
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReengineering the programming of a functional domain of an iterative highly reducing polyketide synthase†
Oliver Piechab and
Russell J. Cox *ab
*ab
aInstitute for Organic Chemistry, Leibniz University of Hannover, Schneiderberg 1B, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bBMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany
First published on 15th May 2020
Abstract
A structural model of the enoyl reductase (ER) catalytic domain of the fungal highly-reducing polyketide synthase squalestatin tetraketide synthase (SQTKS) was developed. Simulated docking of substrates and inhibitors allowed the definition of active site residues involved in catalysis and substrate selectivity. These were investigated in silico with the aim of extending the substrate scope. Residues were identified which limit the substrate selectivity of the SQTKS ER, and these were mutated and the engineered ER domain assayed in vitro. Significant changes to the programming of the mutant SQTKS ER domains were observed allowing the processing of longer and more methylated substrates.
Introduction
The mechanisms underlying the programming of fungal highly-reducing polyketide synthases (HR-PKS) remain poorly understood. Fungal HR-PKS are iterative multifunctional multidomain enzymes which synthesise complex polyketides. They are responsible for the biosynthesis of numerous bioactive fungal metabolites in whole or in part.1 HR-PKS consist of a set of functional domains which perform the same suite of chemical reactions as vertebrate fatty acid synthase (vFAS, Scheme 1A) and which are thought to be arranged in a very similar quaternary structure (Scheme 1B).2 Both vFAS and HR-PKS are iterative – that is they repeatedly use a single set of catalytic domains which, in the case of vFAS, load an acetyl CoA as a starter unit onto the ketosynthase (KS) domain 1 (R = CH3) and condense it with an acyl-carrier protein (ACP)-bound malonyl group 2 to form acetoacetyl ACP 3. This is reduced to an alcohol 4 (ketoreductase, KR), dehydrated (dehydratase, DH) to the α,β-unsaturated thiolester 5 and finally reduced again (enoyl reductase, ER) to form a fully saturated C4 unit 6 which can be extended and reduced again (e.g. 1, R = C3H7, Scheme 1A). vFAS is unprogrammed, and continues extension and full cycles of β-processing until a thiolesterase (TE) recognises and hydrolyses ACP-bound C16 and C18 chains 7.3 HR-PKS, however, are highly programmed. They use the same suite of enzymes, often complemented by an in-cis C-methyltransferase (C-MeT), to produce diverse compounds by controlling the methylation pattern, the level of reduction at the β-carbon, the chain-length (i.e. the number of extensions) and, in some cases, the starter unit itself.4–6 | ||
| Scheme 1 Chain extension and β-processing reactions of vertebrate Fatty Acid Synthase (vFAS). (A) Reactions catalysed by vFAS; (B) structure of vFAS showing position of catalytic domains.7 See text for abbreviations. ΨCMeT = non-functional C-MeT domain. | ||
An example of an HR-PKS is involved in the biosynthesis of squalestatin S1 8, a potent pM inhibitor of squalene synthase, in Phoma sp. C2932.6 The tetraketide sidechain of 8 is biosynthesised by a single iterative HR-PKS known as squalestatin tetraketide synthase (SQTKS, Scheme 2A).8 This system takes acetate as a starter unit and after the first extension methylates it and then fully reduces at the β-carbon. The 2S-diketide intermediate 9 is then extended, methylated and fully reduced again to give the 2S,4S-triketide 10. After the third extension the chain is not methylated, and is only partially reduced to form the 4S,6S,E-α,β-unsaturated acid squalestatin tetraketide 11 (Scheme 2B). The other possible stereoisomers (4R,6S-12, 4R,6R-13 and 4S,6R-14) are not observed. Acid 11 is then activated as a CoA and loaded by a specialised system onto O-6 of the squalestatin core 15 (ref. 9) late in the biosynthetic pathway.6
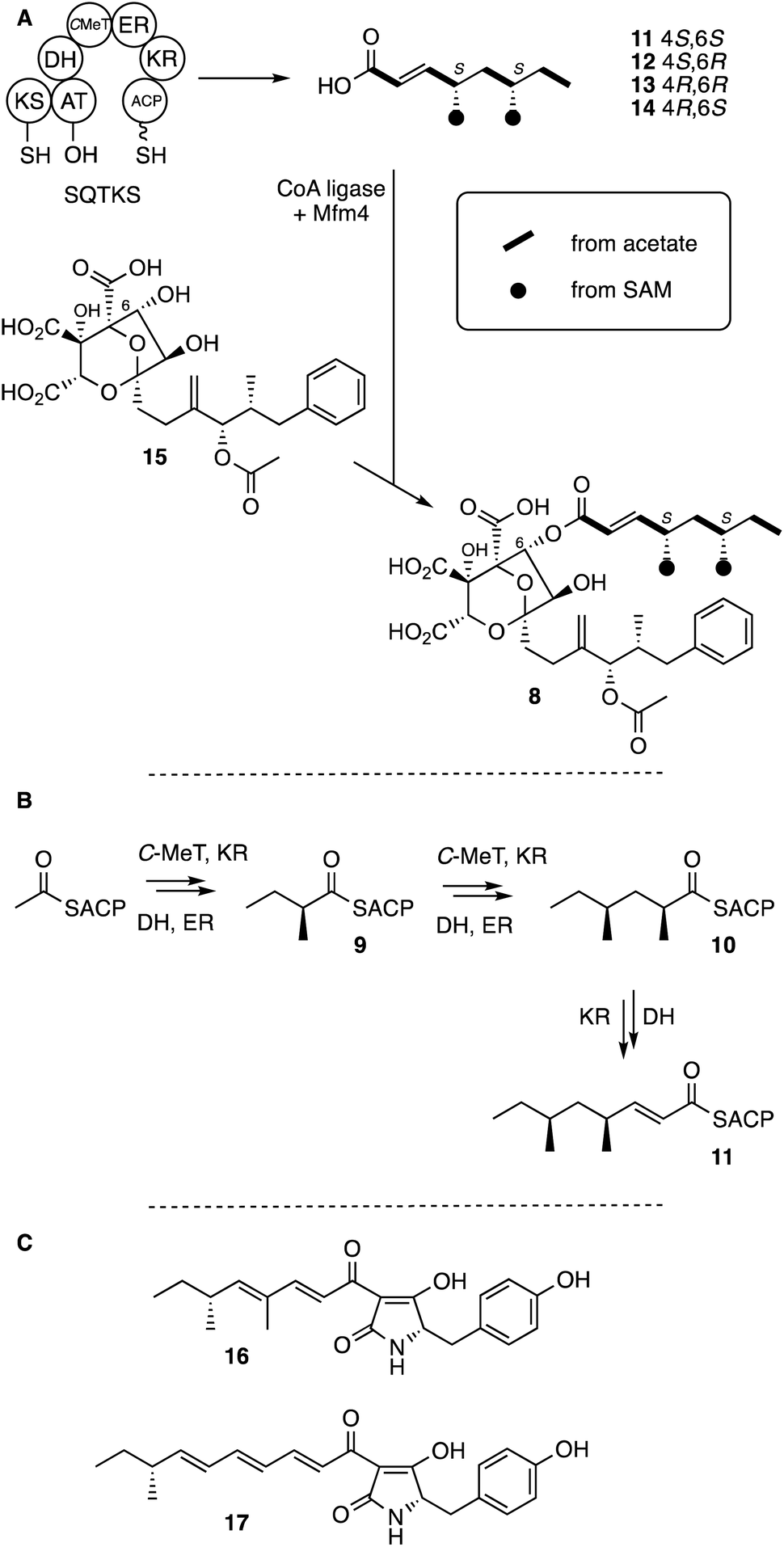 | ||
| Scheme 2 Iterative programmed reactivity of fungal HR-PKS. (A) Overall steps to squalestatin 8; (B) programmed production of 11 by SQTKS showing diketide 9 and triketide 10 intermediates; (C) polyketide–peptide metabolites used in domain swap experiments. | ||
In previous work we have shown that some functional domains of SQTKS can be isolated and studied in vitro. For example the isolated ER domain of SQTKS has been shown to possess very broad substrate selectivity for a range of di- and tri-ketides, but as chain-length and methylation pattern increase the activity diminishes. Indeed, the tetraketide product 11 cannot be processed by the ER, although it can enter the ER active site and it acts as an inhibitor.10,11
The stereoselectivity of hydride transfer at the β-carbon by the isolated SQTKS ER domain was shown to be very high, but the stereoselectivity of reprotonation at the α-carbon is low. This contrasts with the complete PKS which has high stereoselectivity for production of S-methyl branches. Similarly, the SQTKS DH domain has also been studied. It shows very high stereoselectivity for 2R-3R-substrates, but low chain-length selectivity being able to dehydrate di, tri and tetraketides.12,13 In both cases, the stereoselectivities of the DH and ER domains (and by inference the KR domain) of SQTKS have been shown to be identical to that of vFAS. Vederas and coworkers have conducted parallel experiments with isolated KR and C-MeT domains from the lovastatin nonaketide synthase (LNKS)13 and shown that these two domains also display different levels of selectivity.
In a parallel HR-PKS system which synthesises the pentaketide of pretenellin A 16 we have shown that exchange of entire functional domains and sub-domains can reprogramme the HR-PKS, but the results are often imperfect because of loss of fidelity and the creation of mixtures of products.14 For example when swapping the C-MeT domains between the tenellin synthetase (TENS, Scheme 2C) which methylates twice and the desmethylbassianin synthetase (DMBS) which makes predesmethylbassianin 17 and methylates once, mixed products were formed but in which mono-methylation predominated. We reasoned that HR-PKS functional domains must possess a combination of intrinsic and extrinsic selectivities.11 Intrinsic selectivities are those which exist because of direct substrate-specificity by a domain's active site; while extrinsic selectivities arise by protein–protein interactions or by other factors located away from the active site. For example in the pretenellin A 16 system it is clear that the KR exhibits intrinsic selectivity to control chain-length by recognising the length of β-keto intermediates using a specific helix motif in its active site.11 This helix differs in TENS and DMBS and exchange of the helix alone reprogrammes the KR and consequently the entire PKS.11 Contrary to this, the C-MeT domain of TENS appears to control methylation frequency, but detailed modelling suggests there are no significant active-site differences between the TENS and DMBS C-MeT domains.11 Thus the overall programme of a fungal HR-PKS is an emergent property of the interacting intrinsic and extrinsic selectivities of the component catalytic domains.
In the case of SQTKS, chain construction appears to cease because tetraketide 11 cannot be reduced by the ER domain, although unmethylated pentaketides can be reduced.10 The ER thus displays intrinsic selectivity in this case.10 We hypothesised that changing the intrinsic selectivity of the ER could potentially reprogramme SQTKS and as an initial goal we set out to re-engineer the ER to accept 11 as a substrate and also to accept longer intermediates as a first step towards eventually reprogramming a complete HR-PKS. In the absence of crystallographic data for either HR-PKS cis-ER domains15 or complete HR-PKS we attempted to use open-source modelling and docking procedures to build a functional model of SQTKS ER which could allow the design of rational mutations.
Results
SwissModel was selected as a rapid and facile tool for the development of a model of SQTKS ER.16 The ER domain boundaries were approximately located by sequence alignment between SQTKS and vFAS (see ESI†) and the ER domain of CurF (37% identity, PDB 5dp2),17 a trans-AT modular PKS18 involved in the biosynthesis of curacin in cyanobacterium Lyngbya majuscula,19 was selected as the template. An apo-ER model with a QMEAN score of −2.37 and a Cα RMSD of 1.34 Å was rapidly obtained, with most of the structural deviation observed in the peripheral loops.The model consists of three main features: G1887-I2001 (SQTKS numbering) is an N-terminal globular domain which contacts the substrate pantetheine (vide infra); V2002-V2144 is the cofactor-binding domain and includes a Rossmann fold; and D2145-A2207 forms a C-terminal capping and substrate-binding domain (Fig. 1A).
 | ||
| Fig. 1 Modelled structures of SQTKS ER domain: (A) overall fold showing the N-terminal domain (green), the cofactor-binding domain (red) and the C-terminal domain (grey) and NADPH; (B) removal of N-terminal domain showing substrate-binding pocket and F-2157 which blocks its base; (C) triketide substrate 18p docked into the active site pocket; (D) tetraketide inhibitor 11p docked into the active site pocket. | ||
The cofactor NADPH was transferred to the model from PDB 5dp2 and the assembly was minimised using the YASARA NOVA forcefield.20 NADPH contacts S2027, K2055, G2029 (diphosphate), I2119 and V2144 (nicotinamide), and the NADPH binding motif HAASGGVGQA. All these residues are conserved in other PKS ER domains (see ESI†). In particular, the model of the holo-ER indicates that the NADPH exposes its 4′-pro-R hydrogen for reaction (Fig. 1B). This is consistent with vFAS and also with the proven cofactor hydride stereoselectivity of the SQTKS ER domain.10 Ramachandran analysis of all apo and holo models (see ESI†) showed that in all cases, except I1938A bound to 11p, 96.6% of residues are in favoured regions (308/319), 3.1% in allowed regions (10/319) and 0.3% in an outlier region (1/319). In the case of I1938A bound to 11p, residues in outlier regions increased to 2 (0.6%).
Previous in vitro kinetic studies showed that acyl pantetheine (Pant) thiolesters are more effective substrates of the isolated SQTKS ER domain than their cognate N-acetylcysteamine thiolesters (SNAC).10 Therefore pantetheine thiolester substrates were docked into the holo-ER structure using AutoDock Vina,21,22 followed in each case by YASARA minimisation. The diketide 19p and triketide 20p pantetheines are known to be good substrates of the ER domain (e.g. Fig. 1C), while linear (unmethylated) pentaketide pantetheine 24p shows some activity.10 Conversely the 4S,6S-tetraketide 11p acts as an inhibitor rather than a substrate.10 In the case of the triketide 20p the docking results showed that the pantetheine extends parallel to the adenine diphosphate, and locates the thiolester close to the nicotinamide (Fig. 1C). The substrate acyl group extends past the nicotinamide into a substrate-binding pocket lined with hydrophobic residues (vide infra).
The α,β unsaturated system takes up an s-cis conformation and places the reacting β-carbon 3.4 Å away from the reactive 4′-pro-R hydrogen of the cofactor. Importantly the expected Re face at the substrate β-carbon is facing the reactive hydrogen.10 The diketide 19p takes up a similar pose, again with the Re-face of the β-carbon 3.5 Å away from the reactive hydrogen. The pentaketide 24p also locates similarly with an s-cis α,β unsaturated system, although this time more distant from the β-carbon Re face at 4.7 Å. This may explain the poorer activity of this substrate in vitro.
Docking the 4S,6S-tetraketide 11p showed that it can bind in the active site of the SQTKS ER in agreement with its ability to inhibit the enzyme. However it locates with its α,β-unsaturated system in an s-trans conformation (Fig. 1D and 3). Previous work showed that a racemic mixture of stereoisomers of the dimethylated tetraketide pantetheines (i.e. 11p–14p) shows some limited activity when incubated with the isolated ER domain in vitro.10 At least one of the three other possible stereoisomers of the tetraketide (e.g. 12p–14p) must therefore reach a productive conformation for reduction. We therefore docked these three tetraketide stereoisomers and compared the results with the known good substrates 19p, 20p and 21p (Fig. S2.4B†). This showed that the 4S,6R-12p and 4R,6R-13p tetraketide pantetheines bind in the active site with their α,β-unsaturated moieties in s-trans conformations. However, the 4R,6S-14p isomer can reach an s-cis conformation with its β-Re carbon face 3.4 Å from the reactive hydride. These parameters are similar to those of the known good substrates 19p–21p and suggest that the 4R,6S-14p tetraketide is likely to be the stereoisomer responsible for the observed reaction (Fig. 2A).
 | ||
| Fig. 2 Analysis of docked tetraketides in WT and mutant ER domains: (A) 4R,6S-14p docked in the active site of WT SQTKS ER showing hydrophobic residues defining the substrate-binding pocket; (B) 4S,6S-11p docked in the active site of F1941A/I2147A/F2157V SQTKS ER showing hydrophobic residues defining the substrate-binding pocket. NADPH shown in grey; tetraketides shown in green. NADPH-binding substrate surface defined by the cofactor-binding domain and the indicated residues shown as mesh. | ||
Further inspection of these docked models was then performed in an attempt to identify residues lining the active site pocket which could be involved in substrate selectivity. Two factors were considered: first, changes to residues which could allow the 4S,6S-tetraketide 11p to reach a productive reaction conformation; and second, changes to residues which could allow longer substrates to react.
Five residues were identified which appeared to contact either methyl groups or backbone methylenes of 11p–14p: I1938, F1941, L2146 and I2147. F2157 appears to block the bottom of the pocket and prevents access to further volume (Fig. 1B–D and S2.4D†). These five residues were changed to alanine in silico and the resulting models minimised using YASARA. In each case the volume of the active site pocket was estimated using the 3V web server19 (Table 1). Then the 4S,6S-tetraketide 11p was redocked to each structure using the previous methodology. In the case of I1938A, F1941A and F2157A single mutations the volume of the active site pocket appeared to increase as expected (Table 1). However in the cases of L2146A and I2147A the volume appeared to decrease. L2146 and I2147 are more highly conserved amongst HR-PKS ER domains than the other substrate-contacting residues (see ESI†) and may represent structurally important features, holding open the active site pocket. In all cases except for F1941A, the redocked 4S,6S-tetraketide 11p took up an s-trans conformation. However in the case of single or multiple mutations containing the F1941A change, the substrate is able to reach an s-cis conformation and place its β-Re carbon face 2.7 Å from the reactive hydride (Fig. 2B and 3).
 |
 | ||
| Fig. 3 Comparison of tetraketide 11p docked in WT and F1941A/I2147A/F2157V mutant: grey, NADPH; cyan, WT; green, triple mutant. | ||
In order to assess mutations likely to lead to the ability of longer substrates to react we also examined mutations of the five residues to valine, which might prevent a collapse of the active site, as well as combined mutations (Table 1). Examination of the modelled active site pocket of SQTKS revealed an additional void volume beyond the bottom of the pocket, cut off by F2157 (Fig. S2.4D†). In ER domains known to process longer substrates, such as the fumonisin (nonaketide) ER this position is occupied by a smaller residue. We therefore docked pentaketide substrate 24p into various F2157/A and F2157V ER mutants. The results showed that in all cases the pentaketide substrate could reach an s-cis conformation. However in F2157A mutations the β-carbon can approach the cofactor 4′-pro-R hydrogen more closely (ca 3.2 Å) than in the F2157V mutations (ca 4.0 Å).
We previously described the cloning and expression of the SQTKS ER domain and the development of quantitative kinetic assays.10 E. coli optimised DNA sequence encoding the ER was inserted into pET28a allowing expression in E. coli BL21 and purification via the his6-tag (see ESI for details†). Substrates were incubated with enzyme and NADPH, and the consumption of NADPH was observed at 340 nm in a continuous spectrophotometric assay for the WT enzyme (Fig. 4).10
 | ||
| Fig. 4 Enoylpantetheines tested with WT and mutant ER proteins. Kinetic parameters were measured for the reaction shown in which the consumption of NADPH is followed continuously at 340 nm. V/k values shown for each substrate/enzyme combination. Compounds 26p and 27p were inactive vs. all ER variants. | ||
A total of 10 mutations were planned (Table 1). These were introduced into the existing expression system by divergent PCR using mutagenic primers followed by DpnI digestion and recovery in E. coli. Three mutations could not be introduced (L1696A, L1696V and I2147A). In all other cases mutations were correctly introduced and confirmed by sequencing. In the cases of L2146A, L2146V and L2146/I2147 the resultant proteins were insoluble, corresponding to the hypothesis that these residues may have structural importance. However, in the cases of: the single mutants F1941A and F2157A; the double mutants I2147A/F2157V and F1941A/F2157A; and the triple mutant F1941A/I2147A/F2157V, soluble and stable protein could be produced in each case.
The WT and mutant proteins were assayed with a panel of nine prospective substrates including di- (19p), tri- (18p, 20p, 21p), tetra- (11p, 22p, 23p) and penta-ketides (24p, 25p). Heptaketide 26p and cinnamoyl pantetheine 27p were also tested, but were inactive vs. all ER variants. Kinetic parameters were measured in each case (Fig. 4). The WT protein afforded kinetic parameters almost identical to those previously reported.10 For the mutant proteins a range of remarkable changes were observed (Fig. 4).
The F1941A mutant generally showed similar or reduced activity for all substrates. However a significant exception is its activity with 4S,6S-tetraketide 11p which is not a substrate of the WT enzyme. Here good activity was observed as had been suggested by the in silico mutation and docking experiment and is consistent with the mutation allowing the 4S,6S-tetraketide substrate 11p to reach a productive S-cis conformation in the active site.
The F2157A mutation was designed to extend the active site and this aim also seems to have been achieved. While increased activity of this mutation was also observed for shorter substrates, it most noticeably improved activity with the pentaketide substrates 24p and 25p. Notably, it did not allow the 4S,6S-tetraketide 11p to react, again consistent with the in silico experiments which suggest this mutation does not allow 11p to reach an s-cis conformation. The double mutation F1941A/F2157A combines effects of both single mutations: the effects are most noticeable for the 4S,6S-tetraketide 11p and the pentaketides 24p and 25p. In both mutants the docking predicted a shortening of the 4′-pro-R hydrogen to β-carbon distance (from 4.7 Å and 4.6 Å in the WT and F1941A cases to 3.2 Å and 3.3 Å in the single and dual mutants respectively).
The double mutation I1247A/F2157V is the only mutation created which is predicted to reduce the active site volume. Again the effect is most noticeable for the larger substrates which either fail to turn-over (4S,6S-tetraketide 11p) or are no better than WT (pentaketides). Again, the predicted C–H distance of 4 Å correlates well to the observed low activity. Finally the triple F1941A/I2147A/F2157V mutant also appears to combine the effects of other mutations. The mutation of F1941 allows the 4S,6S-tetraketide 11p to be reduced, while the F2157V mutation seems to allow shorter substrates to also react well.
Some changes are unpredictable. The monomethylated triketide 20p is not a natural substrate of the SQTKS ER, but in the case of the WT enzyme it is by far the best substrate tested, being some 3-fold better than the natural triketide 18p. This substrate becomes even more effective in the I2147A/F2157V double mutant where it outcompetes the triketide by a factor of >10. The reasons for this are not yet understood.
Discussion and conclusion
Our results show that despite the lack of structural data for complete HR-PKS or isolated HR-PKS cis-ER domains, it has proven possible to generate a functional structural model for the SQTKS ER which possesses high utility. Numerous features of the model correlate well with measured parameters. These include the stereochemistry of the reduction reaction, for example. The model predicts that structures which can locate the reactive β-carbon within 4 Å of the cofactor 4′-pro-R hydrogen and maintain an s-cis conformation are likely to be substrates, and for simpler substrates such as the pentaketide 24p at least, that predicted C–H distance correlates with measured substrate specificity. This correlates with recent QM/MM studies of the related vFAS ER by Fernandes and coworkers which showed typical β-carbon distances to the reactive hydride of ca 3 Å, although the s-cis vs. s-trans conformation of the bound substrate was not addressed in this study.23In specific cases in silico modelling was verified in vitro – for example prediction that mutation F1941A would specifically convert 4S,6S-tetraketide 11p from an inhibitor in the case of the WT protein to a substrate of the mutant protein was verified. Likewise, mutation F2157A increases the volume of the active site pocket and significantly improves the substrate specificity of longer substrates. Dual mutations appear to have an additive effect.
These are the first reported site directed mutations of an active domain from an HR-PKS which have been rationally designed to change its intrinsic substrate specificity. Previous studies by the group of Leadlay have examined residues involved in stereoselectivity of the enoyl reduction in modular ER systems,24,25 but these non-iterative ER domains need not posses limiting substrate selectivity. By contrast, our results conclusively show, in the case of the SQTKS ER domain at least, that relatively few mutations are required to make a significant change in the substrate specificity of an iterative PKS ER domain. In some cases more than 10-fold changes over WT activity were observed.
Previous experiments to rationally alter the programming of entire HR-PKS have involved domain swaps and have revealed a mechanism in which β-processing domains compete for ACP-bound substrates.11 A combination of intrinsic and extrinsic selectivities is finely balanced to determine the overall HR-PKS programme. In domain swap experiments large changes are made to both the intrinsic and extrinsic selectivities resulting in somewhat unpredictable or uncontrolled changes to the programme and this appears to often result in the production of mixed products or decreased titres. Here, however, we have demonstrated for the first time that very precise mutations of an isolated HR-PKS functional domain can have precise effects on its intrinsic selectivity. Further work will focus on efforts to insert these mutations into a fully functional and complete HR-PKS. However, the great technical challenges involved in placing single mutations in a >250 kDa protein preclude further discussion at this point.
Experimental
All experimental details and characterisation data are contained in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
DFG is thanked for the provision of preparative LCMS and NMR instrumentation (INST 187/621-1, INST 187/686-1). The Leibniz University of Hannover is thanked for the provision of a studentship position (OP). The publication of this article was funded by the Open Access Fund of the Leibniz Universität Hannover.Notes and references
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- Y.-H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- C. Lin and S. Smith, J. Biol. Chem., 1978, 253, 1954–1962 CAS.
- Z. Iqbal, L.-C. Han, A. ares-Sello, R. Nofiani, G. Thormann, A. Zeeck, R. J. Cox, C. L. Willis and T. J. Simpson, Org. Biomol. Chem., 2018, 16, 5524–5532 RSC.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. Lazarus, A. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- S. Jenni, M. Leibundgut, D. Boehringer, C. Frick, B. Mikolásek and N. Ban, Science, 2007, 316, 254–261 CrossRef CAS PubMed.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- K. E. Lebe and R. J. Cox, Chem. Sci., 2018, 10, 1227–1231 RSC.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2016, 8, 1116–1126 RSC.
- X.-L. Yang, S. Friedrich, S. Yin, O. Piech, K. Williams, T. J. Simpson and R. J. Cox, Chem. Sci., 2019, 10, 8478–8489 RSC.
- E. Liddle, A. Scott, L.-C. Han, D. Ivison, T. J. Simpson, C. L. Willis and R. J. Cox, Chem. Commun., 2017, 53, 1727–1730 RSC.
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS PubMed.
- Crystallographic data for trans-acting ER domains is known. But these are structurally unrelated to the cis-acting ER domain discussed here. See for example: B. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. Li, J. C. Vederas, Y. Tang and S. Tsai, Proc. Natl. Acad. Sci. U.S.A., 2010, 109, 11144–11149 CrossRef PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- D. Khare, W. A. Hale, A. Tripathi, L. Gu, D. H. Sherman, W. H. Gerwick, K. Håkansson and J. L. Smith, Structure, 2015, 23, 2213–2223 CrossRef CAS PubMed.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- Z. Chang, N. Sitachitta, J. V. Rossi, M. Roberts, P. M. Flatt, J. Jia, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2004, 67, 1356–1367 CrossRef CAS PubMed.
- E. Krieger, G. Koraimann and G. Vriend, Proteins: Struct., Funct., Bioinf., 2002, 47, 393–402 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- F. E. Medina, M. J. Ramos and P. A. Fernandes, ACS Catal., 2019, 9, 11404–11412 CrossRef CAS.
- D. H. Kwan, Y. Sun, F. Schulz, H. Hong, B. Popovic, J. C. C. Sim-Stark, S. F. Haydock and P. F. Leadlay, Chem. Biol., 2008, 15, 1231–1240 CrossRef CAS PubMed.
- D. H. Kwan and P. F. Leadlay, ACS Chem. Biol., 2010, 5, 829–838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including all experimental details and deposition of all modelled structure coordinates. See DOI: 10.1039/d0ra04026f |
| This journal is © The Royal Society of Chemistry 2020 |