
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnodic SnO2 porous nanostructures with rich grain boundaries for efficient CO2 electroreduction to formate†
Ruizhen Ma‡
b,
Yan-Li Chen‡a,
Yongli Shenb,
Heng Wangb,
Wei Zhang*a,
Su-Seng Pangc,
Jianfeng Huang*d,
Yu Han e and
Yunfeng Zhao*b
e and
Yunfeng Zhao*b
aState Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Taipa, Macau, China. E-mail: wzhang@must.edu.mo
bTianjin Key Laboratory of Advanced Functional Porous Materials, Institute for New Energy Materials & Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: yfzhao@tjut.edu.cn
cFaculty of Information, Macau University of Science and Technology, Taipa, Macau, China
dMulti-scale Porous Materials Center, Institute of Advanced Interdisciplinary Studies, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China. E-mail: jianfeng.huang@cqu.edu.cn
eAdvanced Membranes and Porous Materials Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia
First published on 16th June 2020
Abstract
Formic acid (HCOOH), the acidic form of formate, is an important hydrogen carrier which can be directly used in fuel cells. Development of earth-abundant element-based catalysts to convert carbon dioxide (CO2) into HCOOH or formate with high selectivity and high efficiency has been a vigorous research activity in recent years but remains an unsolved challenge. In this contribution, using one-step anodization, we prepare nanotubular SnO2 porous nanostructures with high surface area (90.1 m2 g−1), large porosity (0.74 cm3 g−1), and rich grain boundaries for electrochemical CO2 reduction (CO2RR). They exhibit stable 95% faradaic efficiency (FE) towards CO2RR and 73% FE for formate at −0.8 VRHE. The notable performance of such SnO2 nanostructures can be attributed to their unique structural and chemical properties, which provide active sites for CO2 adsorption and conversion, and easy access for CO2 to the active sites. The insights gained from the structure/property relationships might be beneficial for designing superior electrocatalysts for CO2 electroreduction into formate.
Introduction
Electrochemical CO2 reduction (CO2RR) to carbon-based fuels is a promising strategy for renewable energy storage and net carbon emission.1–5 Liquid fuels, such as formic acid (HCOOH),6–8 methanol9 and ethanol,10 produced via CO2 electroreduction are actively sought because they can be directly used as energy-intensive carriers for fuel cells. Particularly, formic acid has been identified as an ideal hydrogen carrier owing to its high volumetric density (53.4 g H2 L−1), and low toxicity and volatility.11 However, achieving highly efficient and selective electrocatalysts for converting CO2 into HCOOH is challenging due to the myriad possible reaction pathways and sluggish reaction kinetics in the process of CO2RR.The formation of the intermediate CO2˙− is a rate-determining step for CO2RR in most pathways.12 Metals like Pb, In, Zn, Sn, Pd and Bi, can hardly bind the intermediate CO2˙− and hence are able to produce formic acid (or formate when pH > 3.8) at low overpotentials typically via the outer-sphere mechanism.13 Among these metal-based electrocatalysts, Sn and its derivatives (e.g., SnOx, Sn alloy), as earth-abundant, low-cost and nontoxic materials, have attracted particular research interest, due to their capability of catalyzing CO2 into formate with decent faradaic efficiencies (FEs) at moderate potentials.14–21 For example, Meyer et al. found a maximum FE of 86% for formate at −1.8 VSCE when loading nanoscale SnO2 particles on carbon black.14 Xie et al. reported that Sn quantum sheets confined in graphene exhibited ∼89% FE towards formate at the same potential.15 These results point out that the catalyst size and composition, which are correlated with the number and intrinsic properties of active sites exposed during CO2 reduction, play important roles in determining the formation of formate. In addition, the electrocatalysis of CO2 in aqueous solution is an electrochemical reaction at the three-phase interface of gas (CO2), liquid (electrolyte) and solid (catalyst). The concentration of reactant CO2 on the surface of the catalyst affects the electrocatalytic performance and is governed by at least two factors: (1) the CO2 transport within the catalysts; (2) the CO2 adsorption on the catalysts. Therefore, the structure of the catalyst also matters critically in the formate formation activity. For example, hierarchical mesoporous SnO2 nanosheets were reported to facilitate the mass transfer of CO2, leading to a value of 87% FE for formate at a moderate overpotential (0.88 V).16 SnO2 nanowires with a high density of grain boundaries were proposed to enhance the CO2 reduction into formate (ca. 80% FE at −0.8 VRHE) via reforming the binding energy of the reactant or intermediates.17 Taken together, these works conclude that formate production by CO2RR is sensitive to a variety of parameters, including size, structure and composition etc. However, to date, it remains a challenge to incorporate these parameters to achieve an efficient electrocatalyst for the conversion of CO2 to formate.
In this work, we report the results of an electrochemical study on the CO2 reduction at SnO2 porous nanostructures with integrated characteristics of high surface area, large porosity, and rich grain boundaries that favor the CO2-to-formate conversion. The SnO2 porous nanostructures were prepared by an anodization approach which is a facile yet effective technique for fabricating various porous metal oxides with ordered tubular nanostructure.22 The as-prepared anodic SnO2 porous nanostructures exhibit a tubular structure (pore-size: ∼45 nm) with abundant nanograins within the size range of 3–5 nm. The porous structure provides a large specific surface area (90.1 m2 g−1) and high porosity (0.74 cm3 g−1), while the grain boundaries render a strong CO2 affinity with the heat of adsorption up to 50 kJ mol−1, the upper limit of the physical adsorption. When tested as the electrocatalyst for the CO2RR, the SnO2 aerogels maintain 95% faradaic efficiency (FE) towards CO2RR and 73% FE for HCOOH at −0.8 V vs. RHE for 12 h. Miscellaneous characterizations, including transmission electron microscope (TEM), N2 (77 K) and CO2 (273 and 298 K) adsorption, X-ray photoelectron spectroscopy (XPS) and electrochemistry, combined with theoretical calculations were employed to elucidate the correlations between the structure and composition of the aerogels and their CO2RR performance.
