
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutation-induced change in chignolin stability from π-turn to α-turn†
Yutaka Maruyamaa,
Shunpei Korokub,
Misaki Imaic,
Koh Takeuchic and
Ayori Mitsutake *b
*b
aArchitecture Development Team, FLAGSHIP 2020 Project, RIKEN Center for Computational Science, Kobe 650-0047, Japan
bDepartment of Physics, School of Science and Technology, Meiji University, 1-1-1 Higashi-Mita, Tama-ku, Kawasaki-shi, Kanagawa 214-8571, Japan. E-mail: ayori@meiji.ac.jp
cCellular and Molecular Biotechnology Research Institute, National Institute of Advanced Industrial Science and Technology, 2-3-26 Aomi, Koto, Tokyo 135-0064, Japan
First published on 15th June 2020
Abstract
Chignolin, which consists of 10 amino acids, adopts two stable states in simulations at room temperature at 1 atm: the native and misfolded states. The sequence of chignolin is optimized to form a stable π-turn and thus the native state has a π-turn from Asp3 to Thr8. On the other hand, the misfolded state adopts an α-turn from Asp3 to Gly7. We previously investigated the differences in the stability mechanism of the two states using computational techniques. Our previous detailed energy analysis implied that the native state was stabilized by hydrogen bonding between the side chain atoms of Thr6 and Thr8, and Thr8 was not involved in stabilization of the misfolded state. Thus, we predicted that mutation of Thr8 to a neutral amino acid could stabilize the misfolded structure over the native structure. In the present work, we performed 4 μs molecular dynamics simulations for 19 mutants of the 8th residue. Among them, the T8I, T8F, T8P, T8N, and T8Y mutants, in which the 8th residue was changed to a neutral residue, formed only the misfolded structure at room temperature. Even at high temperature, for the T8P mutant, the native structure was not observed, as the T8P mutant cannot form the native structure because of steric hindrance caused by the distinctive cyclic structure of proline. Interestingly, the T8P mutant at high temperature has trans and cis conformations in the Gly7–Pro8 sequence, with the trans conformation corresponding to the misfolded state. NMR analysis of the T8P mutant supported our results.
1 Introduction
From a molecular perspective, amino acid mutations can alter the stabilities or functions of proteins. Many researchers have altered the structural stabilities or enhanced the functions of proteins by artificially inducing mutations using computational approaches. For example, groups have introduced mutations into membrane proteins to increase their melting temperatures compared to their wild-type counterparts. As a result of these analyses, these membrane proteins had increased stabilities and could be crystallized.1–3 Kuhlman and Baker redesigned the folding pathways of proteins by introducing mutations. Some mutants folded faster than the wild-type proteins, suggesting that natural proteins are not always optimized for folding.4 Thus, studies are being conducted to evaluate the stabilities of protein mutants using computer simulations.5,6When considering protein stability, the contribution of surrounding water must be included because of the competition between the conformational energy of the protein and solvation free energy (SFE). Recently, we investigated the stability of proteins using the three-dimensional reference interaction site model (3D-RISM) theory. The 3D-RISM, which is the statistical mechanics theory for molecular liquid, reproduces the distribution function of the solvent around solute molecules and gives the SFE. To confirm the validity of the total energy given by the sum of the conformational energy of protein and the SFE using 3D-RISM, we analyzed folding simulation data7 for a wide variety of proteins, including a small protein, a β-sheet protein, and α + β protein, and an α-helical protein.8 The structures with the lowest total energy corresponded to the native structures of the proteins. Thus, the total energy is an appropriate energy function for investigating protein stability. For mutation analysis, it is important to estimate the stabilities of individual amino acids. Using the atomic decomposition (AD) method proposed by Chong and Ham,9 the SFE of individual residues can be estimated. Thus, we applied this method to chignolin to investigate the structural stability at the amino acid level.10
Conformational studies of β-hairpin-forming peptides as a model of very early events of protein folding have been examined by many groups.11,12 G-Peptide is a 16-residue peptide corresponding to a dissected fragment of residues 41–56 of the protein G B1 domain. It was the first natural sequence found to fold into a β-hairpin structure in water.12 This peptide is a good model for investigating β-hairpin structure stability in detail. Chignolin, which consists of 10 amino acids, GYDPETGTWG, was designed by Honda et al.13 They designed chignolin to form a stable turn structure using the G-peptide sequence and a database. The amino acid sequence of chignolin is likely to form a π-turn.13–15 This peptide is widely used to test new simulation algorithms and analysis methods.16–29 Chignolin has two stable states at room temperature at 1 atm according to molecular dynamics (MD) simulations: a native state and misfolded state, as shown in Fig. 1.23,25,30–34 Both states have a common β-turn structure from Asp3 to Thr6 but slightly different hydrogen bonding patterns for the backbone atoms. The native state has a π-turn (a π-HB turn (AAAa) in ref. 15) from Asp3 to Thr8, whereas the misfolded state adopts an α-turn (I-αRS turn in ref. 35 and α-turn (AAA) in ref. 14) from Asp3 to Gly7.10 α-turn or π-turn forms hydrogen bonds between backbone atoms of the ith residue and i + 4th or i + 5th residue, respectively (in chignolin, i corresponds to 3). The difference in the dihedral angles from the 3rd to 8th residues of the native and misfolded states involves only Gly7 (see Fig. 4b and ref. 27 and 33). The α-turn of the misfolded state is involved in the π-turn of the native state. Because of the state of the dihedral angles of Gly7, both turn structures were observed in the simulation. As Gly7 has different dihedral angles, the backbone atoms of Thr8 and Phe9 of the misfolded state are reversed from the native state. Thus, the side chains of Tyr2 and Trp9 contact each other in the native state, whereas these residues are on opposite sides in the misfolded state. In addition, the hydrogen bond between the side chains of Thr6 and Thr8 exists in the native state but does not appear in the misfolded state. The conformational transition from the native to misfolded states may occur under high pressure.36
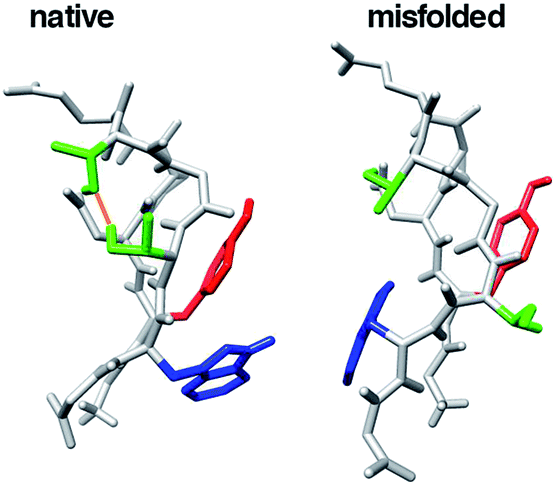 | ||
| Fig. 1 The native and misfolded structures of chignolin. The red and blue regions indicate the side chains of Tyr2 and Trp9, respectively. The green regions indicate the side chains of Thr6 and Thr8. The orange line is the hydrogen bond between the side chains of Thr6 and Thr8. | ||
The denaturation temperature of chignolin is low (315 K), and thus its atomic coordinates were only determined by NMR. To improve its stability, Honda et al. mutated the N- and C-termini. The chignolin mutant, CLN025, contains mutations in two amino acids at both terminals (G1Y and G10Y). Using thermodynamic parameters obtained from fitting calculations,37 the molar fraction of CLN025 in the folded state is estimated to be 99% at 273 K, whereas that of chignolin is 83% at 273 K. The structure of CLN025 was solved by X-ray crystallography and NMR experiments. The crystal structure of CLN025 indicated that it had the same topology as in aqueous solution.37 Even in MD simulations of CLN025, the misfolded structure was not observed.7,8,37,38
Force-field dependence of the native and misfolded states of chignolin was investigated previously.33 The folding thermodynamics of chignolin were investigated by replica-exchange molecular dynamics sampling and four different force fields: AMBER ff03*, AMBER ff03w, AMBER ff99SB, and CHARMM22/CMAP. Chignolin folds to the native structure with all four force fields. However, AMBER ff03*, AMBER ff03w, and AMBER ff99SB also resulted in a population of a misfolded state with a population of 20–50% (depending on the force field parameters for Gly7). Although it was difficult to directly distinguish between the native and misfolded states experimentally because the change between these states occurs on the timescale of a few microseconds, which is much shorter than that on which the NMR signals are averaged, they suggested the existence of a misfolded state in the ensemble by comparing the results of NMR experiments. Based on the difference in the dihedral angle ψ of Gly7, they also proposed the G7K mutant, which preferentially forms the native structure. NMR experiments of the G7K mutant supported their results.
In our previous study, we applied a relaxation mode analysis (RMA) method for simulation of chignolin with structural changes and extracted characteristic states, i.e., the native, misfolded, intermediate, and unfolded states.27,39 RMA approximately estimates slow relaxation modes and rates from trajectories obtained from simulations, and is suitable for investigating a system with large structural changes. Next, we investigated the structural stabilities of the native and misfolded structures at the amino acid level using the AD method.10,39 We examined the relationships between hydrogen bond formation and the energy contribution of each amino acid residue. Even at the amino acid level, competition between the conformational energy and SFE was observed. As a result of the competition between the conformational energy and SFE, hydrogen bonds between atoms of chignolin stabilized the turn structures. We examined the difference in stability between the native and misfolded states. The difference in the dihedral angles from the 3rd to 8th residues of the native and misfolded states was determined for Gly7. The native state is stabilized by the side-chain interaction between Thr6 and Thr8. In contrast, Thr8 does not contribute to stabilization of the misfolded state. We predicted that mutating Thr8 to a neutral amino acid would improve the stability of the misfolded state.
In this study, we confirmed that mutations at Thr8 stabilized the misfolded structure rather than the native structure using MD simulations. We generated mutants in which Thr8 was mutated to 19 other amino acid residues and performed MD simulations at room temperature. The root mean square deviation (RMSD) values and hydrogen-bonds maps were calculated to identify the misfolded and native states. The conformational energy and SFE values of the generated conformations were also calculated to evaluate the stabilities of the states. Based on our detailed analysis, we proposed a mutant in which the misfolded structure was more stable than that in the native structure. NMR experiments of chignolin, CLN025, and T8P mutant were performed to confirm our results.
2 Methodology
2.1 MD simulation details
We generated various structures of chignolin mutants and performed MD simulations with the AMBER package (AMBER 2016)40 using the ff99SB force field for the protein41 and TIP3P model for water.42 The Amber ff99SB force field has been widely used in previous studies.10,22–27,29,32 We generated an extended structure of the chignolin mutant using the leap command and solvated this structure with a 15 Å buffer of water in each direction using the TIP3P model. The chignolin mutants contained 131–148 atoms, and the numbers of water molecules were 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 075–18675. The simulations were performed under neutral pH where Arg, His, and Lys residues are positively charged and Glu and Asp are negatively charged. Several sodium ions (Na+) were included, resulting in a net neutral system. After solvating the chignolin mutants, we minimized the structures with Cα constraints using the steepest descent method (500 steps) and conjugate gradient method (500 steps). After minimizing the structures, the system was heated from 0.1 to 298.15 K over 50 ps, with a 50 ps MD simulation at constant pressure (1 atm) and 298.15 K with Cα constraints to adjust the density of the system. Finally, 4 μs MD simulations for production runs were performed following 500 ps MD simulations for equilibration at 1 atm and 298.15 K. For several mutants, similar calculations were performed near the transition temperature (420 K) to enhance sampling. The time step for the simulation was set to 2 fs. The Langevin thermostat with a friction constant of γ = 2.0 ps−1 was used for temperature control. A cutoff of 8 Å was used to limit the direct space sum while implementing the particle mesh Ewald (PME) algorithm in AMBER. For the equilibration and production runs, the pmemd command was used. For the analysis, the coordinates were saved every 1 ns. The number of samples in each calculation was 4000.
075–18675. The simulations were performed under neutral pH where Arg, His, and Lys residues are positively charged and Glu and Asp are negatively charged. Several sodium ions (Na+) were included, resulting in a net neutral system. After solvating the chignolin mutants, we minimized the structures with Cα constraints using the steepest descent method (500 steps) and conjugate gradient method (500 steps). After minimizing the structures, the system was heated from 0.1 to 298.15 K over 50 ps, with a 50 ps MD simulation at constant pressure (1 atm) and 298.15 K with Cα constraints to adjust the density of the system. Finally, 4 μs MD simulations for production runs were performed following 500 ps MD simulations for equilibration at 1 atm and 298.15 K. For several mutants, similar calculations were performed near the transition temperature (420 K) to enhance sampling. The time step for the simulation was set to 2 fs. The Langevin thermostat with a friction constant of γ = 2.0 ps−1 was used for temperature control. A cutoff of 8 Å was used to limit the direct space sum while implementing the particle mesh Ewald (PME) algorithm in AMBER. For the equilibration and production runs, the pmemd command was used. For the analysis, the coordinates were saved every 1 ns. The number of samples in each calculation was 4000.
For sampling verification, we also performed generalized replica exchange with solute tempering (gREST)43 simulations for several mutants using GENESIS software package.44,45 gREST can reduce the number of replicas compared to the temperature replica-exchange molecular dynamics method. gREST simulations of the mutants adopted all dihedral angle terms as the solute region. We employed four replicas with temperatures of 298.15, 342, 392, and 450 K in gREST. Before production, a 10 ns MD simulation for each replica was conducted to equilibrate the system without any replica exchanges. Next, we performed gREST simulations of the mutants for 400 ns per replica. Exchange of replicas was attempted every 2 ps.
To evaluate the stabilities of the structures obtained by the present simulations, we defined the total energy, G, as the sum of the conformational energy of the protein, E, and SFE, Δμ:8,10,46–48
| G = E + Δμ. | (1) |
We used the Amber Package (Amber 2016) to calculate the conformational energy.40 We also used the 3D-RISM theory49–51 with the reference-modified density functional theory (RMDFT) functional52 to calculate the SFE of the protein in water.
For 3D-RISM calculations, we used the TIP3P model with an additional parameter (σ = 0.4 Å and ε = 0.046 kcal mol−1) for water molecules.53 The conventional 1D-RISM theory was used for the site–site correlation function of water. The number density of water molecules and temperature were 0.033329 molecules per Å3 and 298.15 K, respectively. The 3D-RISM integral equations with the Kovalenko–Hirata closure equations51,54 were solved for a grid of 2563 points in a 64 Å3 cubic cell using graphic processing units.55 A grid space of 0.25 Å was sufficient to calculate the SFE without significant numerical errors. We employed the RMDFT function to obtain exact SFE calculations with the diameter of a hard sphere set to 2.88 Å.
2.2 NMR experiment details
Peptides were purchased at >95% purity from BEX Co., Ltd. For NMR experiments, 1.1, 1.3, and 1.1 mg of chignolin, CLN025, and T8P mutant were dissolved in 500 mL of H2O containing 5% (v/v) D2O adjusted to pH 5.5 with HCl. The final peptide concentration of the sample was 2 mM. NMR spectra were recorded on a Bruker AVANCEIII 600 spectrometer, equipped with a cryogenic probe (Billerica, MA, USA). All two-dimensional NMR experiments, i.e. DQF-COSY, TOCSY, and NOESY, were performed using standard pulse sequences and phase cycling. All experiments were performed at 280 K. In a typical experiment, 1024 (direct) and 1024 (indirect) complexed data points for higher resolution were recorded. The DQF-COSY spectrum was recorded with 2024 (direct) and 1024 (indirect) complexed data points. TOCSY spectra were recorded with a mixing time of 60 ms. The NOESY spectra were recorded with a mixing time of 200 ms. All experiments included the Watergate scheme for water suppression. Processing and analyses of the spectra were performed using the Bruker TOPSPIN and SPARKY programs.3 Results and discussion
3.1 Chignolin
To classify the native and misfolded structures during the simulations, we first determined the differences between the native and misfolded states of chignolin. The time series of the RMSD values of Cα atoms from the native structure (Cα-RMSD) obtained by 4 μs MD simulation of chignolin is shown in Fig. 2. The first coordinate of the solution NMR structure was used as the native structure (PDB 1UAO, model 1). The first-time transition from the unfolded state to the native state (Cα-RMSD ≈ 1.3 Å) occurred at approximately 100 ns. Subsequently, the second-time transition from the native to the misfolded state (Cα-RMSD ≈ 2.2 Å) occurred at approximately 1500 ns. Once it changed to around Cα-RMSD ≈ 2.2 Å, it settled into the misfolded state. The small changes between Cα-RMSD ≈ 2.2 Å and Cα-RMSD ≈ 2.5 Å indicated a difference in the orientation of the side chain of Trp9 in the misfolded state (two values correspond to m1 and m2 classified in the misfolded state, as shown in Fig. 3(c) and (d) of ref. 10.) In the native structure, this rotation of the side chain also occurred, but the change in the backbone was small and does not appear in Fig. 2. During the 4 μs MD simulation, chignolin changed from the native to misfolded states at room temperature.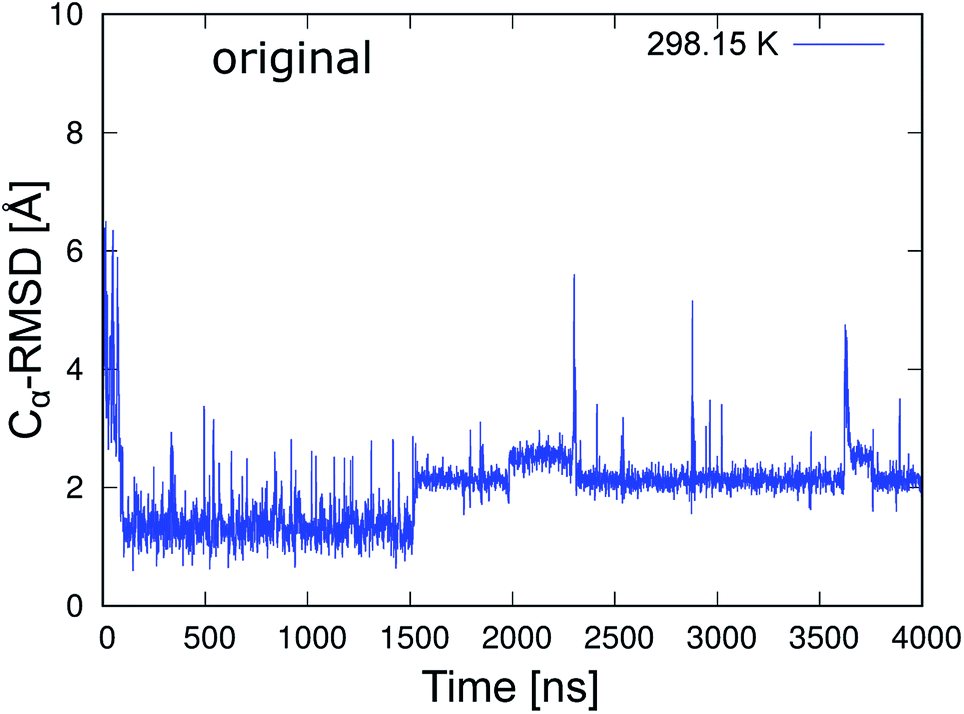 | ||
| Fig. 2 Time series of Cα-RMSD of the original chignolin peptide based on the NMR structure (PDB 1UAO, model 1). | ||
Fig. 3 shows the total energy values of chignolin as a function of the Cα-RMSD. There are two main clusters: one cluster around Cα-RMSD of 1.3 Å and the other around Cα-RMSD of 2.2 Å. The former was classified as the native state and the latter as the misfolded state. In general, structures with the lowest total energy correspond to a small Cα-RMSD. However, the most stable structure of chignolin had a Cα-RMSD of ≈2.1 Å, and the second most stable structure had a value of approximately 1.0 Å. The clusters showed approximately the same energy ranges as indicated by the vertical spreads, suggesting that the native and misfolded states have similar probabilities of existence.
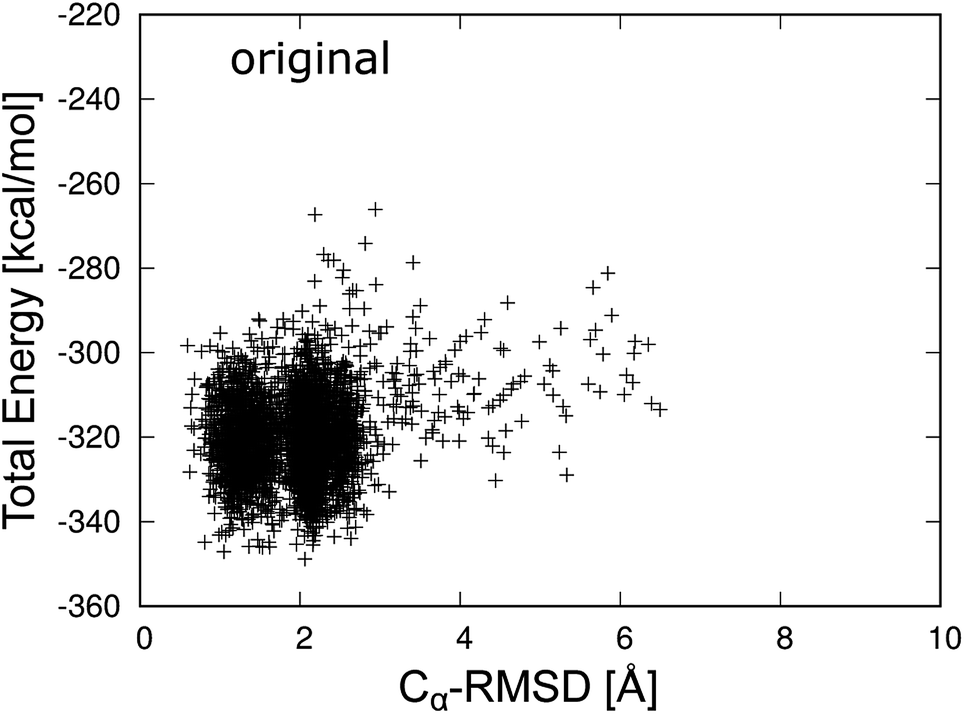 | ||
| Fig. 3 Total energy of the original chignolin peptide as a function of Cα-RMSD. | ||
In addition, there are hydrogen bond patterns in a characteristic quantity that distinguishes the native state from the misfolded state. The native structure forms a hydrogen bond between the amide nitrogen atom on the main-chain of Asp3 (Asp3N) and carbonyl oxygen atom on the main-chain of Thr8 (Thr8O), whereas the misfolded structure forms a hydrogen bond between Asp3N and Gly7O. In previous studies,30 the distance between Asp3N and Gly7O (d(Asp3N–Gly7O)) and that between Asp3N and Thr8O (d(Asp3N–Thr8O)) were used to classify the two states. The distributions of the native and misfolded states, which were classified by principal component analysis,10 are shown in Fig. 4a. The native and misfolded states were clearly separated using these distances. The dihedral angles of Gly7 were also used to distinguish the two states.27,33 Ramachandran plots of Gly7 for the native and misfolded states are also shown in Fig. 4b. The states were clearly distinguished. Some order parameters such as Cα-RMSD, hydrogen bond distances, and the dihedral angles of Gly7 can be used to distinguish the two states. These values resulted in similar classification. Here, we used the Cα-RMSD and hydrogen bond distances to analyze 19 MD simulations of mutants.
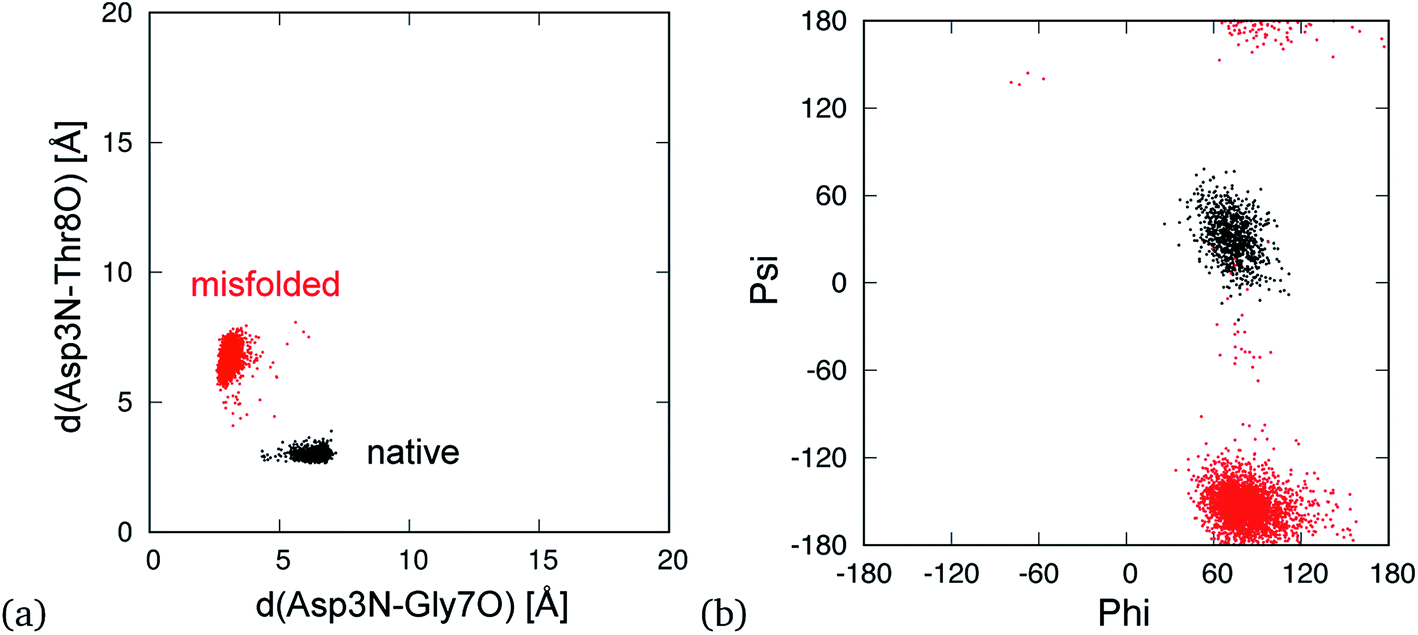 | ||
| Fig. 4 Conformational distribution in plane of d(Asp3N–Gly7O) and d(Asp3N–Thr8O) (a) and the dihedral angles of Gly7 (b). Here, d(X–Y) is distance between X and Y atoms. The black and red points indicate the native and misfolded states, respectively. | ||
3.2 Chignolin mutant
We performed 4 μs MD simulations of 19 mutants at room temperature. Fig. 5 shows the time series of the Cα-RMSD values of mutations for the neutral side chain. The figures of the mutants for charged side chains are shown in Fig. S1 and S2 of the ESI.† The blue line shows the trajectory of the MD simulations at 298.15 K. The Cα-RMSD values of the five mutants, T8I, T8F, T8P, T8N, and T8Y mutants, were stable with a lower limit of approximately 2.0 Å, which corresponds to the misfolded state. These mutants did not show the native structure in the simulations at room temperature. Isoleucine (I), phenylalanine (F), and proline (P) have hydrophobic side chains, whereas asparagine (N) and tyrosine (Y) have polar, uncharged side chains.56 Mutations that cannot form a hydrogen bond between the side chains of residues 6 and 8 stabilized the misfolded structure, as previously predicted.10 However, the T8I mutant did not retain the misfolded structure for long periods of time and instead adopted an extended structure several times. Thus, the denaturation temperature of the T8I mutant may be lower than that of the other mutants. In the remaining 14 mutants, both the native and misfolded states were observed. All the mutations form native and/or misfolded structures. Note that the T8R and T8K mutants, which correspond to positively charged side chains, remain in the native state for longer because they can form a hydrogen bond between the side chains of residues 6 and 8, as shown in Fig. S2.† Thus, these mutants would behave similar to chignolin.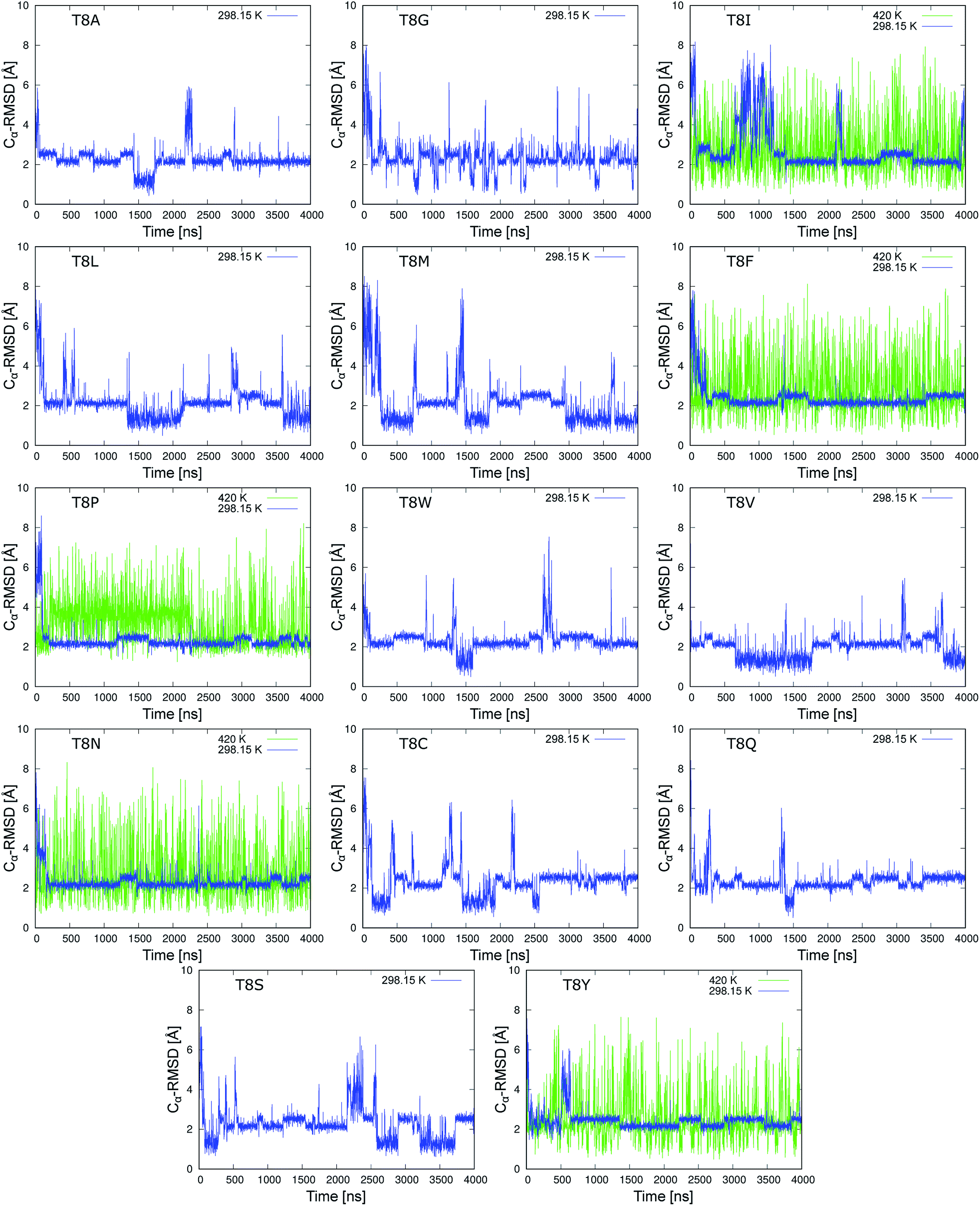 | ||
| Fig. 5 Time series of Cα-RMSD values of the chignolin mutants (neutral side chain) based on the NMR structure (PDB 1UAO model 1). Mutation from the T8A mutant to the T8V mutant correspond to hydrophobic side chains. Mutations from the T8N mutant to the T8Y mutant correspond to a polar uncharged side chain. | ||
In addition, MD simulations at 420 K were performed to increase the sampling of the structures for T8I, T8F, T8P, T8N, and T8Y mutants (green lines in Fig. 5). All five mutants alternated between the compact and extended structures many times, suggesting that sufficient sampling was performed. In four of the mutants (except for the T8P mutant), the native structure with Cα-RMSD values of ≈1 Å appeared. However, the T8P mutant did not display Cα-RMSD values reaching 1.3 Å at either temperature. In contrast, the T8P mutant retained the structure with Cα-RMSD values of ≈4 Å between 200 and 2200 ns. Such behavior was not observed for the other mutants, which is discussed below. Based on these results, the misfolded structure was the most stable state of the T8P mutant.
The total energies of T8I, T8F, T8P, T8N, and T8Y mutants are shown as a function of Cα-RMSD at 298.15 K in Fig. 6. (The results for other mutants are shown in Fig. S3–S6 in the ESI.† The average values of total energy of each state for the mutants and the occurrence rate of each state are also listed in Tables S1 and S2 in ESI.†) For Fig. 6, each mutant formed a cluster indicating the misfolded state near Cα-RMSD = 2.0 Å. No clusters showed the native state of chignolin. The T8I mutant had clusters near Cα-RMSD = 4.0 and 6.0 Å, suggesting that it is less stable compared to the other mutants. Thus, the T8I mutant could adopt intermediate and extended structures. Similarly, the T8N mutant showed a cluster near Cα-RMSD = 3.5 Å. In contrast, the T8P mutant had few distribution points at Cα-RMSD values greater than or equal to 3.0 Å. Thus, the T8P mutant has high structural stability in the misfolded state.
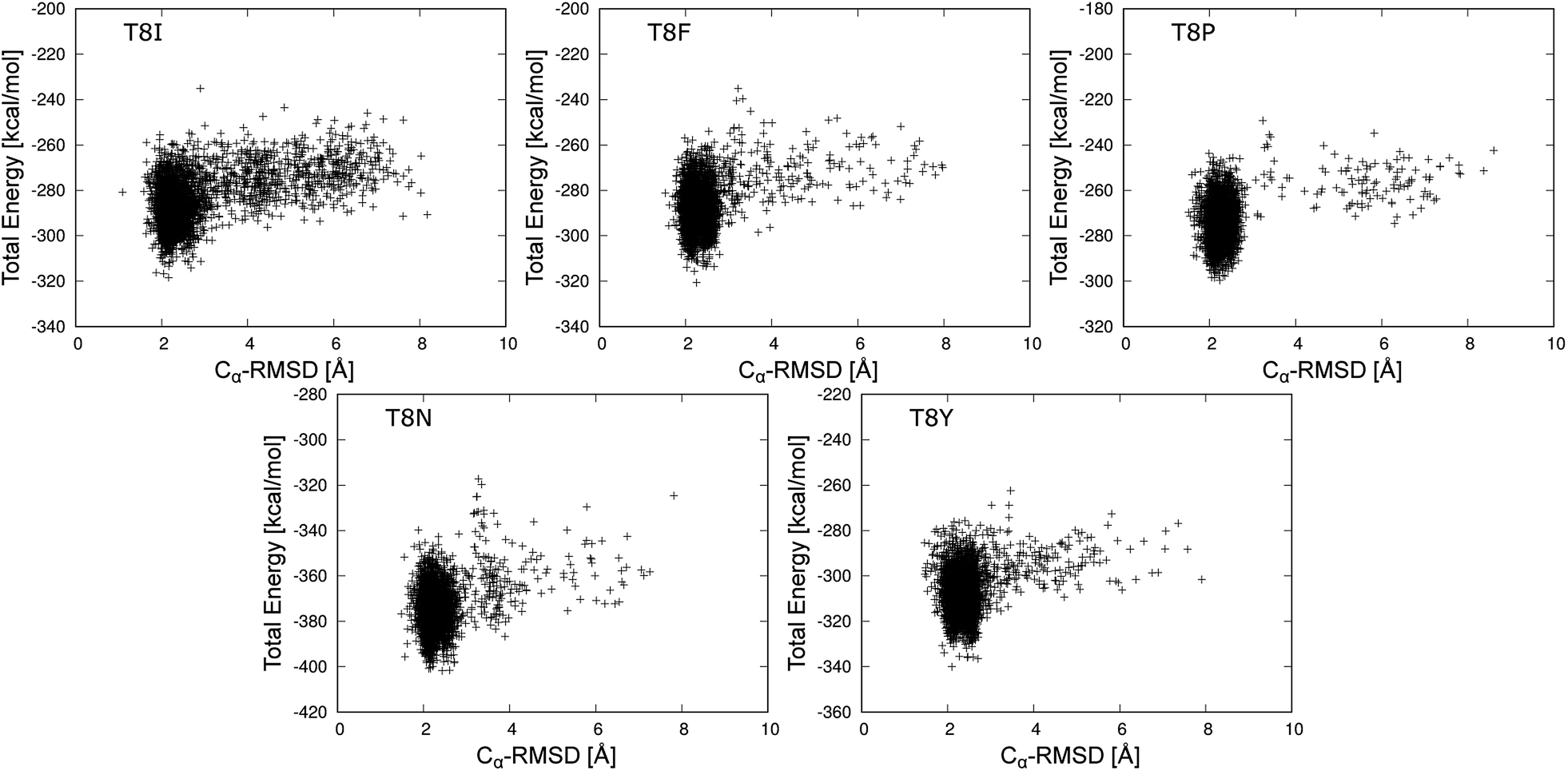 | ||
| Fig. 6 Total energy values of the chignolin mutants as a function of the Cα-RMSD at 298.15 K. | ||
The lowest total energy structures of T8I, T8F, T8P, T8N, and T8Y mutants are shown in Fig. 7. (The lowest total energy structures of the other mutants are shown in Fig. S7–S10 in the ESI.†) As described above, all five mutants formed the misfolded structure. The distances between the side chains of Thr6 and residue 8 were too long to facilitate hydrogen bond formation. The side chains of residue 8 are on the same side as the side chain of Tyr2; however, they were only in contact with each other in the T8F mutant (Tyr2 and Phe8). The presence or absence of this contact did not heavily influence the structural stability of chignolin because of competition between the conformational energy and SFE.10
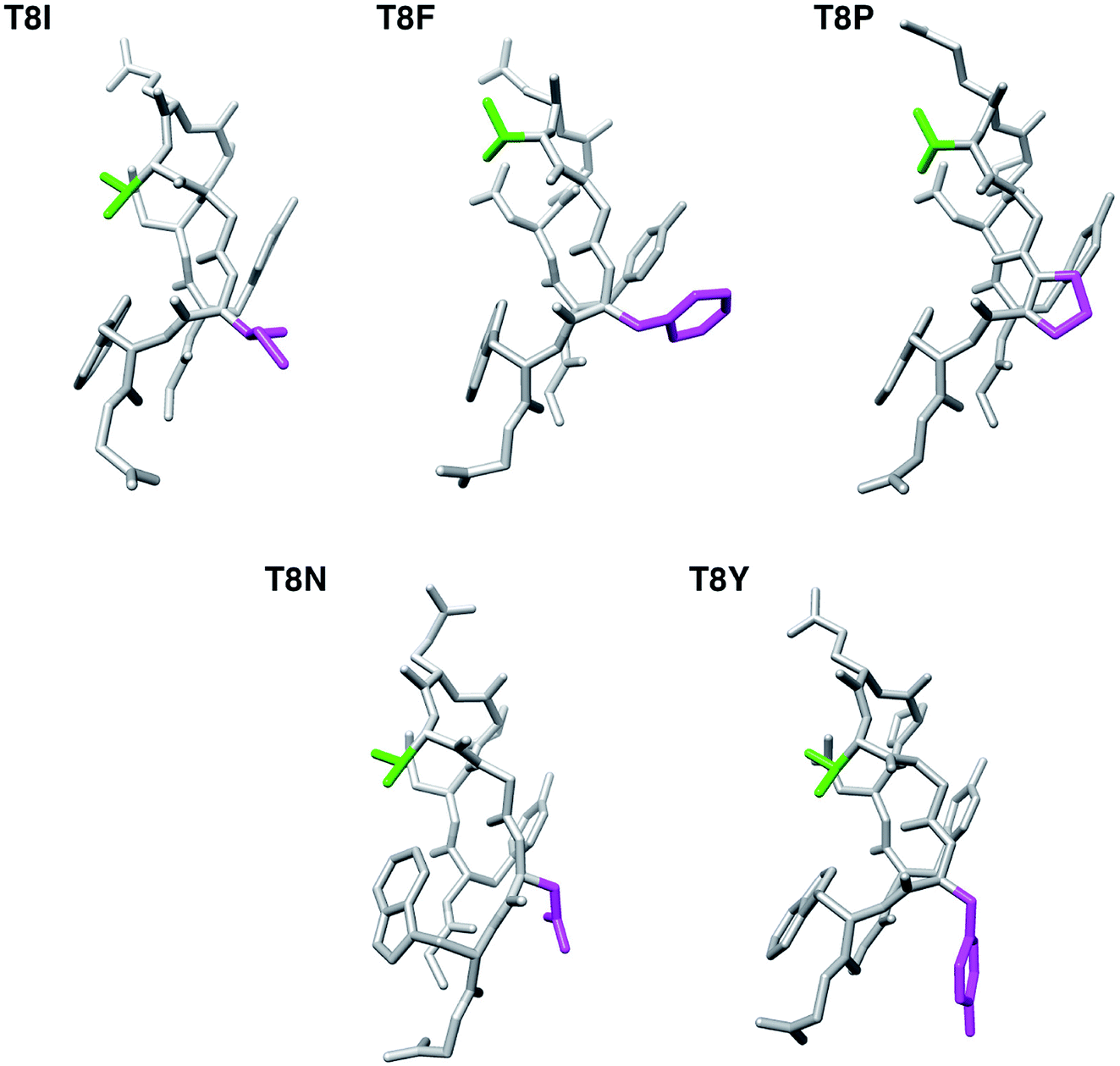 | ||
| Fig. 7 The lowest total energy structures of each chignolin mutant. The green and magenta regions indicate the side chains of Thr6 and residue 8, respectively. | ||
Fig. 8 shows the conformational distributions of mutants projected onto the distances in hydrogen bonds. The distribution of the T8I mutant was widely spread and existed around the native state. This agrees with the results for the T8I mutant shown in Fig. 6. The structure of the T8I mutant was not stable at room temperature. Compared to the T8I mutant, the distribution of the T8F mutant was not widely spread and did not exist around the native state. The T8P mutant quickly became misfolded from the initial extended structure, showing nearly no distribution except for the cluster of the misfolded state. Thus, this mutant stably formed the misfolded structure at room temperature. In the distribution of the T8N mutant, a cluster different from the misfolded state was observed, as shown in Fig. 6. The T8Y mutant showed a cluster that was clearly not the misfolded state. This corresponded to the structures from 3 to 6 Å between 500 and 600 ns in the times series (Fig. 5).
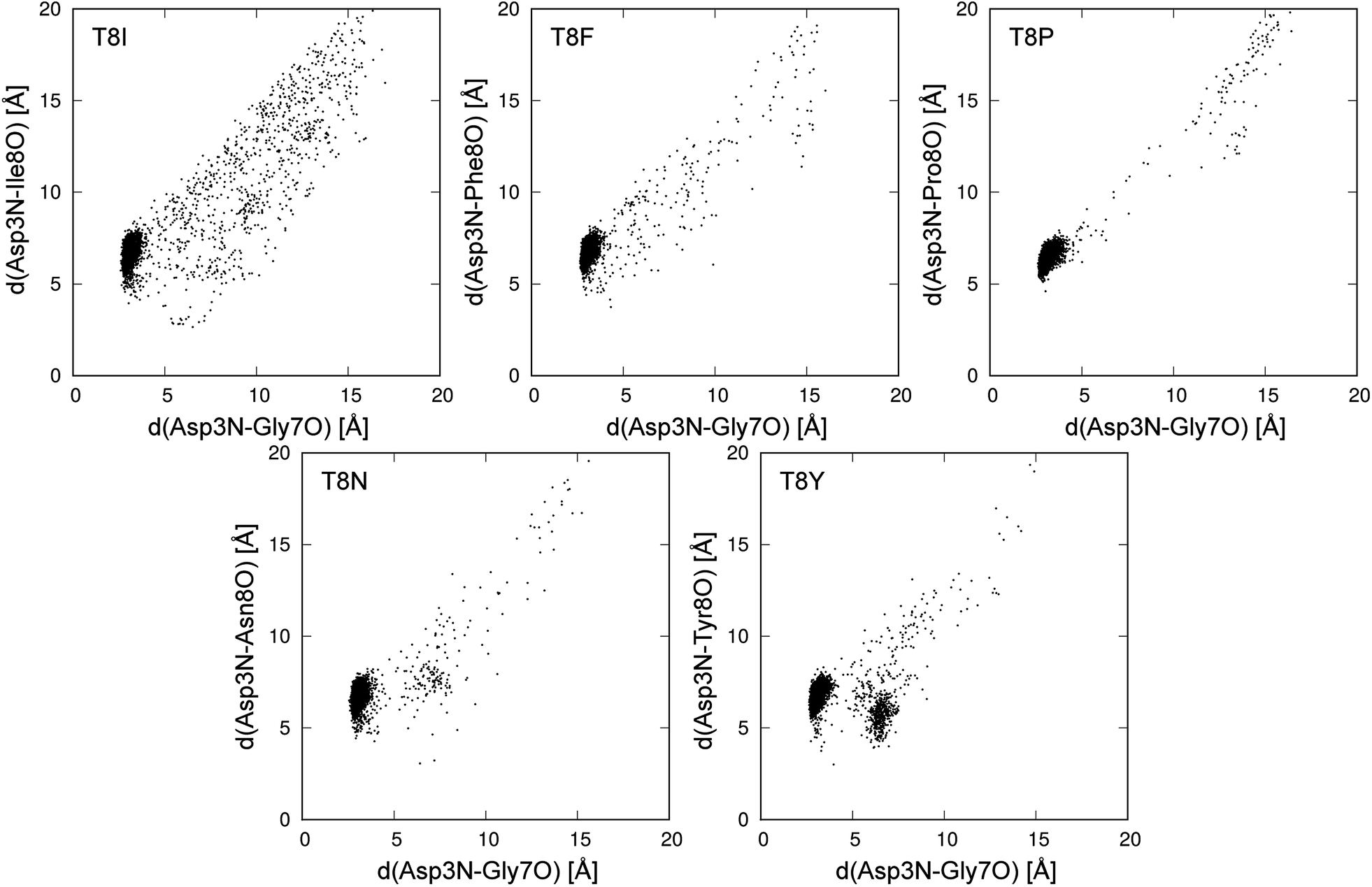 | ||
| Fig. 8 Conformational distribution at 298.15 K in plane of d(Asp3N–Gly7O) and d(Asp3N–Xxx8O). Xxx8 means the mutated 8th amino acid, and d(X–Y) is distance between X and Y atoms. | ||
We also performed gREST simulations of the T8F and T8P mutants to verify the sampling. Fig. 9 shows the conformational distributions of the mutants for the trajectories of the gREST simulations at 298.15 K. The T8F mutant showed a distribution around the native state that did not appear in the MD simulation at 298.15 K. However, the distribution of the T8P mutant extended from the misfolded state to the extended state but does not appear around the native state. Thus, the T8P mutant cannot form the native structure.
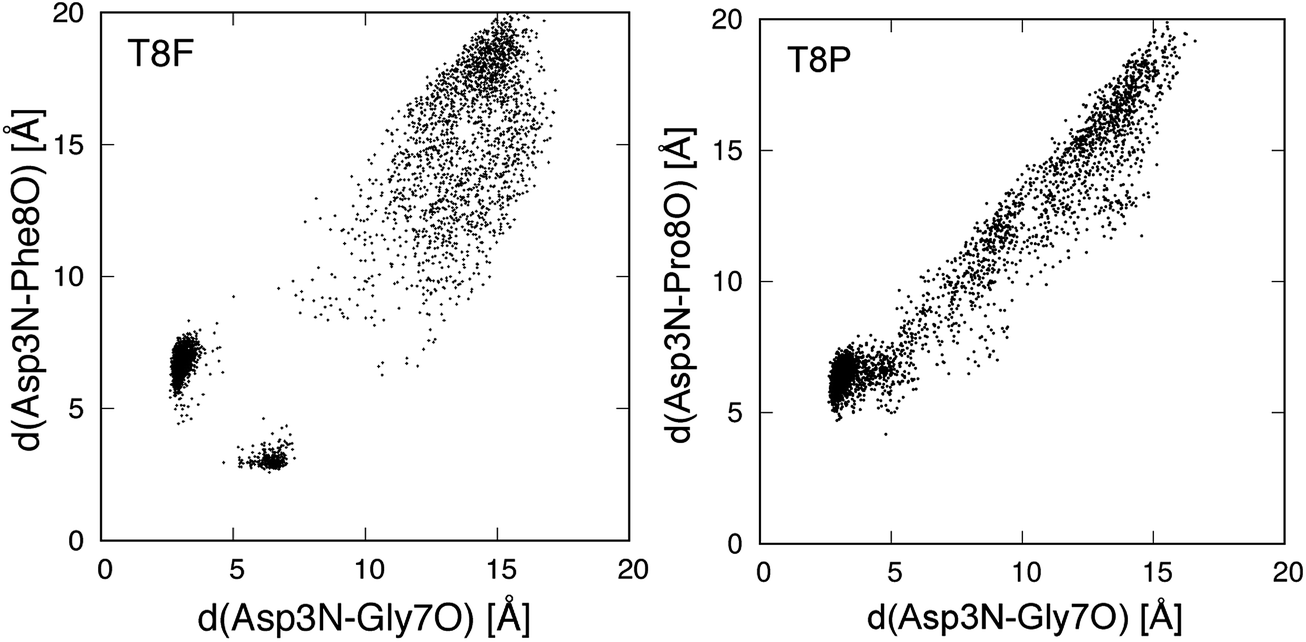 | ||
| Fig. 9 Conformational distribution for gREST simulation in plane of d(Asp3N–Gly7O) and d(Asp3N–Xxx8O). Xxx8 means the mutated 8th amino acid, and d(X–Y) is distance between X and Y atoms. | ||
We examined why the T8P mutant did not form the native structure. Fig. 10 shows the most stable structure of the T8P mutant as a space-filling model. The dihedral angle of Gly7, Ψ, was at approximately −160° in the most stable structure of the T8P mutant. The solid blue line indicates the dihedral angle of Gly7 for the misfolded state. The blue dashed line indicates the dihedral angle when Ψ of Gly7 was approximately 20°, which corresponds to the native structure. As shown in the figure, when Ψ of Gly7 rotated towards the blue dashed line, the side chain of Pro8 interferes with the oxygen atom (shown in red) of Thr6. Furthermore, in the native structure, the side chain of Pro8 interfered with backbone organization. This also prevented hydrogen bond formations in the β-turn. In contrast, the side chains of the other amino acids introduced at residue 8 were more flexible; thus, they were not sterically hindered. Based on these results, the T8P mutant cannot form the native structure.
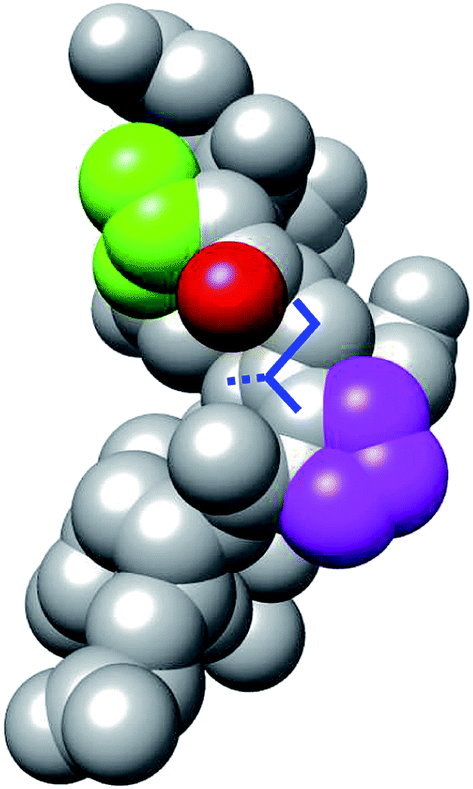 | ||
| Fig. 10 Space-filling model display of the most stable structure of T8P mutant (same angle as in Fig. 7). The green and magenta regions indicate the side chains of Thr6 and Pro8, respectively. The red region indicates the oxygen atom in the main chain of Thr6. The solid blue lines indicate the dihedral angle, Ψ, of Gly7, and the dotted blue line indicates the position of the side chain when rotated approximately 180°. | ||
Below, we discuss the turn structures of chignolin. In general, turn structures are classified into three groups: β-turns, α-turns, and π-turns.15,57–60 These turns form hydrogen bonds between backbone atoms of the ith residue and i + 3rd, i + 4th, or i + 5th residue, respectively. In the original chignolin, the native state has a π-turn from Asp3 to Thr8, whereas the misfolded state adopts an α-turn. The native state also forms a hydrogen bond between the backbone atoms of Asp3 and Thr8, whereas the misfolded state forms a hydrogen bond between Asp3 and Gly7. Furthermore, the hydrogen bond between the side chains of Thr6 and Thr8 primarily stabilizes the π-turn structure in the native state. The mutations of Thr8 destabilize the π-turn structure by removing this hydrogen bond. Dasgupta and Chakrabarti also reported that the (i + 5) position in the π-turn is devoid of proline, as proline at i + 5th residue cannot act as a hydrogen-bond donor.15 In addition, we consider that proline's distinctive cyclic side chain is important for limiting the turn-structures that can be adopted. Thus, a proline residue at position i + 5 renders the α-turn more stable than the π-turn. For example, the position-specific score for the G-peptide-like motif of proline at 8th residue was 0.0 in the analysis by Honda et al.13 This means that the presence of proline at the 8th residue inhibits formation of the chignolin native structure.
We discuss structures with Cα-RMSD values around 4 Å in the MD simulation of T8P at 420 K as shown in Fig. 5. The structure corresponds to the cis conformation in the Gly7–Pro8 sequence, as shown in Fig. 11, whereas the misfolded structure corresponds to the trans conformation. The structures from Tyr2 to Gly7 were similar to each other, as shown in Fig. 7 (T8P) and Fig. 11. The cis conformation of Xaa–Pro is sometimes observed when Xaa is Gly. T8P mutation corresponded to the amino acid sequence. In the simulation at 420 K, the transitions from the trans to cis and from cis to trans conformations were observed at approximately 200 and 2300 ns, respectively (see the green line for the T8P mutant in Fig. 5). When an additional 4 μs simulation at 420 K for T8P mutant was continued, the trans conformation was maintained. The cis type structure showed a higher total energy than that of the misfolded structure, as shown in Fig. S11.† The cis type structure was observed but was more unstable than the misfolded structure.
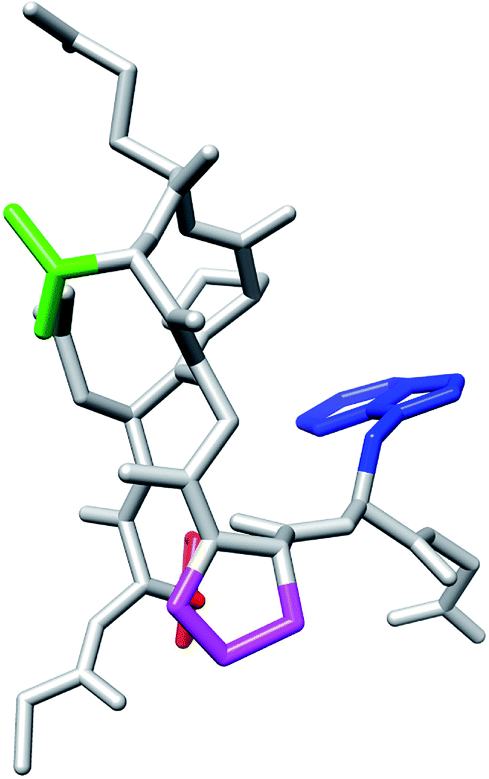 | ||
| Fig. 11 T8P mutant with the cis conformation in the Gly7–Pro8 sequence. The red and blue regions indicate the side chains of Tyr2 and Trp9, respectively. The green and magenta regions also indicate the side chains of Thr6 and Pro8, respectively. | ||
3.3 NMR experiments
Using the thermodynamic parameters obtained from fitting calculations,37 the molar fraction of CLN025 in the folded state was estimated as 99% at 273 K, whereas that of chignolin was 83% at 273 K. Although there was a force field dependency, the native and misfolded states were obtained by MD simulations of chignolin. Simulations of CLN025 did not reveal the misfolded state. To confirm our results, we performed NMR experiments of chignolin, CLN025, and T8P mutant. The NMR spectra were well resolved for chignolin, CLN025, and the T8P mutant. Although the NMR spectra of chignolin and CLN025 corresponded to the single conformational state, those of the T8P mutant revealed the existence of two distinct conformations, which presumably corresponds to a trans and cis conformational equilibrium in the Gly7–Pro8 sequence. The signal intensities of the two conformational states were similar to each other. As the rate of cis–trans interconversion is typically slow, conformational ensemble observed in the NMR experiments can only be observed in MD simulations at high temperature, which may also correspond to longer simulations.To confirm that the two conformational states observed in the NMR experiments originated from the trans and cis conformational equilibrium in the Gly7–Pro8 sequence, we determined the sequential backbone chemical shift assignments of the peptides. From the NOE pattern between Gly7 and Pro8, the two conformational states in the T8P mutant were confirmed to originate from a trans and cis transition at this site (hereafter, the T8P mutant in the trans and cis conformational states are referred to as T8P (trans) and T8P (cis), respectively). The assigned chemical shifts for HN and Hα of chignolin, CLN025, T8P (trans), and T8P (cis) are listed in Tables S3 and S4 of the ESI.† The chemical shifts of HN and Hα of chignolin and CLN025, reported in the PDB (PDB ID 2rvd)37 and BMRB (BMRB code: 5694),13 respectively, are also listed in Tables S3 and S4.† These values were highly correlated with our assignment, although the temperature and buffer conditions were not identical. The correlation of HN and Hα chemical shifts for CLN025, T8P (trans), and T8P (cis) against those of chignolin are shown in Fig. 12. The correlation between CLN025 and chignolin was higher than that for T8P (trans) or T8P (cis). This implies that the T8P mutant has different structures from chignolin, whereas CLN025 has a similar structure.
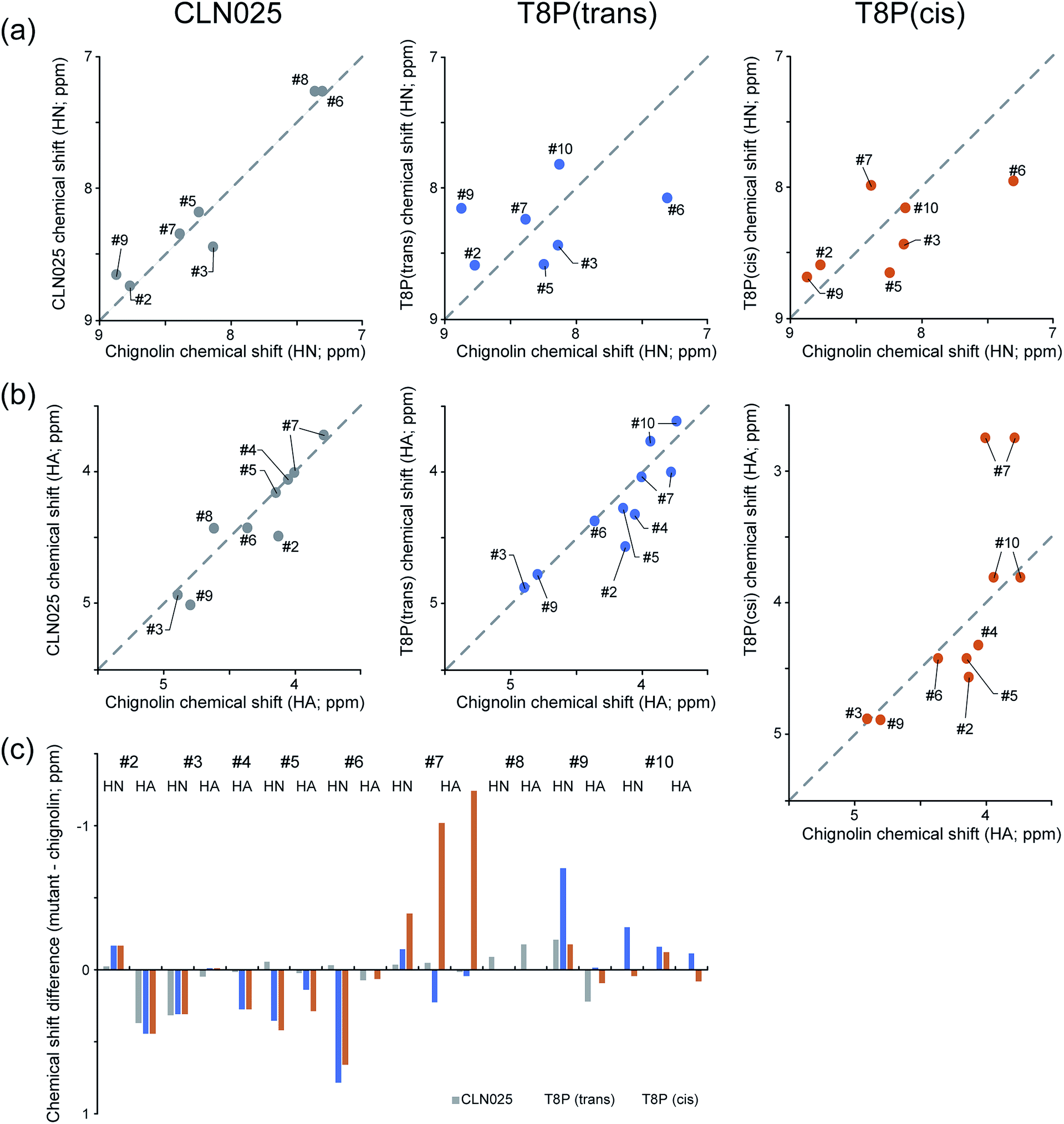 | ||
| Fig. 12 NMR analysis of chignolin, CLN025, and T8P. HN (a) and HA (b) chemical shifts of CLN025 and the T8P mutant (vertical axis) against those of chignolin (horizontal axis). Difference in HN and HA chemical shifts of CLN025 (gray) and the T8P(trans) (blue), T8P(cis) (orange) against chignolin (c). T8P takes trans and cis conformations in the Gly7–Pro8 sequence and these two conformational states are separately analyzed. | ||
The differences in the HN and Hα chemical shifts of CLN025, T8P (trans), and T8P (cis) from those of chignolin are shown in Fig. 12c. For CLN025, the differences in chemical shifts were small, particularly from Hα of Asp3 to Hα of Gly7 (see the gray bars in Fig. 12c). There were differences near the terminal residues. There are three possible reasons for these differences. CLN025 contains mutations in two amino acids at both terminals (G1Y and G10Y). Because tyrosine has an aromatic ring, the chemical shifts of atoms at the terminals, which are located near aromatic compounds, are affected. The second reason is the difference in the fluctuation of terminal residues. (In Fig. S12,† the backbone structures of chignolin (a) and CLN025 (b) obtained in previous NMR experiments (PDB ID 1uao and 2rvd) are shown.13,37) The terminal residues of chignolin fluctuated by more than those of CLN025. In this case, the different results for HN and Hα of Asp3 can also be explained because HN of Asp3 is inside of the structure and thus more affected by residues Thr8 and Phe9 compared to Hα of Aps3. The third possible reason is the difference in the population of the native and misfolded states. Because of the difference in the dihedral angle of Gly7, for the misfolded structure, the orientations of HN and Hα atoms from Thr8 were reversed from the native structure. (In Fig. S13,† the structures without hydrogen atoms of the native and misfolded states obtained by our MD simulations are shown.). The chemical shifts of Tyr2, Thr8, and Phe9 may be influenced by these differences. Although the chemical shifts near the terminal atoms were different, the differences in chemical shifts from Hα of Asp3 to Hα of Gly7 were small.
The structure of CLN025 is similar to that of chignolin. In contrast, the chemical shifts of the T8P mutant and chignolin exhibited large differences, as shown in Fig. 12c (see blue and orange bars). This indicates that both T8P (trans) and T8P (cis) have different structures from chignolin. For the difference between T8P (trans) and T8P (cis), their chemical shifts were similar to those of Tyr2 to Thr6, whereas those of Gly7 to Phe9 were different, supporting our simulation results for T8P as shown in Fig. 7 (T8P) and Fig. 11. Although the misfolded and native structures showed a similar turn structure from Asp3 to Thr6, there were still small differences, and the arrangements of the Tyr2 and Phe9 aromatic rings were different. As the aromatic rings can cause chemical shift changes to rather remote sites, it is possible that the residues from Asp3 to Thr6 have different chemical shifts for the misfolded and native structures. Thus, our experimental results indicate the existence of the misfolded state for the T8P mutant.
4 Conclusions
We previously suggested the possibility of stabilizing the misfolded structure of chignolin by mutating Thr8 to neutral amino acids. Herein, we investigated the effects of mutations on chignolin stability by performing MD simulations. All the mutations resulted in the formation of misfolded and/or native structures. The five mutants (T8I, T8F, T8P, T8N, and T8Y mutants) did not form the native structure, and instead favored the misfolded structure at room temperature. Among them, the T8P mutant formed the misfolded structure even at high temperatures. In the simulation of the T8P mutant at high temperature, structures with the cis conformation in the Gly7–Pro8 sequence were also obtained, whereas those with the trans conformation corresponded to the misfolded state. Energy analysis, in which the total energy was calculated based on the conformational energy and SFE using the 3D-RISM theory with the RMDFT function, also supported the MD simulation results. Mutation of threonine to proline at residue 8 inhibits formation of the native structure because of the distinctive cyclic structure of proline. These five mutations, particularly the T8P mutant, cause chignolin to primarily adopt the misfolded structure.To confirm our simulation results, NMR experiments of chignolin, CLN025, and T8P mutant were performed. The results showed that two conformations with trans and cis configurations in the Gly7–Pro8 sequence were obtained for the T8P mutant. The differences in chemical shifts between chignolin and T8P (trans) were large, whereas those between chignolin and CLN025 were small. This indicates that T8P (trans) has a different structure from the native state. The results of the NMR experiments support our simulation results.
The force field and sampling dependences were used to estimate protein stabilities using computational approaches. In this case, the ff99SB force field may have a higher population of the misfolded state.33 However, a tendency for which stability depends on amino acid mutation may be obtained. Chignolin has a typical stable π-turn structure. Stable turn structures are important in the early folding events of the whole domain and for binding to the other proteins. The stability dependence of the stable π-turn on i + 5th residue, where the ith residue corresponds to Asp3 for chignolin, may be informative for designing turn structures. Moreover, simulations can predict the existence of some possible states. To compare the results of simulations and NMR experiments, both techniques and interpretations of results should be improved.
The structural stabilities of proteins are important for understanding protein-folding mechanisms. Furthermore, studies evaluating how mutations affect structural stability are critical for understanding changes in protein function. Structural information on meta-stable states is useful for altering the relative stability between the native and meta-stable structures and for designing new structures. Recently, it has become possible to perform an extensively long MD simulation. Thus, dynamic analysis methods for identifying characteristic states such as native and meta-stable structures are required. RMA effectively identifies stable and meta-stable structures from simulations with large structural changes. Even when structural information on stable and meta-stable structures is obtained, it is necessary to evaluate the stability at the amino acid level including solvent effects. The energy analysis method using the 3D-RISM theory evaluates the stabilities of characteristic structures including solvent effects at the amino acid level. Using these powerful analysis methods, we suggested possible mutations in chignolin that can stabilize the misfolded structure (i.e., a meta-stable state).10 NMR experiments supported our results.
Identifying misfolded or meta-stable structures is difficult using experimental approaches, as they measure the physical quantities over ensemble properties. Meta-stable structures sometimes provide stability and rate-limiting folding processes. Misfolded and meta-metastable structures can be identified using mutation-based techniques. One strategy for detecting misfolded or meta-stable structures is to make the misfolded state more stable than the native state by introducing mutations, which can be achieved using our computational approaches. In addition, stabilization of the misfolded and meta-stable structures may be useful for designing new structures. In future studies, we will apply these approaches to larger proteins and design new structures based on information of meta-stable structures.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Japan Science and Technology Agency, Precursory Research for Embryonic Science and Technology (JST PRESTO) (JPMJPR13LB, JPMJPR14L5), and the Grant-in-Aid for Scientific Research (B) (20H03230). Numerical calculations were performed in part using Cygnus at the Center for Computational Sciences, University of Tsukuba. Molecular graphics were designed using UCSF Chimera, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.61References
- S. Yasuda, Y. Kajiwara, Y. Takamuku, N. Suzuki, T. Murata and M. Kinoshita, J. Phys. Chem. B, 2016, 120, 3833–3843 CrossRef CAS PubMed.
- Y. Kajiwara, S. Yasuda, Y. Takamuku, T. Murata and M. Kinoshita, J. Comput. Chem., 2017, 38, 211–223 CrossRef CAS PubMed.
- S. Yasuda, Y. Kajiwara, Y. Toyoda, K. Morimoto, R. Suno, S. Iwata, T. Kobayashi, T. Murata and M. Kinoshita, J. Phys. Chem. B, 2017, 121, 6341–6350 CrossRef CAS PubMed.
- B. Kuhlman and D. Baker, Curr. Opin. Struct. Biol., 2004, 14, 89–95 CrossRef CAS PubMed.
- M. Karplus and G. A. Petsko, Nature, 1990, 347, 631–639 CrossRef CAS PubMed.
- P. L. Freddolino, F. Liu, M. Gruebele and K. Schulten, Biophys. J., 2008, 94, L75–L77 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, R. O. Dror and D. E. Shaw, Science, 2011, 334, 517–520 CrossRef CAS PubMed.
- Y. Maruyama and A. Mitsutake, J. Phys. Chem. B, 2017, 121, 9881–9885 CrossRef CAS PubMed.
- S.-H. Chong and S. Ham, J. Chem. Phys., 2011, 135, 034506 CrossRef PubMed.
- Y. Maruyama and A. Mitsutake, J. Phys. Chem. B, 2018, 122, 3801–3814 CrossRef CAS PubMed.
- A. Lewandowska, S. Ołdziej, A. Liwo and H. A. Scheraga, Biophys. Chem., 2010, 151, 1–9 CrossRef CAS PubMed.
- F. Blanco, G. Rivas and L. Serrano, Nat. Struct. Mol. Biol., 1994, 1, 584–590 CrossRef CAS PubMed.
- S. Honda, K. Yamasaki, Y. Sawada and H. Morii, Structure, 2004, 12, 1507–1518 CrossRef CAS PubMed.
- B. Dasgupta, L. Pal, G. Basu and P. Chakrabarti, Proteins, 2004, 55, 305–315 CrossRef CAS PubMed.
- B. Dasgupta and P. Chakrabarti, BMC Struct. Biol., 2008, 8, 39 CrossRef PubMed.
- D. van der Spoel and M. Seibert, Phys. Rev. Lett., 2006, 96, 238102 CrossRef PubMed.
- W. Xu, T. Lai, Y. Yang and Y. Mu, J. Chem. Phys., 2008, 128, 175105 CrossRef PubMed.
- M. Zacharias, J. Chem. Theory Comput., 2008, 4, 477–487 CrossRef CAS PubMed.
- B. L. Kier and N. H. Andersen, J. Am. Chem. Soc., 2008, 130, 14675–14683 CrossRef CAS PubMed.
- S. Roy, S. Goedecker, M. J. Field and E. Penev, J. Phys. Chem. B, 2009, 113, 7315–7321 CrossRef CAS PubMed.
- K. Moritsugu, T. Terada and A. Kidera, J. Chem. Phys., 2010, 133, 224105 CrossRef PubMed.
- T. Yamazaki and A. Kovalenko, J. Phys. Chem. B, 2011, 115, 310–318 CrossRef CAS PubMed.
- R. Harada and A. Kitao, J. Phys. Chem. B, 2011, 115, 8806–8812 CrossRef CAS PubMed.
- H. Okumura, Proteins, 2012, 80, 2397–2416 CrossRef CAS PubMed.
- R. Harada, T. Nakamura, Y. Takano and Y. Shigeta, J. Comput. Chem., 2015, 36, 97–102 CrossRef CAS PubMed.
- R. Harada, Y. Takano and Y. Shigeta, J. Comput. Chem., 2015, 36, 763–772 CrossRef CAS PubMed.
- A. Mitsutake and H. Takano, J. Chem. Phys., 2015, 143, 124111 CrossRef PubMed.
- Y. Nishimoto and D. G. Fedorov, Phys. Chem. Chem. Phys., 2016, 18, 22047–22061 RSC.
- H. Fujisaki, K. Moritsugu, A. Mitsutake and H. Suetani, J. Chem. Phys., 2018, 149, 134112 CrossRef PubMed.
- D. Satoh, K. Shimizu, S. Nakamura and T. Terada, FEBS Lett., 2006, 580, 3422–3426 CrossRef CAS PubMed.
- A. Suenaga, T. Narumi, N. Futatsugi, R. Yanai, Y. Ohno, N. Okimoto and M. Taiji, Chem.–Asian J., 2007, 2, 591–598 CrossRef CAS PubMed.
- A. Kitao, J. Chem. Phys., 2011, 135, 045101 CrossRef PubMed.
- P. Kührová, A. De Simone, M. Otyepka and R. B. Best, Biophys. J., 2012, 102, 1897–1906 CrossRef PubMed.
- Q. Shao, J. Phys. Chem. B, 2015, 119, 3893–3900 CrossRef CAS PubMed.
- V. Pavone, G. Gaeta, A. Lombardi, F. Nastri, O. Maglio, C. Isernia and M. Saviano, Biopolymers, 1996, 38, 705–721 CrossRef CAS PubMed.
- M. Yamauchi and H. Okumura, J. Chem. Phys., 2017, 147, 184107 CrossRef PubMed.
- S. Honda, T. Akiba, Y. S. Kato, Y. Sawada, M. Sekijima, M. Ishimura, A. Ooishi, H. Watanabe, T. Odahara and K. Harata, J. Am. Chem. Soc., 2008, 130, 15327–15331 CrossRef CAS PubMed.
- M. P. D. Hatfield, R. F. Murphy and S. Lovas, J. Phys. Chem. B, 2010, 114, 3028–3037 CrossRef CAS PubMed.
- Y. Maruyama, H. Takano and A. Mitsutake, Biophys. Physicobiol., 2019, 16, 407–429 CrossRef PubMed.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Cohlke, A. W. Goetz, N. Homeyer, et al., AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- W. Jorgensen, J. Chandrasekhar, J. Madura, R. Impey and M. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. Kamiya and Y. Sugita, J. Chem. Phys., 2018, 149, 072304 CrossRef PubMed.
- J. Jung, T. Mori, C. Kobayashi, Y. Matsunaga, T. Yoda, M. Feig and Y. Sugita, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 310–323 CAS.
- C. Kobayashi, J. Jung, Y. Matsunaga, T. Mori, T. Ando, K. Tamura, M. Kamiya and Y. Sugita, J. Comput. Chem., 2017, 38, 2193–2206 CrossRef CAS PubMed.
- T. Imai, Y. Harano, M. Kinoshita, A. Kovalenko and F. Hirata, J. Chem. Phys., 2006, 125, 024911 CrossRef PubMed.
- Y. Maruyama and Y. Harano, Chem. Phys. Lett., 2013, 581, 85–90 CrossRef CAS.
- S.-H. Chong and S. Ham, J. Phys. Chem. B, 2014, 118, 5017–5025 CrossRef CAS PubMed.
- D. Beglov and B. Roux, J. Phys. Chem. B, 1997, 101, 7821–7826 CrossRef CAS.
- A. Kovalenko and F. Hirata, Chem. Phys. Lett., 1998, 290, 237–244 CrossRef CAS.
- A. Kovalenko and F. Hirata, J. Phys. Chem. B, 1999, 103, 7942–7957 CrossRef CAS.
- T. Sumi, A. Mitsutake and Y. Maruyama, J. Comput. Chem., 2015, 36, 1359–1369 CrossRef CAS PubMed.
- B. Pettitt and P. Rossky, J. Chem. Phys., 1982, 77, 1451–1457 CrossRef CAS.
- A. Kovalenko and F. Hirata, J. Chem. Phys., 2000, 112, 10391–10402 CrossRef CAS.
- Y. Maruyama and F. Hirata, J. Chem. Theory Comput., 2012, 8, 3015–3021 CrossRef CAS PubMed.
- J. McMurry and M. Castellion, Fundamentals of Organic and Biological Chemistry, Prentice Hall, New Jersey, 2nd edn, 1999 Search PubMed.
- B. L. Sibanda, T. L. Blundell and J. M. Thornton, J. Mol. Biol., 1989, 206, 759–777 CrossRef CAS PubMed.
- V. Pavone, G. Gaeta, A. Lombardi, F. Nastri and O. Maglio, Biopolymers, 1996, 38, 705–721 CrossRef CAS PubMed.
- K. Gunasekaran, C. Ramakrishnan and P. Balaram, Protein Eng., Des. Sel., 1997, 10, 1131–1141 CrossRef CAS PubMed.
- K. S. Rotondi and L. M. Gierasch, Biopolymers, 2006, 84, 13–22 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01148g |
| This journal is © The Royal Society of Chemistry 2020 |