
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew withanolides from Physalis minima and their cytotoxicity against A375 human melanoma cells†
Meng Zhangab,
Bingyang Zhangb,
Chenxi Guangb,
Benke Jiangb,
Xinya Heb,
Shijie Caob,
Liqin Dingb,
Ning Kangb,
Lixia Chen *a and
Feng Qiu*ab
*a and
Feng Qiu*ab
aSchool of Traditional Chinese Materia Medica, Wuya College of Innovation, Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China. E-mail: fengqiu20070118@163.com; syzyclx@163.com
bSchool of Chinese Materia Medica, Tianjin State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, 10 Poyanghu Road, West Area, Tuanbo New Town, Jinghai District, Tianjin 301617, People's Republic of China
First published on 16th June 2020
Abstract
Seven previously undescribed withanolides, namely physaminilide A–G (1–7), and two artificial withanolides (8–9), along with 10 known analogues (10–19) were isolated from Physalis minima. The structures were established by spectroscopic analysis, including NMR and electronic circular dichroism (ECD) data. Cytotoxicity of all the isolates was evaluated against A375 human melanoma cells. Compounds 2, 5, 8, 10, 11 and 15 exhibited significant cytotoxic activities with IC50 values in the range of 1.2–9.4 μM.
Introduction
Withanolides are a group of C28 steroidal lactones based on the ergostane skeleton that are common in the family Solanaceae.1 Over 900 withanolides have been encountered to date,2 and have captured the attention of researchers due to their diverse structures and pharmacological effects, including antitumor,3–7 antimicrobial,8 anti-inflammatory,9–11 and immunosuppressive activities.12The genus Physalis (Solanaceae), containing about 120 species worldwide, is known to produce a series of withanolides, showed significant antibacterial, anti-inflammatory, and antitumor effects. Five species and two varieties are distributed in mainland China.13,14 Physalis minima L., an annual herb, has been used as a traditional folk medicine for various purposes,15 and it is also utilized to treat analogous conditions in other countries, including Indian, Pakistan, and Japan. Various bioactive withanolides have been isolated from this plant.16–21
As part of a continuing research program on the genus Physalis to discover new withanolides with potential anticancer activities,22–25 the chemical constituents of P. minima were investigated. Seven previously undescribed withanolides (1–7; Fig. 1), two artificial withanolides (8–9; Fig. 1), and 10 known compounds (10–19; Fig. 1) were isolated from a crude EtOAc extract of the aerial parts of this plant. The structures of 1–9 were established by 1D and 2D NMR and electronic circular dichroism (ECD) spectroscopic duly analyses. In addition, all of the isolated withanolides were evaluated for their cytotoxic activities against A375 human melanoma cells.
 | ||
| Fig. 1 Structures of compounds 1–19 from Physalis minima. | ||
Results and discussion
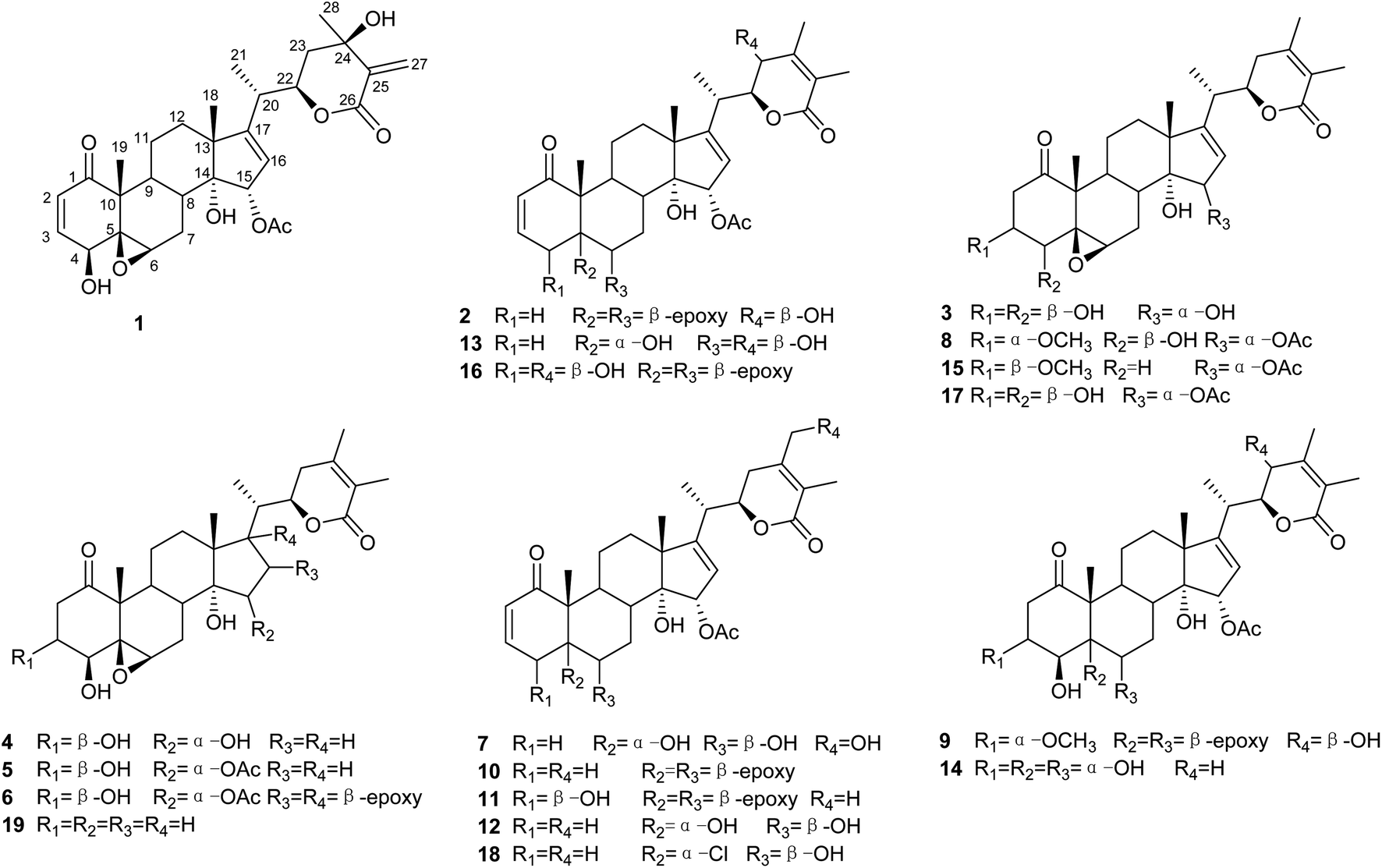
Compound 1 (physaminilide A) was obtained as a white powder. Its molecular formula was determined to be C30H38O9 on the basis of the 13C NMR data and the positive ion peak at m/z 543.2583 ([M + H]+, calcd for C30H39O9, 543.2594) in the HRESIMS, which indicated 12 degrees of unsaturation. Analysis of the NMR data (Tables 1 and 3) clearly indicated that 1 is closely related to the known compound physagulide C,26 with the major differences being due to several signals in the side chain. The 1H NMR spectrum revealed the characteristic signals of H-2, H-3, H-4 and H-6 for a 1-oxo-2-ene-5β,6β-epoxy-4β-hydroxy-moiety at δH 6.53 (1H, d, J = 9.8 Hz), 7.32 (1H, dd, J = 9.8, 6.1 Hz), 4.08 (1H, d, J = 6.1 Hz) and 3.38 (1H, br s), respectively. The substitution pattern of rings A/B was confirmed by the signals at δC 203.2 (C-1), 132.2 (C-2), 146.1 (C-3), 70.7 (C-4), 64.4 (C-5) and 61.3 (C-6) in the 13C