
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel liquid crystalline and fire retardant molecules based on six-armed cyclotriphosphazene core containing Schiff base and amide linking units†
Zuhair Jamain *ab,
Melati Khairuddean*b and
Tay Guan-Sengc
*ab,
Melati Khairuddean*b and
Tay Guan-Sengc
aFaculty of Science and Natural Resources, Universiti Malaysia Sabah (UMS), 88400 Kota Kinabalu, Sabah, Malaysia. E-mail: zuhairjamain@ums.edu.my
bSchool of Chemical Sciences, Universiti Sains Malaysia (USM), 11800 Penang, Malaysia. E-mail: melati@usm.my
cSchool of Industrial Technology, Universiti Sains Malaysia (USM), 11800 Penang, Malaysia
First published on 4th August 2020
Abstract
Nucleophilic substitution reaction between 4-hydroxybenzaldehyde and hexachlorocyclotriphosphazene, HCCP formed hexakis(4-formlyphenoxy)cyclotriphosphazene, 1. Intermediates 2a–e was formed from the alkylation reaction of methyl 4-hydroxybenzoate with alkyl bromide which further reduced to form benzoic acid intermediates. Further reaction of 2a–e and other substituted benzoic acid formed 3a–h, which then reduced to give subsequent amines, 4a–h. Other similar reaction was used to synthesis 4i. Condensation reaction between 1 and 4a–i yielded hexasubstituted cyclotriphosphazene compounds, 5a–i having Schiff base and amide linking units, and these compounds consist of different terminal substituents such as heptyl, nonyl, decyl, dodecyl, tetradecyl, hydroxy, carboxy, chloro, and nitro groups, respectively. Compound 5j with amino substituent at terminal end was formed from the reduction of 5i. All the intermediates and compounds were characterized using Fourier Transform Infrared (FT-IR), Nuclear Magnetic Resonance (NMR) and CHN elemental analysis. Mesophase texture of these compounds were determined using Polarized Optical Microscope (POM) and their mesophase transition were further confirmed using Differential Scanning Calorimetry (DSC). Only compounds 5a–e with alkoxy chains exhibited smectic A phase while other intermediates (1, 2a–e, 3a–h, and 4a–i) and final compounds (5f–j) are found to be non-mesogenic with no liquid crystal behaviour. The confirmation of the identity of the SmA phase was determined using XRD analysis. The study on the structure–properties relationship was conducted in order to determine the effect of the terminal group, length of the chains and linking units to the mesophase behaviour of the compounds. Moreover, the fire retardant properties of these compounds were determined using Limiting Oxygen Index (LOI) testing. Polyester resin with LOI value of 22.53% was used as matrix for moulding in the study. The LOI value increased to 24.71% when this polyester resin incorporated with 1 wt% of HCCP. Generally, all the final compounds showed a positive results with LOI value above 27% and the highest LOI value was belonged to compound 5i with 28.53%. The high thermal stability of the Schiff base molecules and the electron withdrawing group of the amide bonds and nitro group enhanced the fire retardant properties of this compound.
1. Introduction
In the early years, Allcock and his co-workers started the research work on phosphazene compounds which was then developed into the investigation of phosphazene liquid crystalline materials.1,2 Hexachlorocyclotriphosphazene, HCCP (N3P3Cl6) is a ring consisting of alternating phosphorus and nitrogen atoms, whereby six chlorine atoms can be substituted with any selected nucleophiles. Due to the high reactivity of the P–Cl bond, the corresponding substitution method allows the structure–activity studies in the side arm and their multiarmed rigid ring allows the exploration of the discotic structures.3 It was reported that compounds with the cyclotriphosphazene core (N3P3) attached to different substrates exhibited different physical and chemical properties.4 They have been used in many fields such as liquid crystal and fire retardant applications due to their fascinating properties.5,6Liquid crystal (LC) is an anisotropic material with flowing properties (liquid) as well as ordered (solid) and optical properties.7 Since it has the features of both fluid and solid, it became an important intermediate phase or mesomorphic phase.8 LC of organic compounds is divided into two categories which are lyotropic and thermotropic LC. Thermotropic LC are influenced by the change in temperature while lyotropic LC which are composed mainly of the amphiphilic bilayer are concentration and temperature dependant.9,10
The influence of different elements and the extended chemical subunits on the molecules allows the construction of the targeted liquid crystal compounds. The molecular shape and the terminal chain length are the key variables in designing new liquid crystal compounds with specific types of molecular organization in a particular range of temperature.11–13 Linking units are normally structural units that connect one core to another, which maintain the linearity of the core while being compatible with the rest of the structure. A linking unit between ring systems is to increase the length of the molecules and to alter the polarisability and flexibility of the molecules.14 Schiff base is one the interesting linking unit that provide a stepped core structure which can maintain the linearity of the molecules to provide high stability. This enables the mesophase formation whereby the phase transition temperature and the physical properties changes are usually contributed by the linking group.15 On the other hand, the formation of an amide linking unit led to higher rigidity due to the presence of the partial double bond character of the C–N bond which resulted in higher transition and clearing temperatures of the mesogens.16 The partial double bond in C–N bond reduces the coplanarity of the molecule and the broadening of the rigid core, which resulted in the molecules that favoured a lamellar arrangement in the smectic layer.17 The correlation between the molecular structure such as core system, linking units and terminal groups are the most important aspect in liquid crystal field.
Jiménez et al. (2011) reported the synthesis of hexasubstituted cyclotriphosphazene derivatives attached to the side arms with an ester linking unit but different terminal chains. The formation of the mesophase depended on the balance between the rigid core and the flexible terminal chains while the type and stability of the mesophase were determined by the space filling and nanosegregation. The combination of these factors along with tailoring of the interface curvature by the space requirements of the incompatible molecular blocks will determines the difference mesophase morphologies of these cyclotriphosphazene molecules.18 Moreover, He et al. (2013) have synthesized the room temperature columnar mesogens of hexasubstituted cyclotriphosphazenes compounds with Schiff base and ether linking units. They found out that increasing the chain length increased the melting point and decreased the clearing point.19
On the other side, HCCP also known to be used as a molecule to have high fire retardancy. Fire retardant materials play an important role in protecting property from damage. Fire retardation occurred due to the removal of heat from the materials that can burn and the formation of char during the fire which will interrupt the contact from combustion.20 Generally, materials with limiting oxygen index (LOI) value above 26% are considered fire retardant and self-extinguishing behaviour can be observed.21,22 Zhang et al. (2012) have synthesized and studied the fire retardant properties of hexakis(4-nitrophenoxy)cyclotriphosphazene on poly(ethylene terephthalate). The LOI value of poly(ethylene terephthalate) increased from 26.8% to 35.1% when 10 wt% of this compound was added.23 Moreover, Rong et al. have conducted two independent studies on the cyclotriphosphazene core system in 2015 and 2017. Their findings showed that the modification of cyclotriphosphazene core system able to induce the thermal stability, which enhances the fire resistance of the compounds.24,25
Therefore, the interest of the research is to gain a better insight of the structure–properties relationship of these types of compounds. Addition of HCCP into a compound is to increase the resistance of the material towards ignition. Both liquid crystal and fire retardant properties were studied. Thus, this research focused on the preparation of a series of hexasubstituted cyclotriphosphazene derivatives with Schiff base and amide linking units that bore a different terminal end. In this work, polyester resin will be used as a medium for moulding in order to study the fire retardant of the compounds. Only 1 wt% is added in this resin in order to achieve highest fire retardancy with less additive usage. Polyester resin is chosen due to good mechanical properties, fast curing, low cost and more sensitive to elevated temperatures.
2. Results and discussion
In this work, HCCP has been used as a core system to prepare hexasubstituted cyclotriphosphazene compounds consist of Schiff base and amide linking units. Intermediate 1 was synthesized from the reaction of HCCP with 4-hydroxybenzaldehyde. Methyl 4-hydroxybenzoate with different alkyl bromide (heptyl, nonyl, decyl, dodecyl, and tetradecyl) were used to form methyl 4-(alkoxy)benzoates. Without isolation, these methyl 4-(alkoxy)benzoates were further reduced to form subsequent benzoic acid intermediates, 2a–e. Intermediates 3a–h with amide linking unit were afforded from the reaction of 2a–e and some para substituted benzoic acid such as hydroxy, carboxy, and chloro, respectively. These intermediates were then reduced to form 4a–h. Intermediate 4i was prepared using different synthetic route. Finally, condensation reaction of 1 with 4a–i yielded final compounds 5a–i with Schiff base and amide linking units. Further reduction of 5i formed compound 5j with amino group at the terminal end. The structures of these intermediates and final compounds were confirmed using FTIR, NMR and CHN elemental analysis. The overall synthesis pathway for intermediates and hexasubstituted cyclotriphosphazene compounds for this series are described in Schemes 1 and 2.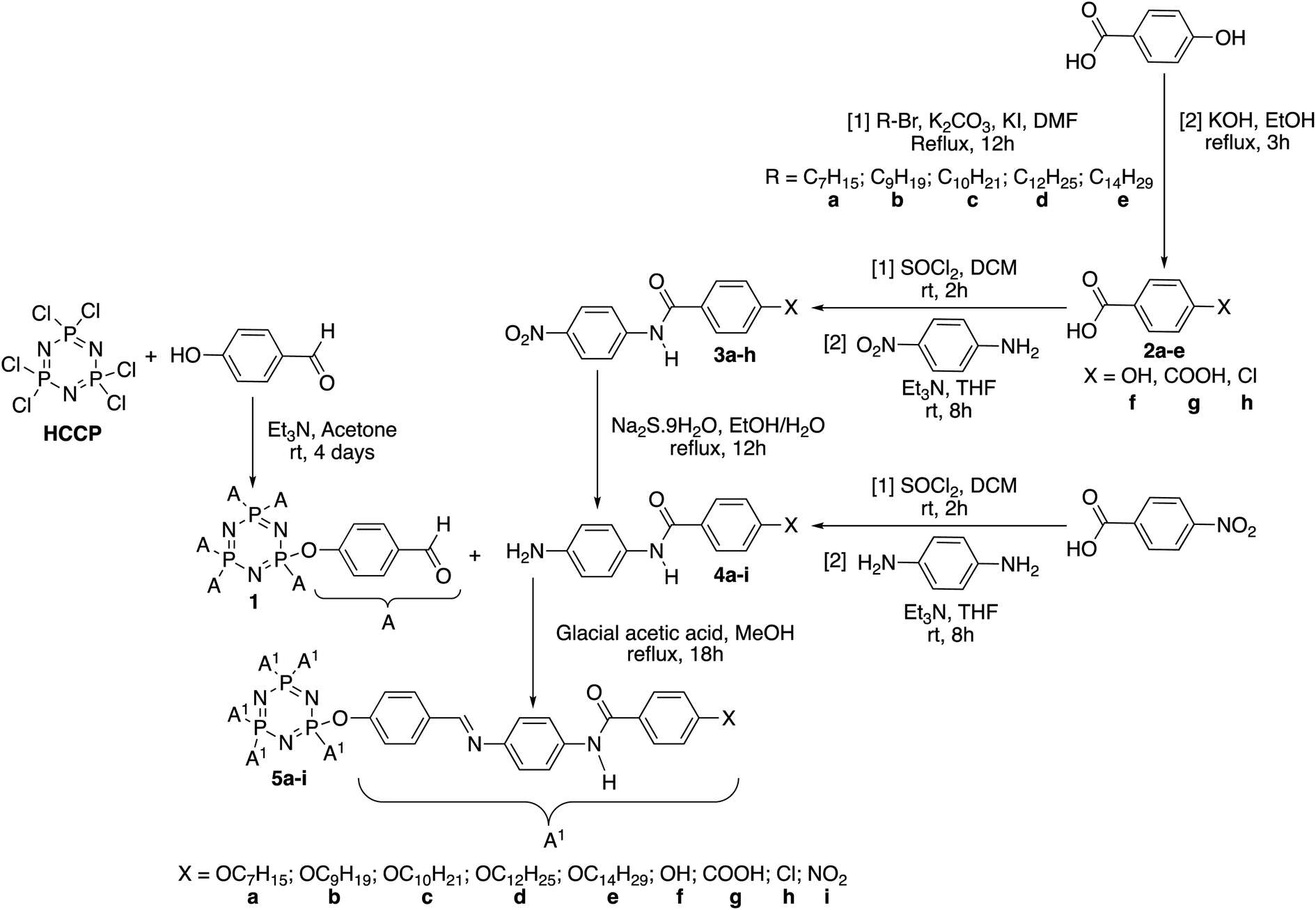 | ||
| Scheme 1 Formation of intermediates (1, 2a–e, 3a–h, and 4a–i) and hexasubstituted cyclotriphosphazene compounds, 5a–i. | ||
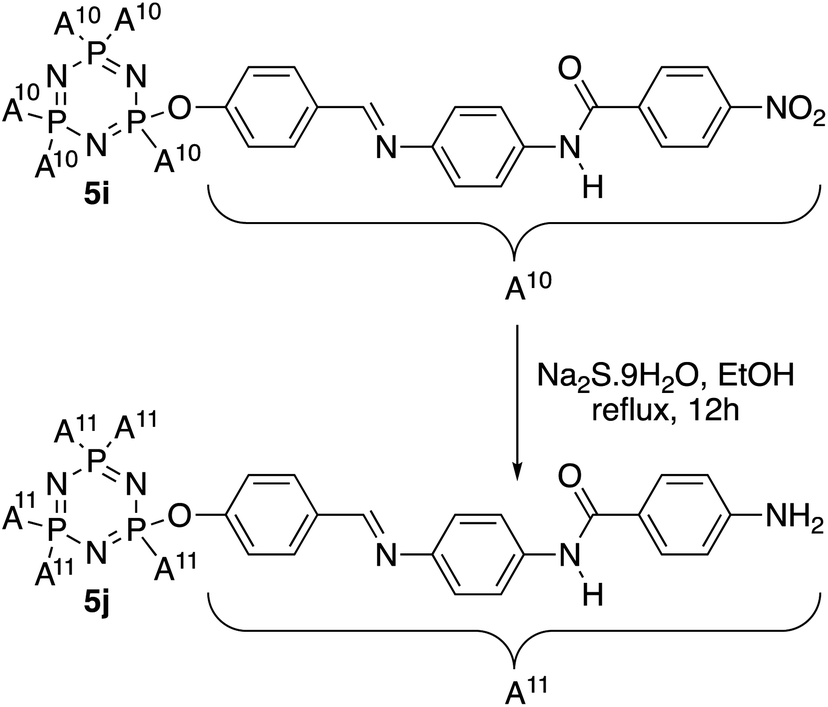 | ||
| Scheme 2 Reduction of compound 5i to 5j. | ||
2.1. FTIR spectral data discussion
Intermediate 1 was successfully synthesized with the disappearance absorption band at 3250 cm−1 for O–H stretching. The other absorption bands were observed at 2730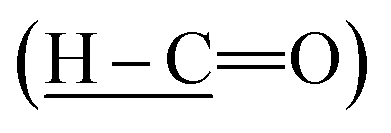 (H–C
(H–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1700 (CO), 1595 (CC), and 1205 cm−1 (C–O). The IR data of intermediates 2a–e showed the absorption bands at 3250 (O–H stretching), 2852 and 2921 (Csp3–H stretching), 1680 (CO stretching), 1605 (CC stretching), and 1250 cm−1 (C–O stretching). Intermediates 3a–h were successfully synthesized as evident from the appearance of a new band at 3300 cm−1 (N–H stretching) of the amide linkage. Further reduction of intermediates 3a–h gave rise to intermediates 4a–h with the appearance of two absorption bands at 3200 and 3400 cm−1 (N–H stretching) in the IR spectra of the latter. Intermediate 4i which was synthesized using other method also showed similar bands at 3207 and 3408 cm−1 (N–H stretching). Other bands at 1690 (CO), 1590 (CC), 1250 (C–O), and 1175 cm−1 (C–N) stretching were observed in both intermediates 3a–h and 4a–i. The diagnostic bands at 3200 and 825 cm−1 were attributable to the O–H stretching and C–Cl bending, respectively in 3f–g and 4f–g.
O), 1700 (CO), 1595 (CC), and 1205 cm−1 (C–O). The IR data of intermediates 2a–e showed the absorption bands at 3250 (O–H stretching), 2852 and 2921 (Csp3–H stretching), 1680 (CO stretching), 1605 (CC stretching), and 1250 cm−1 (C–O stretching). Intermediates 3a–h were successfully synthesized as evident from the appearance of a new band at 3300 cm−1 (N–H stretching) of the amide linkage. Further reduction of intermediates 3a–h gave rise to intermediates 4a–h with the appearance of two absorption bands at 3200 and 3400 cm−1 (N–H stretching) in the IR spectra of the latter. Intermediate 4i which was synthesized using other method also showed similar bands at 3207 and 3408 cm−1 (N–H stretching). Other bands at 1690 (CO), 1590 (CC), 1250 (C–O), and 1175 cm−1 (C–N) stretching were observed in both intermediates 3a–h and 4a–i. The diagnostic bands at 3200 and 825 cm−1 were attributable to the O–H stretching and C–Cl bending, respectively in 3f–g and 4f–g.
The overlay IR spectra of compounds 5a–j with Schiff base and amide linking units are shown in Fig. 1. The reaction between hexasubstituted cyclotriphosphazene benzaldehyde, 1 and intermediates 4a–i gave 5a–i. The appearance of the expected absorption band at 1644 cm−1 for CN stretching confirmed the successful condensation reaction of intermediates 4a–i. Further reduction of compound 5i produced compound 5j with the appearance of two-spike at 3207 and 3416 cm−1 (N–H stretching). Besides, all the compounds showed the absorption band for the N–H stretching of the amide linkage at 3365 cm−1.
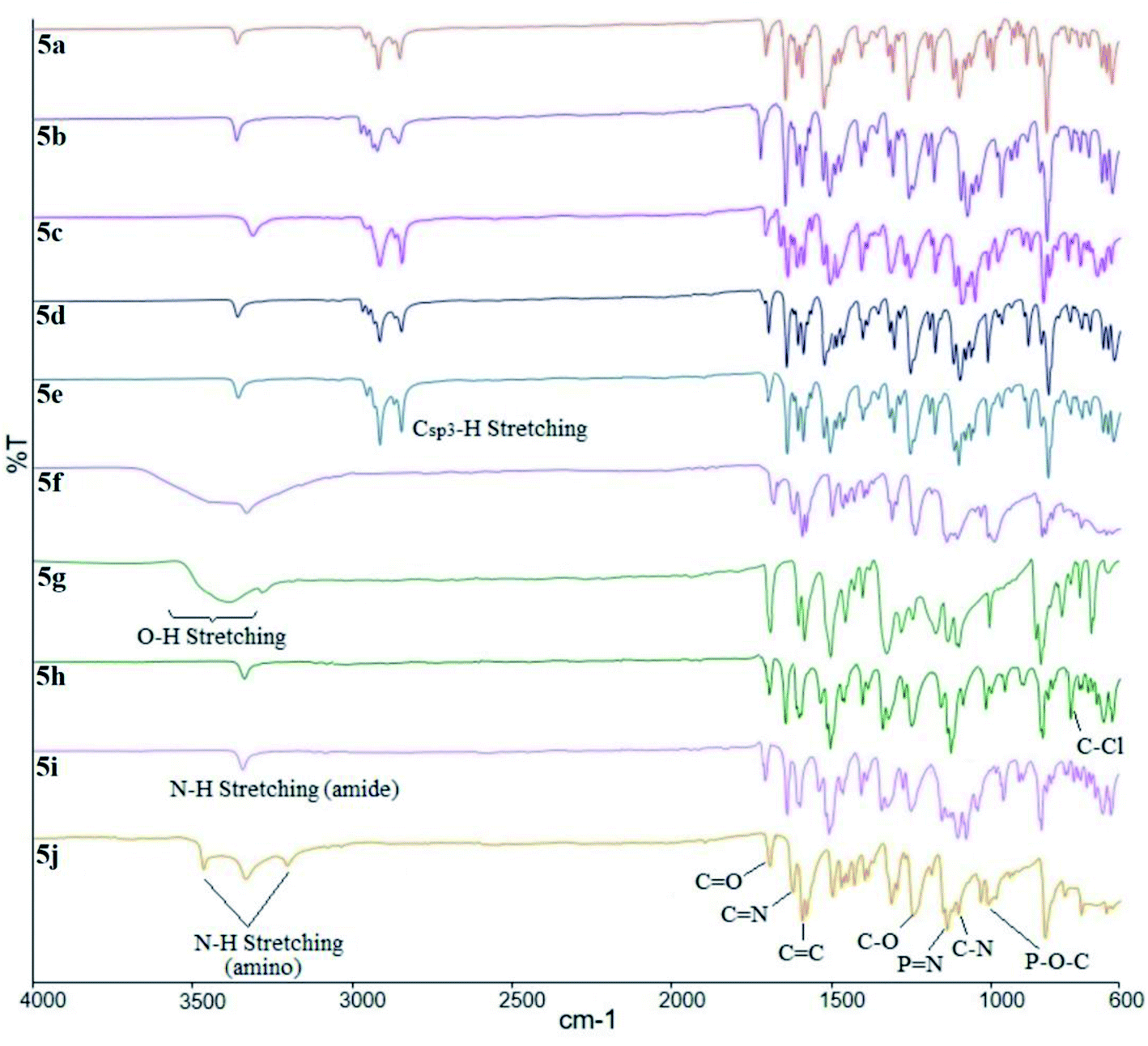 | ||
| Fig. 1 FTIR spectra overlay of compounds 5a–j. | ||
On the other hand, compounds 5f and 5g showed the overlapping of the absorption bands for hydroxy (–OH) and N–H stretching of the amide groups because peak broadening of the O–H stretching. Other absorptions bands were observed at 1691 cm−1 for the carbonyl (CO) stretching, and also at 1592, 1254, and 1178 cm−1 for CC, C–O, and C–N stretching, respectively. As usual, the diagnostic bands at 2851 and 2919 cm−1 which corresponding to the Csp3–H symmetrical and asymmetrical stretching of alkoxy chains can be observed in compounds 5a–e. In addition, the bending at 819 cm−1 was assigned to the C–Cl bending in compound 5h. The absorption bands from the cyclotriphosphazene ring, PN stretching appeared at 1197 cm−1, while P–O–C bending was located at 977 cm.
2.2. NMR spectral data discussion
The absent of the hydroxyl signal in the 1H-NMR spectrum of intermediate 1 confirmed the nucleophilic substitution of the 4-hydroxybenzaldehyde. The singlet at δ 9.90 ppm was assigned to the aldehydic proton and two doublets at δ 7.78 and 7.16 ppm were assigned for two different aromatic protons in the compound. The 13C-NMR data of 1 showed the signal for aldehydic carbon at δ 191.69 ppm and four aromatic signals at δ 153.59, 133.55, 131.46, and 121.03 ppm. The 31P NMR showed a singlet at δ 7.60 ppm, indicated that all the benzaldehyde was successfully attached to the phosphorus atom in the cyclotriphosphazene ring.The 1H-NMR data of 2a–e revealed similar pattern with two doublets assigned for two aromatic protons, while all the peaks in the upfield region were assigned to the aliphatic protons of heptyl, nonyl, decyl, dodecyl, and tetradecyl chains. The 13C-NMR data of 2a–e showed the carbon signals for aromatic ring and aliphatic chains. The signal for the aliphatic carbons displayed similar pattern but only differed in the intensity, which due to the different length in the carbon chains. The carboxy carbon for 2a–e showed a signal at the most downfield region at δ 168.78, 169.36, 168.78, 167.95, and 169.18 ppm, respectively. No signal for the methyl observed in both spectra indicating that the reduction reaction was a success.
Meanwhile, the 1H-NMR data of intermediates 3a–h showed a similar pattern with a singlet corresponded to the amide proton (N–H) in the most downfield region at δ 10.61, 10.22, 10.33, 10.25, 10.25, 9.72, 11.06, and 9.90 ppm, respectively. Both the 1H and 13C-NMR data showed the presence of four doublets of protons and eight carbons signal for the aromatic ring. The reduction of intermediates 3a–h to 4a–h showed the appearance of N–H signals of the amine group at δ 5.00 ppm in the 1H-NMR spectra. For final compound, 5c was used to represent the structure confirmation in the series. The structure of compound 5c with complete atomic numbering is illustrated in Fig. 2.
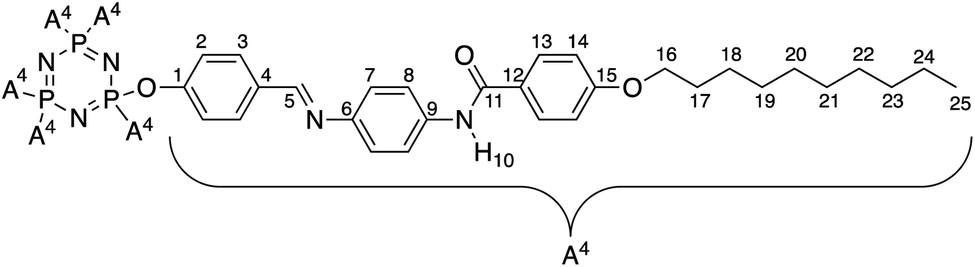 | ||
| Fig. 2 Structure of compound 5c with complete atomic numbering. | ||
In the 1H NMR spectrum of compound 5c (Fig. 3), two singlets in the downfield region at δ 9.82 (H5) and δ 8.65 ppm (H10) were assigned to the amide and azomethine protons, respectively. Five distinguishable doublets located within δ 7.02–7.96 ppm were assigned to the aromatic protons. One of these signals with integrating to four protons were overlapped and appeared as a triplet (H3 and H13).
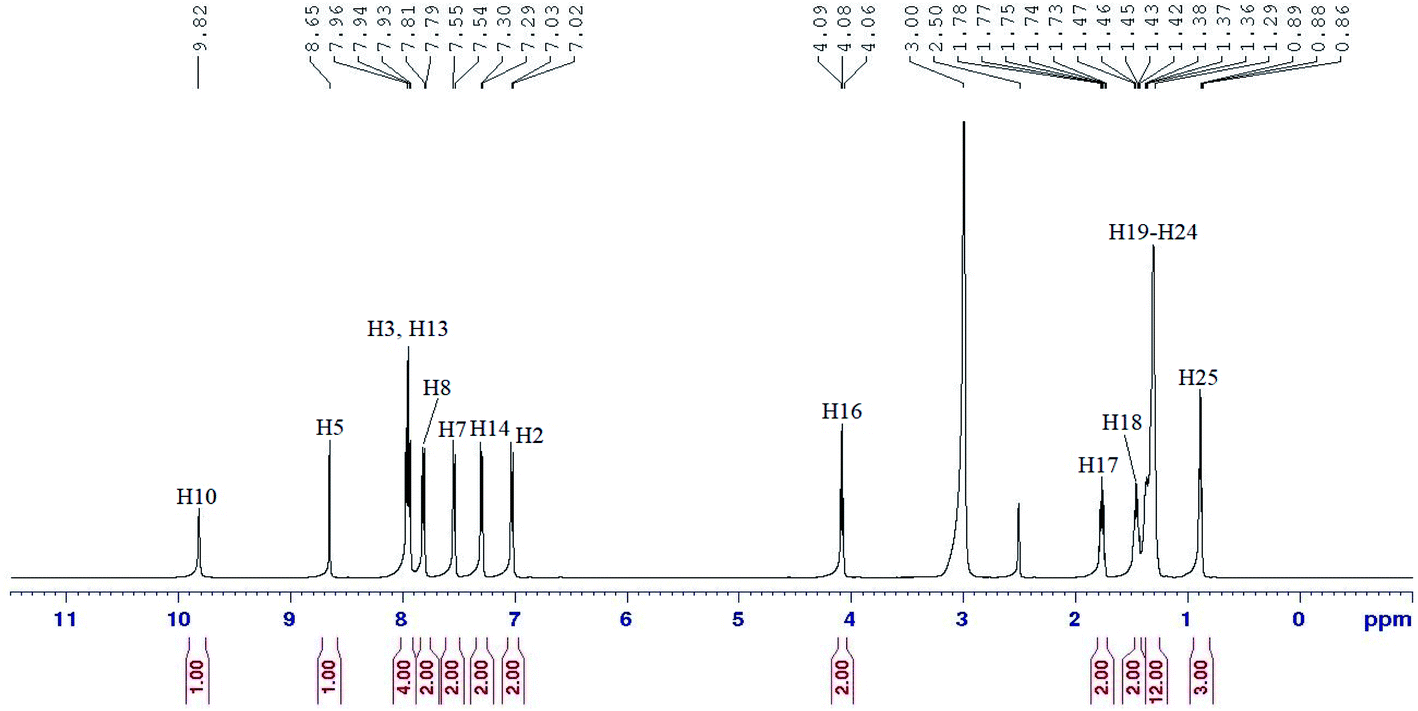 | ||
| Fig. 3 1H NMR spectrum of compound 5c. | ||
However, the effect of the amide group as an electron withdrawing group which caused the electron density of the molecule to decrease, forced H8 to experience more deshielding effect compared to H7 and H14, which was more deshielded than H2. An oxymethylene proton, H16 appeared as a triplet at δ 4.08 ppm and all the methylene protons (H17–H24) were assigned in the region of δ 1.27–1.78 ppm. A triplet in the most upfield region was assigned to H25 at δ 0.88 ppm.
The 13C NMR spectrum of compound 5c (Fig. 4a) showed 23 carbon signals comprising of one azomethine, seven quaternary, six aromatics, one oxymethylene, seven methylene, and one methyl carbons. DEPT 90 (Fig. 4b) and DEPT 135 (Fig. 4c) experiments were conducted to distinguish the types of carbons. In the DEPT 90 spectrum, the azomethine carbon, C5 was observed at δ 158.09 ppm while the aromatic carbons resonated at δ 130.47 (C3), 129.94 (C13), 129.28 (C8), 121.75 (C7), 121.69 (C14), and 114.81 ppm (C2).
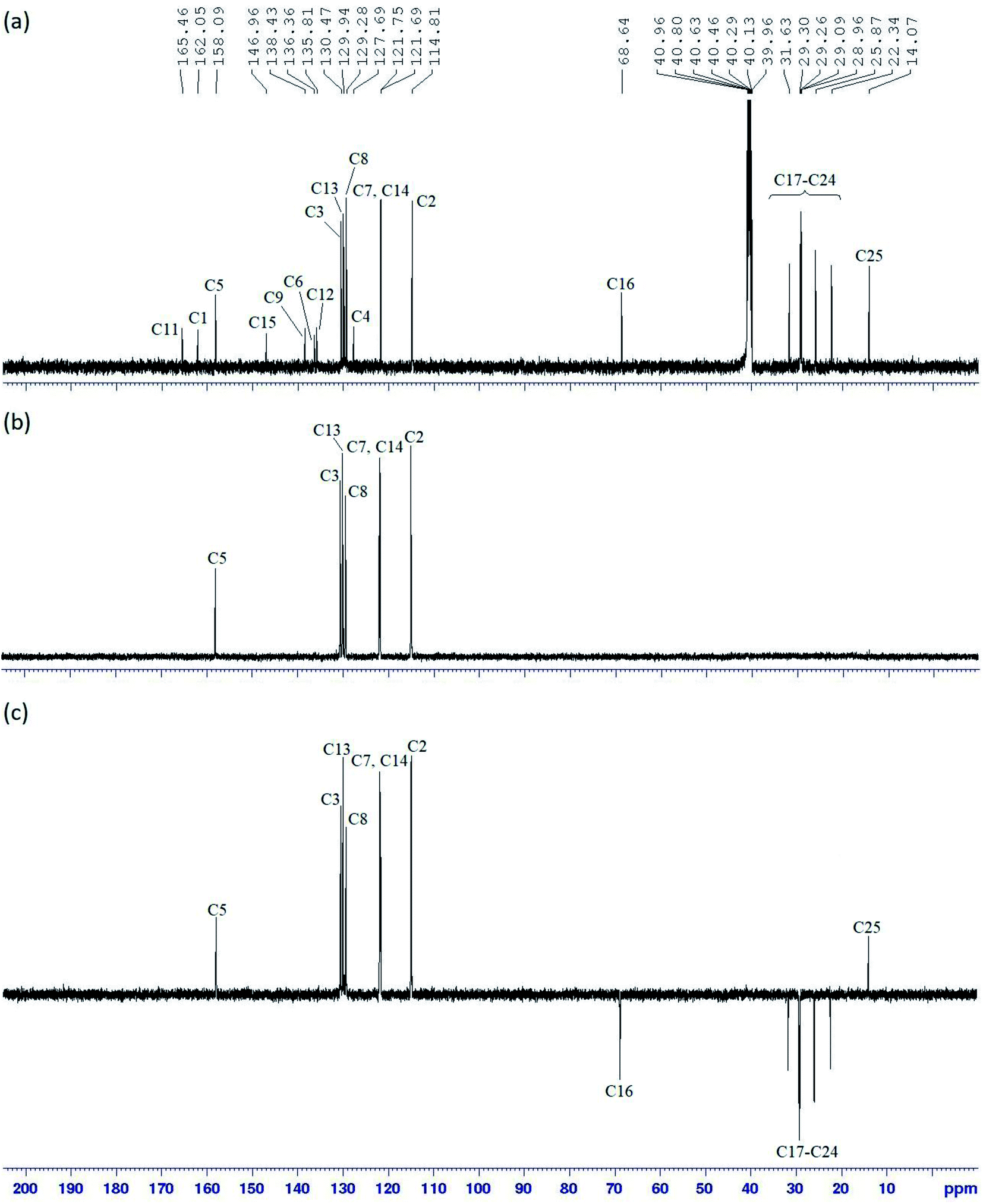 | ||
| Fig. 4 (a) 13C NMR, (b) DEPT 90 and (c) DEPT 135 spectra of compound 5c. | ||
For others homologues, the 1H and 13C NMR spectra of compounds 5a, 5b, 5d, and 5e appeared a similar splitting patterns and chemical shifts as shown in compound 5c but only differed in term of the number of protons and carbons in the alkyl chains. Compounds 5f–j attached with small substituents such as hydroxy, carboxy, chloro, nitro, and amino at the terminal end showed a singlet of amide signal and six doublets for aromatic protons in the 1H NMR spectra.
Both 1H and 13C NMR spectra of these compounds showed a slight difference in the chemical shift values, which due to the chemical environment, electronegativity effect, and bond angles. Meanwhile, compounds 5f and 5g displayed an additional signal for hydroxyl and carboxyl proton peaks in the 1H NMR spectra. Moreover, all the compounds in this series showed a singlet in the 31P NMR spectrum due to hexa-substituted in the arms (Fig. 5).
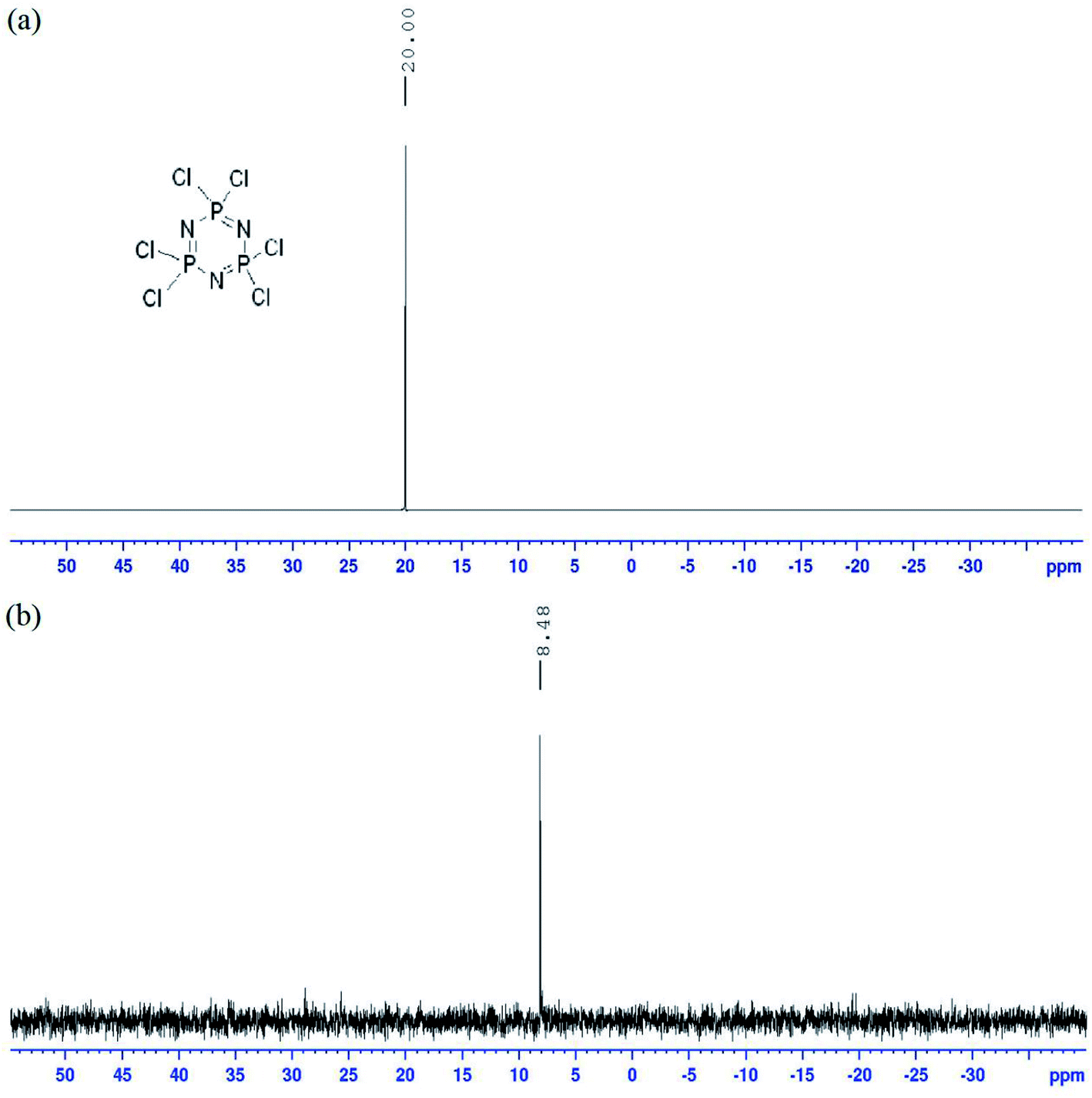 | ||
| Fig. 5 31P NMR spectrum of compound 5c. | ||
2.3. Determination of liquid crystal behaviour using POM
The POM optical photomicrograph was obtained using the Olympus system model bx53 linksys32. In this study, only compounds 5a–e with Schiff base and amide linking unit attached to different chain length at the terminal end were found to be mesogenic. These compounds showed Smectic A (SmA) phase of focal-conic fans in both heating and cooling cycles (Fig. 6). However, other intermediates and compounds did not show any liquid crystal texture, therefore these compounds are known as non-mesogenic. Observation under POM for compounds 5f–j only showed the phase transition of crystal to isotropic in the heating cycle. Similar phase transitions were also observed in the reversed order of the cooling cycle. | ||
| Fig. 6 The optical photomicrographs of compounds 5a–e in the cooling cycle showing the SmA phase, (a) 270.55 °C for compound 5a, (b) 268.77 °C for compound 5b, (c) 268.50 °C for compound 5c, (d) 255.43 °C for compound 5d, and (e) 247.78 °C for compound 5e (magnification scale: 20 × 0.25). | ||
2.4. Determination of thermal transitions using POM
The DSC experiment was conducted to confirm the phase transition observed under POM. Only compounds with liquid crystal were further sent for DSC measurement. The transition temperatures and enthalpy change involved during the transition in the heating and cooling cycles were recorded in the DSC thermogram. The thermal enthalpy, ΔH (kJ mol−1) of each phase transition of compounds 5a–e are summarized in Table 1.| Compound | Mode | Transition temperature (°C), enthalpy, ΔH (kJ mol−1) | ||||
|---|---|---|---|---|---|---|
| a Cr = crystal, SmA = smectic A, I = isotropic. | ||||||
| 5a | Heating | Cr | 188.02, 186.54 | SmA | 286.83, 48.13 | I |
| Cooling | I | 280.47, −58.43 | SmA | 170.05, −212.30 | Cr | |
| 5b | Heating | Cr | 177.09, 173.10 | SmA | 287.87, 68.64 | I |
| Cooling | I | 282.57, −73.98 | SmA | 161.53, −207.61 | Cr | |
| 5c | Heating | Cr | 174.44, 156.52 | SmA | 286.15, 70.96 | I |
| Cooling | I | 278.75, −77.83 | SmA | 158.16, −204.07 | Cr | |
| 5d | Heating | Cr | 171.44, 191.73 | SmA | 281.61, 83.37 | I |
| Cooling | I | 275.76, −88.98 | SmA | 155.73, −211.63 | Cr | |
| 5e | Heating | Cr | 168.58, 125.75 | SmA | 276.25, 76.22 | I |
| Cooling | I | 267.13, −76.78 | SmA | 149.00, −182.89 | Cr | |
The DSC thermograms of compounds 5a–e showed two curves for the transition of crystal to SmA and to isotropic phase in both cycles. All compounds showed high melting and clearing temperature in the heating cycle. From the DSC data, the melting temperature was observed at 188.02, 177.09, 174.44, 171.44, and 168.58 °C, which corresponded to compounds 5a–e, respectively. The DSC thermogram of compound 5c was illustrated in Fig. 7. The melting and clearing temperature of the compounds showed similar pattern whereby the temperature decreased as the number of alkyl chains increased. As reported by Moriya et al. (2006), the melting and clearing temperature decreased with an increased number of alkyl chains.26 Moreover, all the compounds have high enthalpy changes at the clearing temperature. This behaviour was related to the high molecular weight of hexasubstituted cyclotriphosphazene compounds, which effected by their π-energy, hydrogen-bonding and van der Waals forces.27
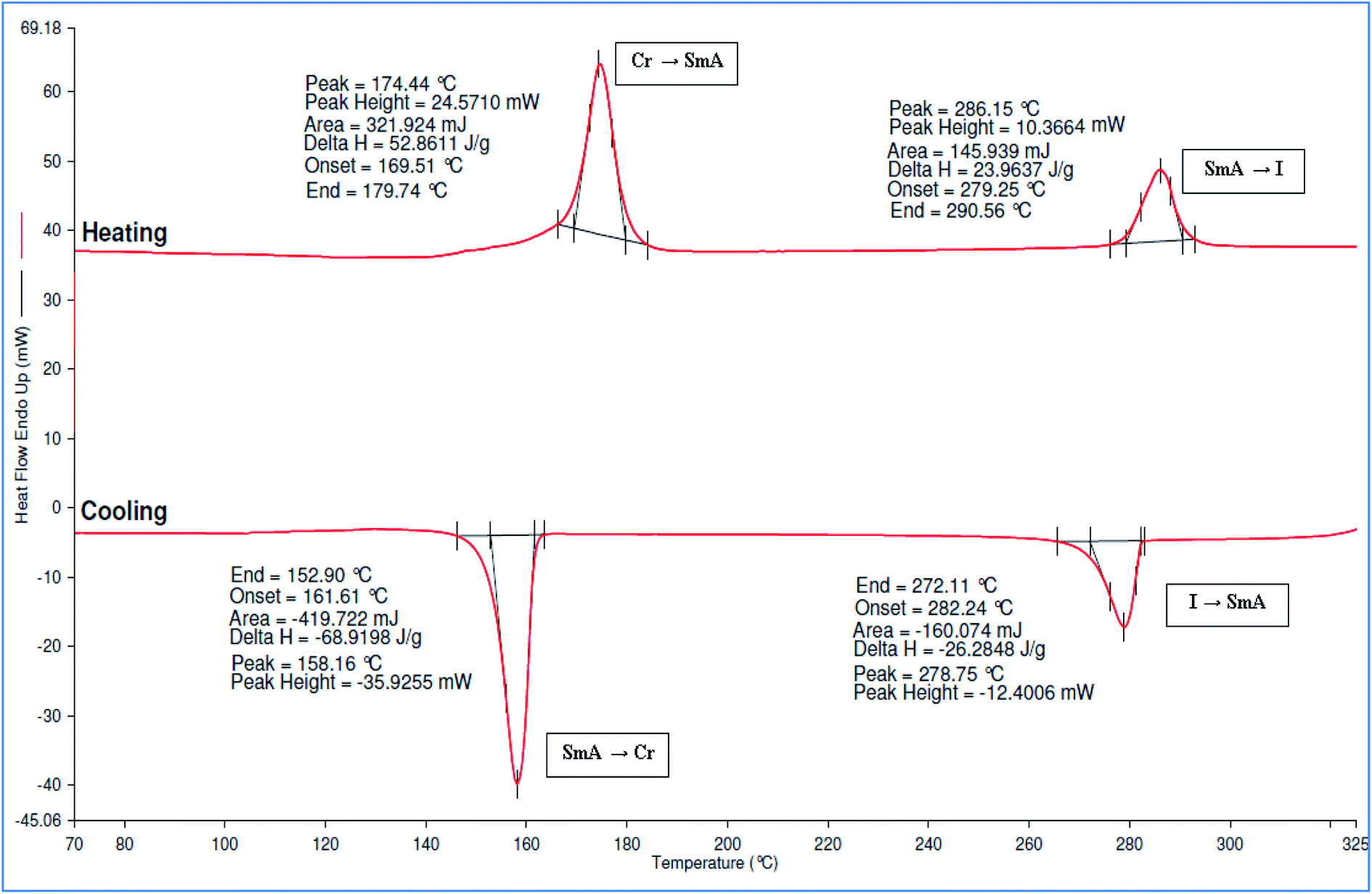 | ||
| Fig. 7 DSC thermogram of compound 5c. | ||
2.5. Determination of liquid crystal using XRD analysis
The confirmation of the SmA phase observed in these series were carried out using XRD analysis. The XRD data of compounds 5c is summarized in Table 2. Compound 5c was used as representative for further discussion. The XRD diffractogram of compound 5c is shown in Fig. 8. The XRD diffractogram of compound 5c showed a single sharp peak at 2θ = 1.19° and a broad peak with wide angle at 2θ = 15–21°, indicating that the molecules favour the smectic layered arrangement.28 Generally, compound exhibited the lamellar structure for smectic liquid crystalline phase showed the sharp and strong peak at low angle (1° < 2θ < 4°), and a broad peak associated with lateral packing at 2θ ≈ 20° in the XRD curve.29 The sharp peak indicated the regular arrangement in spaced layer, while a broad peak for the disordered packing of alkyl chains.30 According to the XRD data, the d-layer spacing and molecular length (L) obtained from the molecular calculation of compound 5c was found to be 45.74 Å and 41.23 Å, respectively. The calculated d/L was 1.11 (d ≈ L), which approximately to 1. This phenomenon showing that the SmA phase observed under POM is monolayer arrangement. This suggested that the side arms which connected to the phosphorus atoms of the cyclotriphosphazene ring were arranged three up and three down. The behaviour of homeotropic alignment caused these side arms aligns perpendicular to the cyclotriphosphazene ring as this shape is suitable for the smectic arrangement.30 The proposed molecular shape and the possible smectic arrangement of compound 5c are shown in Fig. 9.| XRD data analysis | Value |
|---|---|
| 2 theta | 1.93 |
| d-Layer spacing | 45.74 |
| Molecular length (L) | 41.23 |
| Calculated d/L | 1.11 |
| Arrangement of SmA | Monolayer |
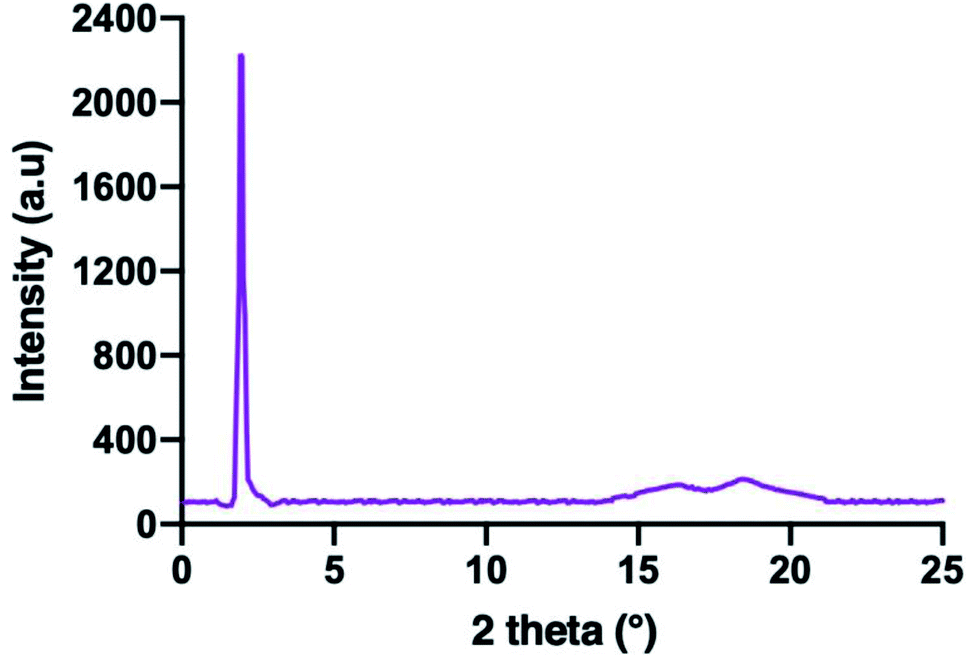 | ||
| Fig. 8 XRD diffractogram of compound 5c. | ||
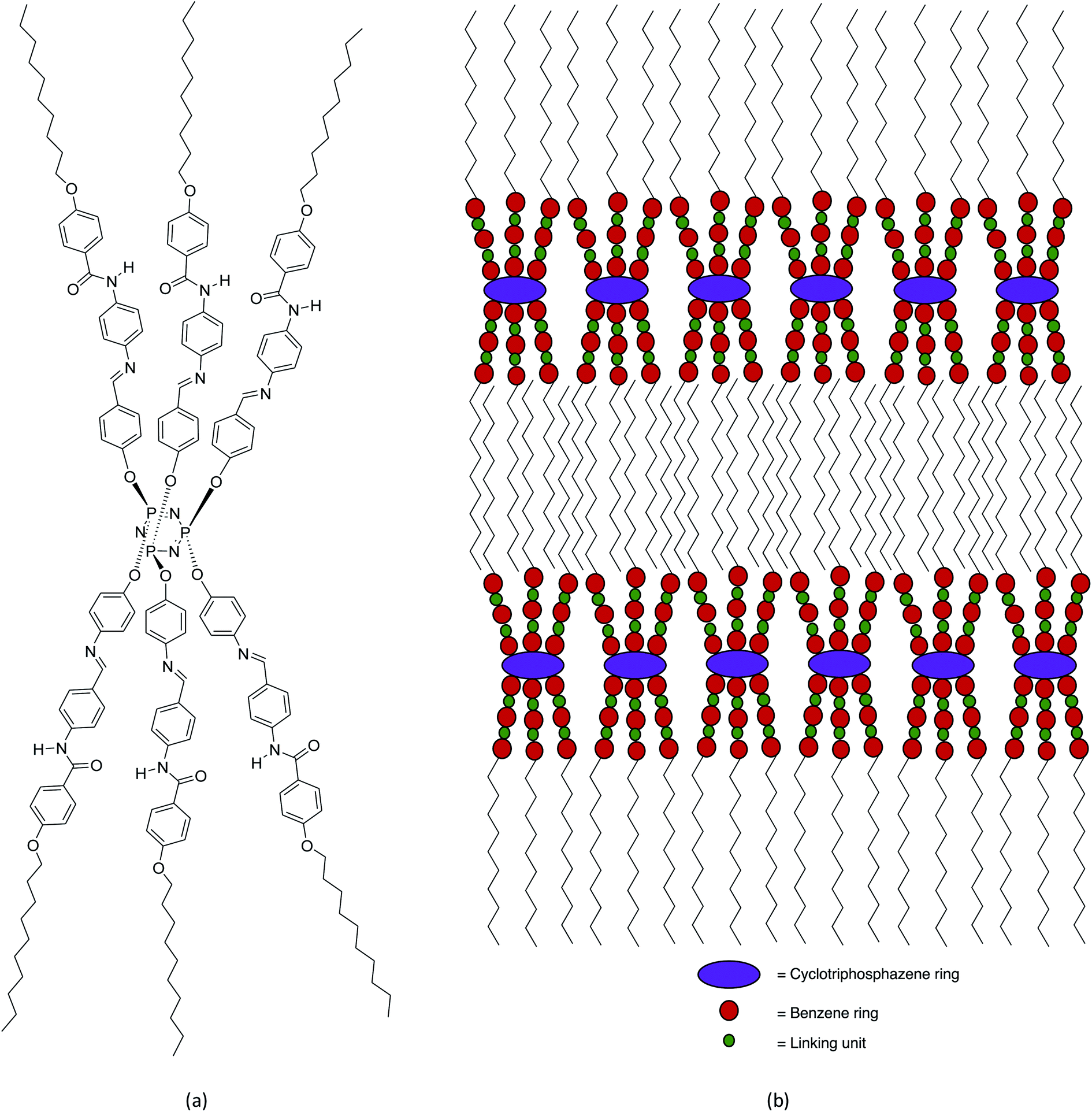 | ||
| Fig. 9 (a) The proposed molecular shape and (b) the possible smectic arrangement of compound 5c. | ||
2.6. Structure–properties relationship
The skeleton structures of a molecule influence the liquid crystal properties. A compound must possess certain requirement in order to exhibit liquid crystal mesophase. The physical properties of even the simplest liquid crystal compound are truly remarkable due to the self-assembly of molecules in an ordered, yet fluid, and liquid crystal mesophase.The main criteria for a molecule to adopt liquid crystal behaviour include the molecular shape which should be relatively thin or flat within rigid molecular frameworks, which usually based on benzene rings.31 In general, terminal group which extends the molecular long axis without increasing the molecular width increase the thermal stability of the mesophase. The nature of the terminal substituents or end groups in the molecule of the mesogen has profound influence on the liquid crystal properties of the compound. There are varieties of terminal groups being employed in the liquid crystal molecules and the common groups vary from a small polar substituent (e.g. chloro or nitro) to a long chain (e.g. alkyl or alkoxy).32 The choice of a terminal group is very important to generate the expected liquid crystal mesophase in a molecule.
Most of the molecules consist of one terminal alkyl chain showed liquid crystal behaviour. This was proven when all the final compounds (5a–e) with the alkoxy chains at the terminal end exhibited the smectic A phase in both cooling and heating cycles. The length of the alkoxy side chain showed strong influence on the mesophase formation.33 Longer alkoxy chains induced the liquid crystal behaviour with broader temperature ranges as well as decreased melting temperatures, Tm.34 In addition, the number of alkoxy side chains greatly affect molecular self-assembly.35 Besides, the long alkyl chains add flexibility to the rigid core and stabilize the molecular interactions needed for the formation of liquid crystal mesophase.34
Conjugation within the Schiff base moiety led to possible of controlling the alignment and orientation of their molecules to generate liquid crystal properties.36 Schiff base linking unit provide a stepped core structure which can maintain the molecular linearity.32 This linearity provides high stability and enabling mesophase formation. However, all compounds (5f–j) with small substituent such as hydroxy, carboxy, chloro, nitro, and amino groups at the terminal end were found to be non-mesogenic. Kelker and Hatz reported the behaviour of the small substituent at the terminal end do not always form liquid crystal phases.33,35 The lone pair of electrons or π bonds in compounds 5f and 5g tend to resonate onto the aromatic ring which resulted in the cancellation of dipole moments of the molecules.36,37 As a result, the formation of the mesophase in the molecules cannot be induced.
Even though compound 5h and 5i attached with polar substituent, which possesses strong dipole moment, the presence of the amide linking unit resulted in high melting temperature and the mesophase cannot be observed.16 Compound 5j did not exhibited any liquid crystal character due to the properties of the NH2 as an electron donating group, which maximise the repulsive interactions between adjacent aromatic rings and caused the tendency for formation of the mesophase was reduced.38 The POM observation of compounds 5a–j are summarized in Table 3.
| Compound | Terminal substituent | POM observation | |
|---|---|---|---|
| 5a | Heptyloxy | (–OC7H15) | Smectic A |
| 5b | Nonyloxy | (–OC9H19) | Smectic A |
| 5c | Decyloxy | (–OC10H21) | Smectic A |
| 5d | Dodecyloxy | (–OC12H25) | Smectic A |
| 5e | Tetradecyl | (–OC14H29) | Smectic A |
| 5f | Hydroxy | (–OH) | Non-mesogenic |
| 5g | Carboxy | (–COOH) | Non-mesogenic |
| 5h | Chloro | (–Cl) | Non-mesogenic |
| 5i | Nitro | (–NO2) | Non-mesogenic |
| 5j | Amino | (–NH2) | Non-mesogenic |
2.7. Determination of fire retardant properties using LOI testing
In this research, polyester resin has been used as a matrix for moulding. Polyester resins are unsaturated synthetic resin and can be considered as a combustible material. This resin produces a lot of heat during combustion due to low thermal stability and becomes a potential to fire risk. Thus, the modification of polyester resin by mixing with hexasubstituted cyclotriphosphazene compounds having fire retardant properties are needed to overcome this problem.The sample was prepared by mixing 1 wt% of the final compound with polyester resin. About 1 wt% of methyl ethyl ketone peroxide (MEKP) curing agent was added to the mixture and stirred until the sample is homogeneous and then poured into the moulds. The samples were cured for 5 hours in an oven at 60 °C and left overnight at room temperature before it was burned using LOI testing. The LOI test was performed using an FTT oxygen index, according to BS 2782: Part 1: Method 141 and ISO 4589 with the dimension of 120 mm × 10 mm × 4 mm.
According to LOI data in Table 4, pure polyester resin has the LOI value of 22.53%. The LOI value of polyester resin was increased to 24.71% when incorporated with 1 wt% of HCCP. Schiff base linking units was found to enhance the properties of fire-retardant. This phenomenon was attributed to the high thermal stability of the Schiff base molecules, which promoted the formation of char on the surface in the condensed phase.39 Interestingly, when Schiff base unit was combined with an amide unit, the LOI values showed a slight increase. This positive result was attributed to the properties of the electron withdrawing group of the amide bond which enhanced the flammability properties of the compounds.40,41 As a result, all the compounds have high LOI value, indicating these compounds have high thermal stability and fire retardancy. Compound 5i showed the highest LOI value compared to other compounds due to the effect of electron withdrawing of nitro group which enhance the synergistic effect of P–N bonds.42 This behaviour caused the compounds exhibited both condensed and gas phase action and thus, prevents the sample from further burning. Hence, these compounds possessed highest LOI value.
| Material | LOI value (%) |
|---|---|
| Pure polyester resin | 22.53 (±0.00) |
| Polyester resin + 1 wt% of HCCP | 24.71 (±0.00) |
| Compound 5a | 27.90 (±0.00) |
| Compound 5b | 27.71 (±0.00) |
| Compound 5c | 27.71 (±0.00) |
| Compound 5d | 27.55 (±0.00) |
| Compound 5e | 27.53 (±0.00) |
| Compound 5f | 28.37 (±0.00) |
| Compound 5g | 27.93 (±0.04) |
| Compound 5h | 28.42 (±0.00) |
| Compound 5i | 28.53 (±0.00) |
| Compound 5j | 27.90 (±0.00) |
3. Experimental
3.1. Chemicals
The chemicals and solvents used in this work are phosphonitrilic chloride trimer, 1-bromoheptane, 1-bromononane, 1-bromodecane, 1-bromododecane, 1-bromotetradecane, 4-hydroxybenzaldehyde, 4-hydroxybenzoic acid, 4-chlorobenzoic acid, 4-nitrobenzoic acid, terephthalic acid, 1,4-phenylenediamine, 4-nitroaniline, sodium sulphide hydrate, potassium carbonate, potassium iodide, potassium hydroxide, sodium hydroxide, glacial acetic acid, thionyl chloride, methanol, ethanol, acetone, dichloromethane, tetrahydrofuran, N,N-dimethylformamide, n-hexane, ethyl acetate, and triethylamine. All the chemicals and solvents were used as received without further purification and purchased from Merck, Qrëc (Asia), Sigma-Aldrich, Acros Organics, Fluka, and BDH (British Drug Houses).3.2. Syntheses
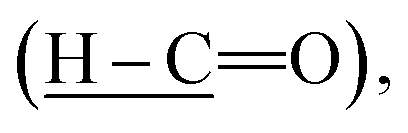 1699 (CO stretching), 1595 (CC stretching), 1205 (C–O stretching), 1151 (PN stretching), 944 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.90 (s, 1H), 7.78 (d, J = 10.0 Hz, 2H), 7.16 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 191.69, 153.59, 133.55, 131.46, 121.03. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 7.60 (s, 1P). CHN elemental analysis: calculated for C42H30N3O12P3: C: 58.55%, H: 3.51%, N: 4.88%; found: C: 58.28%, H: 3.48%, N: 4.83%.
1699 (CO stretching), 1595 (CC stretching), 1205 (C–O stretching), 1151 (PN stretching), 944 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.90 (s, 1H), 7.78 (d, J = 10.0 Hz, 2H), 7.16 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 191.69, 153.59, 133.55, 131.46, 121.03. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 7.60 (s, 1P). CHN elemental analysis: calculated for C42H30N3O12P3: C: 58.55%, H: 3.51%, N: 4.88%; found: C: 58.28%, H: 3.48%, N: 4.83%.4-(Heptyloxy)benzoic acid, 2a. Intermediate 2a was synthesized according to the method reported by Chun-Chieh et al. (2016) with some modifications.43 Methyl-4-hydroxybenzoate (15.20 g, 0.10 mol) and 1-bromoheptane (17.89 g, 0.10 mol) dissolved in 20 mL DMF separately, were mixed in a 100 mL round bottom flask. Potassium carbonate (20.73 g, 0.15 mol) and potassium iodide (1.66 g, 0.01 mol) were added to the mixture and was refluxed for 12 hours. Upon completion, the mixture was poured into 300 mL cold water and the precipitate formed was filtered. The precipitate was mixed with potassium hydroxide, KOH (11.22 g, 0.20 mol) in 150 mL ethanol. The mixture was refluxed for 3 hours. All the reaction progress was monitored by TLC. The mixture was then poured into 300 mL water and a clear solution was formed. HCl was added to the solution and the mixture was stirred slowly until the precipitate was formed. The precipitate was filtered, washed with water and dried overnight to obtain a white powder. The same method was used to synthesis 2b–e. Yield: 20.23 g (85.72%), mp: 105.2–106.5 °C, white powder. FTIR (cm−1): 3250 (O–H stretching), 2919 and 2850 (Csp3–H stretching), 1684 (C
O stretching), 1607 (aromatic CC stretching), 1252 (C–O stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 7.78 (d, J = 5.0 Hz, 2H), 6.73 (d, J = 10.0 Hz, 2H), 3.96 (t, J = 7.5 Hz, 2H), 1.68–1.74 (m, 2H), 1.40–1.46 (m, 2H), 1.27–1.38 (m, 6H), 0.89 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 168.78, 158.91, 135.20, 130.31, 112.80, 67.73, 31.09, 28.83, 28.28, 25.44, 21.82, 13.60. CHN elemental analysis: calculated for C14H20O3: C: 71.16%, H: 8.53%; found: C: 71.01%, H: 8.57%.
4-(Nonyloxy)benzoic acid, 2b. 1-Bromononane (20.69 g, 0.10 mol) was used in the reaction. Yield: 22.31 g (84.51%), mp: 104.8–105.7 °C, white powder. FTIR (cm−1): 3250 (O–H stretching), 2919 and 2849 (Csp3–H stretching), 1694 (C
O stretching), 1606 (aromatic CC stretching), 1252 (C–O stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 7.80 (d, J = 10.0 Hz, 2H), 6.74 (d, J = 5.0 Hz, 2H), 3.97 (t, J = 5.0 Hz, 2H), 1.69–1.74 (m, 2H), 1.41–1.47 (m, 2H), 1.27–1.39 (m, 10H), 0.88 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 169.36, 159.16, 134.83, 130.37, 113.02, 67.95, 31.10, 28.84, 28.76, 28.62, 28.39, 25.46, 21.80, 13.48. CHN elemental analysis: calculated for C16H24O3: C: 72.69%, H: 9.15%; found: C: 71.95%, H: 9.15%.
4-(Decyloxy)benzoic acid, 2c. 1-Bromodecane (22.09 g, 0.10 mol) was used in the reaction. Yield: 23.68 g (85.18%), mp: 104.1–105.3 °C, white powder. FTIR (cm−1): 3253 (O–H stretching), 2920 and 2849 (Csp3–H stretching), 1694 (C
O stretching), 1606 (aromatic CC stretching), 1251 (C–O stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 7.78 (d, J = 10.0 Hz, 2H), 6.73 (d, J = 10.0 Hz, 2H), 3.96 (t, J = 5.0 Hz, 2H), 1.68–1.73 (m, 2H), 1.40–1.46 (m, 2H), 1.26–1.36 (m, 12H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 168.78, 159.00, 134.78, 130.36, 112.83, 67.73, 31.13, 28.82, 28.80, 28.77, 28.63, 28.47, 25.45, 21.85, 13.60. CHN elemental analysis: calculated for C17H26O3: C: 73.35%, H: 9.41%; found: C: 73.14%, H: 9.35%.
4-(Dodecyloxy)benzoic acid, 2d. 1-Bromododecane (24.89 g, 0.10 mol) was used in the reaction. Yield: 26.88 g (87.84%), mp: 104.2–105.6 °C, white powder. FTIR (cm−1): 3250 (O–H stretching), 2920 and 2850 (Csp3–H stretching), 1702 (C
O stretching), 1603 (aromatic CC stretching), 1250 (C–O stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 7.83 (d, J = 5.0 Hz, 2H), 6.84 (d, J = 10.0 Hz, 2H), 4.00 (t, J = 7.5 Hz, 2H), 1.69–1.75 (m, 2H), 1.40–1.44 (m, 2H), 1.26–1.35 (m, 16H), 0.87 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 167.95, 160.55, 134.78, 130.75, 113.53, 67.88, 31.13, 28.83, 28.81, 28.79, 28.78, 28.67, 28.58, 28.47, 25.39, 21.85, 13.59. CHN elemental analysis: calculated for C19H30O3: C: 74.47%, H: 9.87%; found: C: 73.74%, H: 9.98%.
4-(Tetradecyloxy)benzoic acid, 2e. 1-Bromotetradecane (27.73 g, 0.10 mol) was used in the reaction. Yield: 28.12 g (84.19%), mp: 103.8–104.7 °C, white powder. FTIR (cm−1): 3251 (O–H stretching), 2919 and 2850 (Csp3–H stretching), 1701 (C
O stretching), 1607 (aromatic CC stretching), 1252 (C–O stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 7.80 (d, J = 5.0 Hz, 2H), 6.74 (d, J = 10.0 Hz, 2H), 3.97 (t, J = 5.0 Hz, 2H), 1.68–1.74 (m, 2H), 1.40–1.46 (m, 2H), 1.20–1.36 (m, 20H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 169.18, 159.27, 134.27, 130.42, 113.02, 67.91, 31.11, 28.83, 28.82, 28.81, 28.80, 28.79, 28.77, 28.76, 28.59, 28.44, 25.44, 21.80, 13.49. CHN elemental analysis: calculated for C21H34O3: C: 75.41%, H: 10.25%; found: C: 74.51%, H: 10.15%.
4-(Heptyloxy)-N-(4-nitrophenyl)benzamide, 3a. Intermediate 3a was synthesized according to the method reported by Davidson et al. (2006) with some modifications.44 4-Heptyloxybenzoic acid, 2a (7.08 g, 0.03 mol) and thionyl chloride (3.57 g, 0.03 mol) in 40 mL DCM was mixed in 100 mL round bottom flask to form an acid chloride. The mixture was stirred at room temperature for 2 hours to form a clear solution. A solution of 4-nitroaniline (4.14 g, 0.03 mol) in 20 mL THF was added dropwise to the mixture and a white precipitate began to form. Triethylamine, Et3N (1.52 g, 0.015 mol) was finally added and the mixture was stirred for 8 hours. All the reactions progress are monitored by TLC. The precipitate formed was filtered and the filtrate was collected. After it was dried, the product formed was recrystallised from methanol. The same method was used to synthesis 3b–h. Yield: 7.56 g (70.79%), mp: 137.1–139.8 °C, yellow powder. FTIR (cm−1): 3357 (N–H stretching), 2932 and 2854 (Csp3–H stretching), 1660 (C
O stretching), 1598 (aromatic CC stretching), 1248 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.61 (s, 1H), 8.23 (d, J = 5.0 Hz, 2H), 8.03 (d, J = 5.0 Hz, 2H), 7.95 (d, J = 10.0 Hz, 2H), 7.04 (d, J = 10.0 Hz, 2H), 4.03 (t, J = 7.5 Hz, 2H), 1.68–1.74 (m, 2H), 1.36–1.42 (m, 2H), 1.23–1.33 (m, 6H), 0.85 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.15, 162.59, 146.09, 143.42, 130.26, 126.95, 124.78, 120.56, 115.03, 68.82, 31.51, 29.08, 28.66, 25.81, 22.22, 13.94. CHN elemental analysis: calculated for C20H24N2O4: C: 67.40%, H: 6.79%, N: 7.86%; found: C: 66.81%, H: 6.84%, N: 7.77%.
4-(Nonyloxy)-N-(4-nitrophenyl)benzamide, 3b. Intermediate 2b (7.92 g, 0.03 mol) was used in the reaction. Yield: 7.90 g (68.58%), mp: 136.3–138.2 °C, yellow powder. FTIR (cm−1): 3349 (N–H stretching), 2921 and 2851 (Csp3–H stretching), 1660 (C
O stretching), 1601 (aromatic CC stretching), 1248 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.22 (s, 1H), 8.18 (d, J = 10.0 Hz, 2H), 8.00 (d, J = 10.0 Hz, 2H), 7.96 (d, J = 10.0 Hz, 2H), 7.03 (d, J = 10.0 Hz, 2H), 4.09 (t, J = 5.0 Hz, 2H), 1.73–1.78 (m, 2H), 1.42–1.48 (m, 2H), 1.25–1.39 (m, 10H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.26, 162.50, 145.95, 143.26, 130.27, 126.65, 124.87, 120.56, 114.93, 68.66, 31.54, 29.17, 29.00, 28.95, 28.83, 25.77, 22.29, 14.04. CHN elemental analysis: calculated for C22H28N2O4: C: 68.73%, H: 7.34%, N: 7.29%; found: C: 68.03%, H: 7.39%, N: 7.27%.
4-(Decyloxy)-N-(4-nitrophenyl)benzamide, 3c. Intermediate 2c (8.24 g, 0.03 mol) was used in the reaction. Yield: 8.66 g (72.53%), mp: 135.5–137.1 °C, yellow powder. FTIR (cm−1): 3346 (N–H stretching), 2919 and 2851 (Csp3–H stretching), 1657 (C
O stretching), 1598 (aromatic CC stretching), 1246 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.33 (s, 1H), 8.19 (d, J = 10.0 Hz, 2H), 8.00 (d, J = 5.0 Hz, 2H), 7.95 (d, J = 5.0 Hz, 2H), 7.03 (d, J = 10.0 Hz, 2H), 4.07 (t, J = 7.5 Hz, 2H), 1.70–1.76 (m, 2H), 1.39–1.45 (m, 2H), 1.22–1.36 (m, 12H), 0.85 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.18, 162.51, 146.05, 143.24, 130.29, 126.72, 124.89, 120.52, 114.93, 68.66, 31.59, 29.24, 29.20, 29.02, 28.99, 28.92, 25.80, 22.32, 14.07. CHN elemental analysis: calculated for C23H30N2O4: C: 69.32%, H: 7.59%, N: 7.03%; found: C: 69.12%, H: 7.59%, N: 6.99%.
4-(Dodecyloxy)-N-(4-nitrophenyl)benzamide, 3d. Intermediate 2d (9.18 g, 0.03 mol) was used in the reaction. Yield: 9.15 g (71.60%), mp: 133.4–135.8 °C, yellow powder. FTIR (cm−1): 3344 (N–H stretching), 2916 and 2849 (Csp3–H stretching), 1657 (C
O stretching), 1598 (aromatic CC stretching), 1248 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.25 (s, 1H), 8.16 (d, J = 10.0 Hz, 2H), 7.96 (d, J = 5.0 Hz, 2H), 7.92 (d, J = 10.0 Hz, 2H), 7.00 (d, J = 10.0 Hz, 2H), 4.05 (t, J = 5.0 Hz, 2H), 1.68–1.74 (m, 2H), 1.37–1.43 (m, 2H), 1.19–1.34 (m, 16H), 0.82 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.31, 162.52, 146.90, 143.32, 130.24, 126.65, 124.86, 120.61, 114.97, 68.69, 31.54, 29.23, 29.22, 29.18, 29.16, 29.15, 28.93, 28.88, 25.74, 22.27, 13.99. CHN elemental analysis: calculated for C25H34N2O4: C: 70.40%, H: 8.03%, N: 6.57%; found: C: 70.06%, H: 8.03%, N: 6.50%.
4-(Tetradecyloxy)-N-(4-nitrophenyl)benzamide, 3e. Intermediate 2e (10.02 g, 0.03 mol) was used in the reaction. Yield: 9.68 g (71.07%), mp: 131.7–133.6 °C, yellow powder. FTIR (cm−1): 3343 (N–H stretching), 2916 and 2851 (Csp3–H stretching), 1657 (C
O stretching), 1606 (aromatic CC stretching), 1254 (C–O stretching), 1178 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.25 (s, 1H), 8.17 (d, J = 5.0 Hz, 2H), 7.96 (d, J = 5.0 Hz, 2H), 7.92 (d, J = 10.0 Hz, 2H), 7.01 (d, J = 5.0 Hz, 2H), 4.05 (t, J = 5.0 Hz, 2H), 1.69–1.74 (m, 2H), 1.37–1.43 (m, 2H), 1.17–1.36 (m, 20H), 0.82 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.32, 162.53, 146.91, 143.33, 130.25, 126.66, 124.86, 120.61, 114.98, 68.70, 31.55, 29.25, 29.23, 29.22, 29.19, 29.18, 29.16, 29.15, 28.94, 28.89, 25.75, 22.28, 14.00. CHN elemental analysis: calculated for C27H38N2O4: C: 71.34%, H: 8.43%, N: 6.16%; found: C: 70.68%, H: 8.38%, N: 6.12%.
4-(Hydroxy)-N-(4-nitrophenyl)benzamide, 3f. 4-Hydroxybenzoic acid (4.14 g, 0.03 mol) was used in the reaction. Yield: 5.66 g (73.13%), mp: 163.6–165.2 °C, yellow powder. FTIR (cm−1): 3340 (N–H stretching), 3201 (O–H stretching), 1698 (C
O stretching), 1600 (aromatic CC stretching), 1252 (C–O stretching), 1175 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.72 (s, 1H), 8.04 (d, J = 5.0 Hz, 2H), 7.71 (d, J = 10.0 Hz, 2H), 6.90 (d, J = 5.0 Hz, 2H), 6.88 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.77, 162.55, 146.89, 143.12, 130.75, 125.80, 124.47, 120.50, 113.96. CHN elemental analysis: calculated for C13H10N2O4: C: 60.47%, H: 3.90%, N: 10.85%; found: C: 60.10%, H: 3.88%, N: 10.79%.
4-(Carboxy)-N-(4-nitrophenyl)benzamide, 3g. Terephthalic acid (4.98 g, 0.03 mol) was used in the reaction. Yield: 5.12 g (59.67%), mp: 168.7–170.8 °C, light brown powder. FTIR (cm−1): 3341 (N–H stretching), 3100–3270 (O–H stretching), 1700 (C
O stretching), 1606 (aromatic CC stretching), 1248 (C–O stretching), 1177 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 11.06 (s, 1H), 10.07 (s, 1H), 8.10 (d, J = 10.0 Hz, 2H), 8.08 (d, J = 5.0 Hz, 2H), 7.98 (d, J = 10.0 Hz, 2H), 6.90 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 167.03, 164.33, 140.06, 139.29, 136.08, 130.55, 130.34, 129.94, 126.57, 116.19. CHN elemental analysis: calculated for C14H10N2O5: C: 58.75%, H: 3.52%, N: 9.79%; found: C: 58.43%, H: 3.50%, N: 9.80%.
4-(Chloro)-N-(4-nitrophenyl)benzamide, 3h. 4-Chlorobenzoic acid (4.69 g, 0.03 mol) was used in the reaction. Yield: 6.11 g (73.67%), mp: 155.1–157.4 °C, yellow powder. FTIR (cm−1): 3342 (N–H stretching), 1691 (C
O stretching), 1602 (aromatic CC stretching), 1250 (C–O stretching), 1170 (C–N stretching), 827 (C–Cl bending). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.90 (s, 1H), 8.02 (d, J = 10.0 Hz, 2H), 7.83 (d, J = 10.0 Hz, 2H), 7.55 (d, J = 5.0 Hz, 2H), 6.87 (d, J = 5.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.58, 162.51, 146.91, 143.21, 130.69, 125.84, 124.57, 121.22, 117.10. CHN elemental analysis: calculated for C13H9ClN2O3: C: 56.44%, H: 3.28%, N: 12.81%; found: C: 56.27%, H: 3.25%, N: 12.76%.
N-(4-Aminophenyl)-4-(heptyloxy)benzamide, 4a. Intermediate 4a was synthesized according to the method reported by Shaoyu and Jiangbing (2012) with some modifications.45 A solution of intermediate 3a (5.00 g, 0.014 mmol) in 40 mL ethanol and a solution of sodium sulphide hydrate, Na2S·9H2O (0.89 g, 0.01 mol) in 20 mL ethanol and 20 mL water were mixed in 250 mL round bottom flask. The mixture was refluxed for 12 hours. The reaction progress was monitored by TLC. Upon completion, the mixture was cooled in an ice bath. The precipitate formed was filtered, washed with cold ethanol and dried to form a white precipitate. The same method was used to synthesis 4b–h. Yield: 3.55 g (77.53%), mp: 155.3–157.1 °C, white powder. FTIR (cm−1): 3330 (N–H amide stretching), 3403 and 3204 (N–H amine stretching), 2921 and 2854 (Csp3–H stretching), 1696 (C
O stretching), 1609 (aromatic CC stretching), 1254 (C–O stretching), 1173 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.69 (s, 1H), 7.89 (d, J = 5.0 Hz, 2H), 7.34 (d, J = 5.0 Hz, 2H), 7.00 (d, J = 5.0 Hz, 2H), 6.53 (d, J = 10.0 Hz, 2H), 4.87 (s, 2H), 4.02 (t, J = 5.0 Hz, 2H), 1.69–1.75 (m, 2H), 1.38–1.44 (m, 2H), 1.25–1.36 (m, 6H), 0.86 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 164.64, 161.46, 145.46, 129.72, 128.76, 127.64, 122.74, 114.38, 114.17, 68.15, 31.68, 29.05, 28.87, 25.90, 22.50, 14.40. CHN elemental analysis: calculated for C20H26N2O2: C: 73.59%, H: 8.03%, N: 8.58%; found: C: 73.26%, H: 8.08%, N: 8.51%.
N-(4-Aminophenyl)-4-(nonyloxy)benzamide, 4b. Intermediate 3b (5.00 g, 0.013 mol) was used in the reaction. Yield: 3.60 g (78.09%), mp: 153.7–154.9 °C, white powder. FTIR (cm−1): 3340 (N–H amide stretching), 3410 and 3201 (N–H amine stretching), 2919 and 2851 (Csp3–H stretching), 1698 (C
O stretching), 1609 (aromatic CC stretching), 1254 (C–O stretching), 1178 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.69 (s, 1H), 7.88 (d, J = 10.0 Hz, 2H), 7.33 (d, J = 10.0 Hz, 2H), 6.99 (d, J = 10.0 Hz, 2H), 6.53 (d, J = 10.0 Hz, 2H), 4.87 (s, 2H), 4.02 (t, J = 5.0 Hz, 2H), 1.69–1.74 (m, 2H), 1.38–1.43 (m, 2H), 1.22–1.35 (m, 10H), 0.85 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 164.64, 161.46, 145.46, 129.72, 128.75, 127.63, 122.75, 114.38, 114.18, 68.15, 31.72, 29.40, 29.21, 29.09, 29.04, 25.92, 22.55, 14.41. CHN elemental analysis: calculated for C22H30N2O2: C: 74.54%, H: 8.53%, N: 7.90%; found: C: 74.31%, H: 8.54%, N: 7.88%.
N-(4-Aminophenyl)-4-(decyloxy)benzamide, 4c. Intermediate 3c (5.00 g, 0.013 mol) was used in the reaction. Yield: 3.58 g (77.44%), mp: 153.4–154.1 °C, white powder. FTIR (cm−1): 3340 (N–H amide stretching), 3404 and 3213 (N–H amine stretching), 2919 and 2851 (Csp3–H stretching), 1700 (C
O stretching), 1606 (aromatic CC stretching), 1254 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.68 (s, 1H), 7.89 (d, J = 10.0 Hz, 2H), 7.34 (d, J = 10.0 Hz, 2H), 7.00 (d, J = 10.0 Hz, 2H), 6.53 (d, J = 10.0 Hz, 2H), 4.87 (s, 2H), 4.03 (t, J = 7.5 Hz, 2H), 1.69–1.75 (m, 2H), 1.38–1.44 (m, 2H), 1.22–1.35 (m, 12H), 0.86 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 164.57, 161.45, 145.48, 129.72, 128.77, 127.69, 122.73, 114.38, 114.15, 68.15, 31.75, 29.45, 29.40, 29.21, 29.15, 29.05, 25.93, 22.55, 14.42. CHN elemental analysis: calculated for C23H32N2O2: C: 74.96%, H: 8.75%, N: 7.60%; found: C: 74.65%, H: 8.75%, N: 7.57%.
N-(4-Aminophenyl)-4-(dodecyloxy)benzamide, 4d. Intermediate 3d (5.00 g, 0.012 mol) was used in the reaction. Yield: 3.61 g (78.32%), mp: 151.8–153.4 °C, yellow powder. FTIR (cm−1): 3352 (N–H amide stretching), 3402 and 3206 (N–H amine stretching), 2919 and 2851 (Csp3–H stretching), 1698 (C
O stretching), 1606 (aromatic CC stretching), 1254 (C–O stretching), 1178 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.69 (s, 1H), 7.89 (d, J = 10.0 Hz, 2H), 7.34 (d, J = 10.0 Hz, 2H), 7.00 (d, J = 5.0 Hz, 2H), 6.53 (d, J = 10.0 Hz, 2H), 4.87 (s, 2H), 4.02 (t, J = 7.5 Hz, 2H), 1.69–1.75 (m, 2H), 1.38–1.44 (m, 2H), 1.22–1.34 (m, 16H), 0.85 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 164.62, 161.45, 145.47, 129.72, 128.77, 127.65, 122.74, 114.37, 114.16, 68.14, 31.75, 29.48, 29.46, 29.44, 29.43, 29.19, 29.16, 29.04, 25.92, 22.55, 14.41. CHN elemental analysis: calculated for C25H36N2O2: C: 75.72%, H: 9.15%, N: 7.06%; found: C: 75.56%, H: 9.13%, N: 7.01%.
N-(4-Aminophenyl)-4-(tetradecyloxy)benzamide, 4e. Intermediate 3e (5.00 g, 0.011 mol) was used in the reaction. Yield: 3.66 g (78.38%), mp: 150.6–152.8 °C, yellow powder. FTIR (cm−1): 3345 (N–H amide stretching), 3411 and 3203 (N–H amine stretching), 2921 and 2851 (Csp3–H stretching), 1698 (C
O stretching), 1606 (aromatic CC stretching), 1256 (C–O stretching), 1176 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.31 (s, 1H), 7.88 (d, J = 10.0 Hz, 2H), 7.34 (d, J = 10.0 Hz, 2H), 6.98 (d, J = 5.0 Hz, 2H), 6.59 (d, J = 5.0 Hz, 2H), 4.06 (t, J = 7.5 Hz, 2H), 1.73–1.77 (m, 2H), 1.41–1.46 (m, 2H), 1.25–1.38 (m, 20H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 164.94, 161.69, 145.25, 131.67, 129.63, 129.39, 122.94, 114.74, 114.60, 68.65, 31.62, 29.34, 29.33, 29.31, 29.27, 29.26, 29.24, 29.12, 29.06, 28.96, 25.86, 22.31, 14.02. CHN elemental analysis: calculated for C27H40N2O2: C: 76.37%, H: 9.50%, N: 6.60%; found: C: 76.18%, H: 9.47%, N: 6.62%.
N-(4-Aminophenyl)-4-(hydroxy)benzamide, 4f. Intermediate 3f (5.00 g, 0.019 mol) was used in the reaction. Yield: 3.22 g (72.87%), mp: 211.1–213.6 °C, white powder. FTIR (cm−1): 3340 (N–H amide stretching), 3405 and 3211 (N–H amine stretching), 3200 (O–H stretching), 1691 (C
O stretching), 1609 (aromatic CC stretching), 1254 (C–O stretching), 1178 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.73 (s, 1H), 8.24 (d, J = 5.0 Hz, 2H), 8.10 (d, J = 10.0 Hz, 2H), 7.71 (d, J = 10.0 Hz, 2H), 6.89 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.28, 163.72, 150.37, 136.75, 132.54, 131.07, 128.83, 124.02, 116.26. CHN elemental analysis: calculated for C13H12N2O2: C: 68.41%, H: 5.30%, N: 12.27%; found: C: 68.17%, H: 5.27%, N: 12.19%.
N-(4-Aminophenyl)-4-(carboxy)benzamide, 4g. Intermediate 3g (4.50 g, 0.016 mol) was used in the reaction. Yield: 3.10 g (76.96%), mp: 232.6–233.9 °C, white powder. FTIR (cm−1): 3351 (N–H amide stretching), 3401 and 3202 (N–H amine stretching), 3203 (O–H stretching), 1692 (C
O stretching), 1608 (aromatic CC stretching), 1254 (C–O stretching), 1178 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.06 (s, 1H), 8.10 (d, J = 10.0 Hz, 2H), 7.98 (d, J = 5.0 Hz, 2H), 7.92 (d, J = 10.0 Hz, 2H), 6.67 (s, 2H), 6.60 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 167.07, 156.08, 139.27, 136.17, 136.11, 130.34, 129.94, 126.81, 112.86. CHN elemental analysis: calculated for C14H12N2O3: C: 65.62%, H: 4.72%, N: 10.93%; found: C: 65.28%, H: 4.70%, N: 10.85%.
N-(4-Aminophenyl)-4-(chloro)benzamide, 4h. Intermediate 3h (5.00 g, 0.018 mol) was used in the reaction. Yield: 3.05 g (68.43%), mp: 170.4–172.1 °C, white powder. FTIR (cm−1): 3344 (N–H amide stretching), 3473 and 3207 (N–H amine stretching), 1641 (C
O stretching), 1701 (aromatic CC stretching), 1252 (C–O stretching), 1189 (C–N stretching), 825 (C–Cl bending). 1H-NMR (500 MHz, CDCl3) δ, ppm: 10.08 (s, 1H), 8.33 (d, J = 5.0 Hz, 2H), 8.09 (d, J = 10.0 Hz, 2H), 7.88 (d, J = 10.0 Hz, 2H), 6.61 (s, 2H), 6.58 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.87, 163.80, 150.45, 136.76, 132.60, 131.29, 128.90, 124.33, 118.11. CHN elemental analysis: calculated for C13H11ClN2O: C: 63.29%, H: 4.49%, N: 14.37%; found: C: 63.12%, H: 4.50%, N: 14.26%.
O stretching), 1595 (aromatic CC stretching), 1248 (C–O stretching), 1146 (C–N stretching). 1H-NMR (500 MHz, CDCl3) δ, ppm: 9.93 (s, 1H), 7.90 (d, J = 10.0 Hz, 2H), 7.87 (d, J = 5.0 Hz, 2H), 7.59 (d, J = 6.0 Hz, 2H), 6.63 (s, 2H), 6.59 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ, ppm: 166.59, 162.98, 150.10, 136.44, 132.37, 131.10, 128.51, 123.89, 114.70. CHN elemental analysis: calculated for C14H12N2O3: C: 65.62%, H: 4.72%, N: 10.93%; found: C: 65.34%, H: 4.69%, N: 10.82%.Hexakis{((4-(heptyloxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5a. Compound 5a was synthesized according to the method reported by Jamain et al. (2019) and Fareed et al. (2013) with some modifications.32,47 A mixture of intermediate 1 (0.70 g, 0.81 mmol) and intermediate 4a (1.86 g, 5.69 mmol) in 50 mL methanol was mixed in a 100 mL round bottom flask. A few drops of glacial acetic acid were added to the solution. The mixture was refluxed for 18 hours. The reaction progress was monitored by TLC. Upon completion, the precipitate formed was filtered and dried overnight. The product was recrystallised from ethanol. The same method was used to synthesis 5b–h. Yield: 1.47 g (66.74%), mp: 183.6–185.1 °C, yellow powder. FTIR (cm−1): 3365 (N–H stretching), 2921 and 2854 (Csp3–H stretching), 1691 (C
O stretching), 1644 (CN stretching), 1592 (aromatic CC stretching), 1254 (C–O stretching), 1197 (PN stretching), 1178 (C–N stretching), 977 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.83 (s, 1H), 8.65 (s, 1H), 7.95 (d, J = 5.0 Hz, 2H), 7.94 (d, J = 5.0 Hz, 2H), 7.80 (d, J = 10.0 Hz, 2H), 7.55 (d, J = 5.0 Hz, 2H), 7.29 (d, J = 10.0 Hz, 2H), 7.02 (d, J = 10.0 Hz, 2H), 4.08 (t, J = 5.0 Hz, 2H), 1.73–1.79 (m, 2H), 1.42–1.48 (m, 2H), 1.28–1.40 (m, 6H), 0.89 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 165.46, 162.05, 158.11, 146.97, 138.44, 136.35, 135.82, 130.48, 129.94, 129.29, 127.70, 121.74, 121.69, 114.82, 68.65, 31.56, 29.11, 28.71, 25.85, 22.29, 14.06. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.49 (s, 1P). CHN elemental analysis: calculated for C162H174N15O18P3: C: 71.74%, H: 6.47%, N: 7.75%; found: C: 71.57%, H: 6.40%, N: 7.71%.
Hexakis{((4-(nonyloxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5b. Intermediate 4b (2.02 g, 5.69 mmol) was used in the reaction. Yield: 1.53 g (65.41%), mp: 178.1–180.5 °C, yellow powder. FTIR (cm−1): 3365 (N–H stretching), 2919 and 2849 (Csp3–H stretching), 1691 (C
O stretching), 1646 (CN stretching), 1593 (aromatic CC stretching), 1256 (C–O stretching), 1197 (PN stretching), 1178 (C–N stretching), 974 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.82 (s, 1H), 8.65 (s, 1H), 7.96 (d, J = 10.0 Hz, 2H), 7.94 (d, J = 10.0 Hz, 2H), 7.81 (d, J = 10.0 Hz, 2H), 7.54 (d, J = 10.0 Hz, 2H), 7.29 (d, J = 10.0 Hz, 2H), 7.02 (d, J = 10.0 Hz, 2H), 4.08 (t, J = 7.5 Hz, 2H), 1.73–1.79 (m, 2H), 1.42–1.48 (m, 2H), 1.24–1.39 (m, 10H), 0.88 (t, J = 5.0 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 165.44, 162.05, 158.04, 146.95, 138.46, 136.36, 135.82, 130.46, 129.94, 129.27, 127.71, 121.74, 121.69, 114.80, 68.64, 31.63, 29.28, 29.11, 28.93, 25.89, 22.36, 14.08. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.51 (s, 1P). CHN elemental analysis: calculated for C174H198N15O18P3: C: 72.55%, H: 6.93%, N: 7.29%; found: C: 72.04%, H: 6.88%, N: 7.22%.
Hexakis{((4-(decyloxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5c. Intermediate 4c (2.09 g, 5.69 mmol) was used in the reaction. Yield: 1.55 g (64.39%), mp: 175.7–177.6 °C, yellow powder. FTIR (cm−1): 3363 (N–H stretching), 2919 and 2851 (Csp3–H stretching), 1690 (C
O stretching), 1644 (CN stretching), 1593 (aromatic CC stretching), 1259 (C–O stretching), 1197 (PN stretching), 1181 (C–N stretching), 977 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.82 (s, 1H), 8.65 (s, 1H), 7.96 (d, J = 10.0 Hz, 2H), 7.94 (d, J = 5.0 Hz, 2H), 7.80 (d, J = 10.0 Hz, 2H), 7.55 (d, J = 5.0 Hz, 2H), 7.29 (d, J = 10.0 Hz, 2H), 7.03 (d, J = 5.0 Hz, 2H), 4.08 (t, J = 7.5 Hz, 2H), 1.73–1.78 (m, 2H), 1.42–1.48 (m, 2H), 1.24–1.38 (m, 12H), 0.88 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 165.46, 162.05, 158.10, 146.96, 138.43, 136.36, 135.81, 130.47, 129.94, 129.28, 127.69, 121.75, 121.69, 114.81, 68.64, 31.63, 29.30, 29.26, 29.09, 28.97, 25.87, 22.34, 14.07. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.48 (s, 1P). CHN elemental analysis: calculated for C180H210N15O18P3: C: 72.92%, H: 7.14%, N: 7.09%; found: C: 72.76%, H: 7.11%, N: 7.02%.
Hexakis{((4-(dodecyloxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5d. Intermediate 4d (2.25 g, 5.69 mmol) was used in the reaction. Yield: 1.66 g (65.25%), mp: 171.3–173.8 °C, yellow powder. FTIR (cm−1): 3363 (N–H stretching), 2916 and 2851 (Csp3–H stretching), 1695 (C
O stretching), 1644 (CN stretching), 1592 (aromatic CC stretching), 1255 (C–O stretching), 1197 (PN stretching), 1178 (C–N stretching), 978 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.82 (s, 1H), 8.65 (s, 1H), 7.95 (d, J = 10.0 Hz, 2H), 7.94 (d, J = 5.0 Hz, 2H), 7.80 (d, J = 10.0 Hz, 2H), 7.54 (d, J = 10.0 Hz, 2H), 7.29 (d, J = 10.0 Hz, 2H), 7.02 (d, J = 10.0 Hz, 2H), 4.07 (t, J = 7.5 Hz, 2H), 1.72–1.78 (m, 2H), 1.42–1.48 (m, 2H), 1.24–1.37 (m, 16H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 165.46, 162.04, 158.07, 146.96, 138.42, 136.36, 135.80, 130.46, 129.93, 129.27, 127.68, 121.75, 121.68, 114.80, 68.63, 31.63, 29.35, 29.32, 29.29, 29.09, 29.08, 28.98, 25.87, 22.34, 14.06. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.50 (s, 1P). CHN elemental analysis: calculated for C192H234N15O18P3: C: 73.61%, H: 7.53%, N: 6.71%; found: C: 73.23%, H: 7.48%, N: 6.65%.
Hexakis{((4-(tetradecyloxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5e. Intermediate 4e (2.41 g, 5.69 mmol) was used in the reaction. Yield: 1.84 g (68.64%), mp: 168.5–170.5 °C, yellow powder. FTIR (cm−1): 3317 (N–H stretching), 2921 and 2849 (Csp3–H stretching), 1691 (C
O stretching), 1641 (CN stretching), 1599 (aromatic CC stretching), 1259 (C–O stretching), 1189 (PN stretching), 1178 (C–N stretching), 977 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.82 (s, 1H), 8.64 (s, 1H), 7.95 (d, J = 10.0 Hz, 2H), 7.94 (d, J = 5.0 Hz, 2H), 7.81 (d, J = 5.0 Hz, 2H), 7.54 (d, J = 5.0 Hz, 2H), 7.29 (d, J = 10.0 Hz, 2H), 7.02 (d, J = 5.0 Hz, 2H), 4.07 (t, J = 7.5 Hz, 2H), 1.72–1.78 (m, 2H), 1.42–1.47 (m, 2H), 1.22–1.38 (m, 20H), 0.87 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 165.44, 162.04, 158.04, 146.95, 138.44, 136.36, 135.81, 130.45, 129.93, 129.27, 127.69, 121.74, 121.67, 114.79, 68.63, 31.64, 29.37, 29.36, 29.35, 29.34, 29.29, 29.11, 29.09, 28.97, 25.87, 22.34, 14.05. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.47 (s, 1P). CHN elemental analysis: calculated for C204H258N15O18P3: C: 74.22%, H: 7.88%, N: 6.36%; found: C: 73.89%, H: 7.80%, N: 6.31%.
Hexakis{((4-(hydroxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5f. Intermediate 4f (1.30 g, 5.69 mmol) was used in the reaction. Yield: 1.23 g (71.33%), mp: 282.7–285.1 °C, yellow powder. FTIR (cm−1): 3356 (N–H stretching), 3260 (O–H stretching), 1697 (C
O stretching), 1640 (CN stretching), 1590 (aromatic CC stretching), 1253 (C–O stretching), 1190 (PN stretching), 1152 (C–N stretching), 980 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 8.42 (s, 1H), 7.94 (d, J = 5.0 Hz, 2H), 7.73 (d, J = 10.0 Hz, 2H), 7.50 (d, J = 10.0 Hz, 2H), 7.12 (d, J = 10.0 Hz, 2H), 6.88 (d, J = 10.0 Hz, 2H), 6.80 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 167.05, 160.60, 157.36, 156.14, 143.65, 138.25, 131.57, 130.67, 130.23, 129.09, 128.33, 122.59, 116.17, 116.07. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.52 (s, 1P). CHN elemental analysis: calculated for C120H90N15O18P3: C: 67.89%, H: 4.27%, N: 9.90%; found: C: 67.70%, H: 4.23%, N: 9.86%.
Hexakis{((4-(carboxy)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5g. Intermediate 4g (1.46 g, 5.69 mmol) was used in the reaction. Yield: 1.31 g (70.39%), mp: 296.1–298.5 °C, orange powder. FTIR (cm−1): 3336 (N–H stretching), 3290 (O–H stretching), 1699 (C
O stretching), 1642 (CN stretching), 1601 (aromatic CC stretching), 1254 (C–O stretching), 1190 (PN stretching), 1174 (C–N stretching), 978 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 8.61 (s, 1H), 8.21 (d, J = 10.0 Hz, 2H), 8.10 (d, J = 5.0 Hz, 2H), 8.01 (d, J = 10.0 Hz, 2H), 7.94 (d, J = 5.0 Hz, 2H), 7.22 (d, J = 10.0 Hz, 2H), 6.82 (d, J = 5.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 167.47, 166.28, 157.21, 156.28, 150.29, 142.55, 140.52, 136.92, 131.02, 130.09, 128.60, 123.95, 123.19, 116.24. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.57 (s, 1P). CHN elemental analysis: calculated for C126H90N15O24P3: C: 66.05%, H: 3.96%, N: 9.17%; found: C: 65.81%, H: 3.94%, N: 9.09%.
Hexakis{((4-(chloro)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5h. Intermediate 4h (1.40 g, 5.69 mmol) was used in the reaction. Yield: 1.25 g (68.89%), mp: 272.5–275.8 °C, orange powder. FTIR (cm−1): 3360 (N–H stretching), 1695 (C
O stretching), 1644 (CN stretching), 1601 (aromatic CC stretching), 1254 (C–O stretching), 1189 (PN stretching), 1172 (C–N stretching), 1014 (P–O–C stretching), 819 (C–Cl bending). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 8.44 (s, 1H), 7.86 (d, J = 10.0 Hz, 2H), 7.77 (d, J = 10.0 Hz, 2H), 7.41 (d, J = 5.0 Hz, 2H), 7.39 (d, J = 10.0 Hz, 2H), 7.14 (d, J = 10.0 Hz, 2H), 6.79 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 167.36, 156.69, 156.48, 142.59, 138.31, 136.02, 135.30, 131.48, 130.20, 130.05, 129.18, 129.00, 123.00, 116.27. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.53 (s, 1P). CHN elemental analysis: calculated for C120H84Cl6N15O12P3: C: 64.53%, H: 3.79%, N: 9.42%; found: C: 64.13%, H: 3.77%, N: 9.38%.
Hexakis{((4-(nitro)benzamide)methaneyllidene)azaneylylidene}triazaphosphazene, 5i. Intermediate 1 (1.50 g, 1.74 mmol) and intermediate 4i (3.13 g, 0.012 mol) were used in the reaction. Yield: 2.23 g (71.15%), mp: 280.1–283.3 °C, yellow powder. FTIR (cm−1): 3367 (N–H stretching), 1691 (C
O stretching), 1641 (CN stretching), 1598 (aromatic CC stretching), 1252 (C–O stretching), 1187 (PN stretching), 1173 (C–N stretching), 1019 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 9.81 (s, 1H), 8.66 (s, 1H), 8.23 (d, J = 10.0 Hz, 2H), 8.04 (d, J = 10.0 Hz, 2H), 7.26 (d, J = 10.0 Hz, 2H), 6.82 (d, J = 10.0 Hz, 2H), 6.51 (d, J = 10.0 Hz, 2H), 6.46 (d, J = 10.0 Hz, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 157.63, 155.02, 148.82, 148.71, 142.42, 142.05, 140.85, 129.47, 124.29, 123.79, 123.56, 116.32, 116.06, 116.01. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.54 (s, 1P). CHN elemental analysis: calculated for C120H84N21O24P3: C: 62.75%, H: 3.69%, N: 12.81%; found: C: 62.33%, H: 3.65%, N: 12.69%.
O stretching), 1645 (CN stretching), 1591 (aromatic CC stretching), 1254 (C–O stretching), 1196 (PN stretching), 1172 (C–N stretching), 1013 (P–O–C stretching). 1H-NMR (500 MHz, DMSO-d6) δ, ppm: 8.68 (s, 1H), 8.25 (d, J = 5.0 Hz, 2H), 8.06 (d, J = 5.0 Hz, 2H), 7.27 (d, J = 10.0 Hz, 2H), 6.99 (d, J = 5.0 Hz, 2H), 6.84 (d, J = 5.0 Hz, 2H), 6.56 (d, J = 10.0 Hz, 2H), 5.16 (s, 2H). 13C-NMR (125 MHz, DMSO-d6) δ, ppm: 157.67, 155.00, 148.73, 147.96, 142.47, 142.07, 129.64, 129.47, 128.94, 124.29, 123.56, 119.45, 116.32, 115.76. 31P-NMR (500 MHz, DMSO-d6) δ, ppm: 8.55 (s, 1P). CHN elemental analysis: calculated for C120H96N21O12P3: C: 68.08%, H: 4.57%, N: 13.89%; found: C: 67.77%, H: 4.59%, N: 13.79%.4. Conclusion
All the intermediates and hexasubstituted cyclotriphosphazene compounds were successfully synthesized and characterized. POM has been used to determine the mesophase behaviour of these compounds and the results showed that only compounds 5a–e with alkoxy chains have liquid crystal properties. However, other compounds 5f–j were found to be non-mesogenic with no liquid crystal behaviour. The presence of the amide linking unit caused compounds 5f–j with small substituent such as hydroxy, carboxy, chloro, nitro, and amino have high melting temperature and liquid crystal behaviour cannot be induced although the Schiff base provide linearity and thermal stability in the molecules. The XRD data of compound 5c confirmed the formation of SmA phase. The homeotropic alignment was occurred and suggested that the side arms were aligned three up and three down. Further study on the fire retardancy of final compounds was done using LOI testing. All the compounds gave excellent results and compound 5i showed the highest LOI value with 28.53%. The effect of electron withdrawing of nitro group play an important role for the compound to have high fire retardant value as this nitro group can induced the P–N synergistic effect.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the Universiti Malaysia Sabah (UMS) for Grant No. SGA0037-2019 and Universiti Sains Malaysia (USM) for RUI (USM) Grant No. 1001/PKIMIA/811332.References
- H. R. Allcock and R. L. Kugel, J. Am. Chem. Soc., 1965, 87, 4216–4217 CrossRef CAS.
- H. R. Allcock, Chem. Rev., 1972, 72, 315–356 CrossRef CAS.
- K. Moriya, T. Suzuki, S. Yano and M. Kajiwara, Liq. Cryst., 1995, 19, 711–713 CrossRef CAS.
- H. R. Allcock, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 661–671 CrossRef CAS.
- J. Barberá, J. Jiménez, A. Laguna, L. Oriol, S. Pérez and J. L. Serrano, Chem. Mater., 2006, 18, 5437–5445 CrossRef.
- J. M. Khurana, S. Chauhan and G. Bansal, Monatsh. Chem., 2004, 135, 83–87 CrossRef CAS.
- Z. Hussain, F. Qazi, M. I. Ahmed, A. Usman, A. Riaz and A. D. Abbasi, Biosens. Bioelectron., 2016, 85, 110–127 CrossRef CAS PubMed.
- S. Singh and D. Dunmur, Liquid crystals: Fundamentals, World Scientific, 2002, pp. 1–3 Search PubMed.
- P. J. Collings and M. Hird, Introduction to Liquid Crystal: Chemistry and Physic, Taylor & Francis, MIT Press, London, 1997 Search PubMed.
- P. J. Collings, Liquid Crystals: Nature's Delicate Phase of Matter, Princeton Univ. Press, New Jersey, 2nd edn, 2002, p. 204 Search PubMed.
- C. Kim and H. R. Allcock, Macromolecules, 1987, 20, 1726–1727 CrossRef CAS.
- K. Moriya, H. Mizusaki, M. Kato, T. Suzuki, S. Yano, M. Kajiwara and K. Tashiro, Chem. Mater., 1997, 9, 255–263 CrossRef CAS.
- K. Moriya, T. Suzuki, H. Mizusaki, S. Yano and M. Kajiwara, Chem. Lett., 1997, 26, 1001–1002 CrossRef.
- D. Frenkel and B. M. Mulder, Mol. Phys., 1985, 55, 1171–1192 CrossRef CAS.
- Z. Jamain, M. Khairuddean, N. N. Zulbaharen and T. K. Chung, Malaysian Journal of Chemistry, 2019, 21, 73–85 Search PubMed.
- K. Václav, H. Martin, S. Jiří, N. Vladimíra and P. Damian, Liq. Cryst., 2012, 39, 943–955 CrossRef.
- G. W. Gray, Molecular structure and the properties of liquid crystals, Academic Press, London, 1962 Search PubMed.
- L. Jiménez, A. Laguna, A. M. Molter, J. L. Serrano, J. Barbera and L. Oriol, Chem.–Eur. J., 2011, 17, 1029–1039 CrossRef PubMed.
- Q. He, H. Dai, X. Tan, X. Cheng, F. Liu and C. Tschierske, J. Mater. Chem. C, 2013, 1, 7148–7154 RSC.
- Y. Jae Shin, Y. Rok Ham, S. H. Kim, D. H. Lee, S. B. Kim, C. S. Park, Y. M. Yoo, J. G. Kim, S. H. Kwon and J. S. Shin, J. Ind. Eng. Chem., 2010, 16, 364–367 CrossRef.
- Y. L. Liu, Y. C. Chiu and T. Y. Chen, Polym. Int., 2003, 52, 1256–1261 CrossRef CAS.
- K. Faghihi and M. Hagibeygi, Turk. J. Chem., 2007, 31, 65–73 CAS.
- X. Zhang, Y. Zhong and Z. Mao, Polym. Degrad. Stab., 2012, 97, 1504–1510 CrossRef CAS.
- Y. Rong, H. Wentian, X. Liang, S. Yan and L. Jinchun, Polym. Degrad. Stab., 2015, 122, 102–109 CrossRef.
- Y. Rong, W. Bo, H. Xiaofeng, M. Binbin and L. Jinchun, Polym. Degrad. Stab., 2017, 144, 62–69 CrossRef.
- K. Moriya, T. Masuda, T. Suzuki, S. Yano and M. Kajiwara, Mol. Cryst. Liq. Cryst., 2006, 318, 267–277 CrossRef.
- S. Ahuja and N. Jespersen, Modern Instrumental Analysis, Elsevier Science, 2006 Search PubMed.
- D. Pociecha, D. Kardas, E. Gorecka, J. Szydlowska, J. Mieczkowski and D. Guillon, J. Mater. Chem., 2003, 13, 34–37 RSC.
- B. Y. Zhang, Y. G. Jia, D. S. Yao and X. W. Dong, Liq. Cryst., 2004, 31, 339–345 CrossRef CAS.
- D. Davarci, CBU J. Sci., 2016, 12, 535–542 CAS.
- M. Hird, J. W. Goodby, N. Gough and K. J. Toyne, J. Mater. Chem., 2001, 11, 2732–2742 RSC.
- Z. Jamain, M. Khairuddean and S. A. Saidin, J. Mol. Struct., 2019, 1186, 293–302 CrossRef CAS.
- Z. Jamain, M. Khairuddean and T. Guan-Seng, Int. J. Mol. Sci., 2020, 21, 4267 CrossRef PubMed.
- E. L. Heeley, D. J. Hughes, Y. El Aziz, I. Williamson, P. G. Taylor and A. R. Bassindale, Phys. Chem. Chem. Phys., 2013, 15, 5518–5529 RSC.
- H. Kelker and R. Hatz, Handbook of liquid crystals, Verlag Chemie, Weinheim, Germany, Deerfield Beach, Florida, USA, 1980 Search PubMed.
- Z. Jamain, M. Khairuddean and T. Guan-Seng, Molecules, 2020, 25, 2122 CrossRef CAS.
- J. A. Uhood, E. G. Tarik and H. R. Howraa, Molecules, 2010, 15, 5620–5628 CrossRef PubMed.
- Z. Galewski and H. J. Coles, J. Mol. Liq., 1999, 79, 77–87 CrossRef CAS.
- H. S. El-Wahab, Pigm. Resin Technol., 2015, 44, 101–110 CrossRef.
- S. Zahra, J. Nasrin and S. Shahla, Carbohydr. Polym., 2015, 118, 183–198 CrossRef PubMed.
- W. De-Yi, Novel Fire Retardant Polymers and Composite Materials, Woodhead Publishing Series in Composites Science and Engineering: Number 73, 2017 Search PubMed.
- K. A. Salmeia, S. Gaan and G. Malucelli, Polymers, 2016, 8, 319–355 CrossRef PubMed.
- H. Chun-Chieh, C. Yu-Chaing, C. San-Yuan and L. Hong-Cheu, RSC Adv., 2016, 6, 32319–32327 RSC.
- A. H. Davidson, S. J. Davies and D. F. C. Moffat, PCT Int. Appl., 2006, 2006117552 Search PubMed.
- Z. Shaoyu and X. Jiangbing, Faming Zhuanli Shenqing, CN102618062, 2012.
- A. P. Avdeenko and I. L. Marchenko, Russ. J. Org. Chem., 2001, 37, 822–829 CrossRef CAS.
- G. Fareed, M. A. Versiani, N. Afza, N. Fareed, M. A. Kalhoro, S. Yasmeen and M. A. Anwar, J. Chem. Soc. Pak., 2013, 35, 427–431 CAS.
- Z. Deyan, W. Yangyang, J. Jiong, Y. Wenzhu, Q. Baofeng, L. Xia and S. Xuan, ChemComm, 2015, 51, 10656–10659 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. 1–5: DSC thermogram of compounds 5a–e, respectively. See DOI: 10.1039/d0ra03812a |
| This journal is © The Royal Society of Chemistry 2020 |