 Considering these key challenges, optimal multifunctional prodrugs should be explored to overcome the reduced toxicity. Moreover, it is of great importance to clarify the difference in the redox environment from blood vessels to tumor cells. An accurate and detailed understanding of the redox environment in human body is instructive in exploiting disulfide containing linkers. Varied disulfide based bonds exhibit different extracellular stability and intracellular cleavage rate, both of which are key parameters for successful prodrug design. Furthermore, the working mechanism of these prodrugs may be intrinsically different from their parent drugs, it is necessary to study the interactions between cancer cells and these new theranostic prodrugs in terms of intracellular transportation, metabolism and deposition site.
2.3 Disulfide-containing cytotoxic agent–macromolecule conjugates
Because of the strong affinity between antibody and antigens that are highly expressed on cancer cells, antibody–drug conjugates (ADCs), monoclonal antibodies conjugated with cytotoxic small molecule payload through chemical linkers, can specifically and efficiently deliver potent cytotoxic compounds to cancer cells. In recent years, booming research pipelines together with various FDA approved ADCs, such as moxetumomab pasudotox, polatuzumab vedotin and fam-trastuzumab-deruxtecan-nxki, implicate that this drug delivery platform can be used for next-generation of oncotherapy.65,66 Among the three components of ADCs, the linker chemistries determine the therapeutic window. Successful linker technology should prevent the premature release of the toxic payload in bloodstream and at the meantime ensure that the free toxic payload is released at the intended disease site at an adequate rate. Therefore, tremendous effort has been invested in the conjugation and linker chemistries in ADC field.65 Based on different mechanisms for release of the toxic payload, the linker strategies fall into two main classes: cleavable and non-cleavable linkers. Disulfide bond within the linker, due to its relative stability in circulation and efficient free drug release rate at tumor site, is one of the most recognized and attractive strategies for cleavable linkers in ADC design, along with hydrazone. The first disulfide-containing ADC, namely gemtuzumab ozogamicin (Mylotarg®, Table 1),67 was developed by Pfizer and was approved by FDA in 2000 for treatment of patients with relapsed CD33-positive acute myeloid leukemia. Mylotarg® is a pH and reduction dual-sensitive ADC for delivering calicheamicin derivative, a potent antitumor antibiotic. Disulfide and N-acyl hydrazine linkage were employed to achieve reduction and acid-sensitivity, respectively. Mylotarg®'s drug-to-antibody ratio (DAR) value, a key parameter to impact PK/PD and cytotoxicity of ADCs, is 2–3 (typically 3–4 as a major species).68 Tracing back the development of Mylotarg®, the process is long and has experienced a lot of ups and downs. In 2010, Mylotarg® was withdrawn from market due to a higher rate of fatal toxicities compared to standard chemotherapy. Fortunately, Mylotarg® was reapproved as induction therapy for treatment of relapsed or refractory acute myeloid leukemia (AML) in patients 2 years of age and older with a modified fractionated dosing regimen in 2017. The tortuous development process of Mylotarg® highlights the significance of understanding the relationship between premature drug release and a reasonable dosing regimen in early stages of clinical development. Similar to Mylotarg®, this dual-sensitive linker was also employed in inotuzumab ozogamicin (Besponsa®, Table 1), another FDA-approved ADC for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. The structure of Besponsa® involves a CD22-targeting monoclonal antibody for targeting a well-characterized antigen and calicheamicin derivative as the payload that causes DNA double-strand cleavage. The DAR value of Besponsa® is 4–7, a well-controlled range, which not only ensured potency but also prevented the risk of aggregation.68,69 Another approved ADC containing disulfide linker, namely moxetumomab pasudotox (Lumoxiti®, Table 1),70 was developed by MedImmune and its parent company AstraZeneca for relapsed or refractory hairy cell leukemia in 2018. In this system, disulfide bond was used to fuse Pseudomonas exotoxin A (payload) with the recombinant murine anti-CD22 monoclonal antibody. The resultant Lumoxiti® displayed significant clinical benefits in CD22-expressing hairy cell leukemia.| ADC | Target | Cytotoxic payload | Clinical phase | Indications | Sponsor (licensee) | Ref. |
|---|---|---|---|---|---|---|
| a STs, solid tumors. | ||||||
| Gemtuzumab ozogamicin (Mylotarg®) | CD33 | Calicheamicin derivative | Approved | CD33-positive AML; relapsed or refractory AML | Pfizer | 67 |
| Inotuzumab ozogamicin (Besponsa®) | CD22 | Calicheamicin derivative | Approved | Acute lymphoblastic leukaemia | Pfizer | 69 |
| Moxetumomab pasudotox (Lumoxiti®) | CD22 | Pseudomonas exotoxin A | Approved | Relapsed or refractory hairy cell leukemia | AstraZeneca | 70 |