Experimental section
Chemicals and instruments
Sn foil (thickness of 200 μm, purity > 99.99%, Renxin Metal), Nafion solution (5 wt%, Shanghai Hesen), Nafion® 117 cation exchange membrane (H+ form, Shanghai Hesen), carbon black (VULCAN XC72, Cabot), and other chemicals (Analytic Reagent) are used as received. The X-ray diffraction (XRD) was taken from Rigaku Ultima Iv. A scanning electron microscope (SEM, FEI, Verios 460L) and a high-resolution transmission electron microscope (HRTEM, FEI, Talos F200X) were utilized to characterize the morphology and high-resolution structure. X-ray photoelectron spectrometer (XPS) was determined on a Thermo Scientifica (ESCALAB250xi). Electrochemical measurements were carried on an electrochemical workstation (CHI760e, Shanghai CHI). Products in the gas phase were determined by a gas chromatograph (GC, 9790II, Zhejiang Fuli) equipped with thermal conductivity and a flame ionization detector. Products in solution was detected by a nuclear magnetic resonance (NMR, Bruker, 400M).Synthesis of SnO2 tubular array by anodic oxidation
The surface oxide layer was firstly removed by soaking an Sn foil in a KOH solution (0.1 M) for 5 min, and then the Sn foil was ultrasonically cleaned in deionized (DI) water for later use. The anodization process was carried in a two-electrode cell, where the cleaned Sn foil was used as a working electrode and a graphite plate was used as the counter electrode with the distance of 1 cm. A constant voltage (8.0–12.0 V) was applied to the working electrode using a potentiostat instrument in a 0.5 M oxalic acid (H2C2O4·2H2O, 99.5%) electrolyte for 5 min at room condition. After anodization, the Sn foil was repeatedly washed with DI water before ultrasonically stripped.Fabrication of catalytic electrode
The electrode was fabricated through a dropping-coating method. Typically, SnO2 powders (4 mg), carbon black (2 mg) and Nafion® solution (60 μL) were mixed with 1 mL mixture of DI water and ethanol (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in v/v) and followed with ultrasonic treatment for 5 min to form a homogeneous ink. The suspension (80 μL) was dropped onto a glassy carbon electrode with an area of 1 × 1 cm2 to form the catalyst layer with a loading amount of 300 μg cm−2 and the electrode were naturally dried for further electrochemical measurements.
:3 in v/v) and followed with ultrasonic treatment for 5 min to form a homogeneous ink. The suspension (80 μL) was dropped onto a glassy carbon electrode with an area of 1 × 1 cm2 to form the catalyst layer with a loading amount of 300 μg cm−2 and the electrode were naturally dried for further electrochemical measurements.
Electrochemical measurement
All electrochemical CO2 reductions were performed by using a well-sealed H-type cell with standard three-electrodes, in which two chambers were separated with a piece of Nafion® 117 membrane. One side of the H-cell was equipped with inlet and outlet, where CO2 (99.99%) could be bubbled into the reaction cell through an inlet and the product in the gas phase would be introduced into a GC by the outlet. The area of the working electrode was 1 cm × 1 cm. An Ag/AgCl electrode (sat. KCl) was used as a reference electrode, and a Pt foil (99.999%, 1 cm × 1 cm) was used as a counter electrode, which was infiltrated into the electrolyte of 0.5 M KHCO3 aqueous solution for all electrochemical testing. The cyclic voltammetry (CV) under potentials ranging from 0.4 to −1.0 VRHE by varying the scan rate of 50 mV s−1. The time–current curve was obtained by carrying out constant working potential at various potentials −0.7 to −1.2 VRHE. Meanwhile, the average current density was obtained by dividing the total current by the geometric area of the working electrode. The potential values involved in this work were all calibrated to the potential of the reversible hydrogen electrode (RHE) by using eqn (1):23| E(vs. RHE) = E(vs. Ag/AgCl) + 0.197 + 0.0591 × pH | (1) |
The high-purity CO2 was continuously introduced into the electrolyte (0.5 M KHCO3) for 30 min to make the solution reach a saturated state of CO2 (pH = 7.2) before the electrochemical test. The flow rate of CO2 gas was controlled to be 5 mL min−1 and the cathode chamber electrolyte was continuously stirred to accelerate the bubble diffusion on the electrode surface at a rotation speed of 600 rpm during the electrolysis process. All tests for this work were controlled at room temperature and ambient pressure.
Product determination
The gas-phase products were quantified using a GC during 3 h long bulk electrolysis. Under the same test environment, the gas peak area detected by gas chromatography has a certain linear relationship with its actual concentration, so that the gas with known concentration can be used to calibrate and analyze the gas with unknown concentration, which combined with current density to calculate the faradaic efficiency (FE) by using eqn (2)–(4):23| jH2 = ((peak area)/α) × (flow rate) × (2FP0/RT) × (electrode area)−1 | (2) |
| jCO = ((peak area)/β) × (flow rate) × (2FP0/RT) × (electrode area)−1 | (3) |
| FEH2/CO (%) = (iH2/CO/itotal) × 100 = (VH2/CO × Q × (2FP0/RT)) × 100 | (4) |
485 C mol−1), R is ideal gas constant (8.314 J (mol K)−1), P0 = 1.013 bar, T = 273.15 K.
The product in solution was detected and quantified by NMR. The 0.4 mL of post-cathodic reaction mixed with 0.1 mL deuteroxide (D2O) and 0.1 mL dimethyl sulfoxide (DMSO, 100 ppm) as internal standard. The 1H spectrum was measured with water suppression by a presaturation method. The area ratio of the formate peak to the DMSO peak was compared to the standard curve made by using sodium formate and the internal standard DMSO to calculate the amounts of formate products.
Computational details
All Spin-polarized calculations in this work are carried out using the Quickstep24 module of the CP2K program package,24,25 a program developed for electronic structure calculations and molecular dynamics based on the Gaussian and plane waves formalism.26 The generalized-gradient approximation (GGA) with spin-polarized revised PBE functional (revPBE) is selected to describe the exchange-correlation energy. All the structures and energies of the stationary points included in our work are evaluated at the DFT-D3 level with the approximation suggested by Grimme27 added to the revPBE calculated energy. The wavefunctions were expanded in an optimized double-ζ Gaussian basis set28,29 with a cutoff energy of 500 Rydberg.30 Core electrons have been modeled by scalar relativistic norm-conserving pseudo potentials30 with 4, 6, 1 and 4 valence electrons for Sn, O, H and C, respectively. Brillouin zone integration is performed with a reciprocal space mesh consisting of only the gamma point. The convergence criterion for the maximum force is set as 2 × 10−3. Thermochemistry calculation is implemented by TAMkin,31 a toolkit for normal mode analysis A (19.11 Å × 20.10 Å) and 5 O–Sn–O layers thick supercell has been selected to represent the SnO2{110} surface. A vacuum region between repeated slabs is set to be 30 Å, which ensures the negligible interaction between periodic replicas.Results and discussion
Fig. 1 illustrates the typical fabrication of the porous SnO2 nanostructures by the anodization approach wherein an Sn foil and a graphite plate were used as the anode and cathode, respectively, in an oxalic acid solution (0.5 M). After being applied with a specific voltage (i.e., 8, 10 and 12 V) for 5 min, the porous SnO2 structures were formed on the surface of the Sn foil. They are hereafter denoted as SnO2–AOx (x: the anodization voltage). The SnO2–AOx layers can be easily exfoliated from the anode, and then used to construct the catalytic electrode for CO2RR.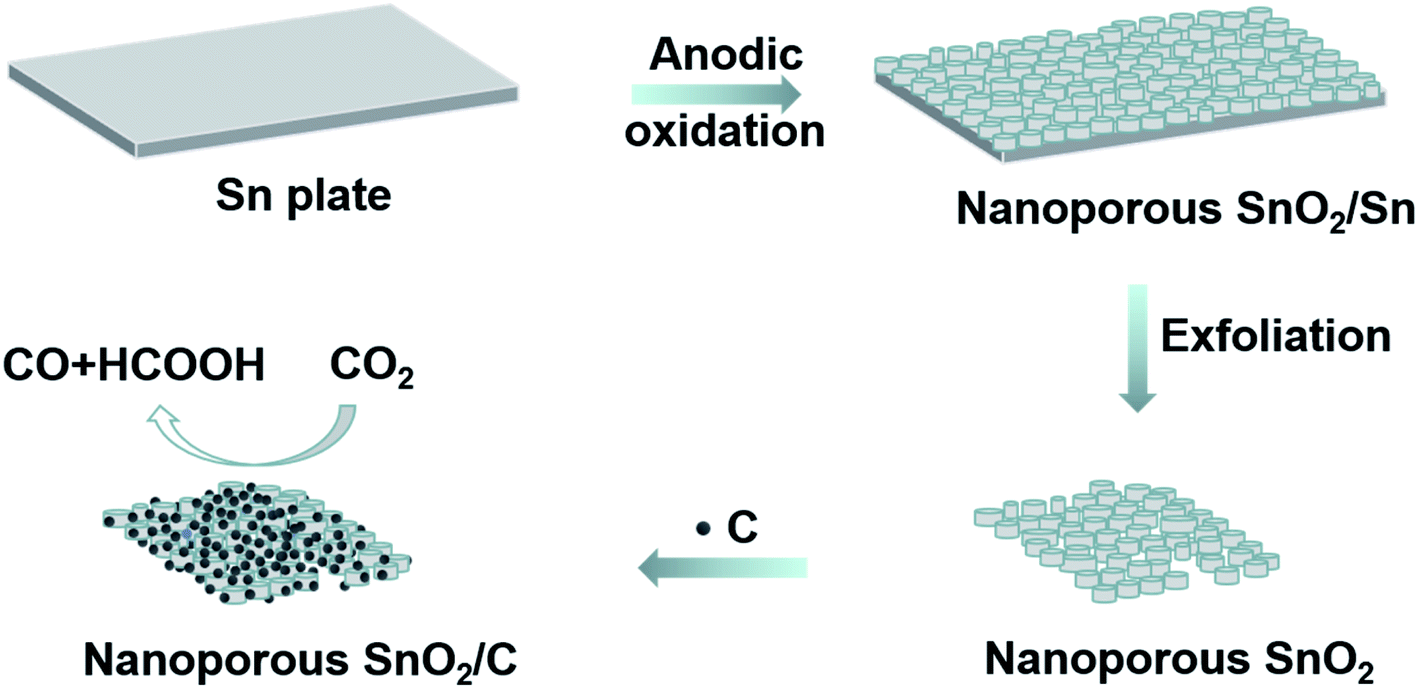 | ||
| Fig. 1 Schematic representation of the fabrication of anodic SnO2–AOx porous nanostructures that are used as the electrocatalysts for CO2RR. | ||
Fig. 2 and S1† report the structural characterization of the as-prepared SnO2–AOx aerogels by scanning electron microscopy (SEM) and transmission electron microscope (TEM). From the SEM of the SnO2–AO10 (Fig. 2a and b), highly ordered porous structures are observed with the average pore size within the range of 40–50 nm (Fig. 2c). Varying the anodization voltage (i.e., 8 and 12 V) leads to similarly porous SnO2–AO8 and SnO2–AO12 nanostructures with only slightly different pore sizes (Fig. S1†). The cross-section SEM image further illustrates that the porous SnO2 layer is comprised of arrays of tubes that are nearly perpendicular to the Sn substrate (Fig. 2d and S2†). Here, the tubes are expected to promote the mass transfer and reaction during CO2RR.14 TEM was additionally employed to study the tubular structure, which shows that the wall thickness of the tubes is 5–10 nm (Fig. 2e). Interestingly, a closer view enabled by the high-resolution TEM (HRTEM) reveals that the tube walls are constituted by many small interconnected nanoparticles (3–5 nm) through grain boundaries (Fig. 2f and S3†). The individual particles have good crystallinity, as indicated by the interplanar distance of 0.33 nm, which corresponds to the d-spacing of the (110) plane of SnO2, and further confirmed by some discrete spots in the corresponding electron diffraction pattern of the HRTEM image (inset of Fig. 2f). In addition, the diffraction rings resulting from many spots very close together demonstrate the polycrystalline nature of the tube wall as a whole. Since ultra-small SnOx nanoparticles and grain boundaries have been found to be active sites for CO2RR,16,17,32 the hierarchical SnO2–AO10 nanostructures that feature tubular structure, ultra-small NPs and abundant grain boundaries show great promise for CO2RR.
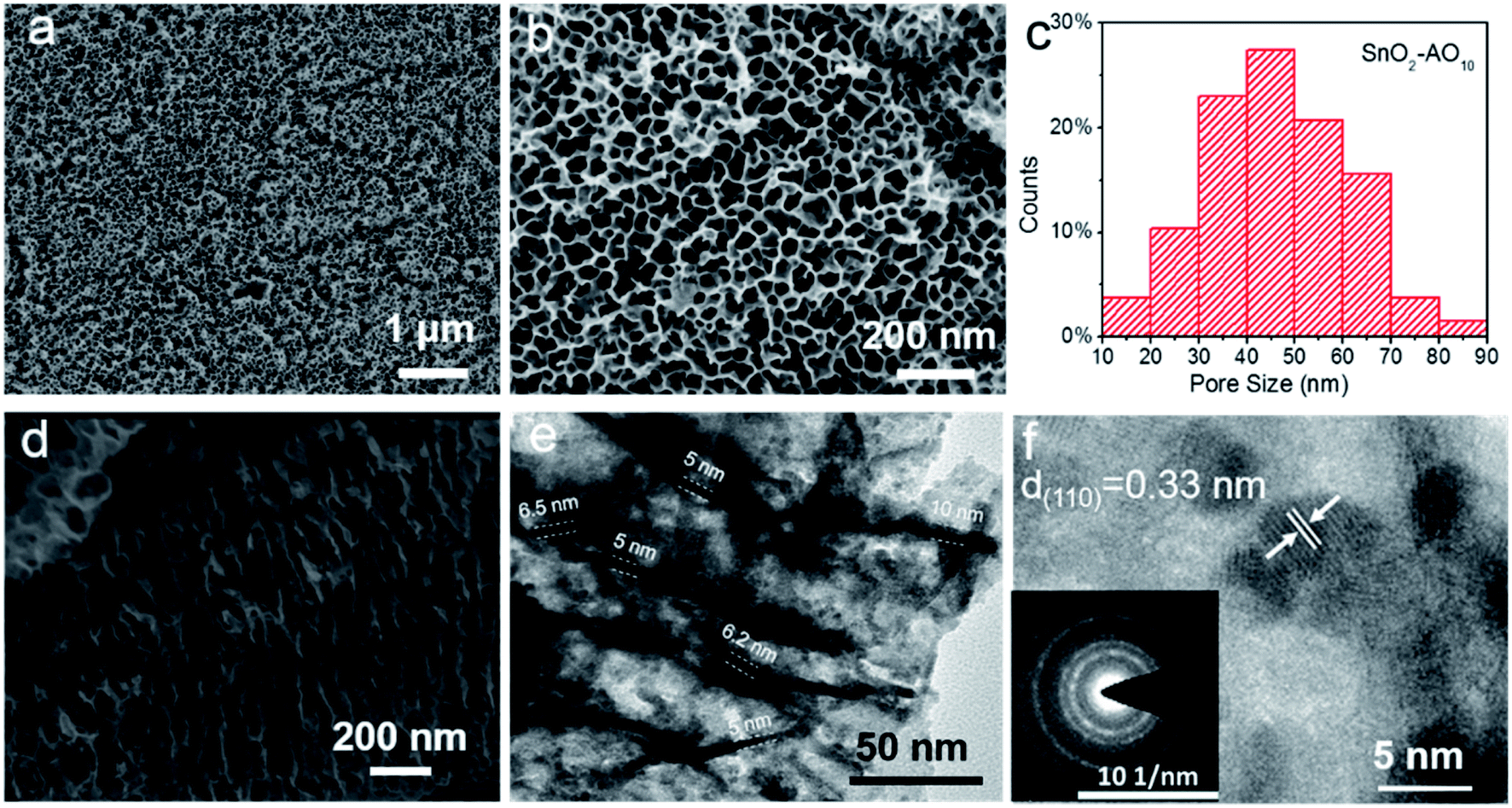 | ||
| Fig. 2 (a and b) SEM images and (c) the corresponding pore size statistical analysis of the surface of SnO2–AO10. (d) SEM and (e) TEM images of the cross-section of SnO2–AO10. (f) High-resolution TEM image and the corresponding electron diffraction pattern (inset) of SnO2–AO10. | ||
To gain more insights into the porous structure, N2 adsorption–desorption isotherms at 77 K were measured to characterize the surface area and porosity of the SnO2–AO10 nanostructures. As shown in Fig. 3a, the N2 uptake increases slowly with the pressure in the low-pressure range then shoots up when P/P0 exceeds 0.9. The isotherm thus belongs to the Type-II, as defined by IUPAC,33 which indicates the presence of both micropores and mesopores in hierarchical porous structures. By applying the Barrett–Joyner–Halenda (BJH) method to the desorption data, the mesopores were found to be within an average size range of 40–50 nm (Fig. 3b), in good agreement with the SEM results (Fig. 2b and c) and reflecting the actual pores created by the anodic oxidation. On the basis of additional data of CO2 adsorption at 273 K, the micropores were found to display a bimodal size distribution at 0.6 and 0.8 nm (Fig. 3c).
