
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUrsolic acid derivatives are potent inhibitors against porcine reproductive and respiratory syndrome virus†
Yang Chen‡
a,
Hui Li‡c,
Li Wuab,
Mingxin Zhanga,
Yarou Gaoa,
Heng Wangab,
Dan Xuab,
Weisan Chend,
Gaopeng Song*bc and
Jianxin Chen *ab
*ab
aGuangdong Provincial Key Laboratory of Veterinary Pharmaceutics Development and Safety Evaluation, College of Veterinary Medicine, South China Agricultural University, Guangzhou 510642, China. E-mail: jxchen@scau.edu.cn; Tel: +86-20-8528-0234
bGuangdong Laboratory for Lingnan Modern Agriculture, South China Agricultural University, Guangzhou 510642, China. Fax: +86-20-8528-0293; Tel: +86-20-8528-0293E-mail: vinsin1021@126.com
cCollege of Materials and Energy, South China Agricultural University, Guangzhou 510642, China
dDepartment of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria 3086, Australia
First published on 15th June 2020
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is one of the most devastating viral pathogens of swine and has a substantial economic impact on the global pork industry. Currently, vaccination strategies provide very limited protection against PRRSV transmission. Therefore, there is an urgent need to develop new antiviral strategies to prevent PRRSV pandemics. In this study, we showed that 3-O-β-chacotriosyl ursolic acid (1) and its ester analogs possessed anti-PRRSV activity in vitro, of which bioisosteric surrogates 7–15 were further generated with the aim of enhancing the selective index. Our results showed that amidation of the 17-COOH group of UA could significantly reduce cytotoxicity and enhance anti-PRRSV activity in MARC-145 cells. Among them, compound 9 displayed the strongest anti-PRRSV activity with the least cytotoxicity. Potent inhibition of representative compounds 9 and 12 on PRRSV infection was observed not only in MARC-145 cells, but also in primary porcine alveolar macrophages, PRRSV-target cells in vivo. Furthermore, compounds 8, 9, 12 and 14 exhibited broad-spectrum inhibitory activities in vitro against high pathogenic type 2 PRRSV NADC30-like and GD-XH strains as well as classical CH-1a and VR2332 strains. Mechanistically, compounds 9 and 12 inhibited PRRSV replication by directly inactivating virions and therefore affecting all tested stages of the virus life cycle, including viral entry, replication and progeny virus release, but did not affect cellular susceptibility to PRRSV. Our findings suggest that compound 9 could be a hit PRRSV inhibitor and deserves further in vivo studies in swine.
1. Introduction
Porcine reproductive and respiratory syndrome (PRRS) is the most economically important infectious disease that causes huge economic losses in the worldwide pig industry. PRRS, caused by porcine reproductive and respiratory syndrome virus (PRRSV), is characterized by reproductive failure in sows, severe respiratory disease and poor growth performance in piglets and growing pigs.1,2 This disease was first observed simultaneously in North America and Europe in the late 1980s, and subsequently became an epidemic disease in North America, Europe, and Asia within the succeeding years.3–6 In 2006, a highly pathogenic PRRSV (HP-PRRSV) strain broke out in China, which presented as high fever, high morbidity, and high mortality among pig farms, resulting in substantial economic losses.7 In 2013, the annual loss caused by PRRS to the American swine industry was approximately 664 million USD.8PRRSV is a small, enveloped virus with a single-stranded, nonsegmented, positive-sense RNA genome that is approximately 15 kb in length containing at least 10 open reading frames (ORFs 1a, 1b, 2a, 2b, 3–7 and the newly identified ORF5a).9–11 The virus has been classified into two major genotypes: type I, or European-like (prototype Lelystad), and type II, or North American-like (prototype VR-2332).12 Although the two genotypes share only approximately 60% sequence identity, the overall disease phenotype and gross clinical symptoms are similar.13,14 Interestingly, PRRSV infection is highly restricted to porcine alveolar macrophages (PAMs), the primary targets of PRRSV in vivo.15 Most importantly, PRRSV has the intrinsic ability to adapt and evolve to form new strains frequently, due to its high mutation rate at 3.29 × 10−3 substitutions per nucleotide site per year.16
Currently, vaccination remains the most prevalent method of controlling PRRSV infections. However, commercially available vaccines only provide limited protection against PRRSV due to antigenic heterogeneity, replication in and destruction of lung alveolar macrophages, antibody-dependent enhancement (ADE) and viral persistence.17 Therefore, the development of new strategies for controlling this infectious disease, especially novel drugs against PRRSV is an urgent need. Previous studies have reported that some natural compounds have antiviral activities against PRRSV infection, including glycyrrhizin,18 flavaspidic acid AB,19 morpholino oligomer,20 proanthocyanidin A2 (ref. 21) and xanthohumo.22 Despite this, no effective drugs are commercially available for treating PRRSV infections.
Pentacyclic triterpenoids (PTs), a class of abundant natural products in plants have received increasing attentions due to their wide spectrum of pharmacological activities, including antiviral, antitumor, immunomodulatory, anti-inflammatory, and hepatoprotective activities.23 Some PTs are already marketed as therapeutic agents or dietary supplements. For example, glycyrrhizin (glycyrrhizic acid) has been used in treating chronic hepatic diseases for over 40 years in China and Japan clinically. Ursolic acid (UA), oleanolic acid (OA) and their derivatives represent the most well-known two classifications of PTs with noticeable biological activities, including antiviral and antibacterial properties. In 1996 Quere et al. found remarkable anti-HIV activity of UA through inhibiting HIV-1 protease.24 In 2013 Kong et al. reported that OA and UA exhibited anti-HCV activity through suppressing HCV NS5B RNA-dependent RNA polymerase activity as noncompetitive inhibitors.25 Recently, we demonstrated that platycodin D (PD), an oleanane type triterpenoid saponin from the root of Platycodon grandiflorum A. DC, possessed potent and broad-spectrum inhibition on PRRSV replication at micromolar concentrations.26 In addition, UA, OA and analogues were also found to possess antibacterial activity. In 2014 Nascimento reported that modification of the UA at C-3 (hydroxyl group) to yield 3 β-formyloxy-urs-12-en-28-oic acid resulted in significant antibacterial and antioxidant properties.27 In 2015 Kim et al. showed that OA can kill L. monocytogenes, E. faecium, and E. faecalis by destroying the bacterial cell membrane.28 Recently, Blanco-Cabra et al. confirmed the antibacterial mechanism of action of OA derivatives through penetrating and damaging the bacterial cell membrane.29
In our previous studies, we discovered a series of 3-O-β-chacotriosyl UA derivatives as novel IAV H5N1 entry inhibitors.30 Recently, we reported that incorporation of two “privileged fragments”, 2-(piperidin-1-yl) ethan-1-amine and 2-(1,3-oxazinan-3-yl)ethan-1-amine, into 3-O-β-chacotriosyl UA resulted in improved activity against IAVs in vitro.31 Structurally, the above UA derivatives belong to the PT saponins, as does PD, with the C-20 methyl group shifted to C-19. Based on their similar structural skeleton and strong anti-H5N1 activity, in this study, we evaluated anti-PRRSV activity of 3-O-β-chacotriosyl UA 1 and its analogs 2–15 (Fig. 1). 3-O-β-Chacotriosyl UA amide derivatives 9 and 12 exhibited potent anti-PRRSV activity in Marc-145 cells and PAMs at micromolar concentrations and minimal cytotoxicity. Their PRRSV inhibiting mechanisms were also investigated. To our knowledge, this is the first report of anti-PPRSV activities of UA derivatives.
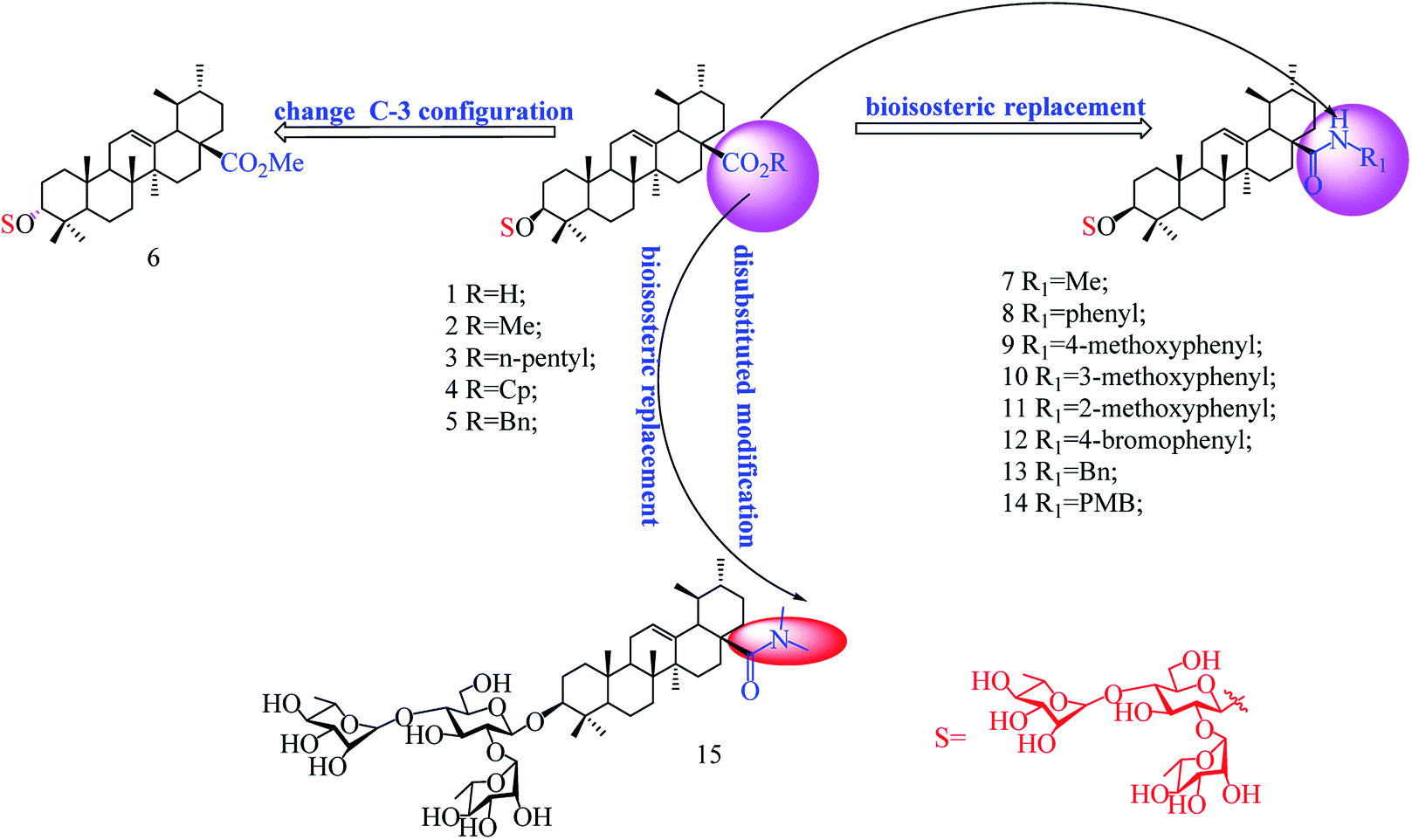 | ||
| Fig. 1 Chemical structure of the compounds used in this study. | ||
2. Materials and methods
2.1 Cell lines and viruses
MARC-145 cells, a PRRSV-permissive cell line derived from African green monkey kidney cell line MA-104 were obtained from the American Type Culture Collection (ATCC) and grown in Dulbecco's minimum essential medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Biological Industries, Israel) and 100 IU mL−1 of penicillin and 100 μg mL−1 streptomycin at 37 °C with 5% CO2.Porcine alveolar macrophages (PAMs) were obtained from the lungs of 4- to 6 week-old PRRSV-negative Large-White piglets (Xinli Pig Farm, Wuzhou, China) by lung lavage according to a previously described method.32 Briefly, the lungs were washed three times with pre-cooled phosphate buffered saline (PBS) solution containing penicillin (300 IU mL−1) and streptomycin (300 μg mL−1). Cells were centrifuged at 800 g for 10 minutes, resuspended in RPMI 1640 supplemented with 10% FBS and 100 IU mL−1 of penicillin and 100 μg mL−1 streptomycin at 1 × 106 cells per ml in 6-well plate, and then incubated at 37 °C for 2 h. The suspending cells (mainly lymphocytes and red blood cells) were removed and adherent cells were PAMs.26
Four type 2 PRRSV strains including classical CH-1a and VR2332 strains, and highly pathogenic NADC30-like and GD-XH strains were propagated in MARC-145 cells in DMEM with 3% FBS (essential medium). Virus preparations were titrated and stored at −80 °C. Virus titers were determined using a microtitration infectivity assay. Briefly, virus preparations were 10-fold serially diluted in essential medium. Confluent monolayers of MARC-145 cells or PAMs prepared in 96-well plates were inoculated in quadruplicates with 100 μL of each sample and incubated for 2 h at 37 °C. The inoculum was then discarded, and the cell monolayer replenished with fresh essential medium and incubated for an additional 72 h and monitored for cytopathic effects (CPE) daily. The titer of each preparation was calculated based on the amount of CPE and expressed as a 50% tissue culture infective dose (TCID50/1 mL).
2.2 Preparation of compounds
Ribavirin, a broad-spectrum antiviral agent, was used as positive control and purchased form Star Lake Bioscience Co., Ltd (Zhaoqing, China). UA and its derivatives and ribavirin were dissolved in dimethyl sulfoxide (DMSO, Sigma, USA) and diluted with essential medium before use. The final concentration of DMSO in the culture medium was less than 0.4%.2.3 Cytotoxicity assay
The cytotoxicity of compounds was evaluated using MTT assay.26 Briefly, for MARC-145 cells, 5 × 104 cells per well were seeded in 96-well plates and grown at 37 °C for 36 h. For PAMs, 2 × 105 cells per well were seeded in 96-well plates and incubated at 37 °C for 12 h. The medium was then replaced with fresh one containing serially diluted compound and the cells were further incubated for 48 h. The culture medium was removed and replaced with 100 μL 3-(4,5-dimethylthiozol-2-yl)-3,5-dipheryl tetrazolium bromide (MTT; Sigma-Aldrich) solution (0.5 mg mL−1 in PBS) and incubated at 37 °C for 4 h. After removal of the supernatant, 150 μL of DMSO was added to all of the wells to dissolve the formazan crystals for 10 min at 37 °C. Cell viability was measured as the absorbance at 490 nm with a microplate reader (Thermo fisher scientific, USA) and expressed as a percentage of the control level. The mean optical density (OD) values from six wells per treatment were used as the cell viability index. The 50% cytotoxic concentration (CC50) was analyzed by GraphPad Prism 7.0 (GraphPad Software, San Diego, CA).2.4 Antiviral activity assay
The antiviral activity assay was performed for compounds to compare their in vitro PRRSV inhibiting capacities. MARC-145 or PAM cell monolayers grown in 96-well plates were infected with PRRSV (0.05 MOI for MARC-145 cells and 0.5 MOI for PAMs) in essential medium at 37 °C for 2 h. Supernatants were removed and fresh DMEM containing different concentrations of each compound was then added. At indicated time points, viral titer in the final supernatant was determined by the end point dilution assay using MARC-145 cells and expressed as log10 TCID50/1 mL.26 Ribavirin was used as a positive antiviral drug control in this study.2.5 Indirect immunofluorescence assay
For immunostaining, the PRRSV-infected or uninfected cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.25% Triton X-100 for 10 min at room temperature (r.t.), blocked with 1% bovine serum albumin (BSA) for 60 min at RT and then incubated with a mouse monoclonal antibody against the N-protein of PRRSV (clone 4A5, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400 dilution, MEDIAN Diagnostics, Korea) at 4 °C overnight. After three washes with PBS, the cells were incubated for 1 h at RT with a goat anti-mouse secondary antibody conjugated with Alexa Fluor® 568 (red) (Cell Signaling Technology, USA) at 1:1000 dilution. Nuclei were counterstained using 50 μL of 4,6-diamidino-2-phenylindole (DAPI, 300 nM; Sigma Chemical Co., USA) (blue). Immunofluorescence was captured using the Leica DMI 4000B fluorescence microscope (Leica, Wetzlar, Germany). Fluorescence optical densities (OD) (blue and red, respectively) of each well were digitized using Software Image J. Normalized OD values (%) from compound-treated samples were compared to those from corresponding DMSO control groups (set as 100%). Protection percentage from compound-treated sample = [(100 − Normalized OD of compound-treated samplered)/Normalized OD of compound-treated sampleblue] × 100%. The EC50 value (the concentration required to protect 50% cells from PRRSV infection) was determined by plotting the protection percentage as a function of compound concentration and calculated by nonlinear regression function with the GraphPad Prism 7.0 software.
:400 dilution, MEDIAN Diagnostics, Korea) at 4 °C overnight. After three washes with PBS, the cells were incubated for 1 h at RT with a goat anti-mouse secondary antibody conjugated with Alexa Fluor® 568 (red) (Cell Signaling Technology, USA) at 1:1000 dilution. Nuclei were counterstained using 50 μL of 4,6-diamidino-2-phenylindole (DAPI, 300 nM; Sigma Chemical Co., USA) (blue). Immunofluorescence was captured using the Leica DMI 4000B fluorescence microscope (Leica, Wetzlar, Germany). Fluorescence optical densities (OD) (blue and red, respectively) of each well were digitized using Software Image J. Normalized OD values (%) from compound-treated samples were compared to those from corresponding DMSO control groups (set as 100%). Protection percentage from compound-treated sample = [(100 − Normalized OD of compound-treated samplered)/Normalized OD of compound-treated sampleblue] × 100%. The EC50 value (the concentration required to protect 50% cells from PRRSV infection) was determined by plotting the protection percentage as a function of compound concentration and calculated by nonlinear regression function with the GraphPad Prism 7.0 software.
2.6 Real-time reverse-transcription PCR (RT-PCR)
Total RNA was extracted from cells or culture supernatants using the total RNA rapid extraction kit (Fastagen, Shanghai, China) by following manufacturer's instructions. RNA was reverse-transcribed into first-strand cDNA using a reverse transcription kit (TaKaRa, Japan). PCR amplification was performed on 1 μL of reverse-transcribed product with primers designed against PRRSV-NSP9 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase, used as the endogenous control). The primers used for PCR amplification are listed in Table 1. Real-time PCR was performed using 2 × RealStar Green Power Mixture (containing SYBR Green I Dye) (Genstar, Beijing, China) on the CFX96 Real-time PCR system (Bio-Rad, USA).Relative mRNA expression was calculated by 2−ΔΔCT method using DMSO-treated infected cells or DMSO-treated mock-infected cells as reference samples for determining PRRSV-NSP9 gene expression, respectively.
2.7 Direct PRRSV-compound interaction
To investigate whether compound directly interact with the virus, 100 μL of PRRSV GD-XH (2 × 106 PFU) was mixed with various concentrations of testing compound in essential medium (0.9 mL total volume) for 1 h at 37 °C. Then PRRSV and the compound were separated by ultrafiltration (7500 g, 10 min) at 4 °C. PRRSV trapped in the ultrafiltration filter were washed twice with essential medium to remove residual compound before being resuspended in essential medium for infecting MARC-145 cells grown in 6-well plates for 2 h. After three washes with PBS, the cells were cultured in fresh medium for an additional 48 h at 37 °C and then subjected to virus titer assessment using the end point dilution assay and viral N protein analysis using IFA.2.8 Statistical analysis
All experiments were performed at least three times. The results were presented as mean ± standard deviation (SD). Statistical significance was determined by Student's t test when only two groups were compared or by one-way analysis of variance (ANOVA) when more than two groups were compared. *P < 0.05, **P < 0.01, and ***P < 0.001 were considered to be statistically significant at different levels.3. Results
3.1 Synthesis and chemical structure characterization of ursolic acid (UA) derivatives
As shown in Scheme 1, benzyl 3-O-β- chacotriosyl oleanolate 22 was prepared by the similar route as that previously reported for compounds 1–6,30 which then was subjected to acetic anhydride, followed by hydrolysis of Bn group in the presence of 10% Pd/C, led to compound 23. The key intermediate 23 was treated with oxalyl chloride to yield 28-acyl chloride, which was then condensed with appropriate amines and finally deprotected with MeONa to yield the corresponding target saponins 7–15, respectively, as shown in Fig. 1 and Supplementary materials.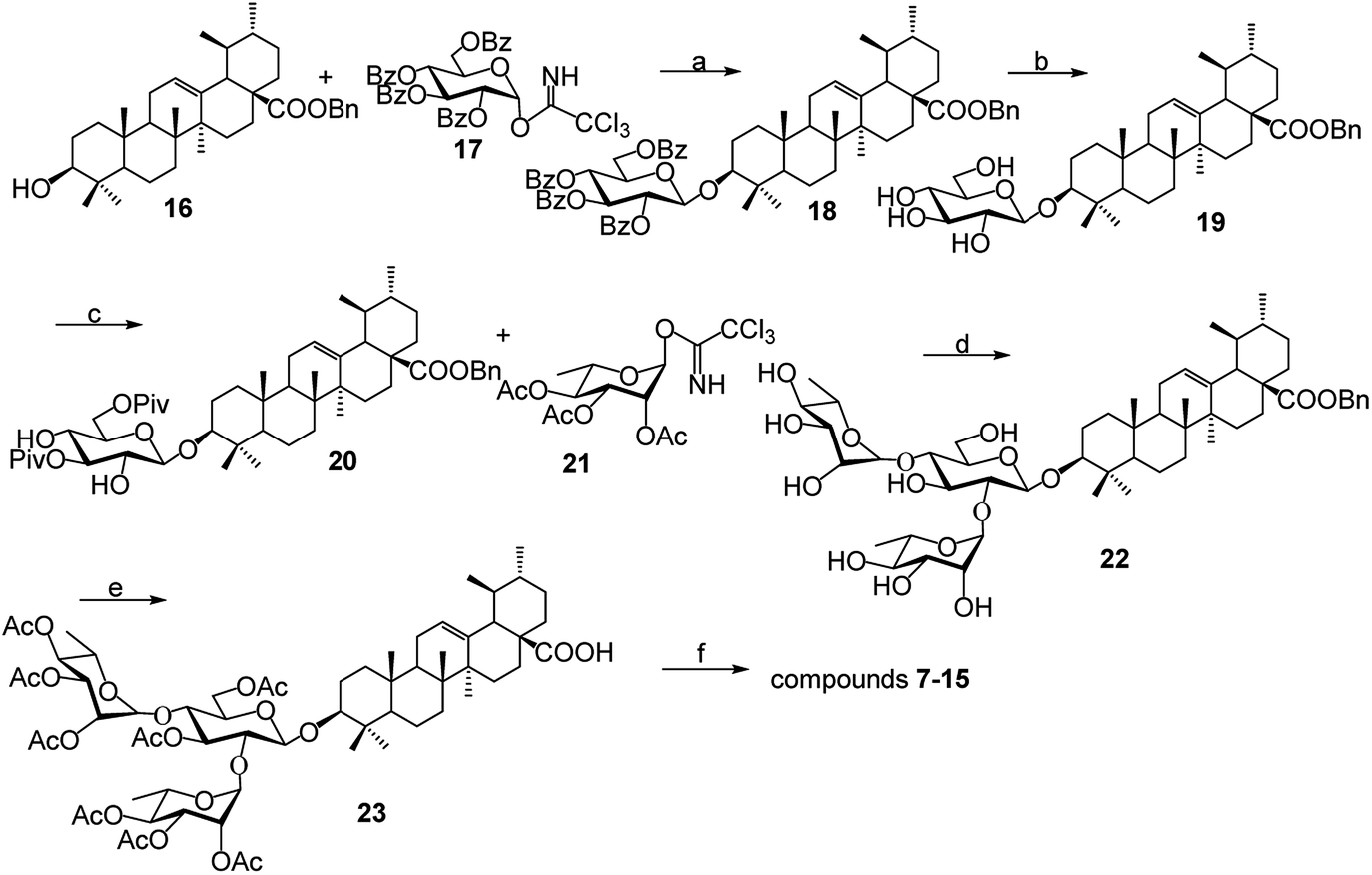 | ||
| Scheme 1 Reagents and conditions: (a) TMSOTf, CH2Cl2; (b) MeONa, CH3OH; (c) PivCl, pyridine; (d) (i) TMSOTf, CH2Cl2, (ii) NaOH, CH3OH; (e) (i) Ac2O, DMAP, pyridine, CH2Cl2, (ii) 10% Pd/C, H2, CH3OH–CH2Cl2; (f) (i) (COCl)2, CH2Cl2 (ii) R1R2NH, Et3N, CH2Cl2; (iii) CH3ONa, CH3OH. | ||
3.2 Relationship of chemical structure and antiviral activity of UA derivatives against PRRSV in MARC-145 cells
Cellular toxicity of UA and compounds 1–15 were firstly examined in MARC-145 cells using MTT assay and was expressed as relative cell viability of the viable cells in the DMSO control (set up as 100%).26 Antiviral activity and EC50 value (the concentration required to protect 50% cells from virus infection) for UA and compounds 1–15 against PRRSV infection in MARC-145 cells were assessed using indirect immunofluorescence assay (IFA).26 The CC50 values (the 50% cytotoxic concentration of compounds on MARC-145 cells) and EC50 values of compounds against PRRSV GD-XH strain are shown in Table 2. Among the tested compounds, 9 exhibited the most potent inhibition on PRRSV GD-XH (EC50 = 2.03 μM) and the lowest cytotoxicity on MARC-145 cells (CC50 > 160 μM). Ribavirin, a well-known inhibitor of viral RNA polymerase, was used as a positive antiviral drug control in this study. Our results showed that ribavirin exhibited a weaker inhibition (EC50 = 23.7 μM) on PRRSV infection compared to compound 9 in the same assays, but with a much less cytotoxicity (CC50 > 640 μM). Results of compound 9's cytotoxicity and antiviral activity reflected by IFA, as well as those of another representative compound 12, are shown in Fig. 2.| Compound | bCC50 (μM) | cEC50 (μM) | dSelectivity index (SI) |
|---|---|---|---|
| a UA, ursolic acid.b CC50, the 50% cytotoxic concentrations of compounds on MARC-145 cells.c EC50, the 50% effective concentrations for keeping cells from PRRSV infection.d SI (selectivity index) is the ratio of CC50 to EC50. Data are presented as means ± SD of results from three independent experiments. | |||
| 1 | 29.0 ± 1.8 | 3.51 ± 0.42 | 8.3 |
| 2 | 36.5 ± 2.1 | 8.98 ± 0.55 | 4.1 |
| 3 | 81.3 ± 3.9 | 6.21 ± 0.47 | 13 |
| 4 | 41.6 ± 2.7 | 5.29 ± 0.31 | 7.9 |
| 5 | 81.4 ± 3.3 | 5.02 ± 0.37 | 16 |
| 6 | >160 | 28.3 ± 1.9 | >5.7 |
| 7 | 109 ± 5.6 | 4.01 ± 0.29 | 27 |
| 8 | >160 | 3.89 ± 0.33 | >41 |
| 9 | >160 | 2.03 ± 0.25 | >79 |
| 10 | >160 | 12.1 ± 0.94 | 13 |
| 11 | 88.3 ± 4.1 | 3.18 ± 0.39 | 28 |
| 12 | >160 | 3.88 ± 0.40 | >41 |
| 13 | >160 | 5.91 ± 0.44 | >27 |
| 14 | >160 | 4.89 ± 0.50 | >33 |
| 15 | >160 | 33.5 ± 2.6 | >4.8 |
| UA | 65.3 ± 3.2 | 8.03 ± 0.93 | 8.1 |
| Ribavirin | >640 | 23.7 ± 3.1 | >27 |
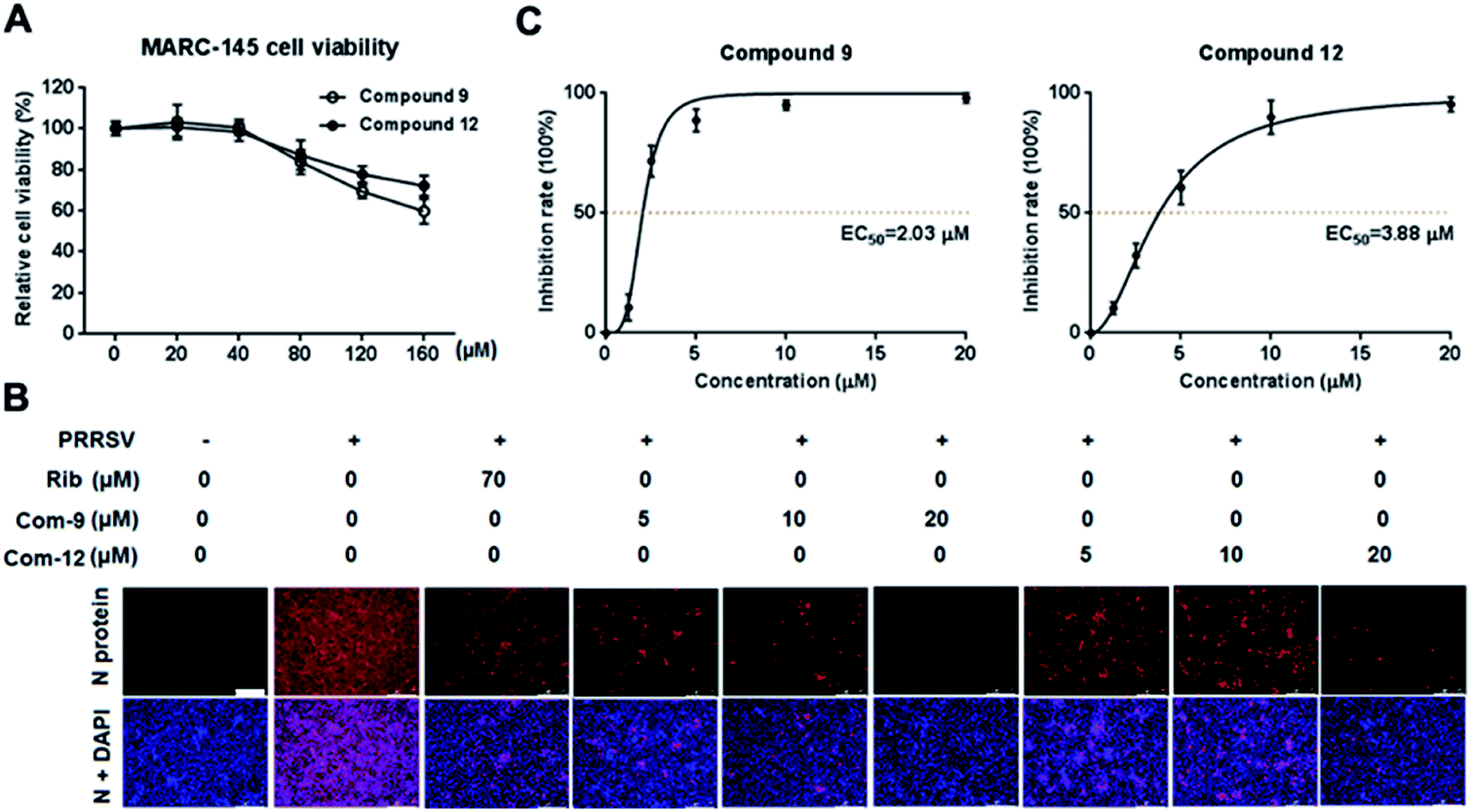 | ||
| Fig. 2 The cellular toxicity and antiviral activity of compounds 9 and 12 against PRRSV GD-XH in MARC-145 cell cultures. (A) Cytotoxicity of 9 and 12 was examined in MARC-145 cells using MTT assay at 48 h post compound treatment and results are shown as relative cell viability of the viable cells in the absence of the compound (set as 100%). (B and C) Antiviral activity of compounds 9 (Com-9) and 12 (Com-12) against PRRSV infection in MARC-145 cells was examined using immunofluorescence assay (IFA) at 48 hpi. Cells grown in 96-well plates were infected with PRRSV GD-XH (0.05 MOI) for 2 h at 37 °C and then cultured in fresh medium containing various concentrations of 9 or 12. IFA for the N protein of PRRSV was performed at 48 hpi using Alexa Fluor 568-conjugated goat anti-mouse secondary antibody (red). Nuclei were counterstained using 4,6-diamidino-2-phenylindole (DAPI) (blue). The EC50 value (concentration of compound required to protect 50% cells from infection) was determined based on IFA as described in the Methods. (B) Representative IFA images from one of the three independent experiments. Scale bar: 250 μm. (C) Dose–response curves for 9 and 12 from the three independent IFA experiments. | ||
The results in Table 2 indicated that all the 3-O-β-chacotriosyl UA analogues 1–15 had different antiviral activity, suggesting that the antiviral activity of the saponins is sensitive to the change of their structures. Comparing the antiviral activity of compounds 2–5 with that of 1, a remarkable reduction in the inhibitory activity was served despite the fact esterification of the 17-COOH group of UA was helpful to reduce cytotoxicity against MARC-145 cells. These results demonstrated that the hydroxyl group of the COOH group was indispensable for anti-PRRSV activity. Of the two 3-hydroxyl group isomers, the 3α-form 6 displayed a significant loss of antiviral activity compared to the 3β-form 2, probably due to the decreased active conformation of aglycone toward the potential acceptor, which indicated that configuration at C-3 was a crucial factor for the antiviral activity. Encouragingly, amide derivatives 7–14, bioisosteric surrogates of UA esters 2–5, exhibited increased selectivity index while maintaining similar or slightly enhanced inhibitory activity compared with 1. Notably, 9 showed the strongest anti-PRRSV activity and the most enhanced selective index (SI > 79) among this set of compounds. While there was not enough information to fully understand the effect of the substituted groups in the benzene ring on the anti-PRRSV activity, the location of the individual methoxy group had an impact on the bioactivity. For instance, 9 or 14 exhibited a slight better inhibition than 8 or 13, respectively, suggesting methoxy group modification at para-position could enhance antiviral activity. However, the shifting the methoxy group from position 4 to position 3 (10) decreased inhibition, while shifting to position 2 (11) resulted in increased cytotoxicity. Moreover, the attachment of bromine group at the para-position of the phenyl ring gave rise to 12, which had a similar antiviral activity as 8. These results revealed subtle modifications of the phenyl moiety with electron-donating groups, such as methoxy group at para-position of the phenyl ring, was beneficial to antiviral activity. It was found that a disubstituted amide structure 15 showed lower activity than monosubstituted 7, owing to the absence of the polar NH group, which further supported the fact that keeping a polar group at the 17-COOH position was critical for antiviral activity.
3.3 UA and its derivatives show broad-spectrum anti-PRRSV activity
To explore whether 3-O-β-chacotriosyl ursolic acid analogues possess broad inhibitory effects on different PRRSV subtypes, four representative derivatives (8, 9, 12, 14) with SI > 30, as well as UA, were evaluated for their antiviral activities against three other common PRRSV strains (CH-1a, VR2332 and NADC30-like) using IFA on infected MARC-145 cells at 48 hpi. As shown in Table 3, the four compounds exhibited inhibitions on PRRSV CH-1a, VR2332 and NADC30-like strains similar to or even slightly stronger than those on PRRSV GD-XH strain, indicating that 3-O-β-chacotriosyl ursolic acid analogues possess broad-spectrum anti-PRRSV activities. Among evaluated compounds, 9 always showed the most potent inhibition on the three different PRRSV strains' replication. Compound 12 is the bromine substitution at the para-position of the phenyl ring of 8, which also exhibited considerable inhibition on PRRSV replication. Thus, 9 and 12 were selected for further antiviral and mechanism studies.| Compound | CH-1a | VR2332 | NADC30-like | |||
|---|---|---|---|---|---|---|
| EC50 (μM) | SI | EC50 (μM) | SI | EC50 (μM) | SI | |
| 8 | 6.02 ± 0.46 | >27 | 2.47 ± 0.31 | >65 | 2.03 ± 0.25 | >79 |
| 9 | 3.89 ± 0.30 | >41 | 1.30 ± 0.22 | >123 | 1.28 ± 0.18 | >125 |
| 12 | 4.38 ± 0.37 | >36 | 1.56 ± 0.20 | >102 | 1.28 ± 0.16 | >125 |
| 14 | 15.7 ± 1.1 | >10 | 4.02 ± 0.36 | >40 | 3.73 ± 0.28 | >43 |
| UA | 11.3 ± 0.78 | 5.8 | 5.66 ± 0.52 | 11 | 5.03 ± 0.35 | 13 |
| Ribavirin | 31.5 ± 2.8 | >20 | 22.3 ± 3.1 | >29 | 16.8 ± 2.1 | >38 |
3.4 Compounds 9 and 12 show outstanding inhibition on PRRSV replication in MARC-145 cells
Among tested UA and the 15 derivatives, 9 and 12 showed stronger inhibition on PRRSV GD-XH replication and less cytotoxicity in MARC-145 cells, with SI values of 79 and 41, respectively. To confirm the anti-PRRSV effects of 9 and 12, we further examined the inhibition of the two compounds on PRRSV GD-XH strain in MARC-145 cells using virus titration and RT-PCR at 48 hpi. As shown in Fig. 3A, treatment with 9 or 12 resulted in a significant reduction of PRRSV titer in a dose-dependent manner. Treatment with 20 μM of 9 or 12 led to a 6.4log or 6.3log reduction in progeny virus production, respectively, compared to that in DMSO-treated control (Fig. 3A). Similar inhibitions of 9 and 12 on PRRSV replication were reflected by reduced viral NSP9 RNA level in a dose-dependent manner (Fig. 3B). Ribavirin, a well-known inhibitor of viral RNA polymerase, was used as a positive antiviral drug control in this study. Our results showed that the treatment of 70 μM ribavirin led to a similar reduction of progeny virus titer to those treated with 5 μM of 9 or 10 μM of 12. We further studied the PRRSV inhibition kinetics from 24 to 72 hpi by the two compounds. As expected, the addition of 20 μM of 9 or 12 significantly inhibited progeny virus titers at all time-points (Fig. 3C).
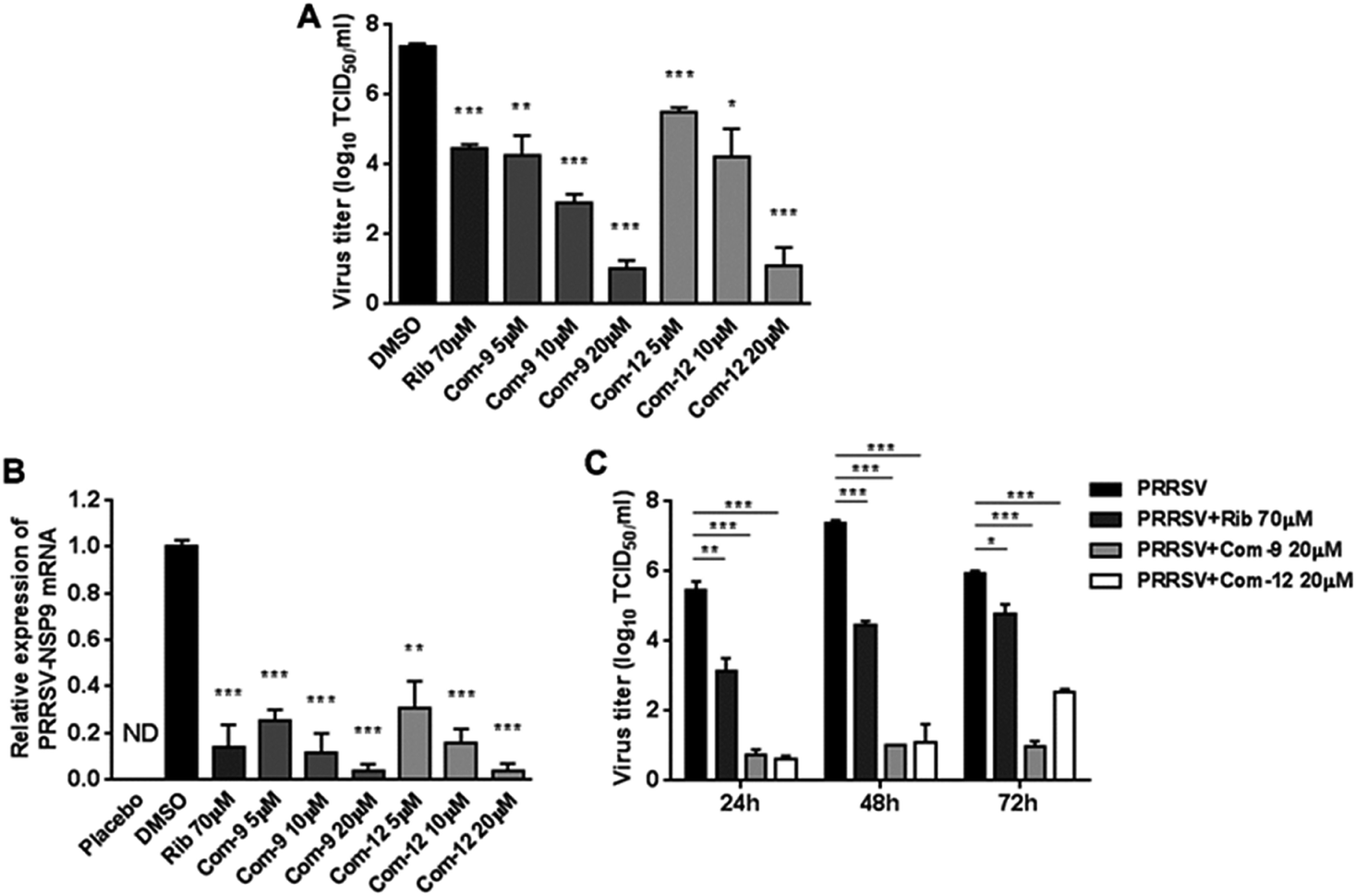 | ||
| Fig. 3 Assessing antiviral activities of compounds 9 and 12 in PRRSV-infected MARC-145 cells by virus titer and PCR. Cells grown in 6-well plates were infected with PRRSV GD-XH (0.05 MOI) for 2 h at 37 °C and then cultured in fresh medium containing various concentrations of 9 or 12. At 48 h ((A) and (B)) or indicated time-points (C) post infection, the samples were subjected to viral titer, or RT-PCR analysis. ((A) and (C)) The PRRSV titer was determined after treatment with compounds for 48 h (A) or indicated time-points (C) using the end point dilution assay and expressed as log10 TCID50/1 mL. (B) Relative PRRSV NSP9 mRNA level was analyzed using real-time RT-PCR after 48 h compound treatment. Expression of GAPDH was shown as a loading control, and samples with solvent DMSO addition were used as no inhibition control (set as 1). Statistical significances are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001 compared to DMSO-treated control. | ||
3.5 Compounds 9 and 12 inhibit PRRSV replication in PAMs
Since porcine alveolar macrophages (PAMs) were identified to be the major target cell type of PRRSV infection in pigs in vivo, we questioned whether 9 and 12 were also able to inhibit PRRSV replication in ex vivo PAMs. We initially assessed cytotoxicity of 9 and 12 on PAMs using MTT assay at 48 h post compound treatment. As shown in Fig. 4A, compound 12 at concentrations of ≤160 μM and 9 at concentrations of ≤120 μM did not exhibit any cytotoxicity on PAMs, indicating minimal cytotoxicity of 9 and 12 to PRRSV targeting cells. Next, we evaluated the antiviral effects of 9 and 12 against NADC30-like PRRSV infection in PAMs through determining virus titration at 24 and 48 hpi. As shown in Fig. 4B and C, compound 9, as well as 12, showed significant PRRSV suppression at both 24 and 48 hpi. Treatments with 20 μM of 9 or 12 resulted in a 4.8log or 4.6log reduction, respectively, in progeny virus production at 48 hpi when compared to that in the DMSO control (Fig. 4C). These results demonstrated that 9 and 12 also effectively inhibited PRRSV infection in ex vivo PAMs with minimal cytotoxicity.
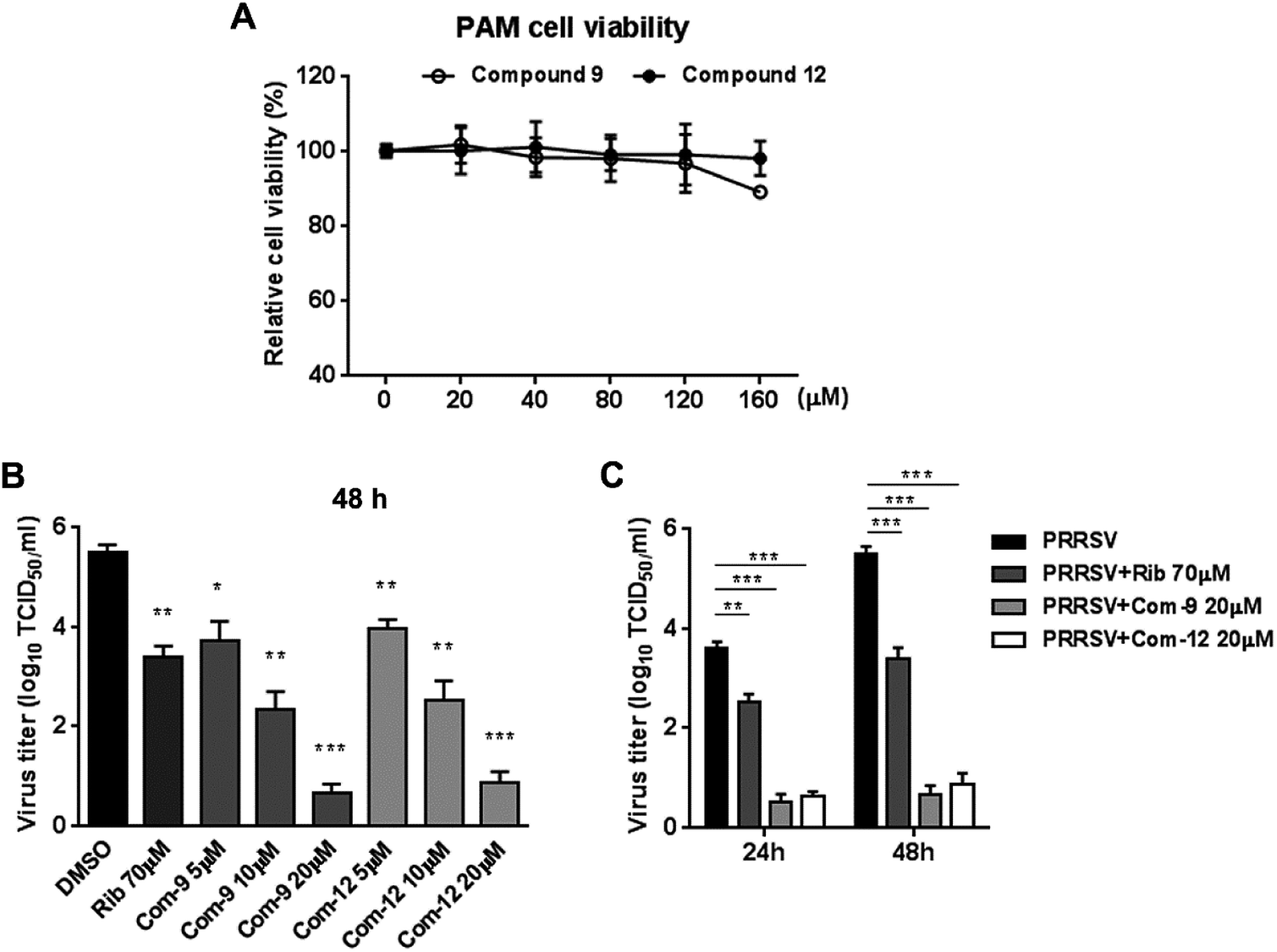 | ||
| Fig. 4 Cytotoxicity and anti-PRRSV activity of compounds 9 and 12 in PAM cells. (A) Cellular toxicity of 9 and 12 was examined in PAMs at 48 hpi using MTT assay. (B) and (C) PAMs grown in 24-well plates were infected with NADC30-like PRRSV (0.5 MOI) for 2 h at 37 °C and then treated with various concentrations of 9 or 12. 24 or 48 h later, the samples were submitted to viral titer determination using the end point dilution assay and expressed as log10 TCID50/1 mL. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to that of the DMSO control. | ||
3.6 Compounds 9 and 12 inhibit PRRSV at multiple stages of its replication cycle
To identify whether 9 and 12 inhibited PRRSV infection by targeting host cells or by targeting the virus itself, we performed time course studies for the inhibitory effects of the two compounds using IFA. MARC-145 cells were treated with 9 or 12 for 2 h prior to virus infection (Pre-treatment), or for 2 h during the viral infection (Co-treatment), or for 48 h after 2 h virus infection and removal (Post-treatment), as shown in Fig. 5A. Results in Fig. 5B and C showed that pre-treatment of 9 or 12 did not reduce viral NP production, indicating that 9 and 12 do not impair the susceptibility of MARC-145 cells to PRRSV infection. However, in the co-treatment and post-treatment modes, compounds 9 and 12 significantly reduced PRRSV N protein production in a dose-dependent manner, indicating the two compounds potently inhibited PRRSV replication.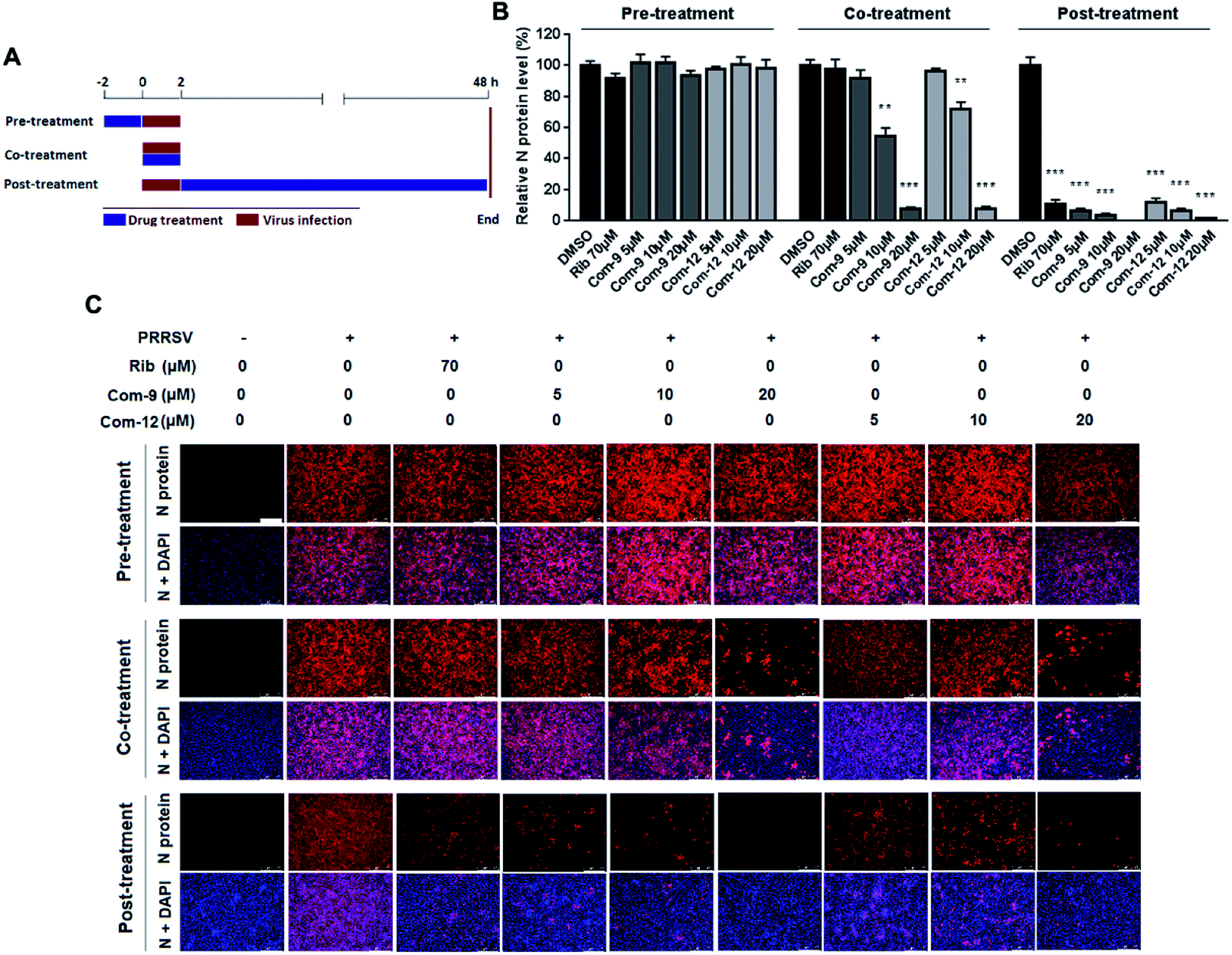 | ||
| Fig. 5 Compounds 9 and 12 suppressed PRRSV replication in co- and post-treatment modes. MARC-145 cells grown in 24-well plates were treated with indicated compound for 2 h prior to virus infection (Pre-treatment), or for 2 h during viral infection (Co-treatment), or for 48 h after 2 h virus infection and removal (Post-treatment) (A). For the three treatment modes, 0.05 MOI of PRRSV GD-XH was used for infecting cells for 2 h. At 48 hpi, the cells were subjected to viral N protein analysis using IFA (B) and (C). Results shown in (B) are normalized N protein levels based on the fluorescence optical densities (OD) of the images from three independent experiments. Software Image J was used to digitize image OD. Results from compound-treated samples were compared to those from corresponding DMSO control groups (set as 100%) (B). Representative IFA images of one of the three independent experiments are shown in (C). Scale bar: 250 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the DMSO control. | ||
In order to explore the mechanism of the two compounds mediated PRRSV inhibition, we first examined the effects of 9 and 12 on virus entry through cell attachment and subsequent internalization. Marc-145 cells were infected with a higher dose of RRSV GD-XH (0.5 MOI) in the presence or absence of 9 and 12 at 4 °C, which allows virus binding but not cellular internalization (Fig. 6A, treatment B). As shown in Fig. 6B, 9 or 12 treatment at 10 μM significantly reduced viral mRNA levels, suggesting that 9 and 12 directly exerted their inhibitory effects on PRRSV binding to Marc-145 cells. Kreutz and Nauwynck have previously reported that PRRSV is internalized from the surface of Marc-145 cells within 3 hpi.33 Thus, to examine whether 9 and 12 might also affect the internalization of PRRSV, PRRSV infected Marc-145 cells were treated with 9 or 12 during 2–5 hpi (Fig. 6A, treatment C). As shown in Fig. 6C, virus replication was also significantly inhibited when infected cells were treated with 10 μM of 9 or 12 during this time period, suggesting that 9 and 12 also inhibited PRRSV internalization.