| Mirvetuximab soravtansine (IMGN853) | FRα | DM4 | Phase III | Ovarian cancer | ImmunoGen | 71 |
| Coltuximab ravtansine (SAR3419) | CD19 | DM4 | Phase II | Diffuse large B-cell lymphoma | Sanofi | 73 and 74 |
| Lorvotuzumab mertansine (IMGN901) | CD56 | DM1 | Phase II stopped | Small cell lung cancer | ImmunoGen | 78 |
| AVE9633 | CD33 | DM4 | Phase I stopped | AML | Sanofi | 79 |
| Indatuximab ravtansine (BT-062) | CD138 | DM4 | Phase I | Multiple myeloma | Biotest | 80 |
| Anetumab ravtansine (Bay-94-9343) | Mesotherin | DM4 | Phase II | Pancreatic cancer | Bayer HealthCare | 81 |
| SAR-566658 | CA6 | DM4 | Phase I | CA6-positive advanced STsa | Sanofi | 82 |
| SAR408701 | CEACAM5 | DM4 | Phase I | Advanced STsa | Sanofi | 83 |
| SAR428926 | LAMP1 | DM4 | Phase I | Advanced STsa | Sanofi | 84 |
| HKT288 | Cadherin-6 | DM4 | Phase I | Epithelial ovarian cancer | Novartis Pharmaceuticals | 85 |
| Cantuzumab mertansine | CanAg | DM1 | Phase I stopped | CanAg-expressing advanced STsa | ImmunoGen | 86 |
| IMGN242 | CanAg | DM4 | Phase II | Gastric or gastroesophageal (GE) junction cancer | ImmunoGen | 87 |
| IMGN388 | Integrin αV | DM4 | Phase I | STsa | ImmunoGen | 88 |
| BIIB015 | Cripto | DM4 | Phase I | Relapsed/refractory STsa | Biogen | 89 |
Promoted by the clinical success of Mylotarg®, Besponsa® and Lumoxiti®, a growing number of disulfide-containing ADCs have been tested clinically. Here, we only present some typical ADCs with disulfide bond, which are being explored in clinical stage. Maytansinoids are an extremely potent class of antitubulin agents. Compared with traditional chemotherapy molecules, such as DOX, MTX, and vincristine, the cytotoxicity of maytansinoids are 100 to 1000 fold greater.71 Consequently, an enormous effort has been invested to develop ADCs that use maytansinoids as payloads. For example, mirvetuximab soravtansine (IMGN853) utilizes a potent maytansine analog DM4 and a disulfide linker to target folate receptor-α (FRα). Ongoing phase III monotherapy and Phase Ib/II combination trials have revealed that IMGN853 had good tolerance and potent efficacy on patients with platinum-resistant ovarian cancer.72 Coltuximab ravtansine (SAR3419), an anti-CD19-DM4 immunoconjugate with a cleavable disulfide linker, is also being evaluated in phase II studies for the treatment of relapsed or refractory diffuse large B-cell lymphoma.73,74 Moreover, by changing the antibodies, many other antibody–disulfide–maytansine complexes have also entered various clinical trials. The list of disulfide-containing ADCs being explored in the clinical stage is summarized in Table 1. Further explorations of this traceless disulfide technology are also carried out in preclinical studies. Aromatic rings and methyl groups are often employed adjacent to the disulfide bond. These ADCs exhibited a delicate balance between extracellular stability and intracellular cleavage efficiency. These types of linkers have been employed in marketed prodrug Mylotarg®, and more recently, in several promising candidates in preclinical or clinical trials.75–77
With recent advances in comprehension of the tumor microenvironment, a number of stimuli-responsive nanomedicines have been developed to enhance drug delivery efficiency and antitumor efficacy.90–96 Among them, reduction-sensitive nanomedicines are widely studied. By incorporating disulfide linkers into the polymeric backbone or side chains, many disulfide-containing reduction-responsive polymeric nanocarriers can be obtained. As an example, Xu et al.97 developed a heterotargeted nanococktail (PPPDMA). In the multidrug delivery system, DOX and PTX were connected to the side chains of the polymer through disulfide bonds. Once the PPPDMA was internalized into tumor cells, the released DOX and PTX would synergistically enhance the anticancer efficacy, and the disulfide linker ensured selective drug release, which alleviated the systemic toxicity of the drug cocktail. Similarly, numerous reduction-sensitive nanomedicines have been developed, as summarized by several excellent reviews.98–106 In particular, based on hydroxyethyl starch (HES), which has been widely used as blood plasma volume expander in clinical settings, our group has prepared a variety of HES-ss-drug conjugates for tumor targeting delivery (Fig. 3).107–110 For example, in a recent study, we prepared a theranostic nanoparticle DHP, which was composed of HES-ss-PTX conjugate and NIR cyanine fluorophore DiR.107 To our knowledge, DiR can be used for photoacoustic imaging and PTT. In DHP, the fluorescence of DiR is quenched by the aggregation caused quenching (ACQ) effect. After incubation with tumor cells with overexpressed GSH, we observed a simultaneous PTX release and DiR fluorescence recovery. These suggested that this disulfide based theranostic nanoparticle can be used as in vivo probe for both fluorescent and photoacoustic imaging and chemo–photothermal combination therapy. However, most of these disulfide based nanotherapeutics have been evaluated in laboratories so far, and there is still a large gap for clinical translation.