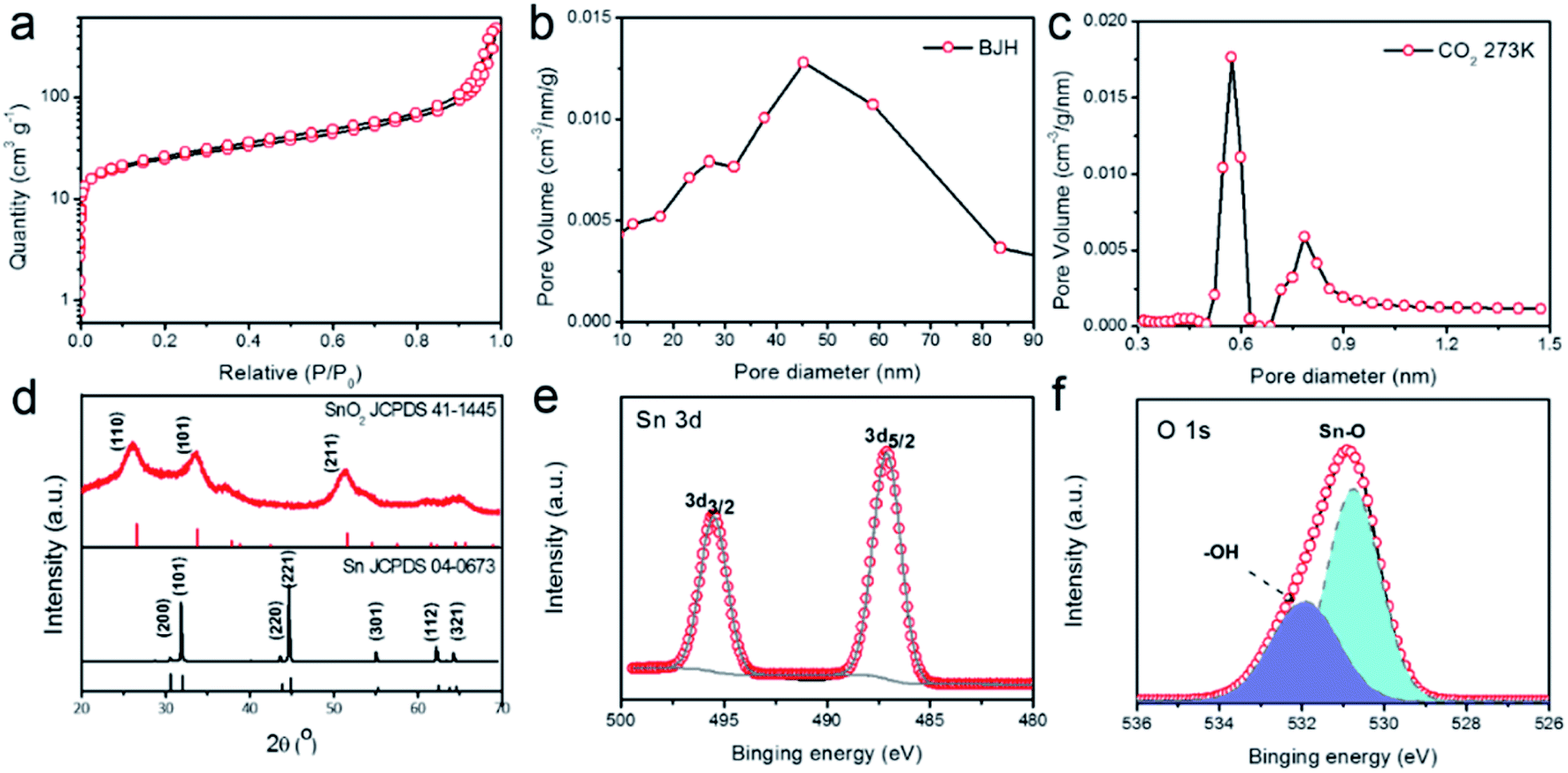 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherm (at 77 K), (b) the corresponding pore size distribution calculated based on the desorption data of the isotherm, and (c) pore size distribution derived from CO2 adsorption at 273 K, (d) XRD patterns of SnO2–AO10 and Sn foil, (e) XPS Sn3d and (f) XPS O1s spectrum of SnO2–AO10. | ||
These micropores may arise from the gaps between nanoparticles in the tube walls, as revealed by the HRTEM image of the structure (Fig. 2f). Thanks to these pores of micro- and meso-sizes, the SnO2–AO10 nanostructures exhibit an extremely high BET surface area (90.1 m2 g−1) and porosity (0.74 cm3 g−1). As a result, fast mass transport and easy access of reactants to the active sites during CO2RR can be expected.
Finally, we investigated the crystal phase and chemical structure of the SnO2–AO10 aerogels by X-ray diffraction (XRD, Fig. 3d) and X-ray photoelectron spectroscopy (XPS, Fig. 3e and f). From the XRD pattern, the diffraction peaks that can be unambiguously indexed to rutile tetragonal SnO2 (JCPDS card no. 41-1445) are observed, confirming the pure SnO2 phase and crystallinity of SnO2–AO10 nanostructures and demonstrating the successful conversion of pure Sn to SnO2.34 As a comparison, SnO2–AO8 and SnO2–AO12 nanostructures exhibit similar pure SnO2 phase but much poorer crystallinity (Fig. S4†). However, the mechanism as to how the anodization voltage affects the crystallinity is unclear at this moment, which will be examined in the future. Concerning the compositional information, the two peaks at 495.5 and 487.1 eV in the XPS spectrum are the characteristic peaks that can be assigned to Sn 3d3/2 and 3d5/2 ionizations, respectively, confirming the Sn4+ valence state of Sn element in the tin oxide aerogels (Fig. 3e).34 Additionally, the XPS spectrum of O 1 s was collected (Fig. 3f). The peak fitting analysis verifies the oxygen bonding either with Sn as Sn–O bond (530.8 eV) or with H as –OH (531.9 eV) adsorbed on the aerogels surface.35
To evaluate the CO2RR performance, we first examined the catalytic activity of SnO2–AOx nanostructures and an Sn plate by the Cyclic Voltammetry (CV) (Fig. 4a and S5†). In the N2-saturated 0.5 M KHCO3 solution, the marked increase in the cathodic current at ∼−0.80 V vs. RHE is associated with the hydrogen evolution reaction (HER), a parasite and competing reaction in aqueous solution against CO2RR. By contrast, a more drastic current increase occurring at a much lower potential (−0.6 VRHE) was observed in the CO2-saturated 0.5 M KHCO3 solution, meaning that the CO2RR is catalytically more favored over HER on the SnO2–AO10 catalysts. It is worth noting that the two cathodic peaks correspond to the reduction of SnO2 to SnO (at −0.26 VRHE) and SnO to Sn (at −0.34 VRHE), while the anodic peaks arise from the oxidation of Sn to SnO (at −0.1 V) and SnO to SnO2 (at 0.05 V).34 As control samples, SnO2–AO8, SnO2–AO12 and Sn electrode exhibit slightly more negative onset potentials (∼−0.7 VRHE) for the CO2RR and much lower current density at each specific potential (Fig. S6,† for example, SnO2–AO8: ∼6 mA cm−2, SnO2–AO12: ∼3 mA cm−2, Sn: ∼2 mA cm−2, SnO2–AO10: ∼10 mA cm−2 at −0.8 VRHE). The lower activity of the other two SnO2 nanostructures (i.e., SnO2–AO8 and SnO2–AO12) might be due to their poorer crystallinity (Fig. S4†) which limits the electrical conductivity, as corroborated by the Electrochemical Impedance Spectroscopy (EIS) analysis of the electrodes made of the three SnO2–AOx nanostructures (Fig. S7†). Overall, the CV results demonstrate the superior CO2RR activity of SnO2–AO10 aerogels to those of the Sn plate and the SnO2–AO8 and SnO2–AO12nanostructures. We further assessed the selectivity (FE) of the SnO2–AOx nanostructures and Sn plate toward each product (i.e., H2, CO, formate) at potentials ranging from −0.7 to −1.1 VRHE, the results of which are summarized in Fig. 4b and separately presented in Fig. S8–S12.† Consistent with previous reports,12 the main products detected by NMR and GC during the electrolysis on the Sn plate are H2 and formate (Fig. 4b, S8 and S9†). The FE for formate progressively increases with the potential from ∼20% at −0.7 VRHE until reaching a maximum of ∼48% at −1.1 VRHE. Conversely, H2 dominates in the lower potential range (FE: ∼65% at −0.7 VRHE).
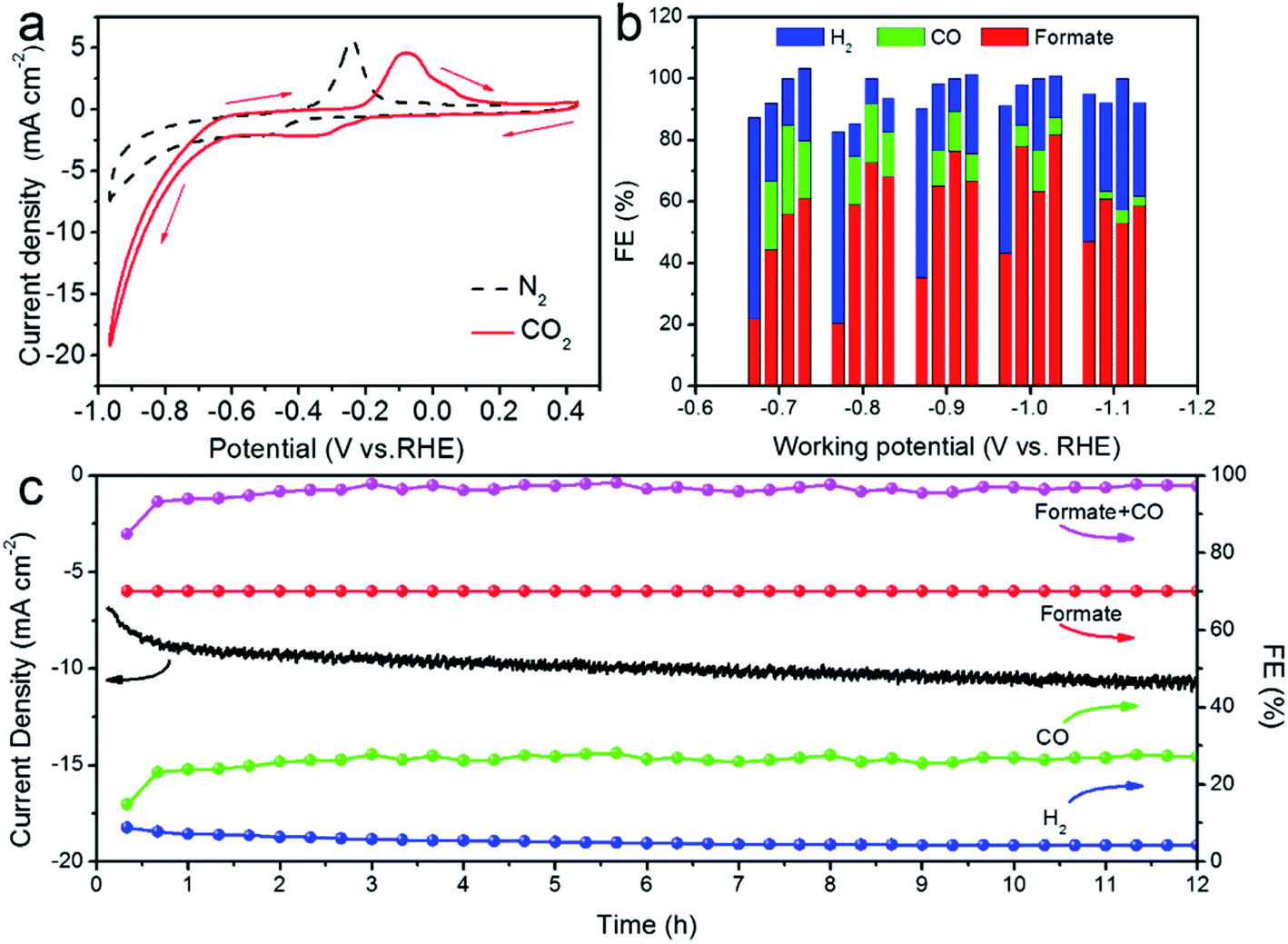 | ||
| Fig. 4 (a) CV curves of SnO2–AO10 in N2- and CO2-saturated 0.5 M KHCO3 solution, scan rate: 50 mV s −1. (b) FE of Sn plate, SnO2–AO8, SnO2–AO10, and SnO2–AO12 (from left to right at each potential) for H2, CO and formate. (c) Temporal evolution of the current density and FE toward H2, CO, formate and CO + formate of the SnO2–AO10 catalyst at −0.8 VRHE for 12 h. | ||
Although the FE for H2 drops as the potential becomes more negative, it sustains at a high level of ∼50% at −1.1 VRHE, indicative of strong competition of HER against CO2RR on the Sn plate. Interestingly, the anodization of the Sn plate resulted in a dramatic improvement in the CO2RR. As illustrated in Fig. 4b and S10–S12,† utilizing SnO2–AOx nanostructures as the electrocatalyst generates an additional product, i.e. CO. The FE toward CO, which decreases with the increasingly negative potential, can be as high as ∼25% at low potentials of −0.7 and −0.8 VRHE. Moreover, regardless of the potential, the formation of formate has been significantly boosted. For example, the highest FE achieved on SnO2–AO12 (∼82% at −1.0 VRHE) corresponds to a ∼1.8-fold enhancement over that on the Sn plate (∼46% at −1.0 VRHE) and is even comparable with those state-of-the-art results ever reported (Table S1†). Concomitantly, HER is substantially suppressed. In particular, the FE for H2 obtained over SnO2–AO10 is critically below ∼5% at −0.8 VRHE, in stark contrast with the ∼60% over the Sn plate. If we consider the total carbon conversion, the best performance is achieved over the SnO2–AO10 aerogels which display ∼95% total FE (CO: ∼22%; formate: ∼73%) towards the CO2RR at −0.8 VRHE. Essentially, such a superior electrocatalytic performance can be well maintained, as evidenced by the stable current density and the C1 product (CO + formate) conversion, as well as the constantly low FE toward H2 (<∼5%) over a long-term electrolysis of 12 h (Fig. 4c). A plausible explanation for the high stability is good preservation of the porous structure, grain boundaries and the chemical state of the Sn species (i.e., Sn4+), as unravelled by the SEM, XRD and depth-dependent XPS characterizations on the post-mortem SnO2–AO10 catalyst (Fig. S13–S15†). These structural and compositional parameters achieved by simply anodizing the Sn plate are thus the key to understanding the mechanism by which the anodic SnO2 nanostructures promote pronouncedly the CO2RR.
As suggested by earlier studies, porous structures are highly beneficial for CO2RR because they provide a large surface area and facilitate the mass transfer.36 Our SnO2 aerogels should be particularly advantageous in this aspect, because of their unique porous structure. As a matter of fact, the BET surface area (90.1 m2 g−1) for the SnO2–AO10 nanostructures is considerably higher than that attained by most porous SnO2 nanostructures (e.g., SnO2 porous nanowires: 35 m2 g−1,17 mesoporous tin oxide: 69.2 m2 g−1,36) and reaches a comparable level to that of hierarchical mesoporous SnO2 nanosheets (93.6 m2 g−1).16 Also, the EIS conducted on the SnO2–AO10 electrode indeed shows a much-improved electrolyte (containing CO2 in the form of KHCO3) transfer over that on the Sn plate (Fig. S7†). In addition to the accessibility to the reactant, another important factor in determining the catalytic behavior is the intrinsic active sites the catalyst owns. As mentioned above, the SnO2 aerogels present a high density of grain boundaries. Grain boundaries have been proposed to be enhanced active sites for CO2RR due to their favorable electronic and chemical properties which tune the binding energy of the reaction intermediates.37,38 Here, we further propose that the grain boundary might also benefit the CO2 adsorption on the catalyst, the first step of CO2RR. In an experiment of CO2 adsorption–desorption on the SnO2–AO10 nanostructures at 298 K (Fig. 5a), the isosteric heats of adsorption (Qst) at zero coverage is found to be as high as 50 kJ mol−1 (Fig. 5b), which is the upper boundary of the CO2 physical adsorption heat (25–50 kJ mol−1) and approaches the chemical adsorption heat (60–90 kJ mol−1).39,40 Lastly, the chemical state of the Sn species should be also relevant, especially with the production of formate. Tin oxide has displayed remarkable activity and selectivity towards formate.41,42 The Sn4+ species that can mostly survive (∼85%, Fig. S14†) during CO2RR are thus believed to play a crucial role in the formation of formate on SnO2–AO10 aerogels. Advancing one step, we performed first-principles calculations to assess the key energy pathways of the formation of H2, CO and formate on the SnO2 (110) surface (see details in the ESI†). The free energy diagram in Fig. 5c clearly shows that the production of formic acid is energetically more favorable than either the evolution of H2 or the formation of CO, thus confirming the importance of the tin oxide in driving the FE toward formate.
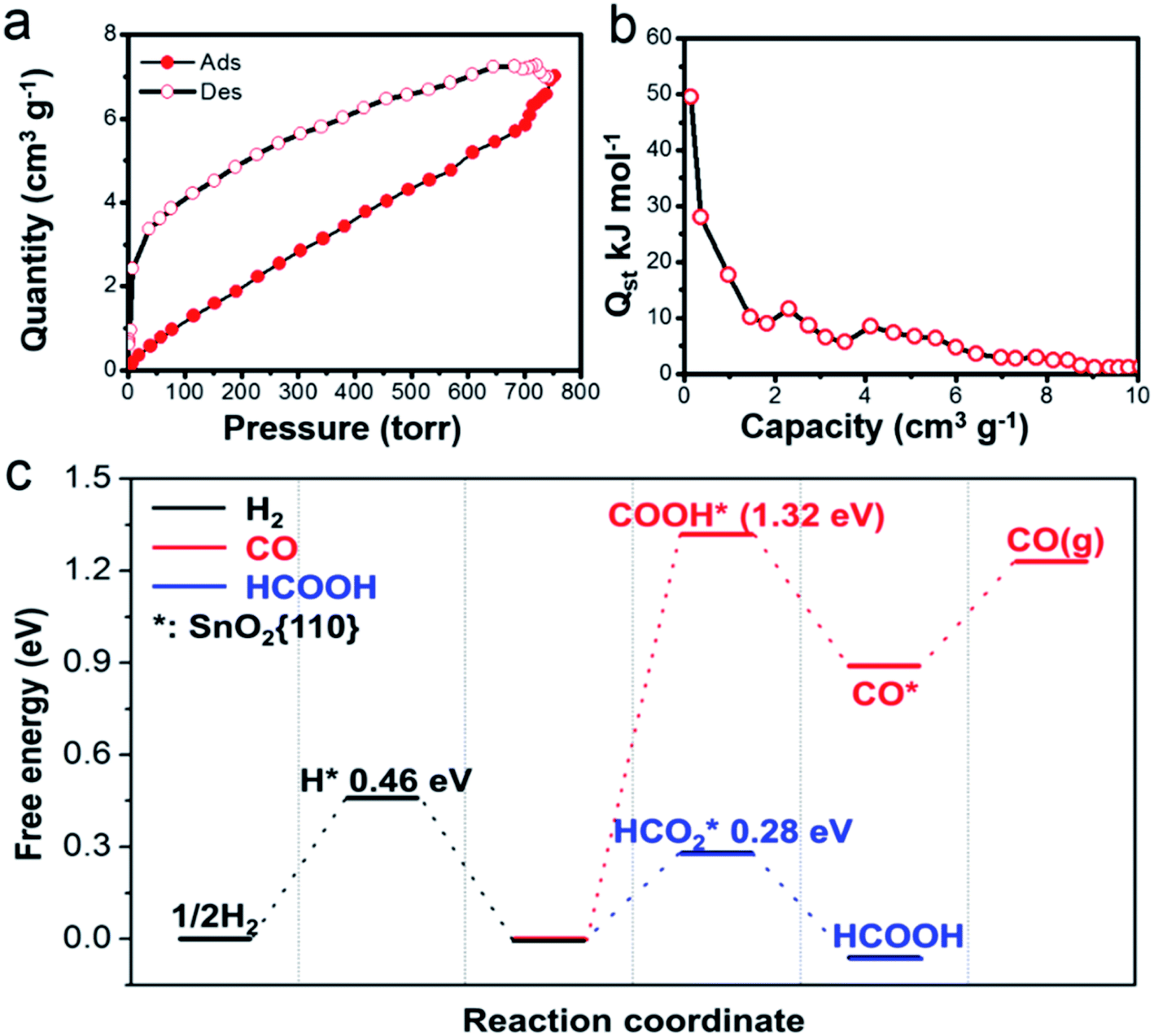 | ||
| Fig. 5 (a) CO2 adsorption–desorption isotherm at 298 K and (b) the corresponding heat of CO2 adsorption derived from the isotherm by employing the Clausius–Clapeyron equation. (c) Free energy diagrams for the reaction pathways to H2, CO and HCOOH on SnO2 (110). | ||
Conclusions
In summary, we have fabricated tubular SnO2 nanostructures via facile anodization of an Sn plate. When tested as electrocatalysts for CO2RR, the SnO2 nanostructures can achieve a relatively high current density at moderate potentials for selective and stable production of formate. The outstanding performance can be attributed to a combination of factors related to the structural and compositional properties: (1) the high specific surface area (90.1 m2 g−1) and large porosity (0.74 cm3 g−1) enable easy access for CO2 to the catalytic sites by amply exposing the active sites and advancing the electrolyte transfer; (2) the grain boundaries offer active sites for enhancing the CO2 adsorption and tuning the binding energies of intermediates; (3) the Sn4+ species together with its good preservation under CO2 conditions intrinsically dictates the selectivity for formate and inhibits the H2 evolution. We expect this study to provide useful guidelines for developing porous electrocatalysts for efficient CO2 electroreduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Development Fund, Macau SAR (File no. 002/2017/AFJ), the National Natural Science Foundation of China (NSFC-FDCT: 51761165012), the Fundamental Research Funds for the Central Universities (2020CDJQY-A072), and the Thousand Talents Program for Distinguished Young Scholars.Notes and references
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- J. F. Huang and R. Buonsanti, Chem. Mater., 2019, 31, 13–25 CrossRef CAS.
- F. J. Yu, P. H. Wei, Y. Yang, Y. H. Chen, L. M. Guo and Z. Q. Peng, Nano Mater. Sci., 2019, 1, 60–69 CrossRef.
- J. F. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De-Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2019, 9, 3117 CrossRef PubMed.
- Y. K. Chen, K. J. Chen, J. W. Fu, A. Yamaguchi, H. M. Li, H. Pan, J. H. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020 DOI:10.1016/j.nanoms.2019.10.006.
- R. Kortlever, I. Peters, S. Koper and M. T. M. Koper, ACS Catal., 2015, 5, 3916–3923 CrossRef CAS.
- T. N. Huan, P. Simon, G. Rousse, I. Genois, V. Artero and M. Fontecave, Chem. Sci., 2017, 8, 742–747 RSC.
- S. B. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arguer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nature Rev. Mater., 2016, 1, 16059 CrossRef CAS.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Israel J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136(5), 1734–1737 CrossRef CAS PubMed.
- F. C. Lei, W. Liu, Y. F. Sun, J. Q. Xu, K. T. Liu, L. Liang, T. Yao, B. C. Pan, S. Q. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- S. Liu, F. Pang, Q. Zhang, R. Guo, Z. Wang, Y. Wang, W. Zhang and J. Ou, Appl. Mater. Today, 2018, 13, 135–143 CrossRef.
- X. Wang, J. Lv, J. X. Zhang, X. L. Wang, C. Z. Xue, G. Q. Bian, D. S. Li, Y. Wang and T. Wu, Nanoscale, 2020, 12, 772–784 RSC.
- Y.-W. Choi, F. Scholten, I. Sinev and B. R. Cuenya, J. Am. Chem. Soc., 2019, 141, 5261–5266 CrossRef CAS PubMed.
- J. Wang, Y. Ji, Q. Shao, R. Yin, J. Guo, Y. Li and X. Huang, Nano Energy, 2019, 59, 138–145 CrossRef CAS.
- H. C. Shin, J. Dong and M. Liu, Adv. Mater., 2004, 16, 237–240 CrossRef CAS.
- K. Jiang, Y. F. Huang, G. S. Zeng, F. M. Toma, W. A. Goddard III and A. T. Bell, ACS Energy Lett, 2020, 5, 1206–1214 CrossRef CAS.
- J. Vandevondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- CP2K version 4.1, CP2K is freely available from, http://www.cp2k.org.
- B. Geraldlippert and J. Huttermicheleparrinello, Mol. Phys., 1997, 92, 477–488 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- M. Fronzi, S. Piccinin, B. Delley, E. Traversa and C. Stampfl, Phys. Chem. Chem. Phys., 2009, 11, 9188–9199 RSC.
- V. Shapovalov and H. Metiu, J. Catal., 2007, 245, 205–214 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B, 1995, 54, 1703–1710 CrossRef PubMed.
- A. Ghysels, T. Verstraelen, K. Hemelsoet, M. Waroquier and S. V. Van, J. Chem. Inf. Model., 2010, 50, 1736–1750 CrossRef CAS PubMed.
- J. Gu, F. Heroguel, J. Luterbacher and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 2943–2947 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- R. Daiyan, X. Lu, W. H. Saputera, Y. H. Ng and R. Amal, ACS Sustainable Chem. Eng., 2018, 6, 1670–1679 CrossRef CAS.
- L. Fan, X. Li, B. Yan, J. Feng, D. Xiong, D. Li, L. Gu, Y. Wen, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502057 CrossRef.
- H. Ge, Z. Gu, P. Han, H. Shen, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2018, 531, 564–569 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- K.-S. Kim, W. J. Kim, H.-K. Lim, E. K. Lee and H. Kim, ACS Catal., 2016, 6, 4443–4448 CrossRef CAS.
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- T. Watabe and K. Yogo, Sep. Purif. Technol., 2013, 120, 20–23 CrossRef CAS.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J.-j. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03152f |
| ‡ Ruizhen Ma and Yan-Li Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |