
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic core–shell Fe3O4@Cu2O and Fe3O4@Cu2O–Cu materials as catalysts for aerobic oxidation of benzylic alcohols assisted by TEMPO and N-methylimidazole†
Binyu Xua,
Samuthirarajan Senthilkumarb,
Wei Zhong*b,
Zhongquan Shenb,
Chunxin Lub and
Xiaoming Liu *ab
*ab
aSchool of Chemistry, Nanchang University, Nanchang, Jiangxi, China. E-mail: xiaoming.liu@mail.zjxu.edu.cn
bCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, Zhejiang, China. E-mail: weizhong@mail.zjxu.edu.cn
First published on 10th July 2020
Abstract
In this work, core–shell Fe3O4@Cu2O and Fe3O4@Cu2O–Cu nanomaterials for aerobic oxidation of benzylic alcohols are reported with 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and N-methylimidazole (NMI) as the co-catalysts. To anchor Cu2O nanoparticles around the magnetic particles under solvothermal conditions, the magnetic material Fe3O4 was modified by grafting a layer of L-lysine (L-Lys) to introduce –NH2 groups at the surface of the magnetic particles. With amine groups as the anchor, Cu(NO3)2 was used to co-precipitate the desired Cu2O by using ethylene glycol as the reducing agent. Prolonging the reaction time would lead to over-reduced forms of the magnetic materials in the presence of copper, Fe3O4@Cu2O–Cu. The nanomaterials and its precursors were fully characterized by a variety of spectroscopic techniques. In combination with both TEMPO and NMI, these materials showed excellent catalytic activities in aerobic oxidation of benzylic alcohols under ambient conditions. For most of the benzylic alcohols, the conversion into aldehydes was nearly quantitative with aldehydes as the sole product. The materials were recyclable and robust. Up to 7 repeat runs, its activity dropped less than 10%. The over-reduced materials, Fe3O4@Cu2O–Cu, exhibited slightly better performance in durability. The magnetic properties allowed easy separation after reaction by simply applying an external magnet.
1. Introduction
The selective oxidation of alcohols to aldehydes is an important organic transformation in the pharmaceutical industry as well as in other organic synthesis.1 Of the aldehydes, aromatic aldehydes are often used for the synthesis of drugs, vitamins, dyes, spices, and resins and so on.2 To date, many homogeneous as well as heterogeneous catalysts have been developed for the oxidation of alcohols to prepare aldehydes or ketones.3–7 Despite the progress achieved so far, the research in this area remains intensive due to existing challenges in this conversion such as harsh conditions, expensive metals, reusability of the catalysts. In nature, copper-based catalysis plays vital roles in biological systems.8 One of the examples is the copper-containing enzyme, galactose oxidase (GOase), which catalyses the aerobic oxidation of primary alcohols in some species of fungi under physiological conditions.9 Tremendous efforts have been taken to mimic the enzyme in both approaches, structurally and functionally mimicking based on various copper complexes.10–12 Homogeneous system reported by Semmelhack13 et al., Cu(I)/TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxyl), contributed significantly to this copper-based catalysis. Later on, Sheldon14 et al., Stahl and co-workers15–17 and other research groups reported a wide range of homogeneous copper catalysts which were based on the combination of various nitroxyl radicals and ligands for the oxidation of various alcohols.18–20 Some of the catalytic systems showed promising catalytic activity in aerobic oxidation of alcohols including aliphatic alcohols.21,22 One such example was CuI/TEMPO/NMI system which could effectively catalyse the aerobic oxidation of 1-octanol.23 However, there have been disadvantages for these systems one way or another, for example, tedious synthesis when specific ligand is needed, problems in catalyst recovery and reusability. To address these issues, a variety of heterogeneous catalysts supported on matrices have been developed. A wide range of materials such as clay, zeolite, metal organic frameworks, polymers, activated carbon, silicates, magnetic materials etc., have been employed as such a matrix.24,25About two decades ago, a pioneering work by Ley et al. demonstrated a polymer-supported perruthenate for the aerobic oxidation of alcohols.26 Further, they found that mesoporous silicate (MCM-41) could be an alternative support for the catalyst.27 Polyoxometalates was also turned out a good carrier to support palladium nanocatalyst for the aerobic oxidation of secondary alcohols in aqueous medium and the catalyst was recoverable.28 Polystyrene was reported as a matrix to support gold–platinum nanoparticles for the aerobic oxidation of 1-phenylethanol under mild conditions.29 Although the noble metal catalysts such as Ru, Pt, Pd and Au supported on matrices showed excellent efficiency and selectivity for this aerobic oxidation,30 it is desirable to develop non-noble metal catalysts in terms of both cost and sustainability. Recent developments in this regard have suggested that employing non-precious metals to assemble catalytic systems is possible. For example, copper could be supported on MOF for aerobic oxidation of alcohols under various conditions.31,32 Montmorillonite was also used to support mettaloporphyrins (Co, Fe, Mn) for efficient catalysis on the oxidation of both aromatic and aliphatic alcohols.33
Transition metal oxides, on one hand, could be the matrix to support catalysts. On the other hand, the metal sites of the oxides could be promising catalysts with adjustable electronic structures widely used in various organic transformations involving electron transfer reactions.34,35 Albadi et al. reported the first row transition-metals and its oxides that could be able to oxidize alcohols in presence of molecular oxygen under reflux conditions.36 For example, copper, the active site in galactose oxidase catalysing aerobic oxidation of primary alcohols into aldehydes, was employed in a number of heterogeneous catalysts such as Au–CuO,37 CuO–rectorite,38 Cu–MnOx39 and Cu–TiO2 (ref. 40) which catalysed effectively the selective oxidation of benzyl alcohols into aldehydes.
In viewing of the low cost of copper and its biological relevance, cuprous oxide combining with its magnetisation was considered to further to be exploited in heterogeneity as heterogeneous catalysts. Of the many magnetic materials,41–44 Fe3O4 is the most favourable option due to easy functionalization and low cost. Grafting a layer of organic modifiers or inorganic linkers such as polymers, organosilanes, aminoacids etc., onto its surface would help to protect the magnetite and allow easy access for the coordination of metal catalysts.45–47 Fe3O4@Cu2O showed photocatalytic activity in degradation of organic dyes as well as the synthesis of quinazolines.48,49 However, to our best knowledge, the employment of such system in aerobic oxidation of alcohols into aldehydes have not been reported. Being inspired by the copper chemistry on aerobic oxidation of alcohols in biology and the easy recovery property of the Fe3O4-based cuprous oxide, herein, we report the preparation of Fe3O4@Cu2O as well as its over-reduced form, Fe3O4@Cu2O–Cu, their characterisations and catalytic activity on selective aerobic oxidation of benzylic alcohols into benzylic aldehydes. With the presence of TEMPO and N-methylimidazole (NMI), the materials could oxidise nearly quantitatively most of the benzylic alcohols examined in this work into corresponding aldehydes at ambient temperature. The two materials exhibited easy separation and robustness. The loss in converting yields for two materials were less than 10% up to 7 runs with the over-reduced catalyst showing slightly better durability.
2. Results and discussion
2.1 Preparation and characterization of the nanomaterials
The preparation of core–shell nanomaterials is illustrated in Scheme 1. First, Fe3O4 nanoparticles with hydroxyl groups on their surfaces were grafted with L-Lys to introduce –NH2 groups that were used as anchors for the immobilization of the in situ formed Cu2O nanoparticles. The formation of materials of Fe3O4, Fe3O4-L-Lys and Fe3O4-L-Lys@Cu2O were stepwise confirmed using FTIR spectroscopy as shown in Fig. 1. The presence of hydroxyl groups on the surface of magnetite (Fe3O4) was evidenced by the strong absorption peak at 3423 cm−1 and a sharp peak at 616 cm−1 corresponding to the stretching vibration of Fe–O bond as shown in Fig. 1b. The L-lysine modified Fe3O4 was proved by the presence of a sharp peak at 1619 cm−1 (COO−), and a broad peak at 3435 (NH2) and 607 cm−1 (Fe–O), respectively, as shown in Fig. 2c. Finally, the in situ formed Cu2O on the surface of L-lysine modified Fe3O4 was confirmed by the presence of an additional sharp peak at 629 cm−1 which is assigned to the stretching vibrations of Cu–O as shown in Fig. 2d. These results suggested that the in situ formed Cu2O was successfully anchored to the surface of the magnetic particles through the coordinating interaction of the L-lysine with copper. EDX analysis showed additional evidence for the presence of Fe, Cu, N, C, O in the core–shell Fe3O4@Cu2O nanomaterials and the weight of copper present in the catalyst was about 49%, equivalent to 3.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in molar ratio for Cu:Fe (Table S1†).
:1 in molar ratio for Cu:Fe (Table S1†).
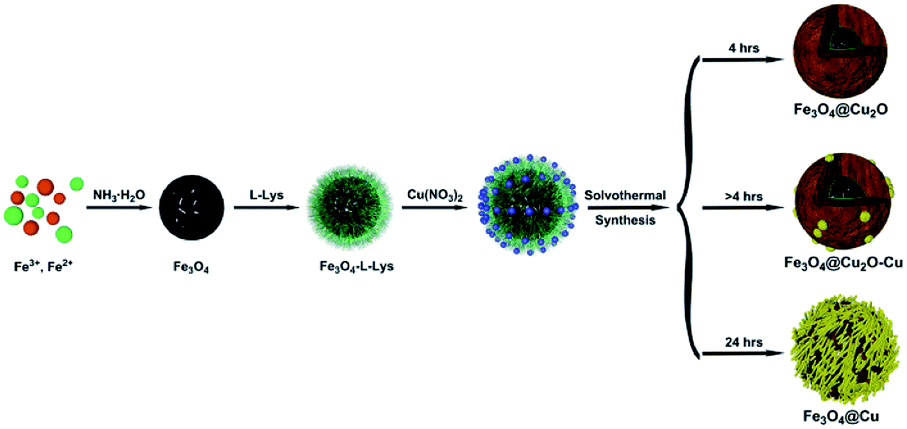 | ||
| Scheme 1 Synthetic route for the preparation of core–shell nanomaterials. | ||
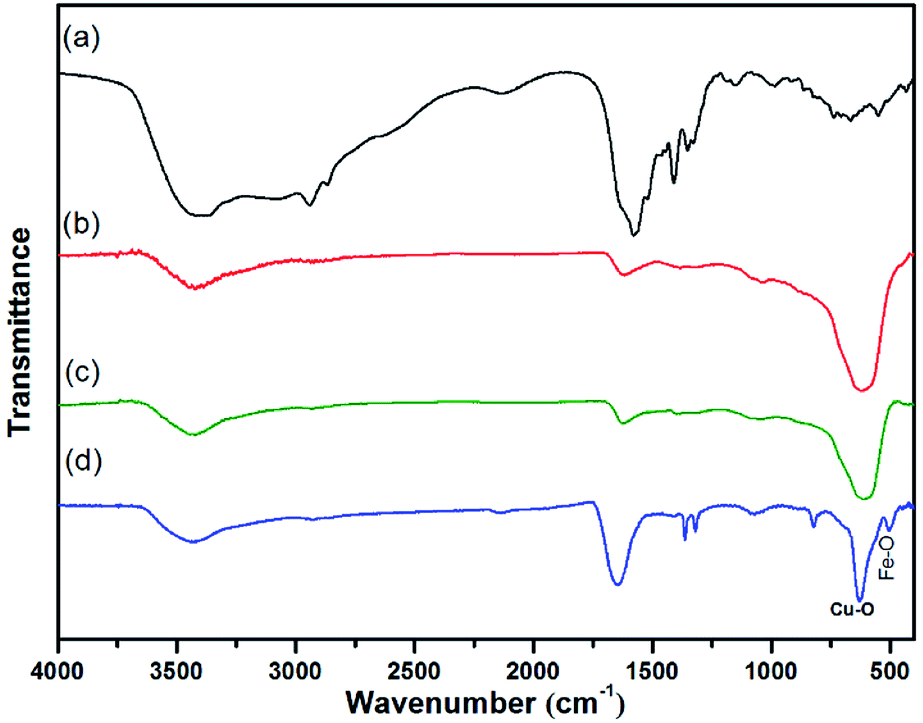 | ||
| Fig. 1 FT-IR spectra of (a) L-Lys, (b) Fe3O4, (c) Fe3O4–L-Lys and (d) Fe3O4@Cu2O. | ||
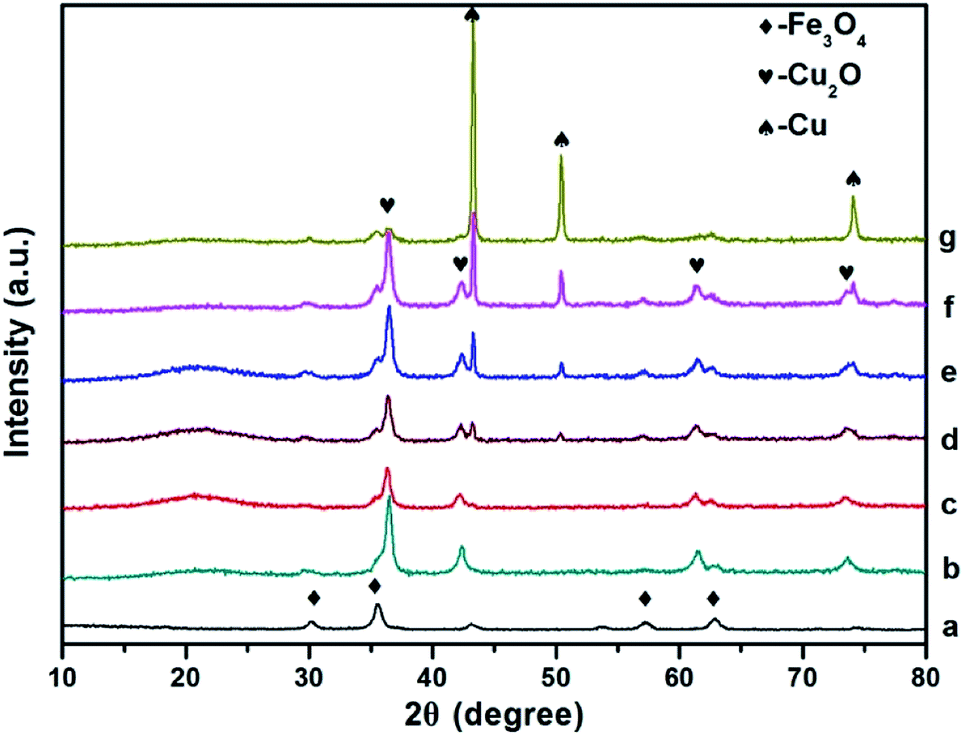 | ||
| Fig. 2 Comparison of p-XRD spectra of Fe3O4 (a), Fe3O4@Cu2O ((b), 4 h), Fe3O4@Cu2O–Cu nanomaterials ((c), 5 h; (d), 7 h; (e), 9 h; (f), 12 h) and Fe3O4@Cu ((g), 24 h). | ||
The powder XRD patterns of the as prepared Fe3O4@Cu2O/Fe3O4@Cu2O–Cu and the magnetic precursors are shown in Fig. 2. The observed peaks for the formation of Cu2O@Fe3O4 nanomaterials at 4 h are clearly matched with the PDF Card No. 19-0629 of Fe3O4 and PDF Card No. 05-0667 of Cu2O nanoparticles, respectively. Furthermore, no other peaks are observed around 20°, indicating that the products were purely face-centred cubic Fe3O4 crystalline phase. Prolonging the reaction time produced the over-reduced materials Fe3O4@Cu2O–Cu and Fe3O4@Cu, as indicated by the additional three diffraction peaks (Fig. 2, c–g) which belong to the planes of a cubic phase of Cu crystals (PDF Card No. 04-0836). Quantitative analysis to determine the ratio of Cu/Cu2O was carried out using calibration curve (Fig. S1†) established by employing the peak intensities at 36.4° and 43.3° from the powder XRD spectra of the standard samples, mechanically mixed Cu2O with Cu at various ratios. For example, the ratios of Cu/Cu2O were estimated as 0.32 mol% and 1.01 mol% for Fe3O4@Cu2O-5 and Fe3O4@Cu2O–Cu-7 materials, respectively (Table S2†), which suggest that the content of Cu increased continuously as the reaction time increased.
As shown in Fig. 3a, Fe3O4 exhibited a rough surface of many irregular spherical structures with an average diameter of 385 nm that changed remarkably after the deposition of Cu2O, Fig. 3b. The TEM images of the nanomaterial reveals much ordered structures of core–shell Fe3O4 which was surrounded by a layer of Cu2O nanospheres with an average thickness of 150 nm, Fig. 3c and d. The HR-TEM images confirm the coexistence of Fe3O4 and Cu2O (Fig. 3e) in which the pattern corresponds to the planes of Fe3O4 (220) and Cu2O (111), respectively. Moreover, the core–shell Fe3O4@Cu2O nanomaterial has an average diameter of about 700 nm that can be evidenced from the particle size distribution shown in Fig. 3f. The average particle size of Cu2O in the nanospheres on the surface of the core–shell Fe3O4 was estimated as 16 nm by using Scherrer's formula.42
 | ||
| Fig. 3 SEM images of the as prepared Fe3O4 (a), core–shell Fe3O4@Cu2O (b), HR-TEM images of core–shell Fe3O4@Cu2O nanomaterial (c–e) and particle size distribution analysis for the core–shell Fe3O4@Cu2O nanomaterial (f). | ||
In order to reveal the oxidation states of the components and their interactions with each other, XPS study was carried out for Fe3O4@Cu2O (Fig. 4a–c). In Fig. 4a, the presence of Fe, Cu and O elements in the core–shell nanomaterials were confirmed. The two bands at 711.4 and 723.7 eV shown in Fig. 4b are assigned to Fe2p3/2 and Fe2p1/2, respectively, corresponding to Fe2+ (FeO) and Fe3+ (Fe2O3) peaks.45 Intense core-level lines of Cu are shown as a Cu2p doublet (Cu2p1/2 and Cu2p3/2) between 920–970 eV (Fig. 4c). It shows two distinct peaks at 952.2 eV and 932.2 eV belonging to Cu2p3/2 and Cu2p1/2 of cuprous oxide, respectively.48 There is a main peak at 532 eV assigned to the lattice O in cuprous oxide. The existence of cuprous oxide was also proved by Auger spectroscopy,18 showing a sharp peak in Cu LMM triplet kinetic energy at 570.2 eV. Also, the characteristic peaks for the Fe3O4 and Cu2O in the over-reduced catalyst of Fe3O4@Cu2O–Cu-7 was clearly coincident with the XPS data of Fe3O4@Cu2O. In addition to those commonly observed peaks, a peak of weak intensity at 918.7 eV showed the existence of metallic Cu in the nanomaterial of Fe3O4@Cu2O–Cu-7 (4).
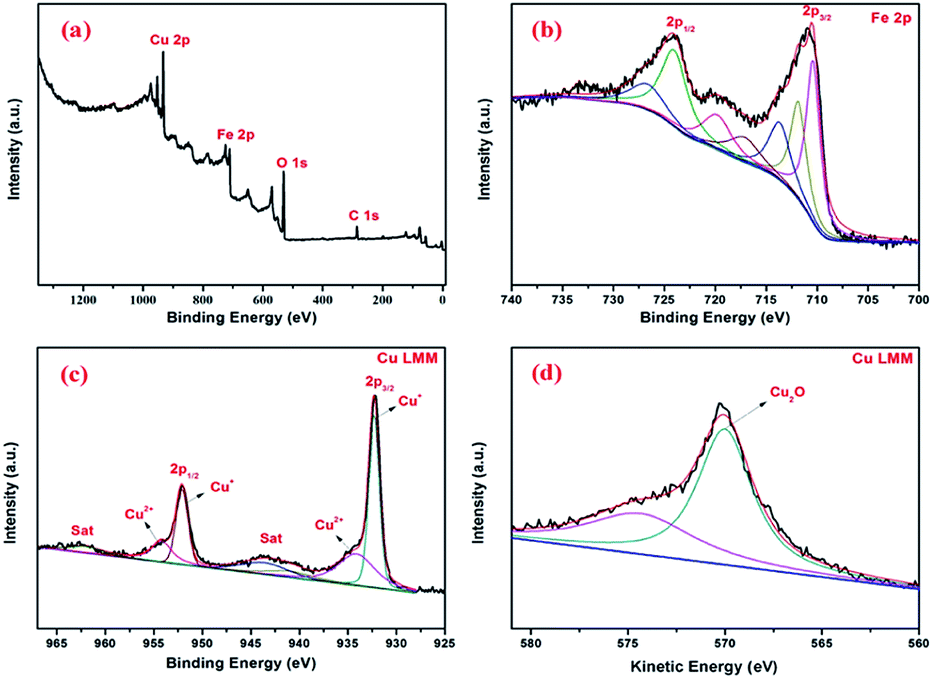 | ||
| Fig. 4 XPS spectra of core shell Fe3O4@Cu2O nanomaterial: (a) survey spectrum; (b) Fe2p; (c) Cu2p; (d) Cu LMM Auger spectra. | ||
The magnetic properties of the core–shell Fe3O4@Cu2O and its precursor was measured via the vibrating sample magnetometer (VSM). Fig. 5 shows the magnetic hysteresis curves of the Fe3O4 and core–shell Fe3O4@Cu2O nanomaterials, the saturation magnetization (Ms) values are 72.1 emu g−1 and 22.5 emu g−1, respectively. The low saturation magnetization value for Fe3O4@Cu2O was due to the increased content of the Cu2O nanospheres around the core–shell Fe3O4. This magnetic property allowed its easy separation in performing catalysis (Fig. 5, inset).
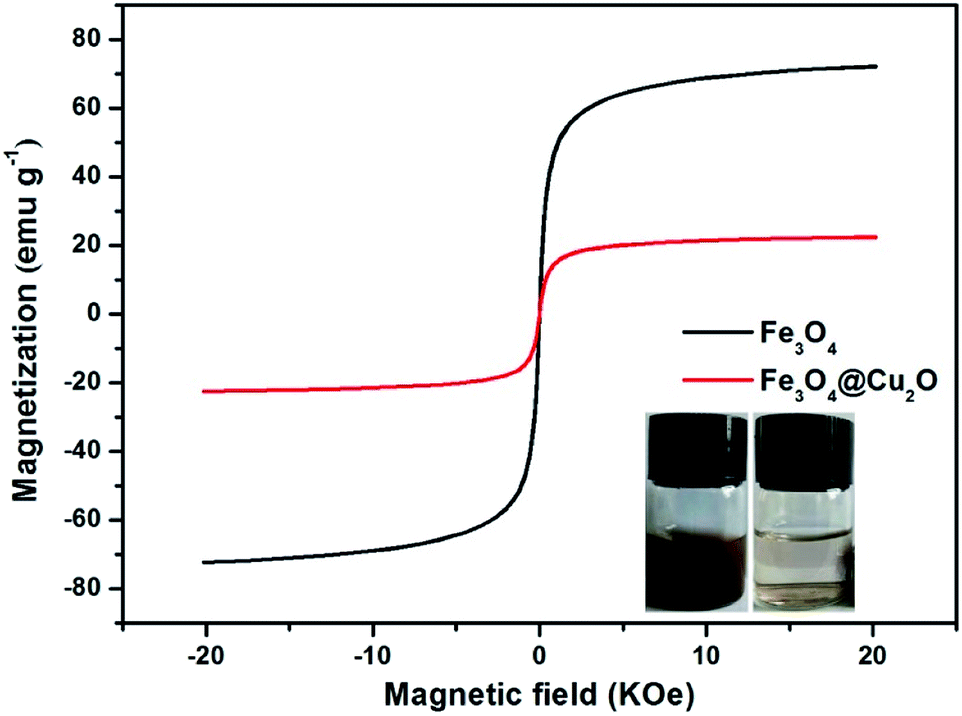 | ||
| Fig. 5 Magnetization curves of Fe3O4 and Fe3O4@Cu2O. | ||
2.2 Catalytic activity on the aerobic oxidation of alcohols and its possible mechanism
The aerobic oxidation of benzyl alcohol was chosen as a probing reaction to investigate the catalytic efficiency of the core–shell nanomaterials. The optimization studies were carried out with various factors and the results are shown in Table 1. It was clear that either NMI alone or its combination with TEMPO without the core–shell materials did not make the reaction proceed (Table 1, entries 1–4). In presence of copper catalysts such as Cu2O and metallic copper along with NMI and TEMPO, the reaction proceeds well and afforded good yields (Table 1, entries 5 & 6). These results suggested that the copper catalysts played a major role as an active site in the present catalytic system. The magnetic Fe3O4@Cu2O catalyst along with NMI and TEMPO yielded quantitatively benzaldehyde as the sole product (Table 1, entry 7). Replacing N-methylimidazole (NMI) with 1-butyl and 1-tert-butylimidazoles did not affect substantially the yield but the reaction rate slowed down (Table 1, entries 8 & 9). Compared to the Fe3O4@Cu2O, the over-reduced catalysts of Fe3O4@Cu2O–Cu with different contents of Cu showed essentially the same activities as Fe3O4@Cu2O (Table 1, entries 10–13). But in reaction rate, the presence of metallic copper made some differences. It seems that the increase in the ratio of metallic copper retarded the reaction at its initial stage although at the end of the reaction, the yield was not affected significantly. Further, the presence of the metal copper seems to enhance its robustness (vide infra). By increasing the temperature to 82 °C (Table 1, entry 15), the reaction rate was increased substantially in the initial stage and the reaction time can be shorted to 12 h (Fig. S3†).| Entry | Catalysts | TEMPO (mmol) | Ligand | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: alcohol (5 mmol), catalyst (15 mg), TEMPO (0.25 mmol), NMI (0.25 mmol), acetonitrile (5 mL), 25 °C, 18 h. For the sample coding, the suffix number x of the codes (entries 10–13) in Fe3O4@Cu2O–Cu-x denotes the reaction time in the catalyst preparation.b Results by GC analysis.c 0.14 mmol.d 0.07 mmol.e Yield at 12 h.f At 82 °C for 12 h. | ||||
| 1 | — | — | NMI | — |
| — | 0.25 | NMI | 1 | |
| 3 | Fe3O4 | 0.25 | NMI | 1 |
| 4 | Fe3O4@Cu2O | 0.25 | — | 1 |
| 5 | Cuc | 0.25 | NMI | 94 |
| 6 | Cu2Od | 0.25 | NMI | 90 |
| 7 | Fe3O4@Cu2O | 0.25 | NMI | 80e/>99 |
| 8 | Fe3O4@Cu2O | 0.25 | 1-Butyl imidazole | 99 |
| 9 | Fe3O4@Cu2O | 0.25 | 1-tert-Butyl imidazole | 96 |
| 10 | Fe3O4@Cu2O–Cu-5 | 0.25 | NMI | 78e/>99 |
| 11 | Fe3O4@Cu2O–Cu-7 | 0.25 | NMI | 68e/>99 |
| 12 | Fe3O4@Cu2O–Cu-9 | 0.25 | NMI | 51e/>99 |
| 13 | Fe3O4@Cu2O–Cu-12 | 0.25 | NMI | 46e/98 |
| 14 | Fe3O4@Cu | 0.25 | NMI | 60e/91 |
| 15 | Fe3O4@Cu2Of | 0.25 | NMI | >99 |
In order to check the influence of a solvent on the catalysis, various protic and aprotic solvents were employed under similar conditions and the results are presented in Table 2. The conversion results are more favourable in less-polar solvents like Toluene, THF, DCM and CH3CN than polar solvents such as H2O, DMF, and DMSO (Table 2, entries 1–7). Especially, in acetonitrile, the reaction proceeded well and afforded excellent yield (>99%), which agrees with what was observed in homogenous catalysis by CuI/NMI/TEMPO.23 Based on these results, acetonitrile was employed as the solvent medium throughout the study.
2.3 Heterogeneity and reusability of the catalysts
To check the heterogeneity of the catalyst, a set of normal reaction with optimized conditions were performed and the progress of the reaction was monitored for every 2 h. After 10 hours, the catalyst was removed from one of the experiments to allow the reaction to continue under the same conditions. At the end of the reaction, there was a slight increase in the yield without the presence of the catalyst, which was contributed to the bleached copper in the solution (Fig. 6).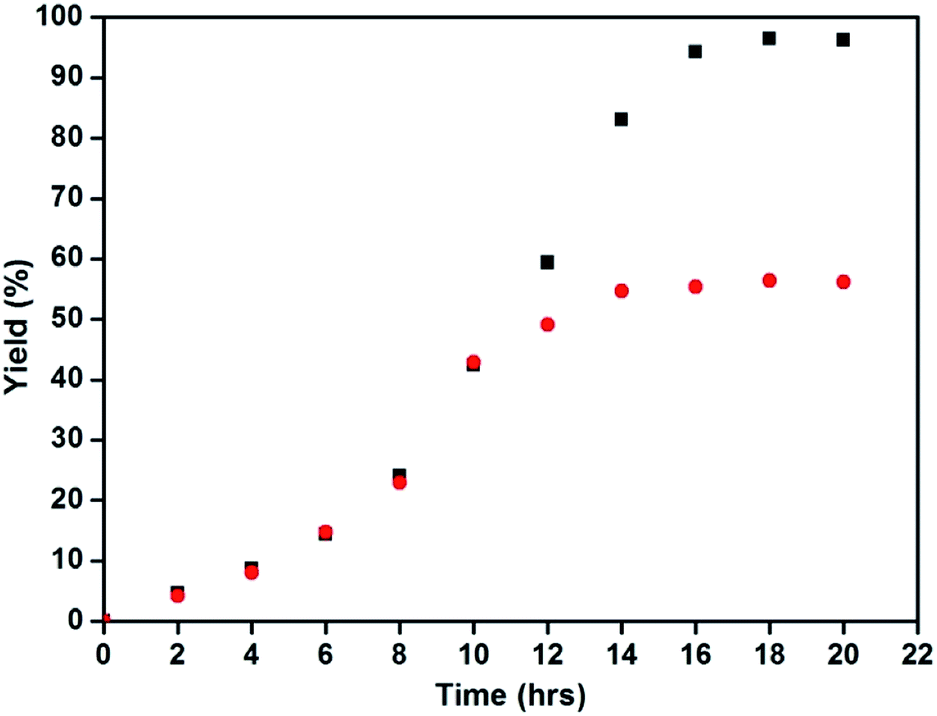 | ||
| Fig. 6 Heterogeneity test of the Fe3O4@Cu2O catalyst (black: reaction proceeded as usual and red: reaction proceeded after Fe3O4@Cu2O was removed at 10 h). | ||
The reusability of the catalyst was examined for the selective oxidation of benzyl alcohol. After the completion of reaction, the catalyst was separated by applying an external magnet and washed three times with acetonitrile, dried under vacuum at 60 °C for 12 hours before being used for next run under the same reaction conditions. Fig. 7 (left) shows the reusability of Fe3O4@Cu2O catalyst and it remained substantially active for over eight times. A slight decrease in catalytic activity after five repetitive runs was due to the loss of Cu2O via chemically bleaching. ICP analysis suggested that the copper being bleached into the solution accounted for 5.8% of the total copper content. As shown in Fig. 7 (right), the over-reduced catalysts, for example, Fe3O4@Cu2O–Cu-7, exhibited much better stability. But at this stage, we do not understand how the co-existence of minimum amount of metallic copper in the catalyst could enhance its robustness. The p-XRD and XPS studies of the fresh and recycled catalysts of Fe3O4@Cu2O–Cu-7 did not surrender much information on this matter (Fig. S4 and S5†).
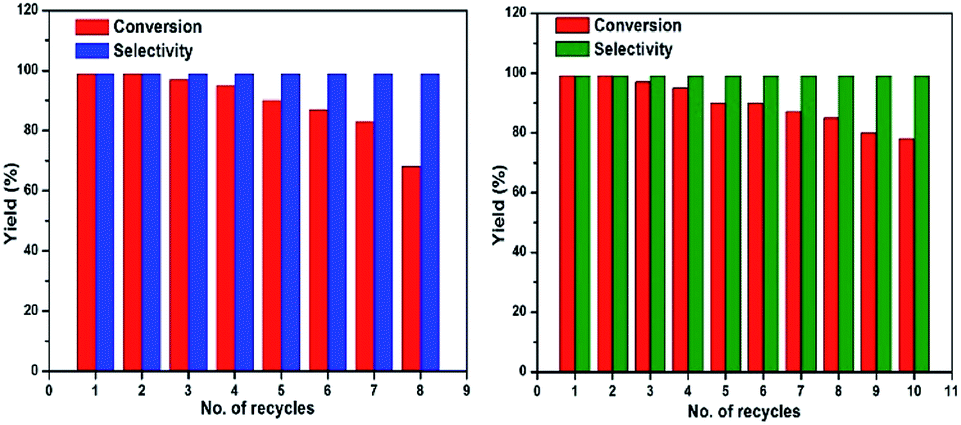 | ||
| Fig. 7 Reusability of Fe3O4@Cu2O (left) and reusability of Fe3O4@Cu2O–Cu-7 (right). | ||
Based on the previous reported literatures,14,23 a simplified mechanism for this catalytic system was proposed in Scheme 2. First, O2 is activated by Cu(I) and the metal site is oxidised to Cu(II). The involvement of NMI may enhance the reducing capability via coordination to the metal site and acts probably as a base as well. Then, the substrate is adsorbed on the surface at CuII species, followed by deprotonation of the bound alcohol. This process is similar to the reported homogenous catalytic system, in which the hydroxyl group formed from the reduction of O2 acted as an internal base and deprotonated readily the proton of the bound substrate to form alkoxide-bound adduct.15,17,23 Finally, the abstraction of α-hydrogen from the substrate by TEMPO led to the desired carbonyl compound and regenerated Cu(I) species. Meanwhile, the production of TEMPOH could act as hydrogen source for next catalytic cycle supported by the evidence that the employment of TEMPOH, the reduced form of TEMPO, as the co-catalyst showed better catalytic activity than TEMPO in the catalysis at the initial stage (Fig. S6†).
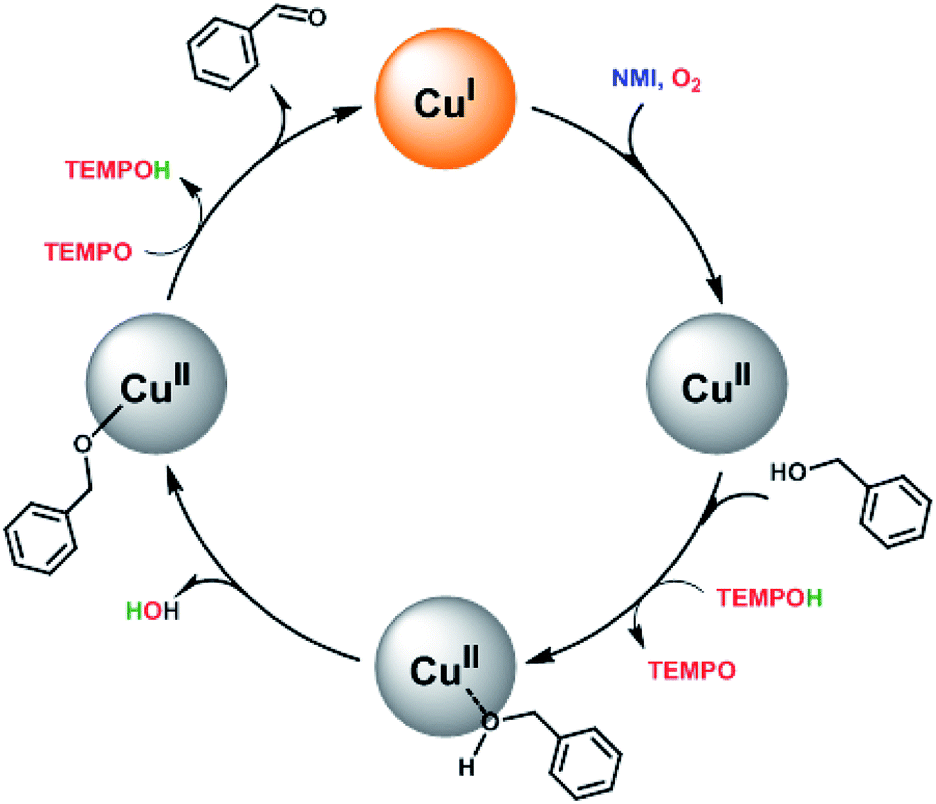 | ||
| Scheme 2 A possible mechanism for the aerobic oxidation of benzylic alcohols catalyzed by the magnetic core–shell materials with both TEMPO and NMI as the co-catalysts. | ||
This proposed mechanism was further confirmed by the results obtained from p-XRD and XPS studies of the recycled catalysts. The p-XRD data of the recycled catalysts (Fig. S2†) shown that the peak intensity of Cu2O was weakened, hence there were no characteristic peaks of CuO in the recycled catalysts (4th and 7th run). But the XPS spectra of the recycled catalysts of Fe3O4@Cu2O (4th & 7th recycle) revealed the formation of CuO by the presence of two additional peaks with binding energies of 934.20 eV and 954.14 eV respectively (Fig. 8, top left). It was further supported by the Cu LMM auger spectrum with the presence of additional peak at 568.5 eV for CuO along with Cu2O (Fig. 8, Top right).
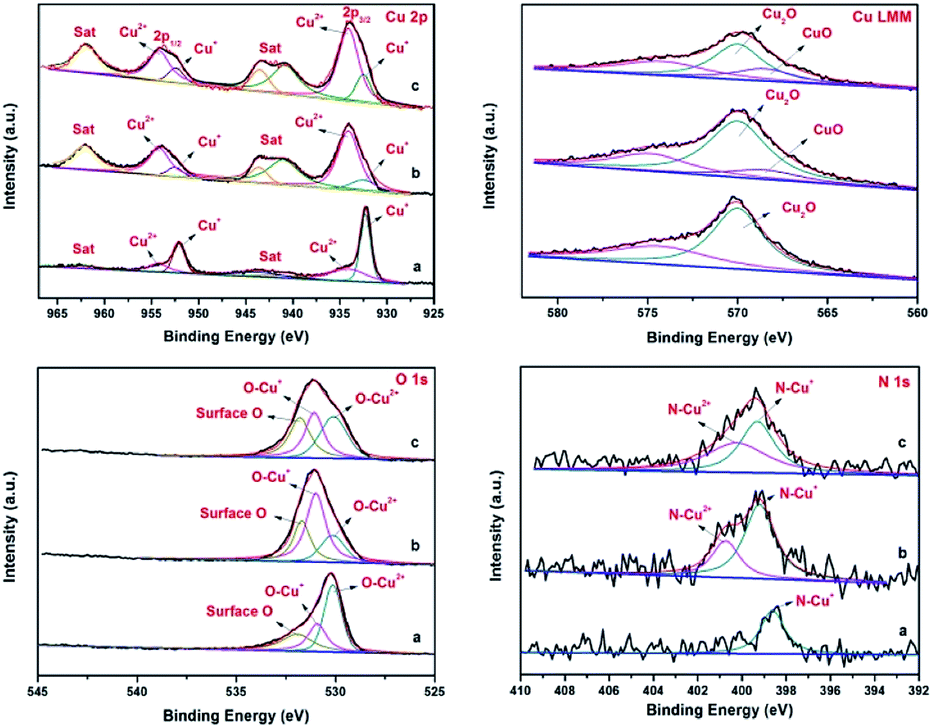 | ||
| Fig. 8 Comparison of XPS spectra of fresh and recycled Fe3O4@Cu2O catalysts: Cu2p (Top left), Cu LMM (Top right), O1s (Bottom left) and N1s (Bottom right) spectrum; (a) fresh catalyst, (b) 4th recycling and (c) 7th recycling. | ||
In the O1s core level, there is an increase in peak areas of O–Cu2+ and surface-oxygen were observed for consecutive runs due to the surface oxidation of cuprous oxide (Fig. 8, bottom left). Analogously, a slight increase in the binding energy of N1s was also observed with the formation of N–Cu2+ at 400.7 eV in the recycled catalysts (Fig. 8, bottom right). The XPS studies of the recycled Fe3O4@Cu2O–Cu-7 also reveals the above same information except the formation of instable intermediate Cu(OH)2 species (Fig. S7,† top right). In the O1s spectrum, the intensity of O–Cu+ and O–Cu2+ was slightly lower than that of surface oxygen (Fig. S7,† bottom left). Perhaps, the oxidation of metallic copper present in the catalyst might be a reason for the enhancement of reusability and robustness of the over-reduced Fe3O4@Cu2O–Cu-7 catalyst.
2.4 Substrate scope
To explore the applicable scope of the catalysts, various alcoholic substrates were tested under the optimized reaction conditions and the results are summarized in Table 3. In most cases, selective oxidation of benzyl alcohols into corresponding aldehydes proceeded quantitatively with aldehydes as the sole products. Electron-donating groups like methyl-substituted benzyl alcohols showed slightly differentiated results among para-, meta- and ortho-positions (Table 3, entries 2–4), which could be contributed to both electronic and steric reasons. However, it is surprising that electron-withdrawing groups like chloro and nitro substituted benzyl alcohols did not show any negative effect on the reaction (Table 3, entries 5–8). In fact, on the contrary, it seems that the electron-withdrawing group promoted the catalysis. It is well known that during the conversion of an alcohol into its aldehyde, a number of steps are involved, for example, deprotonating the bound substrate before the first electron transfer and the abstraction of the hydrogen atom from the methylene group which accompanies the transfer of the second electron to complete the oxidation. These observations may imply that in the catalytic reaction, the deprotonation is one of the rate-determining steps and the abstraction of hydrogen atom from the methylene group is not, since electron-withdrawing group would increase the acidity of the substrate. For allylic alcohols, the catalyst showed also excellent efficiency (Table 3, entries 9 & 10). Furthermore, the catalyst was effective on some of the aerobic oxidation of heterocyclic alcohols (Table 3, entry 11). Interestingly, for pyridin-2-yl-methanol the activity dropped drastically (Table 3, entry 12). This may be due to that the nitrogen atom on the pyridyl group competed with the NMI for coordination which might block the catalytic sites and resulted in a very low yield, which agrees with what was observed in homogenous catalysis.23 Finally, it must be said that its activity on aerobic oxidation of aliphatic alcohols and secondary alcohols was low (Table 3, entries 14–16).| Entry | Substrate | Product | Selectivity (%) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: alcohol (5 mmol), Fe3O4@Cu2O (15 mg), TEMPO (0.25 mmol, 39.1 mg), NMI (0.25 mmol, 20 mg), CH3CN (5 mL), 25 °C, 18 h.b Confirmed by GC-MS analysis. | ||||
| 1 | 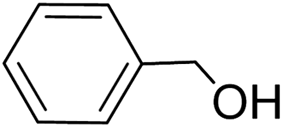 |
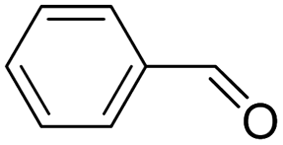 |
>99 | >99 |
| 2 | 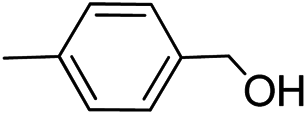 |
 |
>99 | >99 |
| 3 |  |
 |
>99 | 92 |
| 4 |  |
 |
>99 | 96 |
| 5 |  |
 |
>99 | >99 |
| 6 |  |
 |
>99 | >99 |
| 7 |  |
 |
>99 | >99 |
| 8 |  |
 |
>99 | >99 |
| 9 |  |
 |
>99 | >99 |
| 10 |  |
 |
>99 | >99 |
| 11 | 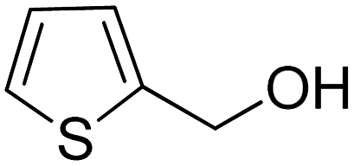 |
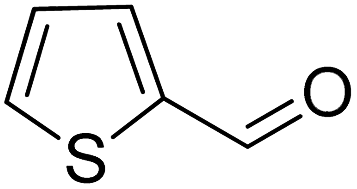 |
>99 | >99 |
| 12 | 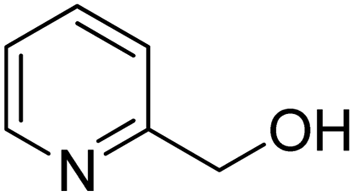 |
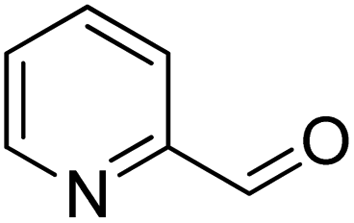 |
>99 | 14 |
| 13 | 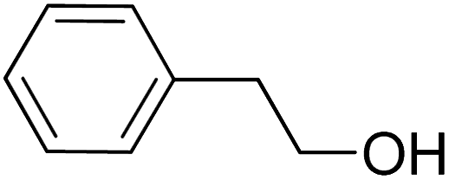 |
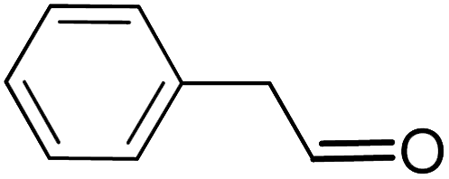 |
16.5 | 9 |
| 14 | 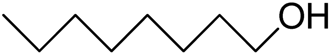 |
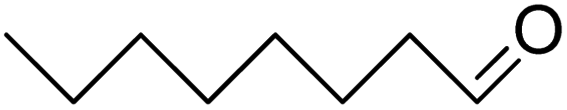 |
>99 | 5 |
| 15 | 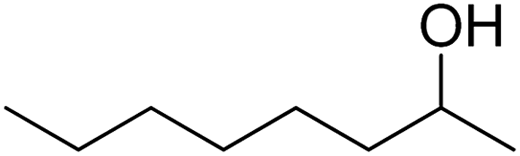 |
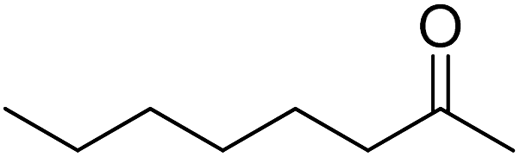 |
>99 | 3 |
| 16 | 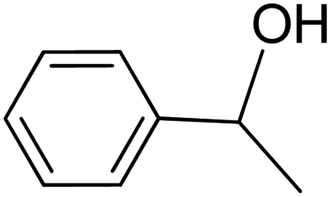 |
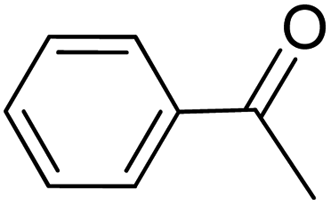 |
— | — |
Compared to those catalytic systems reported in the literatures (Table 4), the present catalytic system has the advantages, easy separation, functional group tolerance, using atmospheric air as the oxidant, operation at room temperature, good durability and reusability.
| Entry | Reaction conditions | Selectivity (%) | Yield (%) | Catalyst recycle | Ref. |
|---|---|---|---|---|---|
| 1 | Fe3O4@Cu2O, NMI, TEMPO, air, 25 °C, 18 h | >99 | 99 | Magnetisation | This work |
| 2 | Fe2O4–Co, H2O2, 110 °C, 5 h | 100 | 99 | Magnetisation | 42 |
| 3 | Fe3O4/Cys–Pd, O2, 55 °C, 1.5 h | >99 | 85 | Magnetisation | 43 |
| 4 | Fe3O4/MPA–PHEA-Cr, O2, 45 °C, 4 h | 99 | 60 | Magnetisation | 44 |
| 5 | CuO–rectorite, TEMPO, K2CO3, O2, 50 °C, 24 h | >99 | 85 | Centrifugation | 38 |
| 6 | Cu–MnOx, TEMPO, O2, 120 °C, 6 h | 98 | 98 | Centrifugation | 39 |
| 7 | Cu3(BTC)2, TEMPO, Na2CO3, O2, 75 °C, 22 h | 98 | 89 | Centrifugation | 31 |
| 8 | Ru-kaolin, TBHP, O2, 100 °C, 3 h | 100 | 97 | Centrifugation | 30 |
3. Conclusions
In summary, we have reported the preparation and characterisation of Fe3O4@Cu2O magnetic materials and its over-reduced analogues. Alongside with TEMPO and NMI additive, the magnetic materials showed excellent catalytic efficiency towards the aerobic oxidation of benzylic alcohols under ambient conditions. The electron-withdrawing groups seem to promote the reaction with no differentiation in their substituting position on the phenyl ring. On the contrary, the electron-donating groups such as methyl group did show subtle decrease in activity when the methyl group was at ortho- and para-positions. These observations may suggest that the deprotonation step in the catalysis might be important and be one of the rate-determining steps since an electron-withdrawing group would certainly increase the acidity of the alcohol and thus facilitate its deprotonation. In addition to its easy separation, the materials showed robustness in repetitive applications with no significant loss in activity. The over-reduced materials, Fe3O4@Cu2O–Cu-7, showed even better durability. Further work has been undertaken to improve its catalytic activity for the aerobic oxidation of aliphatic alcohols and explore how the presence of metallic copper in the materials improved the robustness.4. Experimental
4.1 Materials and instrumentation
All commercial reagents, substrates were purchased from Aldrich, Macklin or Aladdin and used as received without further purification. The as-prepared core–shell Fe3O4@Cu2O nanomaterial and its precursors were characterized by different spectroscopic methods. IR (KBr) spectra were recorded on a Perkin-Elmer 781 spectrophotometer. X-ray diffraction (XRD) measurements were performed with a Philips powder diffractometer type PW 1373 goniometer. It was equipped with a graphite monochromator crystal. The X-ray wavelength was 1.5405 Å and the diffraction patterns were recorded in the 2θ range (10–80) with scanning speed of 2° min−1. Morphology and particle dispersion was investigated by scanning electron microscopy (SEM) (Cam scan MV2300). The chemical composition of the as prepared nanomaterial was measured by EDS (Energy Dispersive X-ray Spectroscopy) performed in SEM. The shape and size of the Fe3O4@Cu2O nanomaterial were identified by transmission electron microscope (TEM) using a Philips EM208 microscope operating at an accelerating voltage of 90 kV. XPS analysis was used to investigate the oxidation states of elements by measuring the binding energy of that element. XPS experiments were performed with an Al KR monochromator X-ray source operating at 15 kV using Thermo Fisher Scientific, USA. During spectrum acquisition, the test chamber pressure is maintained above 10–6 mbar for using low-energy electrons to expel electron guns to neutralize possible surface roughness. Magnetic properties of the NPs were detected by a superconducting quantum interference device of vibrating sample magnetometer (SQUID, Quantum Design MPMS).4.2 Synthesis of Fe3O4 nanomaterials
The magnetite nanomaterials (Fe3O4) were synthesized by the co-precipitation method according to the reported literature.49 About 5.84 g of FeCl3·6H2O and 2.15 g of FeCl2·4H2O (Fe3+:Fe2+ = 2:1) were dissolved in 100 mL of deionized water under N2 atmosphere at 85 °C. Then, 10 mL of 25% NH4OH was added and stirred for 2 h. Now, the colour of the bulk solution turned to black, the precipitates were separated and washed several times with deionized water and ethanol using magnetic decantation. After that, Fe3O4 nanomaterials were treated with 0.1 M L-lysine solution for 30 min, which were then separated with a magnet and washed several times with deionized water and dried under vacuum at 60 °C for 10 h.
4.3 Synthesis of core–shell Fe3O4@Cu2O nanomaterials
The synthesis of core–shell Fe3O4@Cu2O nanomaterials was based on the reported literature method.49 About 0.18 g of Cu(NO3)2·3H2O was added to a mixture containing 17 mL of ethanol and 8.5 mL of ethylene glycol, followed by vigorous stirring. Then 25 mg of as-prepared magnetite was added to the above solution and sonicated for 30 min. After that, it was sealed in a Teflon-lined stainless-steel autoclave (100 mL capacity) and heated to 160 °C for 4 h. Then, the resultant solid was magnetically separated by using external magnet and washed three times with deionized water and ethanol to eliminate inorganic and organic impurities. Finally, the obtained core–shell Fe3O4@Cu2O nanomaterial was dried under vacuum at 60 °C for 5 h. The over-reduced materials were analogously prepared by prolonging the reaction time.4.4 Aerobic oxidation of alcohols using core–shell Fe3O4@Cu2O nanomaterials
All reactions were carried out on a reaction tube (25 mL) in which benzyl alcohol (5 mmol), catalyst (15 mg), TEMPO (0.25 mmol), NMI (0.25 mmol) and acetonitrile (5 mL) were added respectively and stirred at room temperature. The aliquots of the reaction mixture were used for product analysis at regular interval by gas chromatic technique. The retention times for various compounds were determined by injecting pure compounds under identical GC conditions. Conversion of substrates was calculated from GC data and the oxidation products were identified from GC-MS results. At the end of the reaction, the catalyst was collected using an external magnet and reused for next run where applicable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (Grant no. 21571083), the Natural Science Foundation of Zhejiang Province (Grant no. LY18B010007, LY19B010001), Science and Technology Program of Jiaxing (Grant No. 2018AY11003, 2018AY11010), the Government of Zhejiang Province (Qianjiang Professorship for XL) and Jiaxing University (Summit Program of Jiaxing University for Leading Talents) for supporting this work.Notes and references
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- J. P. Klinman, Chem. Rev., 1996, 96, 2541–2562 CrossRef CAS PubMed.
- M. M. Whittaker and J. W. Whittaker, Biophys. J., 1993, 64, 762–772 CrossRef CAS PubMed.
- E. T. T. Kumpulainen and A. M. P. Koskinen, Chem.–Eur. J., 2009, 15, 10901–10911 CrossRef CAS PubMed.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- M. E. Ali, M. M. Rahman, S. M. Sarkar and S. B. A. Hamid, J. Nanomater., 2014, 2014, 1–23 Search PubMed.
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed.
- P. Chaudhuri, M. Hess, U. Flörke and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 2217–2220 CrossRef CAS.
- M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS.
- P. Chaudhuri, M. Hess, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 1999, 38, 1095–1098 CrossRef CAS PubMed.
- Y. Sasano, N. Kogure, S. Nagasawa, K. Kasabata and Y. Iwabuchi, Org. Lett., 2018, 20, 6104–6107 CrossRef CAS PubMed.
- G. Zhan, W. Zhong, Z. Wei, Z. Liu and X. Liu, Dalton Trans., 2017, 46, 8286–8297 RSC.
- M. F. Semmelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends, G.-J. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, ACS Catal., 2013, 3, 2599–2605 CrossRef CAS PubMed.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- D. Zhai and S. Ma, Org. Chem. Front., 2019, 6, 3101–3106 RSC.
- L. Marais and A. J. Swarts, Catalysts, 2019, 9, 395–422 CrossRef.
- M. M. Hossain and S.-G. Shyu, Adv. Synth. Catal., 2010, 352, 3061–3068 CrossRef CAS.
- A. Ochen, R. Whitten, H. E. Aylott, K. Ruffell, G. D. Williams, F. Slater, A. Roberts, P. Evans, J. E. Steves and M. J. Sanganee, Organometallics, 2019, 38, 176–184 CrossRef CAS.
- Z. Liu, Z. Shen, N. Zhang, W. Zhong and X. Liu, Catal. Lett., 2018, 148, 2709–2718 CrossRef CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- Y.-Z. Chen, R. Zhang, L. Jiao and H.-L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- J. Habermann, S. V. Ley and J. S. Scott, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 1253–1256 RSC.
- A. Bleloch, B. F. G. Johnson, S. V. Ley, A. J. Price, D. S. Shephard and A. W. Thomas, Chem. Commun., 1999, 1907–1908 RSC.
- L. Chen, T. Feng, P. Wang, Z. Chen, R. Yan, B. Liao and Y. Xiang, Appl. Catal., A, 2016, 523, 304–311 CrossRef CAS.
- Y. Li, Y. Gao and C. Yang, Chem. Commun., 2015, 51, 7721–7724 RSC.
- B. Zadam, D. Obaid, A. Mayoufi, P. Beaunier, F. Launay and F. Z. El Berrichi, Res. Chem. Intermed., 2019, 45, 1281–1293 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS.
- S.-C. Chen, S.-N. Lu, F. Tian, N. Li, H.-Y. Qian, A.-J. Cui, M.-Y. He and Q. Chen, Catal. Commun., 2017, 95, 6–11 CrossRef CAS.
- X. Zhou and H. Ji, Chin. J. Catal., 2012, 33, 1906–1912 CrossRef CAS.
- G. Zhao, F. Yang, Z. Chen, Q. Liu, Y. Ji, Y. Zhang, Z. Niu, J. Mao, X. Bao, P. Hu and Y. Li, Nat. Commun., 2017, 8, 14039 CrossRef PubMed.
- R. Poreddy, C. Engelbrekt and A. Riisager, Catal. Sci. Technol., 2015, 5, 2467–2477 RSC.
- J. Albadi, A. Alihoseinzadeh and A. Mansournezhad, Synth. Commun., 2015, 45, 877–885 CrossRef CAS.
- H. Wang, W. Fan, Y. He, J. Wang, J. N. Kondo and T. Tatsumi, J. Catal., 2013, 299, 10–19 CrossRef CAS.
- W. Liu, J. Yang and J. Cai, Res. Chem. Intermed., 2019, 45, 549–561 CrossRef CAS.
- R. Ali, K. Nour, A. Al-warthan and M. R. H. Siddiqui, Arabian J. Chem., 2015, 8, 512–517 CrossRef CAS.
- J. Fan, Y. Dai, Y. Li, N. Zheng, J. Guo, X. Yan and G. D. Stucky, J. Am. Chem. Soc., 2009, 131, 15568–15569 CrossRef CAS PubMed.
- Q. Zhang, X. Yang and J. Guan, ACS Appl. Nano Mater., 2019, 2, 4681–4697 CrossRef CAS.
- M. Nasrollahzadeh, M. Bagherzadeh and H. Karimi, J. Colloid Interface Sci., 2016, 465, 271–278 CrossRef CAS PubMed.
- F. Zamani and S. M. Hosseini, Catal. Commun., 2014, 43, 164–168 CrossRef CAS.
- F. Zamani, S. M. Hosseini and S. Kianpour, Dalton Trans., 2014, 43, 3618–3625 RSC.
- L. Geng, B. Zheng, X. Wang, W. Zhang, S. Wu, M. Jia, W. Yan and G. Liu, ChemCatChem, 2016, 8, 805–811 CrossRef CAS.
- R. Azuma, S. Nakamichi, J. Kimura, H. Yano, H. Kawasaki, T. Suzuki, R. Kondo, Y. Kanda, K.-i. Shimizu, K. Kato and Y. Obora, ChemCatChem, 2018, 10, 2378–2382 CrossRef CAS.
- M. Tang, S. Zhang, X. Li, X. Pang and H. Qiu, Mater. Chem. Phys., 2014, 148, 639–647 CrossRef CAS.
- Z.-P. Li, Y.-Q. Wen, J.-P. Shang, M.-X. Wu, L.-F. Wang and Y. Guo, Chin. Chem. Lett., 2014, 25, 287–291 CrossRef CAS.
- A. B. Raut and B. M. Bhanage, ChemistrySelect, 2017, 2, 10055–10060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details for the materials and TEMPOH. See DOI: 10.1039/d0ra04064a |
| This journal is © The Royal Society of Chemistry 2020 |