NMR spectrum. The 1H NMR and HSQC spectrum revealed that the signals at δH 6.65 (1H, br s) and 6.32 (1H, br s) could be assigned to the terminal double bond carbon at δC 125.7. Five methyl group signals [δH 1.35 (3H, s), 1.87 (3H, s), 1.22 (3H, d, J = 7.0 Hz), 1.55 (3H, s), and 2.01 (3H, s)] were observed for δC 16.7 (C-18), 17.3 (C-19), 18.3 (C-21), 29.3 (C-28), and 21.6 (-OAc), respectively. The degrees of unsaturation and the chemical shifts of C-24 (δC 69.7), C-25 (δC 146.5) and C-27 (δC 125.7) suggested that 1 possesses a six-membered double bond migration δ-lactone moiety in side chain, with the Δ24,25 double bond shifted to Δ25,27, and a hydroxy group linked to C-24.27 These deductions were supported by the HMBC correlations from CH3-28 (δH 1.55) to C-23/C-24/C-25, H-27a (δH 6.65) to C-24/C-25/C-26, H-27b (δH 6.32) to C-24/C-26, H-23a (δH 2.36) to C-22/C-24/C-28, and H-23b (δH 2.27) to C-24/C-25/C-28, and 1H–1H COSY correlations between H-22 (δH 4.63) and H-23a (δH 2.36)/H-23b (δH 2.27)/H-20 (δH 2.64) (Fig. 2). Since the NMR signals of rings B-D of compound 1 were in good agreement with those of physagulide C, the relative stereochemistry of 1 was supposed to be the same as physagulide C. Therefore, the hydroxy group linked to C-14 was proposed as α-oriented. The NOESY correlations of H-7a (δH 2.94) with H-8 (δH 2.37) and H-15 (δH 5.82) confirmed that the acetoxy group is α-oriented, and these of H-6 (δH 3.38) with H-4 (δH 4.08) and H-7b (δH 1.99) consistent with the hydroxy group at C-4 and 5,6-epoxy moiety being β-oriented. The NOESY correlations of the CH3-28 (δH 1.55) with H-22 (δH 4.63) and H-23b (δH 2.27), and H-23b (δH 2.27) with H-22 (δH 4.63) implied that the hydroxy group at C-24 is β-oriented (Fig. 2).28 The 20S,22R configurations of 1 were determined based on the biogenetic considerations and the characteristic small coupling pattern (3.4 Hz) of two gauche conformation protons, H-20 and H-22.3 The absolute configuration of C-22 was further established as R based on the ECD positive Cotton effect at 250 nm.25 Thus, the structure of 1 was assigned as (20S,22R)-15α-acetoxy-5β,6β-epoxy-4β,14α,24β-trihydroxy-1-oxowitha-2,16,25-trienolide, and has been named physaminilide A.| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Assignments based on HSQC, HMBC, and NOESY data. | |||||
| 2 | 6.53 d (9.8) | 6.19 dd (10.0, 2.7) | 3.31 dd (15.0, 7.5) | 3.28 dd (15.0, 7.5) | 3.43 dd (15.7, 7.5) |
| 3.28 dd (15.0, 2.5) | 3.24 br d (15.0) | 3.09 br dd (15.7) | |||
| 3 | 7.32 dd (9.8, 6.1) | 6.83 ddd (10.0, 6.0, 2.2) | 4.72 m | 4.70 m | 4.72 m |
| 4 | 4.08 d (6.1) | 3.00 br d (19.0) | 4.05 br s | 4.03 br s | 3.99 s |
| 1.95 br s | |||||
| 6 | 3.38 br s | 3.23 br s | 3.89 br s | 3.88 br s | 3.71 s |
| 7 | 2.94 m | 2.84 br d (14.5) | 3.24 m | 3.13 m | 3.02 br d (13.4) |
| 1.99 m | 1.93 m | 3.14 m | 2.93 m | 2.12 m | |
| 8 | 2.37 m | 2.40 m | 2.43 m | 2.34 m | 2.31 m |
| 9 | 2.06 m | 2.39 m | 2.91 m | 2.73 m | 2.30 m |
| 11 | 2.22 m | 2.41 m | 1.87 m | 1.86 m | 1.74 m |
| 2.04 m | 1.55 m | 1.64 m | 1.67 m | 1.58 m | |
| 12 | 1.79 m | 1.84 m | 2.11 m | 2.61 br t (13.0) | 2.10 m |
| 1.62 m | 1.78 m | 1.82 m | 1.82 m | 1.74 m | |
| 15 | 5.82 d (2.5) | 5.88 d (2.6) | 5.05 m | 4.64 br s | 5.57 br s |
| 16 | 6.09 d (2.5) | 6.39 d (2.6) | 6.13 m | 2.36 m | 2.41 m |
| 1.91 m | 1.96 m | ||||
| 18 | 1.35 s | 1.47 s | 1.35 s | 1.37 s | 1.34 s |
| 19 | 1.87 s | 1.37 s | 1.85 s | 1.85 s | 1.75 s |
| 20 | 2.64 m | 3.13 m | 2.63 m | 2.37 m | 2.32 m |
| 21 | 1.22 d (7.0) | 1.36 d (7.0) | 1.27 d (7.0) | 1.12 d (7.0) | 1.09 d (7.0) |
| 22 | 4.63 dt (12.0, 3.4) | 4.47 br s | 4.43 br dd (13.0, 3.0) | 4.55 br dd (13.0, 3.0) | 4.54 dd (13.0, 3.0) |
| 23 | 2.36 m | 4.46 br s | 2.48 m | 2.29 m | 2.31 m |
| 2.27 m | 2.07 m | 1.88 m | 1.96 m | ||
| 27 | 6.65 br s | 1.85 s | 1.84 s | 1.91 s | 1.93 s |
| 6.32 br s | |||||
| 28 | 1.55 s | 1.82 s | 1.50 s | 1.60 s | 1.60 s |
| OAc | 2.01 s | 2.00 s | 2.21 s | ||
 | ||
| Fig. 2 Key 1H–1H COSY, HMBC and NOESY correlations for compounds 1. | ||
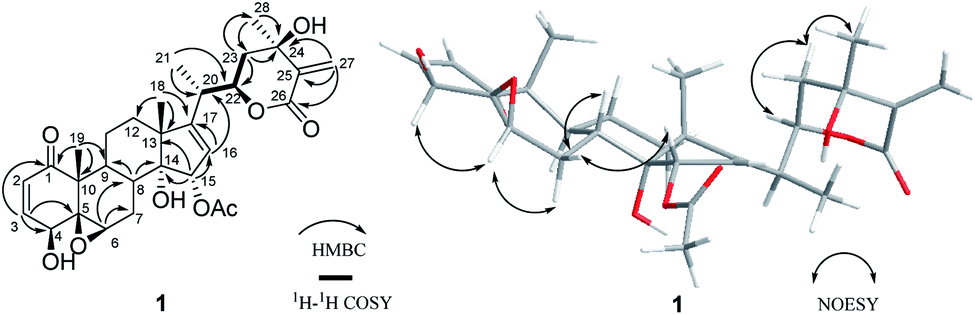
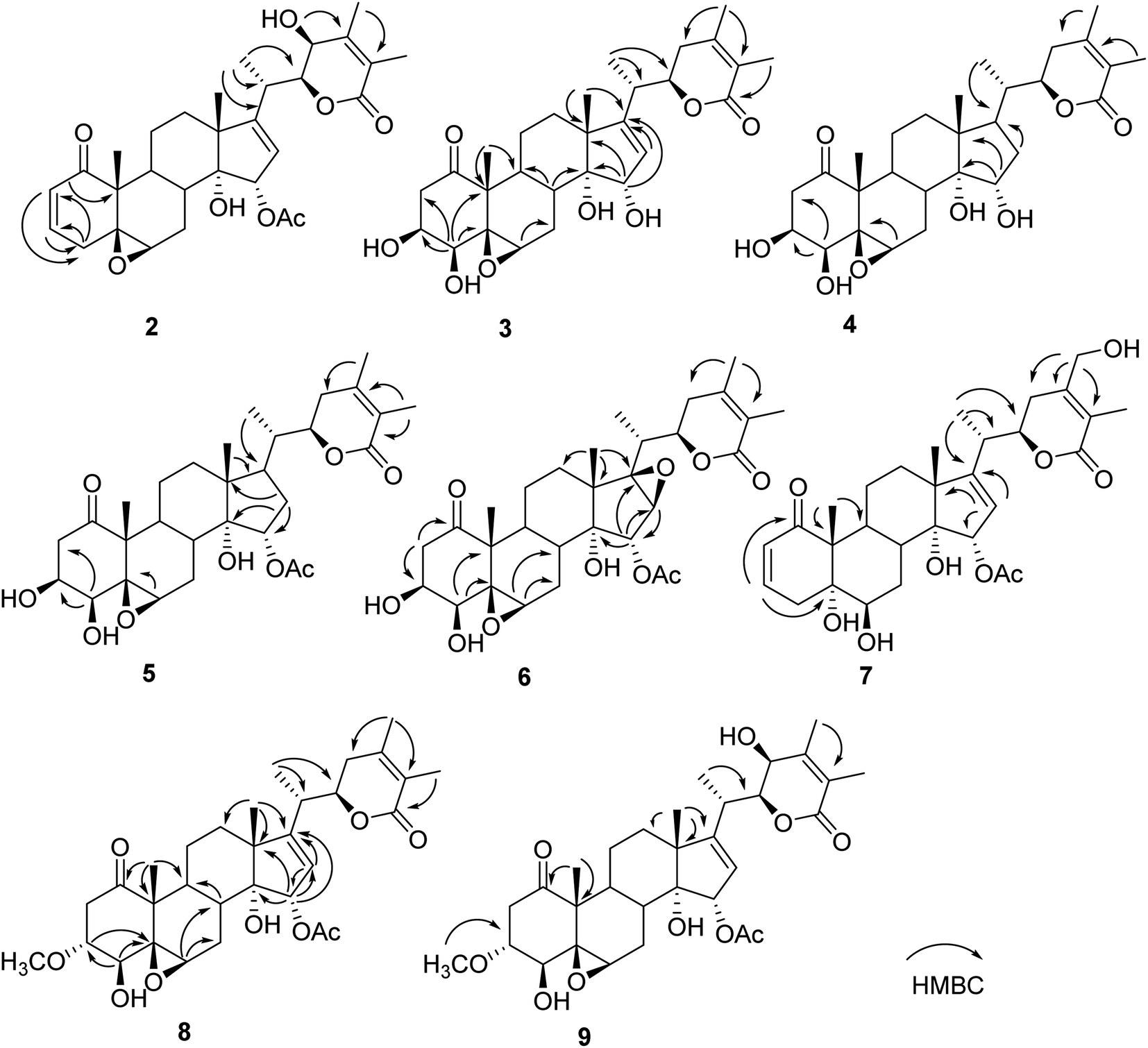
Compound 2 (physaminilide B) was assigned the molecular formula, C30H38O8, based on its HRESIMS (m/z 527.2623 [M + H]+, calcd for C30H39O8, 527.2645) and 13C NMR data. Detailed comparison of the NMR data of 2 (Tables 1 and 3) with those of physagulide D (16)26 showed these compounds to share the same substitution pattern in rings B–D and the side chain. The upfield shift of C-4 (−37.2 ppm) at δC 33.6, and the assignment of δH 3.00 (1H, br d, J = 19.0 Hz), δH 1.95 (1H, br s) to H2-4, suggested the absence of a hydroxy group at C-4 in 2, which was corroborated by the HMBC correlations from H-2 to C-4/C-5, and H-4a to C-2/C-3/C-5/C-6 (Fig. 3). The NOESY correlations of H-8 with H-15 confirmed an α-orientation of the acetoxy group on C-15 (Fig. 4). Based on the positive Cotton effect at 250 nm in the ECD spectrum, a 22R configuration was suggested.25 Thus, the structure of 2 was proposed as (20S,22R)-15α-acetoxy-5β,6β-epoxy-14α,23β-dihydroxy-1- oxowitha-2,16,24-trienolide.
 | ||
| Fig. 3 Key HMBC correlations for compounds 2–9. | ||
 | ||
| Fig. 4 Key NOESY correlations for compounds 2–9. | ||
The molecular formula of 3 (physaminilide C) was determined as C28H38O8 from its 13C NMR and HRESIMS (m/z 503.2647 [M + H]+, calcd for C28H39O8, 503.2645) data. Analysis of the NMR spectra suggested the structure of 3 (Tables 1 and 3) to be closely related to that of physaminimin F (17),29 except for the replacement of an acetoxy group at C-15 by a hydroxy group. The HMBC correlations from H-16 to C-13/C-14/C-15/C-17 were consistent with the location of a hydroxy group at C-15 (Fig. 3). The NOESY correlations of H-4 with H-3/H-6, H-7a (δH 3.24) with H-6/H-9, and H-15 with H-7b (δH 3.14) confirmed that the hydroxy group at C-15 is α-oriented, the hydroxy groups at C-3, C-4 are β-oriented, and the 5,6-epoxy moiety is β-oriented (Fig. 4). The positive Cotton effect at 250 nm in the ECD spectrum indicated a 22R configuration.25 Thus, compound 3 was established as (20S,22R)-5β,6β-epoxy-3β,4β,14α,15α-tetrahydroxy-1-oxowitha-16,24-dienolide.
The molecular formula of 4 (physaminilide D) was determined as C28H40O8 on the basis of the HRESIMS (m/z 505.2800 [M + H]+, calcd for C28H41O8, 505.2801) and 13C NMR data. The NMR spectra of 4 (Tables 1 and 3) were closely comparable to those of compound 3, except for the lack of any signals for a 16,17-double bond. This was corroborated by the HMBC correlations from CH3-18 to C-17, and CH3-21 to C-17 (Fig. 3). The NOESY correlations of H-15 with H-8/CH3-18, confirmed that the hydroxy group at C-15 is α-oriented (Fig. 4). The orientation of 14-OH was proposed as α-oriented by the similarity of the NMR signals to those of 3. The absolute configuration of C-22 was established as R through the ECD Cotton effect observed at 250 nm.25 Thus, compound 4 was assigned as (20S,22R)-5β,6β-epoxy-3β,4β,14α,15α-tetrahydroxy-1-oxowitha-24-enolide.
Compound 5 (physaminilide E) gave a molecular formula of C30H42O9 from its HRESIMS (m/z 547.29332 [M + H]+, calcd for 547.2907) and 13C NMR data. Analysis of the NMR data of 5 (Tables 1 and 3) showed close correlations to the data for physaminimin F (17).29 The main difference was the absence of a 16,17-double bond in 6. This was corroborated by the HMBC correlations from H-16a (δH 1.96) to C-13/C-14/C-15, CH3-18 to C-17, and CH3-21 to C-17 (Fig. 3). The α-orientation of OAc-15 was supported by the NOESY correlations H-7a (δH 3.02) with H-15/H-8 (Fig. 4). The absolute configuration of C-22 was established as R through the ECD Cotton effect observed at 250 nm.25 Accordingly, compound 5 was established as (20S,22R)-15α-acetoxy-5β,6β-epoxy-3β,4β,14α-trihydroxy-1-oxowitha-16,24-dienolide.
Compound 6 (physaminilide F) gave a molecular formula of C30H40O10 from its HRESIMS (m/z 561.2686 [M + H]+, C30H41O10, calcd for 561.2700) and 13C NMR data. Comparison of the NMR data of 7 (Tables 2 and 3) with those of physaminimin F (17)29 indicated their identical A–C rings and the side chain. The key differences arose from the downfield shifts of C-16 (δC 59.4) and C-17 (δC 76.7) in 6 suggested the presence of a 16,17-epoxy group, which were confirmed by the HMBC correlations from H-15 to C-13/C-14/C-16/C-17, H-16 to C-14/C-15, and CH3-18 to C-12/C-13/C-14/C-17 (Fig. 3). Its β-orientation was established by the NOESY correlations of H-16 with CH3-21, H-12a (δH 1.67) with H-16/H-9 (Fig. 4). The absolute configuration of C-22 was established as R through the ECD Cotton effect observed at 250 nm.25 Thus, the structure of 6 was established as (20S,22R)-15α-acetoxy-5β,6β:16β,17β-diepoxy-3β,4β,14α-trihydroxy-1-oxowitha-24-enolide.
| Position | 6 | 7 | 8 | 9 |
|---|---|---|---|---|
| a Assignments based on HSQC, HMBC, and NOESY data. | ||||
| 2 | 3.45 dd (16.0, 7.2) | 6.17 dd (10.0, 2.5) | 3.26 dd (16.0, 8.0) | 3.25 dd (16.0, 8.0) |
| 3.04 dd (16.0, 2.5) | 2.96 m | 2.95 dd (16.0, 3.0) | ||
| 3 | 4.70 m | 6.70 ddd (10.0, 5.0, 2.1) | 3.96 m | 3.95 br dd (8.0, 3.0) |
| 4 | 3.97 br s | 3.78 dt (19.7, 2.5) | 3.94 m | 3.93 br s |
| 2.40 dd (19.7, 5.0) | ||||
| 6 | 3.64 br s | 4.21 br s | 3.51 br s | 3.49 br s |
| 7 | 2.91 br d (14.3) | 2.81 m | 2.98 m | 2.97 m |
| 1.95 m | 2.68 m | 2.12 m | 2.10 m | |
| 8 | 2.03 m | 2.77 m | 2.28 m | 2.28 m |
| 9 | 2.25 m | 3.43 m | 2.26 m | 2.24 m |
| 11 | 1.78 m | 2.71 m | 1.69 m | 1.69 m |
| 1.43 m | 1.57 m | 1.48 m | 1.49 m | |
| 12 | 1.67 br d (13.3) | 2.07 m | 1.75 m | 1.80 m |
| 1.60 br d (13.3) | 1.94 m | 1.58 m | 1.63 m | |
| 15 | 5.43 br s | 6.21 d (2.6) | 5.89 d (2.5) | 5.93 d (2.6) |
| 16 | 3.80 br s | 6.02 d (2.6) | 6.12 d (2.5) | 6.41 d (2.6) |
| 18 | 1.31 s | 1.44 s | 1.32 s | 1.44 s |
| 19 | 1.70 s | 1.70 s | 1.70 s | 1.66 s |
| 20 | 2.55 m | 2.69 m | 2.62 m | 3.13 dd (14.0, 7.0) |
| 21 | 1.00 d (7.0) | 1.28 d (7.0) | 1.23 d (7.0) | 1.36 d (7.0) |
| 22 | 4.49 br dd (12.6, 3.5) | 4.50 dt (13.0, 3.9) | 4.43 dt (13.0, 3.7) | 4.48 br s |
| 23 | 2.30 m | 2.87 m | 2.42 m | 4.48 br s |
| 2.13 m | 2.63 m | 2.01 m | ||
| 27 | 1.90 s | 1.93 s | 1.85 s | 1.85 s |
| 28 | 1.73 s | 4.45 d (14.6) | 1.51 s | 1.84 s |
| 4.34 d (14.6) | ||||
| OAc | 2.32 s | 2.18 s | 2.21 s | 2.19 s |
| OMe | 3.30 s | 3.29 s | ||
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| a Assignments based on HSQC, HMBC, and NOESY data. | |||||||||
| 1 | 203.2 | 204.1 | 211.2 | 211.2 | 210.9 | 211.1 | 205.5 | 209.9 | 209.8 |
| 2 | 132.2 | 129.2 | 44.4 | 44.4 | 44.3 | 44.1 | 129.5 | 41.4 | 41.4 |
| 3 | 146.1 | 146.2 | 70.1 | 70.0 | 69.3 | 69.1 | 142.7 | 78.9 | 78.9 |
| 4 | 70.7 | 33.6 | 79.5 | 79.6 | 78.8 | 78.5 | 37.3 | 74.5 | 74.6 |
| 5 | 64.4 | 62.2 | 65.3 | 65.2 | 65.1 | 64.8 | 77.7 | 64.8 | 64.7 |
| 6 | 61.3 | 64.2 | 60.7 | 60.6 | 60.3 | 60.3 | 75.3 | 59.3 | 59.3 |
| 7 | 25.7 | 25.9 | 25.7 | 27.5 | 27.2 | 26.5 | 28.9 | 25.6 | 25.5 |
| 8 | 36.8 | 36.6 | 36.6 | 37.5 | 36.9 | 36.1 | 37.9 | 36.6 | 36.5 |
| 9 | 40.9 | 40.5 | 39.5 | 40.4 | 39.7 | 38.4 | 37.0 | 39.0 | 39.0 |
| 10 | 48.8 | 49.1 | 51.6 | 51.8 | 51.8 | 51.3 | 53.0 | 51.3 | 51.3 |
| 11 | 21.8 | 24.5 | 21.9 | 21.9 | 21.7 | 20.3 | 24.7 | 21.4 | 21.5 |
| 12 | 38.0 | 39.2 | 38.2 | 42.4 | 41.7 | 31.8 | 39.7 | 37.8 | 38.6 |
| 13 | 53.2 | 53.4 | 52.8 | 46.5 | 47.3 | 47.2 | 53.7 | 53.3 | 53.6 |
| 14 | 81.8 | 81.4 | 82.2 | 85.1 | 84.8 | 82.2 | 82.4 | 81.9 | 81.9 |
| 15 | 84.7 | 84.9 | 82.8 | 78.8 | 81.7 | 78.5 | 83.8 | 84.8 | 84.9 |
| 16 | 122.3 | 123.5 | 127.6 | 38.3 | 35.1 | 59.4 | 122.6 | 122.7 | 123.4 |
| 17 | 162.5 | 162.6 | 157.9 | 52.9 | 52.8 | 76.7 | 162.7 | 162.5 | 162.7 |
| 18 | 16.7 | 17.5 | 17.0 | 15.8 | 18.3 | 15.7 | 17.6 | 16.6 | 17.3 |
| 19 | 17.3 | 15.7 | 15.8 | 18.4 | 15.6 | 15.7 | 15.8 | 15.5 | 15.4 |
| 20 | 36.6 | 32.8 | 35.5 | 38.6 | 38.6 | 34.6 | 35.8 | 35.6 | 32.8 |
| 21 | 18.3 | 20.9 | 18.4 | 15.5 | 15.7 | 14.4 | 18.6 | 18.2 | 20.9 |
| 22 | 79.6 | 85.4 | 79.6 | 79.2 | 78.9 | 77.3 | 80.4 | 79.1 | 85.4 |
| 23 | 41.1 | 67.0 | 32.8 | 30.5 | 30.9 | 33.3 | 28.2 | 32.7 | 67.0 |
| 24 | 69.7 | 154.3 | 150.0 | 149.9 | 149.9 | 149.7 | 153.7 | 149.9 | 154.3 |
| 25 | 146.5 | 121.7 | 122.0 | 122.1 | 122.1 | 122.2 | 121.3 | 122.1 | 121.8 |
| 26 | 165.9 | 165.2 | 166.8 | 167.1 | 166.9 | 166.4 | 166.9 | 166.6 | 165.2 |
| 27 | 125.7 | 13.5 | 13.0 | 13.0 | 13.0 | 13.0 | 12.5 | 12.9 | 13.5 |
| 28 | 29.3 | 16.1 | 20.1 | 20.4 | 20.3 | 20.4 | 61.2 | 20.5 | 16.1 |
| OAc | 170.2 | 170.3 | 170.5 | 170.4 | 171.2 | 170.5 | 170.4 | ||
| OAc | 21.6 | 21.6 | 21.9 | 21.4 | 21.9 | 21.6 | 21.6 | ||
| OMe | 57.2 | 57.2 | |||||||
Compound 7 (physaminilide G) was isolated as a white powder. Its molecular formula was determined to be C30H40O9 based on the positive HRESIMS data at m/z 545.2723 [M + H]+ (calcd for C30H41O9, 545.2751) combined with the 13C NMR data.
Its 1H and 13C NMR data (Tables 2 and 3) were similar to those of the known compound withaminimin (12),30 with the only major difference being a set of oxygenated methylene protons observed for 7 at δH 4.34 (1H, d, J = 14.6 Hz) and δH 4.45 (1H, d, J = 14.6 Hz), which were assigned to a hydroxymethyl substituent at C-28 based on the HMBC correlations of H-28 (δH 4.34, δH 4.45) with C-23/C-24/C-25 (Fig. 3). From all the evidence obtained, compound 7 was determined as (20S,22R)-15α-acetoxy-5α,6β,14α,28-tetrahydroxy-1-oxowitha-2,16,24-trienolide.
Compound 8 (3-methoxy-2,3-dihydrowithangulatin A) gave a molecular formula of C31H42O9 from the HRESIMS (m/z 559.2891 [M + H]+, C31H43O9, calcd for 559.2907) and 13C NMR data. The NMR spectroscopic data of 8 (Tables 2 and 3) showed a close similarity with those of physaminimin F (17),29 except for the presence of additional signals at δH 3.96 (1H, m) and δH 3.30 (3H, s), indicating the presence of a methoxy group in 8. The HMBC correlations between OCH3-3 [δH 3.30 (3H, s)] and C-3 (δC 78.9), suggested that the methoxy group was located at C-3 (Fig. 3). The NOESY correlations of H-3 with CH3-19, H-16 with H-15/CH3-18, H-6 with H-4/H-7a (δH 2.98), and H-7a with H-9 confirmed that the acetoxy group is α-oriented, the methoxy group at C-3 is α-oriented, and the hydroxy group at C-4 and the 5,6-epoxy moiety are β-oriented (Fig. 4). The ECD spectrum exhibited a positive Cotton effect at 250 nm, suggesting a 22R configuration.25 Based on the above evidence, the structure of 8 was established as (20S,22R)-15α-acetoxy-5β,6β-epoxy-4β,14α-dihydroxy-3α-methoxy-1-oxowitha-16,24-dienolide.
The HRESIMS analysis of compound 9 (3-methoxy-2,3-dihydrophysagulide D) provided the molecular formula of C31H42O10 (m/z 575.2850 [M + H]+, calcd for C31H43O10, 575.2856). The NMR spectra suggested that the structure of 9 (Tables 2 and 3) is closely related to that of physagulide D,26 except for the absence of two olefinic protons of the α,β-unsaturated ketone in ring A, and the presence of signals at δH 3.95 (1H, br dd, J = 8.0, 3.0 Hz), δH 3.29 (3H, s) attributable to H-3 and OCH3-3 in 9. The methoxy group was determined to be located at C-3 (δC 78.9) by HMBC correlations between OCH3-3 and C-3 (Fig. 3). The NOESY correlations of H-2a (δH 3.25) with H-3/CH3-19, CH3-18 with H-15, H-6 with H-4/H-7a (δH 2.97), and H-7a with H-9 confirmed that the acetoxy group at C-15 and the methoxy group at C-3 are α-oriented, the hydroxy group at C-4 and the 5,6-epoxy moiety are β-oriented (Fig. 4). The positive Cotton effect at 250 nm in ECD spectrum indicated a 22R configuration.25 Therefore, 9 was identified as (20S,22R)-15α-acetoxy-5β,6β-epoxy-4β,14α,23β-trihydroxy-3α-methoxy-1-oxowitha-16,24-dienolide.
Compounds 8–9 were hypothesized to be intermolecular Michael-type addition adducts of polar protic solvent nucleophiles like methanol or ethanol,31,32 the former solvent established compound 8, named as 3-methoxy-2,3-dihydrowithangulatin A, while the latter solvent gave compound 9, obtained as 3-methoxy-2,3-dihydrophysagulide D. Compound 11 (10 mg) was heated with methanol (5 mL) for 8 h to afford compound 8. The methanol solution of compound 11 was examined at four time intervals (0, 2, 4, and 8 h) by HPLC analysis. The results showed the artifact 8 was increased in a time-dependent manner (Fig. S74†). Therefore, the use of methanol or ethanol should be cautious in the extraction and purification processes of withanolides with the 1-oxo-2-ene system in ring A.
By comparing their analytical data with those reported in the literature, the known compounds were identified as physagulin A (10),33 withangulatin A (11),34 withaminimin (12),30 physagulin M (13),33 physaliolide C (14),11 physagulin N (15),33 physagulide D (16),26 physaminimin F (17),29 physagulin B (18),35 27-deoxy-14-hydroxywithaferin A (19).36
Cytotoxicity of all the isolates was evaluated against A375 human melanoma cells. Among them, compounds 1, 3–4, 6–7, 9, 12–14, and 16–19 were inactive for A375 human melanoma cells used (IC50 > 10 μM). Meanwhile, Compounds 2, 5, 8, 10, 11 and 15 exhibited significant cytotoxic activities with IC50 values in the range of 1.2–9.4 μM. These results indicated that withanolides with 5β,6β-epoxy possessed significant cytotoxic activities. The 16,17-double bond in ring D was essential for cytotoxicity. Furthermore, the 15-acetoxy group also had a slight influence on their cytotoxicity.
Experimental
General experimental procedures
Optical rotations were measured with a PerkinElmer 241 polarimeter. UV spectra were collected in a Shimadzu UV 2201 spectrophotometer. ECD spectra were recorded on a Bio-Logic Science MOS-450 spectrometer. IR spectra were obtained on a Bruker IFS 55 spectrometer. NMR experiments were performed on Bruker AV-400 and AV-600 spectrometers. Chemical shifts are reported as δ (ppm) related to the solvent pyridine-d5 (δH 7.58 and δC 135.91) as references, and coupling constants (J values) are given in Hz. HRESIMS data were obtained on an Agilent 6210 TOF mass spectrometer. Silica gel GF254, obtained from Qingdao Marine Chemical Factory, was used for TLC. Sephadex LH-20 for gel-permeation chromatography was obtained from Pharmacia. Column chromatography (CC) was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Factory) and octadecyl silica gel (Merck Chemical Company Ltd., Darmstadt, Germany). RP-HPLC separations were conducted using an LC-6AD liquid chromatograph, SPD-20A UV detector (Shimadzu, Kyoto, Japan), equipped with a YMC Pack ODS-A column (250 × 20 mm, 120 Å, 5 μm).Plant material
Physalis minima was collected from Fujian Province, China, in September 2014, and identified by Professor Jincai Lu, Shenyang Pharmaceutical University. A voucher specimen (PM-2014) has been deposited in the herbarium of Shenyang Pharmaceutical University.Extraction and isolation
The air-dried entire plant materials of P. minima (9.0 kg) were extracted three times with EtOH/H2O (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 v/v) (100 L × 2 h). The resulting extract (840 g) was concentrated under a vacuum, suspended in H2O (5.0 L), and partitioned successively with petroleum ether (3 × 5.0 L), EtOAc (3 × 5.0 L) to generate the EtOAc fraction. The EtOAc fraction (200 g) was subjected to silica gel CC eluted with a gradient of CH2Cl2/MeOH (100:1 to 0:1 v/v) to give eight subfractions (E1–E8). Fraction E3 (40.0 g) yielded four subfractions by silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1). Subfraction E33 (15.0 g) was separated on a silica gel CC eluted with petroleum ether/EtOAc (100:1 to 0:1) to produce five subfractions (E331–E335), subfraction E332 (392.5 mg) was applied to Sephadex LH-20 column eluted with CH2Cl2/MeOH (1:1) and separated by HPLC (MeOH/H2O, 70:30) to yield 12 (40.0 mg, tR = 14.0 min). Subfraction E333 (142.3 mg) was separated by HPLC (MeOH/H2O, 70:30) to afford 10 (200.0 mg, tR = 26.0 min). Subfraction E336 (1.0 g) was purified by HPLC (MeOH/H2O, 70:30) to yield 15 (4.0 mg, tR = 34.0 min). Subfraction E34 (15.0 g) was subjected to silica gel CC using CH2Cl2/MeOH (100:1 to 0:1) as eluent to produce five subfractions (E341–E345). Subfraction E341 (4.0 g) and E342 (7.0 g) were chromatographed on silica gel column eluted with petroleum ether/acetone (100:1 to 0:1) and then separated using HPLC (MeOH/H2O, 70:30) to obtain 11 (100.0 mg, tR = 22.0 min) and 18 (5.0 mg, tR = 30.0 min). Subfraction E345 (0.62 g) was separated by HPLC (MeOH/H2O, 70:30) to produce 1 (3.0 mg, tR = 27.0 min). Fraction E4 (30.0 g) was separated by silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to yield two subfractions (E41–E42). Subfraction E42 (20.0 g) was separated on a silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to yield two subfractions (E421–E422). Subfraction E421 (12.0 g) and E422 (5.0 g) were chromatographed on a silica gel column eluted with petroleum ether/EtOAc (100:1 to 0:1) and then separated by HPLC (MeOH/H2O, 70:30) to produce 2 (5.0 mg, tR = 34.0 min), 8 (20.0 mg, tR = 39.0 min), and 19 (3.0 mg, tR = 45.0 min). Fraction E6 (30.0 g) was subjected to silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to produce five subfractions (E61–E65). Subfraction E64 (15.0 g) was subjected to a silica gel CC eluted with petroleum ether/acetone (100:1 to 0:1) to produce E641 (1.53 g), Subfraction E641 was applied to a Sephadex LH-20 column eluted with CH2Cl2/MeOH (1:1), and then purified by HPLC (MeOH/H2O, 70:30) to yield 6 (16.2 mg, tR = 27.0 min), 9 (30.0 mg, tR = 25.0 min), and 16 (18.2 mg, tR = 21.0 min). Fraction E7 (18.0 g) and E8 (13.0 g) were subjected to silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to produce seven subfractions (E71–E77) and four subfractions (E81–E84), respectively. Subfraction E76 (7.3 g) was separated on a silica gel CC eluted with CH2Cl2/acetone (100:1 to 0:1) to yield three subfractions (E761–E763). Subfraction E762 (2.3 g) was subjected to passage over an ODS silica gel CC eluted with MeOH/H2O (1:9 to 0:1) and then purified by HPLC (MeOH/H2O, 70:30) to yield 5 (4.5 mg, tR = 31.0 min) and 17 (5.0 mg, tR = 27.0 min). Subfraction E763 (1.0 g) and E77 (1.8 g) were purified by HPLC (MeOH/H2O, 70:30) to yield 7 (3.0 mg, tR = 25.0 min), 13 (50.0 mg, tR = 24.0 min), and 14 (42.0 mg, tR = 26.0 min). Subfraction E83 (2.0 g) was chromatographed on a Sephadex LH-20 column eluted with CH2Cl2–MeOH (1:1) to yield E832, subfraction E832 (1.1 g) was subjected to ODS silica gel CC, eluted with MeOH/H2O (1:9 to 0:1), and then purified by HPLC (MeOH/H2O, 55:45) to yield 3 (25.1 mg, tR = 23.0 min), 4 (6.7 mg, tR = 26.0 min).
ε) 206 (3.55) nm; ECD (c 4.6 × 10−4 M, MeOH) λmax (Δε) 249 (+1.75), 289 (−1.27) nm; IR (KBr) νmax 3385, 2918, 1714, 1373, 1234, 1023 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 543.2583 [M + H]+ (calcd for C30H39O9, 543.2594).ε) 206 (3.49) nm; ECD (c 9.5 × 10−4 M, MeOH) λmax (Δε) 250 (+2.00), 300 (−0.51) nm; IR (KBr) νmax 3307, 2918, 1710, 1377, 1243, 1021 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 527.2623 [M + H]+ (calcd for C30H39O8, 527.2645).ε) 212 (3.77) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 210 (+7.51), 250 (+4.35), 290 (−3.48) nm; IR (KBr) νmax 3394, 2920, 2840, 1697, 1647, 1468, 1384, 1130 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 503.2647 [M + H]+ (calcd for C28H39O8, 503.2645).ε) 205 (3.19), 225 (3.22) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 251 (+3.73), 290 (−3.31) nm; IR (KBr) νmax 3386, 2918, 1685, 1398, 1140, 1031 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 505.2800 [M + H]+ (calcd for C28H41O8, 505.2801).ε) 205 (3.02), 228 (3.11) nm; ECD (c 9.1 × 10−4 M, MeOH) λmax (Δε) 253 (+2.51), 289 (−2.00) nm; IR (KBr) νmax 3391, 2913, 1701, 1378, 1255, 1139, 1031 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 547.2933 [M + H]+ (calcd for C30H43O9, 547.2907).ε) 205 (3.01), 226 (3.11) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 249 (+3.33), 292 (−1.97) nm; IR (KBr) νmax 3374, 2915, 1705, 1375, 1224, 1025 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 561.2686 [M + H]+ (calcd for C30H41O10, 561.2700).ε) 218 (3.76) nm; ECD (c 9.2 × 10−4 M, MeOH) λmax (Δε) 259 (+2.58), 330 (−1.00) nm; IR (KBr) νmax 3394, 3189, 3008, 2920, 2849, 1646, 1468, 1419, 1261, 1119 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 545.2723 [M + H]+ (calcd for C30H41O9, 545.2751).ε) 212 (3.78) nm; ECD (c 1.8 × 10−3 M, MeOH) λmax (Δε) 250 (+2.81), 290 (−2.36) nm; IR (KBr) νmax 3394, 3189, 2920, 2849, 1646, 1467, 1418, 1253, 1114 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 559.2891 [M + H]+ (calcd for C31H43O9, 559.2907).ε) 203 (3.45) nm; IR (KBr) νmax 3392, 2917, 1809, 1714, 1375, 1236, 1096 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 575.2850 [M + H]+ (calcd for C31H43O10, 575.2856).
:25 v/v) (100 L × 2 h). The resulting extract (840 g) was concentrated under a vacuum, suspended in H2O (5.0 L), and partitioned successively with petroleum ether (3 × 5.0 L), EtOAc (3 × 5.0 L) to generate the EtOAc fraction. The EtOAc fraction (200 g) was subjected to silica gel CC eluted with a gradient of CH2Cl2/MeOH (100:1 to 0:1 v/v) to give eight subfractions (E1–E8). Fraction E3 (40.0 g) yielded four subfractions by silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1). Subfraction E33 (15.0 g) was separated on a silica gel CC eluted with petroleum ether/EtOAc (100:1 to 0:1) to produce five subfractions (E331–E335), subfraction E332 (392.5 mg) was applied to Sephadex LH-20 column eluted with CH2Cl2/MeOH (1:1) and separated by HPLC (MeOH/H2O, 70:30) to yield 12 (40.0 mg, tR = 14.0 min). Subfraction E333 (142.3 mg) was separated by HPLC (MeOH/H2O, 70:30) to afford 10 (200.0 mg, tR = 26.0 min). Subfraction E336 (1.0 g) was purified by HPLC (MeOH/H2O, 70:30) to yield 15 (4.0 mg, tR = 34.0 min). Subfraction E34 (15.0 g) was subjected to silica gel CC using CH2Cl2/MeOH (100:1 to 0:1) as eluent to produce five subfractions (E341–E345). Subfraction E341 (4.0 g) and E342 (7.0 g) were chromatographed on silica gel column eluted with petroleum ether/acetone (100:1 to 0:1) and then separated using HPLC (MeOH/H2O, 70:30) to obtain 11 (100.0 mg, tR = 22.0 min) and 18 (5.0 mg, tR = 30.0 min). Subfraction E345 (0.62 g) was separated by HPLC (MeOH/H2O, 70:30) to produce 1 (3.0 mg, tR = 27.0 min). Fraction E4 (30.0 g) was separated by silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to yield two subfractions (E41–E42). Subfraction E42 (20.0 g) was separated on a silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to yield two subfractions (E421–E422). Subfraction E421 (12.0 g) and E422 (5.0 g) were chromatographed on a silica gel column eluted with petroleum ether/EtOAc (100:1 to 0:1) and then separated by HPLC (MeOH/H2O, 70:30) to produce 2 (5.0 mg, tR = 34.0 min), 8 (20.0 mg, tR = 39.0 min), and 19 (3.0 mg, tR = 45.0 min). Fraction E6 (30.0 g) was subjected to silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to produce five subfractions (E61–E65). Subfraction E64 (15.0 g) was subjected to a silica gel CC eluted with petroleum ether/acetone (100:1 to 0:1) to produce E641 (1.53 g), Subfraction E641 was applied to a Sephadex LH-20 column eluted with CH2Cl2/MeOH (1:1), and then purified by HPLC (MeOH/H2O, 70:30) to yield 6 (16.2 mg, tR = 27.0 min), 9 (30.0 mg, tR = 25.0 min), and 16 (18.2 mg, tR = 21.0 min). Fraction E7 (18.0 g) and E8 (13.0 g) were subjected to silica gel CC eluted with CH2Cl2/MeOH (100:1 to 0:1) to produce seven subfractions (E71–E77) and four subfractions (E81–E84), respectively. Subfraction E76 (7.3 g) was separated on a silica gel CC eluted with CH2Cl2/acetone (100:1 to 0:1) to yield three subfractions (E761–E763). Subfraction E762 (2.3 g) was subjected to passage over an ODS silica gel CC eluted with MeOH/H2O (1:9 to 0:1) and then purified by HPLC (MeOH/H2O, 70:30) to yield 5 (4.5 mg, tR = 31.0 min) and 17 (5.0 mg, tR = 27.0 min). Subfraction E763 (1.0 g) and E77 (1.8 g) were purified by HPLC (MeOH/H2O, 70:30) to yield 7 (3.0 mg, tR = 25.0 min), 13 (50.0 mg, tR = 24.0 min), and 14 (42.0 mg, tR = 26.0 min). Subfraction E83 (2.0 g) was chromatographed on a Sephadex LH-20 column eluted with CH2Cl2–MeOH (1:1) to yield E832, subfraction E832 (1.1 g) was subjected to ODS silica gel CC, eluted with MeOH/H2O (1:9 to 0:1), and then purified by HPLC (MeOH/H2O, 55:45) to yield 3 (25.1 mg, tR = 23.0 min), 4 (6.7 mg, tR = 26.0 min).
ε) 206 (3.55) nm; ECD (c 4.6 × 10−4 M, MeOH) λmax (Δε) 249 (+1.75), 289 (−1.27) nm; IR (KBr) νmax 3385, 2918, 1714, 1373, 1234, 1023 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 543.2583 [M + H]+ (calcd for C30H39O9, 543.2594).ε) 206 (3.49) nm; ECD (c 9.5 × 10−4 M, MeOH) λmax (Δε) 250 (+2.00), 300 (−0.51) nm; IR (KBr) νmax 3307, 2918, 1710, 1377, 1243, 1021 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 527.2623 [M + H]+ (calcd for C30H39O8, 527.2645).ε) 212 (3.77) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 210 (+7.51), 250 (+4.35), 290 (−3.48) nm; IR (KBr) νmax 3394, 2920, 2840, 1697, 1647, 1468, 1384, 1130 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 503.2647 [M + H]+ (calcd for C28H39O8, 503.2645).ε) 205 (3.19), 225 (3.22) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 251 (+3.73), 290 (−3.31) nm; IR (KBr) νmax 3386, 2918, 1685, 1398, 1140, 1031 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 505.2800 [M + H]+ (calcd for C28H41O8, 505.2801).ε) 205 (3.02), 228 (3.11) nm; ECD (c 9.1 × 10−4 M, MeOH) λmax (Δε) 253 (+2.51), 289 (−2.00) nm; IR (KBr) νmax 3391, 2913, 1701, 1378, 1255, 1139, 1031 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 1 and 3; HRESIMS m/z 547.2933 [M + H]+ (calcd for C30H43O9, 547.2907).ε) 205 (3.01), 226 (3.11) nm; ECD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 249 (+3.33), 292 (−1.97) nm; IR (KBr) νmax 3374, 2915, 1705, 1375, 1224, 1025 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 561.2686 [M + H]+ (calcd for C30H41O10, 561.2700).ε) 218 (3.76) nm; ECD (c 9.2 × 10−4 M, MeOH) λmax (Δε) 259 (+2.58), 330 (−1.00) nm; IR (KBr) νmax 3394, 3189, 3008, 2920, 2849, 1646, 1468, 1419, 1261, 1119 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 545.2723 [M + H]+ (calcd for C30H41O9, 545.2751).ε) 212 (3.78) nm; ECD (c 1.8 × 10−3 M, MeOH) λmax (Δε) 250 (+2.81), 290 (−2.36) nm; IR (KBr) νmax 3394, 3189, 2920, 2849, 1646, 1467, 1418, 1253, 1114 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 559.2891 [M + H]+ (calcd for C31H43O9, 559.2907).ε) 203 (3.45) nm; IR (KBr) νmax 3392, 2917, 1809, 1714, 1375, 1236, 1096 cm−1; 1H NMR (600 MHz, pyridine-d5) and 13C NMR (150 MHz, pyridine-d5) data, see Tables 2 and 3; HRESIMS m/z 575.2850 [M + H]+ (calcd for C31H43O10, 575.2856).Cytotoxicity assays
The cytotoxic activity of these compounds for A375 human melanoma cells was determined using an MTT assay.24 The A375 cells were obtained from ATCC (Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) in a humidified atmosphere (37 °C, 5% CO2). Cells (3 × 103 cells per well) were placed in 96-well plates for 24 h. The test compounds with different concentrations were applied to the 96-well plates, incubated for 48 h. 5-Fluorouracil was used as the positive control. Then, MTT (5 mg mL−1) was added to the cells and maintained for 2.5 h. The cells were resolved with DMSO (150 μL) after removed from the medium. All assays were performed in triplicate (Table 4).| Compoundb | IC50 (μM) |
|---|---|
| A375 | |
| a Results for the compounds and positive control are expressed as IC50 values in μM.b Compounds 1, 3–4, 6–7, 9, 12–14, and 16–19 were inactive for A375 cells used (IC50 > 10 μM).c Positive control. | |
| 2 | 3.4 ± 0.9 |
| 5 | 2.0 ± 1.1 |
| 8 | 9.4 ± 3.3 |
| 10 | 1.9 ± 0.1 |
| 11 | 1.2 ± 0.4 |
| 15 | 7.3 ± 2.1 |
| 5-Fluorouracilc | 18.5 ± 0.7 |
Conclusions
In conclusion, 19 withanolides were isolated from Physalis minima, which included eleven previously undescribed withanolides, physaminilide A–G (1–7) and two artificial withanolides (8–9), together with ten known ones (10–19). The structures of the new compounds were elucidated by a combined analysis of the IR, UV, HRESIMS, NMR and electronic circular dichroism (ECD) spectra. All the isolates were assayed cytotoxic activities against human melanoma A375 cells. Compounds 2, 5, 8, 10, 11 and 15 exhibited significant cytotoxic activities with IC50 values in the range of 1.2–9.4 μM.Conflicts of interest
The authors declare no conflict interest.Acknowledgements
This research was funded by the National Natural Science Foundation of China (NSFC) (Grant No. 81773594 and 21472138), National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” (2017ZX09301012).Notes and references
- L. X. Chen, H. He and F. Qiu, Nat. Prod. Rep., 2011, 28, 705–740 RSC.
- B. Y. Yang, G. Y. Xia, J. Pan, Y. Liu, Q. H. Wang and H. X. Kuang, Phytochem. Rev., 2016, 15, 771–797 CrossRef CAS.
- L. X. Chen, G. Y. Xia, H. He, J. Huang, F. Qiu and X. L. Zi, RSC Adv., 2016, 6, 52925–52936 RSC.
- C. M. Cao, X. Wu, K. Kindscher, L. Xu and B. N. Timmermann, J. Nat. Prod., 2015, 78, 2488–2493 CrossRef CAS.
- H. P. Zhang, A. K. Samadi, R. J. Gallagher, J. J. Araya, X. Tong, V. W. Day, M. S. Cohen, K. Kindscher, R. Gollapudi and B. N. Timmermann, J. Nat. Prod., 2011, 74, 2532–2544 CrossRef CAS PubMed.
- L. X. Chen, G. Y. Xia, F. Qiu, C. L. Wu, P. D. Andria and X. L. Zi, Sci. Rep., 2016, 6, 32582 CrossRef CAS PubMed.
- H. I. F. Magalhaes, M. R. Torres, L. V. Costa-Lotufo, M. O. De Moraes, C. Pessoa, M. L. Veras, E. R. Silveira, O. D. L. Pessoa and A. P. N. N. Alves, J. Pharm. Pharmacol., 2006, 58, 235–241 CrossRef CAS PubMed.
- A. M. Abou-Douh, Arch. Pharm., 2002, 335, 267–276 CrossRef CAS.
- B. Y. Yang, R. Guo, T. Li, J. J. Wu, J. Zhang, Y. Liu, Q. H. Wang and H. X. Kuang, Steroids, 2014, 87, 26–34 CrossRef CAS PubMed.
- L. Qiu, F. Zhao, Z. H. Jiang, L. X. Chen, Q. Zhao, H. X. Liu, X. S. Yao and F. Qiu, J. Nat. Prod., 2008, 71, 642–646 CrossRef CAS PubMed.
- J. P. Wu, X. Li, J. P. Zhao, R. J. Wang, Z. F. Xia, X. R. Li, Y. L. Liu, Q. M. Xu, I. A. Khan and S. L. Yang, Phytochemistry, 2018, 155, 164–170 CrossRef CAS PubMed.
- B. Y. Yang, G. Y. Xia, Y. Liu, L. Li, H. Jiang, L. Yang, Q. H. Wang and H. X. Kuang, Phytochem. Lett., 2014, 8, 92–96 CrossRef CAS.
- W. N. Zhang and W. Y. Tong, Chem. Biodiversity, 2016, 13, 48–65 CrossRef PubMed.
- Institute of Botany of the Chinese Academy of Sciences, and Kunming Institute of Botany of the Chinese Academy of Sciences, Chinese Flora (Zhongguo Zhiwu Zhi), Science Press, Beijing, 1978, vol. 67, pp. 50–59 Search PubMed.
- State Administration of Traditional Chinese Medicine of the People's Repulic of China, Zhonghua Bencao (Chinese Herbal Medicine), Shanghai Science and Technology House, Shanghai, 1999, vol. 7, pp. 292–293 Search PubMed.
- M. I. Choudhary, S. Yousaf, S. Ahmed, Samreen, K. Yasmeen and Atta-Ur-Rahman, Biodiversity, 2005, 2, 1164–1173 CrossRef CAS PubMed.
- M. I. Choudhary, S. Yousuf, Samreen, S. Ahmed and Atta-Ur-Rahman, Nat. Prod. Res., 2007, 21, 877–883 CrossRef CAS.
- G. Sen and H. D. Pathak, Phytochemistry, 1995, 39, 1245–1246 CrossRef CAS.
- M. Kawai, B. Makino, H. Yamamura and Y. Butsugan, Phytochemistry, 1996, 43, 661–663 CrossRef CAS.
- L. Ma, X. W. Gan, Q. P. He, H. Y. Bai, M. Arfan, F. C. Lou and L. H. Hu, Helv. Chim. Acta, 2007, 90, 1406–1419 CrossRef CAS.
- M. Sahai and I. Kirson, J. Nat. Prod., 1984, 47, 527–529 CrossRef CAS PubMed.
- H. He, L. H. Zang, Y. S. Feng, L. X. Chen, N. Kang, T. Shinichi, O. Satoshi, F. Qiu and I. Takashi, J. Ethnopharmacol., 2013, 148, 544–555 CrossRef CAS.
- H. He, L. H. Zang, Y. S. Feng, J. Wang, W. W. Liu, L. X. Chen, N. Kang, S. Tashiro, S. Onodera, F. Qiu and I. Takashi, J. Nat. Prod., 2013, 76, 880–888 CrossRef CAS PubMed.
- G. Y. Xia, Y. Li, J. W. Sun, L. Q. Wang, X. L. Tang, B. Lin, N. Kang, J. Huang, L. X. Chen and F. Qiu, Steroids, 2016, 115, 136–146 CrossRef CAS PubMed.
- C. P. Sun, C. Y. Qiu, T. Yuan, X. F. Nie, H. X. Sun, Q. Zhang, H. X. Li, L. Q. Ding, F. Zhao, L. X. Chen and F. Qiu, J. Nat. Prod., 2016, 79, 1586–1597 CrossRef CAS PubMed.
- T. Ma, W. N. Zhang, L. Yang, C. Zhang, R. Lin, S. M. Shan, M. D. Zhu, J. G. Luo and L. Y. Kong, RSC Adv., 2016, 6, 53089–53100 RSC.
- H. P. Zhang, J. Bazzill, R. J. Gallagher, C. Subramanian, P. T. Grogan, V. W. Day, K. Kindscher, M. S. Cohen and B. N. Timmermann, J. Nat. Prod., 2012, 76, 445–449 CrossRef PubMed.
- E. Maldonado, A. L. Perez-Castorena, C. Garces and M. Martinez, Steroids, 2011, 76, 724–728 CrossRef CAS PubMed.
- Y. Z. Guan, S. M. Shan, W. Zhang, J. G. Luo and L. Y. Kong, Steroids, 2014, 82, 38–43 CrossRef CAS PubMed.
- E. G. Hugo, M. Cojocaru, S. C. Sinha, M. Saha, A. Bagchi, A. Ali and A. B. Ray, Phytochemistry, 1987, 6, 1801–1804 Search PubMed.
- C. M. Cao, H. Zhang, R. J. Gallagher and B. N. Timmermann, J. Nat. Prod., 2013, 76, 2040–2046 CrossRef CAS PubMed.
- S. B. Wang, D. R. Zhu, B. Nie, J. Li, Y. J. Zhang, L. Y. Kong and J. G. Luo, Bioorg. Chem., 2018, 81, 396–404 CrossRef CAS PubMed.
- F. Abe, S. Nagafuji, M. Okawa and J. Kinjo, Chem. Pharm. Bull., 2006, 54, 1226–1228 CrossRef CAS PubMed.
- M. Mizuno, M. Kato, N. Hosoi, M. Iinuma, T. Tanaka, A. Kimura, H. Ohashi, H. Sakai and T. Kajita, Heterocycles, 1990, 31, 1409–1412 CrossRef CAS.
- B. Makino, M. Kawai, T. Ogura, M. Nakanishi, H. Yamamura and Y. Butsugan, J. Nat. Prod., 1995, 58, 1668–1674 CrossRef CAS.
- S. Chatterjee and S. K. Chakraborti, Antonie van Leeuwenhoek, 1980, 46, 59–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, CD, HRESIMS and NMR spectra of 1–9. See DOI: 10.1039/d0ra04106h |
| This journal is © The Royal Society of Chemistry 2020 |