
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultistage antiplasmodial activity of hydroxyethylamine compounds, in vitro and in vivo evaluations†
Neha
Sharma
a,
Yash
Gupta
b,
Meenakshi
Bansal
 a,
Snigdha
Singh
a,
Prateek
Pathak
c,
Mohd
Shahbaaz
cd,
Raman
Mathur
b,
Jyoti
Singh
e,
Mohammad
Kashif
e,
Maria
Grishina
c,
Vladimir
Potemkin
c,
Vinoth
Rajendran
f,
Poonam
cg,
Prakasha
Kempaiah
b,
Agam Prasad
Singh
*e and
Brijesh
Rathi
*ac
a,
Snigdha
Singh
a,
Prateek
Pathak
c,
Mohd
Shahbaaz
cd,
Raman
Mathur
b,
Jyoti
Singh
e,
Mohammad
Kashif
e,
Maria
Grishina
c,
Vladimir
Potemkin
c,
Vinoth
Rajendran
f,
Poonam
cg,
Prakasha
Kempaiah
b,
Agam Prasad
Singh
*e and
Brijesh
Rathi
*ac
aLaboratory for Translational Chemistry and Drug Discovery, Department of Chemistry, Hansraj College University Enclave, University of Delhi, Delhi 110007, India. E-mail: brijeshrathi@hrc.du.ac.in
bDepartment of Medicine, Stritch School of Medicine, Loyola University Chicago, 2160 South 1st Avenue, Maywood, IL 60153, USA
cSouth Ural State University, Laboratory of Computational Modelling of Drugs, 454080, Russia
dSouth African National Bioinformatics Institute, University of the Western Cape, Private Bag X17, Bellville, Cape Town 7535, South Africa
eInfectious Diseases Laboratory, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110067, India. E-mail: singhap@nii.ac.in
fDepartment of Microbiology, School of Life Sciences, Pondicherry University, Puducherry 605014, India
gDepartment of Chemistry, Miranda House, University of Delhi, Delhi 110007, India
First published on 25th September 2020
Abstract
Malaria, a global threat to the human population, remains a challenge partly due to the fast-growing drug-resistant strains of Plasmodium species. New therapeutics acting against the pathogenic asexual and sexual stages, including liver-stage malarial infection, have now attained more attention in achieving malaria eradication efforts. In this paper, two previously identified potent antiplasmodial hydroxyethylamine (HEA) compounds were investigated for their activity against the malaria parasite's multiple life stages. The compounds exhibited notable activity against the artemisinin-resistant strain of P. falciparum blood-stage culture with 50% inhibitory concentrations (IC50) in the low micromolar range. The compounds' cytotoxicity on HEK293, HepG2 and Huh-7 cells exhibited selective killing activity with IC50 values > 170 μM. The in vivo efficacy was studied in mice infected with P. berghei NK65, which showed a significant reduction in the blood parasite load. Notably, the compounds were active against liver-stage infection, mainly compound 1 with an IC50 value of 1.89 μM. Mice infected with P. berghei sporozoites treated with compound 1 at 50 mg kg−1 dose had markedly reduced liver stage infection. Moreover, both compounds prevented ookinete maturation and affected the developmental progression of gametocytes. Further, systematic in silico studies suggested both the compounds have a high affinity towards plasmepsin II with favorable pharmacological properties. Overall, the findings demonstrated that HEA and piperidine possessing compounds have immense potential in treating malarial infection by acting as multistage inhibitors.
Introduction
Malaria, the deadliest disease, continues to be a threat for the human population worldwide, inordinate in endemic tropical and Sub-Saharan Africa regions. Based on the World Malaria Report 2019, at least 228 million cases were noted in 2018, with an estimated number of 405![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths globally. In 2018, 67% of all deaths reported were of children under the age of five, making them the most vulnerable group affected by malaria.1 This highlights the current burden of malaria and the advances achieved in disease control. Malaria is caused by eukaryotic protozoans belonging to Plasmodium species. Out of the five species, Plasmodium falciparum, is accountable for pathogenic human malaria.2 The complex life cycle of P. falciparum consists of multiple forms, i.e., asexual (liver and blood-stage) and sexual (gametocyte and transmission stages). All these life stages of the parasite development offer potential drug targets individually.3
000 deaths globally. In 2018, 67% of all deaths reported were of children under the age of five, making them the most vulnerable group affected by malaria.1 This highlights the current burden of malaria and the advances achieved in disease control. Malaria is caused by eukaryotic protozoans belonging to Plasmodium species. Out of the five species, Plasmodium falciparum, is accountable for pathogenic human malaria.2 The complex life cycle of P. falciparum consists of multiple forms, i.e., asexual (liver and blood-stage) and sexual (gametocyte and transmission stages). All these life stages of the parasite development offer potential drug targets individually.3
Although several approaches viz. insecticide spraying, insecticide-treated bed nets, and availability of effective chemotherapeutic drugs like artemisinin-based combination therapies (ACTs),4 are available to control malaria. It is still considered a lethal infectious pathogen, particularly in the developing part of the world.5 Therefore, ACTs remain the most effective treatments for P. falciparum malaria.6,7 However, the emergence of increasing resistance to these front-line treatments has severely hindered current efforts to reduce disease burden and reduce mortality rates.8–10 This scenario creates the possibility of not having even a single reliable effective drug capable of treating the P. falciparum malaria, particularly in endemic regions.10 Therefore, generic ACTs are the only efficient treatments available in these regions with no personalized malarial drug resistance testing being done.11 Moreover, the non-artemisinin drugs have also shown reduced efficiency against the P. falciparum infection.9,12 More importantly, artemisinin resistance in P. falciparum is an unprecedented spread occurring in South-East Asia, eastern India and Central African countries.13,14 It poses a higher risk to the control and eradication effort. Notably, the licensed RTS,S vaccine, Mosquirix, was introduced into clinical trials among the children aged 5–17months with the efficacy of ∼35%.15 Altogether, more efficacious therapeutics are in demand to overcome the issue of clinical resistance to the frontline drugs as well as develop new molecules that exhibit multistage activity.
Current therapeutics are selectively efficacious during the asexual blood-stage infection and fail to inhibit the sexual and liver stages. This eventually results in the development of clinical drug resistance in other life cycle stages of parasites.16,17 The renewed guidelines for malaria eradication emphasize on identifying new compounds with multistage antimalarial activity i.e., able to inhibit liver, asexual and sexual blood stages.18 This approach is highly essential as one of the critical paths for the elimination of malaria. Therefore, active drug molecules whose potency is not restricted to a spectrum of pathogenic asexual blood stages19 are needed to overcome the issue of increasing resistance. The clinically approved antimalarial therapeutics, namely atovaquone (ATQ) and pyronaridine, displayed significant multistage activity to discover bioactive entities.16,20 On a particular note, the discovery of drugs active against the liver stage and the sexual blood stage (gametocyte) was hampered partly due to the limited availability of established screening assays.21,22 Precisely, the liver stage is a necessary progressive step that leads to the symptomatic asexual blood-stage, representing an attractive molecular drug-targets. Previously, we reported that hydroxyethylamine (HEA) analogs inhibited cultured blood-stage parasites at sub-micromolar range,23 explicitly supporting the two compounds, 1 and 2 (Fig. 1). Encouraged by these initial results, we intended to evaluate antiplasmodial potency against various life-stages of P. falciparum in culture condition and mice model of P. berghei infection to establish their multistage activity. Therefore, we assessed the efficacy of both the compounds against the blood stage of the artemisinin (ART) resistant field isolate (in vitro), liver stage, ookinete, and gametocyte stages. We have also systematically performed in silico studies to gain insight into the Quantitative-Structure–Activity Relationship (QSAR) and identify the possible targets.
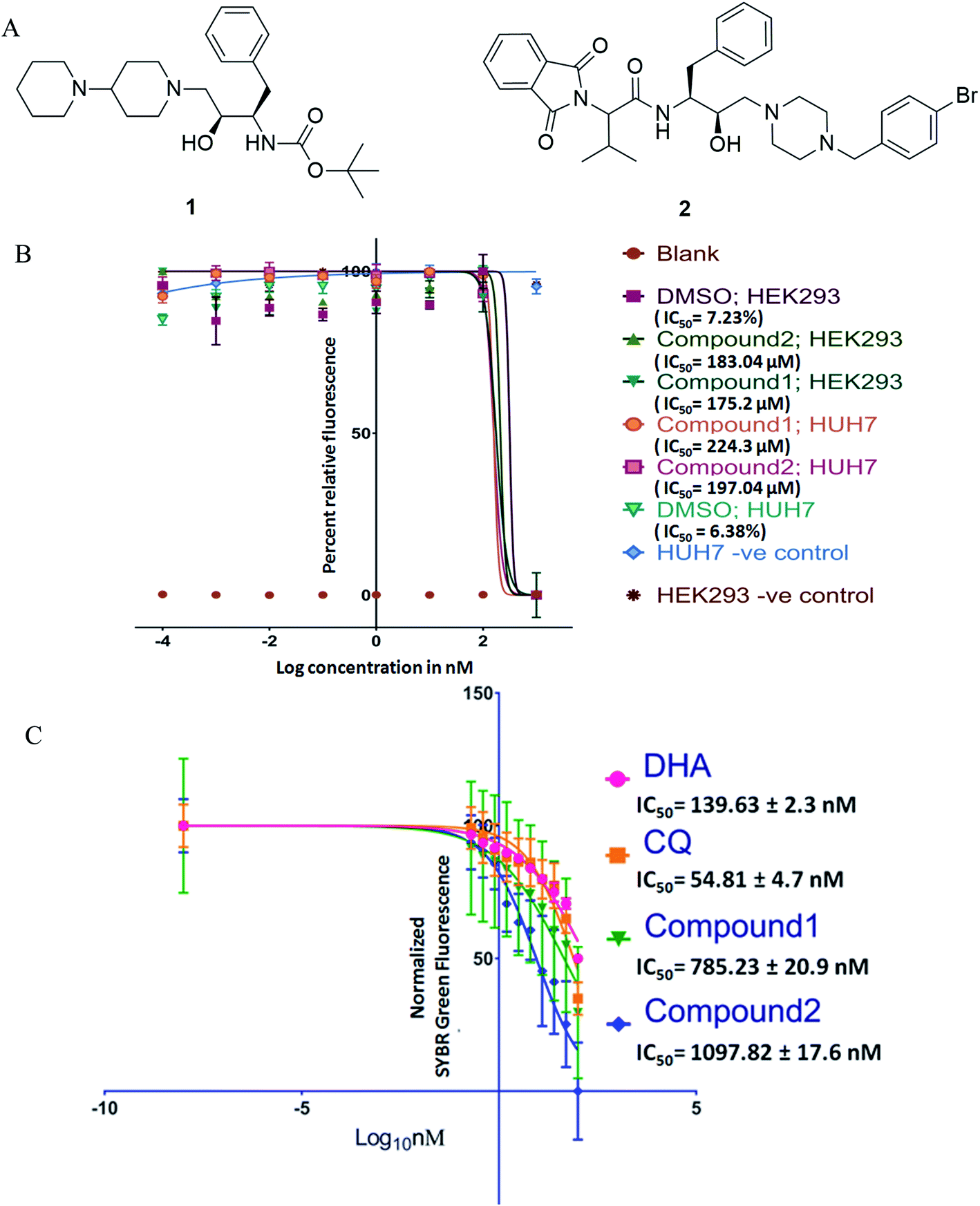 | ||
| Fig. 1 (A) Structure of compounds 1 and 2; (B) cytotoxicity data for Alamar blue assay (IC50) were transformed, X = log(10), normalized between 0% and 100% and fitted with sigmoidal dose–response curves; (C) anti-plasmodial activity of compounds against ART-resistant strain, data (relative SYBR green-1 fluorescence) were transformed, X = log(10), normalized between 0% and 100% and fitted with sigmoidal dose–response curves. DHA – dihydroartemisinin, CQ – chloroquine. | ||
Results and discussions
Assessment of blood stage antiplasmodial activity and cytotoxicity
Antimalarial drug discovery is now partly shifted towards the therapeutics with multistage activity, probably due to a part of the malaria eradication agenda.16,24 Encouraged by the blood stage antiplasmodial activity of compounds23 (1 and 2; Fig. 1A), we decided to explore their potency against various life forms of P. falciparum. These two compounds were synthesized following the reported procedures.23 As the first part of the investigation, we evaluated the cytotoxicity of the compounds by exposing HEK293 (Kidney) and HepG2/Huh 7.1 (liver) cells to varying drug concentrations corresponding to exponentially higher than the blood-stage IC50 values. This was to establish if the observed antiplasmodial effects of the compounds are due to non-specific inhibitory activity. As shown in Fig. 1B, both compounds were non-toxic at pharmacological concentration ranges. Testing these compounds on kidney and liver cells showed no observable toxic effects when used up to 100 times higher concentration required to kill 50% of blood-stage parasites, indicating the specific killing ability of the compounds.Interestingly, compounds are required in high micromolar concentrations (>170 μM) to cause toxicity to mammalian cells, whereas the IC50 for P. falciparum strains found to be in the submicromolar range (∼1 μM). The compounds (Fig. 1) possess HEA scaffold23,25 that is crucial to achieving the potent antiplasmodial activity.24 Both compounds 1 and 2 displayed IC50 values of 1.16 μM and 1.25 μM, respectively against P. falciparum (3D7) strain and ∼3 μM against P. falciparum (7GB) strain after 48 h of treatment as reported earlier.23 While testing against the ART-resistant P. falciparum [MRA-1241 (IPC4912, ART-resistant), BEI-MR4],26 both compounds had substantial inhibitory activity (Fig. 1C). The IC50 testing was performed with ART resistant strain to determine cross-resistance with compounds tested as shifts in IC50s. The compounds markedly inhibited the growth of ART resistant strain with IC50, 785.23 ± 20.9 nM for compound 1 and 1097.82 ± 17.6 nM for compound 2 after 72 h of treatment. Our results clearly showed that the compounds have anti-plasmodial activities against P. falciparum (CQ and ART sensitive and resistant strains) in culture. Drug resistance is a complex phenotype comprising all over changes in the mutant parasites such as overexpressing efflux pumps and various cellular components affecting a range of physiological characteristics. The consistent efficacy of compounds against resistant parasites shows no cross-resistance with the inherent resistance of CQ and ART resistant strains and against non-specific resistant phenotype. This also negates the possibility of observed efficacy to be reference strain 3D7 specific anomaly.
To further evaluate the in vivo efficacy of compounds, a mouse model of malaria infected with Plasmodium berghei NK-65 was developed. The results of these studies are presented in Fig. 2A and B. There was a dose-dependent suppression in blood-stage parasites with no signs of toxicity. It was observed that subcutaneous administration of four consecutive doses of compound 1 at 100 mg kg−1 significantly reduced the blood-stage parasites on day 5 (P < 0.05), 8 (P < 0.01), and 15 (P < 0.01) as compared to the untreated control. We found that mice treated with 1 exhibited a significant decline in the parasite load compared to 2 (Fig. 2A). The mean survival time of the treated group was slightly higher than in untreated ones (Fig. 2B). Notably, compound 1 could not eliminate the blood-stage infection at both the dosages. We reasoned that this effect is probably due to the difference in the drug distribution and varied uptake towards infected RBC. Our findings also indicate that partial clearance of blood parasites is due to shorter half-life (less than 4 h) and the rapid elimination of 1 from the circulation. Our results demonstrated that 1 exhibited inhibitory action on the growth of P. berghei infection in mice.
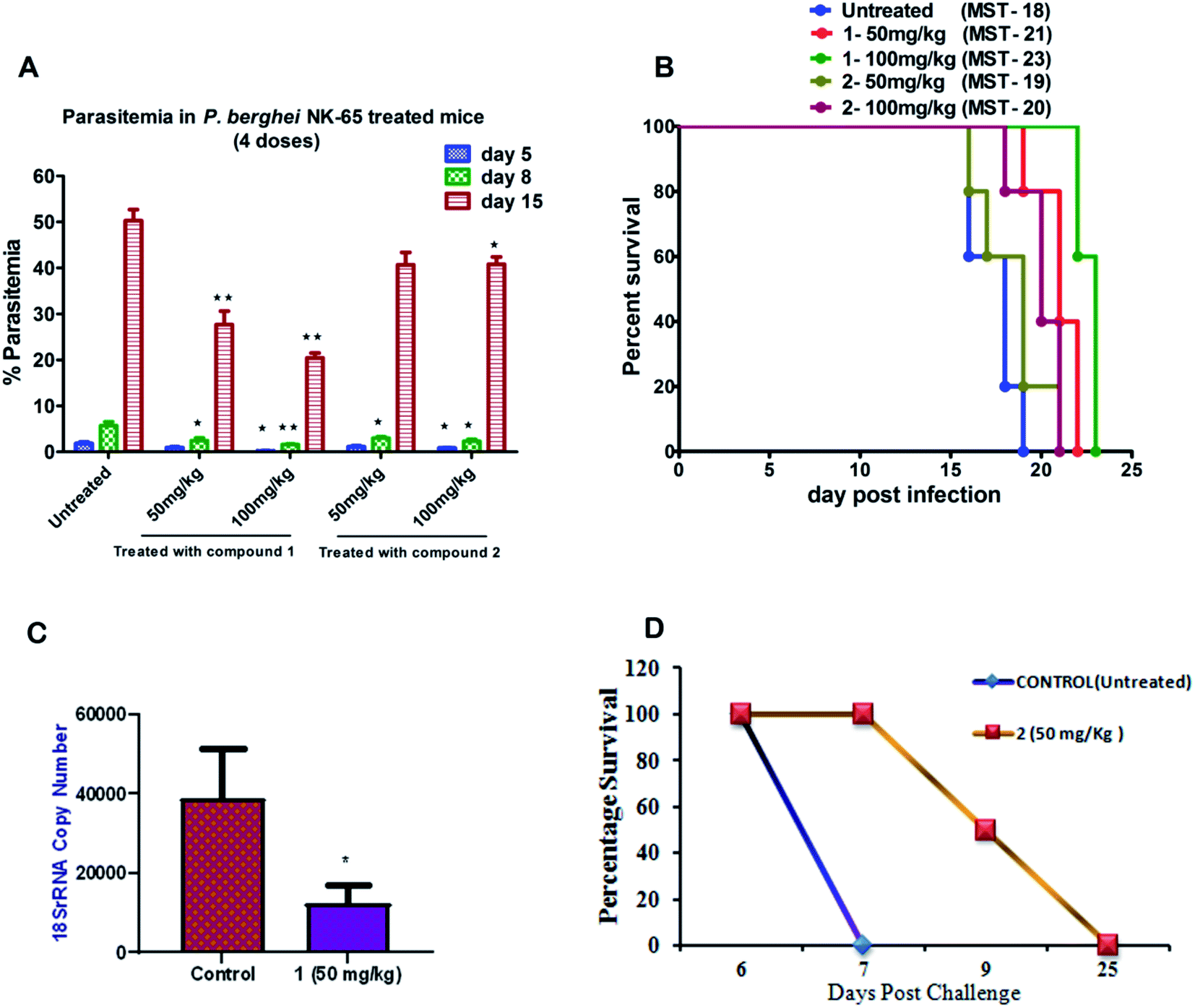 | ||
| Fig. 2 Assessment of the efficacy of compounds (1 and 2) against P. berghei NK-65 infection in the mouse model; (A) the mean percentage parasitemia of treated group versus untreated ones on days 5, 8, and 15 post-treatment. The data represent means ± SD using five animals (n = 5) in each group; (B) the survival response of mice during blood-stage infection and mean survival time (MST) (in days) treated versus untreated ones; (C) efficacy of compound 1 at 50 mg kg−1 (i.p) against P. berghei liver stage infection was compared with control mice using unpaired t-test (P < 0.02); and (D). The survival analysis of mice in liver-stage infection was monitored for compound 2 until day 30 post-infection (n = 4 per group). | ||
Assessment of inhibition potential of compounds during P. berghei liver-stage infection
Based on the results from blood-stage inhibition assays and cytotoxicity assays, the inhibitory activity of compounds was tested against the liver stage P. berghei infection under in vitro and in vivo condition. Both compounds displayed an inhibitory effect against liver-stage infection. Since the parasite density is very low in the liver stage as compared to other developmental stages, this represents one of the potential targets for the antimalarial drugs that prohibit blood-stage infection and subsequent transmission to mosquitoes can also be quenched.27–29 Therefore, HepG2 cells infected P. berghei sporozoites were treated at 1, 10 and 100 μM concentrations of both the compounds in culture. The parasite load was systematically quantified in control and the treated cells. The moderate efficacy (44% inhibition) was noted at the 1 μM concentration of compound 1. Compound 1 showed a significant inhibition (∼99%) upon treating cells with 10 μM. However, the efficacy was reduced when the cells were treated at 100 μM, possibly due to compounds precipitation at higher concentration. The IC50 value of compound 1 was 1.89 μM, whereas compound 2 did not show significant activity at a lower concentration, with an IC50 value of 30 μM.We also tested the liver stage activity of both compounds in mice infected with P. berghei sporozoites through mosquito bites, which result in the maturation of merozoites in ∼55 hours. The infected mice were treated with a fixed-dose, 50 mg kg−1 of compounds 1 and 2 for three days. Interestingly, compound 1 showed 67% inhibition, as shown in Fig. 2C. For compound 2, the starting of the blood-stage parasite infection was monitored after three days post-challenge by Giemsa staining. One day delay was observed at the onset of the prepatent period. Single-day delay in blood-stage infection represents ∼10-fold decrease in liver parasite burden.24 Also, survival and parasitemia for the infected mice were monitored after the treatment. Control mice showed a mean survival time (MST) of ∼10 days, whereas the treated mice displayed an improved MST of ∼16.5 days in the case of compound 2 (Fig. 2D). One day delay decreases the load of the parasite in the liver that could probably contribute to the survival benefits. The parasitemia percentage remained low until the day 7 post-sporozoite challenge. The percent of parasitemia increased after day 7 until the death of all the animals. The results indicated that both the molecules exhibited comparable activity against P. berghei liver stage infection in mice models.
Assessment of plasma clearance of compound 1 in mice
It is evident from our findings that compound 1 showed better efficacy than 2 on the parasite growth in P. berghei infection. Therefore, we investigated the half-life of compound 1 in plasma after a single, subcutaneous dose (20 mg kg−1) at different time intervals using a validated HPLC method. We found that compound 1 showed the maximum plasma concentration (Cmax) of 0.2 μg mL−1, the area under the curve (AUC) was 0.5 μg mL−1 h−1. These results indicated that compound 1 remained in the blood circulation with less than 4 h, which significantly correlates with a therapeutic effect in suppressing the blood-stage and liver-stage infection in mice. The developed compound retained in the blood circulation for a shorter time resulting in less exposure to the blood parasites causing the suppression in parasitemia.Anti-gametocyte activity
Next, we evaluated the effect of both the compounds on the development of sexual erythrocytic stages of Pf3D7 cultures. Stage I and II gametocytes were incubated for 48 h with drug concentrations (0.5 μM) corresponding to the median IC50 values of compounds at asexual stage assays. The counts of dead or deformed gametocytes, were recorded at stipulated time points.Treatment with the compounds resulted in a decreased proportion of stage III to V gametocytes (Fig. 3A), and morphologically distorted gametocytes compared to DMSO control (Fig. 3B). Enumeration of the counts showed survival of only 30–45% gametocytes after 24 h of treatment; however, at later time points, there were no observable mature gametocytes (Fig. 3B). Aside from this, we observed that the untreated gametocytes displayed a distinct morphological appearance like ghost cells (remnant RBCs) due to the total consumption of hemoglobin by parasites, whereas the treated ones failed to show.
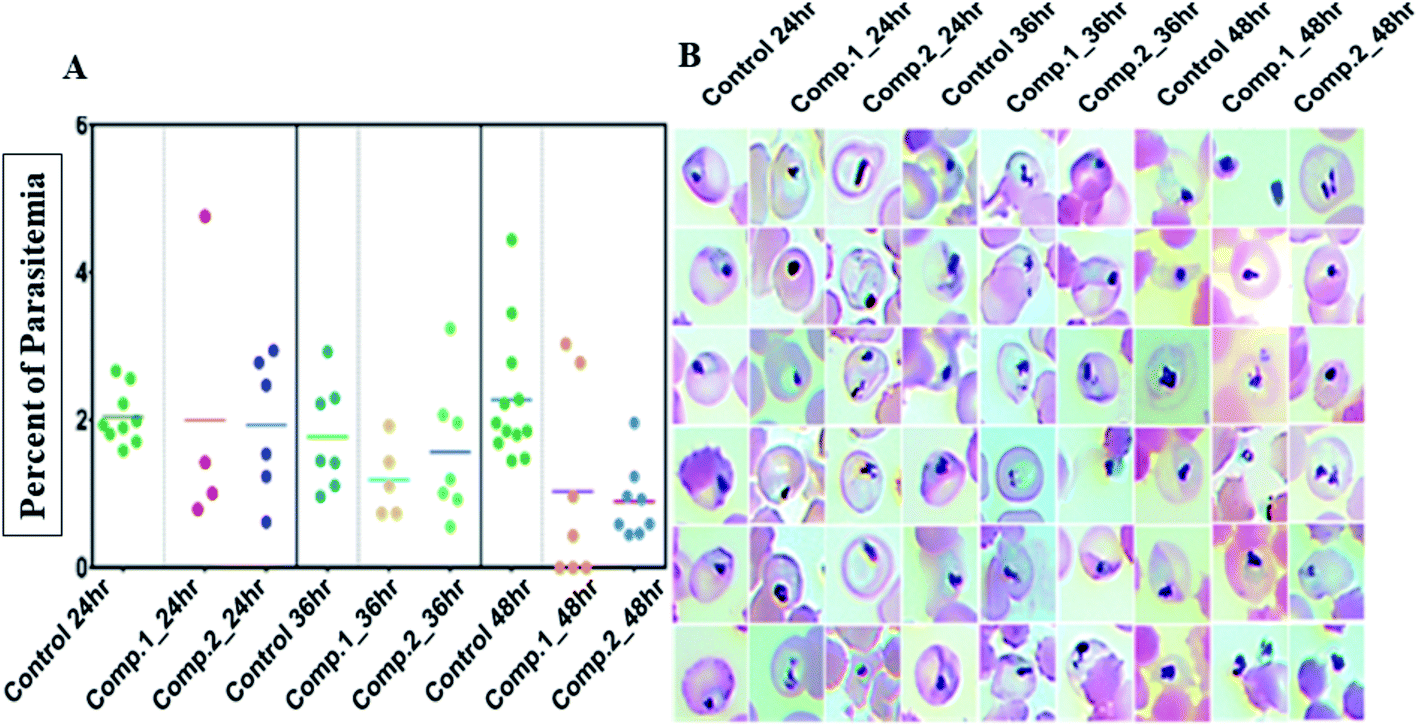 | ||
| Fig. 3 (A) Each scatters plot column represents different sets of samples from different time points and treatments. A 100 μL 3rd, 4th and 5th-day gametocyte rich culture was seeded in microtiter plates in triplicates for each treatment (0.5 μM) and control and three wells each was fixed and stained by the Giemsa staining. Both compounds had an apparent reduction of intact gametocytes compared to control; (B) the most representative morphologies were documented. Both compounds had an evident detrimental effect on the normal morphology to the extent that none of the treated samples had any mature gametocytes even though with 50% viability in SYBR green screenings. | ||
Activity against ookinete
Next, efficacy for both compounds was tested against the transmission stage of the parasite because widely available antimalarial agents selectively target blood-stage infection.The drug molecules with the capability to intervene in the transmission stages of the parasites are considered as high-value partners for the combination treatments due to their potential role in achieving malaria eradication.30Plasmodium transmission relies upon gametocytes capacity to convert into ookinetes, the prominent type of the parasite that attacks and develops a malarial infection in the mosquito midgut. By blocking this transmission stage, further malarial infection rates can be minimized to a greater extent.31 We observed that both the compounds, 1 and 2 showed significant inhibition against ookinete with the IC50 values of 0.704 ± 0.054 μM and 0.7025 ± 0.004 μM, respectively. An increase in the concentration of the compounds reduced the parasites growth, leading to deformed parasites (Fig. 4).
 | ||
| Fig. 4 Structural changes in the sexual stage of malaria. Efficacy of compound 1 (A) and 2 (B) against ookinete formation; (C) inhibition curve for both the compounds; (D) representative images of the DMSO control (healthy) ookinete and treatment with both the compounds at different concentrations. The photomicrograph of smears was stained with Giemsa shown at 1000× magnification. | ||
Computational studies
P value i.e., 3.29. However, compound 2 found to be slightly above lipophilic/hydrophilic balance, i.e., 6.11. Compound 1 follows the Lipinski's rule of five more stringently, whereas compound 2 showed one violation.32 The oral absorption prognosis stated that both compounds have maximum tendency to absorb through the gastrointestinal tract. It is also noticeable that 1 tends to cross the blood–brain barrier. However, compound 2 lacks these characteristics. The metabolism study of the compounds on various sites of cytochrome P450 revealed that compound 1 has lower metabolism score at CYP450-3A4 and CYP450-2D6 (0.43 and 0.22 probability score, respectively), however, it does not have a metabolic tendency at the site of cytochrome CYP450 1A2, CYP450 2C19, and CYP450 2C9. Compound 2 is more likely to metabolize on cytochrome CYP450 3A4 (i.e. 0.91 probability score). However, this was reported a nearly negligible metabolic tendency on the site of cytochrome CYPP450 2D6, CYP450 1A2, and CYP450 2C9. At the same time low cytotoxicity profiles with Huh-7 cells were observed, a liver carcinoma line with a xenobiotic degrading enzyme, negates the possibility of a toxic metabolite formed by these degradations. The overall ADME result showed that the compounds have substantial efficacy for further investigations.
| SN | logP |
TPSA | GI Abs | BBB permeability | Metabolism at the site of CYP450 | Lip. vio | TOX | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A2 | C19 | 2C9 | 3A4 | 2D6 | |||||||
|
a logP is the logarithm of the octanol–water partition coefficient; TPSA: total polar surface area; GI abs: gastrointestinal absorption; BBP: blood–brain barrier permeability; metabolism CYP450: the probability of metabolism on site of cytochrome-450; lip. vio: total number of violations of lipinski rule of five; tox: the probability of maximum toxicity of the compounds.
|
|||||||||||
| 1 | 3.29 | 65.04 | High | Yes | No | No | No | 0.43 | 0.22 | 0 | 0.76 |
| 2 | 6.11 | 94.88 | High | No | No | Yes | No | 0.91 | 0.01 | 1 | 0.82 |
The distribution of pharmacophore, anti-pharmacophore (an atom or group of atoms that reduce biological activity, providing, for example, steric hindrances), and ballast part in 1 and 2 is shown in the Fig. 5A and B. The result showed that the ‘–OH’ group present in 1 have an anti-pharmacophore property and need to be replaced by an amide group. However, compound 2 doesn't show any pharmacophoric and anti-pharmacophoric part. The toxicity studies showed that the oxygen atom present in side chains 1 and 2 has a role in toxicity and needs to be replaced by an atom or group that reduces the toxicity of the compound, such as an amino group shown in Fig. 5C and D. Such a replacement, according to the 3D QSAR prediction, will lead to a decrease in the probability of toxicity by 0.28 and 0.32 for compounds 1 and 2, correspondingly. At large, 3D QSAR study indicated that oxygen atom is connected to the basic skeleton of the compounds act as an anti-pharmacophoric skeleton. To increase the anti-malarial activity of both the compounds, the hydroxyl group (bridged chain) should be replaced by suitable electrophile.
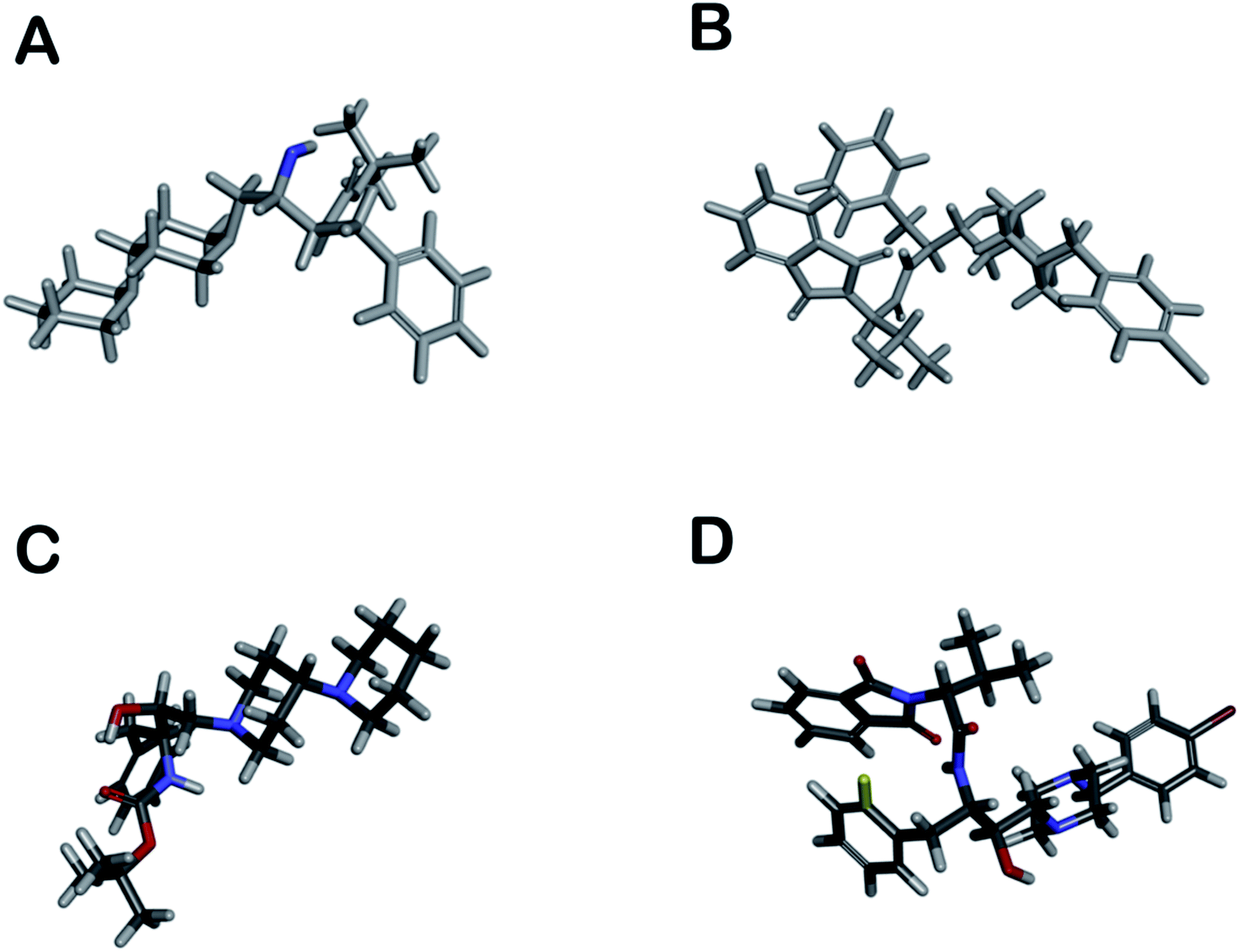 | ||
| Fig. 5 (A and B) shows pharmacophoric (red, not seen), antipharmacophore (blue), and ballast part (grey) in compounds; however, (C and D) show toxicophoric (red) and non-toxicophoric (blue) part in compounds. | ||
Proteins – inhibitors interaction analysis
Both compounds were docked into the active site of the studied proteins, and the resulted outcomes are shown in Table 2. The highest binding affinity of both compounds was observed for plasmepsin-II [PLM II], malarial aspartyl protease. Compound 2 showed the highest combination of free energy of binding and inhibition constant of −8.49 kcal mol−1 and 600.03 nM, respectively. On the other hand, compound 1 showed the interaction energy of −7.56 with an inhibition constant to be predicted around 10.55 μM. Apart from PLM II, both the compounds showed relatively higher binding energy for CDK as compared to the other proteins (Table 2).| Target proteins | Compound 1 | Compound 2 | ||||
|---|---|---|---|---|---|---|
| Selected pose | BE | IC | Selected pose | BE | IC | |
| a GST – glutathione S-transferase Q8MU52; CYB – cytochrome b Q02768; MPEXP – malaria protein EXP-1 P04926; PLM – plasmepsin-II P46925; DPS – dihydropteroate synthetase Q27738; CDK – calcium-dependent protein kinase 1 P62344; GR – glutathione reductase Q94655; DOD – dihydroorotate dehydrogenase (quinone), mitochondrial Q08210; FR – ferredoxin-NADP reductase, apicoplast C6KT68; DRTS – bifunctional dihydrofolate reductase-thymidylate synthase P13922. | ||||||
| GST | 30 | −3.77 | 1.72 mM | 9 | −1.69 | 57.82 mM |
| CYB | 19 | −2.75 | 9.68 mM | 33 | −1.3 | 111.76 mM |
| MPEXP | 19 | −4.63 | 401.46 μM | 27 | −5.47 | 98.28 μM |
| PLM | 41 | −7.56 | 10.55 μM | 39 | −8.49 | 600.03 nM |
| DPS | 35 | −4.22 | 805.09 μM | 26 | −4.7 | 358.79 μM |
| CDK | 40 | −6.77 | 10.94 μM | 3 | −5.47 | 97.91 μM |
| GR | 43 | −4.05 | 1.07 mM | 23 | −2.56 | 13.18 mM |
| DOD | 26 | −5.52 | 89 μM | 27 | 12.54 | N/A |
| FR | 3 | −3.96 | 1.25 mM | 25 | −4.25 | 768.57 μM |
| DRTS | 6 | −5.99 | 40.75 μM | 2 | −2.64 | 11.7 mM |
The analyses of the active site of the crystal structure of PLM (PDB ID – 1LEE) showed that the residues Val78 and Ser79 are the flap residues while the Asp34 and Asp214 form the catalytic dyad, which is surrounded by Ser218 and Gly36 residues.33 Compound 2 was observed to have interactions with Asp34, Asn39, Val78, Ser79, Ile123, Leu131 and Gly216 (Fig. 6A). Moreover, the interaction pocket of 1 residue contained Ile32, Asp34, Gly36, Tyr77, Val78, Ile123, Asp214, Thr217, Leu292 and Ile300 (Fig. 6B). These analyses indicated that 2 could inhibit the functions of the multiple protein targets with a relatively higher affinity than compound 1.
 | ||
| Fig. 6 Docked images. (A) Compound 1 with PLM 2; (B) compound 2 with PLM 2. | ||
MD simulations (100 ns each) were performed on generated PLM II docked complexes with both the compounds for further conformational assessments and validations. The interaction patterns between the PLM II and compounds in the generated trajectories were analyzed in the form of the distance deviations from initial docking and hydrogen bonding dynamic patterns during MD simulations. The computed distances between the PLM II and the studied inhibitors were found to be fluctuating between 0.15−0.25 nm, indicating that all the inhibitors were present in the interacting pocket throughout during MD simulations (Fig. 7). The different patterns of the hydrogen bonds (H-bonds) were observed for the studied system. Compound 1 showed the presence of up to five hydrogen bonds with PLM (Fig. 7), while with compound 2, the protein only forms up to four hydrogen bonds during MD simulations. While observing the generated pattern of H-bonds, compound 1 even showed relatively higher repeat in the H-bonds followed by compound 2.
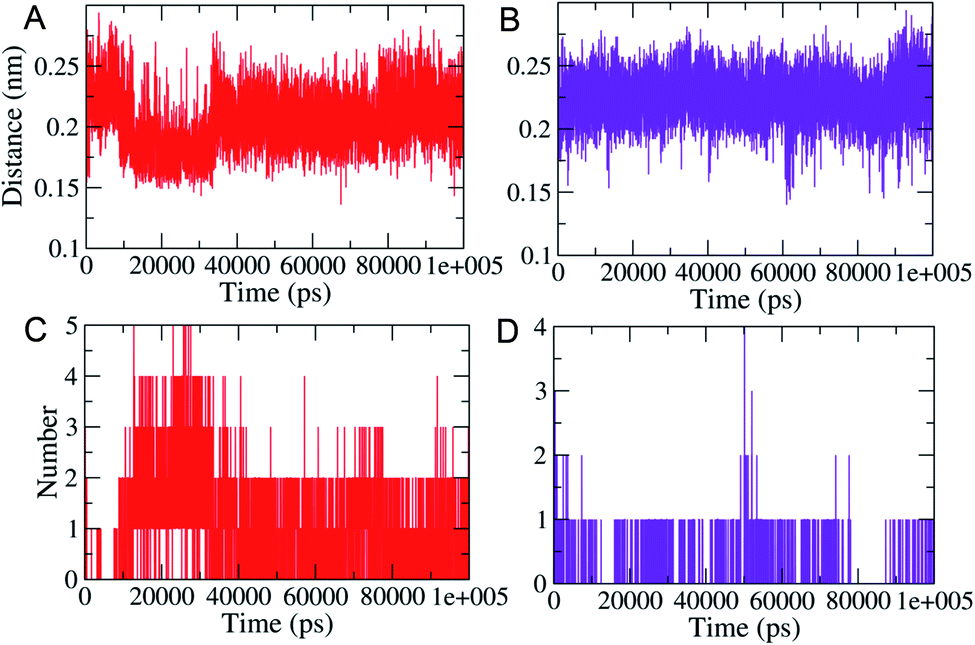 | ||
| Fig. 7 MD simulations based generated plots showing the calculated distances between the PLM and the designed inhibitors (A; top, left) and (B; top, right). Highlighted the changes in hydrogen bonds (C and D). Red and violet color represent compound 1 and 2, respectively. | ||
The compactness of the studied compounds was further assessed in terms of Rg values, which showed that the docked complex of 1 showed a relatively lower Rg value of around 1.95 nm (Fig. 8). Furthermore, both the systems showed comparatively similar stability as indicated from the RMSD values, which are fluctuating between 0.25–4.2 nm (Fig. 8). Moreover, the MM/PBSA based algorithm was used to calculate the interaction energies between the PLM II and the studied inhibitors (Fig. 9). The energy analysis showed that the primary contributor for the total energy is electrostatic energy instead of Van der Waals forces in all the studied systems. The highest average total energy of the interaction of −1780.61 kJ mol−1 seen between PLM II and compound 1. While compound 2 showed similar patterns in which total energy was observed around −1474.49 kJ mol−1. These outcomes provided a more in-depth insight into the interaction of both compounds with PLM II and validated that they bind to the protein efficiently. Still, the interactions for compound 1 were more favorable as assessed from the MD simulation analyses.
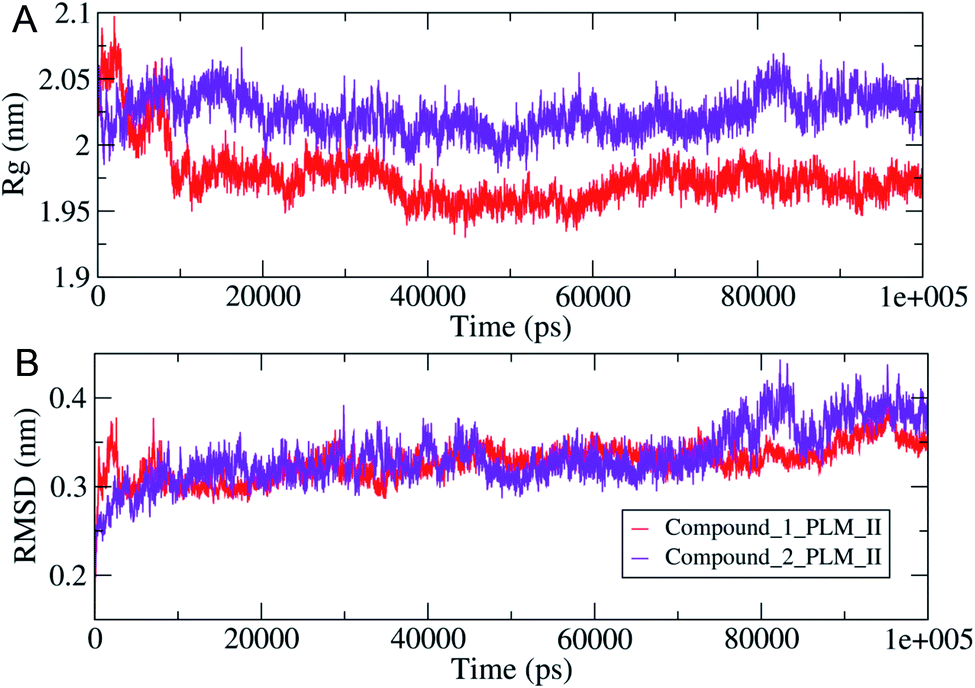 | ||
| Fig. 8 The observed structural changes with (A, top) were showing the variation in the system compactness in terms of Rg values (B, lower), illustrating the changes in the RMSD values observed during 100 ns MD simulations. | ||
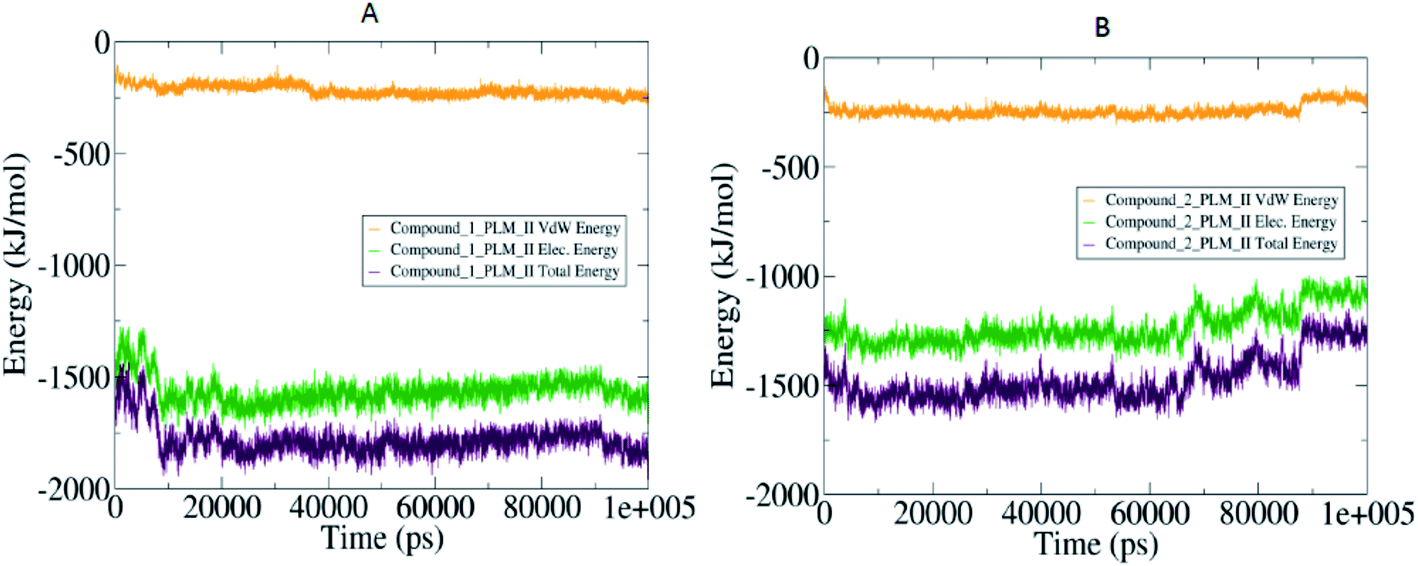 | ||
| Fig. 9 The MM/PBSA based dynamic pattern of calculated interaction energies highlighted in the form of the 2-D curves with (A) observed variation in the compound 1 and PLM II complex: (B) quantified values for compound 2 and PLM II system. | ||
Conclusion
Regardless of significant advances in controlling and ultimately eradicating malaria, it remains a global health problem, particularly in endemic countries. Hydroxyethylamine based compounds were selected for their potency against drug-resistant strains, including multistage activity in culture and in animal models. Both compounds showed potent blood-stage inhibitory activity against ART-resistant strain, anti-gametocyte action probably through the reduced capability to digest hemoglobin (due to inhibition of PLMs) and thereby failed to mature. The compounds were effective against the blood stage of the P. berghei infected mice and mainly compound 1 reduced the parasitemia load more efficiently. For better efficacy, compound 1 could be administered in conjunction with standard partner drugs with greater half-life and that acts synergistically. Our findings also suggested that compound 1 is equally potent against blood-stage and liver-stage infection in mice and inhibited the progression of the gametocyte stage. The in silico studies suggested PLM II as a potential target for both compounds based on their high binding affinities. However, the preliminary screening resulted in partial inhibitory activity against PLM as reported earlier,23 suggesting a possibility of other drug targets through which compound inhibits parasite growth. Notably, neither of these compounds were found toxic to human cells at therapeutic dosages. Taken together, our data provide new information on compound 1 as a multistage inhibitor against all life forms of the Plasmodium. This could be a potential choice for further chemical modifications and the generation of derivatives with the improved biochemical and pharmacokinetic profile.Experimental
Compounds were synthesized and purified following the previously reported procedures.23 NMR details are depicted in ESI.†Animals
Experiments were performed in accordance with the standard protocols approved by Institutional Animal Ethics Committee (IAEC) of the National Institute of Immunology, New Delhi (Dr Agam P Singh's lab), under the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. The animals used were 6 to 8 weeks old and housed under standard controlled conditions at 25 °C with a 12 h light–dark cycle. Animals had access to standard laboratory pellet diet and water available at all times.In vitro anti-plasmodial activity against ART-resistant P. falciparum
Determination of blood stages and percent parasitemia was performed by Giemsa-stained thin smears from parasite cultures. Antimalarial inhibition activity (IC50) was measured using the SYBR Green I assay, as reported by our group24,25 early ring-stage parasites (>80% of rings) from ART resistant (IPC5202, MRA-1240, MR4) cultures were incubated in the presence of 1:2 serial dilution of each compound tested for 72 h, in concentrations ranging from 100000–195.31 nM. Fluorescence intensity was determined using the SpectraMax® M5 Multi-Mode Microplate Reader, with excitation and emission wavelengths of 485 and 530 nm, respectively, and analyzed by nonlinear regression with Normalization & Baseline-corrections using software GraphPad Prism to determine IC50 values.
The in vivo antimalarial study
The mouse model of malaria infection was developed by intraperitoneal administration of 1 × 107Plasmodium berghei-NK65 infected RBC to healthy C57BL6 mice. The in vivo efficacy of compounds was performed in accordance to a slightly modified version of the ‘Peters four day suppressive test’.34 For experimental infections, each animal was injected with 1 × 107 infected RBC through intraperitoneal injection. After 48 h of post-infection, mice were treated with 50 or 100 mg kg−1 body weight of compounds suspended in vehicle control (10% DMSO) for four consecutive days through subcutaneous route. The control group was treated with only vehicle control. The efficacy of the treatment was determined by measuring the parasitemia and survival of mice on days 5, 8, and 15 by making thin smears of blood, from the tail vein of mice, stained with 10% Giemsa and examined at 1000× magnification. The percent parasitemia was measured by counting infected and non-infected RBCs from randomly selected optical fields and expressed as the number of infected RBCs per 100 RBCs. The survival rate of mice (mean survival time, MST) was determined after treatment until 30 days post-infection. The reduction in the number of parasitemia was taken as the index for the therapeutic activity of the test compounds. The percentage of parasitemia was calculated manually using Cell Counting Aid software35 using the formula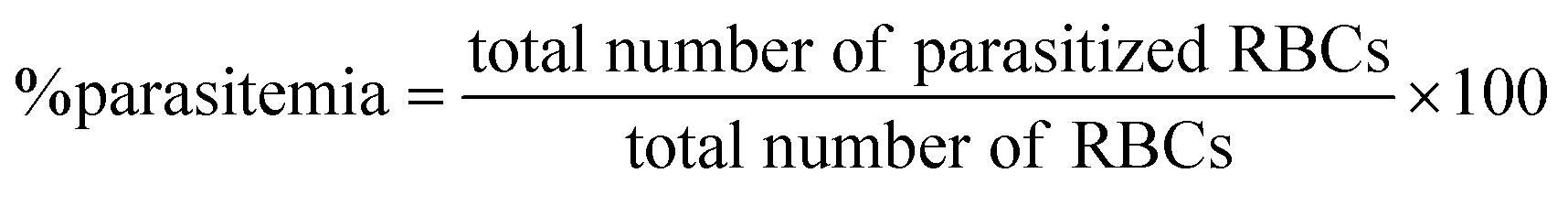
Pharmacokinetic study
A pharmacokinetic study was performed for compound 1 using Swiss albino mice weighing 25 to 30 g. The experimental animals were separated into two groups consisting of three animals each. The mice of the first group were given subcutaneous administration of 1 (20 mg kg−1) suspended in DMSO:PEG-400:5% glucose (2:10:88) (vehicle control) while the second group was administrated with vehicle control (without test compound). At different time intervals (0.25, 0.5, 1, 2, 4, 8, and 24 h), a volume of 0.2 mL of blood was withdrawn from retro-orbital plexus and collected in tubes containing heparin and subjected to centrifugation at 5000 rpm for 15 min for separating the plasma. The concentrations in plasma were determined using Gilson (USA) HPLC instrument. Various pharmacokinetic parameters were determined by plotting drug concentration versus time.
In vitro liver-stage infection in HepG2 cells
Salivary glands of Anopheles stephensi mosquitoes infected with GFP-expressing P. berghei ANKA (Insectaries at National Institute of Immunology, New Delhi, India) were dissected out. Sporozoites were perched in inoculation medium (DMEM with 10% FBS, 3% P/S), and allowed to infect HepG2 cells. The cells were treated with compounds before the infection at various concentrations. The plate was centrifuged at 3000 rpm for 5 minutes to ensure the sporozoites settled down in the plate and incubated at 37 °C for 3 h. The medium was replaced with pre-warmed medium (DMEM, 10% FBS, 3% P/S, and 0.1% fungizone). After two days, the cells were harvested in trizol reagent and freezed at −80 °C. RNA isolation followed by quantitative RT-PCR was performed to estimate the parasite load in the infected cells.Liver-stage infection in mice, treatment and survival assay
GFP-expressing P. berghei ANKA was maintained in Anopheles stephensi mosquitoes (Insectaries at the National Institute of Immunology (New Delhi, India)). Sporozoite count per salivary gland and infectivity was measured before the experimental infection of mice. Briefly, anesthetized mice were exposed to a cage of starved mosquitoes having sporozoites (100/5 mouse) for 15 minutes with intermittent disturbing of mosquito every 2–3 minutes to encourage the biting process. Biting was done at 22 + 1 °C and further maintained in dark conditions in a controlled condition. The first dose (50 mg kg−1) was administered 24 h before the infection, second dose was given 2 h post sporozoite challenge (biting) through the intraperitoneal route.For calculating parasite load, the liver of experimental mice was isolated 50 h post-infection. The mice were anesthetized, sterilized the abdominal area with 70% ethanol and dissected the liver and homogenized it in a precold denaturating solution. RNAs were isolated from the homogenized sample followed by real-time PCR. Survival rate was monitored on a daily basis with a subsequent monitoring on blood stage infection on day 3 onwards. Blood smears were obtained from the tail vein of mice and stained with 10% Giemsa and the parasites were counted using a 100× oil objective.
Cytotoxicity assays in human liver and kidney cells in culture
Cytotoxicity of test compounds was evaluated by assessing the cell viability of Huh 7.1 and HEK293 cells using the Alamar Blue assay.36 Cells (1 × 103 per well) were exposed to different concentrations of test compounds for 24 h in 96-well plate format along with 10/100 μL Alamar Blue solution (10X = 4.8 mM in phosphate buffer) per well. Cell viability was measured using the SpectraMax® M5 Multi-Mode Microplate Reader, at 530 nm excitation wavelength and 590 nm emission wavelength for metabolically reduced resazurin dye. The percent cell viability were expressed as CC50 at which 50% of the cells died/metabolically slowed down due to drug exposure compared to the non-treated and vehicular control. The average minima and maxima were added for normalizations with negative control and blanked respectively to retain their relative global trend. The toxicities observed in part were contributed by DMSO vehicle's cytostatic effect as seen in vehicular controls, although the volume was 1:100 (Vehicle to Media).
In vitro gametocyte culture of P. falciparum
Gametocyte cultures were initiated as described previously.24 Briefly, on day 0, cultures were synchronized at the ring stage by lysis of the flask pellet with 5 volumes of 5% sorbitol for 10 min at 37 °C and maintained at 0.2% parasitemia and 4% hematocrit. The culture was grown in RPMI 1640 medium (Gibco) supplemented with hypoxanthine (Sigma-Aldrich), sodium bicarbonate and 1.5% (wt/vol) AlbuMAX II. Cultures with >8% gametocytes (on day 12 normally) were treated with 50 mM N-acetyl-D-glucosamine (GLcNA, Sigma-Aldrich) and 50 ng mL−1 of bistratene A for 3–5 days to remove asexual forms. The percentage of asexual forms and gametocytes were calculated on Giemsa-stained smears for performing anti-gametocyte assays.In vitro gametocyte parasite growth inhibition assay
Aliquots 100 μL of cultures containing gametocytes at the indicated stages (preferably stage I) were transferred to 96-well plate in duplicates and incubated at 37 °C in the presence of 5% CO2. The culture medium was changed after 48 h and the appropriate concentration of test compounds was maintained. Blood smears of parasites were prepared and stained with Giemsa and the parasitemia was monitored using oil immersion microscopic analysis at 1000× magnification.Activity against ookinete
To study whether the test compound blocks the transmission stage, the sexual stage of the malaria parasite (PbGFPcon) was used to check its ability to inhibit ookinete development in vitro. For this, 1.2 mg phenylhydrazine (Sigma-Aldrich, St. Louis, USA) in 0.9% NaCl was intraperitoneally (i.p.) injected into BALB/c mice 3 days before P. berghei infection. The treated mice were then inoculated with 2 × 106P. berghei-infected erythrocytes intraperitoneally to initiate the blood-stage infection. Parasitemia was allowed to reach ∼5–10% at three days post-infection, and the gametocytemia was observed to be 1–2%. The mice were anesthetized and the blood was withdrawn in heparin containing tubes and added to the ookinete culture medium. The parasites were grown in the medium with a blood/medium volume ratio of 1:10 at 19 °C for 24 h. After 24 h, the culture was Giemsa-stained and analyzed for ookinetes morphology. The number of ookinetes of both normal and abnormal morphology was calculated.
Theoretical calculations
3D QSAR studies were performed via the “Cinderella Shoe”37 methods and within the CoMIn algorithm.38 The pharmacophore part (in red), antipharmacophore skeleton (blue), and inactive parts (grey color) of the synthesized compounds (1 and 2) are displayed in Fig. 5A and B. The grey part of the ring does not participate in pharmacological volubility but liable for structure configuration and matrix function to provide the necessary mutual arrangement of pharmacophoric fragments. However, the toxicity prognosis was shown in Fig. 5C and D (toxicophoric red and non-toxicophoric blue). The ADME profile like logP, toxicity, metabolism at CYP450:3A4, CYP450:2D6, and predicted anti-inflammatory activity was carried out using the website www.chemosophia.com.39 However, TPSA, Lipinski violation, H donor, H acceptor, water solubility score, GI absorption score, and the possibility of metabolism at CYP1A2, CYP2C19, and CYP2C9 measured by http://www.swissadme.ch and Cresset software.40,41
Target selection and molecular modelling
To identify the possible receptors for the designed inhibitors, a target based search was performed in the DrugBank,42 which resulted in the identification of 10 potential protein molecules in P. falciparum. These are listed as “approved” drug targets in DrugBank and their structural information was obtained from the PDB and Uniprot databases. Moreover, the structures of calcium-dependent protein kinase 1 (CDK), cytochrome B (CYB), dihydropteroate synthetase (DPS) and malaria protein EXP-1 (MPEXP) were predicted by satisfying the spatial restraints using homology modelling. The available crystal structures of the proteins such as bifunctional dihydrofolate reductase thymidylate synthase (DRTS), dihydroorotate dehydrogenase quinone mitochondrial (DOD), ferredoxin NADP reductase apicoplast (FR), glutathione reductase (GR), glutathione-S-transferase (GST), plasmepsin II (PLM II), were further optimized using the “Prepare protein” module of Schrodinger suite. The structures of studied inhibitors were generated using the “2-D sketcher” of Schrodinger and then converted to three dimensional (3-D) coordinates, which were further optimized utilizing the “LigPrep” module.Molecular docking studies
Consequently, the generated optimized structures of the respective proteins and ligands were subjected to molecular docking based interaction analyses using AutoDock 4 package.43 The available protocol resulted in the generation of bound protein-ligand conformations, based on free energy calculations, which were performed based on of the empirical force field as well as the Lamarckian genetic algorithms.44 Primarily, the grid boxes of varied dimensions along the XYZ directions were created using the AutoGrid module with a spacing of 0.375 Å. The efficiency of the predictions was further enhanced by setting the docking parameters coupled with the Lamarckian genetic algorithm to their maximum values, which involved setting the number of individuals in the population to 250 as well as the maximum number of energy assessments to a “longer” interval. The 100 bound conformations were generated for each protein and ligand system (Total 60 systems) which were further grouped according to the RMSD tolerance of 2.0 Å. The free energy of binding was used as a parameter for assessing the efficiency of interactions, was calculated on the basis of the given equation.44Here, ΔGvdW symbolizes the van der Waals, ΔGH-bond represents hydrogen bonding while ΔGelec and ΔGsol stand for the free energy of electrostatics and solvation, respectively. The re-scoring of the generated docked complexes was performed using DrugScoreX server,45 which increases the efficiency of the predictions by incorporating knowledgebase scoring functions.
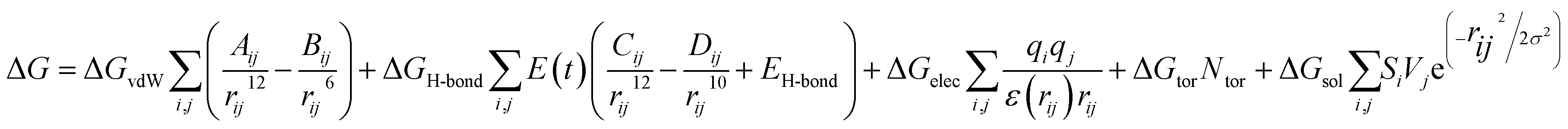
Molecular dynamics simulations
The docked complexes of PLM were subjected to MD simulations using GROMACS46 (version – 2018-2) for the validation of the interaction studies. Primarily, the topologies of the protein structure in different complexes were generated using the GROMOS96-53a6 force field46 and for the ligands topology generation, the PRODRG server47 was used, as GROMACS does not possess the topology generation ability for natural ligands. The PRODRG server lacks the functionality of generating the partial charges, therefore, the correction was performed using the DFT method implemented in GAUSSIAN which utilized the B3LYP6-31G(d,p) basis set and the CHELPG program.48 After successful topology generation, all the docked complexes were solvated by immersion in the SPC/E water model49 and in order to generate the stable systems, neutralization was performed by adding the counterions. The Na+ and Cl− ions were added to neutralize the systems. Consequently, the solvated and neutralized systems were subjected to energy minimization step by using combined steepest descent and conjugate gradient algorithms, with a convergence criterion of 0.005 kcal mol−1. The position restraints were applied to the structure of the minimized system ligands before the equilibration phase.The equilibration step was performed in a combination of NVT (constant volume) and NPT (constant pressure) ensemble conditions, each at 100 ps time scale. The temperature of 300 K was maintained for the system using Berendsen weak coupling method and pressure of 1 bar was maintained utilizing Parrinello-Rahman barostat in the equilibration stage. The final conformational production stage was carried out using the LINCS algorithm for 100 ns timescale and trajectories were generated, which were analyzed in order to understand the behavior of each complex in the explicit water environment. The changes in the H-bonds, as well as distances between the PLM II and designed inhibitors in the complex systems, were analyzed. Furthermore, the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) protocols implemented in g_mmpbsa package50 were used for the calculation of free energy of binding, which provides deeper insights into the interaction mechanisms of the protein and the ligand molecules.
Author contributions
BR, PK, APS and Poonam jointly conceptualized the idea, analyzed data and executed the study; NS, MB and SS performed synthesis and characterization; VR tested in vivo blood stage efficacy; YG and RM performed cytotoxicity and antiplasmodial efficacy against resistant strain and gametocyte activity; PP and MS performed the in silico analysis. NS, JS, MK undertook testing experiments on liver stage (in vitro and in vivo), and ookinete stage. All the authors contributed to writing, independent reviewing, and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BR is grateful to the Science and Engineering Research Board, New Delhi, for financial support (ECR/2015/000448). The work was supported by Act 211 Government of the Russian Federation, contract 02.A03.21.0011 and by the Ministry of Science and Higher Education of Russia (Grant FENU-2020-0019) to VP. APS is grateful to DBT, Govt of India, for financial support (BT/PR21569/NNT/28/1234/2017). Authors sincerely thank the Department of Medicine, Loyola University Medical Center and Stritch School of Medicine for providing the funding support for the Drug Discovery Program. NS is grateful to CSIR, Govt of India.References
- WHO, World malaria report, World Health Organization, Geneva, 2019 Search PubMed.
- M. A. Phillips, J. N. Burrows, C. Manyando, R. H. van Huijsduijnen, W. C. Van Voorhis and T. N. C. Wells, Malaria, Nat. Rev. Dis. Primers, 2017, 3, 17050, DOI:10.1038/nrdp.2017.50.
- A. F. Cowman, J. Healer, D. Marapana and K. Marsh, Malaria: Biology and Disease, Cell, 2016, 167, 610–624, DOI:10.1016/j.cell.2016.07.055.
- B. Galatas, Q. Bassat and A. Mayor, Malaria Parasites in the Asymptomatic: Looking for the Hay in the Haystack, Trends Parasitol., 2016, 32, 296–308, DOI:10.1016/j.pt.2015.11.015.
- T. N. C. Wells, R. H. van Huijsduijnen and W. C. Van Voorhis, Malaria medicines: a glass half full?, Nat. Rev. Drug Discovery, 2015, 14, 424–442, DOI:10.1038/nrd4573.
- N. Y. Gorobets, Y. V. Sedash, B. K. Singh, Poonam and B. Rathi, An overview of currently available antimalarials, Curr. Top. Med. Chem., 2017, 17, 2143–2157, DOI:10.2174/1568026617666170130123520.
- S. Bhatt, D. J. Weiss, E. Cameron, D. Bisanzio, B. Mappin, U. Dalrymple, K. E. Battle, C. L. Moyes, A. Henry, P. A. Eckhoff, E. A. Wenger, O. Briët, M. A. Penny, T. A. Smith, A. Bennett, J. Yukich, T. P. Eisele, J. T. Griffin, C. A. Fergus, M. Lynch, F. Lindgren, J. M. Cohen, C. L. J. Murray, D. L. Smith, S. I. Hay, R. E. Cibulskis and P. W. Gething, The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015, Nature, 2015, 526, 207–211, DOI:10.1038/nature15535.
- D. Menard and A. Dondorp, Antimalarial drug resistance: a threat to malaria elimination, Cold Spring Harbor Perspect. Med., 2017, 7, 1–24, DOI:10.1101/cshperspect.a025619.
- M. Imwong, T. Jindakhad, C. Kunasol, K. Sutawong, P. Vejakama and A. M. Dondorp, An outbreak of artemisinin resistant falciparum malaria in Eastern Thailand, Sci. Rep., 2015, 5, 1–7, DOI:10.1038/srep17412.
- A. P. Phyo, S. Nkhoma, K. Stepniewska, E. A. Ashley, S. Nair, R. McGready, C. L. Moo, S. Al-Saai, A. M. Dondorp, K. M. Lwin, P. Singhasivanon, N. P. J. Day, N. J. White, T. J. C. Anderson and F. Nosten, Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study, Lancet, 2012, 379, 1960–1966, DOI:10.1016/s0140-6736(12)60484-x.
- N. M. Pazhayam, J. Chhibber-Goel and A. Sharma, New leads for drug repurposing against malaria, Drug Discovery Today, 2019, 24, 263–271, DOI:10.1016/j.drudis.2018.08.006.
- N. J. White, Does antimalarial mass drug administration increase or decrease the risk of resistance?, Lancet Infect. Dis., 2017, 17, e15–e20, DOI:10.1016/s1473-3099(16)30269-9.
- S. Das, B. Saha, A. K. Hati and S. Roy, Evidence of Artemisinin-Resistant Plasmodium falciparum Malaria in Eastern India, N. Engl. J. Med., 2018, 379, 1962–1964, DOI:10.1056/nejmc1713777.
- CDC, https://www.cdc.gov/malaria/malaria_worldwide/impact.html, CDC Malaria Impact, https://www.cdc.gov/malaria/malaria_worldwide/impact.html, 2018.
- S.C.T.P. RTS, Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa : final results of a phase 3 , individually randomised , controlled trial, Lancet, 2017, 386, 31–45, DOI:10.1016/s0140-6736(15)60721-8.
- Poonam, Y. Gupta, N. Gupta, S. Singh, L. Wu, B. S. Chhikara, M. Rawat and B. Rathi, Multistage inhibitors of the malaria parasite: emerging hope for chemoprotection and malaria eradication, Med. Res. Rev., 2018, 38, 1511–1535, DOI:10.1002/med.21486.
- F.-J. Gamo, L. M. Sanz, J. Vidal, C. de Cozar, E. Alvarez, J.-L. Lavandera, D. E. Vanderwall, D. V. S. Green, V. Kumar, S. Hasan, J. R. Brown, C. E. Peishoff, L. R. Cardon and J. F. Garcia-Bustos, Thousands of chemical starting points for antimalarial lead identification, Nature, 2010, 465, 305–310, DOI:10.1038/nature09107.
- E. G. Tse, M. Korsik and M. H. Todd, The past, present and future of anti-malarial medicines, Malar. J., 2019, 18, 93 CrossRef PubMed.
- S. Yahiya, A. Rueda-Zubiaurre, M. J. Delves, M. J. Fuchter and J. Baum, The antimalarial screening landscape—looking beyond the asexual blood stage, Curr. Opin. Chem. Biol., 2019, 50, 1–9, DOI:10.1016/j.cbpa.2019.01.029.
- M. Delves, D. Plouffe, C. Scheurer, S. Meister, S. Wittlin, E. A. Winzeler, R. E. Sinden and D. Leroy, The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites, PLoS Med., 2012, 9, 1–14, DOI:10.1371/journal.pmed.1001335.
- R. Raphemot, D. Posfai and E. R. Derbyshire, Current therapies and future possibilities for drug development against liver-stage malaria, J. Clin. Invest., 2016, 126, 2013–2020, DOI:10.1172/jci82981.
- M. J. Delves, C. Miguel-Blanco, H. Matthews, I. Molina, A. Ruecker, S. Yahiya, U. Straschil, M. Abraham, M. L. León, O. J. Fischer, A. Rueda-Zubiaurre, J. R. Brandt, Á. Cortés, A. Barnard, M. J. Fuchter, F. Calderón, E. A. Winzeler, R. E. Sinden, E. Herreros, F. J. Gamo and J. Baum, A high throughput screen for next-generation leads targeting malaria parasite transmission, Nat. Commun., 2018, 9, 1–13, DOI:10.1038/s41467-018-04928-9.
- A. K. Singh, S. Rathore, Y. Tang, N. E. Goldfarb, B. M. Dunn, V. Rajendran, P. C. Ghosh, N. Singh, N. Latha, B. K. Singh, M. Rawat and B. Rathi, Hydroxyethylamine based phthalimides as new class of plasmepsin hits: design, synthesis and antimalarial evaluation, PLoS One, 2015, 10, 1–20, DOI:10.1371/journal.pone.0139347.
- S. Singh, V. Rajendran, J. He, A. K. Singh, A. O. Achieng, Vandana, A. Pant, A. S. Nasamu, M. Pandit, J. Singh, A. Quadiri, N. Gupta, Poonam, P. C. Ghosh, B. K. Singh, L. Narayanan, P. Kempaiah, R. Chandra, B. M. Dunn, K. C. Pandey, D. E. Goldberg, A. P. Singh and B. Rathi, Fast-Acting Small Molecules Targeting Malarial Aspartyl Proteases, Plasmepsins, Inhibit Malaria Infection at Multiple Life Stages, ACS Infect. Dis., 2019, 5, 184–198, DOI:10.1021/acsinfecdis.8b00197.
- A. K. Singh, V. Rajendran, S. Singh, P. Kumar, Y. Kumar, A. Singh, W. Miller, V. Potemkin, Poonam, M. Grishina, N. Gupta, P. Kempaiah, R. Durvasula, B. K. Singh, B. M. Dunn and B. Rathi, Antiplasmodial activity of hydroxyethylamine analogs: synthesis, biological activity and structure activity relationship of plasmepsin inhibitors, Bioorg. Med. Chem., 2018, 26, 3837–3844, DOI:10.1016/j.bmc.2018.06.037.
- B. Witkowski, C. Amaratunga, N. Khim, S. Sreng, P. Chim, S. Kim, P. Lim, S. Mao, C. Sopha, B. Sam, J. M. Anderson, S. Duong, C. M. Chuor, W. R. J. Taylor, S. Suon, O. Mercereau-Puijalon, R. M. Fairhurst and D. Menard, Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in cambodia: in vitro and ex vivo drug-response studies, Lancet Infect. Dis., 2013, 13, 1043–1049, DOI:10.1016/s1473-3099(13)70252-4.
- Y. Sato, S. Ries, W. Stenzel, S. Fillatreau and K. Matuschewski, The Liver-Stage Plasmodium Infection Is a Critical Checkpoint for Development of Experimental Cerebral Malaria, Front. Immunol., 2019, 10, 2554, DOI:10.3389/fimmu.2019.02554.
- N. Sharma, A. Verma, Poonam, P. Kempaiah and B. Rathi, Chemical libraries targeting Liver Stage Malarial infection, Chem. Biol. Lett., 2019, 6, 14 CAS.
- S. Singh, N. Sharma, C. Upadhyay, S. Kumar and B. Rathi, Poonam. Small Molecules Effective Against Liver and Blood Stage Malarial Infection, Curr. Top. Med. Chem., 2018, 18, 2008–2021, DOI:10.2174/1568026619666181129143623.
- R. N. Rabinovich, C. Drakeley, A. A. Djimde, B. F. Hall, S. I. Hay and J. Hemingway, et al., malERA: an updated research agenda for malaria elimination and eradication, PLoS Med., 2017, 14, e1002456, DOI:10.1371/journal.pmed.1002456.
- R. E. Sinden, Developing transmission-blocking strategies for malaria control, PLoS Pathog., 2017, 13, e1006336, DOI:10.1371/journal.ppat.1006336.
- V. Potemkin and M. Grishina, Principles for 3D/4D QSAR classification of drugs, Drug Discovery Today, 2008, 13, 952–959, DOI:10.1016/j.drudis.2008.07.006.
- O. A. Asojo, E. Afonina, S. V. Gulnik, B. Yu, J. W. Erickson, R. Randad, D. Medjahed and A. M. Silva, Structures of Ser205 mutant plasmepsin II from Plasmodium falciparum at 1.8 a in complex with the inhibitors rs367 and rs370, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 2001–2008 CrossRef PubMed.
- W. Peter, H. Portus and L. Robinson, The four day suppressive in vivo antimalarial test, Ann. Trop. Med. Parasitol., 1995, 69, 155–171, DOI:10.1080/00034983.1975.11686997.
- C. Ma, P. Harrison, L. Wang and R. L. Coppel, Automated estimation of parasitaemia of Plasmodium yoelii-infected mice by digital image analysis of Giemsa-stained thin blood smears, Malar. J., 2010, 9, 348, DOI:10.1186/1475-2875-9-348.
- T. L. Riss, et al., Cell viability assays, Assay Guidance Manual, Eli Lilly & Company and the National Centre for Advancing Translational Sciences, 2016, https://www.ncbi.nlm.nih.gov/books/NBK144065 Search PubMed.
- V. Potemkin, O. Galimova and M. Grishina, Cinderella's Shoe for virtual drug discovery screening and design, Drugs Future, 2010, 35, 14–15 Search PubMed.
- A. Potemkin, M. Grishina and V. Potemkin, Grid-based Continual Analysis of Molecular Interior for Drug Discovery, QSAR and QSPR, Curr. Drug Discovery Technol., 2017, 14, 181, DOI:10.2174/1570163814666170207144018.
- ChemoSophia, Chemosophia s.r.o., Prague, Czech Republic, 2018, http://www.chemosophia.com Search PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation, J. Chem. Inf. Model., 2006, 46, 665–676, DOI:10.1021/ci050357s.
- D. S. Wishart, C. Knox, A. C. Guo, D. Cheng, S. Shrivastava, D. Tzur, B. Gautam and M. Hassanali, DrugBank: a knowledgebase for drugs, drug actions and drug targets, Nucleic Acids Res., 2008, 36, D901–D906, DOI:10.1093/nar/gkm958.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- G. Neudert and G. Klebe, DSX: a knowledge-based scoring function for the assessment of protein-ligand complexes, J. Chem. Inf. Model., 2011, 51, 2731–2745, DOI:10.1021/ci200274q.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854, DOI:10.1093/bioinformatics/btt055.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6, J. Comput. Chem., 2004, 25, 1656–1676, DOI:10.1002/jcc.20090.
- A. W. Schuttelkopf and D. M. van Aalten, PRODRG: a tool for high-throughput crystallography of protein-ligand complexes, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363, DOI:10.1107/s0907444904011679.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, et al., Gaussian 2009, 09, Gaussian, Inc Search PubMed.
- J. Zielkiewicz, Structural properties of water: comparison of the SPC, SPCE, TIP4P, and TIP5P models of water, J. Chem. Phys., 2005, 123, 104501, DOI:10.1063/1.2018637.
- R. Kumari, R. Kumar and A. Lynn, g_mmpbsa – a GROMACS tool for high-throughput MM-PBSA calculations, J. Chem. Inf. Model., 2014, 54, 1951–1962, DOI:10.1021/ci500020m.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03997g |
| This journal is © The Royal Society of Chemistry 2020 |