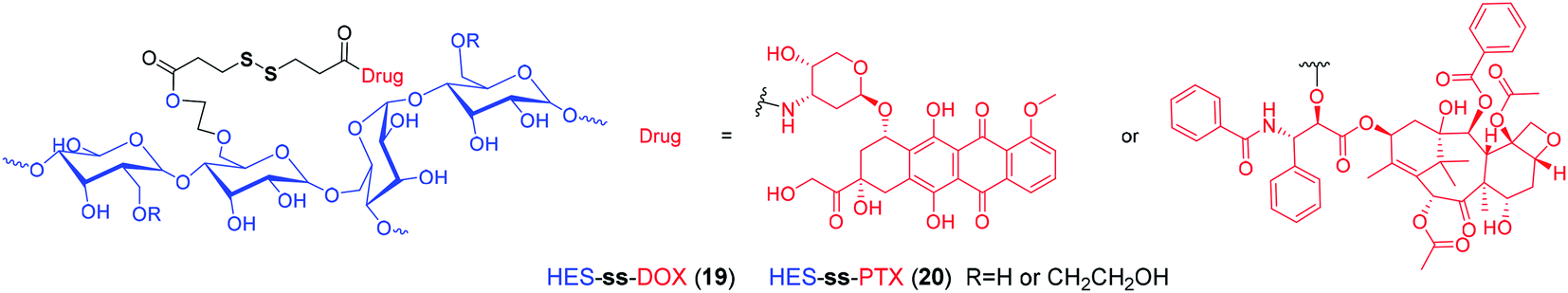 | ||
| Fig. 3 Structures of reduction-sensitive prodrugs based on HES. | ||
3. Optimization of disulfide linkers
For disulfide-containing prodrugs to be selective and potent, the cleavable disulfide linker should be optimized to achieve two key properties: (a) high stability in circulation to avoid premature drug release. And (b) rapid self-immolation rate in tumor cells to ensure adequate parent drug release. However, only a limited number of disulfide based targeting delivery systems have undergone stability evaluation in vivo. Most of them was only tested in buffer containing varied concentrations of GSH. This puts the widely-accepted notion that the cleavable disulfide linkers respond selectively in tumor cells into question. In fact, the redox environment in different compartments is an external factor that affects the selective drug release. However, due to the complex thiol pools in vivo and concentrations of these thiol pools ranging from μM to mM in different compartments, it is challenging to develop stable and potent disulfide-containing prodrugs for tumor targeting delivery.The structure of the prodrug itself constitutes an internal factor that affects the self-immolation kinetics of disulfide bonds. While drug release from disulfide-containing prodrugs could occur in vivo via enzymatic reaction or hydrolysis in low pH environment, the plausible mechanism for disulfide cleavage and immolation is proposed and presented in Fig. 4. Initially, an intracellular deprotonated thiol such as Cys-S− or GS− would attack the disulfide bond on the prodrug (I) to give the protonated or non-protonated intermediates (II, IV) and thiol-cysteine adduct (III). Then, IV may preferentially undergo cyclization reaction to give the free drug 1 (VI) (Path a), in which thiolate anion moiety of substrate IV reacts with its adjacent carbonyl group. Alternatively, the cyclization and subsequent decarboxylation can give the final product VI (Path b). Thiol-cysteine adduct (III), the other product of I after disulfide exchange reaction, can be reduced into anion V, via a nucleophilic attack of deprotonated thiol. Finally, VII is obtained through the cyclization (Path A) or hydrolysis catalysed by abundant hydrolytic enzymes or weak acidity in tumor microenvironment (Path B).77,111
 | ||
| Fig. 4 Plausible catabolism and free drug release mechanism of disulfide-containing prodrugs. RSH = Cys or GSH; RSSR = CySS or GSSG; R1 = cytotoxic agent 1; R2 = cytotoxic agent 2 or other functional small molecular (target agent, photosensitizer, immunomodulatory agent or peptide) or macromolecule (polymer or antibody); X = O or NH. | ||
Given the complex redox environment and multiple prodrug structures, which constitute the external and internal factors for the selective drug release, respectively, it is clear that the thiol–disulfide exchange reactions are greatly influenced by electronics, steric hindrance and substituent position of disulfide linker. Next, we will discuss these parameters one by one.
In 2018, Sun et al.112 demonstrated that the substituent position of disulfide bond exerted great impact on drug release and cytotoxicity. Three PTX-ss-citronellol (CIT) conjugates were designed and synthesized, namely α-PTX-ss-CIT (21), β-PTX-ss-CIT (22), and γ-PTX-ss-CIT (23), in which the sulfur atoms were located the α-, β-, or γ-position of the ester bond, respectively (Fig. 5A). The nanoassemblies of α-PTX-ss-CIT and γ-PTX-ss-CIT underwent rapid free drug release in the presence of 10 mM DTT which was similar to reductive potential in cytoplasm. In contrast, under the same condition, β-PTX-ss-CIT showed only 45% PTX release within 24 h. It was also found that disulfide bonds with different substituent positions had significantly affected cytotoxicity. Among all three conjugates, α-PTX-ss-CIT exhibited the most cytotoxic, which may be attributed to the rapid drug release when exposed to the redox environment of cancer cells. Nevertheless, the adjacent benzamide nitrogen of PTX may participate in the hydrolysis reaction by serving as a nucleophilic hydrogen bond/proton acceptor.113 A more detailed and quantitative evaluation is necessary to extend to other cytotoxic agents, such as DOX and CPT, and to clearly demonstrate whether the sulfur atoms on the α- of the ester bond is beneficial to parent drugs release.
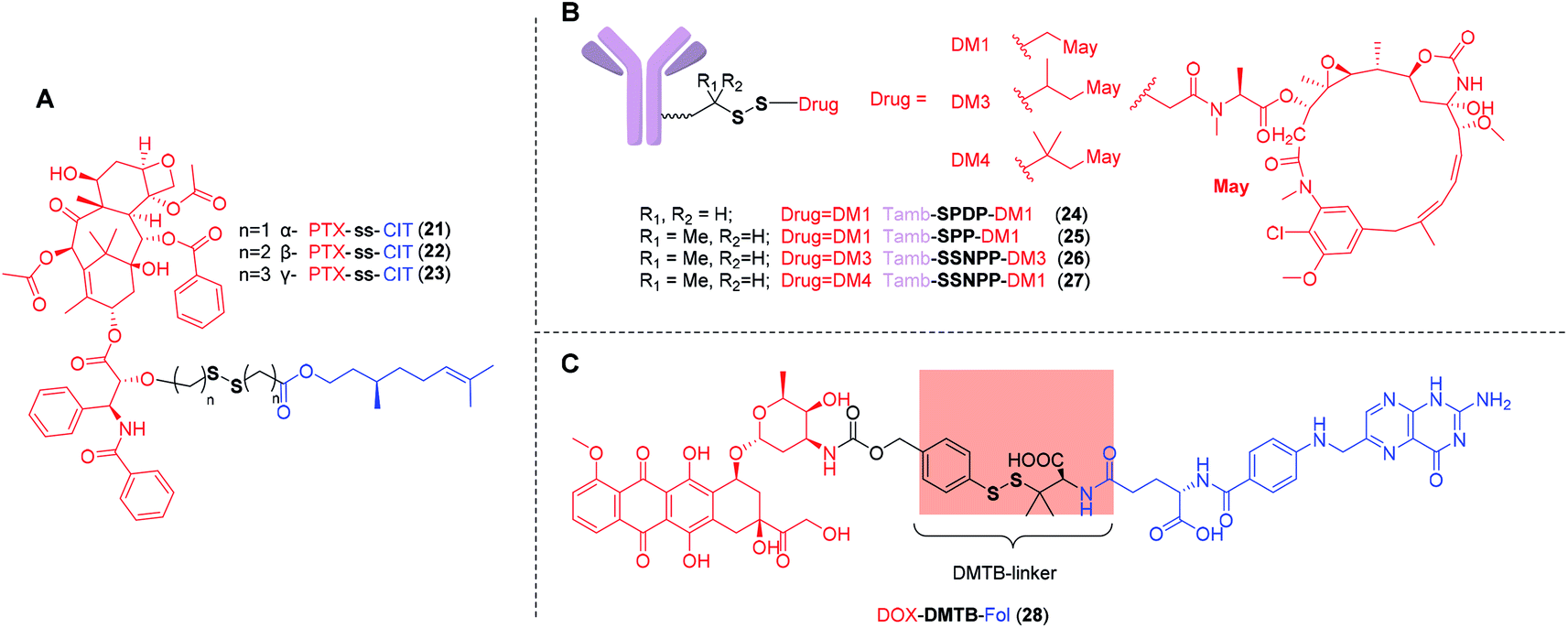 | ||
| Fig. 5 (A) Structures of PTX-ss-CIT with different substituent positions of disulfide bond containing carbon chain. (B) Tamb-ss-DM1 with different sterically hindered disulfide bonds. (C) Structure of DOX-DTMB-Fol with aromatic disulfide linker. | ||
Compared with relatively simplistic, unhindered disulfide linker, the disulfide bonds with steric protection are less sensitive to the redox environment in vivo, which is, in fact, beneficial to avoid premature release of drugs. However, the substituents on either side of the disulfide should not be too large. Phillips et al.114 reported a series of trastuzumab ADCs with different sterically hindered disulfide bonds (Fig. 5B). Pharmacokinetic analyses showed that Tamb-SPDP-DM1 with no adjacent methyl groups (24) showed the fastest plasma clearance rate and was undetectable by day 3. In contrast, Tamb-SPP-DM1 with one adjacent methyl group (25), Tamb-SSNPP-DM3 with two adjacent methyl groups (26), and Tamb-SSNPP-DM4 with three adjacent methyl group (27) displayed good stability and remained in the circulation after one week. Among them, a large amount of 27 could still be detected in the blood after 7 days, indicating that excessively large steric hindrance was also not conducive to maytansinoid release. In vivo efficacy evaluation showed that compared with 25 and 27, 26 displayed most potent efficacy in a resistant MMTV-HER2 Fo5 mammary tumour model, which may be ascribed to the moderate steric hindrance. These results underscore the necessity of optimizing the linkers to ensure the extracellular stability and cleavage efficiency in targeted cells. A suitable disulfide-containing linker can neither be crowded nor unhindered near the disulfide.
In another work, Wu et al.115 developed a new linker, α, α-dimethyl-substituted p-dithiobenzyl urethane (DMTB), which resulted in both increased extracellular disulfide stability and rapid intracellular self-immolation kinetics. Because the disulfide exchange reactions are initiated by nucleophilic attack of a deprotonated thiol (Fig. 4), the acidity is not conducive to deprotonation of thiol. One of the grand challenges for disulfide-containing prodrugs to release parent drugs is that the self-immolation can be slow under acidic sites such as tumor interstitium (pH ∼ 6.8) and endocytic or lysosomal compartments (pH 4–6). Compared with aliphatic thiol, aromatic thiol with the lower pKa (∼6) can be used to promote the exchange of thiol–disulfide. On the other hand, the stability of these resultant prodrugs was dramatic increased in GSH buffer by incorporating two methyl groups on one side of p-thiobenzyl disulfide linker. Conjugate DOX-DTMB-Fol (28) is a promising agent comprising a DOX, the cleavable α,α-dimethyl-substituted aromatic disulfide linker and a Fol (Fig. 5C). It was found 28 was completely converted into DOX within 5 h in the presence of 10 mM GSH at pH 7.4, suggesting that, with the DTMB linker, the cleavage efficiency was effective in the cytosol. In contrast, only 8% of free DOX was detected after 6 h incubation in 0.2 mM GSH buffer. The reduced GSH in buffer simulated the redox environment in blood circulation. However, compared with the simple simulated redox environment in vitro, a more detailed and quantitative evaluation in vivo is required to verify the high stability in plasma and rapid self-immolation kinetics at tumor site. Results from in vivo studies will help reveal the full potential of this novel disulfide-containing linker.
4. Summary and outlook
Driven by the successful disulfide based ADCs, including Mylotarg®, Besponsa® and Lumoxiti®, many more disulfide based prodrugs have been developed during the past decades. Compared with conventional chemotherapeutics, these prodrugs have the capability of selectively delivering therapeutic reagents into tumor cells. Moreover, by coupling fluorescent reporter units, some of these multifunctional prodrugs have achieved real-time drug tracking during circulation, which is of great significance to clarify their biodistribution. Development of the optimal prodrug may, however, need to account for the extracellular environment, including toxicity to normal tissues and stability in circulation. The premature free drug release in blood circulation or slow cleavage efficiency in tumor cells present clinical translation hurdles for disulfide based prodrugs. With the exponential growth in disulfide containing multifunctional prodrugs, the rational design and optimization of disulfide bonds should be followed up to overcome these obstacles. To this end, we systematically discuss the effects of the spatial structure and electronics around disulfide bonds on drug release in vivo, and propose the optimization direction of disulfide based bonds for selective self-immolation. These discussions hold great significance to the rational design of redox responsive prodrugs.Suitable linker technology is a prerequisite for safe and effective disulfide based prodrugs. Understanding the relationship between the linker and prodrug transformation is key to addressing the conflicting requirements in extracellular and intracellular conditions. The disulfide containing prodrugs should maintain integrity in plasma and delivery processes, but should be cleaved instantly for traceless parent drug release within tumor cells. Although it is believed that the increased thiol pools in tumor cells are conductive to selective drug release, the acidic environment in tumor interstitium or in endocytic and lysosomal compartments can slow down the rate of thiol–disulfide exchanges. Therefore, due to the differences in redox potential and pH from the blood vessel to the intracellular environment, it can be expected that the relatively simplistic, unhindered disulfide linkers may not meet the requirements for selective drug release. Based on the disulfide-containing prodrugs summarized in this review, it is evident that the extracellular stability and intracellular cleavage efficiency are critically regulated by the electronics, steric hindrance and substituent position of disulfide linker. Owing to the reasonable requirement for stability in circulation and rapid self-immolation kinetics in tumor cells, the next generation of disulfide linkers may have moderate steric protection.
Overall, due to the heterogeneous redox environments in different tissues, there remains a considerable unmet need to develop suitable disulfide linkers. With the insights obtained from the recent disulfide bond containing prodrugs, we are going to design a series of novel disulfide linkers and evaluate their redox selectivity both in vitro and in vivo for enhanced cancer chemotherapy.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by grants from National Science Foundation of China (31972927, and 31700867), Scientific Research Foundation of Huazhong University of Science and Technology (3004170130), and Program for HUST Academic Frontier Youth Team (2018QYTD01).References
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- V. Abet, F. Filace, J. Recio, J. Alvarez-Builla and C. Burgos, Eur. J. Med. Chem., 2017, 127, 810–827 CrossRef CAS PubMed.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- D. Bhosle, S. Bharambe, N. Gairola and S. S. Dhaneshwar, Indian J. Pharm. Sci., 2006, 68, 286–294 CrossRef CAS.
- A. Nudelman and A. Rephaeli, J. Med. Chem., 2000, 43, 2962–2966 CrossRef CAS PubMed.
- W. Kim, D. Kim, S. Jeong, S. Ju, H. Lee, S. Kim, J.-W. Yoo, I.-S. Yoon and Y. Jung, Pharmaceutics, 2019, 11, 585 CrossRef CAS PubMed.
- K. Harrap, P. Riches, E. Gilby, S. Sellwood, R. Wilkinson and I. Konyves, Eur. J. Cancer, 1977, 13, 873–881 CrossRef CAS PubMed.
- I. Niculescu-Duvaz, A. Cambanis and E. Tarnauceanu, J. Med. Chem., 1967, 10, 172–174 CrossRef CAS PubMed.
- B. Baltzer, E. Binderup, W. Von Daehne, W. Godtfredsen, K. Hansen, B. Nielsen, H. Sorensen and S. Vangedal, J. Antibiot., 1980, 33, 1183–1192 CrossRef CAS PubMed.
- S. Wang, P. Huang and X. Chen, Adv. Mater., 2016, 28, 7340–7364 CrossRef CAS PubMed.
- R. Li, F. Peng, J. Cai, D. Yang and P. Zhang, Asian J. Pharm. Sci., 2019 DOI:10.1016/j.ajps.2019.06.003.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- S. F. Betz, Protein Sci., 1993, 2, 1551–1558 CrossRef CAS PubMed.
- P. Zhang, J. Wu, F. Xiao, D. Zhao and Y. Luan, Med. Res. Rev., 2018, 38, 1485–1510 CrossRef CAS PubMed.
- L. Turell, H. Botti, S. Carballal, R. Radi and B. Alvarez, J. Chromatogr. B, 2009, 877, 3384–3392 CrossRef CAS PubMed.
- A. J. Stewart, C. A. Blindauer, S. Berezenko, D. Sleep, D. Tooth and P. J. Sadler, FEBS J., 2005, 272, 353–362 CrossRef CAS PubMed.
- L. Brulisauer, M. A. Gauthier and J. C. Leroux, J. Controlled Release, 2014, 195, 147–154 CrossRef CAS PubMed.
- Z. Li, C. Xiao, T. Yong, Z. Li, L. Gan and X. Yang, Chem. Soc. Rev., 2020, 49, 2273–2290 RSC.
- Z. Li, C. Di, S. Li, X. Yang and G. Nie, Acc. Chem. Res., 2019, 52, 2703–2712 CrossRef CAS.
- Y. Wang, D. Liu, Q. Zheng, Q. Zhao, H. Zhang, Y. Ma, J. K. Fallon, Q. Fu, M. T. Haynes, G. Lin, R. Zhang, D. Wang, X. Yang, L. Zhao, Z. He and F. Liu, Nano Lett., 2014, 14, 5577–5583 CrossRef CAS.
- Y. Wang, X. Wang, F. Deng, N. Zheng, Y. Liang, H. Zhang, B. He, W. Dai, X. Wang and Q. Zhang, J. Controlled Release, 2018, 279, 136–146 CrossRef CAS PubMed.
- Z. Fan, G. Liu, Y. Li, J. Ma, J. Lin, F. Guo, Z. Hou and L. Xie, RSC Adv., 2016, 6, 82949–82960 RSC.
- P. Pradeepkumar, A. M. Elgorban, A. H. Bahkali and M. Rajan, New J. Chem., 2018, 42, 10366–10375 RSC.
- O. Y. Zolotarskaya, L. Xu, K. Valerie and H. Yang, RSC Adv., 2015, 5, 58600–58608 RSC.
- P. Laskar, S. Somani, S. J. Campbell, M. Mullin, P. Keating, R. J. Tate, C. Irving, H. Y. Leung and C. Dufès, Nanoscale, 2019, 11, 20058–20071 RSC.
- L. Qiu, Q. Liu, C.-Y. Hong and C.-Y. Pan, J. Mater. Chem. B, 2016, 4, 141–151 RSC.
- M. Rajan, P. Krishnan, P. Pradeepkumar, M. Jeyanthinath, M. Jeyaraj, M. P. Ling, P. Arulselvan, A. Higuchi, M. A. Munusamy, R. Arumugam, G. Benelli, K. Murugan and S. S. Kumar, RSC Adv., 2017, 7, 46271–46285 RSC.
- Y. Ma, Q. Mou, X. Zhu and D. Yan, Mater. Today Chem., 2017, 4, 26–39 CrossRef.
- Q. Pei, X. Hu, Z. Li, Z. Xie and X. Jing, RSC Adv., 2015, 5, 81499–81501 RSC.
- K. Cai, X. He, Z. Song, Q. Yin, Y. Zhang, F. M. Uckun, C. Jiang and J. Cheng, J. Am. Chem. Soc., 2015, 137, 3458–3461 CrossRef CAS PubMed.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CAS.
- Q. Pei, X. Hu, S. Liu, Y. Li, Z. Xie and X. Jing, J. Controlled Release, 2017, 254, 23–33 CrossRef CAS PubMed.
- Q. Pei, X. Hu, L. Wang, S. Liu, X. Jing and Z. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 26740–26748 CrossRef CAS PubMed.
- Q. Pei, X. Hu, J. Zhou, S. Liu and Z. Xie, Biomater. Sci., 2017, 5, 1517–1521 RSC.
- B. Feng, F. Zhou, B. Hou, D. Wang, T. Wang, Y. Fu, Y. Ma, H. Yu and Y. Li, Adv. Mater., 2018, 30, 1803001 CrossRef PubMed.
- X. Zheng, L. Wang, S. Liu, W. Zhang, F. Liu and Z. Xie, Adv. Funct. Mater., 2018, 28, 1706507 CrossRef.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef PubMed.
- C. Lan and S. Zhao, J. Mater. Chem. B, 2018, 6, 6685–6704 RSC.
- A. Sharma, M. G. Lee, M. Won, S. Koo, J. F. Arambula, J. L. Sessler, S. G. Chi and J. S. Kim, J. Am. Chem. Soc., 2019, 141, 15611–15618 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed. Engl., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- F. Zhang, G. Zhu, O. Jacobson, Y. Liu, K. Chen, G. Yu, Q. Ni, J. Fan, Z. Yang, F. Xu, X. Fu, Z. Wang, Y. Ma, G. Niu, X. Zhao and X. Chen, ACS Nano, 2017, 11, 8838–8848 CrossRef CAS PubMed.
- M. Hou, P. Xue, Y.-E. Gao, X. Ma, S. Bai, Y. Kang and Z. Xu, Biomater. Sci., 2017, 5, 1889–1897 RSC.
- W. He, X. Hu, W. Jiang, R. Liu, D. Zhang, J. Zhang, Z. Li and Y. Luan, Adv. Healthcare Mater., 2017, 6, 1700829 CrossRef PubMed.
- S. Dong, J. He, Y. Sun, D. Li, L. Li, M. Zhang and P. Ni, Mol. Pharm., 2019, 16, 3770–3779 CrossRef CAS.
- P. Huang, G. Wang, Y. Su, Y. Zhou, W. Huang, R. Zhang and D. Yan, Theranostics, 2019, 9, 5755–5768 CrossRef CAS.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS.
- Y. Tang, Y. Li, R. Xu, S. Li, H. Hu, C. Xiao, H. Wu, L. Zhu, J. Ming, Z. Chu, H. Xu, X. Yang and Z. Li, Nanoscale, 2018, 10, 17265–17274 RSC.
- G. Vite, I. Vlahov, S. Howard, S.-H. Kim, P. Kleindl, H. Santhapuram, Y. Wang, M.-L. Wen, C. Leamon and F. Lee, Cancer Res., 2008, 68, 4155 Search PubMed.
- P. P. Peethambaram, L. C. Hartmann, D. J. Jonker, M. de Jonge, E. R. Plummer, L. Martin, J. Konner, J. Marshall, G. D. Goss, V. Teslenko, P. L. Clemens, L. J. Cohen, C. M. Ahlers and L. Alland, Invest. New Drugs, 2015, 33, 321–331 CrossRef CAS PubMed.
- R. W. Naumann, R. L. Coleman, R. A. Burger, E. A. Sausville, E. Kutarska, S. A. Ghamande, N. Y. Gabrail, S. E. DePasquale, E. Nowara, L. Gilbert, R. H. Gersh, M. G. Teneriello, W. A. Harb, P. A. Konstantinopoulos, R. T. Penson, J. T. Symanowski, C. D. Lovejoy, C. P. Leamon, D. E. Morgenstern and R. A. Messmann, J. Clin. Oncol., 2013, 31, 4400–4406 CrossRef CAS PubMed.
- R. W. Naumann, L. Gilbert, A. Habbe, H. Ma, S. Ghamande and I. B. Vergote, Gynecol. Oncol., 2014, 133, 177 Search PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS.
- M. Hou, S. Li, Z. Xu and B. Li, Chem.–Asian J., 2019, 14, 3840–3844 CrossRef CAS.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS.
- X. Li, J. Kim, J. Yoon and X. Chen, Adv. Mater., 2017, 29, 1606857 CrossRef PubMed.
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271 RSC.
- X. Song, X. Han, F. Yu, X. Zhang, L. Chen and C. Lv, Theranostics, 2018, 8, 2217–2228 CrossRef CAS PubMed.
- M. H. Lee, A. Sharma, M. J. Chang, J. Lee, S. Son, J. L. Sessler, C. Kang and J. S. Kim, Chem. Soc. Rev., 2018, 47, 28–52 RSC.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668–12674 CrossRef CAS PubMed.
- S. Bhuniya, S. Maiti, E. O. Kim, H. Lee and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469–4474 CrossRef CAS PubMed.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- T. Kim, H. M. Jeon, H. T. Le, T. W. Kim, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 7690–7693 RSC.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS.
- K. J. Norsworthy, C. W. Ko, J. E. Lee, J. Liu, C. S. John, D. Przepiorka, A. T. Farrell and R. Pazdur, Oncologist, 2018, 23, 1103–1108 CrossRef CAS.
- I. Pysz, P. J. M. Jackson and D. E. Thurston, in Cytotoxic Payloads for Antibody–Drug Conjugates, The Royal Society of Chemistry, 2019, pp. 1–30, 10.1039/9781788012898-00001.
- S. Thota and A. Advani, Eur. J. Haematol., 2017, 98, 425–434 CrossRef CAS.
- S. Dhillon, Drugs, 2018, 78, 1763–1767 CrossRef CAS.
- W. Widdison, S. Wilhelm, K. Veale, J. Costoplus, G. Jones, C. Audette, B. Leece, L. Bartle, Y. Kovtun and R. Chari, Mol. Pharm., 2015, 12, 1762–1773 CrossRef CAS PubMed.
- K. N. Moore, L. P. Martin, D. M. O'Malley, U. A. Matulonis, J. A. Konner, I. Vergote, J. F. Ponte and M. J. Birrer, Future Oncol., 2017, 14, 123–136 CrossRef PubMed.
- H. M. Kantarjian, B. Lioure, S. K. Kim, E. Atallah, T. Leguay, K. Kelly, J.-P. Marolleau, M. Escoffre-Barbe, X. G. Thomas, J. Cortes, E. Jabbour, S. O'Brien, P. Bories, C. Oprea, L. Hatteville and H. Dombret, Clin. Lymphoma, Myeloma Leuk., 2016, 16, 139–145 CrossRef PubMed.
- M. Trneny, G. Verhoef, M. J. S. Dyer, D. Ben Yehuda, C. Patti, M. Canales, A. Lopez, F. T. Awan, P. G. Montgomery, A. Janikova, A. M. Barbui, K. Sulek, M. J. Terol, J. Radford, A. Guidetti, M. D. I. Nicola, L. Siraudin, L. Hatteville, S. Schwab, C. Oprea and A. M. Gianni, Haematologica, 2018, 103, 1351–1358 CrossRef CAS PubMed.
- J. D. Sadowsky, T. H. Pillow, J. Chen, F. Fan, C. He, Y. Wang, G. Yan, H. Yao, Z. Xu, S. Martin, D. Zhang, P. Chu, J. dela Cruz-Chuh, A. O'Donohue, G. Li, G. Del Rosario, J. He, L. Liu, C. Ng, D. Su, G. D. Lewis Phillips, K. R. Kozak, S.-F. Yu, K. Xu, D. Leipold and J. Wai, Bioconjugate Chem., 2017, 28, 2086–2098 CrossRef CAS.
- D. Zhang, T. H. Pillow, Y. Ma, J. D. Cruz-Chuh, K. R. Kozak, J. D. Sadowsky, G. D. Lewis Phillips, J. Guo, M. Darwish, P. Fan, J. Chen, C. He, T. Wang, H. Yao, Z. Xu, J. Chen, J. Wai, Z. Pei, C. E. Hop, S. C. Khojasteh and P. S. Dragovich, ACS Med. Chem. Lett., 2016, 7, 988–993 CrossRef CAS PubMed.
- D. Zhang, A. Fourie-O’Donohue, P. S. Dragovich, T. H. Pillow, J. D. Sadowsky, K. R. Kozak, R. T. Cass, L. Liu, Y. Deng, Y. Liu, C. E. C. A. Hop and S. C. Khojasteh, Drug Metab. Dispos., 2019, 47, 1156–1163 CrossRef CAS PubMed.
- M. A. Socinski, F. J. Kaye, D. R. Spigel, F. J. Kudrik, S. Ponce, P. M. Ellis, M. Majem, P. Lorigan, L. Gandhi, M. E. Gutierrez, D. Nepert, J. Corral and L. P. Ares, Clin. Lung Cancer, 2017, 18, 68–76 CrossRef CAS PubMed.
- S. Lapusan, M. B. Vidriales, X. Thomas, S. de Botton, A. Vekhoff, R. Tang, C. Dumontet, R. Morariu-Zamfir, J. M. Lambert, M.-L. Ozoux, P. Poncelet, J. F. San Miguel, O. Legrand, D. J. DeAngelo, F. J. Giles and J.-P. Marie, Invest. New Drugs, 2012, 30, 1121–1131 CrossRef CAS PubMed.
- S. Jagannath, T. Heffner, D. Avigan, R. Lutz and K. A. Anderson, Blood, 2011, 118, 305 CrossRef.
- H. L. Kindler, S. Novello, D. Fennell, G. Blumenschein, A. Bearz, G. Ceresoli, J. Aerts, J. Spicer, P. Taylor, A. Greystoke, K. Nackaerts, L. Calabro, S. Burgers, R. Jennens, A. Sporchia, A. Walter, J. Siegel, B. Childs, C. Elbi and R. Hassan, J. Thorac. Oncol., 2017, 12, S1746 CrossRef.
- V. Boni, O. Rixe, D. Rasco, C. Gomez-Roca, E. Calvo, J. C. Morris, A. W. Tolcher, S. Assadourian, H. Guillemin and J.-P. Delord, Mol. Cancer Ther., 2013, 12, A73 Search PubMed.
- A. Gazzah, N. Stjepanovic, M. H. Ryu, J. Tabernero, J. C. Soria, P. Bedard, Y. K. Kang, R. Bahleda, H. Guillemin-Paveau, C. Henry, L. Hatteville, C. Zilocchi, B. Demers and C. Hierro, Eur. J. Cancer, 2016, 69, S14–S15 CrossRef.
- Y. Baudat, B. Cameron, T. Dabdoubi, A. M. Lefebvre, A. Merino-Trigo, C. Thomas, V. Pecheux, B. Genet, L. Calvet and L. Blot, Cancer Res., 2016, 76, 1198 Search PubMed.
- C. U. Bialucha, S. D. Collins, X. Li, P. Saxena, X. Zhang, C. Durr, B. Lafont, P. Prieur, Y. Shim, R. Mosher, D. Lee, L. Ostrom, T. Hu, S. Bilic, I. L. Rajlic, V. Capka, W. Jiang, J. P. Wagner, G. Elliott, A. Veloso, J. C. Piel, M. M. Flaherty, K. G. Mansfield, E. K. Meseck, T. Rubic-Schneider, A. S. London, W. R. Tschantz, M. Kurz, D. Nguyen, A. Bourret, M. J. Meyer, J. E. Faris, M. J. Janatpour, V. W. Chan, N. C. Yoder, K. C. Catcott, M. A. McShea, X. Sun, H. Gao, J. Williams, F. Hofmann, J. A. Engelman, S. A. Ettenberg, W. R. Sellers and E. Lees, Cancer Discovery, 2017, 7, 1030–1045 CrossRef CAS PubMed.
- A. W. Tolcher, L. Ochoa, L. A. Hammond, A. Patnaik, T. Edwards, C. Takimoto, L. Smith, J. de Bono, G. Schwartz, T. Mays, Z. L. Jonak, R. Johnson, M. DeWitte, H. Martino, C. Audette, K. Maes, R. V. J. Chari, J. M. Lambert and E. K. Rowinsky, J. Clin. Oncol., 2003, 21, 211–222 CrossRef CAS PubMed.
- L. W. Goff, K. Papadopoulos, J. A. Posey, A. T. Phan, A. Patnaik, J. G. Miller, S. Zildjian, J. J. O'Leary, A. Qin and A. Tolch, Veterinary Surgery, 2009, 27, 438–444 CrossRef.
- J. Bendell, K. Moore, A. Qin, D. Johnson and A. W. Tolcher, EJC Suppl., 2010, 8, 152 CrossRef.
- P. Sapra, A. T. Hooper, C. J. O'Donnell and H.-P. Gerber, Expert Opin. Invest. Drugs, 2011, 20, 1131–1149 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- H.-J. Li, J.-Z. Du, X.-J. Du, C.-F. Xu, C.-Y. Sun, H.-X. Wang, Z.-T. Cao, X.-Z. Yang, Y.-H. Zhu, S. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. Li, H. Yang, J. Yao, H. Yu, X. Chen, P. Zhang and C. Xiao, Colloids Surf., B, 2018, 169, 273–279 CrossRef CAS PubMed.
- M. Wu, J. Li, X. Lin, Z. Wei, D. Zhang, B. Zhao, X. Liu and J. Liu, Biomater. Sci., 2018, 6, 1457–1468 RSC.
- B. Sui, C. Cheng, M. Wang, E. Hopkins and P. Xu, Adv. Funct. Mater., 2019, 29, 1906433 CrossRef CAS.
- X. Zhang, L. Han, M. Liu, K. Wang, L. Tao, Q. Wan and Y. Wei, Mater. Chem. Front., 2017, 1, 807–822 RSC.
- X. Guo, Y. Cheng, X. Zhao, Y. Luo, J. Chen and W.-E. Yuan, J. Nanobiotechnol., 2018, 16, 74 CrossRef.
- S. A. Sufi, S. Pajaniradje, V. Mukherjee and R. Rajagopalan, Antioxid. Redox Signaling, 2019, 30, 762–785 CrossRef CAS PubMed.
- L. Han, X.-Y. Zhang, Y.-L. Wang, X. Li, X.-H. Yang, M. Huang, K. Hu, L.-H. Li and Y. Wei, J. Controlled Release, 2017, 259, 40–52 CrossRef CAS PubMed.
- H. Sun, Y. Zhang and Z. Zhong, Adv. Drug Delivery Rev., 2018, 132, 16–32 CrossRef CAS.
- Z. Jiang and S. Thayumanavan, Isr. J. Chem., 2020, 60, 132–139 CrossRef CAS.
- E. Ghassami, J. Varshosaz and S. Taymouri, Curr. Pharm. Des., 2018, 24, 3303–3319 CrossRef CAS PubMed.
- R. Bej, P. Dey and S. Ghosh, Soft Matter, 2020, 16, 11–26 RSC.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97–126 RSC.
- Y. Li, Y. Wu, J. Chen, J. Wan, C. Xiao, J. Guan, X. Song, S. Li, M. Zhang, H. Cui, T. Li, X. Yang, Z. Li and X. Yang, Nano Lett., 2019, 19, 5806–5817 CrossRef CAS PubMed.
- Y. Li, H. Hu, Q. Zhou, Y. Ao, C. Xiao, J. Wan, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 19215–19230 CrossRef CAS PubMed.
- C. Yu, C. Liu, S. Wang, Z. Li, H. Hu, Y. Wan and X. Yang, Cancers, 2019, 11, 207 CrossRef CAS PubMed.
- H. Hu, Y. Li, Q. Zhou, Y. Ao, C. Yu, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 30833–30844 CrossRef CAS PubMed.
- A. K. Jain, M. G. Gund, D. C. Desai, N. Borhade, S. P. Senthilkumar, M. Dhiman, N. K. Mangu, S. V. Mali, N. P. Dubash, S. Halder and A. Satyam, Bioorg. Chem., 2013, 49, 40–48 CrossRef CAS PubMed.
- B. Sun, C. Luo, H. Yu, X. Zhang, Q. Chen, W. Yang, M. Wang, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- W. A. Klis, J. G. Sarver and P. W. Erhardt, Tetrahedron Lett., 2001, 42, 7747–7750 CrossRef CAS.
- G. D. Lewis Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons, E. Mai, W. A. Blattler, J. M. Lambert, R. V. Chari, R. J. Lutz, W. L. Wong, F. S. Jacobson, H. Koeppen, R. H. Schwall, S. R. Kenkare-Mitra, S. D. Spencer and M. X. Sliwkowski, Cancer Res., 2008, 68, 9280–9290 CrossRef CAS PubMed.
- Y. Zheng, Y. Shen, X. Meng, Y. Wu, Y. Zhao and C. Wu, ChemMedChem, 2019, 14, 1196–1203 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |