
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceK2S2O8-mediated radical cyclisation of 2-alkynylthioanisoles or -selenoanisoles: a green and regioselective route to 3-nitrobenzothiophenes and benzoselenophenes†
Shi-Chao Lu ab,
Botao Wuc,
Shi-Peng Zhanga,
Ya-Ling Gonga and
Shu Xu*a
ab,
Botao Wuc,
Shi-Peng Zhanga,
Ya-Ling Gonga and
Shu Xu*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, and Beijing Key Laboratory of Active Substance Discovery and Druggability Evaluation, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 2A NanWei Road, Xicheng District, Beijing 100050, P. R. China. E-mail: xushu@imm.ac.cn
bState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P. R. China
cCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China
First published on 19th May 2020
Abstract
An acid, transition-metal, and chromatography-free radical nitration/cyclisation of 2-alkynylthioanisoles or -selenoanisoles has been developed. This is the first example of the use of highly unstable 2-nitrovinyl radicals for C–S bond formation. This facile route efficiently produces 3-nitrobenzothiophenes and benzoselenophenes, which are difficult to access via classical methods. Density functional theory (DFT) calculations were carried out to probe the reaction mechanism. The resulting products were tested for their in vitro anti-tuberculosis activity, and compounds 2d and 2l showed significant activities against sensitive and drug-resistant strains.
The nitro group is considered an important and unique functional group within medicinal chemistry, owing to its strong electron withdrawing ability and hydrogen bonding ability. The introduction of a nitro group to (hetero)arenes typically has a pronounced effect on the parent molecule's biological activity and generally causes marked changes in their physical (e.g., lipophilicity and solubility) and chemical properties.1 As the need for more novel drugs becomes paramount, medicinal chemists have begun to revaluate the use of nitro groups within clinical candidates, and thus their synthesis remains an important area of research. As part of an ongoing program within our group to design and synthesise novel anti-tuberculosis drugs containing nitro groups,2 we became very interested in 3-nitrobenzothiophenes due to the fact that these scaffolds are privileged structures within drug discovery.3 A literature search for these compounds showed that the synthetic methods to access 3-nitrobenzothiophenes are not well-developed (Fig. 1). The majority of reported strategies rely on the nitration of benzothiazoles through electrophilic substitution reactions in fuming nitric acid.4 Although successful, this approach has several major disadvantages including poor regioselectivity, low yield, and large amounts of harmful by-products. In particular, the regioselective nitration of benzothiazoles containing other electron-rich (hetero)arenes is difficult, impeding their application in further biological studies (Fig. 1a).4 Thus, the development of novel, green, and efficient methods for construction of 3-nitrobenzothiophenes and their derivatives is highly desirable.
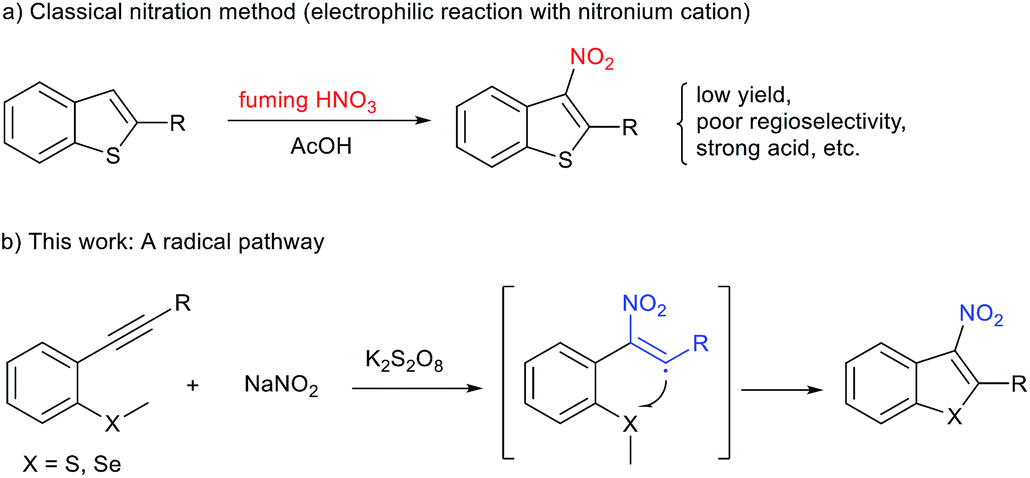 | ||
| Fig. 1 Methods for the synthesis of 3-nitrobenzothiophenes. | ||
Recently, significant progress in the synthesis of nitroalkenes via radical addition to alkenes has been achieved in an atom- and step-economical manner.5 However, methods for constructing nitroalkenes via 2-nitrovinyl species generated from the addition of a reactive nitrogen dioxide radical to alkynes is much less common. This is almost certainly due to the difficulty of trapping out these 2-nitrovinyl intermediates (σ sp2 radicals) due to their inherent instability and subsequent short lifetime.6 To date, only few examples have described the use of 2-nitrovinyl radicals in the difunctionalisation of alkynes through subsequent C–I, C–O, or C–C bond formation.6–8 However, to the best of our knowledge, 2-nitrovinyl radicals have not been used for C–S bond-forming reactions. Based on our previous experience in radical cyclisation reactions and preparing nitro-containing heterocycles,9 we envisaged the possibility of trapping these 2-nitrovinyl radicals using sulfur. This approach would provide the community with ready access to 3-nitrobenzothiophenes in a highly regioselective manner (Fig. 1b). At the outset of this study, we envisaged two major challenges to overcome. Firstly, we have to control the reactivity of the NO2 radical, allowing it to selectivity attack the alkyne. This then produces our desired vinyl radical that undergoes cyclisation and afford the desired product. Secondly, we had to ensure that we used a solvent that inhibited the unproductive oxidation of thioether.
We began our investigation employing methyl(2-(phenylethynyl)phenyl)sulfane (1a) as the model substrate in the presence of 2.5 equiv. of t-BuONO5c in N,N-dimethylformamide (DMF) under air at 110 °C for 12 h (entry 1, Table 1). We obtained 2a in 55% yield with its structure confirmed by X-ray crystallography.10 To get an insight into the reaction, we performed some mass balance experiments and observed large quantities of unwanted sulfoxide by-products. To avoid this, we screened other NO2 sources in an attempt to optimize the reaction. After some experimentation we observed that treatment of substrate 1a with the heterogeneous nitration system (KNO2/K2S2O8 or NaNO2/K2S2O8) in CH3CN,11 2a was produced in 80% and 87%, respectively (Table 1, entries 2 and 3). The reaction proceeded equally well under an argon atmosphere. The investigation of the reaction temperature showed that 110 °C was the optimum temperature for this transformation (Table 1, entries 4 and 5). We then examined the effect of other solvent systems on the model reaction (Table 1, entries 6–11) and found that CH3CN the optimum choice. Replacing K2S2O8 with other oxidants, such as Na2S2O8, (NH4)2S2O8, and O2 decreased the yield (Table 1, entries 12–14). In addition, decreasing the amount of K2S2O8 or NaNO2 led to a lower yield (Table 1, entries 15–17). Consequently, the optimised conditions were 2.0 equiv. of NaNO2 and 2.0 equiv. of K2S2O8 in CH3CN under air at 110 °C for 12 h.
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (equiv.) | NO2 source (equiv.) | Solvent | Temp. (°C) | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), oxidant (0.4 mmol), nitrate (0.4 mmol), and solvent (1.5 mL) under air at indicated temperature for 12 h.b Under Ar. | |||||
| 1 | — | t-BuONO (2.5) | DMF | 110 | 55 |
| 2 | K2S2O8 (2) | KNO2 (2) | CH3CN | 110 | 80 |
| 3 | K2S2O8 (2) | NaNO2 (2) | CH3CN | 110 | 87(86b) |
| 4 | K2S2O8 (2) | NaNO2 (2) | CH3CN | 120 | 78 |
| 5 | K2S2O8 (2) | NaNO2 (2) | CH3CN | 100 | 50 |
| 6 | K2S2O8 (2) | NaNO2 (2) | H2O | 110 | 56 |
| 7 | K2S2O8 (2) | NaNO2 (2) | C2H5OH | 110 | 0 |
| 8 | K2S2O8 (2) | NaNO2 (2) | EtOAc | 110 | 54 |
| 9 | K2S2O8 (2) | NaNO2 (2) | H2O/CH3CN | 110 | 38 |
| 10 | K2S2O8 (2) | NaNO2 (2) | H2O/EtOH | 110 | 0 |
| 11 | K2S2O8 (2) | NaNO2 (2) | H2O/EtOAc | 110 | 52 |
| 12 | Na2S2O8 (2) | NaNO2 (2) | CH3CN | 110 | 64 |
| 13 | (NH4)2S2O8 (2) | NaNO2 (2) | CH3CN | 110 | 16 |
| 14 | O2 | NaNO2 (2) | CH3CN | 110 | 0 |
| 15 | K2S2O8 (1.5) | NaNO2 (2) | CH3CN | 110 | 67 |
| 16 | K2S2O8 (1) | NaNO2 (2) | CH3CN | 110 | 44 |
| 17 | K2S2O8 (2) | NaNO2 (1.5) | CH3CN | 110 | 58 |
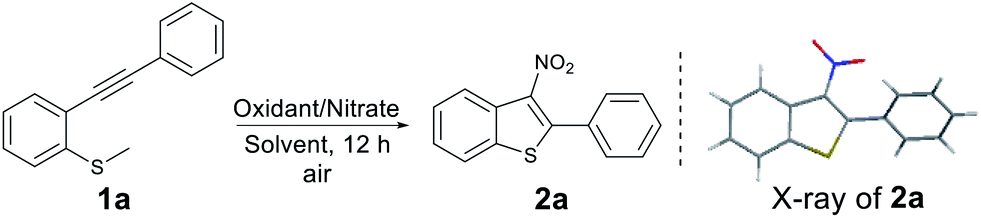
With the optimised conditions in hand, we turned our attention to the scope and generality of the reaction using various 2-alkynylthioanisoles as substrates (Table 2). As shown the method was amenable to a variety of 2-alkynylthioanisoles with an aryl group at the R1 position, affording the desired products in moderate to good yields (Table 2, 2a–2o). Among them, halogens at various positions on the phenyl rings were compatible, allowing further transformations through traditional cross-coupling reactions. In addition, stronger electron-withdrawing substituents such as –CN or –NO2 on the phenyl ring gave yields of 55% and 46%, respectively (Table 2, 2j and 2k). The radical cyclisation reaction was also suitable for heterocyclic 2-alkynylthioanisoles and produced the corresponding products in acceptable yields (Table 2, 2l–2o). It is important to note here that in these examples, classical aromatic electrophilic nitration would provide the products with poor selectivity. We also explored the effect the substitution pattern on the aromatic ring of the thiomethyl group had on the reaction. Once again, as shown, these were tolerated, providing the products in synthetically useful yields. (Table 2, 2p–2s). However, substrates bearing n-butyl or trimethylsilyl groups failed to afford the desired products under our optimised conditions (Table 2, 2t and 2u). It is assumed that the resulting vinyl radicals from these two substrates could not be delocalized around the phenyl ring's π orbital, and thus is highly unstable.
| a Reaction conditions: 1a (0.2 mmol), oxidant (0.4 mmol), nitrate (0.4 mmol), and CH3CN (1.5 mL) under air at 110 °C for 12 h. |
|---|
 |
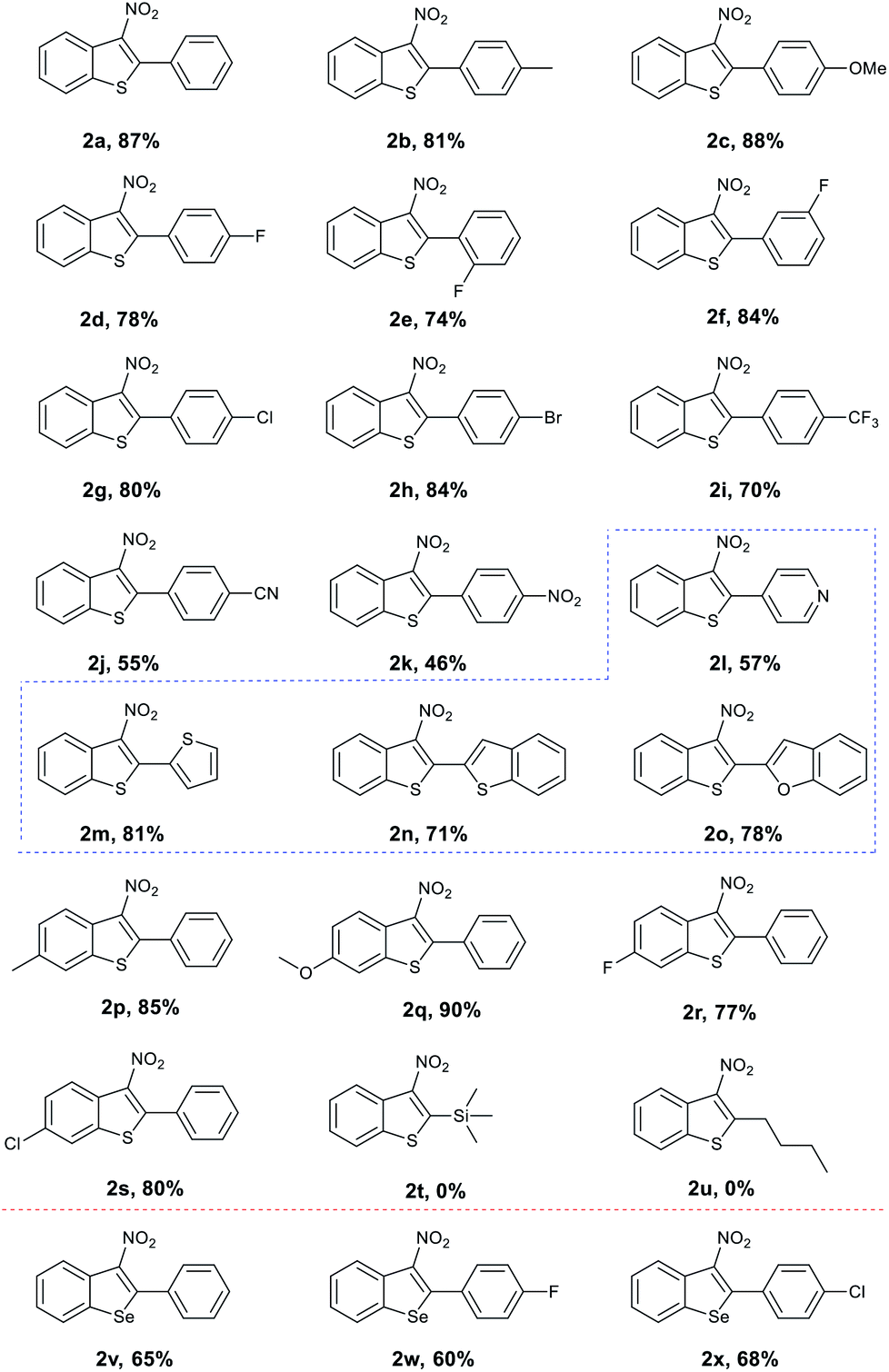 |
Encouraged by these results, we explored the radical cyclisation reaction of 2-alkynylselenoanisoles (Table 2, 2v–2x). We were surprised to find that upon treating the seleno variants to our conditions, 3-nitrobenzoselenophenes were obtained in moderate yields. In addition, we evaluated the reactivity of more substrates such as 1-methoxy-2-(phenylethynyl)benzene and N,N-dimethyl-2-(phenylethynyl)aniline, toward the present reaction conditions. However, these substrates failed to afford the desired products.
The reaction with the model substrates was easily scaled up to 5 mmol, affording 2a in 75% yield. To investigate structure–activity relationships of their anti-tuberculosis activity, further synthetic transformation of the 3-nitrobenzothiophenes were performed (Scheme 1). The NO2 group was easily reduced to afford 2-phenylbenzo[b]thiophen-3-amine (3) in 92% yield, and 2a was transformed to 4 with m-CPBA as an oxidant in 78% yield.
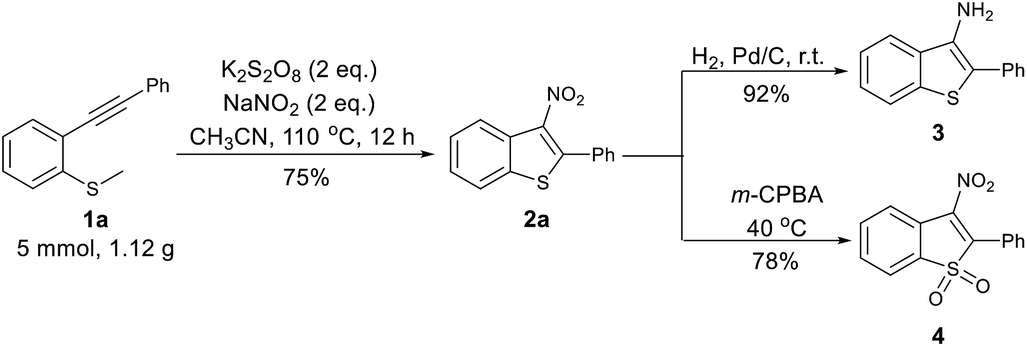 | ||
| Scheme 1 Gram scale-up and the transformation of product. | ||
To gain insights into the reaction mechanism, some control experiments were performed (Scheme 2). To rule out the possibility of 2-phenylbenzo[b]thiophene 5 as an intermediate in the reaction we subjected 2-phenylbenzo[b]thiophene 5 to our standard reactions conditions. We observed no product and recovered the majority of starting material. Furthermore, when radical scavengers butylated hydroxytoluene (BHT) or (2,2,6,6-tetramethylpiperdin-1-yl)oxyl (TEMPO) were added to the reaction mixture, the reaction was inhibited (Scheme 2b), and Me-TEMPO (6) was detected by high-resolution mass spectrometry. These results indicate that the tandem cyclisation reaction proceeded through a radical mechanism and generation of a methyl radical. When the methyl group on the sulfur atom was changed to a phenyl group, 2a was also obtained in a yield of 32%, and benzene was detected by GC-MS (Scheme 2c). This indicates that the released phenyl radical underwent hydrogen abstraction from the reaction mixture. A cationic mechanism involving electrophilic addition of NO2+ to alkynes can therefore be excluded.
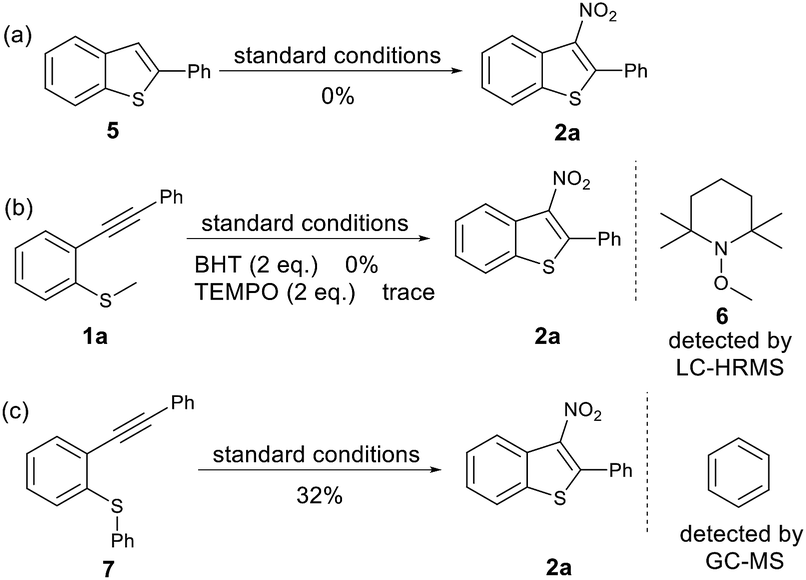 | ||
| Scheme 2 Control experiments. | ||
Based on these control experiments and previous studies,6c,12 a plausible reaction mechanism for this radical cyclisation reaction is proposed (Scheme 3). Initially, NaNO2 is converted into a NO2 radical (A) in the presence of K2S2O8. Subsequently, radical A is selectively added to the C–C triple bond of 1a, which provides the vinyl radical (B) that undergoes intramolecular cyclisation with the SMe moiety. Finally, the desired product 2a is obtained along with the release of the methyl radical that further reacts via H-abstraction from the reaction mixture.
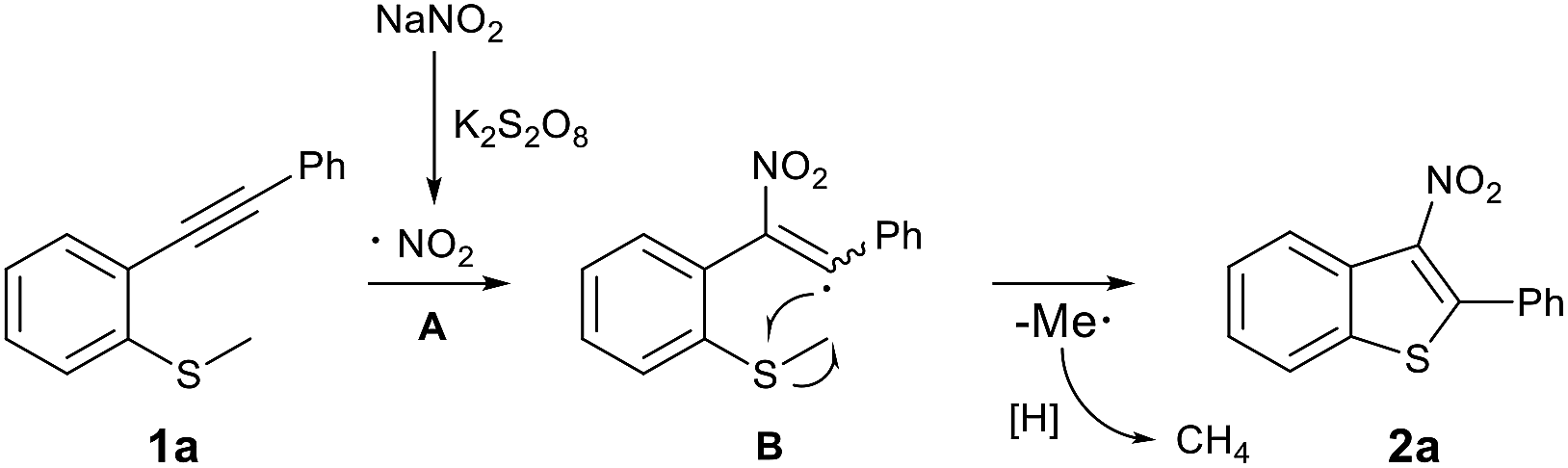 | ||
| Scheme 3 Proposed mechanism. | ||
In order to confirm if the proposed mechanism is correct, a series of density functional theory (DFT) calculations were carried out. The reaction pathway with the intermediates and transition states are shown in Fig. 2. Nitrite can be converted to NO2 radical by a single-electron oxidation process.12 Based on the B3LYP(D3BJ)/6-311G** level of theory13 in Gaussian 09 program,14 we have found that NO2 radical is regioselectively added to the C–C triple bond of 1a via TS1 with a barrier of 17.93 kcal mol−1, providing the vinyl-typed radical B. An unreactive NO2 radical attacking an alkyne to produce a highly reactive vinyl radical is somewhat disfavoured. To overcome this barrier, elevated temperature is required, as per our optimised reaction conditions. The resulting vinyl radical B could be delocalised into the phenyl ring π orbital system, and the ΔG of system drops to 14.16 kcal mol−1. Obviously, the substrates bearing aryl groups are favoured, as observed during our investigation which is borne out in our experimental observations that the desired products 2t and 2u could not be obtained under the present conditions. Subsequently, intermediate B undergoes intramolecular cyclisation via TS2 with the SMe moiety, and the ΔG‡ is increased to 20.12 kcal mol−1. With the formation of methyl radical and a benzo-thiophene ring, the ΔG of system drops to −3.03 kcal mol−1 due to the decisive thermodynamic stabilisation by the aromatic effect of π109.
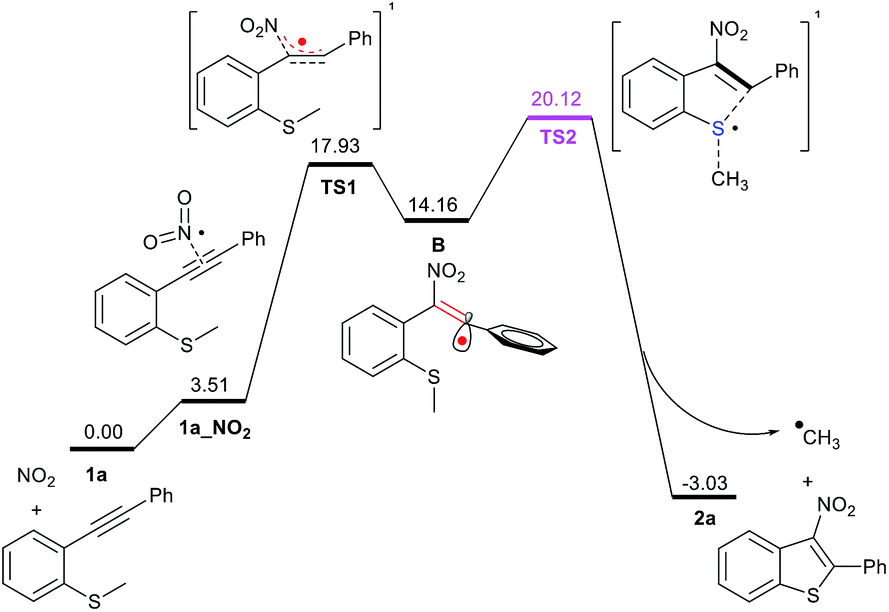 | ||
| Fig. 2 The relative free energies (ΔG, in black numbers) are given in kcal mol−1. Calculated at the B3LYP(D3BJ)/6-311G** level. | ||
With this library of compounds in hand, we investigated their anti-tuberculosis activity using the microplate alamarBlue assay.15 The details can be found in the ESI (Table S2†). The results showed compounds 2d and 2l exhibited the highest inhibitory activities against sensitive and drug-resistant Mycobacterium tuberculosis (Mtb) strains, with MIC90 values of 2–4 μg mL−1 3-Nitrobenzoselenophenes 2w also showed the potent anti-tuberculosis activity against drug-resistant Mtb strains, with a MIC90 value of 2 μg mL−1. Moreover, the necessity of the nitro group was supported by the amino-substituted compound 3, which led to complete loss of activity. These results makes it possible for us to develop novel anti-Mtb agents.
In conclusion, we have developed a green and regioselective route to 3-nitrobenzothiophenes and benzoselenophenes through NaNO2/K2S2O8-mediated radical cyclisation. Compared to the nitration with fuming nitric acid, this strategy features excellent selectivity, no chromatography, mild reaction conditions, and broad functional group tolerance. The applications of our methodology and further structural modifications for developing the novel anti-tuberculosis drug are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is financially supported by the National Natural Science Foundation of China (21602256 and 21672262), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-3-009 and 2017-I2M-2-004), the Drug Innovation Major Project (2018ZX09711001-005-001), the Fundamental Research Funds for CAMS/PUMC (2018RC350006), and the State Key Laboratory of Natural and Biomimetic Drugs (K20170205). We thank Hairong Huang, Beijing Chest Hospital for her assistance with the microplate alamarBlue assay.Notes and references
- K. Nepali, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62, 2851–2893 CrossRef CAS PubMed.
- (a) P. Zhang, S. Somersan-Karakaya, S. Lu, J. Roberts, M. Pingle, T. Warrier, D. Little, X. Guo, S. J. Brickner, C. F. Nathan, B. Gold and G. Liu, J. Med. Chem., 2014, 57, 3755–3772 CrossRef PubMed; (b) Y. Gong, S. Somersan-Karakaya, X. Guo, P. Zheng, B. Gold, Y. Ma, D. Little, J. Roberts, T. Warrier, X. Jiang, M. Pingle, C. F. Nathan and G. Liu, Eur. J. Med. Chem., 2014, 75, 336–353 CrossRef CAS.
- (a) R. S. Keri, K. Chand, S. Budagumpi, S. S. Balappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed; (b) J. Xu, X. Yu, J. Yan and Q. Song, Org. Lett., 2017, 19, 6292–6295 CrossRef CAS PubMed; (c) T. Cai, J. Liu, H. Zhang, X. Wang, J. Feng, R. Shen and Y. Gao, Org. Lett., 2019, 21, 4605–4608 CrossRef CAS; (d) X. Gong, M. Wang, S. Ye and J. Wu, Org. Lett., 2019, 21, 1156–1160 CrossRef CAS PubMed; (e) W. Liu, Y. Q. Hu, X. Y. Hong, G. X. Li, X. B. Huang, W. X. Gao, M. C. Liu, Y. Xia, Y. B. Zhou and H. Y. Wu, Chem. Commun., 2018, 54, 14148–14151 RSC; (f) Z. Zhang and W. Tang, Acta Pharm. Sin. B, 2018, 8, 721–732 CrossRef PubMed.
- K. E. Chippendale, B. Iddon and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1972, 1, 2023–2030 RSC.
- (a) S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355–3358 CrossRef CAS PubMed; (b) Y. Liu, J.-L. Zhang, R.-J. Song, P.-C. Qian and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 9017–9020 CrossRef CAS PubMed; (c) T. Shen, Y.-Z. Yuan and N. Jiao, Chem. Commun, 2014, 50, 554–556 RSC; (d) V. Motornov, V. M. Muzalevskiy, A. A. Tabolin, R. A. Novikov, Y. V. Nelyubina, V. G. Nenajdenko and S. L. loffe, J. Org. Chem., 2017, 82, 5274–5284 CrossRef CAS PubMed; (e) Y. Chen, Y. Ma, L. Li, H. Jiang and Z. Li, Org. Lett., 2019, 21, 1480–1483 CrossRef CAS PubMed.
- For C–O bond formation, see (a) U. Dutta, S. Maity, R. Kancherla and D. Maiti, Org. Lett., 2014, 16, 6302–6305 CrossRef CAS PubMed; (b) H. Yan, G. Rong, D. Liu, Y. Zheng, J. Chen and J. Mao, Org. Lett., 2014, 16, 6306–6309 CrossRef CAS PubMed; (c) T.-S. Zhang, L. Yang, P.-J. Cai, S.-J. Tu and B. Jiang, Chem. - Asian J., 2019, 14, 4383–4388 CrossRef CAS PubMed.
- For C–I bond formation, see (a) M. S. Yusubov, I. A. Perederina, V. D. Filimonov, T.-H. Park and K.-W. Chi, Synth. Commun., 1998, 28, 833–836 CrossRef CAS; (b) S. Hlekhlai, N. Samakkanad, T. Sawangphon, M. Pohmakotr, V. Reutrakul, D. Soorukram, T. Jaipetch and C. Kuhakarn, Eur. J. Org. Chem., 2014, 2014, 7433–7442 CrossRef CAS; (c) X. Xu, X. Li, Y. Fan, B. Zhou, K. Chen and B. Wang, Synlett, 2017, 28, 1657–1659 CrossRef.
- For C–C bond formation, see (a) X. H. Hao, P. Gao, X. R. Song, Y. F. Qiu, D. P. Jin, X. Y. Liu and Y. M. Liang, Chem. Commun., 2015, 51, 6839–6842 RSC; (b) X.-H. Yang, X.-H. Ouyang, W.-T. Wei, R.-J. Song and J.-H. Li, Adv. Synth. Catal., 2015, 357, 1161–1166 CrossRef CAS; (c) W. Liu, Y. Zhang and H. Guo, J. Org. Chem., 2018, 83, 10518–10524 CrossRef CAS PubMed.
- (a) S. C. Lu, P. R. Zheng and G. Liu, J. Org. Chem., 2012, 77, 7711–7717 CrossRef CAS PubMed; (b) S. C. Lu, Y. L. Gong and D. M. Zhou, J. Org. Chem., 2015, 80, 9336–9341 CrossRef CAS PubMed; (c) S. C. Lu, H. S. Li, Y. L. Gong, S. P. Zhang, J. G. Zhang and S. Xu, J. Org. Chem., 2018, 83, 15415–15425 CrossRef CAS PubMed.
- It is difficult to determine its structure by NMR, because compound 2a contains three continuous quaternary carbon centers, and the known data in the literature4 is incomplete.
- S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085–5144 CrossRef CAS.
- Y.-M. Li, X.-H. Wei, X.-A. Li and S.-D. Yang, Chem. Commun., 2013, 49, 11701–11703 RSC.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Mont-gomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Nor-mand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- Z. Zong, W. Jing, J. Shi, S. Wen, T. Zhang, F. Huo, Y. Shang, Q. Liang, H. Huang and Y. Pang, Antimicrob. Agents Chemother., 2018, 62, e00165-18 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1914313. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03894f |
| This journal is © The Royal Society of Chemistry 2020 |