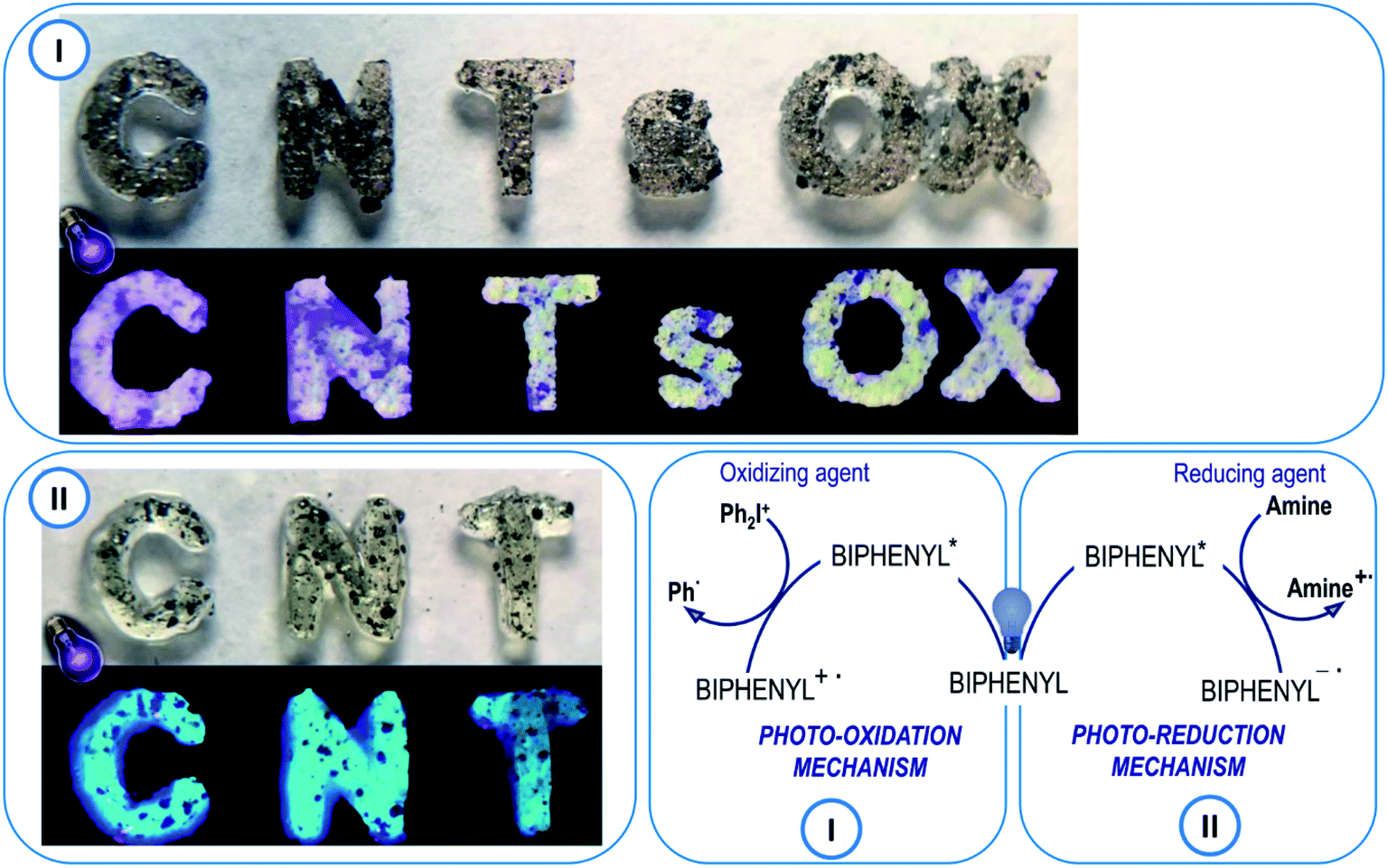DOI:
10.1039/D0RA04146G
(Paper)
RSC Adv., 2020,
10, 32162-32182
Multifunctional biphenyl derivatives as photosensitisers in various types of photopolymerization processes, including IPN formation, 3D printing of photocurable multiwalled carbon nanotubes (MWCNTs) fluorescent composites†
Received
8th May 2020
, Accepted 19th August 2020
First published on 28th August 2020
Abstract
A series of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were investigated in terms of photosensitisation in various photopolymerization processes in UV-A and vis light conditions. A full spectroscopic analysis of the tested compounds was performed. In addition to excellent spectroscopic properties, these compounds enable highly efficient photopolymerization processes, including free-radical, cationic and hybrid photopolymerization. As proven by a real-time FTIR study, these photosensitisers allow the formation of both thin and thick layers from different monomers. Finally, the investigated 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were used to obtain multiwalled carbon nanotubes (MWCNTs) composites for which the degree of conversion was determined using real-time FT-IR and Photo-Differential Scanning Calorimetry (Photo-DSC). Selected derivatives were applied as photosensitisers in two-component photoinitiating systems, operating according to the mechanism of photo-oxidation and photo-reduction, for the preparation of photo-cured MWCNTs composites. The importance of the quantity of multiwalled carbon nanotubes (MWCNTs) added to the polymeric matrix on the curing degree is also discussed in this study. The structures of the MWCNTs composites were analysed using an optical and fluorescence microscope. Moreover, this study also examines the applicability of new photoinitiator systems for printing nanocomposites by vat photopolymerization, which has gained increasing attention in recent years. Therefore, photocurable nanocomposite resin based on methacrylates was used for 3D printing in room temperature and atmospheric conditions, under a visible LED with emission at 405 nm, in order to obtain fluorescent photocurable patterns.
Introduction
Photopolymerization processes are a well-known method for manufacturing polymeric materials in various fields of the chemical industry.1–6 Numerous advantages, such as conducting a reaction at room temperature, low heat consumption, lack of solvents and fast reaction, make light-induced polymerization suitable for use in the solvent-free paint industry,7 optoelectronics,8 3D printing9–18 and many others.8,19–25 Photopolymerization is also a technique applied to obtain composite26 and nanocomposite materials,27,28 thanks to the afforded temporal and spatial control of this process. Composites are a well-developed class of multifunctional materials that are formed by combining an organic polymer with an inorganic filler, often at nanometric scale.29 In recent years, polymer composites have gained popularity due to their numerous advantages such as: high strength, corrosion resistance and the possibility to control their properties.30 The addition of nanofillers changes the final properties of the composites, e.g. by increasing their heat resistance, changing mechanical properties and sometimes vesting the composites with conductive properties.31 One of the methods for producing nanocomposites is photocuring of a given composition,32–34 which allows to transform a liquid monomer into a solid polymer quickly, and is particularly useful for hybrid compositions.35,36 The morphology of so obtained composites is significant: the more homogeneously dispersed is the nanofiller in the polymer matrix, the higher is the reinforcement of the polymer.37 There are two common methods for producing polymeric nanocomposites. The in situ technique requires the dispersion of a nanoadditive in a monomeric mixture containing an initiator or an initiating system.38 Such a composition is then exposed to light and polymerized. The second method, the ex situ approach, involves the addition of a filler to the dissolved or melted polymer, homogenizing the components and evaporating the solvent or cooling the mixture. Compared to the ex situ method, the in situ technique does not require the use of any solvents and provides better dispersion of the nanoadditive in the polymer matrix. Additionally, the use of light-induced polymerization with the in situ technique creates the opportunity to discover new materials or to improve well-known products manufactured by other methods.39 The advantages of photopolymerization are undoubtedly its high speed (down to a few seconds), the possibility to control the polymerization process,40 the absence of solvents and the use of a small amount of energy for the reaction to occur. The photo-induced polymerization of nanocomposites enables not only the production of polymers in mass, but also the creation of laminates41 and films with irregular shapes.42
According to the above, various types of photocurable polymer nanocomposites are currently being studied, especially nanocomposites containing silica nanoparticles. These compositions mainly contain acrylate43 or methacrylate44 monomers, which photopolymerize according to the free-radical mechanism. Other useful reinforcement additives for polymer resins are carbon nanotubes (CNTs).45 Since their discovery in 1991, CNTs have been valued for their unique properties, including mechanical and electrical properties.46,47 The compositions with added carbon nanotubes or graphite, based mainly on epoxy resins are cured by the cationic polymerization mechanism.48,49 Usually, small quantities of CNTs, generally between 0.1% and 5.0% (w/w), are added to the polymeric matrix to improve not only the mechanical but also the thermal properties. For these reasons, obtaining composite materials of this type is extremely attractive. In the literature, also the nanocomposites containing: clays,50 natural51 and polymeric fibres,52 oxides (e.g. aluminium oxides),53 polyhedral oligomeric silsesquioxanes,54,55 silica,56–59 and nitrides (i.e. boron or silicon nitrides)60 are described. The effectiveness in improving material properties by incorporation of such nanoadditives into polymers makes the photocured polymeric composites useful, for example, in adhesives,61 coatings,62 electronics63 etc.
In this article, we present a complete cycle of research: from the synthesis of new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives through their application as photosensitisers of light-induced polymerization processes and the determination of their full spectroscopic characteristics, to the investigation of two-component initiating systems containing these derivatives in terms of their applicability for photopolymerization of example monomers, such as 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE) and trimethylolpropane triacrylate (TMPTA). In our previous paper we studied 2-amino-4-methyl-6-phenyl-benzene-1,3-dicarbonitrile derivatives as photosensitizers for iodonium salts used in cationic photopolymerization processes.64 On the basis of those research we have modified previously tested structures in order to improve their numerous parameters – increase the range of absorption, increase reactivity, and improve photostability (in the absence of a photoinitiator). The 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives presented in this article have fulfilled the requirements for photosensitizers and eliminated some deficiencies that we noticed in the previously examined groups of biphenyl derivatives. The new photosensitizers feature an extended absorption spectrum, which increases the range of light sources that can be used when using commercial initiators with a narrow absorption range reaching UV (for example iodonium salts). The 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives enable the use of safe irradiation sources such as light-emitting diodes emitting in near UV or visible range. Another attribute of these compounds is their exceptional light stability in a precursor state. In addition, the applicability of the 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives for preparation of reinforced photocurable composites containing different amounts of MWCNTs was investigated. Finally, 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were identified as highly efficient photosensitisers for iodonium salts used as photoinitiators in photoinitiating systems, and as type II photoinitiators, when combined with an appropriate amine. Both photoinitiating system were applied for the preparation of photocurable carbon nanotube (CNTs) composites. The analysis of the kinetics of polymerization for these composites was performed via real-time FT-IR and photo-DSC. Lastly, the possibility of 3D printing using the designed compositions was checked. The use of these biphenyl photosensitizers accelerates the photopolymerization reaction and increases its efficiency. Therefore, their use in 3D printing, allows to reduce the amount of applied light, while providing excellent optical resolution and in some cases, transparency of the final printout.
Experiments
Materials
A series of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were investigated in terms of photosensitisation in various photopolymerization processes. The following compounds were tested: 2-(diethylamino)-4-(1-ethylpropyl)-6-phenylbenzene-1,3-dicarbonitrile (BI-PH), 2-(diethylamino)-4-(1-ethylpropyl)-6-(4-methylphenyl)benzene-1,3-dicarbonitrile (BI-PH-CH3), 2-(diethylamino)-6-(1-ethylpropyl)-4-(4-methoxyphenyl) benzene-1,3-dicarbonitrile (BI-PH-O-CH3), 4-(4-cyanophenyl)-2-(diethylamino)-6-(1-ethylpropyl)benzene-1,3-dicarbonitrile (BI-PH-CN), 2-(diethylamino)-4-(1-ethylpropyl)-6-(4-methylsulphonylphenyl)benzene-1,3 dicarbonitrile (BI-PH-SO2-CH3), 2-(diethylamino)-4-(1-ethylpropyl)-6-(4-methylsulphanylphenyl)benzene-1,3 dicarbonitrile (BI-PH-S-CH3), 2-(diethylamino)-4-(1-ethylpropyl)-6-[4-(trifluoromethyl)phenyl]benzene-1,3 dicarbonitrile (BI-PH-CF3), 2-(diethylamino)-4-(1-ethylpropyl)-6-(1-naphthyl)benzene-1,3-dicarbonitrile (BI-1-NPH), 2-(diethylamino)-4-(1-ethylpropyl)-6-(2-naphthyl)benzene-1,3-dicarbonitrile (BI-2-NPH), 4-(9-anthryl)-2-(diethylamino)-6-(1-ethylpropyl)benzene-1,3-dicarbonitrile (BI-1-AN). The synthesis pathways of the analysed derivatives studied are described in detail in ESI,† which also includes the results of NMR and LC-MS analysis. Structures of the photosensitizers studied are shown in Fig. 1.
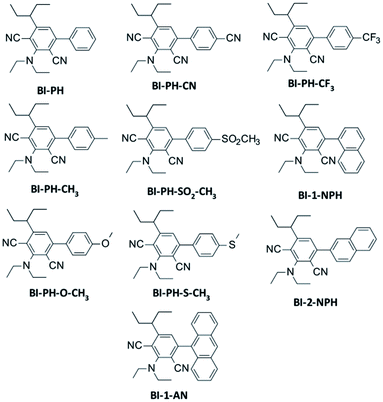 |
| | Fig. 1 Structures of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives studied. | |
The commercially available photoinitiator bis-(4-t-butylphenyl)iodonium hexafluorophosphate (Speedcure 938, Lambson Ltd, Wetherby, UK) was used. Ethyl 4-(dimethylamino)benzoate (EDB, from Sigma Aldrich) was used as an electron donor for the type II photoinitiating systems based on 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives. The basic monomers used were: trimethylolpropane triacrylate (TMPTA, from Sigma Aldrich) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE, Lambson Ltd, Wetherby, UK) for free-radical polymerization (FRP) and cationic polymerization (CP) respectively. Additional systems were formed from a mixture of methacrylate monomers: bisphenol A-glycidyl methacrylate (BisGMA, from Sigma Aldrich) and triethylene glycol dimethacrylate (TEGDMA, from Sigma Aldrich) for FRP, and 2,2-bis[4-(glycidyloxy)phenyl]propane (DGEBA, from Sigma Aldrich) and 1,2-epoxy-3-phenoxypropane (EPXPROP, from Sigma Aldrich) for CP. For the formation of interpenetrating polymer networks (IPNs), the following monomers were used: 3,4-epoxycyclohexylmethyl methacrylate (Cyclomer M100, from Daicel Corporation, Japan), trimethylolpropane tris(3-mercaptopropionate) (TRITHIOL, from Sigma Aldrich), and tris[4-(vinyloxy)butyl] trimellitate (TRIVINYL, from Sigma Aldrich). For the formation of photocurable carbon nanotube compositions we used thin multiwalled carbon nanotubes (MWCNTs) NC7000™ kindly provided by Nanocyl S.A. (Sambreville, Belgium). The average diameter and length, evaluated by high-resolution transmission electron microscopy (HRTEM), was 10−9 m and 1.5 μm respectively. The specific surface area of MWCNTs, estimated by the Brunauer–Emmett–Teller (BET) method, was around 250–300 m2 g−1. The carbon purity and metal oxide percentage, calculated by thermogravimetric analysis (TGA), was >95.0 and <5.0 respectively. The primary applications of MWCTNs NC7000™ are for coatings and polymer composites, which have a low electrical percolation threshold. Therefore, from a practical point of view, industrial NC7000™ carbon nanotubes are used in various markets, including transportation (automotive, aeronautic, marine), electronics (for electronic packaging) and energy (e.g. for lithium-ion), and industrial applications including dynamic rubber parts, coatings and heating elements. The structures of all used compounds are shown in Fig. S20 in ESI.†
Computations
In order to calculate the energy gap between first triplet (T1) and ground state (S0) for each 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivative, the Gaussian 09 package was used. The density functional theory (DFT) method at a B3LYP/6-31G (d, p) level was applied to optimise the molecules in their first triplet excited state (T1) and the ground state (S0). The frontier molecular orbital properties were evaluated and visualised using GaussView 5.0. All computations were performed using open-source software available in PLGrid Infrastructure.
Determination of electrochemical properties
The electrochemical determination of oxidation (Eox vs. Ag/AgCl) and reduction (Ered vs. Ag/AgCl) potentials of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were determined in acetonitrile by cyclic voltammetry using tetrabutylammonium hexafluorophosphate (0.1 M) (from Sigma Aldrich) as a supporting electrolyte and M161 Electrochemical Analyzer with M164 Electrode Stand, from MTM-ANKO, Cracow, Poland. Other parameters were as follows: scan rate was 0.1 V s−1, ferrocene was used as an internal standard, a platinum disk was used as a working electrode and a silver/silver chloride electrode (Ag/AgCl) was the reference. The Gibbs free energy change ΔGet for an electron transfer occurring in two component initiating systems was determined from eqn (1):| |
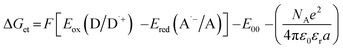 | (1) |
where Eox (D/D˙+) – oxidation potential of electron donor, Ered (A˙−/A) – reduction potential of electron acceptor, E00 – excited state energy, NA – Avogadro number, e – electron charge, ε0 – dielectric permittivity of vacuum, εr – dielectric constant of the solvent, a – effective distance between the radical cation (D˙+) and radical anion (A˙−) within an ion pair. The last term in eqn (1) which represents electrostatic interaction energy between the ionic species formed upon the electron transfer, is usually small and becomes practically equal to zero in polar solvents, where the radical ions pairs are dissociated, or in the case of electron transfer between an ion and an unchanged molecule, where no new ion pairs are generated.
The singlet excited state energy was calculated from the excitation and emission spectra using the Quanta Master™ 40 spectrofluorometer (from Photon Technology International (PTI), currently part of Horiba).
Spectroscopic characteristics
The absorbance properties of the 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were examined using SilverNova TEC-X2 spectrometer (from StellarNet, Inc., Tampa, FL, USA) with the range of 190–1100 nm. The spectrometer was equipped with a broadband UV/vis deuterium–halogen light source (SL5, from StellarNet, Inc., Tampa, FL, USA), which covers the 190–2500 nm emission range.
Fluorescence measurements were carried out using the same miniature spectrometer. The spectral characteristics of the derivatives were measured in acetonitrile at room temperature (25 °C) using 10 mm thick quartz cuvettes. The extinction source of light was UV-LED-320 with λmax = 320 nm (UVTOP315-BL-TO39, Roithner Laser Technik GmbH, Wien, Austria).
The fluorescence quantum yield of biphenyl derivatives (φf) was determined by comparision with the fluorescence quantum yield of Coumarin 1 standard in anhydrous ethanol, with spectroscopic grade (λex = 366 nm, ϕ = 0.64).9 The fluorescence quantum yields of for the tested derivatives were determined based on the formula:65
| |
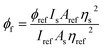 | (2) |
where
ϕref – was the fluorescence quantum yield of Coumarin 1,
ns and
nref are the solvents' refractive indices of the solvent used for sample (biphenyl derivative) and the reference, respectively,
As and
Aref were absorbance values of the sample and the reference at the excitation wavelength,
Is and
Iref were the integrated fluorescence intensities of the sample and the reference.
Fluorescence quenching
For the investigation of fluorescent quenching of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives by Speedcure 938, the Quanta Master™ 40 (from Photon Technology International (PTI), currently part of Horiba) was used. Small amounts of Speedcure 938 were added to the solution of the tested compound in acrylonitrile and the emission spectra ware measured individually. The maximal concentration of the quencher was about 2.7 × 10−2 mol dm−3 The polarity difference between monomer and acetonitrile is negligible.
Fluorescence lifetime
EasyLife™ (Fluorescence Lifetime Fluorometer, from Photon Technology International (PTI), currently part of Horiba) was used to record the fluorescence decay curves under exposure of a pulsed LED excitation source emitting at a wavelength of 310 nm. A solution of colloidal silica LUDOX® (from Sigma Aldrich), highly diluted in water, was used as a reference (IRF). The fluorescence lifetimes (τ) of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were determined by fitting decay curves, after a deconvolution with the Fluorescence Decay Analysis Software.
Steady state photolysis
The photolysis of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in acetonitrile was carried out under exposure to UV-LED-365 (M365L2, from Thorlabs Inc., Tampa, FL, USA), emitting wavelength λmax = 365 nm (∼190 mW cm−2, at 700 mA current) for 30 min. The photolysis of biphenyl derivatives in the presence of Speedcure 938 (2.7 × 10−2 mol dm−3) was studied using the same parameters, but for 10 min. The source of light was powered by a DC2200 regulated power supply (from Thorlabs Inc., Tampa, FL, USA). During the photolysis the UV-vis spectra were recorded with a weak broadband beam from a UV/vis deuterium–halogen light source (SL5, from StellarNet, Inc., Tampa, FL, USA), which was perpendicular to the irradiation beam used for the photolysis.
Real-time FT-IR experiments
The photopolymerization progress was measured by real-time FT-IR using an FT-IR i10 NICOLET™ spectrometer with a horizontal adapter (from Thermo Scientific, Waltham, MA, USA). The light source was turned on 10 s after the start of the polymerization monitoring procedure. Photocurable compositions were prepared by dissolving appropriate amounts of photoinitiator and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in different monomers. The weight ratio of the photoinitiating system was calculated from the monomer content. All compositions were prepared under the appropriate conditions, such as using dark glass, weighing the components in a room with minimal ambient light and mixing them in the dark until all ingredients were completely dissolved. Excellent solubility in different monomers was observed for all biphenyl derivatives.
The decrease of absorbance in the absorption range of polymerized groups is proportional to the number of groups that have polymerized. Therefore, the degree of conversion was calculated by taking measurements of the areas under the peak corresponding to a polymerizing group or bond using eqn (3):
| |
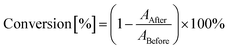 | (3) |
where
ABefore – area of the absorbance peak corresponding to a given group or bond in the investigated monomer prior to photopolymerization, and
AAfter – area of the same absorbance peak, at the given polymerization time. The locations of the characteristic absorbance peak for the studied monomers are given below for each type of photopolymerization.
Free-radical photopolymerization of acrylate monomers. A thin-layer composition was prepared by dissolving the photosensitiser and iodonium salt in trimethylolpropane triacrylate (TMPTA) monomer in such proportions as to obtain the concentrations of ∼0.1 wt% (3.69 × 10−3 mol dm−3) of on appropriate 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives and ∼1 wt% of Speedcure 938 (1.9 × 10−2 mol dm−3). A drop of the mixture was placed between two polypropylene films to avoid the negative impact of oxygen on the polymerization process (the thickness of the composition was around 25 μm). The laminated samples were deposited on a horizontal holder for the FT-IR spectrometer and were irradiated for 400 s. The decay of the double bond absorbance of the acrylate monomer content was continuously monitored at 1635 cm−1.
Performance of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in free-radical photopolymerization of methacrylate monomers as photoinitiating systems. The photocurable composition consisted of the following monomers: bisphenol A glycidyl methacrylate (BisGMA) and triethylene glycol dimethacrylate (TEGDMA) (50%/50% w/w) in the presence of the two component photoinitiating system based on the biphenyl derivatives (0.1 wt%) and Speedcure 938 (1 wt%) (cationic photoinitiating system) or the EDB ∼1.5% by weight (7.7 × 10−2 mol dm−3) (type II free-radical photoinitiating system). The composition was placed in the ring form (1500 μm thickness and 12 mm diameter) in a horizontal adapter. The content of double bond of methacrylate was continuously monitored in air at about 6165 cm−1 for 600 s.
Cationic photopolymerization of epoxide monomers. The mixture was made of 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE) and appropriate quantities of Speedcure 938 (1 wt%) and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.1 wt%) calculated according to the quantity of the used monomer. A drop of the composition was placed on the BaF2 pellet (the thickness of the composition was around 25 μm). Epoxy content was continuously followed in air at about 790 cm−1 for 800 s.
Cationic photopolymerization of glycidyle monomers. The composition was composed of two monomers: 2,2-bis[4-(glycidyloxy)phenyl]propane (DGEBA) and 1,2-epoxy-3-phenoxypropane (EPXPROP) (70%/30% w/w), 2 wt% (7.38 × 10−3 mol dm−3) of the appropriate 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivative and 0.2 wt% of Speedcure 938 (3.8 × 10−2 mol dm−3). Prior to the measurement, one drop of the solution was placed on a BaF2 pellet and spread form ∼25 μm layer. The glycidyl group content was continuously monitored in air at about 915 cm−1 for 600 s.
Formation of interpenetrating polymer network. For this purpose, the following compositions were prepared: (1) 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE) and trimethylolpropane triacrylate (TMPTA) at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio together with a photoinitiating system based on the biphenyl derivative (0.1 wt%) and Speedcure 938 (1.0 wt%); (2) 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE), trimethylolpropane triacrylate (TMPTA) and 3,4-epoxycyclohexylmethyl methacrylate (M100) at 1:1:1 weight ratio containing a photoinitiating system based on the biphenyl derivative (0.1 wt%) and Speedcure 938 (1.0 wt%). The polymerization process was carried out under three measurement conditions: in air (on a BaF2 pellet; sample thickness ∼ 25 μm), in air (in the ring form; sample dimension: 1.5 mm thickness and 12 mm diameter), in laminate (between two polypropylene films, sample thickness ∼ 25 μm). The decay of the double bond absorbance of acrylate monomer was continuously monitored at about 1635 cm−1 and epoxy content was continuously monitored in air at about 790 cm−1 for 600 s.
:1 weight ratio together with a photoinitiating system based on the biphenyl derivative (0.1 wt%) and Speedcure 938 (1.0 wt%); (2) 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane-carboxylate (CADE), trimethylolpropane triacrylate (TMPTA) and 3,4-epoxycyclohexylmethyl methacrylate (M100) at 1:1:1 weight ratio containing a photoinitiating system based on the biphenyl derivative (0.1 wt%) and Speedcure 938 (1.0 wt%). The polymerization process was carried out under three measurement conditions: in air (on a BaF2 pellet; sample thickness ∼ 25 μm), in air (in the ring form; sample dimension: 1.5 mm thickness and 12 mm diameter), in laminate (between two polypropylene films, sample thickness ∼ 25 μm). The decay of the double bond absorbance of acrylate monomer was continuously monitored at about 1635 cm−1 and epoxy content was continuously monitored in air at about 790 cm−1 for 600 s.
3D printing experiments
A Laser Engraver Printer machine (NEJE DK-8-KZ) was used to obtain the laser write printout under laser intensity of 150 mW cm−2 at 405 nm (spot diameter, ∼75 μm). For the 3D printing experiments the composition consisted of DGEBA/EPXPROP (70%/30% w/w) in the presence 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives/Speedcure 938 (0.2/2% w/w) was selected. The generated 3D objects were observed in an optical stereo microscope (Bresser Advance ICD 10–160 Zoom Stereo-Microscope, Bresser GmbH, Germany) and DSX1000 kindly provided by OLYMPUS.
Formation of nanocomposites with MWCNTs by photopolymerization
The mixture was obtained by dissolving ∼0.1 wt% (3.69 × 10−3 mol dm−3) of the appropriate 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivative and ∼1 wt% of the Speedcure 938 (1.9 × 10−2 mol dm−3) or ∼1.5 wt% of the EDB (7.7 × 10−2 mol dm−3) in the following monomers: bisphenol A glycidyl methacrylate (BisGMA) and triethylene glycol dimethacrylate (TEGDMA) (50%/50% w/w). After complete dissolution of all components, appropriate amounts of carbon nanotubes were added to the composition and stirred for several minutes. The contents of MWCNTs were as follows: 0.1%, 0.25% and 0.5% by weight. For both: real-time FT-IR and photo-DSC experiments, a pure composition (without carbon nanotubes) was also analysed for comparison.
Real-time FT-IR experiments. A drop of composition was placed between two layers of polypropylene films. The thickness of the composition was ∼25 μm and the sample was deposited on a horizontal holder in a real-time FT-IR spectrometer. The free-radical photopolymerization of methacrylate monomers was monitored in laminate conditions by following the decrease of the double bond absorbance at 1635 cm−1.
Photo-differential scanning calorimetry (photo-DSC) experiments. Photo-DSC was used to investigate kinetics of photopolymerization processes of the proposed photocurable CNT compositions. The photo-DSC studies were conducted with a Photo-DSC 204 F1 Phoenix® from Netzsch – Gerätebau GmbH (Germany) equipped with a 365 nm LED light spot cure system, Bluepoint LED eco (product of Hönle UV Technology, Germany) was set to 5.45 [W cm−2] at the surface of the sample. The light intensity was measured by an OmniCure® R2000 radiometer (product of Excelitas Technologies®, Ontario, Canada) at the tip of the light guide (product of Excelitas Technologies®, Ontario, Canada). After that, the intensity of the light at the guide tip was converted to the intensity at the sample surface. All measurements were conducted in an inert atmosphere (nitrogen flow of 20 [ml min−1]).The photopolymerization measurements of photocurable CNTs compositions were carried out in isothermal mode at 25 °C and 70 °C. A sample of 2 ± 0.5 mg (sample thickness ∼ 120 μm) was placed in an aluminium crucibles from Netzsch – Gerätebau GmbH (Germany), covered with a clear quartz disc and subjected to 10 s of isothermal conditioning before and 30 s after light exposure. An empty pan was used as the reference. All measurements were performed in duplicate. The heat flow of the reaction was recorded as a function of time.
The determination of the conversion (Cphoto-DSC) and the rate of polymerization (Rp) involve the molecular weight, density and theoretical enthalpy per mol of the polymerizing functional group of the monomer (ΔH0) (for the methacrylate monomers this was 56.6 kJ mol−1).66 The determination of the conversion (Cphoto-DSC) and the rate of polymerization (Rp) involve the molecular weight, density and theoretical enthalpy per mol of the polymerizing functional group of the monomer (ΔH0) (for the methacrylate monomers this was 56.6 kJ mol−1). The heat liberated during the polymerization reaction was directly proportional to the number of polymerized double bonds in the monomers system. By integrating the area under the exothermic peak, the conversion of the bonds (Cphoto-DSC) could be calculated with eqn (4):
| |
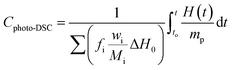 | (4) |
where
Mi – molecular mass of i-monomer [g mol
−1];
fi – functionality of i-monomer;
wi – percentage of i-monomer; Δ
H0 – enthalpy of polymerization the methacrylate monomers [J mol
−1] (per one functional group);
H – heat flow [W];
mp – sample mass [g].
The polymerization rate (Rp) was also calculated from eqn (5):
| |
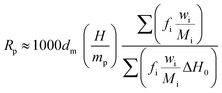 | (5) |
where
dm – density of monomers composition [g ml
−1]; Δ
H0 – enthalpy of polymerization the methacrylate monomers [J mol
−1] (per one functional group);
H – heat flow [W];
Mi – molecular mass of i-monomer [g mol
−1];
fi – functionality of i-monomer;
wi – percentage of i-monomer;
mp – sample mass [g].
A differential photocalorimeter was used for the determination of additional thermal polymerization processes, such as the thermal post-curing of reinforced photocured polymer composites. The samples (taken after the light-cure procedure at both 25 °C and 70 °C) were placed into aluminium pans and the DSC signal was measured in two cycles from 25 °C to 250 °C at 10 K min−1, held for 1 min at 250 °C and subsequently cooled to 25 °C at 10 K min−1.
Irradiation sources
The light sources used for real-time FT-IR experiments were as follows: 365 nm M365L2 UV-LED diode (I0 = 3.77 mW cm−2, Thorlabs Inc., Tampa, FL, USA), 405 nm M405L2 (I0 = 8.13 mW cm−2, Thorlabs Inc., Tampa, FL, USA), 365 nm M365LP1 UV-LED diode (I0 = 16.27 mW cm−2, Thorlabs Inc., Tampa, FL, USA), 405 nm M405LP1 (I0 = 21.23 mW cm−2, Thorlabs Inc., Tampa, FL, USA) and 365 nm high-power LED SOLIS-365C (Thorlabs Inc., Tampa, FL, USA) powered by a DC2200 regulated power supply (Thorlabs Inc., Tampa, FL, USA). In all cases, the power of the light source (I0) was corrected for the distance between the tip of the light guide and the sample.
Results and discussion
Spectroscopic characteristics
In order to obtain the full spectroscopic characteristics of the new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives, the absorption and fluorescence spectra were measured and spectroscopic parameters were determined (Table 1). The UV-vis spectra of the investigated compounds were recorded in acetonitrile and, on this basis, the maximum value of the molar extinction coefficient in the long-wavelength range was determined (Fig. 2). As shown in Table 1, all biphenyl derivatives are characterised by a sufficiently high molar extinction coefficient, reaching values ∼750–4250 [dm3 mol−1 cm−1], to be used as photosensitisers. In addition, the extinction coefficient values were determined for the wavelength corresponding to the used light sources (i.e. UV-A LED with maximum emission at 365 nm and vis-LED with maximum emission at 405 nm). All investigated derivatives absorb in the UV-A and the visible range. Therefore these derivatives can be used as photosensitisers in the processes of light-induced polymerization in those ranges.
Table 1 Spectral characteristics of the 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives studied in acetonitrile
| Acronym |
λmax-aba [nm] |
ε @ λmax-aba [dm3 mol−1 cm−1] |
ε @ 365 nm [dm3 mol−1 cm−1] |
ε @ 405 nm [dm3 mol−1 cm−1] |
ϕf |
| For the longest wavelength absorption band. |
| BI-PH |
358 |
2120 |
2050 |
370 |
0.045 |
| BI-PH-CH3 |
357 |
1770 |
1680 |
190 |
0.050 |
| BI-PH-O-CH3 |
349 |
1750 |
1590 |
260 |
0.041 |
| BI-PH-CN |
369 |
1370 |
1360 |
610 |
0.059 |
| BI-PH-SO2-CH3 |
364 |
1420 |
1400 |
440 |
0.044 |
| BI-PH-S-CH3 |
310 |
8120 |
1720 |
230 |
0.043 |
| BI-PH-CF3 |
361 |
1620 |
1600 |
440 |
0.052 |
| BI-1-NPH |
357 |
830 |
730 |
70 |
0.050 |
| BI-2-NPH |
365 |
1830 |
1830 |
310 |
0.012 |
| BI-1-AN |
366 |
4240 |
4220 |
1050 |
0.158 |
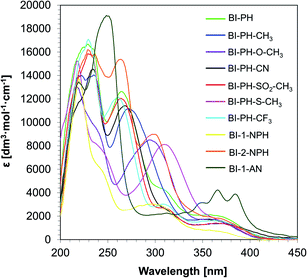 |
| | Fig. 2 UV-visible absorption spectra of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in acetonitrile. | |
The fluorescence spectra of the new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were also measured in acetonitrile. The maximum luminescence intensity and the corresponding wavelength were determined from the obtained spectra. The results are shown in Table 1.
Determination of electrochemical properties
The new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives are proposed as photosensitisers of iodonium salts. In such a system, the biphenyl derivatives initiate a photoinduced electron transfer reaction from its excited state to the iodonium salt (i.e. bis-(4-t-butylphenyl)iodonium hexafluorophosphate – Speedcure 938). The biphenyl molecule is an electron donor and, after light absorption, it is oxidised during the electron transfer process. The second molecule, which in this case is Speedcure 938, is an electron acceptor which is reduced in the electron transfer process. An important advantage of such a two-molecular initiating system is that it can be universally used for different types of polymerization. During the reduction of iodonium salt, phenyl radicals are generated, which quickly decay into secondary radicals capable for the initiation of free-radical polymerization reactions. Conversely, the radical-cations generated by the oxidation of the biphenyl derivative, combined with the anions derived from the onium salt, can decay into strong protonic acids, thus enabling the initiation of cationic photopolymerization. Several studies have been carried out to verify this mechanism. First of all, the oxidation potentials of the new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives were determined by cyclic voltammetry. The obtained potential values were used to calculate the free energy change of the electron transfer (ΔGet) between the components of the initiating system by applying the classical Rehm–Weller equation. Negative free energy change values were obtained, which confirmed the feasibility of oxidation of the biphenyl derivative by Speedcure 938, from a thermodynamic point of view. Such calculations were carried out for the singlet and triplet state of the investigated biphenyl derivatives. Singlet state energy was determined from the excitation and emission spectra, while triplet excited states energy was established by molecular orbital calculations using the density functional theory (DFT) method at a B3LYP/6-31G (d, p) level of theory. The summary of the calculation results can be found in Table 2, while contour plots of HOMOs and LUMOs, optimised by the B3LYP/6-31G (d, p) level of theory, are given in ESI.†
Table 2 Electrochemical and thermodynamic properties of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in the photo-oxidation mechanism
| Acronym |
Eox vs. Ag/AgCl [V] |
ES1 [eV] |
ΔGet(S1)a [eV] |
ET1 [eV] |
ΔGet(T1)a [eV] |
τ(S1) [ns] |
Ksv [M−1] |
kq [M−1 s−1] |
Φet(S1) |
Calculated from the classical Rehm–Weller equation: 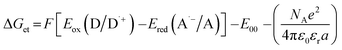 , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor, Ered(A˙−/A) – electrochemically determined reduction potential of the electron acceptor (−0.64 V for the diphenyliodonium salt vs. Ag/AgCl),67,68 ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[Speedcure 938]/(1 + KSV[Speedcure 938]) for the concentration of iodonium salt [Speedcure 938] = 0.021 mol dm−3. , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor, Ered(A˙−/A) – electrochemically determined reduction potential of the electron acceptor (−0.64 V for the diphenyliodonium salt vs. Ag/AgCl),67,68 ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[Speedcure 938]/(1 + KSV[Speedcure 938]) for the concentration of iodonium salt [Speedcure 938] = 0.021 mol dm−3. |
| BI-PH |
1.728 |
3.02 |
−0.61 |
2.63 |
−0.26 |
2.70 |
26.68 |
9.88 × 109 |
0.36 |
| BI-PH-CH3 |
1.561 |
3.04 |
−0.80 |
2.62 |
−0.42 |
2.06 |
17.32 |
8.40 × 109 |
0.27 |
| BI-PH-O-CH3 |
1.541 |
3.05 |
−0.83 |
2.60 |
−0.42 |
1.72 |
23.55 |
1.37 × 1010 |
0.33 |
| BI-PH-CN |
1.561 |
2.82 |
−0.58 |
2.51 |
−0.30 |
4.59 |
7.27 |
1.59 × 109 |
0.13 |
| BI-PH-SO2-CH3 |
1.555 |
2.89 |
−0.66 |
2.63 |
−0.43 |
4.86 |
10.03 |
2.06 × 109 |
0.17 |
| BI-PH-S-CH3 |
1.573 |
3.02 |
−0.77 |
2.60 |
−0.39 |
1.89 |
24.05 |
1.27 × 1010 |
0.34 |
| BI-PH-CF3 |
1.635 |
2.94 |
−0.63 |
2.55 |
−0.28 |
2.88 |
10.09 |
3.50 × 109 |
0.17 |
| BI-1-NPH |
1.579 |
3.08 |
−0.82 |
2.47 |
−0.25 |
2.44 |
18.07 |
7.40 × 109 |
0.28 |
| BI-2-NPH |
1.597 |
2.99 |
−0.71 |
2.56 |
−0.32 |
2.61 |
25.39 |
9.71 × 109 |
0.35 |
| BI-1-AN |
1.570 |
3.05 |
−0.80 |
1.78 |
0.43 |
3.36 |
27.11 |
8.07 × 109 |
0.36 |
An additional measurement of the investigated mechanism was the performance of the steady state photolysis of the derivatives in the presence of iodonium salt (Speedcure 938). During the light exposure of the biphenyl derivatives solution without any additive, the solution absorbance did not change significantly, which confirmed photo-stability of the derivatives studied (Fig. S71–S90, ESI†). The light exposure of the derivative in the presence of diphenyliodonium salt was quite different; there was a strong interaction between the components of the two-molecular photoinitiating system, which caused rapid decrease of the absorbance of the biphenyl derivative in the range of its maximum absorption. Additionally, there was a noticeable increase of the absorbance in the long-wavelength range, which corresponded to the appearance of photolysis decay products. The photolysis results are given in ESI.†
In addition, the reactivity of the proposed two-component photoinitiating system was determined by fluorescence quenching experiments. Correlations between the fluorescence intensity of the derivatives and the amount of added quencher were determined on the basis of the fluorescence spectra of the derivatives, tested during the addition of iodonium salt. The intensity of the fluorescence decreased with increased the amount of added quencher, which confirmed that the electron transfer process proceeded properly. Based on these results, the Stern–Volmer coefficients (KSV) were determined. Then, the rate constant of the electron transfer between 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives and diphenyliodonium salt (Speedcure 938) was determined from Stern–Volmer equation:
| |
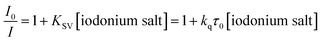 | (6) |
where
I0 and
I – fluorescence intensity of photosensitiser in the absence and presence of the iodonium salt (Speedcure 938) as the quencher respectively;
τ0 – lifetime of the excited state of the biphenyl photosensitiser in the absence of quencher.
Finally, the quantum efficiency of the electron transfer from the excited singlet state (Φet(S1)) in the process of photo-oxidation was calculated for the initial concentration of the quencher using the following equation:
| |
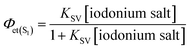 | (7) |
Additionally, the use of the new 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives is also proposed in the free-radical type II initiating systems. In this type of photoinitiating system, the biphenyl derivatives act as the electron acceptor, while the radicals initiating the polymerization reaction are generated by oxidation of the co-initiator molecule. Tertiary amines are generally used as such co-initiators, as they exhibit relatively low oxidation potential e.g. ethyl 4-(dimethylamino)benzoate (EDB). EDB is a typical amine widely used with camphorquinone as a visible light free-radical photoinitiating system for dental compositions. In this system, EDB behaves as an electron donor and becomes an amino alkyl radical able to initiate the free-radical photopolymerization reaction. Therefore, EDB was applied with the biphenyl derivatives was used as a photoinitiating system for free-radical photopolymerization processes in further experiments. Gibbs' free energy was calculated for both the singlet and triplet states of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives. The obtained free energy change of electron transfer (ΔGet) proves that the electron transfer process in the system (amine-biphenyl derivative) is thermodynamically feasible for all derivatives, except for BI-PH-CF3 containing the trifluoromethyl group. The calculations indicate that the photo-reduction process may take place from the excited singlet state, as confirmed by the negative ΔGet values for this state, while it is not allowed from the triplet state (Table 3).
Table 3 Electrochemical and thermodynamic properties of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in the photo-reduction mechanism
| Acronym |
Ered vs. Ag/AgCl [V] |
ES1 [eV] |
ΔGet(S1)a [eV] |
ET1 [eV] |
ΔGet(T1)a [eV] |
Calculated from the equation: 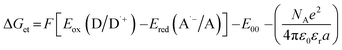 , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor (−1.058 V for amine EDB vs. Ag/AgCl)69, Ered (A˙−/A) – electrochemically determined reduction potential of the electron acceptor, ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[EDB]/(1 + KSV[EDB]) for the concentration of amine [EDB] = 0.086 mol dm−3. , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor (−1.058 V for amine EDB vs. Ag/AgCl)69, Ered (A˙−/A) – electrochemically determined reduction potential of the electron acceptor, ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[EDB]/(1 + KSV[EDB]) for the concentration of amine [EDB] = 0.086 mol dm−3. |
| BI-PH |
−1.854 |
3.02 |
−0.09 |
2.63 |
0.28 |
| BI-PH-CH3 |
−1.840 |
3.04 |
−0.13 |
2.62 |
0.28 |
| BI-PH-O-CH3 |
−1.927 |
3.05 |
−0.05 |
2.60 |
0.39 |
| BI-PH-CN |
−1.515 |
2.82 |
−0.23 |
2.51 |
0.07 |
| BI-PH-SO2-CH3 |
−1.604 |
2.89 |
−0.21 |
2.63 |
0.03 |
| BI-PH-S-CH3 |
−1.771 |
3.02 |
−0.18 |
2.60 |
0.23 |
| BI-PH-CF3 |
−1.997 |
2.94 |
0.13 |
2.55 |
0.50 |
| BI-1-NPH |
−1.813 |
3.08 |
−0.19 |
2.47 |
0.40 |
| BI-2-NPH |
−1.720 |
2.99 |
−0.20 |
2.56 |
0.22 |
| BI-1-AN |
−1.735 |
3.05 |
−0.24 |
1.78 |
1.01 |
Real-time FT-IR experiments
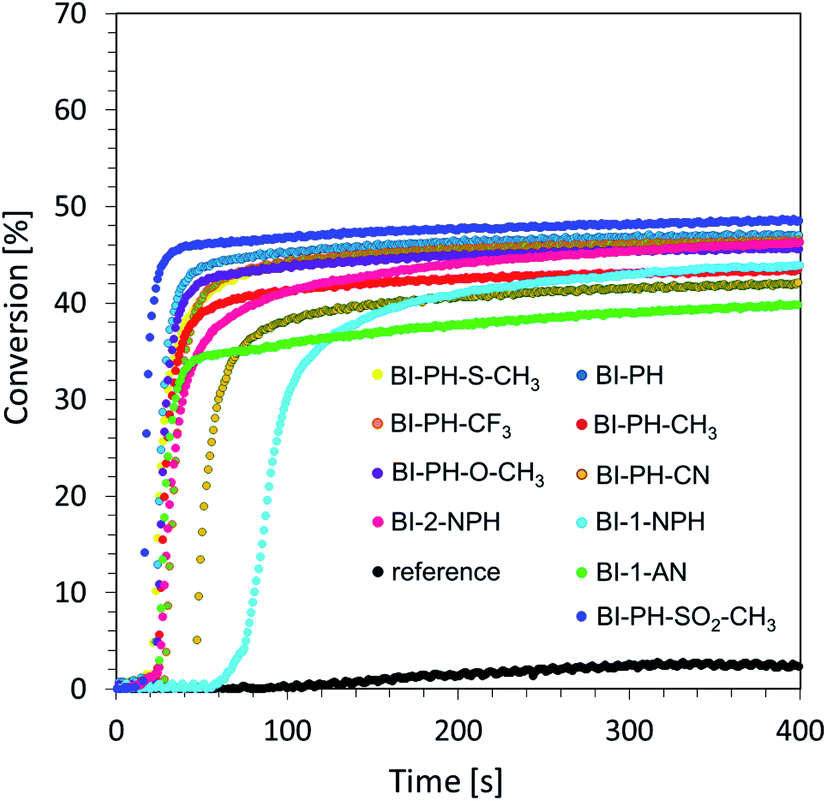
Free-radical photopolymerization of acrylate monomers. In industrial application, the most common method of photopolymerization is free-radical photopolymerization, which is based on the photochemical generation of free radicals, which initiate the polymerization process of acrylate and methacrylate monomers. The wide industrial use of free-radical photopolymerization results directly from the number of advantages of this type of photoinduced process: free-radical photopolymerization is very fast, its kinetics can be controlled by light, it exhibits compatibility with a wide variety of monomers based on acrylate and methacrylate groups, and its tolerance to water allows the development of emulsion and suspension techniques that can be used on an industrial scale. Accordingly, we investigated the novel 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives as photosensitisers in a two-component initiating system, with bis-(4-t-butylphenyl)iodonium hexafluorophosphate, in order to initiate efficiently free-radical photopolymerization of trimethylolpropane triacrylate monomer (TMPTA). A mixture of TMPTA with iodonium hexafluorophosphate (1.0% by weight) was used as a reference. Light-induced photopolymerization was carried out in laminate conditions to prevent the negative influence of oxygen, and the process was controlled by monitoring the decrease of the bond at 1635 cm−1 corresponding to the double-bond content. Conversion-time profiles under exposure to vis-LED @ 405 nm are given in Fig. 3.
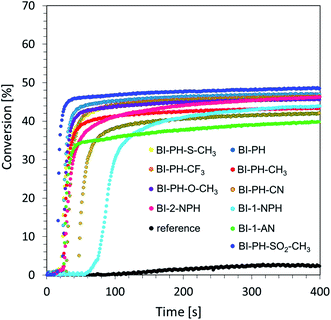 |
| | Fig. 3 Free-radical photopolymerization profiles (double bond conversion vs. irradiation time) initiated by a two-component photoinitiating system based on diphenyliodonium salt (1% wt) and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.1% wt) under irradiation at 405 nm (8.13 mW cm−2). The reference photoinitiator was bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1% wt) alone. | |
From the obtained data, it is clearly seen that the proposed bimolecular initiating system containing 1 wt% of iodonium salt and 0.1% of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives can be used to initiate free-radical photopolymerization. The summary of the results obtained for the investigated derivatives at irradiation of the photocurable system with UV-LED @ 365 nm and vis-LED @ 405 nm is included in Table 4.
Table 4 Summary of final functional group conversions of different monomers polymerized in various photopolymerization processes, with the use of a two-component initiating system containing 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.1 wt%) and iodonium salt (1 wt%)
| Acronym |
Conversion [%] |
| Cationic polymerization |
Free-radical polymerization |
| CADE monitored at ∼790 cm−1 |
Glycidyl monomers monitored at ∼915 cm−1 |
TMPTA monitored at ∼1635 cm−1 |
| @ 365a nm |
@ 405b nm |
@ 365c nm |
@ 365a nm |
@ 405b nm |
| LED @ 365 nm ∼ 3.77 mW cm−2. LED @ 405 nm ∼ 8.13 mW cm−2. LED @ 365 nm ∼ 16.27 mW cm−2. |
| BI-PH |
32.4 |
32.0 |
84.5 |
47.0 |
47.0 |
| BI-PH-CH3 |
38.8 |
31.5 |
81.3 |
43.5 |
43.5 |
| BI-PH-O-CH3 |
34.1 |
31.8 |
84.0 |
45.5 |
45.5 |
| BI-PH-CN |
9.3 |
20.5 |
66.4 |
42.1 |
42.1 |
| BI-PH-SO2-CH3 |
37.6 |
41.0 |
80.3 |
47.5 |
48.5 |
| BI-PH-S-CH3 |
43.1 |
27.6 |
68.7 |
46.5 |
46.5 |
| BI-PH-CF3 |
40.0 |
29.5 |
82.0 |
46.5 |
46.5 |
| BI-1-NPH |
24.6 |
11.4 |
75.3 |
39.9 |
43.8 |
| BI-2-NPH |
33.9 |
17.0 |
79.6 |
44.6 |
46.2 |
| BI-1-AN |
26.6 |
23.6 |
79.2 |
47.7 |
39.7 |
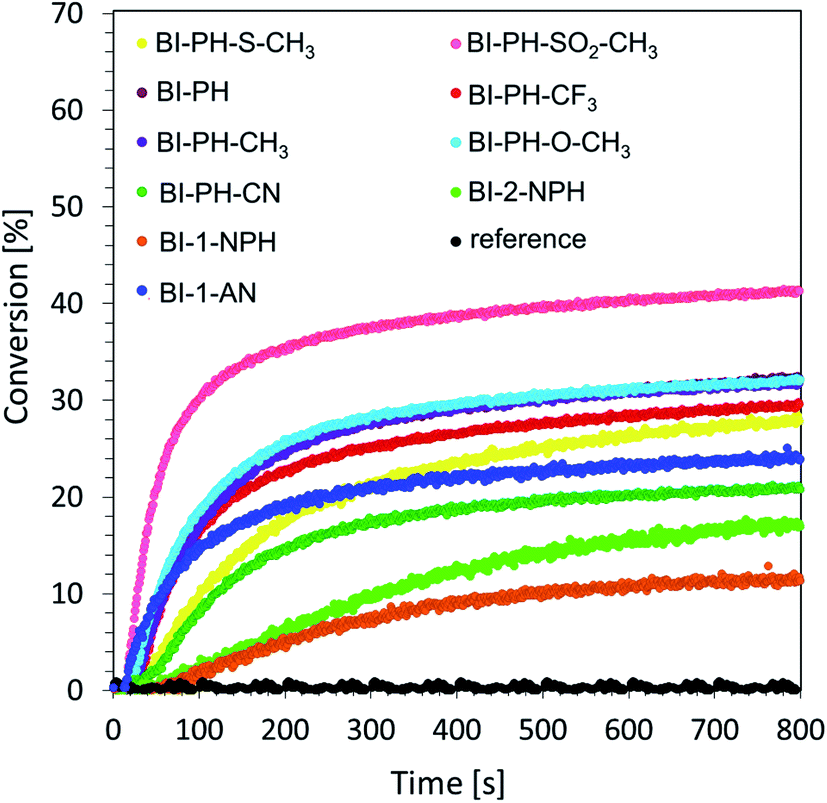
Cationic photopolymerization of epoxide monomers. Currently, much attention is paid to the development of cationic photopolymerization technologies, due to the resistance of such systems to the phenomenon of oxygen inhibition. The process of cationic polymerization has many advantages, including its living character, which ensures that the reaction remains effective even after the irradiation source has been switched off. As a result, it is possible to achieve a high degree of conversion, which plays an extremely important role in industrial practice. The following monomers undergo this type of reaction: epoxides, vinyl ethers, siloxanes, oxetans, cyclic acetals etc. At this stage of the research, we investigated the cationic photopolymerization of 3,4-epoxycyclohexylmethyl-3,4-epoxy-cyclohexane-carboxylate monomer (CADE) using a two-component initiating system consisting of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives and iodonium salt (Speedcure 938) (0.1%/1% w/w). A mixture of the monomer CADE with diphenyliodonium salt alone (1.0% by weight) was used as a reference. The measurements were carried out in air, under exposure to vis-LED @ 405 nm and UV-LED @ 365 nm. The kinetic profiles of the photopolymerization reaction during the exposure of the composition to visible light are shown in Fig. 4.
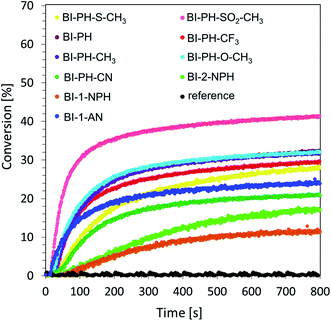 |
| | Fig. 4 Cationic photopolymerization profiles (epoxy function conversion vs. irradiation time) of CADE monomers, initiated by a two-component photoinitiating system based on diphenyliodonium salt (1% wt) and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.1% wt) under irradiation at 405 nm (8.13 mW cm−2).The reference was bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1% wt) without sensitiser. | |
A summary of the results obtained for 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives under irradiation with UV-LED @ 365 nm and vis-LED @ 405 nm is shown in Table 4.
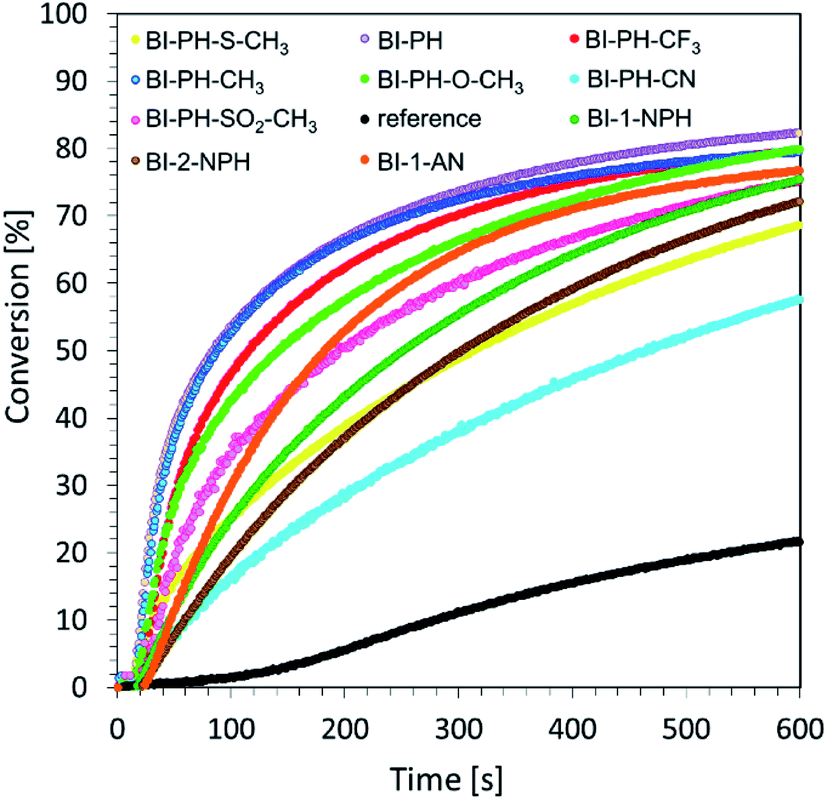
Photoactivated cationic ring-opening photopolymerization of glycidyl monomers. An interesting monomer from an application perspective is the 2,2-bis[4-(glycidyloxy)phenyl]propane (DEGBA). Due to its high viscosity, however, it is most commonly investigated by the photo-DSC method. Therefore, we combined this monomer with a second one, 1,2-epoxy-3-phenoxypropane (EPXPROP), which has much lower viscosity and the same reactive functional group in order to examine its photopolymerization reaction by means of real-time FT-IR method. We prepared a two-component initiating system based on Speedcure 938 (2 % wt) and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.2% wt). A mixture of the monomers DEGBA/EPXPROP (70%/30% w/w) with bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1.0% wt) was used as a reference. The measurements were carried out in air, under irradiation with vis-LED @ 365 nm. The values of conversion were in the range of up to ∼80% (Table 4), which allowed to obtain a highly cross-linked polymer network. Photopolymerization profiles of the investigated composition are given in Fig. 5.
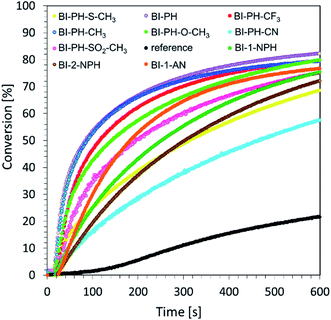 |
| | Fig. 5 Photopolymerization profiles (glycidyl group conversion vs. irradiation time) of DEGBA/EPXPROP (70%/30% w/w) monomers in the presence of a photoinitiating system based on biphenyl derivatives and bis-(4-t-butylphenyl)iodonium hexafluorophosphate (0.1/1% w/w) under irradiation of vis-LED @ 365 nm (16.27 mW cm−2). | |
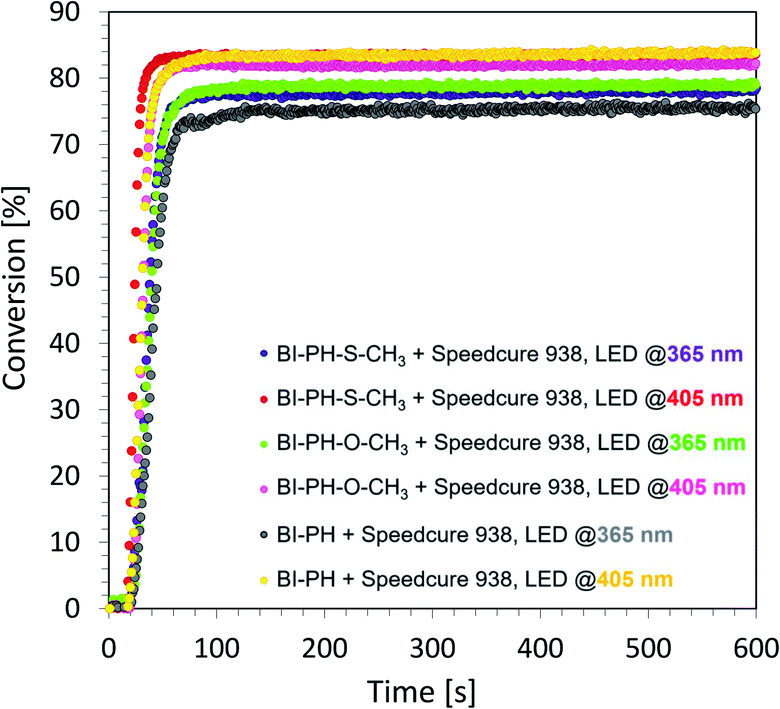
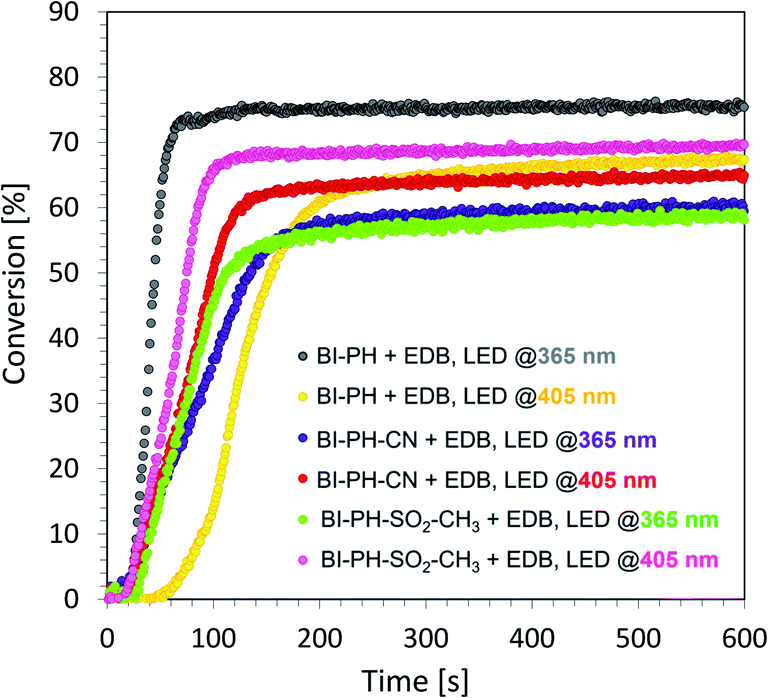
Evaluation of biphenyl derivatives as candidates for type II photoinitiating systems for free-radical polymerization of methacrylate monomers
According to electrochemical analysis, the investigated 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives exhibit the ability to initiate light-induced free-radical polymerization processes according to two types of photoinitiation mechanisms: photo-oxidation and photo-reduction. The effectiveness of the photoinitiation of the polymerization processes by these two mechanisms has been verified on the example of two biphenyls with electron-donating substituents (i.e. BI-PH-S-CH3 and BI-PH-O-CH3) and two with the electron-withdrawing substituents (i.e. BI-PH-CN and BI-PH-SO2-CH3). The base molecule (BI-PH) was chosen as a reference for comparison in both types of initiation reactions of free-radical polymerization. For both types of the initiation mechanism, the photopolymerization reactions of methacrylate monomers (BisGMA)/(TEGDMA) (50%/50% w/w) was examined using: LED @ 365 nm (16.27 mW cm−2), LED @ 405 nm (21.23 mW cm−2) as the light sources and carried out the reactions in air in thick-layer sample.
To study the photooxidation mechanism, a system containing a mixture of monomers BisGMA/TEGDMA (50%/50% w/w), iodonium salt (1 wt%) and the selected biphenyl derivatives (0.1 wt%) was prepared. In this case, a bimolecular photoinitiating system was formed, in which the biphenyl molecule, after absorbing the light, became an electron donor and was oxidised as the result of electron transfer, while the iodonium salt was reduced. It has been found that this two-component initiating systems are also very effective photoinitiating systems not only for monomers, photopolymerizable by cationic polymerization, but are also good for initiation of free-radical photopolymerization of methacrylate monomers at both UV-A LED (with emission at 365 nm) and visible LED (with emission at 405 nm) (Fig. 6). Conversely, for evaluation of the effectiveness of the photoreduction mechanism the composition containing the methacrylate monomers BisGMA/TEGDMA (50%/50% w/w) and a two-component initiating system, composed of EDB (1.5 wt%) as the electron donor, and selected biphenyl derivatives (1 wt%) as electron acceptors was prepared. Such two-component initiating systems have proven to be able to initiate the free-radical photopolymerization reaction also effectively, affording maximum conversion values up to 75% (Fig. 7). All obtained monomer conversions are summarised in Table 5. In addition to the high efficiency of the proposed two types of initiating systems, it has been proven that the use of biphenyl derivatives in free-radical photopolymerization in a thick-layer sample is very effective, and the influence of atmospheric oxygen on the obtained conversion was not significant.
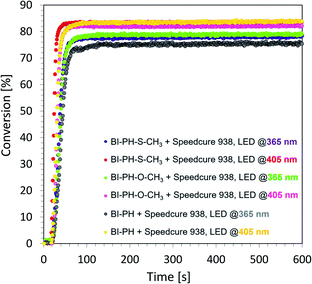 |
| | Fig. 6 Free-radical photopolymerization profiles for BisGMA/TEGDMA (50%/50% w/w) mixture, containing iodonium salt (1 wt%) and biphenyl derivatives (0.1 wt%) as the initiating system, and irradiated at 405 nm (16.27 mW cm−2). | |
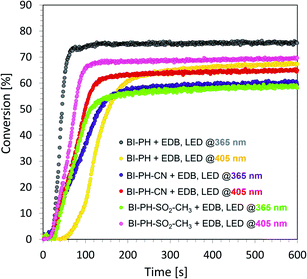 |
| | Fig. 7 Free-radical photopolymerization profiles for BisGMA/TEGDMA (50%/50% w/w) mixture, containing EDB (1.5 wt%) and biphenyl derivatives (0.1 wt%) as the initiating system, and irradiated at 405 nm (21.23 mW cm−2). | |
Table 5 Functional group conversions of the BisGMA/TEGDMA (50%/50% w/w) composition polymerizing according to the photo-oxidising or photo-reducing mechanisms, when exposed to light of different wavelengths
| Conversion [%] |
| Photo-oxidation mechanism |
Photo-reduction mechanism |
| Acronym |
BisGMA/TEGDMA (50%/50% w/w) with diphenyliodonium salt (1 wt%) |
Acronym |
BisGMA/TEGDMA (50%/50% w/w) with EDB (1.5 wt%) |
| @ 365a nm |
@ 405b nm |
@ 365a nm |
@ 405b nm |
| LED @ 365 nm ∼ 3.77 mW cm−2. LED @ 405 nm ∼ 8.13 mW cm−2. |
| BI-PH |
75.0 |
83.8 |
BI-PH |
75.6 |
64.1 |
| BI-PH-O-CH3 |
78.8 |
82.1 |
BI-PH-SO2-CH3 |
58.8 |
69.8 |
| BI-PH-S-CH3 |
78.5 |
83.6 |
BI-PH-CN |
60.2 |
65.3 |
Performance of 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives in preparation of interpenetrating polymer networks (IPNs)
An interpenetrating polymer network (IPN) is a polymer that consists of at least two polymer networks, where each network is the result of separate polymerization reaction. IPN formation has many advantages. First of all, it decreases the unfavourable influence of atmospheric oxygen on the polymerization of acrylates or methacrylates by mixing these monomers with other monomers polymerized by cationic mechanism (e.g. epoxy monomers). Conversely, combining monomers capable to form IPN type networks leads to final products with different properties than those obtained from individual components. In many cases, products of hybrid polymerization are characterised by higher degrees of conversion than single components, increased hardness and better mechanical stability. Finally, creating this type of networks allows to discover new polymeric products.
As shown in previous studies, the proposed 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives can be used with iodonium salt (bis-(4-t-butylphenyl)iodonium hexafluorophosphate) as two-component photoinitiating systems for both free radical and cationic polymerization processes. Encouraged by the high values of the obtained monomer conversions, hybrid polymerization was carried out, leading to the formation of IPN networks. Three biphenyl derivatives (BI-PH-S-CH3, BI-PH and BI-PH-O-CH3) were selected for this study, as they were the most effective as photosensitisers in both free radical and cationic light-induced polymerizations. Each of the tested systems consisted of a suitable mixture of monomers, ∼0.1 wt% (3.69 × 10−3 mol dm−3) of an appropriate 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives and ∼1 wt% of the iodonium salt (1.9 × 10−2 mol dm−3). For comparison, polymerization reactions were carried out in various conditions: in laminate, between two polypropylene films (the thickness of the composition was around 25 μm), and in air: (1) on a BaF2 pellet (thickness ∼ 25 μm), or (2) in a ring form (1.5 mm thickness and 12 mm diameter).
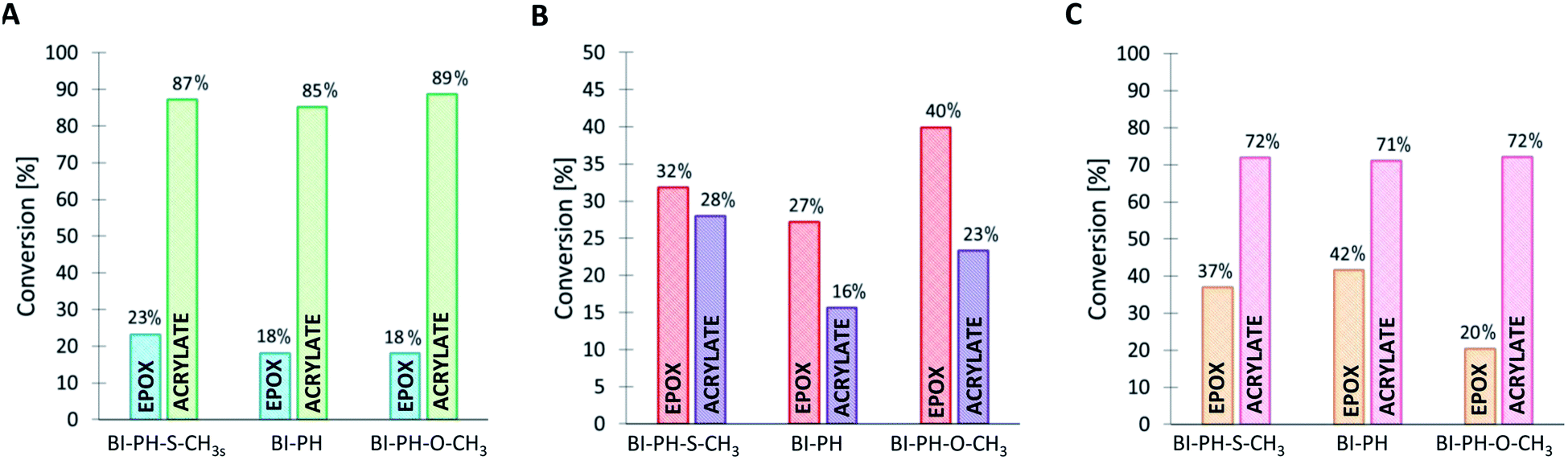
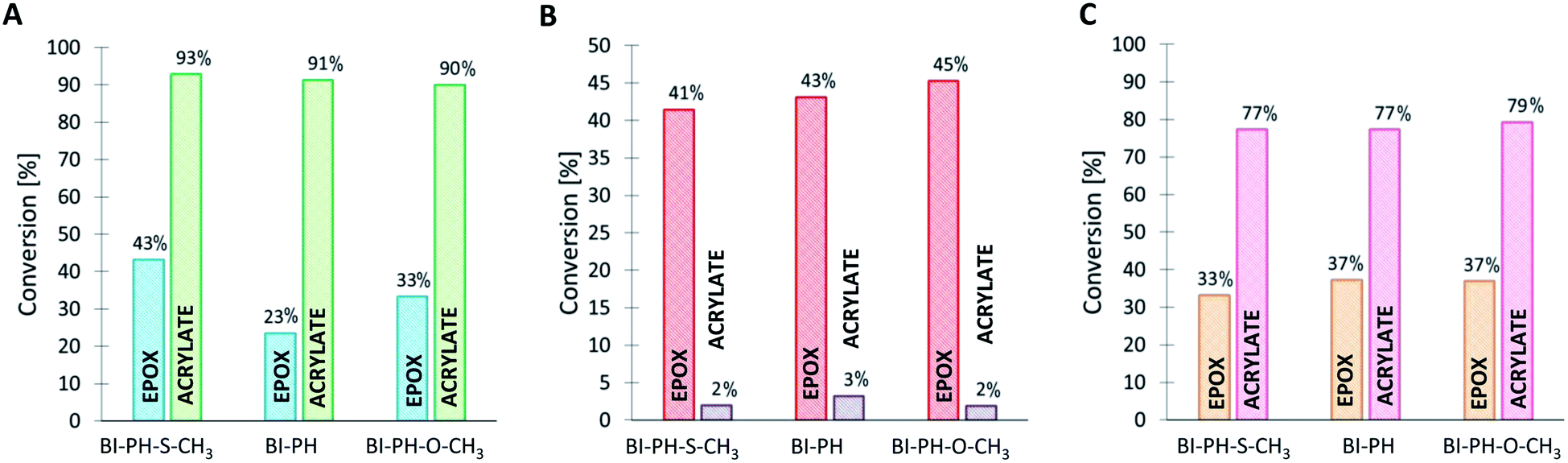
The following compositions were investigated using the real-time FT-IR method: CADE/TMPTA (1:1% w/w) and CADE/TMPTA/M100 (1:1:1% w/w/w). The disappearance of the double bond of acrylate monomer was continuously monitored at 1635 cm−1 for thin layers and at 6165 cm−1 for thick layers, while the ring opening processes were monitored at 790 cm−1 for thin layers and at 3700 cm−1 for thick layers. The final conversion values obtained after irradiation with LED @ 405 nm are shown in Fig. 8 for the CADE/TMPTA system and in Fig. 9 for the CADE/TMPTA/M100 system.
 |
| | Fig. 8 Functional group conversions in IPNs formed from CADE/TMPTA (1:1% w/w). (A) Thick layer at @ 405 nm, (B) air at @ 405 nm, (C) laminate at @ 405 nm (8.13 mW cm−2). | |
 |
| | Fig. 9 Functional group conversions in IPNs formed from CADE/TMPTA/M100 (1:1:1% w/w/w). (A) Thick layer at @ 405 nm, (B) air at @ 405 nm, (C) laminate at @ 405 nm (8.13 mW cm−2). | |
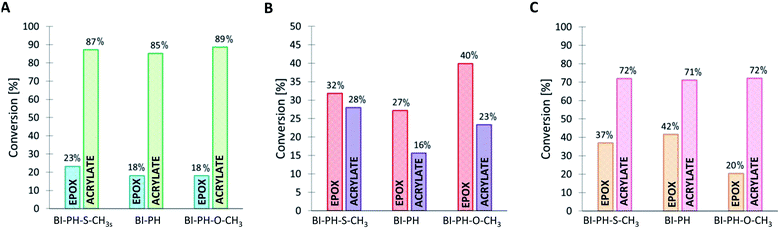
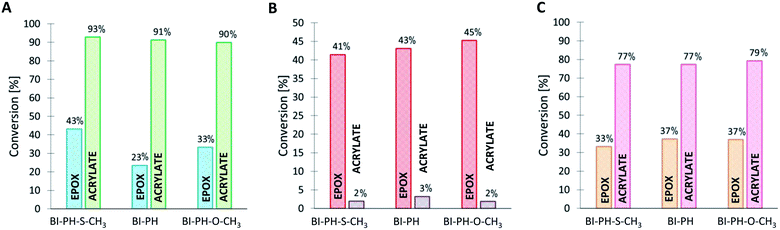
It has been proven that the use of the proposed photoinitiating systems leads to high final conversions during the formation of IPN networks. Depending on the conditions of the polymerization reaction, different degrees of conversion were obtained for different monomers. Thus, when the sample was exposed to oxygen (reaction in air), the polymerization process occurred for both monomers, but significantly higher values of conversion were obtained for the functional group polymerizing by the cationic polymerization mechanism. The opposite is noticeable when the polymerization reaction was carried out in laminate (Fig. 8 and 9). Acrylate monomers showed excellent conversions, whereas the values obtained for epoxy groups were much lower due to the faster free-radical polymerization of acrylates than the cationic polymerization of the epoxides under anaerobic conditions. Interestingly, very good results were obtained for the polymerization of a thick layer of the composition in air, for both the cationic and free-radical processes. For each case, the applicability of the real-time FT-IR technique for monitoring the formation of IPNs has been proven. The list of all final conversions obtained for particular functioned groups under different photopolymerization conditions, is presented in Table 6.
Table 6 Functional group conversions obtained in the process of photo-formation of various types of IPN networks, using photoinitiating systems based on bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1 wt%) and 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives (0.1 wt%), determined by real-time FT-IR
| Composition |
Experimental conditions and monitoring wavelengths |
Functional group conversion |
| BI-PH-S-CH3 |
BI-PH |
BI-PH-O-CH3 |
| Exposure to LED light at @ 365 nm; I0 = 3.77 mW cm−2 |
| CADE/TMPTA (1/1 w/w) |
Laminate |
EPOX at 790 cm−1 |
36.9 |
32.4 |
20.4 |
| ACRYLATE at 1635 cm−1 |
71.9 |
71.1 |
72.2 |
| Air thin layer |
EPOX at 790 cm−1 |
31.8 |
27.2 |
36.9 |
| ACRYLATE at 1635 cm−1 |
28.0 |
15.6 |
23.3 |
| Air thick layer |
EPOX at 3700 cm−1 |
48.1 |
38.8 |
42.9 |
| ACRYLATE at 6165 cm−1 |
87.1 |
85.6 |
86.4 |
| CADE/TMPTA/M100 (1/1/1 w/w/w) |
Laminate |
EPOX at 790 cm−1 |
24.0 |
22.9 |
23.4 |
| ACRYLATE at 1635 cm−1 |
77.1 |
75.5 |
75.8 |
| Air thin layer |
EPOX at 790 cm−1 |
41.0 |
43.1 |
45.3 |
| ACRYLATE at 1635 cm−1 |
3.2 |
3.3 |
3.5 |
| Air thick layer |
EPOX at 3700 cm−1 |
38.8 |
17.8 |
31.8 |
| ACRYLATE at 6165 cm−1 |
90.0 |
86.8 |
86.0 |
| TRITHIOL/TRIVINYL (0.21/0.79 w/w) |
Air thin layer |
THIOL at 2575 cm−1 |
98.2 |
96.2 |
94.6 |
| VINYL at 1635 cm−1 |
99.0 |
97.3 |
96.9 |
|
| Exposure to LED light at @ 405 nm; I0 = 8.13 mW cm−2 |
| CADE/TMPTA (1/1 w/w) |
Laminate |
EPOX at 790 cm−1 |
36.9 |
41.5 |
20.4 |
| ACRYLATE at 1635 cm−1 |
71.9 |
71.1 |
72.2 |
| Air thin layer |
EPOX at 790 cm−1 |
31.8 |
27.2 |
36.9 |
| ACRYLATE at 1635 cm−1 |
28.0 |
15.6 |
23.3 |
| Air thick layer |
EPOX at 3700 cm−1 |
23.1 |
18.0 |
18.0 |
| ACRYLATE at 6165 cm−1 |
87.1 |
88.7 |
85.2 |
| CADE/TMPTA/M100 (1/1/1 w/w/w) |
Laminate |
EPOX at 790 cm−1 |
33.2 |
37.4 |
36.7 |
| ACRYLATE at 1635 cm−1 |
78.7 |
77.2 |
79.2 |
| Air thin layer |
EPOX at 790 cm−1 |
41.9 |
43.5 |
45.2 |
| ACRYLATE at 1635 cm−1 |
2.3 |
3.3 |
1.8 |
| Air thick layer |
EPOX at 3700 cm−1 |
44.0 |
23.9 |
33.2 |
| ACRYLATE at 6165 cm−1 |
92.1 |
91.1 |
90.3 |
| TRITHIOL/TRIVINYL (0.21/0.79 w/w) |
Air thin layer |
THIOL at 2575 cm−1 |
96.5 |
96.7 |
94.7 |
| VINYL at 1635 cm−1 |
96.9 |
95.9 |
93.3 |
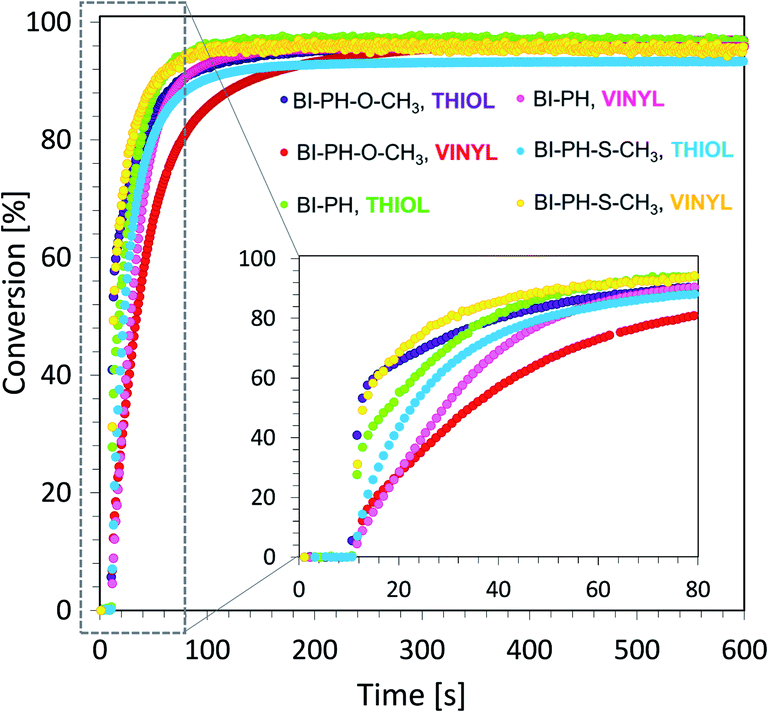
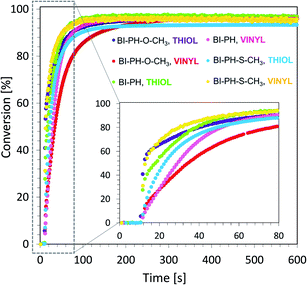
In addition, two new compositions composed of trimethylolpropane tris(3-mercaptopropionate) (TRITHIOL) and tris[4-(vinyloxy)butyl] trimellitate (TRIVINYL) (0.21:0.79 w/w) were tested to create fast polymerizing crosslinked IPNs with a high degree of conversion of the component monomers. The reaction was carried out on a BaF2 pellet (the thickness of the composition was around 25 μm). The vinyl groups content was continuously monitored in air at 1635 cm−1, while the thiol group delay was measured at 2575 cm−1 for 600 s. As shown in Fig. 10, the polymerization reaction in such a network occurred up to 90% conversion for both thiol and vinyl monomers. The final values of the obtained conversions are given in Table 6.
 |
| | Fig. 10 Changes of conversion particular functional groups during photopolymerization of TRITHIOL and TRIVINYL monomers (0.21:0.79 w/w) in air at @ 405 nm (19.82 mW cm−2). | |
3D printing experiments
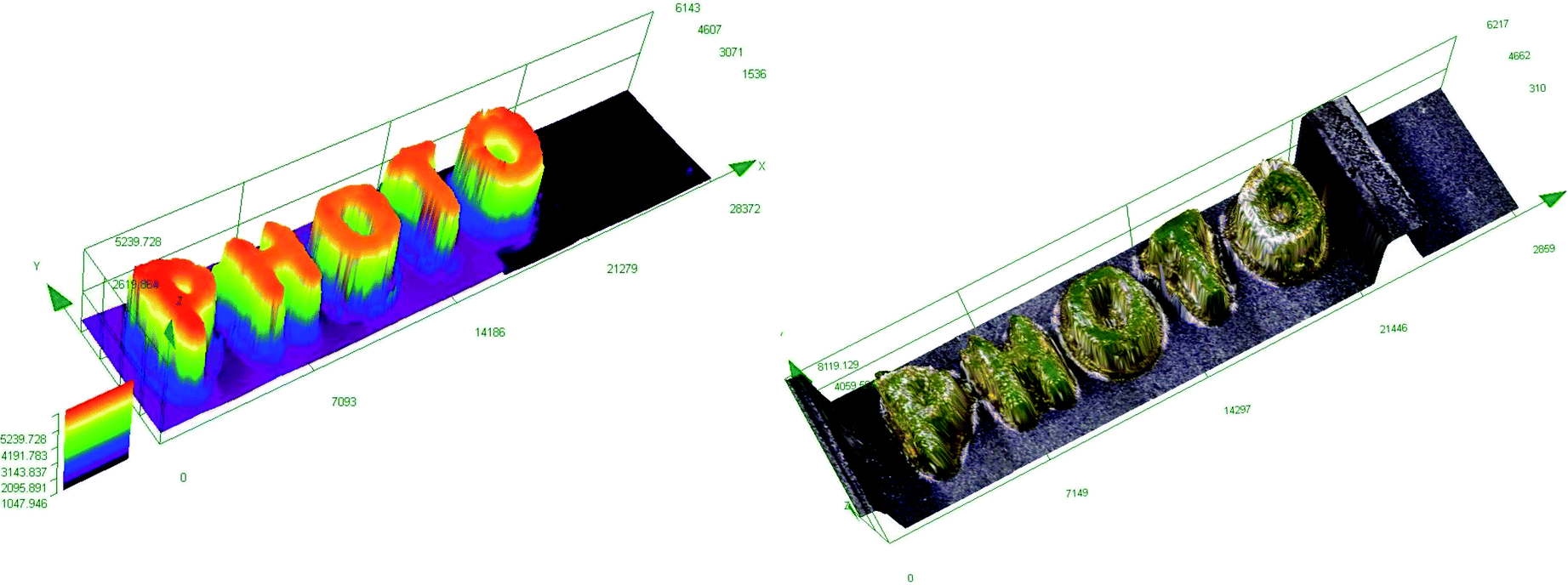
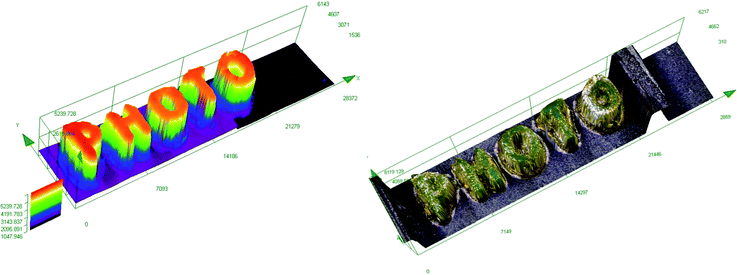
In order to obtain a high-resolution, thick printout, the biphenyl derivative BI-PH-S-CH3 was selected for the 3D printing experiments for the ring-opening cationic photopolymerization of a composition based on diglycidyl ether of bisphenol A (DGEBA). The use of diglycidyl ethers of bisphenol A (DEGDBA) with high molecular weight for the 3D printing process requires addition of low viscosity reactive diluent to reduce viscosity of the DEGDBA monomer. For this reason, a low viscosity glycidyl ether was combined with DEGDBA not only to reduce viscosity of the composition but also to improve the photopolymerization rate. Therefore, the composition consisting of DGEBA/EPXPROP (70%/30% w/w) containing the biphenyl derivative/iodonium salt (0.2/2 %w/w/) was used for the 3D printout. The 3D inscription “PHOTO” was easily obtained, and the printout had high thickness (∼5 mm) and a good quality, as shown in Fig. 11.
 |
| | Fig. 11 3D inscription made by the cationic photopolymerization of monomer mixtures: DEGBA/EPXPROP composition (70%/30% w/w) in the presence of a two-component photoinitiating system based on BI-PH-S-CH3 (0.2 wt%) and bis-(4-t-butylphenyl)iodonium hexafluorophosphate (2 wt%). | |
Preparation of nanocomposites with multiwalled carbon nanotubes (MWCNTs) by photopolymerization
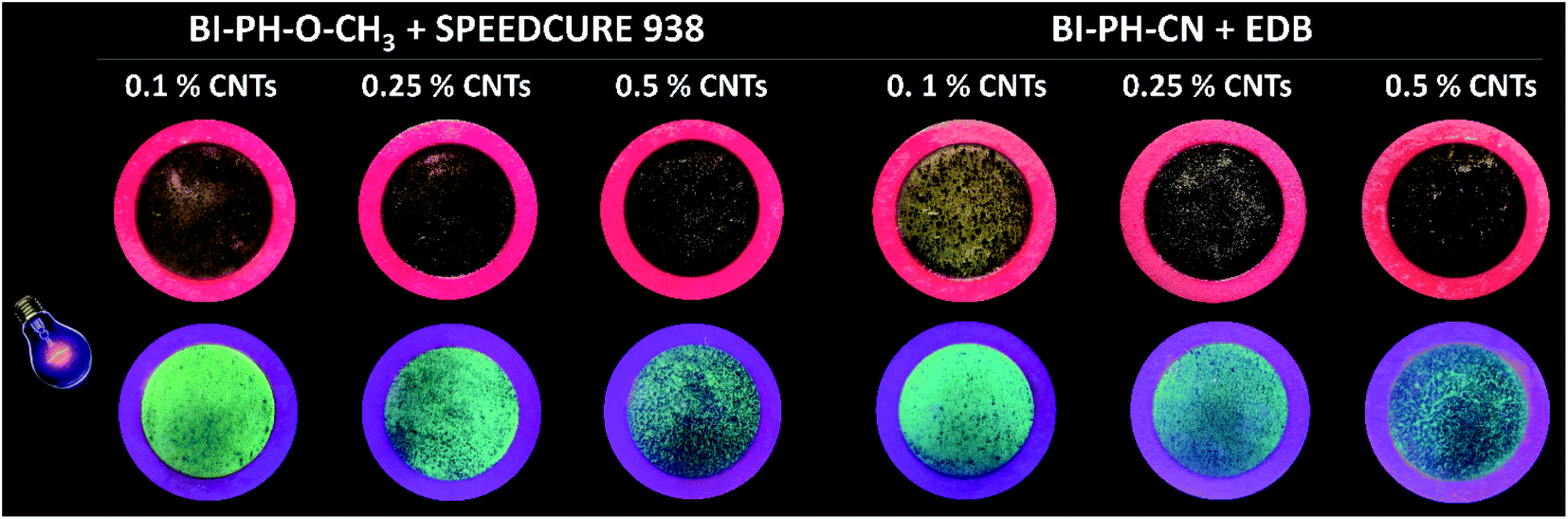
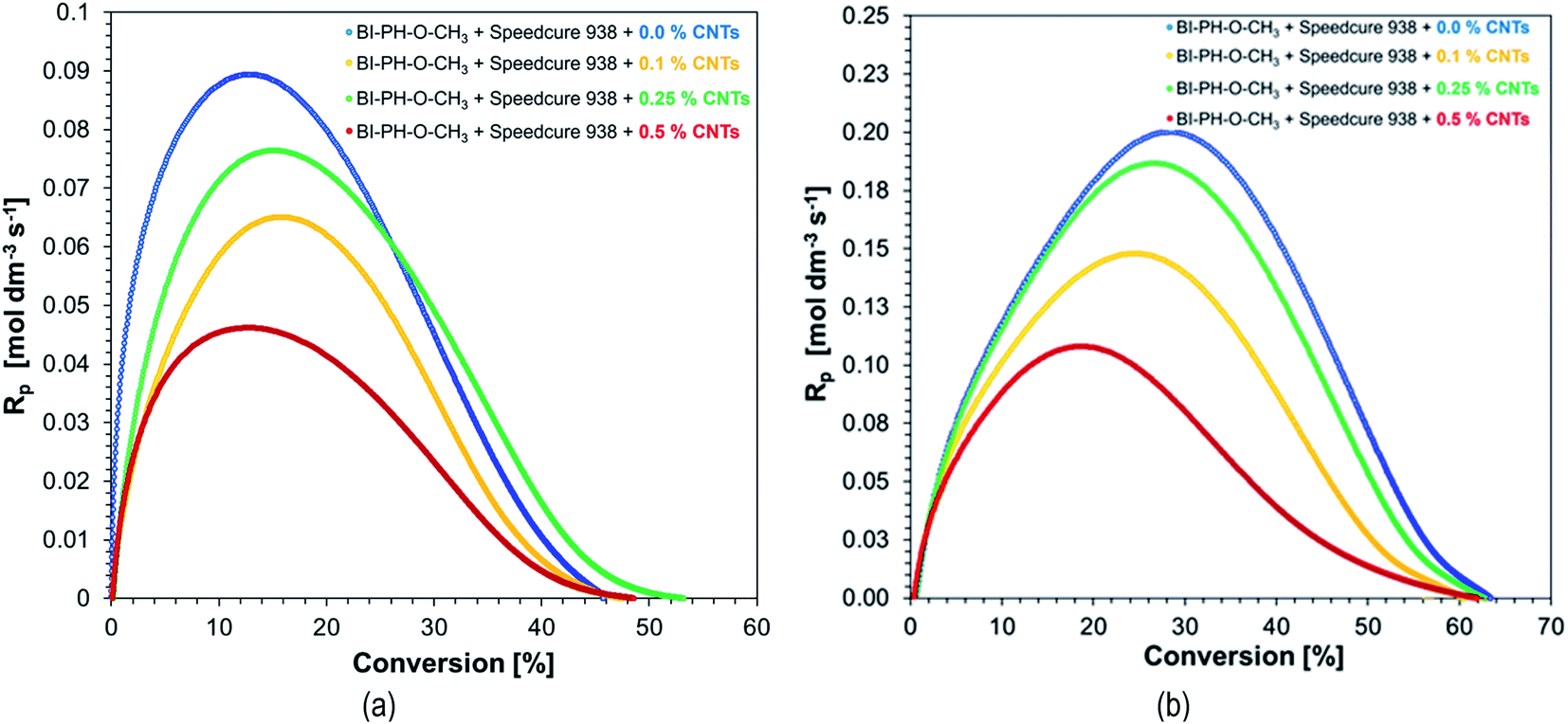
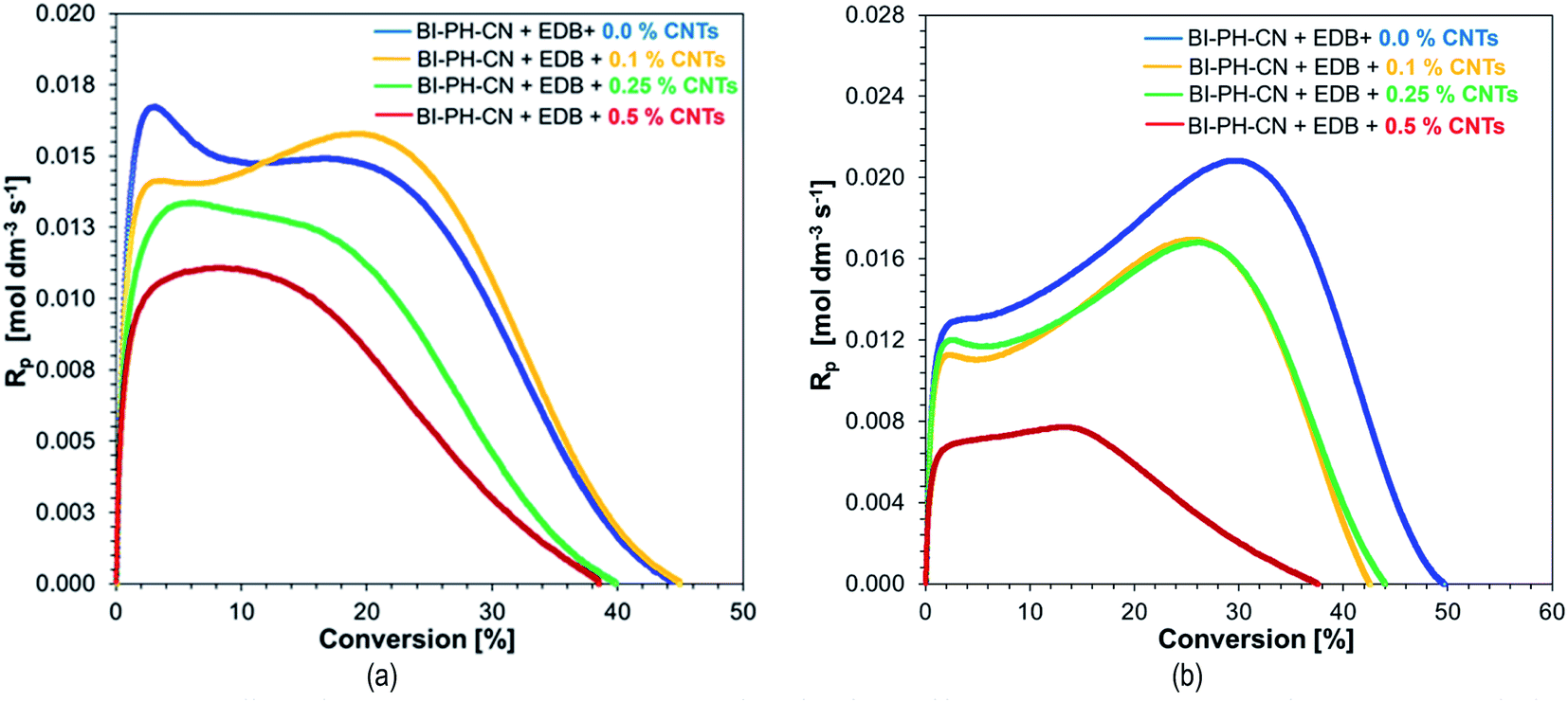
Due to high interest in polymeric nanocomposites in recent years, we have decided to examine the two-component initiation systems, based on the biphenyl derivatives, for the preparation of photocured multiwalled carbon nanotube (MWCNTs) composites (Fig. 12). Two types initiating systems were selected for the compositions of composition based on BisGMA/TEGDMA (50%/50% w/w): (1) BI-PH-O-CH3 (0.1 wt%) with bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1 wt%) for the photopolymerization initiated by the photo-oxidation mechanism; (2) BI-PH-CN (0.1 wt%) with EDB (1.5 wt%) for the reaction initiated by the photo-reduction mechanism. For both systems, the polymerization of pure composition without MWCNTs, as well as the compositions containing the following weight concentrations of the carbon nanotubes: 0.1%, 0.25%, 0.5% were tested. The conversions were determined by real-time FT-IR and photo-DSC. Furthermore, the compositions were tested at elevated temperature (70 °C).
 |
| | Fig. 12 Photographs of MWCNTs/methacrylate composites, obtaining using the initiating systems based on biphenyl derivatives working by the photo-oxidation or photo-reduction mechanisms. (A) photographs of nanocomposites under sunlight; (B) photographs of nanocomposites under excitation 365 nm. | |
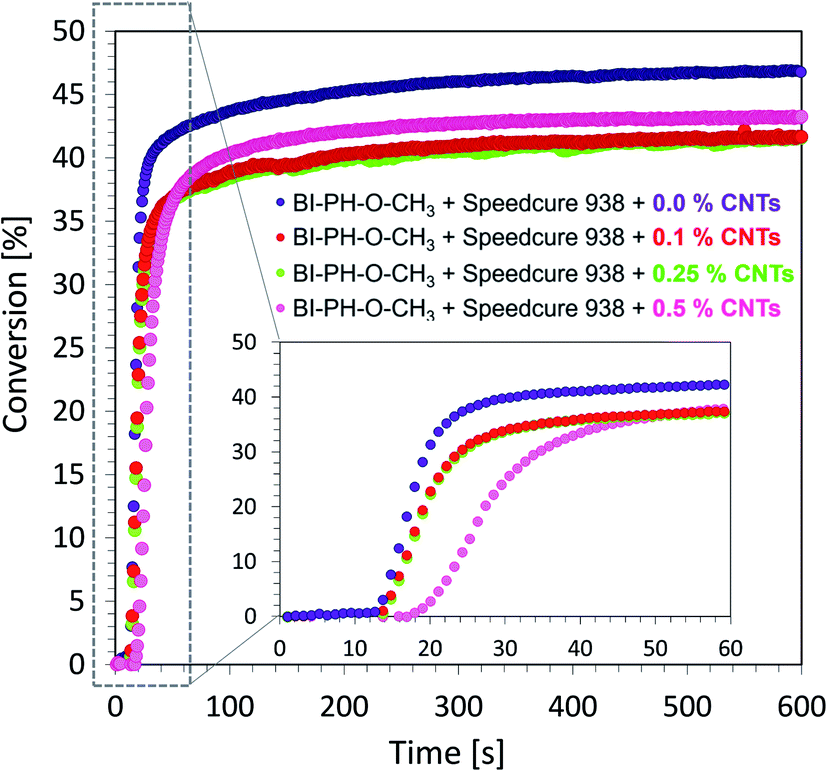
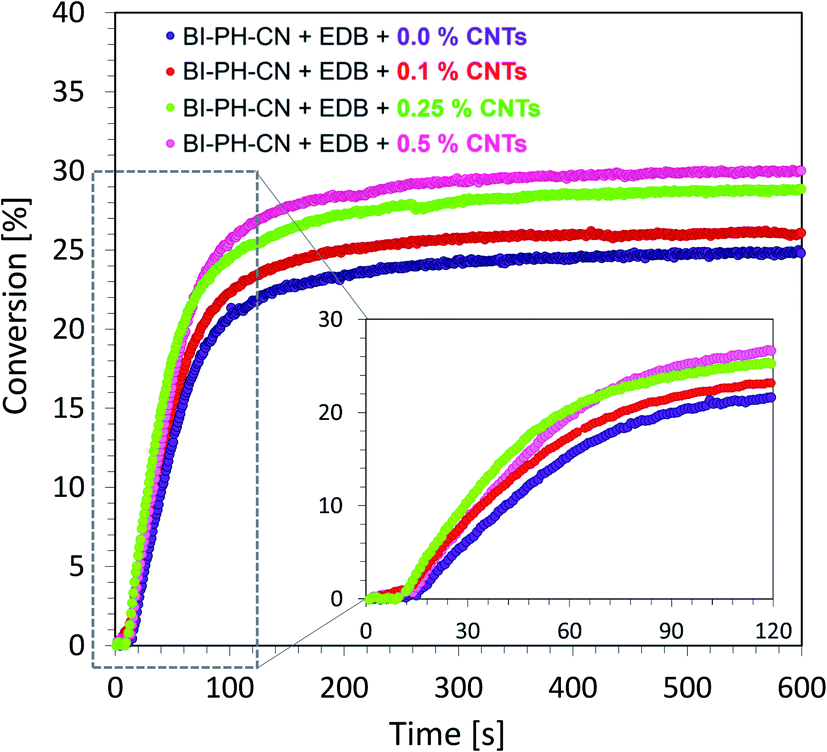
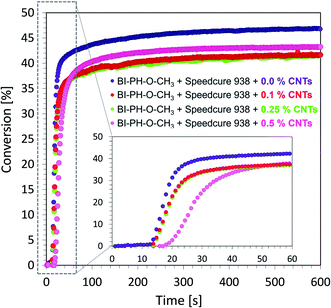
Monitoring of free-radical photopolymerization of the nanocomposites with MWCNT methacrylate compositions by real-time FT-IR. A drop of composition was placed between two polypropylene films (the thickness of the composition was around 25 μm). The sample was exposed to two types of light sources: LED @ 365 nm (16.27 mW cm−2) and LED @ 405 nm (21.23 mW cm−2). Methacrylate groups content was continuously monitored in air at 1635 cm−1 for 600 s. The functional groups conversion profiles are shown in Fig. 13 and 14. Table 7 contains the results obtained for the compositions studied. The investigated biphenyl derivatives in combination with either Speedcure 938 (iodonium salt usually used as the typical cationic photoinitiator) or EDB (usually used as a co-initiator in free-radical type II photoinitiating systems) enabled the free-radical polymerization of the photocurable methacrylate composites containing multiwalled carbon nanotubes. It has been proven that the photopolymerization of very thin coatings (about 25 μm) of such nanocomposites can be monitored using real-time FT-IR, while the addition of nanotubes does not significantly affect the achieved conversions.
 |
| | Fig. 13 Free-radical photopolymerization profiles for the photocurable composites: BisGMA/TEGDMA (50%/50% w/w) with bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1% wt) and BI-PH-O-CH3 (0.1% wt), polymerized at 365 nm (16.27 mW cm−2). | |
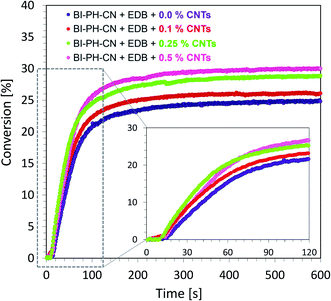 |
| | Fig. 14 Free-radical photopolymerization profiles for the photocurable composites: BisGMA/TEGDMA (50%/50% w/w) with EDB (1.5% wt) and BI-PH-CN (0.1% wt), polymerized at 365 nm (16.27 mW cm−2). | |
Table 7 Final conversions of methacrylate groups in the MWCNT/methacrylate composites based on BisGMA/TEGDMA (50%/50% w/w) monomers and the photoinitiating systems working by the photo-oxidation or photo-reduction of the biphenyl derivatives, determined by real-time FT-IR
| Photo-oxidation mechanism |
Photo-reduction mechanism |
| Biphenyl derivative |
BisGMA/TEGDMA (50%/50% w/w) with iodonium salt (1% wt) |
Biphenyl derivative |
(BisGMA)/(TEGDMA) (50%/50% w/w) with EDB (1.5% wt) |
| Content of CNTs [wt%] |
Conversion in [%] |
Content of CNTs [wt%] |
Conversion in [%] |
| @ 365a nm |
@ 405b nm |
@ 365a nm |
@ 405b nm |
| Real-time FT-IR source of light LED @ 365 nm ∼ 16.27 mW cm−2. Real-time FT-IR source of light LED @ 405 nm ∼ 21.23 mW cm−2. |
| BI-PH-O-CH3 |
0 |
46.7 |
41.0 |
BI-PH-CN |
0 |
24.9 |
30.0 |
| 0.1 |
41.6 |
42.6 |
0.1 |
26.1 |
23.2 |
| 0.25 |
41.5 |
42.5 |
0.25 |
28.9 |
28.2 |
| 0.5 |
43.5 |
36.6 |
0.5 |
30.0 |
26.9 |
On the other hand, the monitoring the photopolymerization of thick layers of these compositions was not possible using the FT-IR technique. Therefore, it was decided to use photo-DSC technique to monitor the photopolymerization reaction of the composite materials in thicker layers than those used for the FT-IR technique.
Post-curing experiments with the photocurable MWCNTs/methacrylate composites. Composites obtained both at room temperature and at 70 °C were exposed to post-curing experiments. The samples were heated twice from 0 °C to 250 °C and cooled twice. On the basis of the first heating, the content of the polymerizable groups remaining after photopolymerization was determined. This study yielded interesting results for the photocured resin and the composites. An exothermic peak on the DSC curves appeared in the temperature range 100 °C to 200 °C for all investigated samples. This means that additional radicals were generated by thermal energy without light irradiation, which led to exothermic reactions during the post-cure heat treatment. Appearance of the exotherm on the DSC curve indicated that the photopolymerization at 25 °C was not completed by the irradiation with 5.45 mW cm−2 intensity for 1200 seconds, which confirmed that at that temperature the polymerization stopped at some point for steric reason, due to solidification of the composition and the excess of free unreacted functional groups in the cured polymer are characteristic for fast reacting systems like the free-radical photopolymerization processes of acrylates and methacrylates.The additional functional group conversions achieved during the post-cure treatment are included in Table 9. It is noteworthy that the total methacrylate groups conversion in the BisGMA/TEGDMA (50%/50%, w/w) monomer system, which is the sum of the conversions achieved during the photopolymerization and the post-cure treatment, was approximately the same, regardless of the nanotube contents (i.e., within an experimental error) in the case of the samples photopolymerized using the BI-PH-O-CH3/Speedcure 938 initiating system the total conversion was on average 63.9% (with standard deviation:2.3%). Whilst in the case of the BI-PH-CN/EDB initiating system, the average total conversion was 54.3% (with standard deviation: 4.2%). This means that the initiating system working by the photo-oxidation mechanism was more effective than the initiating system working by the photo-reduction mechanism. Moreover, these data indicate, that the methacrylate groups conversion of the order of 65% is the maximal conversion achievable in the BisGMA/TEGDMA(50%/50%, w/w) monomer system under given measurement conditions. Further polymerization of the functional groups in the crosslinked polymer that was formed, did not occur for steric reasons. The additional of small amount of MWCNT (up to 0.5 wt%) did not affect significantly that conversion.
Table 9 Summary of photo-DSC obtained during post-curing of selected systems: BI-PH-O-CH3 (0.1% wt) + BisGMA/TEGDMA (50%/50% w/w) with bis-(4-t-butylphenyl)iodonium hexafluorophosphate (1% wt) and BI-PH-CN (0.1% wt) + BisGMA/TEGDMA (50%/50% w/w) EDB (1.5% wt)
| Post-curing effect for the sample with BisGMA/TEGDMA (50%/50% w/w) with iodonium salt (1% wt) at 25 °C with photo-DSC source of light: UV-LED @ 365 nm ∼ 5.45 mW cm−2 |
Post-curing effect for the sample with BisGMA/TEGDMA (50%/50% w/w) with EDB (1.5% wt) at 25 °C with photo-DSC source of light: UV-LED @ 365 nm ∼ 5.45 mW cm−2 |
| Acronym |
Content of CNTs [wt%] |
ΔH [mJ mg−1] |
T at Hmax [°C] |
Hmax [W g−1] |
Acronym |
Content of CNTs [wt%] |
ΔH [mJ mg−1] |
T at Hmax [°C] |
Hmax [W g−1] |
| BI-PH-O-CH3 |
0 |
44.82 |
193.4 |
0.716 |
BI-PH-CN |
0 |
24.02 |
165.3 |
0.749 |
| 0.1 |
43.90 |
184.5 |
0.786 |
0.1 |
18.77 |
162.9 |
0.605 |
| 0.25 |
35.77 |
168.4 |
0.846 |
0.25 |
33.21 |
162.9 |
0.723 |
| 0.5 |
55.97 |
168.9 |
0.930 |
0.5 |
25.02 |
166.4 |
0.692 |
| Post-curing effect for the sample with BisGMA/TEGDMA (50%/50% w/w) with Speedcure 938 (1% wt) at 70 °C with photo-DSC source of light: UV-LED @ 365 nm ∼ 5.45 mW cm−2 |
Post-curing effect for the sample with BisGMA/TEGDMA (50%/50% w/w) with EDB (1.5% wt) at 70 °C with photo-DSC source of light: UV-LED @ 365 nm ∼ 5.45 mW cm−2 |
| Acronym |
Content of CNTs [wt%] |
ΔH [mJ mg−1] |
T at Hmax [°C] |
Hmax [W g−1] |
Acronym |
Content of CNTs [wt%] |
ΔH [mJ mg−1] |
T at Hmax [°C] |
Hmax [W g−1] |
| BI-PH-O-CH3 |
0 |
No post-curing effect |
BI-PH-CN |
0 |
30.32 |
169.4 |
0.644 |
| 0.1 |
No post-curing effect |
0.1 |
46.39 |
168.0 |
0.706 |
| 0.25 |
No post-curing effect |
0.25 |
36.80 |
167.2 |
0.752 |
| 0.5 |
No post-curing effect |
0.5 |
39.54 |
163.2 |
0.560 |
Fluorescent microscopic visualisation of photocurable nanocomposites with MWCNTs
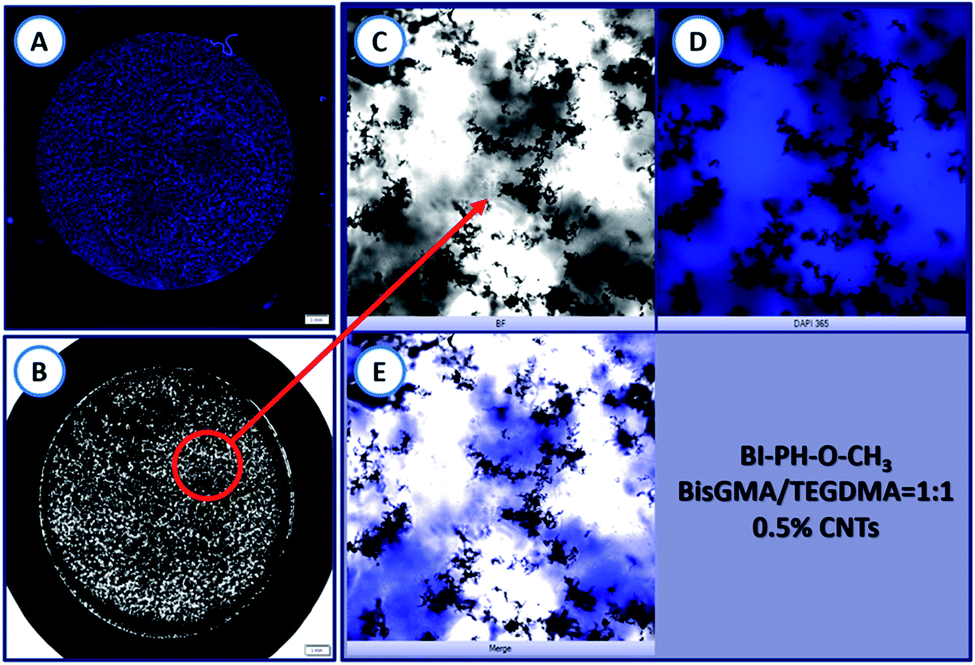
The morphology of the nanocomposites obtained in the ring form (1.5 mm thickness and 12 mm in diameter) was analysed using an inverted fluorescence microscope Olympus IX83 (from OLYMPUS) equipped with X-line lenses and a monochrome camera (Photometrics Prime BSI). On the basis of the recorded images, it was clearly observed that the carbon nanotubes in the composite were visibly aggregated. Because of the high surface energy, due to van der Waals forces, CNTs are extremely difficult to disperse in the polymer matrix and, they agglomerate very easily. In this case, the agglomeration led to the non-uniformity of the photocured samples (Fig. 17).
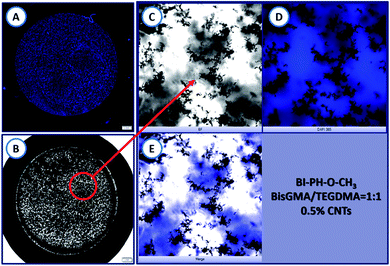 |
| | Fig. 17 Morphology of the composite obtained by photopolymerization of a photocurable composition containing: 0.5 wt% of MWCNTs (NC7000™) in BisGMA/TEGDMA monomer mixture (50%/50% w/w), at room temperature in air. The photopolymerization was initiated with a two-component initiating system composed of BI-PH-O-CH3 (0.1% wt) and iodonium salt (1% wt) and a high-power 365 nm UV light (SOLIS-365C) was used. The UV light intensity at the sample surface was 280 mW cm−2 at room temperature and under atmospheric conditions, the irradiation time was 360 s. (A) The sample seen in 365 nm UV light; (B) the sample seen in daylight; (C) a bright-field microscopy image of the sample illumination by white light; (D) a bright-field microscopy image of the sample illuminated with 365 nm UV-LED light; (E) merge of (C) and (D) images for better visualisation of the MWCNT agglomerates. | |
Therefore, the mixing process is a crucial pre-processing step to disperse homogeneously the nanofiller in the resin and to avoid agglomeration. During preparation of the samples containing, mechanical magnetic stirring was used, which turned out to be not good enough to disperse the nanotubes uniformly. Based on the obtained data, it is suggested that more effective homogenisation of the sample is required for preparing these types of compositions. Alternatively, the agglomeration effect can be prevented using carbon nanotubes with chemical functionalization of their walls.
3D printing experiments with MWCNTs composites
Encouraged by the good results of the conversions obtained for the tested composites, we have decided to examined the possibility of 3D printing from a resin containing a dispersed nanofiller, namely the thin multiwalled carbon nanotubes (MWCNTs) NC7000™. For this purpose, the compositions based on BisGMA/TEGDMA methacrylate monomers were tested. As shown in Fig. 18, the 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives can be successfully applied as photosensitisers for iodonium salts in the photo-oxidation mechanism of initiation process, and as components of bimolecular photoinitiating system with amines as co-initiators, for the 3D photo-printing of nanocomposite materials. As a result of laser irradiation, the printout “CNTs OX” was obtained, from MWCNTs/methacrylate composition containing 0.1% by weight of carbon nanotubes, using the initiating system based on diphenyliodonium salt + BI-PH-O-CH3. This system initiated the polymerization to the photo-oxidation mechanism. Conversely, the “CNT” printout was obtain from the same composition using the initiating system based on EDB + BI-PH-CN. This system initiated the polymerization reaction by the photoreduction of the biphenyl derivative. In both cases, it was demonstrated that the investigated biphenyl derivatives can effectively initiated the process of photopolymerization of the methacrylates containing carbon nanotubes, and proceeded 3D printouts of the nanocomposite with good optical resolution and significant thickness 1.5 mm.
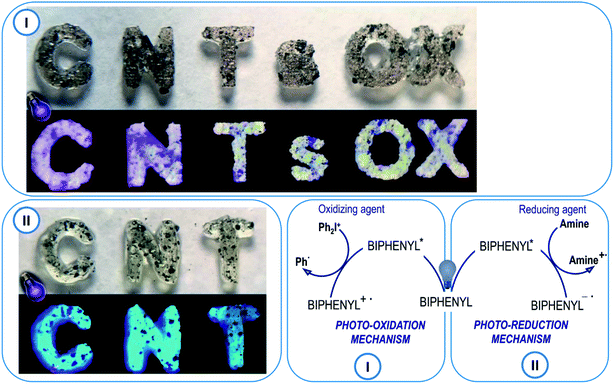 |
| | Fig. 18 3D inscriptions made by free-radical photopolymerization of monomer BisGMA/TEGDMA (50%/50% w/w) mixtures in the presence of a two-component photoinitiating system based on (I) BI-PH-O-CH3 (0.1 wt%) + iodonium salt (1 wt%), (II) BI-PH-CN (0.1 wt%) + EDB (1.5 wt%) at room temperature and under atmospheric conditions. (A) photocurable MWCNT nanocomposites seen in sunlight; (B) photocurable MWCNT nanocomposites seen under excitation 365 nm UV-LED light source. | |
Conclusions
New 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives have been discovered and applied as highly effective photosensitisers for iodonium salt. Moreover these derivatives can be used in combination with amines in two-component photoinitiating systems for UV-A and visible light-initiated polymerization processes. Good spectroscopic properties and favourable electrochemical and thermodynamic properties of these derivatives guarantee a high degree of conversion in ring-opening cationic photopolymerization processes of epoxy and glycidyl monomers and free-radical photopolymerization processes of methacrylate and acrylate monomers. Moreover, the 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives are suitable for initiating system for preparation of interpenetrating polymer networks (IPNs) by photopolymerization method. The high efficiency of these initiating system allowed to obtain IPNs in various conditions: in air or in laminates as thin layers, and in air in thick layers. Additionally, these derivatives are able to initiate the free-radical photopolymerization processes of acrylate monomers by the photo-oxidation or photo-reduction mechanism, depending on the type of co-initiator. The initiating systems containing the biphenyl derivatives in combination with iodonium salts (e.g. Speedcure 938) generate initiating radicals by photo-oxidation of the biphenyl derivative, while the systems containing tertiary amine (e.g. EDB) generate radicals by photo-reduction of the biphenyl derivative. Furthermore, the initiating system based on 2-(diethylamino)-4-(1-ethylpropyl)-6-phenyl-benzene-1,3-dicarbonitrile derivatives are suitable for preparation of photocured multiwalled carbon nanotube (MWCNT) composites. The analysis of images of cross-linked nanocomposites from the fluorescence microscope allowed the conclusion that carbon nanotube reinforcement in the obtained methacrylate composites was agglomerated due to insufficient homogenisation of the sample. Despite this, however, satisfactory conversion rates were obtained during the polymerization of the samples containing 0.1%, 0.25% and 0.5% w/w of MWCNTs, which was confirmed by real-time FT-IR and photo-DSC tests. Finally, the developed initiating systems are suitable for 3D printing of nanocomposites. The results presented in this paper are the first step towards further research into the development of photocurable nanoreinforced compositions for use in 3D printing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Foundation for Polish Science (Warsaw, Poland) TEAM TECH project Grant No. TEAM TECH/2016-2/15 (POIR.04.04.00-00-204B/16-00). Computations were performed with open source software from PLGrid Infrastructure. The authors are also grateful to the Olympus Company and more specifically Mr Mateusz Pernak for the optical microscope DSX1000 and Dr Wojciech Brutkowski for the inverted fluorescence microscope Olympus IX83, both of which were used extensively in this research.
References
- A. Polykarpov and A. Tiwari, in Photocured Materials, ed. A. Tiwari and A. Polykarpov, Royal Society of Chemistry, Cambridge, 1st edn, 2015, Photocured materials: a general perspective, pp. 1–14 Search PubMed.
- I. Kamińska, J. Ortyl and R. Popielarz, Polym. Test., 2016, 55, 310–317 CrossRef.
- D. Nowak, J. Ortyl, I. Kamińska-Borek, K. Kukuła, M. Topa and R. Popielarz, Polym. Test., 2017, 64, 313–320 CrossRef CAS.
- D. Nowak, J. Ortyl, I. Kamińska-Borek, K. Kukuła, M. Topa and R. Popielarz, Polym. Test., 2018, 67, 144–150 CrossRef CAS.
- K. Kostrzewska, J. Ortyl, R. Dobosz and J. Kabatc, Polym. Chem., 2017, 8, 3464–3474 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- P. Glöckner, S. Struck, T. Jung and K. Studer, Radiation Curing: Coatings and Printing Inks: Technical Basics, Applications and Trouble Shooting, Vincentz Network, Hannover, 2008 Search PubMed.
- P. Chang, H. Mei, S. Zhou, K. G. Dassios and L. Cheng, J. Mater. Chem. A, 2019, 7, 4230–4258 RSC.
- J. Olmsted, J. Phys. Chem., 1979, 83, 2581–2584 CrossRef CAS.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593–611 CrossRef CAS.
- M. Layani, X. Wang and S. Magdassi, Adv. Mater., 2018, 30, e1706344 CrossRef PubMed.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641–647 RSC.
- J. Zhang and P. Xiao, Polym. Chem., 2018, 9, 1530–1540 RSC.
- E. Hola, J. Ortyl, M. Jankowska, M. Pilch, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin and J. Lalevée, Polym. Chem., 2020, 11, 922–935 RSC.
- E. Hola, M. Topa, A. Chachaj-Brekiesz, M. Pilch, P. Fiedor, M. Galek and J. Ortyl, RSC Adv., 2020, 10, 7509–7522 RSC.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- Z. Wang, W. Huang, P. Peng and D. E. Fennell, Chemosphere, 2010, 78, 147–151 CrossRef CAS PubMed.
- W. Tomal, M. Pilch, A. Chachaj-Brekiesz, M. Galek, F. Morlet-Savary, B. Graff, C. Dietlin, J. Lalevée and J. Ortyl, Polym. Chem., 2020, 11, 4604–4621 RSC.
- W. A. Green, Industrial Photoinitiators: A technical Guide, CRC Press, Boca Raton, 2010 Search PubMed.
- Z. Czech, A. Kowalczyk, J. Ortyl and J. Swiderska, Pol. J. Chem. Technol., 2013, 15, 12–14 CAS.
- K. Nakamura, Photopolymers: Photoresist materials, processes, and applications, CRC Press, Boca Raton, 2014 Search PubMed.
- U. Poth, Automotive coatings formulation : Chemistry, physics und practices, Vincentz Network, Hannover, 2008 Search PubMed.
- R. Schwalm, UV Coatings, Elsevier, Amsterdam, 2007 Search PubMed.
- W. Tomal and J. Ortyl, Polymers, 2020, 12, 1073 CrossRef CAS PubMed.
- J. Ortyl, J. Wilamowski, P. Milart, M. Galek and R. Popielarz, Polym. Test., 2015, 48, 151–159 CrossRef CAS.
- R. L. Sakaguchi, W. H. Douglas and M. C. Peters, J. Dent., 1992, 20, 183–188 CrossRef CAS PubMed.
- A. Tiwari, A. Shukla, Lalliansanga, D. Tiwari and S. M. Lee, J. Environ. Manage., 2018, 220, 96–108 CrossRef CAS PubMed.
- M. Sangermano, S. Pegel, P. Pötschke and B. Voit, Macromol. Rapid Commun., 2008, 29, 396–400 CrossRef CAS.
- D. Hull and T. W. Clyne, An Introduction to Composite Materials, Cambridge University Press, Cambridge, 1996 Search PubMed.
- M. N. dos Santos, C. V. Opelt, F. H. Lafratta, C. M. Lepienski, S. H. Pezzin and L. A. F. Coelho, Mater. Sci. Eng., A, 2011, 528, 4318–4324 CrossRef.
- Y. Li, M. L. Swartz, R. W. Phillips, B. K. Moore and T. A. Roberts, J. Dent. Res., 1985, 64, 1396–1401 CrossRef CAS PubMed.
- A. Endruweit, M. S. Johnson and A. C. Long, Polym. Compos., 2006, 27, 119–128 CrossRef CAS.
- D. Bomze, P. Knaack, T. Koch, H. Jin and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3751–3759 CrossRef CAS.
- M. Sangermano, A. D'Anna, C. Marro, N. Klikovits and R. Liska, Composites, Part B, 2018, 143, 168–171 CrossRef CAS.
- J.-D. Cho, H.-T. Ju and J.-W. Hong, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 658–670 CrossRef CAS.
- N. Klikovits, R. Liska, A. D'Anna and M. Sangermano, Macromol. Chem. Phys., 2017, 218, 1700313 CrossRef.
- B. Hasiaoui, A. Ibrahim, G. L'Hostis, K. Gautier, X. Allonas, C. Croutxé-Barghorn, B. Durand and F. Laurent, Polym. Adv. Technol., 2019, 30, 902–909 CrossRef CAS.
- F. Li, S. Zhou, B. You and L. Wu, J. Appl. Polym. Sci., 2006, 99, 1429–1436 CrossRef CAS.
- C. M. Chung, J. G. Kim, M. S. Kim, K. M. Kim and K. N. Kim, Dent. Mater., 2002, 18, 174–178 CrossRef CAS PubMed.
- X. Zhang, Y. Duan, X. Zhao and D. Li, J. Compos. Mater., 2016, 50, 1395–1401 CrossRef CAS.
- J. Rayss, W. M. Podkościelny, A. Gorgol, J. Widomski and J. Ryczkowski, J. Appl. Polym. Sci., 1995, 57, 1119–1125 CrossRef CAS.
- G. C. Xu, A. Y. Li, L. De Zhang, G. S. Wu, X. Y. Yuan and T. Xie, J. Appl. Polym. Sci., 2003, 90, 837–840 CrossRef CAS.
- T. Zhao, R. Yu, X. Li, Y. Zhang, X. Yang, X. Zhao and W. Huang, J. Mater. Sci., 2019, 54, 5101–5111 CrossRef CAS.
- E. Andrzejewska and M. Sadej, Polimery, 2008, 53, 321–323 CrossRef CAS.
- W. Khan, R. Sharma and P. Saini, Carbon Nanotube-Based Polymer Composites: Synthesis, Properties and Applications, in Carbon Nanotubes: Current Progress
of their Polymer Composites, ed. M. Reda Berber and I. Hazzaa Hafez, InTech, Rijeka, 1st edn, 2016, pp. 1–46 Search PubMed.
- R. Andrews and M. C. Weisenberger, Curr. Opin. Solid State Mater. Sci., 2004, 8, 31–37 CrossRef CAS.
- M. N. Dos Santos, C. V. Opelt, S. H. Pezzin, S. C. Amico, C. E. Da Costa, J. C. Milan, F. H. Lafratta and L. A. F. Coelho, Mater. Res., 2013, 16, 367–374 CrossRef.
- M. Martin-Gallego, M. Hernández, V. Lorenzo, R. Verdejo, M. A. Lopez-Manchado and M. Sangermano, Polymer, 2012, 53, 1831–1838 CrossRef CAS.
- M. Sangermano, I. Antonazzo, L. Sisca and M. Carello, Polym. Int., 2019, 68, 1662–1665 CrossRef CAS.
- Y.-Y. Wang and T.-E. Hsieh, Chem. Mater., 2005, 17, 3331–3337 CrossRef CAS.
- S. M. Sapuan and N. Bin Yusoff, The Relationship Between Manufacturing and Design for Manufacturing in Product Development of Natural Fibre Composites, in Manufacturing of Natural Fibre Reinforced Polymer Composites, ed. M. S. Salit, M. Jawaid, N. Bin Yusoff and M. E. Hoque, Springer International Publishing, Cham, 1st edn, 2015, pp. 1–15 Search PubMed.
- E. Andrzejewska, M. Andrzejewski, J. Jeczalik and T. Sterzyński, Polimery, 2009, 54, 382–385 CrossRef CAS.
- M. Sadej and E. Andrzejewska, Prog. Org. Coat., 2016, 94, 1–8 CrossRef CAS.
- D. Prządka, A. Marcinkowska and E. Andrzejewska, Prog. Org. Coat., 2016, 100, 165–172 CrossRef.
- A. Marcinkowska, D. Prządka and E. Andrzejewska, J. Coat. Technol. Res., 2019, 16, 167–178 CrossRef CAS.
- M. Sadej, H. Gojzewski and E. Andrzejewska, J. Polym. Res., 2016, 23, 116 CrossRef.
- H. Gojzewski, M. Sadej, E. Andrzejewska and M. Kokowska, Eur. Polym. J., 2017, 88, 205–220 CrossRef CAS.
- H. Gojzewski, M. Sadej, E. Andrzejewska and M. Kokowska, Data Brief, 2017, 12, 528–534 CrossRef PubMed.
- M. Sadej-Bajerlain, H. Gojzewski and E. Andrzejewska, Polymer, 2011, 52, 1495–1503 CrossRef CAS.
- M. Sadej, H. Gojzewski, P. Gajewski, G. J. Vancso and E. Andrzejewska, eXPRESS Polym. Lett., 2018, 12, 790–807 CrossRef CAS.
- P. Jojibabu, Y. X. Zhang and B. G. Prusty, Int. J. Adhes. Adhes., 2020, 96, 102454 CrossRef CAS.
- T. C. Huang, J. M. Yeh and C. Y. Lai, Polymer Nanocomposite Coatings, in Advances in Polymer Nanocomposites: Types and Applications, ed. F. Gao, Woodhead Publishing Limited, Cambridge, 1st edn, 2012, pp. 605–638 Search PubMed.
- W. E. Jones, J. Chiguma, E. Johnson, A. Pachamuthu and D. Santos, Material, 2010, 3, 1478–1496 CrossRef CAS.
- W. Tomal, M. Pilch, A. Chachaj-Brekiesz and J. Ortyl, Catalysts, 2019, 9, 827 CrossRef CAS.
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS.
- S. Ban and J. Hasegawa, Dent. Mater. J., 1984, 3, 85–92 CrossRef CAS PubMed , 129.
- P. P. Romańczyk and S. S. Kurek, Electrochim. Acta, 2017, 255, 482–485 CrossRef.
- B. Strehmel, Z. Phys. Chem., 2014, 228, 129–153 CAS.
- E. Andrzejewska, D. Zych-Tomkowiak, M. Andrzejewski, G. L. Hug and B. Marciniak, Macromolecules, 2006, 39, 3777–3785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04146g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Anna Chachaj-Brekiesz
a,
Anna Chachaj-Brekiesz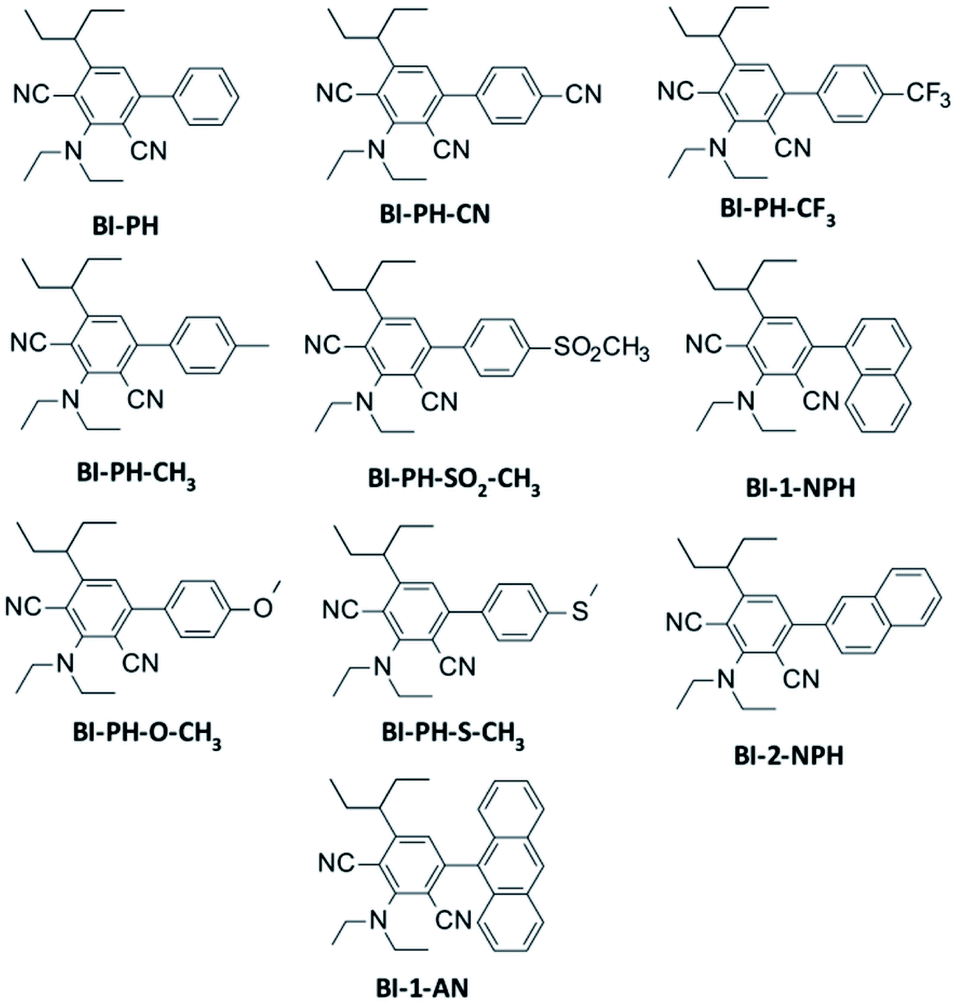
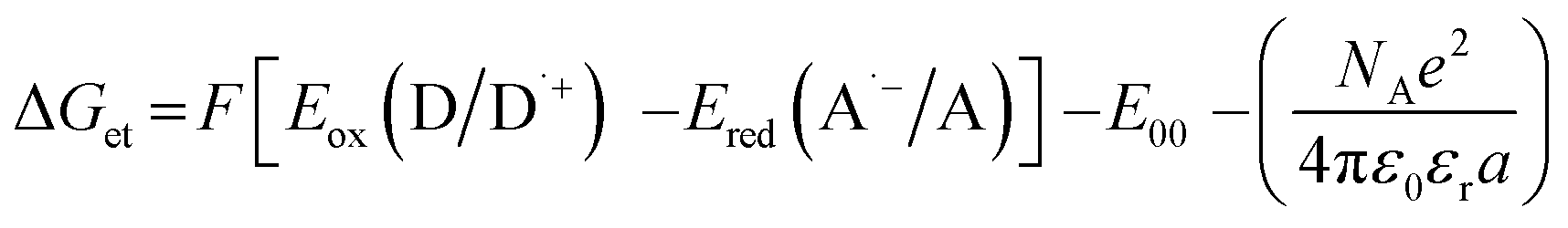
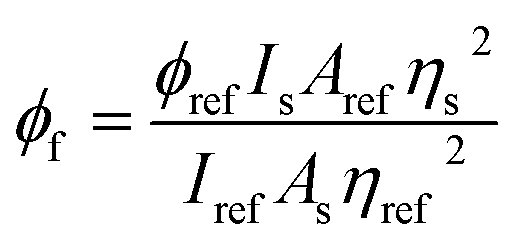
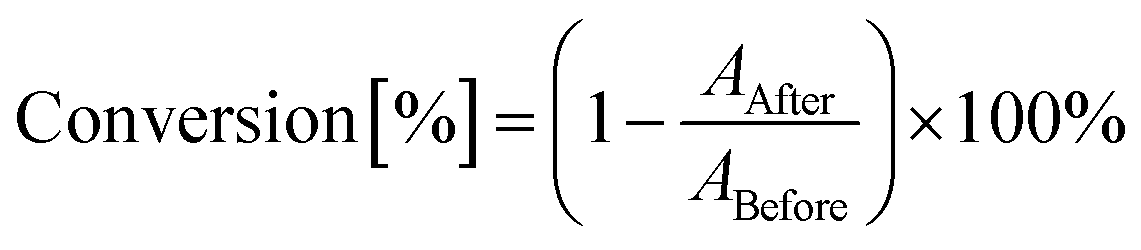
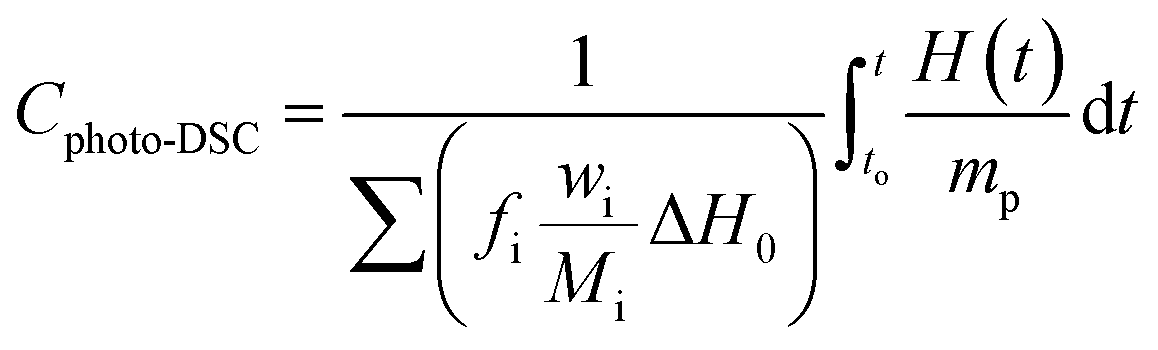
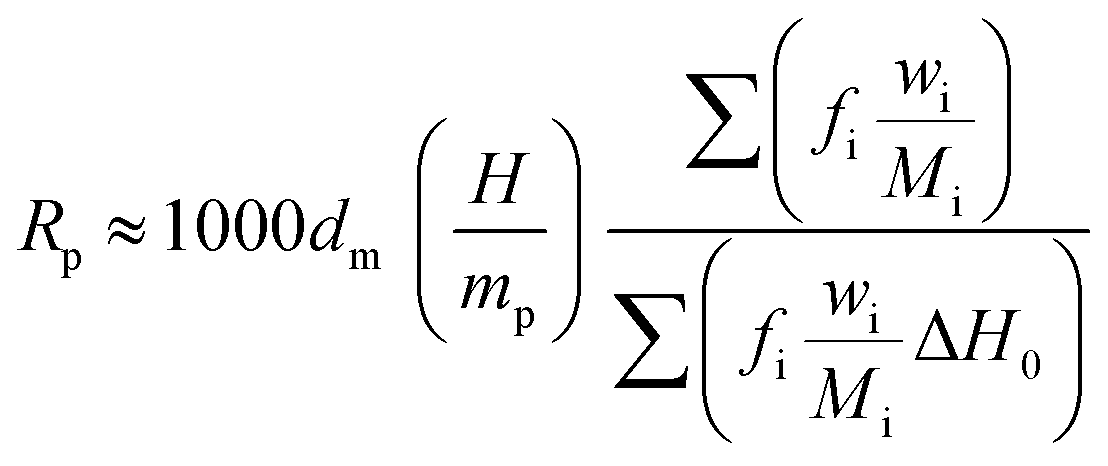
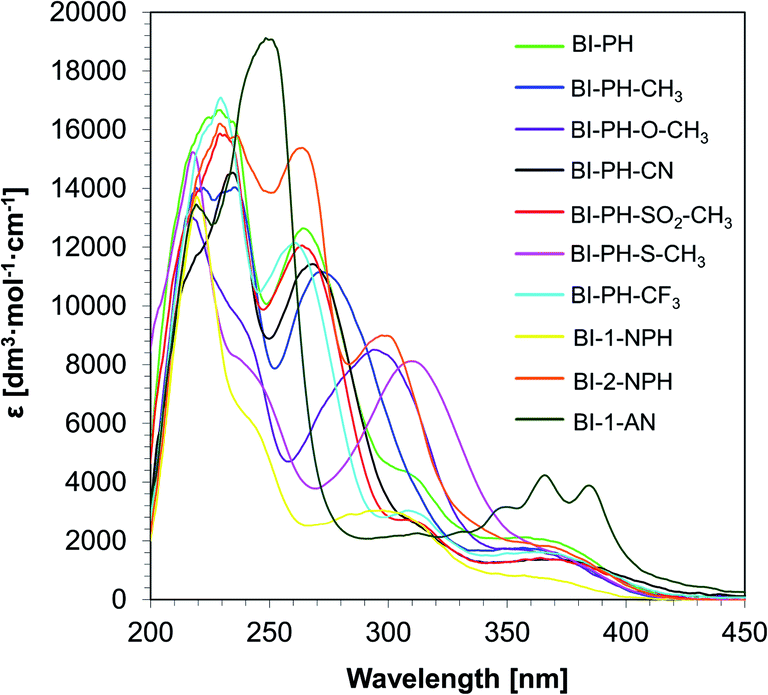
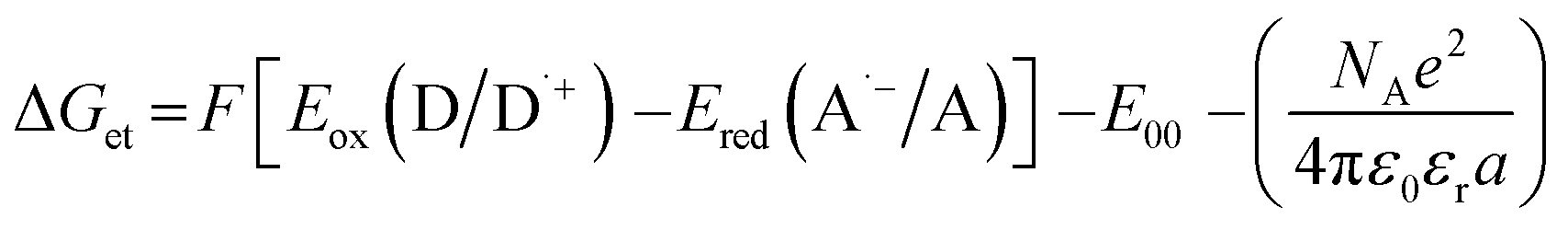 , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor, Ered(A˙−/A) – electrochemically determined reduction potential of the electron acceptor (−0.64 V for the diphenyliodonium salt vs. Ag/AgCl),67,68 ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[Speedcure 938]/(1 + KSV[Speedcure 938]) for the concentration of iodonium salt [Speedcure 938] = 0.021 mol dm−3.
, Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor, Ered(A˙−/A) – electrochemically determined reduction potential of the electron acceptor (−0.64 V for the diphenyliodonium salt vs. Ag/AgCl),67,68 ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[Speedcure 938]/(1 + KSV[Speedcure 938]) for the concentration of iodonium salt [Speedcure 938] = 0.021 mol dm−3.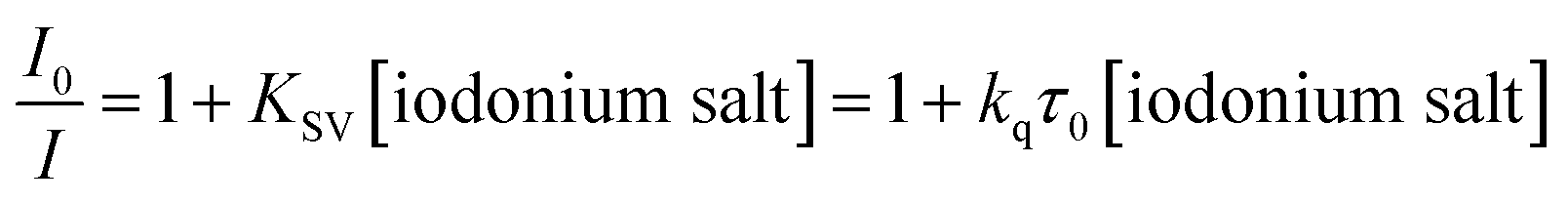
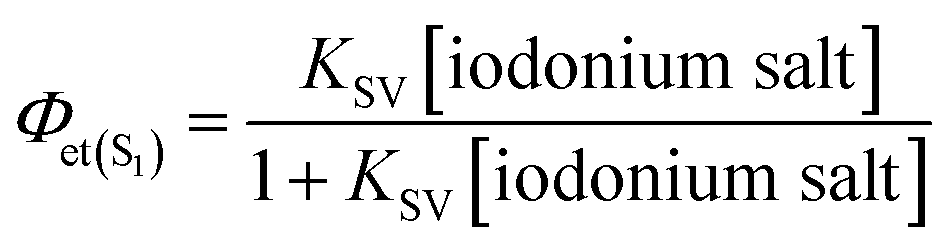
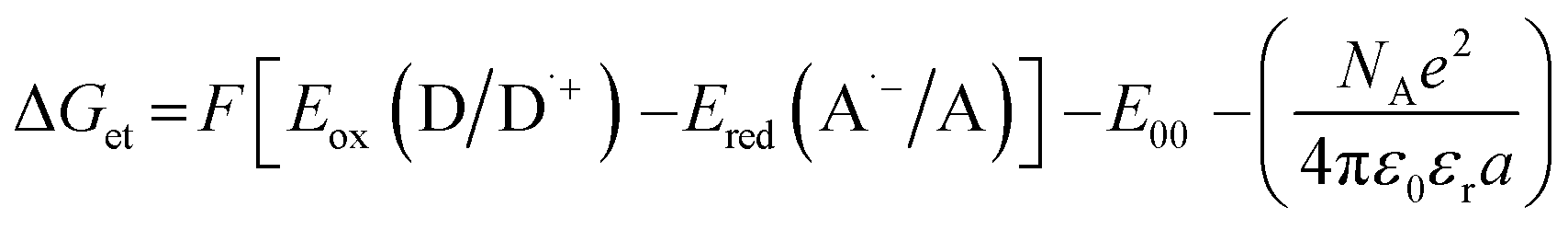 , Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor (−1.058 V for amine EDB vs. Ag/AgCl)69, Ered (A˙−/A) – electrochemically determined reduction potential of the electron acceptor, ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[EDB]/(1 + KSV[EDB]) for the concentration of amine [EDB] = 0.086 mol dm−3.
, Eox(D/D˙+) – electrochemically determined oxidation potential of the electron donor (−1.058 V for amine EDB vs. Ag/AgCl)69, Ered (A˙−/A) – electrochemically determined reduction potential of the electron acceptor, ES1 – singlet state energy of the sensitiser determined based on excitation and emission spectra, ET1 – triplet state energy calculated from molecular orbital calculations (uB3LYP/6-31G* level of theory), Φet – quantum yield of electron transfer, Φet = KSV[EDB]/(1 + KSV[EDB]) for the concentration of amine [EDB] = 0.086 mol dm−3.