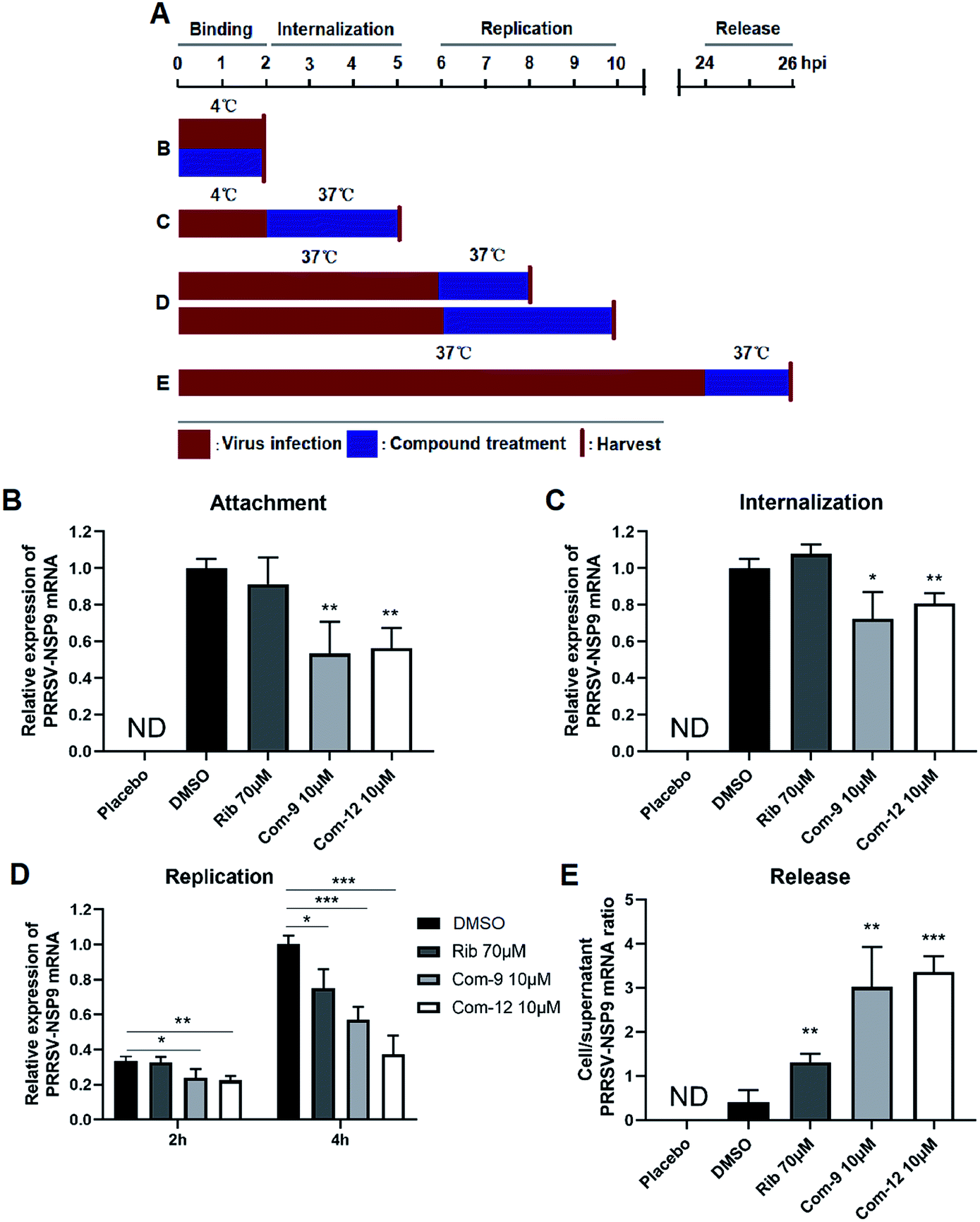 | ||
| Fig. 6 Compounds 9 and 12 inhibited PRRSV at multiple stages of its replication cycle. Marc-145 cells were infected with the PRRSV GD-XH strain at 0.5 or 0.05 MOI. The infected cells were cultured in the presence or absence of 9 or 12 and collected at the indicated time-points post infection to determine viral NSP9 mRNA level by RT-PCR. Cellular GAPDH mRNA was used as a loading control and DMSO sample (set as 1) was used as an uninfected control. (A) Different 9 or 12 treatment schemes. (B) Compound treatment during PRRSV binding. (C) Compound treatment during PRRSV internalization. (D) Compound treatment during PRRSV RNA replication. (E) Compound treatment during PRRSV release. For (B) and (C), 0.5 MOI PRRSV were used to infect cells. For (D) and (E), 0.05 MOI PRRSV were used to infect cells. Statistical significances are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
Next, we examined whether 9 and 12 could also affect PRRSV replication and assembly. Marc-145 cells were infected at 0.05 MOI of PRRSV GD-XH for 6 h at 37 °C to allow normal virus replication and assembly. The infected cells were then cultured in fresh medium containing 10 μM of 9 or 12 to investigate whether virus replication as affected. Cells were collected at 8 and 10 hpi and subjected to RT-PCR analysis for viral RNA contents (Fig. 6A, treatment D). As shown in Fig. 6D, 9 or 12 treatment significantly reduced the viral RNA levels, suggesting that 9 and 12 inhibited PRRSV RNA replication. As expected, positive control ribavirin, an inhibitor of viral RNA polymerase, also exhibited inhibition on PRRSV replication at 10 hpi.
Previous studies have demonstrated that PRRSV progeny viruses are released by 8 hpi.33 Our previous study of PRRSV proliferation dynamics showed that viral mRNA level and titer increased from 12 hpi to 48 hpi.26 To explore whether 9 and 12 directly inhibits viral release, Marc-145 cells were infected with 0.05 MOI of PRRSV GD-XH for 24 h at 37 °C and the cells were then cultured in fresh medium containing 10 μM of 9 or 12 for another 2 h. Subsequently, the cells and their culture supernatants were collected separately and the NSP9 RNA were quantified by RT-PCR (Fig. 6A, treatment E). As shown in Fig. 6E, progeny virus release (viral RNA in the supernatant) is inversely proportional to the ratio of cellular and supernatant viral RNA levels. A lower cell/supernatant viral RNA ratio corresponded to more progeny virus release. Consequently, the addition of 10 μM of 9 or 12 was found to significantly inhibit PRRSV virus release from Marc-145 cells.
3.7 Compounds 9 and 12 directly interact with PRRSV
As shown above, 9 and 12 was able to inhibit PRRSV at multiple stages of its infection cycle, which led us to wonder whether the two compounds were able to directly interact with PRRSV. We therefore mixed the virus with 9 or 12 at various concentrations in essential medium for 1 h at 37 °C, and then separated PRRSV from either compound via ultrafiltration as shown in Fig. 7A. PRRSV left in the ultrafiltration filter were resuspended in essential medium and used for infecting Marc-145 cells. At 48 hpi, virus titers in the supernatants were determined using the end point dilution assay and viral N protein expression in these cells was analyzed using IFA. As shown in Fig. 7B, co-incubation of 9 or 12 (5, 10 and 20 μM) with virus significantly reduced virus titer in Marc-145 cells in a dose-dependent manner. Similar reductions of viral N protein levels were also observed in 9 or 12-treated samples, as shown in Fig. 7C and D. These results demonstrate that 9 and 12 did directly interact with PRRSV particles.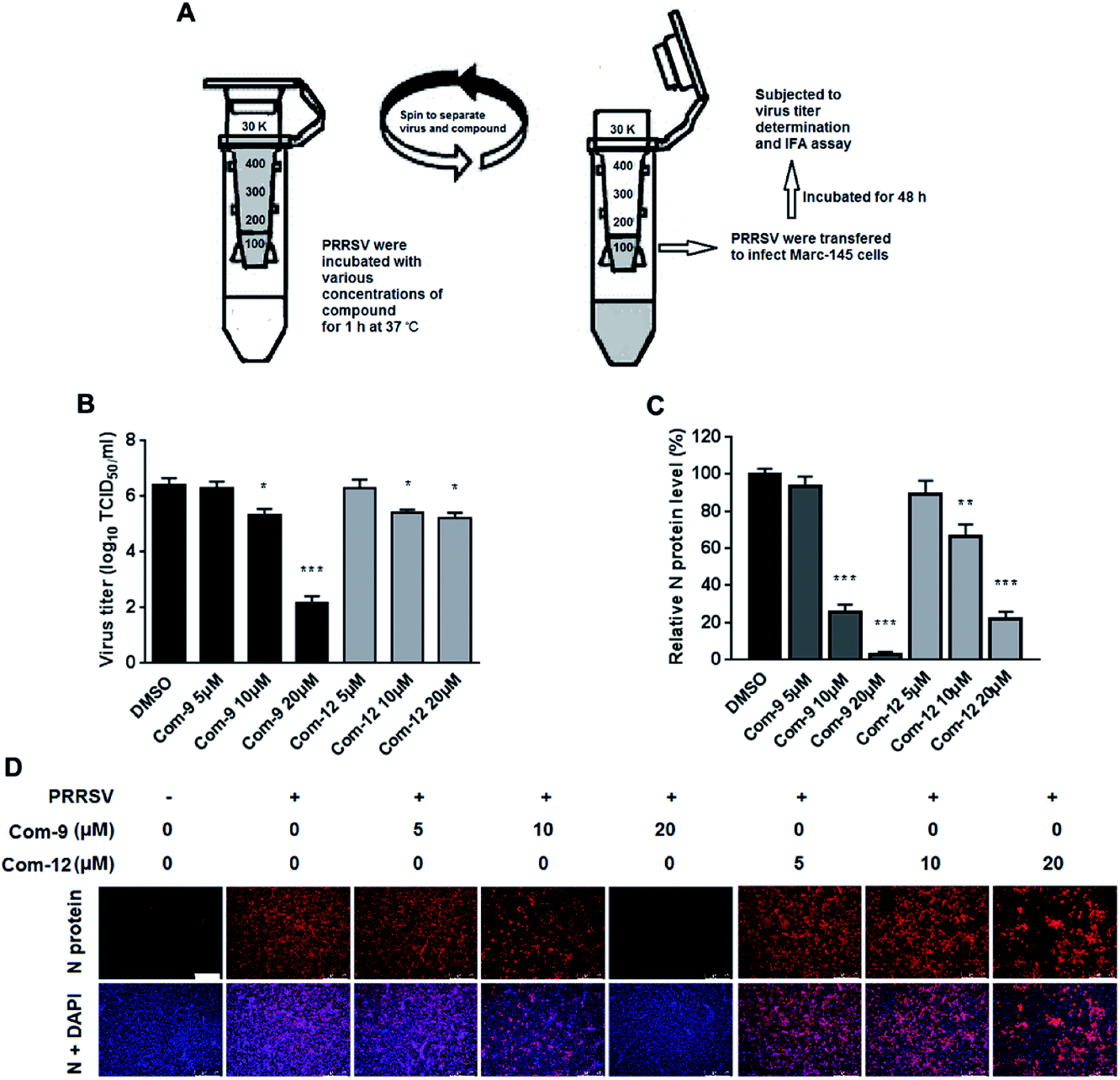 | ||
| Fig. 7 Compounds 9 and 12 interacted with PRRSV directly. 100 μL of PRRSV GD-XH (2 × 106 PFU) was mixed with various concentrations of 9 or 12 in essential medium (0.9 mL total volume) for 1 h at 37 °C. Then PRRSV and unbound compound were separated by ultrafiltration, as shown in the schematics (A). Recovered PRRSV were resuspended to infect MARC-145 cells. At 48 hpi, the virus titers in the supernatants were determined using the end point dilution assay (B), and the viral N protein expression in these cells was analyzed using IFA (C) and (D). Results shown in (C) are normalized N protein levels based on the fluorescence optical densities (OD) of the images from three independent experiments. Representative IFA images of three independent experiments are shown in (D). Scale bar: 250 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the DMSO-treated control. | ||
4. Discussion
Ursolic acid (UA) and its analogs, a group of pentacyclic triterpenoids (PTs) abundantly existing in plants, have received increasing attentions due to their wide activities such as anti-inflammatory, anti-oxidant, anti-apoptotic, and anti-tumour effects.34 UA and its analogs were also reported to possess antiviral activities, including HIV, HBV and human enterovirus.35 In our previous study, we discovered a series of 3-O-β-chacotriosyl ursolic acid derivatives as novel IAV H5N1 entry inhibitors by targeting hemagglutinin (HA).30 Recently, we further demonstrated that introduction of certain amide structures at the 17-COOH of 3-O-β-chacotriosyl ursolic acid could significantly enhance both their anti-H5N1 activity and selective index.36 Therefore, we wonder whether UA and analogs possess anti-PRRSV activities. In this study, we evaluated anti-PRRSV activity of 3-O-β-chacotriosyl ursolic acid 1 and its alkyl ursolate analogs 2–6. Our results showed that 1 exhibited strong antiviral activity but high cytotoxicity against MARC-145 cells, while 2–6 exhibited lower cytotoxicity with slightly reduced antiviral activity. Based on these results, we wanted to further explore a series of 3-O-β-chacotriosyl ursolic acid amide derivatives 7–15 by bioisosteric replacement to generate novel PRRSV inhibitors with enhanced activity and reduced toxicity. Encouragingly, amide derivatives 7–14 exhibited increased selectivity index while maintaining similar or slightly enhanced inhibitory activity compared with that of 1. Notably, 9 showed the strongest anti-PRRSV activity (EC50 = 2.03 μM) among this set of compounds and remarkably enhanced selective index (SI > 79) compared to 1. Compound 12 showed the second highest SI (>41). These results reveal that subtle modifications of phenyl moiety with electron-donating groups such as methoxy or bromine group at para-position of the phenyl ring can be beneficial to enhancing antiviral activity. Potent PRRSV inhibitions by 9 and 12 were observed not only in MARC-145 cells, but also in primary porcine alveolar macrophages, PRRSV-target cells in vivo. Furthermore, 8, 9, 12 and 14 exhibited broad-spectrum inhibitory activities in vitro against high pathogenic type 2 PRRSV NADC30-like and GD-XH strains as well as classical CH-1a and VR2332 strains.One PRRSV infection cycle is orderly composed of virus binding, internalization, RNA replication and viral protein synthesis, assembly, and release from infected cells.37 Through the time course inhibition experiments, we found that 9 and 12 was capable of interfering multiple stages of PRRSV infection cycle, including virus binding, internalization, RNA replication, protein synthesis, assembly and release (Fig. 6). The lack of viral inhibition by compound pre-treatment indicates that 9 or 12 does not affect cell susceptibility to PRRSV (Fig. 5) and suggests that their antiviral might be mediated through direct compound–PRRSV interaction. Indeed, when 9 or 12 and PRRSV were directly mixed and then separated by ultrafiltration, direct interaction was confirmed as 9 or 12 was found to significantly impair virus activity reflected by reduced progeny PRRSV titer (Fig. 7). However, the binding site of 9 and 12 on PRRSV remains unclear. As 9 and 12 could inhibit PRRSV attachment to the MARC-145 cells, it is likely that 9 and 12 binds to one of the PRRSV surface molecules, including E and GP2 to GP5, directly involved in PRRSV binding to its entry receptors including heparan sulphate, sialoadhesin, CD163 and others.38 Moreover, as we have shown that 3-O-β-chacotriosyl ursolic acid was able to inhibit IAV infection via blocking viral membrane fusion,31,36 we cannot exclude the possibility that 9 and 12 may also interfere with PRRSV membrane fusion. Further studies are required to identify the fine anti-PRRSV mechanisms of the two compounds.
In summary, our data demonstrates amidation of 17-COOH group of UA significantly reduces cytotoxicity and enhances anti-PRRSV activity. Potent and broad-spectrum inhibitions of representative compounds 9 and 12 on PRRSV infection are observed not only in MARC-145 cells, but also in primary porcine alveolar macrophages. Mechanistically, 9 and 12 inhibit PRRSV replication by directly interacting with virions therefore inactivating PRRSV. Compounds 9 or 12 could potentially be used as a novel strategy to treat PRRSV infection, which necessitates further studies, especially its antiviral activity in swine.
5. Supplementary materials
5.1. Preparation and identification of compounds 23 and 7–15
Solvents were purified in a conventional manner. Thin layer chromatography (TLC) was performed on precoated E. Merck silica gel 60 F254 plates. Flash column chromatography was performed on silica gel (200–300 mesh, Qingdao, China). 1H NMR and 13C NMR spectra were taken on a JEOL JNM-ECP 600 spectrometer with tetramethylsilane as an internal standard, and chemical shifts are recorded in ppm values. Mass spectra were recorded on a Q-TOF Global mass spectrometer.5.2. 3β-O-[2,4-Di-O-(2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl)-β-(3,6-two-O-acetyl)-D-glucopyranosyl]-12-en-28-oic acid (23)
Compound 22 (341 mg, 0.34 mmol) was dissolved in pyridine (8 mL), Ac2O (0.57 mL, 6.0 mmol) and DMAP (24 mg, 0.19 mmol) was then added, and the mixture was stirred at room temperature (r.t.) for 8 h. The mixture was concentrated in vacuo and dissolved in CH2Cl2 (50 mL), then washed with 1 M HCl (3 × 20 mL), saturated NaHCO3 (3 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, filtered, and concentrated in vacuo to furnish the residue. A mixture of the above residue and 10% Pd/C (60 mg) in MeOH (10 mL) was stirred at r.t. under H2 at atmospheric pressure for 5 h. Then the above mixture was filtered through Celite, and the insoluble substance was washed with MeOH (3 mL × 3). The filtrate was concentrated in vacuo to produce the residue, which was purified by flash chromatography (EtOAc–petroleum ether, 1:2) to give 23 as a white solid (512 mg, 83% for two steps); 1H NMR (CDCl3): δ 5.23–5.26 (m, 3H), 5.18 (d, 1H, J = 10.1, 3.1 Hz, Rha-H-3), 5.11 (d, 1H, J = 3.2, 17 Hz, Rha-H-2), 5.01–5.06 (m, 4H), 4.81 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.53 (d, 1H, J = 7.3 Hz, H-1′), 4.49 (d, 1H, J = 12.1 Hz, H-6′-1), 4.22–4.28 (m, 2H), 3.85–3.88 (m, 1H), 3.77 (t, 1H, J = 9.3 Hz), 3.68 (t, 1H, J = 8.3 Hz), 3.60–3.62 (m, 1H), 3.16 (d, 1H, J = 11.5, 4.1 Hz, H-3), 2.55 (d, 1H, J = 12.4 Hz, H-18), 2.14, 2.13, 2.12, 2.11, 2.05, 2.02, 1.99, 1.98, (each s, each 3H, each CH3CO), 1.18 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.16 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.09, 1.03, 0.92, 0.81, 0.77 (each s, each 3H, CH3), 0.95 (d, 3H, J = 6.0 Hz, CH3), 0.86 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CDCl3): (δ (182.8 (C-28), 170.5, 170.2, 170.1 (two), 170.0 (three), 169.6, 137.8 (C-13), 125.8 (C-12), 103.6 (C-1′), 99.4 (Rha-C-1), 97.0 (Rha-C-1), 90.0, 77.9, 75.7, 75.4, 72.1, 71.1, 70.6, 69.9, 69.7, 68.6, 68.5, 67.9, 66.7, 62.1, 55.9, 52.6, 47.9, 47.6, 42.0, 39.5, 39.0, 38.8, 36.7, 32.9, 30.6, 28.0, 27.8, 26.0, 24.1, 23.6, 23.3, 21.4, 21.2, 20.9 (two), 20.8 (two), 20.7 (two), 18.2, 17.2, 17.1, 17.0, 16.9, 16.2, 15.6; HRESIMS calcd for C64H94O24Na 1269.6033; found 1269.6042.
To prepare compounds 7–15, a solution of compound 23 (1 mmol) in 20 mL dried CH2Cl2 was added to oxalyl chloride (4 mmol). The mixture was stirred at room temperature for 24 h under argon and then concentrated to dryness under reduced pressure. Hexane (3 × 10 mL) was added to the residue, then concentrated under vacuum to dryness. To a dried CH2Cl2 (20 mL) solution of different aniline hydrochloride (1.5 mmol) was added to the above acid chloride in the presence of triethylamine (3.0 mmol). The reaction mixture was stirred at r.t. for 3 h under argon and then concentrated. The obtained residue was dissolved in 2:1 MeOH/CH2Cl2 (15 mL) and then NaOMe was added until pH = 10. After stirred at r.t. for 12 h, the solution was neutralized with Dowex 50 × 8 (H+) resin until pH = 7, filtered and concentrated under vacuum. Then the residue was purified by silica gel column chromatography (CH2Cl2–MeOH, 6:1) to yield compounds 7–15, respectively.
5.3. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-methylamine (7)
1H NMR (CD3OD): δ 5.36 (d, 1H, J = 1.2 Hz, Rha-H-1), 5.32 (t, 1H, J = 3.4 Hz, H-12), 4.84 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 3.95–3.98 (m, 2H), 3.88–3.92 (m, 1H), 3.84 (dd, 1H, J = 3.1, 1.8 Hz, Rha-H-2), 3.79 (dd, 1H, J = 12.1, 1.7 Hz, H-6-2′), 3.75 (dd, 1H, J = 9.5, 3.4 Hz, Rha-H-3), 3.62–3.67 (m, 2H), 3.58 (t, 1H, J = 8.7 Hz), 3.53 (t, 1H, J = 9.3 Hz), 3.43 (t, 1H, J = 8.5 Hz), 3.36–3.40 (m, 2H), 3.30–3.32 (m, 1H), 3.16 (dd, 1H, J = 11.5, 4.1 Hz, H-3), 2.64 (s, 3H, NHCH3), 2.09 (d, 1H, J = 11.0 Hz, H-18), 1.27 (d, 3H, J = 6.0 Hz, CH3), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.10, 1.04, 0.96, 0.95, 0.84, 0.76 (each s, each 3H, CH3), 0.89 (d, 3H, J = 6.2 Hz, CH3); 13C NMR (CD3OD): 179.6 (C-28), 138.6 (C-13), 125.8 (C-12), 104.0 (C-1′), 101.5 (Rha-C-1), 100.6 (Rha-C-1), 89.1, 78.8, 78.0, 76.6, 75.0, 72.5, 72.3, 71.0, 70.7, 70.6, 69.2, 68.7, 63.8, 60.6, 55.8, 52.7, 41.8, 39.4 (two), 38.8, 38.7, 37.1, 36.4, 32.6, 30.5, 29.3, 27.5, 27.2, 25.8, 23.9, 23.0, 22.8, 20.2, 17.9, 16.6, 16.5, 16.4, 16.1, 15.8, 14.7; HRESIMS calcd for C49H81O15NNa 946.5504; found 946.5512.5.4. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-aniline (8)
1H NMR (CD3OD): δ 7.46 (dd, 2H, J = 7.9, 1.1 Hz, NH-Ar-H-2, NH-Ar-H-6), 7.30 (t, 2H, J = 7.9 Hz, NH-Ar-H-3, NH-Ar-H-5), 7.10 (tt, 1H, J = 7.4, 1.1 Hz, NH-Ar-H-4), 5.46 (t, 1H, J = 3.9 Hz, H-12), 5.37 (d, 1H, J = 1.6 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.95 (m, 2H), 3.95–3.88 (m, 1H), 3.85 (dd, 1H, J = 3.3, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6-1′), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.1, 4.1 Hz, H-6-2′), 3.64 (dd, 1H, J = 9.5, 3.4 Hz, Rha-H-3), 3.59 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.1 Hz), 3.47–3.41 (m, 2H), 3.39 (t, 1H, J = 9.0 Hz), 3.34–3.30 (m, 1H), 3.18 (dd, 1H, J = 11.8, 4.3 Hz, H-3), 2.34 (d, 1H, J = 10.5 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.17, 1.06, 0.93, 0.85, 0.75 (each s, each 3H, CH3), 1.01 (d, 3H, J = 6.3 Hz, CH3), 0.96 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CD3OD): δ 177.2 (C-28), 138.8 (C-13), 138.2, 128.2 (two), 125.9 (C-12), 124.0, 121.0 (two), 104.0 (C-1′), 101.7 (Rha-C-1), 100.6 (Rha-C-1), 88.9, 79.1, 77.8, 76.7, 75.0, 72.5, 72.3, 71.0, 70.7, 70.6, 69.4, 68.6, 60.6, 55.9, 52.8, 42.0, 39.5 (two), 38.8 (three), 36.9, 36.4, 32.7, 30.5, 27.7, 27.1, 25.8, 23.9, 23.1, 22.8, 20.2, 17.8, 16.6, 16.5, 16.4, 16.3, 15.8, 14.8; HRESIMS calcd for C54H83O15NNa 1008.5660; found 1008.5662.5.5. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-4-methoxyaniline (9)
1H NMR (CD3OD): δ 7.33 (d, 2H, J = 9.1 Hz, NH-Ar-H-2, NH-Ar-H-6), 6.87 (d, 2H, J = 9.1 Hz, NH-Ar-H-3, NH-Ar-H-5), 5.44 (t, 1H, J = 3.7 Hz, H-12), 5.38 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.8 Hz, H-1′), 4.02–3.96 (m, 2H), 3.95–3.88 (m, 1H), 3.85 (dd, 1H, J = 3.3, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6-1′), 3.78 (s, 3H, OCH3), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.0, 4.1 Hz, H-6-2′), 3.64 (dd, 1H, J = 9.4, 3.3 Hz, Rha-H-3), 3.60 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.1 Hz), 3.48–3.41 (m, 2H), 3.40 (t, 1H, J = 8.6 Hz), 3.34–3.30 (m, 1H), 3.18 (dd, 1H, J = 11.8, 4.2 Hz, H-3), 2.32 (d, 1H, J = 10.6 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.16, 1.06, 0.94, 0.86, 0.78 (each s, each 3H, CH3), 1.00 (d, 3H, J = 6.3 Hz, CH3), 0.96 (d, 3H, J = 7.4 Hz, CH3); 13C NMR (CD3OD): δ 177.1 (C-28), 156.7, 138.8 (C-13), 131.1, 125.9 (C-12), 122.9 (two), 113.4 (two), 104.1 (C-1′), 101.6 (Rha-C-1), 100.5 (Rha-C-1), 88.9, 79.1, 77.9, 76.7, 75.1, 72.5, 72.3, 71.0, 70.8, 70.6, 69.4, 68.6, 61.3, 60.6, 55.9, 52.8, 42.0, 39.6, 38.9, 38.8 (two), 37.0, 36.4, 34.4, 32.8, 30.6, 27.7 (two), 25.8, 23.9, 23.1, 22.8, 20.2, 18.6, 17.9, 16.6 (two), 16.5, 16.4, 15.9, 14.8, 12.8; HRESIMS calcd for C55H85O16NNa 1038.5766; found 1038.5769.5.6. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-3-methoxyaniline (10)
1H NMR (CD3OD): δ 7.20 (t, 1H, J = 2.2 Hz, NH-Ar-H-2), 7.19 (t, 1H, J = 8.2 Hz, NH-Ar-H-5), 6.99 (dd, 1H, J = 8.0, 2.0, Hz, NH-Ar-H-6), 6.67 (dd, 1H, J = 8.3, 2.6 Hz, NH-Ar-H-4), 5.46 (t, 1H, J = 3.8 Hz, H-12), 5.37 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.8 Hz, H-1′), 4.02–3.94 (m, 2H), 3.96–3.88 (m, 1H), 3.85 (dd, 1H, J = 3.4, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6-1′), 3.78 (s, 3H, OCH3), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.1, 4.1 Hz, H-6-2′), 3.64 (dd, 1H, J = 9.4, 3.3 Hz, Rha-H-3), 3.59 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.1 Hz), 3.49–3.39 (m, 2H), 3.39 (t, 1H, J = 9.1 Hz), 3.35–3.29 (m, 1H), 3.18 (dd, 1H, J = 11.7, 4.3 Hz, H-3), 2.33 (d, 1H, J = 10.7 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.16, 1.05, 0.93 0.85, 0.75 (each s, each 3H, CH3), 1.01 (d, 3H, J = 6.4 Hz, CH3), 0.96 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CD3OD): δ 177.2 (C-28), 160.0, 139.3, 138.8 (C-13), 128.9, 126.0 (C-12), 113.0, 109.6, 106.6, 104.1 (C-1′), 101.7 (Rha-C-1), 100.6 (Rha-C-1), 88.9, 79.1, 77.9, 76.7, 75.0, 72.6, 72.3, 71.0, 70.8, 70.6, 69.4, 68.6, 60.6, 55.9, 54.3, 52.8, 42.0, 39.6, 39.5, 38.8 (two), 36.8, 36.4, 32.7, 30.6, 27.7, 27.2, 25.8, 24.0, 23.1, 22.8, 20.2, 17.9, 16.6, 16.5, 16.4 (two), 15.9, 14.8, 13.1; HRESIMS calcd for C55H85O16NNa 1038.5766; found 1038.5771.5.7. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-2-methoxyaniline (11)
1H NMR (CD3OD): δ 8.05 (dd, 1H, J = 8.2, 1.6 Hz, NH-Ar-H-6), 7.44 (dd, 1H, J = 8.1, 1.5 Hz, NH-Ar-H-3), 7.29 (dd, 1H, J = 8.3, 1.5 Hz, NH-Ar-H-4), 7.16–7.10 (m, 1H, NH-Ar-H-5), 5.48 (t, 1H, J = 3.7 Hz, H-12), 5.37 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.8 Hz, H-1′), 4.02–3.94 (m, 2H), 3.96–3.88 (m, 1H), 3.85 (dd, 1H, J = 3.4, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6-1′), 3.75 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.1, 4.1 Hz, H-6-2′), 3.64 (dd, 1H, J = 9.4, 3.3 Hz, Rha-H-3), 3.59 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.1 Hz), 3.48–3.41 (m, 2H), 3.39 (t, 1H, J = 9.4 Hz), 3.36 (s, 3H, OCH3), 3.35–3.29 (m, 1H), 3.18 (dd, 1H, J = 11.8, 4.3 Hz, H-3), 2.23 (d, 1H, J = 11.0 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.18, 1.06, 0.93 0.85, 0.74 (each s, each 3H, CH3), 1.01 (d, 3H, J = 6.3 Hz, CH3), 0.97 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CD3OD): δ 177.2 (C-28), 138.1 (C-13), 134.7, 129.1, 127.2, 127.1, 125.3 (C-12), 125.0, 123.4, 104.0 (C-1′), 101.7 (Rha-C-1), 100.6 (Rha-C-1), 88.9, 79.1, 77.8, 76.8, 75.1, 72.6, 72.3, 71.1, 70.8, 70.6, 69.4, 68.6, 60.6, 55.8, 53.5, 42.0, 39.7, 39.5, 38.9, 38.8, 38.8, 37.2, 36.4, 32.7, 30.5, 27.6, 27.1, 25.8, 24.4, 23.0, 22.8, 20.2, 17.8, 16.6, 16.5, 16.4, 16.3, 15.8, 14.8; HRESIMS calcd for C55H85O16NNa 1038.5766; found 1038.5768.5.8. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-4-bromoaniline (12)
1H NMR (CD3OD): δ 7.41–7.45 (m, 4H, Ar-H), 5.43 (t, 1H, J = 3.7 Hz, H-12), 5.37 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.95 (m, 2H), 3.95–3.89 (m, 1H), 3.85 (dd, 1H, J = 3.3, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 2.0 Hz, H-6-1′), 3.76 (dd, 1H, J = 9.6, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.1, 4.2 Hz, H-6-2′), 3.64 (dd, 1H, J = 9.4, 3.3 Hz, Rha-H-3), 3.60 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.1 Hz), 3.48–3.40 (m, 2H), 3.39 (t, 1H, J = 8.7 Hz), 3.34–3.31 (m, 1H), 3.17 (dd, 1H, J = 11.7, 4.3 Hz, H-3), 2.33 (d, 1H, J = 10.7 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.16, 1.05, 0.93, 0.85, 0.72 (each s, each 3H, CH3), 1.00 (d, 3H, J = 6.3 Hz, CH3), 0.95 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CD3OD): δ 177.2 (C-28), 138.7 (C-13), 137.6, 131.2 (two), 125.9 (C-12), 122.6 (two), 116.3, 104.1 (C-1′), 101.6 (Rha-C-1), 100.5 (Rha-C-1), 88.9, 79.1, 77.9, 76.7, 75.0, 72.6, 72.3, 71.1, 70.8, 70.6, 69.4, 68.6, 60.6, 55.9, 52.7, 41.9, 39.5 (two), 38.8 (two), 36.8, 36.4, 32.7, 30.5, 27.7, 27.2, 25.8, 23.9, 23.1, 22.9, 20.2, 17.9, 16.7, 16.5, 16.4 (two), 15.9, 14.8; HRESIMS calcd for C54H82BrO15NNa 1086.4766; found 1086.4772.5.9. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-benzylamine (13)
1H NMR (CD3OD): δ 8.09 (t, 1H, J = 8.5 Hz, Ar–H), 7.95 (t, 1H, J = 8.6 Hz, Ar–H), 7.65–7.62 (m, 1H, Ar–H), 7.29–7.27 (m, 1H, Ar–H), 7.25–7.22 (m, 1H, Ar–H), 5.39 (d, 1H, J = 1.0 Hz, Rha-H-1), 5.30 (t, 1H, J = 3.6 Hz, H-12), 4.87 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.8 Hz, H-1′), 4.31 (t, 1H, J = 5.0 Hz), 4.02–3.98 (m, 2H), 3.96–3.92 (m, 1H), 3.86 (dd, 1H, J = 3.3, 1.1 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.2, 2.1 Hz, H-6-1′), 3.74 (dd, 1H, J = 9.7, 3.4 Hz, Rha-H-3), 3.68–3.64 (m, 4H), 3.60 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.4 Hz), 3.47–3.41 (m, 2H), 3.39 (t, 1H, J = 8.8 Hz), 3.24–3.20 (m, 1H), 3.18 (dd, 1H, J = 11.8, 4.3 Hz, H-3), 2.17 (d, 1H, J = 9.7 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.11, 1.05, 0.92, 0.86, 0.59 (each s, each 3H, CH3), 0.98 (d, 3H, J = 6.3 Hz, CH3), 0.91 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (CD3OD): δ 178.6 (C-28), 138.8 (C-13), 138.5, 129.2 (C-12), 128.0 (two), 127.5 (two), 126.7, 125.8, 104.0 (C-1′), 101.6 (Rha-C-1), 100.5 (Rha-C-1), 88.9, 79.0, 77.9, 76.7, 75.0, 72.5, 72.3, 71.0, 70.8, 70.6, 69.3, 68.6, 60.6, 55.8, 53.0, 51.5, 48.5, 46.5, 42.9, 41.9, 39.5, 38.9, 38.8 (two), 37.3, 36.4, 32.8, 31.7, 30.5, 29.3, 27.5, 27.1, 25.8, 23.9, 23.0, 22.6, 22.3, 20.2, 17.8, 16.6, 16.5 (two), 16.3, 15.8, 14.7; HRESIMS calcd for C55H85O15NNa 1022.5817; found 1022.5822.5.10. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-4-methoxybenzylamine (14)
1H NMR (CD3OD): δ 7.19 (d, 2H, J = 8.7 Hz, NH-Ar-H-2, NH-Ar-H-6), 6.83 (d, 2H, J = 9.1 Hz, NH-Ar-H-3, NH-Ar-H-5), 5.36 (d, 1H, J = 1.8 Hz, Rha-H-1), 5.26 (t, 1H, J = 3.7 Hz, H-12), 4.84 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.8 Hz, H-1′), 4.26 (d, 1H, J = 14.4 Hz, NHCH2-1′), 4.18 (d, 1H, J = 14.4 Hz, NHCH2-2′), 4.01–3.93 (m, 2H), 3.94–3.86 (m, 1H), 3.83 (dd, 1H, J = 3.4, 1.8 Hz, Rha-H-2), 3.79 (dd, 1H, J = 12.0, 2.0 Hz, H-6-1′), 3.76 (s, 3H, OCH3), 3.74 (dd, 1H, J = 9.6, 3.3 Hz, Rha-H-3), 3.65 (dd, 1H, J = 11.9, 4.0 Hz, H-6-2′), 3.62 (dd, 1H, J = 9.4, 3.5 Hz, Rha-H-3), 3.58 (t, 1H, J = 8.7 Hz), 3.53 (t, 1H, J = 9.1 Hz), 3.48–3.35 (m, 3H), 3.33–3.27 (m, 1H), 3.15 (dd, 1H, J = 11.7, 4.4 Hz, H-3), 2.12 (d, 1H, J = 10.6 Hz, H-18), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.09, 1.04, 0.90, 0.85, 0.53 (each s, each 3H, CH3), 0.96 (d, 3H, J = 6.2 Hz, CH3), 0.89 (d, 3H, J = 6.6 Hz, CH3); 13C NMR (CD3OD): δ 179.9 (C-28), 160.4, 140.0 (C-13), 132.2, 130.4, 130.0, 127.2 (C-12), 119.4, 116.7, 114.8, 105.5 (C-1′), 103.1 (Rha-C-1), 102.0 (Rha-C-1), 90.3, 80.5, 79.3, 78.2, 76.5, 74.0, 73.7, 72.5, 72.2, 72.1, 70.8, 70.0, 62.0, 57.3, 55.7, 54.5, 43.9, 43.8, 43.3, 40.9 (two), 40.3 (two), 40.2, 38.7, 37.8, 34.3, 32.0, 29.0, 28.5, 27.3, 25.3, 24.4, 24.0, 21.6, 19.3, 18.0, 17.9, 17.7, 17.3, 16.2; HRESIMS calcd for C56H87O16NNa 1052.5923; found 1052.5925.5.11. N-{3β-O-[2,4-Di-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl]-urs-12-en-28-oyl}-dimethylamine (15)
Analogously, 13 was prepared as a white solid in 93.6% yield for three steps; Rf 0.45 (3:1, CHCl3–MeOH); 1H NMR (DMSO-d6): δ 5.18 (s, 1H, Rha-H-1), 5.05 (brs, 1H, H-12), 4.67 (s, 1H, Rha-H-1), 4.26 (d, 1H, J = 7.7 Hz, H-1′), 3.77–3.81 (m, 3H), 3.60 (dd, 1H, J = 3.1, 1.7 Hz, Rha-H-2), 3.56 (d, 1H, J = 11.1 Hz, H-6-2′), 3.47 (dd, 1H, J = 9.5, 3.1 Hz, Rha-H-3), 3.40–3.42 (m, 2H), 3.36–3.37 (m, 2H), 3.27 (t, 1H, J = 7.7 Hz), 3.18 (t, 1H, J = 9.3 Hz), 3.17 (t, 1H, J = 9.4 Hz), 3.13–3.15 (m, 1H), 3.02 (dd, 1H, J = 10.8, 2.9 Hz, H-3), 2.87 (s, 6H, CH3), 2.36 (d, 1H, J = 10.5 Hz, H-18), 1.09 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.05 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.00, 0.92, 0.83, 0.72, 0.62 (each s, each 3H, CH3), 0.88 (d, 3H, J = 6.0 Hz, CH3), 0.80 (d, 3H, J = 6.0 Hz, CH3); 13C NMR (DMSO-d6): δ 175.7 (C-28), 137.8 (C-13), 124.3 (C-12), 104.2 (C-1′), 101.0 (Rha-C-1), 100.3 (Rha-C-1), 88.5, 77.6, 76.7, 75.5, 72.3, 72.1, 71.0, 70.7, 70.5 (two), 69.1, 68.6, 63.8, 60.5, 55.8, 48.3, 47.5, 39.0, 38.8, 36.6, 32.6, 30.4, 29.3, 27.9, 27.8, 26.2, 23.3, 21.5, 18.2, 18.1, 17.8, 16.7 (two), 16.5, 16.4, 15.7; HRESIMS calcd for C50H83O15NNa 960.5660; found 960.5672.
Author contributions
Yang Chen and Hui Li performed experiments. Mingxin Zhang and Yarou Gao participated in method development and data analysis. Li Wu, Heng Wang and Dan Xu provided some resources. Gaopeng Song and Weisan Chen participated study design and thoroughly revised the manuscript. Jianxin Chen designed study and wrote the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by the National Natural Science Foundation of China (grant numbers 31872521, 31941019 and 31572565), and the Basic Research & Applying Basic Research Foundation of Guangdong Province (grant numbers 2019B1515210007).Notes and references
- A. Botner, B. Strandbygaard, K. J. Sorensen, P. Have, K. G. Madsen, E. S. Madsen and S. Alexandersen, Vet. Rec., 1997, 141, 497–499 CrossRef CAS PubMed.
- S. H. Done and D. J. Paton, Vet. Rec., 1995, 136, 32–35 CAS.
- T. Baron, E. Albina, Y. Leforban, F. Madec, H. Guilmoto, J. Plana Duran and P. Vannier, Ann. Rech. Vet., 1992, 23, 161–166 CAS.
- K. Hanada, Y. Suzuki, T. Nakane, O. Hirose and T. Gojobori, Mol. Biol. Evol., 2005, 22, 1024–1031 CrossRef CAS PubMed.
- J. K. Lunney, D. A. Benfield and R. R. Rowland, Virus Res., 2010, 154, 1–6 CrossRef CAS PubMed.
- K. Tian, X. Yu, T. Zhao, Y. Feng, Z. Cao, C. Wang, Y. Hu, X. Chen, D. Hu, X. Tian, D. Liu, S. Zhang, X. Deng, Y. Ding, L. Yang, Y. Zhang, H. Xiao, M. Qiao, B. Wang, L. Hou, X. Wang, X. Yang, L. Kang, M. Sun, P. Jin, S. Wang, Y. Kitamura, J. Yan and G. F. Gao, PLoS One, 2007, 2, e526 CrossRef PubMed.
- Y. Li, X. Wang, K. Bo, X. Wang, B. Tang, B. Yang, W. Jiang and P. Jiang, Vet. J., 2007, 174, 577–584 CrossRef CAS PubMed.
- Z. Jiang, X. Zhou, J. J. Michal, X. L. Wu, L. Zhang, M. Zhang, B. Ding, B. Liu, V. S. Manoranjan, J. D. Neill, G. P. Harhay, M. E. Kehrli Jr and L. C. Miller, PLoS One, 2013, 8, e59229 CrossRef CAS PubMed.
- A. E. Firth, J. C. Zevenhoven-Dobbe, N. M. Wills, Y. Y. Go, U. B. R. Balasuriya, J. F. Atkins, E. J. Snijder and C. C. Posthuma, J. Gen. Virol., 2011, 92, 1097–1106 CrossRef CAS PubMed.
- M. Han and D. Yoo, Vet. Microbiol., 2014, 174, 279–295 CrossRef CAS PubMed.
- J. J. Meulenberg, M. M. Hulst, E. J. de Meijer, P. L. Moonen, A. den Besten, E. P. de Kluyver, G. Wensvoort and R. J. Moormann, Virology, 1993, 192, 62–72 CrossRef CAS PubMed.
- C. Burkard, T. Opriessnig, A. J. Mileham, T. Stadejek, T. Ait-Ali, S. G. Lillico, C. B. A. Whitelaw and A. L. Archibald, J. Virol., 2018, 92, e00415-18 CrossRef PubMed.
- J. E. Collins, D. A. Benfield, W. T. Christianson, L. Harris, J. C. Hennings, D. P. Shaw, S. M. Goyal, S. McCullough, R. B. Morrison and H. S. Joo, et al., J. Vet. Diagn. Invest., 1992, 4, 117–126 CrossRef CAS PubMed.
- H. Mardassi, S. Mounir and S. Dea, J. Gen. Virol., 1994, 75, 681–685 CrossRef CAS PubMed.
- J. K. Lunney, Y. Fang, A. Ladinig, N. Chen, Y. Li, B. Rowland and G. J. Renukaradhya, Annu. Rev. Anim. Biosci., 2016, 4, 129–154 CrossRef CAS PubMed.
- J. Song, D. Shen, J. Cui and B. Zhao, Virus Genes, 2010, 41, 241–245 CrossRef CAS PubMed.
- T. Du, Y. Nan, S. Xiao, Q. Zhao and E. M. Zhou, Trends Microbiol., 2017, 25, 968–979 CrossRef CAS PubMed.
- E. Duan, D. Wang, L. Fang, J. Ma, J. Luo, H. Chen, K. Li and S. Xiao, Antiviral Res., 2015, 120, 122–125 CrossRef CAS PubMed.
- Q. Yang, L. Gao, J. Si, Y. Sun, J. Liu, L. Cao and W. H. Feng, Antiviral Res., 2013, 97, 66–73 CrossRef CAS PubMed.
- T. Opriessnig, D. Patel, R. Wang, P. G. Halbur, X. J. Meng, D. A. Stein and Y. J. Zhang, Antiviral Res., 2011, 91, 36–42 CrossRef CAS PubMed.
- M. Zhang, Q. Wu, Y. Chen, M. Duan, G. Tian, X. Deng, Y. Sun, T. Zhou, G. Zhang, W. Chen and J. Chen, PLoS One, 2018, 13, e0193309 CrossRef PubMed.
- X. Liu, J. Bai, C. Jiang, Z. Song, Y. Zhao, H. Nauwynck and P. Jiang, Vet. Microbiol., 2019, 238, 108431 CrossRef CAS PubMed.
- P. Dzubak, M. Hajduch, D. Vydra, A. Hustova, M. Kvasnica, D. Biedermann, L. Markova, M. Urban and J. Sarek, Nat. Prod. Rep., 2006, 23, 394–411 RSC.
- L. Quere, T. Wenger and H. J. Schramm, Biochem. Biophys. Res. Commun., 1996, 227, 484–488 CrossRef CAS PubMed.
- L. Kong, S. Li, Q. Liao, Y. Zhang, R. Sun, X. Zhu, Q. Zhang, W. Jun, X. Wu, X. Fang and Y. Zhu, Antiviral Res., 2013, 98, 44–53 CrossRef CAS PubMed.
- M. Zhang, T. Du, F. Long, X. Yang, Y. Sun, M. Duan, G. Zhang, Y. Liu, E. M. Zhou, W. Chen and J. Chen, Viruses, 2018, 10, 657 CrossRef CAS PubMed.
- P. G. do Nascimento, T. L. Lemos, A. M. Bizerra, Â. M. Arriaga, D. A. Ferreira, G. M. Santiago, R. Braz-Filho and J. G. Costa, Molecules, 2014, 19, 1317–1327 CrossRef PubMed.
- S. Kim, H. Lee, S. Lee, Y. Yoon and K. H. Choi, PLoS One, 2015, 10, e0118800 CrossRef PubMed.
- N. Blanco-Cabra, K. Vega-Granados., L. Moya-Anderico, M. Vukomanovic, A. Parra, L. Álvarez de Cienfuegos and T. Eduard, ACS Infect. Dis., 2019, 5, 1581–1589 CrossRef CAS PubMed.
- G. Song, X. Shen, S. Li, Y. Li, Y. Liu, Y. Zheng, R. Lin, J. Fan, H. Ye and S. Liu, Eur. J. Med. Chem., 2015, 93, 431–442 CrossRef CAS PubMed.
- L. Hui, L. Chen, S. Li, Y. Liao, L. Wang, Z. Liu, S. Liu and G. Song, Bioorg. Med. Chem. Lett., 2019, 29, 2675–2680 CrossRef PubMed.
- T. Ait-Ali, A. D. Wilson, D. G. Westcott, M. Clapperton, M. Waterfall, M. A. Mellencamp, T. W. Drew, S. C. Bishop and A. L. Archibald, Viral Immunol., 2007, 20, 105–118 CrossRef CAS PubMed.
- H. J. Nauwynck, X. Duan, H. W. Favoreel, P. Van Oostveldt and M. B. Pensaert, J. Gen. Virol., 1999, 80, 297–305 CrossRef CAS PubMed.
- S. Mlala, A. O. Oyedeji, M. Gondwe and O. O. Oyedeji, Molecules, 2019, 24, 2751 CrossRef PubMed.
- S. Xiao, Z. Tian, Y. Wang, L. Si, L. Zhang and D. Zhou, Med. Res. Rev., 2018, 38, 951–976 CrossRef PubMed.
- Y. Liao, L. Chen, S. Li, Z. N. Cui, Z. Lei, H. Li, S. Liu and G. Song, Bioorg. Med. Chem., 2019, 27, 4048–4058 CrossRef CAS PubMed.
- N. Vanderheijden, P. L. Delputte, H. W. Favoreel, J. Vandekerckhove, J. Van Damme, P. A. van Woensel and H. J. Nauwynck, J. Gen. Virol., 2003, 77, 8207–8215 CrossRef CAS PubMed.
- W. Van Breedam, P. L. Delpotte, H. Van Gorp, G. Misinzo, N. Vanderheijden, X. Duan and H. J. Nauwynck, J. Gen. Virol., 2010, 91, 1659–1667 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04070c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |