
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe selection of highly specific and selective aptamers using modified SELEX and their use in process analytical techniques for Lucentis bioproduction†
Tanu Bhardwaja,
Anurag S. Rathore b and
Sandeep Kumar Jha*a
b and
Sandeep Kumar Jha*a
aCentre for Biomedical Engineering, Indian Institute of Technology, Hauz Khas, New Delhi-110016, India. E-mail: sandeepkjha@gmail.com
bDepartment of Chemical Engineering, Indian Institute of Technology, Hauz Khas, New Delhi-110016, India
First published on 4th August 2020
Abstract
Aptamers for Lucentis were selected using 10 rounds of a modified and highly stringent SELEX process. Affinity column chromatography was used for the binding, partitioning, and elution steps, and the regeneration of ssDNA was performed via asymmetric PCR in the SELEX process. The interaction of aptamers with Lucentis was studied by means of the HADDOCK web server docking program. In addition, the secondary structures of aptamers were interrogated using the mfold web server to check common regions responsible for better affinity towards Lucentis. The two best aptamers for Lucentis (aptamers 1 and 25) were found to have dissociation constant (Kd) values between 23 and 35 nM by means of thermofluorimetric and non-faradaic impedance spectroscopy (NFIS) analysis. The low dissociation constants in the nanomolar range showed the high specificities of the aptamers for Lucentis. Selectivity tests were also performed using both aptamers with different proteins in which negligible responses were obtained from interfering proteins with respect to Lucentis. Although neither of the two aptamers showed prominent responses to the interfering proteins, slightly better selectivity was shown by aptamer 1. The same aptamers were tested for their application in the detection of Lucentis in spiked and real media broth samples. For this detection test, interdigitated (IDT) gold electrodes on a glass substrate were fabricated using standard photolithography and thermal deposition techniques. NFIS measurements were used for the label-free detection of Lucentis in samples. The linear ranges of detection for aptamers 1 and 25 were found to be 22–100 nM and 40–100 nM, respectively. The LODs for aptamers 1 and 25 were calculated to be 22 nM and 40 nM, respectively, which were significantly better than the values from a HPLC-based detection method (about 240 nM). The real sample analysis results were cross-checked via a standard HPLC method, and better correlation was found between the HPLC and aptamer 1 results than the aptamer 25 results; hence, aptamer 1 can be further analyzed and tested for use in affinity column chromatography and detection-kit/chip-based PAT for Lucentis bioproduction.
Introduction
Wet age-related macular degeneration (AMD) involves the abnormal growth of blood vessels underneath the macula due to enhanced levels of vascular endothelial growth factor A (VEGF-A). Lucentis, or ranibizumab, is a monoclonal antibody fragment (Fab) of recombinant humanized monoclonal antibodies and acts as an anti-VEGF-A therapeutic drug. It is injected into the eyes of patients for the treatment of this disease.1,2 This anti-VEGF drug cancels out the effects of VEGF-A on the macula and improves vision.3 Unfortunately, despite the outstanding medical applications of this drug, the low production yields of Lucentis in bioreactors along with the absence of a suitable low-cost high-affinity biomolecule for process analytical techniques (PAT) makes treatment very expensive.4 Not limited to this, there have been delays in clone selection processes and initial optimization studies for enhancing the yield of Lucentis, resulting in the deployment of tedious and expensive offline analytical techniques like high-pressure liquid chromatography (HPLC).5 Due to scarcity of high-affinity biomolecules for Lucentis, Lucentis purification processes are also hampered. Hence, high-affinity biomolecule-based PAT for its bioproduction would be highly desirable.In the last couple of decades, aptamers (single-stranded nucleic acids) have emerged as alternatives to antibodies and other ligands for affinity assays and purification processes due to the possibility for their in vitro bulk production using polymerase chain reaction (PCR) technology, their easy modification without loss of affinity, and their better stability under adverse conditions. They have been proven in some cases to be better candidates than antibodies, with comparable binding affinities towards target analytes, resulting in the replacement of some antibody-affinity tests with aptamer-affinity tests.6–10 Moreover, between ssDNA and RNA aptamers, ssDNA aptamers are found to be superior due to their resistance to nucleases.11 These aptamers are generated for target analytes using systematic evolution of ligands by exponential enrichment (SELEX). With this method, a mixture of aptamers is incubated with a target analyte and specific high-affinity aptamers are purified from the mixture via partitioning non-binding low-affinity aptamers. SELEX comprises of a repetitive cycle involving five primary steps: the binding of aptamers with the target analyte; the partitioning of non-binding low-affinity aptamers from high-affinity aptamers using affinity methods; the elution of high-affinity aptamers; the amplification of the eluted aptamers; and the regeneration of aptamers for the next round of SELEX.10,12 SELEX has already been employed for the generation of aptamers for many target analytes.9,10,13–19 To complete SELEX within a few cycles and get high-affinity aptamers, the stringency of each cycle is found to play a crucial role. The stringency is increased with each SELEX cycle to lift the level of competition amongst aptamers, and only high-affinity aptamers are going to win out under highly adverse conditions.20
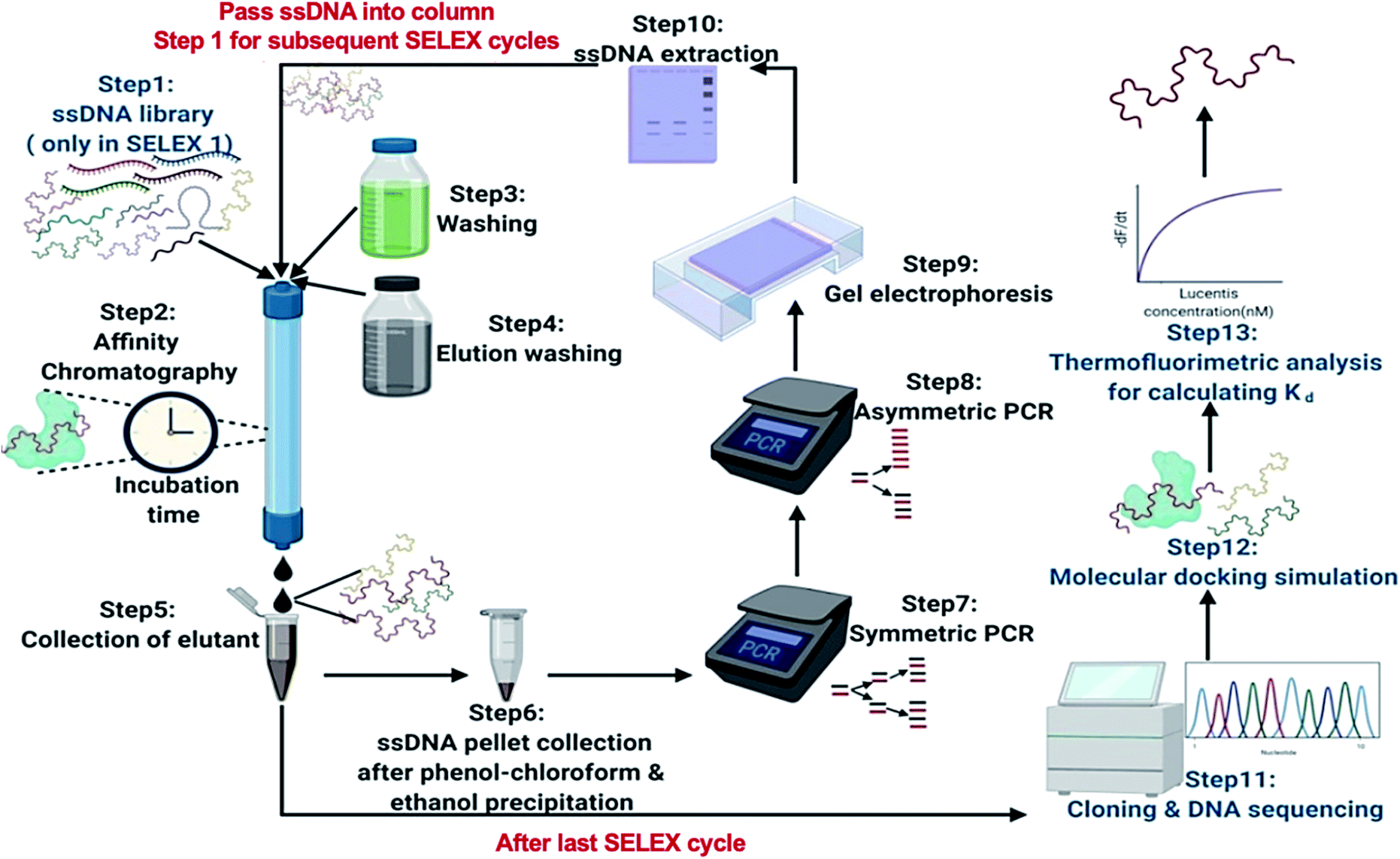 | ||
| Fig. 1 A schematic illustration of the steps involved in the SELEX process. | ||
In this research, we reported the generation of highly specific and selective ssDNA aptamers (Fig. 1) for Lucentis using only 10 rounds of SELEX via employing highly stringent techniques together in each cycle. Then, the dissociation constant (Kd) values of the aptamers were found using thermofluorimetric and non-faradaic impedance spectroscopy (NFIS) methods. The docking of aptamers with Lucentis and the secondary structures were studied using the HADDOCK and mfold web servers. In order to check the application of these aptamers for the detection of Lucentis during different phases of its bioproduction (PAT), spiked and real media broth samples were analysed using aptamer-immobilized gold interdigitated electrodes and NFIS.
Results and discussion
The optimization of symmetric and asymmetric PCR for the regeneration of ssDNA
After the elution of the ssDNA aptamers in each SELEX cycle, PCR amplification was used to generate dsDNA. Hence, every subsequent SELEX cycle demanded the regeneration of ssDNA aptamers to accomplish binding with target analyte, which was done in our work using asymmetric PCR.21 First, the number of cycles and annealing temperature for symmetric PCR were optimized to get a better yield of dsDNA for carrying out asymmetric PCR. Different numbers of PCR cycles (10, 15, 20, 25, 30, and 35) were used for the amplification of the ssDNA aptamers, and the products were seen on 4% agarose after gel electrophoresis. It can be interpreted from the gel images shown in Fig. S1(a)† that an 80 bp dsDNA band originated in lanes 2–7 with a faint non-specific band at a low bp position. After 10 PCR cycles, the 80 bp dsDNA band was extremely light; it became prominent for 15 to 35 PCR cycles, with the existence of a faint non-specific band. Along with this, non-specific bands were observed in lanes 5–7 in negative-control PCR samples (Fig. S1(b)†) for 25–35 cycles, as the risk of opportunistic mispairing increases with a higher number of cycles. Hence, it can be interpreted that 20 PCR cycles provided the most intense dsDNA band at the 80 bp location with the least non-specific bands (no band in the negative control). Similarly, different annealing temperatures of 45, 50, 55, 60, and 65 °C were tested, and the products can be seen in the gel images in Fig. S2.† The intensity of the dsDNA band increased upon increasing the annealing temperature from 45 to 55 °C and decreased at higher temperatures. Hence, it can be concluded that the maximum yield of dsDNA at the 80 bp location was seen at an annealing temperature of 55 °C with minimum levels of non-specific products. Therefore, 20 PCR cycles and an annealing temperature of 55 °C were chosen as the final conditions for symmetric PCR.Then, for the regeneration of ssDNA aptamers from dsDNA, different primer ratios (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 20:1, 100:1, and 500:1 (reverse:forward)) were used, and it can be observed in the gel image in Fig. 2 that an additional band near 50 bp appeared along with the 80 bp dsDNA band after asymmetric PCR (lanes 5–8). This band originated due to ssDNA aptamers, which travelled faster than dsDNA due to their low molecular weight. From a primer ratio of 10:1 to 20:1, the yield of ssDNA aptamers (high-intensity band) exceeded that of dsDNA, but this decreased at primer ratios of 100:1 and 500:1. In our case, the most intense ssDNA aptamer band was obtained at a primer ratio of 20:1 (reverse:forward) in lane 6 with the lowest yield of dsDNA; hence, for the regeneration of ssDNA aptamers in the SELEX process, this primer ratio was used. Further, using this primer ratio, the number of PCR cycles used for asymmetric PCR was also optimized, testing 10, 15, 20, 25, 30, 35, and 40 PCR cycles. A gel image is shown in Fig. S3(a).† It can be seen that the ssDNA aptamer band intensity increased upon going from 10 to 30 cycles and beyond this there was a dip in the band intensity. Meanwhile, as seen in Fig. S3(b),† from 25 to 40 cycles, the formation of non-specific bands was seen in negative controls due to a higher number of PCR cycles being used. Hence, we can conclude that 20 PCR cycles during asymmetric PCR resulted in better yields of ssDNA aptamers in comparison to dsDNA, with no non-specific band in the negative control. The same annealing temperature was used for asymmetric PCR as the primers were the same for both symmetric and asymmetric PCR.
:1, 20:1, 100:1, and 500:1 (reverse:forward)) were used, and it can be observed in the gel image in Fig. 2 that an additional band near 50 bp appeared along with the 80 bp dsDNA band after asymmetric PCR (lanes 5–8). This band originated due to ssDNA aptamers, which travelled faster than dsDNA due to their low molecular weight. From a primer ratio of 10:1 to 20:1, the yield of ssDNA aptamers (high-intensity band) exceeded that of dsDNA, but this decreased at primer ratios of 100:1 and 500:1. In our case, the most intense ssDNA aptamer band was obtained at a primer ratio of 20:1 (reverse:forward) in lane 6 with the lowest yield of dsDNA; hence, for the regeneration of ssDNA aptamers in the SELEX process, this primer ratio was used. Further, using this primer ratio, the number of PCR cycles used for asymmetric PCR was also optimized, testing 10, 15, 20, 25, 30, 35, and 40 PCR cycles. A gel image is shown in Fig. S3(a).† It can be seen that the ssDNA aptamer band intensity increased upon going from 10 to 30 cycles and beyond this there was a dip in the band intensity. Meanwhile, as seen in Fig. S3(b),† from 25 to 40 cycles, the formation of non-specific bands was seen in negative controls due to a higher number of PCR cycles being used. Hence, we can conclude that 20 PCR cycles during asymmetric PCR resulted in better yields of ssDNA aptamers in comparison to dsDNA, with no non-specific band in the negative control. The same annealing temperature was used for asymmetric PCR as the primers were the same for both symmetric and asymmetric PCR.
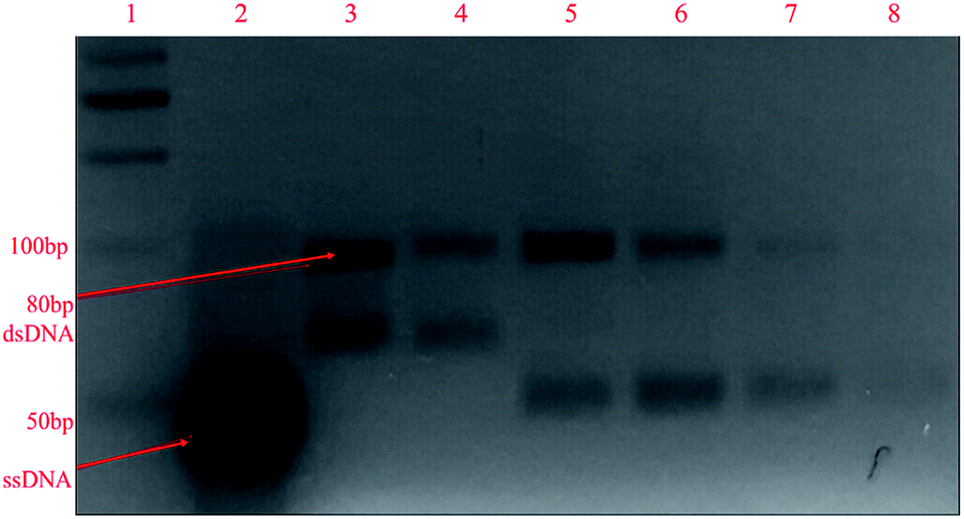 | ||
| Fig. 2 The gel electrophoresis of asymmetric PCR products at different primer ratios (left to right): lane 1: 50 bp ladder; lane 2: pure ssDNA library; lanes 3–4: PCR products of ssDNA library; lanes 5–8: asymmetric PCR products at primer ratios of 10:1, 20:1, 100:1, and 500:1, respectively, on 4% agarose gel. | ||
The in vitro selection of aptamers via SELEX
For the selection of specific aptamers for Lucentis, an N40 ssDNA random library was used, flanked by the constant priming regions required for PCR amplification during the SELEX cycles. Aptamers were selected using Lucentis-immobilized Sepharose beads as the fixed matrix in affinity chromatography. The immobilization of Lucentis on Sepharose beads was confirmed using Fourier-transform infrared spectroscopy (FTIR). FTIR spectra from Sepharose beads before and after the immobilization of Lucentis were recorded and are shown in Fig. S4† and discussed in Section S1 in the ESI.†In SELEX, the number of cycles used directly depends on the strategy used for selection and the stringency of the cycle; for example, by decreasing the concentration of the immobilized target and the incubation time, increasing the number of washing steps, and expanding the elution procedure, higher stringency is obtained. Our SELEX procedure consisted of three phases: a flexible phase, a medium stringency phase, and a highest stringency phase. The purpose of the first flexible phase was to: (i) boost the ssDNA library with aptamers that have affinity, lower or higher, for Lucentis; and (ii) remove the large number of unwanted non-binding aptamers from the ssDNA library. In the flexible phase, the concentration of immobilized Lucentis in the column was 2.3 mg, the incubation time was 60 min, the volume of washing buffer was low, and a single elution technique was used, i.e., 3 M NaCl. In the first SELEX cycle, a single copy of each aptamer was present in the library, and the elutant also contained single copies of the aptamers that have affinity for Lucentis. Hence, this cycle was the most crucial for not missing any highly specific aptamers and, therefore, was kept the easiest. This can be interpreted from the elutant yield from the first cycle; only 1.86% of the aptamers in the ssDNA library showed some affinity for Lucentis and non-binding aptamers were partitioned during the washing step (Fig. 3). Next, two cycles were used to boost this ssDNA library with aptamers from low to high affinity under the least stringent conditions. The improvement in the ssDNA library during these two cycles could be seen via the increase in the elutant yield (from 10 to 16.2%), as seen in Fig. 3. This fact proved that the number of copies or ssDNA aptamers showing some affinity for Lucentis increased in these two cycles.
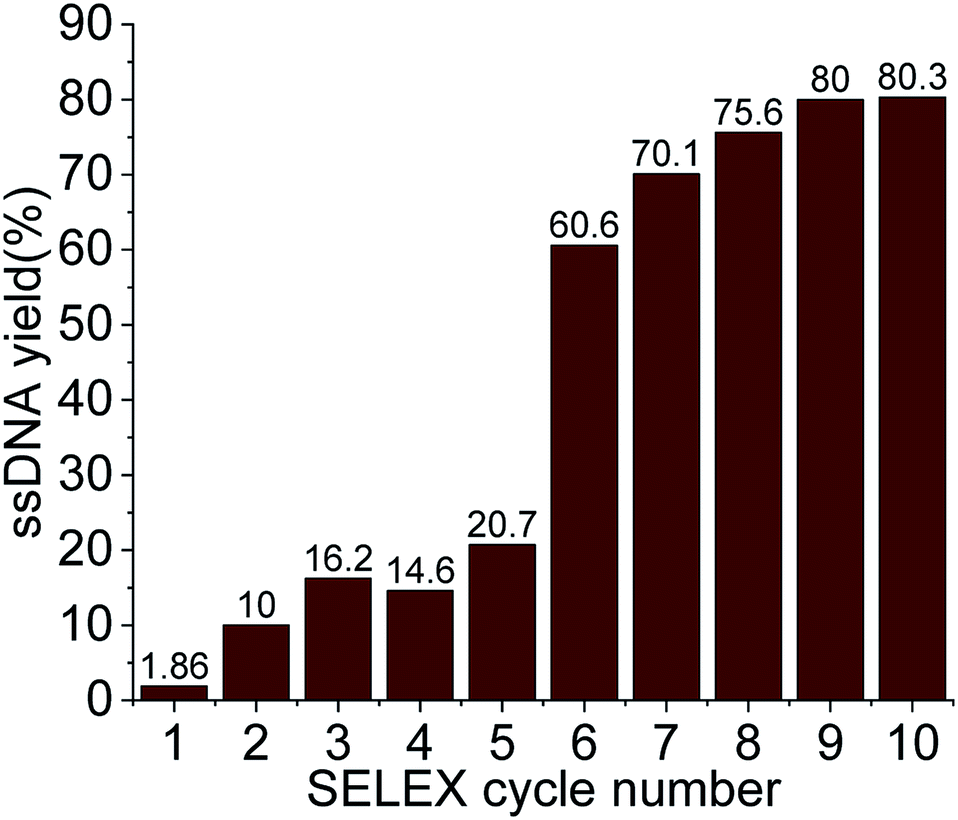 | ||
| Fig. 3 The ssDNA yield eluted in each SELEX cycle for Lucentis. | ||
After obtaining a sufficient pool of aptamers in the first flexible phase, the medium stringency phase (4–6th cycle) started, in which the concentration of immobilized Lucentis and the incubation time were reduced to 0.25 mg and 30 minutes, along with an increase in the volume of washing buffer. The purpose of this phase was to: (i) remove low-to medium-affinity aptamers from the library; and (ii) boost the library with high-affinity aptamers via providing stringent conditions that can be tolerated only by highly specific aptamers for Lucentis. In the fourth SELEX cycle, a decrease was seen in the amount of eluted aptamers due to the abrupt increase in stringency (14.6%). In the next two cycles (5–6th), the aptamer yield in the elutant increased (from 20.7 to 60.6%) showing an increase in the number of aptamers with high affinity for Lucentis retained in the column that tolerated the existence of stringent conditions (Fig. 3). Meanwhile, an increase in the number of washing steps reduced the number of low-to medium-affinity aptamers in the elutant.
Then, the next four cycles (high stringency phase) of SELEX were made more stringent to create competition between the high-affinity aptamers to find the best binding aptamer for Lucentis that can withstand adverse conditions, like decreases in the concentration of Lucentis and incubation time to 0.1 mg and 15 minutes, an increase in the number of washing steps, and the application of three different elution techniques together. In this phase, the aptamers yield increased from 70 to 80% from the seventh to the ninth SELEX cycle, even after applying the most stringent conditions, as shown in Fig. 3. This fact proved that the column retained only highly specific aptamers that could tolerate the adverse conditions that exponentially increased in stringency in this last phase of SELEX. In the last two cycles (9–10th), we did not notice any increase in the yield of aptamers, which suggested that we had reached a pool that contained highly specific aptamers for Lucentis and that there are no further requirements for enrichment. The overall strategy used for SELEX is summarized and shown in Table S1.†
Sequence analysis and docking simulation after SELEX
The 25 specific ssDNA aptamers for Lucentis shown in Table 1 were obtained after cloning and a DNA sequencing study of the elutant upon the completion of the 10th SELEX cycle. Protein Data Bank (PDB) format files of the chains of Lucentis (heavy and light) were obtained from the RCSB PDB website. The active and passive sites of the heavy and light chains of Lucentis were discovered and analyzed using the I-TASSER web server. Then, each aptamer was docked with both chains of Lucentis (heavy and light) using the HADDOCK web server, and the root-mean-square deviation (RMSD) of each docking pair was checked and compared, as shown in Table 2. Out of the top ten docking pair models/clusters provided by the HADDOCK web server, the top model was selected due to its highest reliability and negative Z-score according to the analysis. Two main criteria for the selection of an aptamer were as follows. (i) The RMSD should be less than 1.5 Å for two specifically bound molecules.22 However, to achieve high-affinity models in our case, the RMSD cut-off for selection was 1 Å. Only those docking pairs that had a RMSD less than 1 Å would be selected for further analysis (i.e. criteria (ii)) (Table 2). (ii) As only passive sites were free for interactions with aptamers in an intact molecule of Lucentis, the aptamers that were found to interact with the active sites of heavy or light chains (already occupied with each other) of Lucentis were directly eliminated (interactions of sequences 3, 7, 10, 15, 17, 20, 22, 23, and 24 with the heavy chain; sequences 4, 7, 15, and 17 with the light chain) (Fig. S5†). Upon applying these two conditions to all 25 aptamers, aptamers 1, 9, 14, and 25 were found to exhibit high affinity towards the heavy chain of Lucentis and, hence, were suitable for further experimental analysis. At the same time, they did not bind to the light chain of Lucentis, which indirectly showed the specificity and selectivity of these aptamers for the heavy chain only.| Selected ssDNA aptamer sequences (5′ to 3′) | |
|---|---|
| 1 | AGGGAAGAGAAGGACATATGATGGGGGCTAGTATCGACGTCTGCCTATGTTGTTAGTCGGTGTTGACTAGTACATGACCACTTG |
| 2 | CAAGTGTTTTATGTACTAGTCAACTCGTGACCACATGAAATGTACACCCAACACCTCGCCTTGATCATATGTCCTTCTCTTCCCT |
| 3 | CAAGTGCCTCTTGTACTAGTCAACCATGCCATGACTGTGTGACCTAGGGTACTAGATGCCTCGATCATATGTCCTTCTCTTCCCT |
| 4 | AGGGAAGATAAGGACATATGATCACAGACTGCCCATTGTCACTTTCGATAGTCGGAATGTGCTTGACTAGTACATGACCACTTG |
| 5 | CAAGTGGTTTTTGTACTAGTCAACACGCTTAACTCGGACGTTGTCTAACCAGTCGTTCCCCTCAATCATATGTCCTTCTCTTCCCT |
| 6 | AGGGAAGAGAAGGACATATGATCACCAGCACGTTGCACCAAATCCACAACAGTTCCCCCGCGTTGACTAGTACATGACCACTTG |
| 7 | AGGGAAGAGAAGGACATATGATGACGGCTGGATCTTTCAGAACCTAGGTAACATGTACTGGCTTGACTAGTACATGACCACTTG |
| 8 | AGGGAAGAGAAGGACATATGATTGTGCAATACGAGACCACTCACAATGATAATAAGATCCGTTTGACTAGTACATGACCACTTG |
| 9 | CAAGTGGTCATGTACTAGTCAATGCAAGTGTAATTGACACCGGCTCACCAGCACAGGACCGCATCATATGTCCTTCTCTTCCC |
| 10 | CAAGCTGGTCATGTACTAGTCAACCATGCCATGACTGTGTGACCTAGGGTACTAGATGCCTCGATCATATGTCCTTCTCTTCCCT |
| 11 | ACGTAAGAGAAGGACATATGATGGCCATCGACCCTATCGACCCATCACCGTACTTAGCGTGCTTGACTAGTACATGACCACTTG |
| 12 | CAAGTGTGTCATGTACTAGTCAACACAACGGAATCAAGGTAACTCGATACGCACTTGGTACGCATCATATGTCCTTCTCTTCCCT |
| 13 | AGGGAAGTAGAAGGACATATGATGACACATTGAGACCTATCTACTCGATTCCCTAAGTTGTGGTTGACTAGTACATGACCACTTG |
| 14 | CAAGTGGTCATGTACTAGTCAATCCGCAAGGTATGGGCAGGACGCGACAAGTCCCAGTGGTGATCATATGTCCTTCTCTTCCCT |
| 15 | CAAGTGGTCATGTACTAGTCAATGCAAGTGTAATTGACACCGGCTCACCAGCACAGGACCGCATCATATGTCCTTCTCTTCCCT |
| 16 | CAAGTGGTCATGTACTAGTCAATCCGCAAGGTATGCGCAGGACGCGACAAGTCCCAGTGGTGATCATATGTCCTTCTCTTCCCT |
| 17 | CACGTTGTTCATGTACTAGTCAATCCGCAAGGTATGGGCAGGACGCGACAAGTCCCAGTGGTGATCATATGTCCTTCTCTTCCCT |
| 18 | AGGGAAGTAGAAGGACATATGATGACACATTGAGACCTATCTACTCGATTCCCTAAGTTGTGGTTGACTAGTACATGACCACTTG |
| 19 | CAAGTGTTTCATGTACTAGTCAATGATCCGCTCCTGGTAGTTTCTCGCACCTTGGTGTTCCTGATCATATGTCCTTCTCTTCCCT |
| 20 | ACTGGTAAGAGAAGGACATATGATGCAGCAGGTTGCACGTTTACCGTAGGTCGTCTAGTCCTTATTGACTAGTACATGAC |
| 21 | CAAGTGGTCATGTACTAGTCAACGGCCGGCACTGGACGTTCTAGCTCTATCCTGACGAGCTCATCATATGTCCTTCTCTTCCCT |
| 22 | ATGTTAATAGAAGGACATATGATGGGCGGGCAACGTAGGGTCTATACCCCTTGTCATGGTGCGTTGACTAGTACATGACCACTTG |
| 23 | ACGTTAATAGAAGGACATATGATCGCCCCCTGTACGAAGCGGACGGATGCAAGCGCACCTCCTTTGACTAGTACATGACCACTTG |
| 24 | AGGGAAGAGAAGGACATATGATGACTACTCGCAGTCTCACCATTGATATTATGGGGCGTTCATTGACTAGTACATGACCACTTG |
| 25 | AGGGAAGAGAAGGACATATGATAAGGCTCTGAACGTGCTAAGAGATCGTTGTAGTGAGCTGGTTGACTAGTACATGACCACTTG |
| Seq. | RMSD for heavy chain and ssDNA (Å) | RMSD for light chain and ssDNA (Å) |
|---|---|---|
| 1 | 0.8 ± 0.5 | 1.9 ± 1.6 |
| 2 | 1.6 ± 0.9 | 33.4 ± 0.2 |
| 3 | 1.0 ± 0.6 | 30.5 ± 0.5 |
| 4 | 10.1 ± 0.3 | 1.2 ± 0.7 |
| 5 | 1.5 ± 0.8 | 8.3 ± 0.6 |
| 6 | 2.0 ± 1.5 | 32.9 ± 0.1 |
| 7 | 0.9 ± 0.6 | 0.8 ± 0.5 |
| 8 | 28.5 ± 0.1 | 3.2 ± 1.0 |
| 9 | 0.8 ± 0.4 | 32.7 ± 0.6 |
| 10 | 0.8 ± 0.5 | 1.8 ± 1.0 |
| 11 | 1.7 ± 1.2 | 3.6 ± 2.1 |
| 12 | 1.2 ± 0.8 | 31 ± 0.1 |
| 13 | 6.2 ± 0.8 | 11.5 ± 0.5 |
| 14 | 0.8 ± 0.5 | 1.9 ± 1.1 |
| 15 | 1.4 ± 1.0 | 0.9 ± 0.6 |
| 16 | 1.5 ± 1.0 | 28.9 ± 0.1 |
| 17 | 1.1 ± 0.6 | 0.9 ± 0.5 |
| 18 | 1.9 ± 1.5 | 19.5 ± 0.1 |
| 19 | 30.3 ± 0.1 | 32.6 ± 0.2 |
| 20 | 1.0 ± 0.6 | 1.2 ± 0.8 |
| 21 | 11.8 ± 0.5 | 1.3 ± 0.9 |
| 22 | 0.7 ± 0.4 | 23.8 ± 0.5 |
| 23 | 27.7 ± 0.5 | 13.0 ± 0.5 |
| 24 | 1.0 ± 0.6 | 2.0 ± 1.2 |
| 25 | 0.9 ± 0.6 | 1.4 ± 1.0 |
Aptamer–Lucentis binding assays and the evaluation of dissociation constant (Kd) values using thermofluorimetric and non-faradaic impedance (NFIS) analysis
While docking simulations are required to minimize the need to experimentally evaluate the binding affinities of all the generated aptamers, binding assays are necessary to confirm the specificity and affinity of an aptamer towards the target analyte. Hence, the four selected aptamers were tested for their binding affinities towards Lucentis using thermofluorimetric analysis. In this method, the melting peaks of the aptamers and aptamer–Lucentis complexes were recorded using real-time PCR thermal scanning.23 The greater the difference between the aptamer and aptamer–Lucentis complex melting peak, the higher the stability of the complex and, hence, the affinity of the aptamer for Lucentis (Fig. 4). The results of the binding assays are discussed in Section S2 in the ESI.† Aptamers 1 and 25 showed the maximum differences in the temperatures of the melting peaks; hence, Kd values were estimated only for these aptamers (Fig. S6†). Additionally, thermofluorimetric analysis also helped to establish that the four aptamers obtained after docking simulations, three were found to have good binding affinity for Lucentis, i.e. the docking simulation results showed 75% accuracy. Different concentrations of Lucentis were analyzed with aptamers 1 and 25 to estimate Kd for both aptamers toward Lucentis using thermofluorimetric analysis.24 Calibration curves showing changes in the melting peak height of the aptamer–Lucentis complex versus the concentration of Lucentis for aptamers 1 and 25 are shown in Fig. S7.† The melting peak height change was linear for both aptamers up to a Lucentis concentration of around 110 nM, but above this, the aptamer binding site got saturated with higher concentrations of Lucentis. The Kd values for aptamers 1 and 25 were estimated to be 23.21 ± 9.67 nM and 29.22 ± 6.32 nM, respectively, from thermofluorimetric analysis.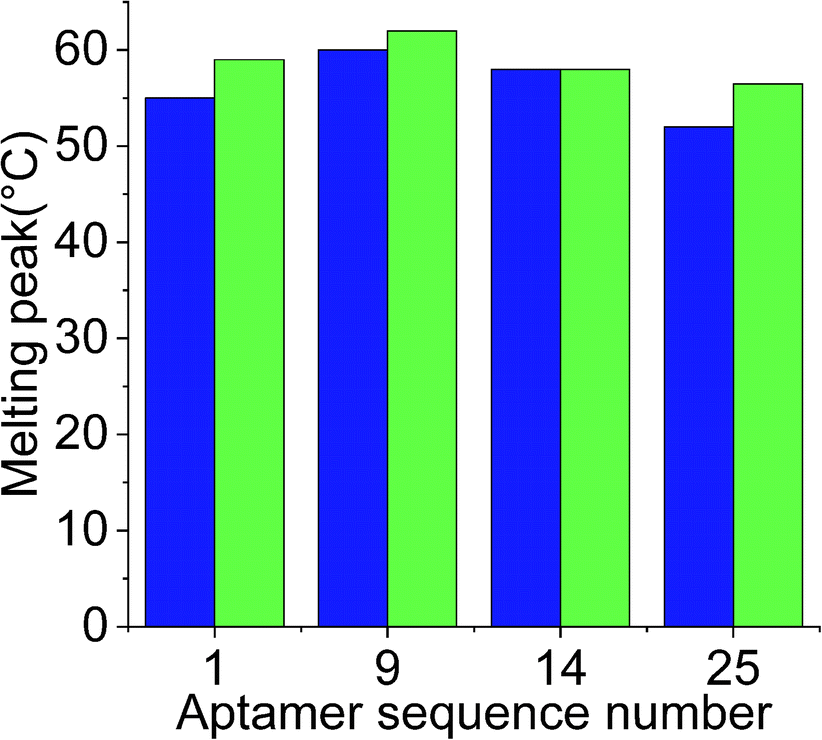 | ||
Fig. 4 Chart showing the difference between the melting peak height of the aptamer ( ) and the aptamer–Lucentis complex ( ) and the aptamer–Lucentis complex ( ), obtained using thermofluorimetric analysis. ), obtained using thermofluorimetric analysis. | ||
These two aptamers, 1 and 25, were again tested for their binding affinities using NFIS. In this test, a known concentration of Lucentis was immobilized on interdigitated (IDT) gold electrodes and then the electrodes were dipped in a 100 nM solution of either aptamer 1 or 25. After the immobilization of Lucentis and upon a reaction between the aptamer and Lucentis, the impedance of the chip increased, as seen through a Nyquist plot. The capacitive component (−Im(Z)/ohm) of the impedance increased with respect to the resistive component (Re(Z)/ohm) which can be seen in Fig. 5(a and b). This behaviour proved the dominance of the capacitive nature of the electrode, showing an almost linear close-to-vertical response. As this is not an ideal capacitor, a straight line parallel to the capacitive component (−Im(Z)/ohm) was not observed in the Nyquist plot. A resistive component was always present parallel to the capacitance plot due to leakage current at defect sites.25 Because the capacitive component (−Im(Z)/ohm) showed more variation with respect to the resistive component (Re(Z)/ohm), we selected the capacitive component (−Im(Z)/ohm) for further data analysis, which was due to the double-layer capacitance of the electrode/electrolyte interface. As the maximum signal was obtained at a frequency of 4.64 Hz, calibration curves showing the change in the capacitive component of impedance (−ΔIm(Z)/ohm) versus the concentration of Lucentis for both aptamers, 1 and 25, were plotted at same frequency (Fig. 5(c)). The Kd values for aptamers 1 and 25 for Lucentis were calculated to be 25.72 ± 4.2 nM and 34.20 ± 5.05 nM, respectively, via this technique. We can see that the Kd values for both aptamers towards Lucentis calculated using two different binding assay methods were very close to each other. This not only cross-verified the results but also confirmed the low Kd values (in the nanomolar range) of the aptamers 1 and 25, indicating the significant binding affinities (specificity) of the aptamers towards Lucentis. Hence, we can say that these two aptamers can be utilized further analysis like selectivity test and for different applications, like the detection of Lucentis.
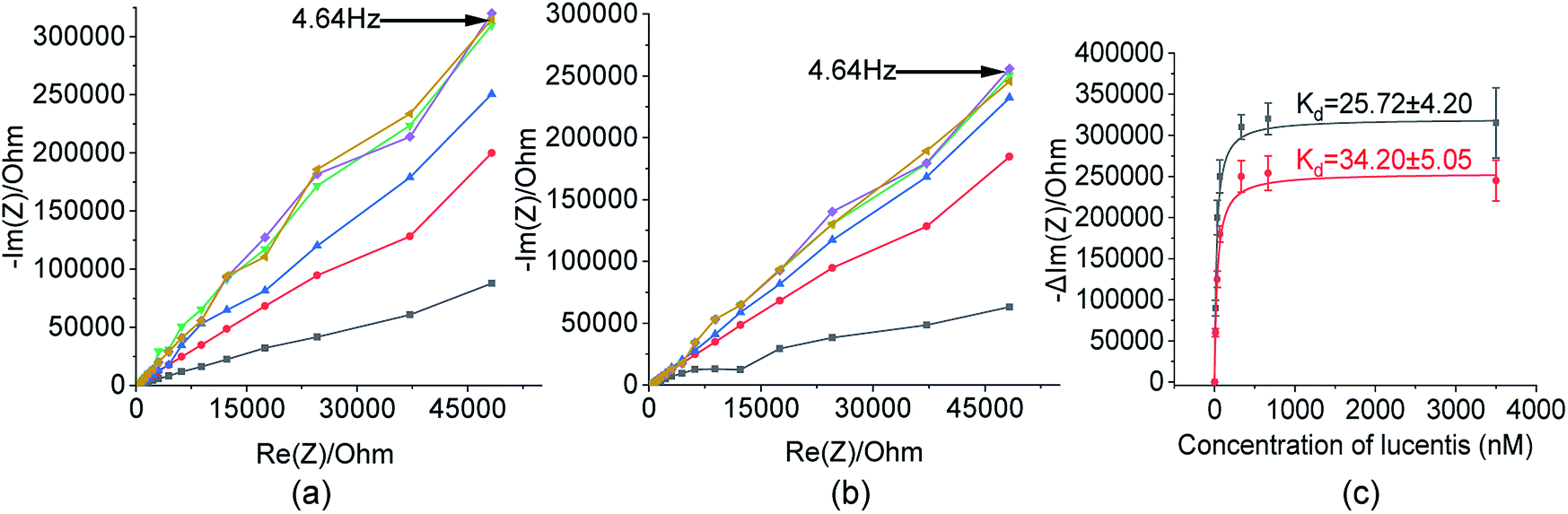 | ||
Fig. 5 Nyquist plots toward 13 nM ( ), 33 nM ( ), 33 nM ( ), 66 nM ( ), 66 nM ( ), 333 nM ( ), 333 nM ( ), 666 nM ( ), 666 nM ( ), and 3500 nM ( ), and 3500 nM ( ) Lucentis for (a) aptamer 1 and (b) aptamer 25. (c) Calibration curves between the capacitive component and concentration of Lucentis for the calculation of dissociation constants for aptamers 1 ( ) Lucentis for (a) aptamer 1 and (b) aptamer 25. (c) Calibration curves between the capacitive component and concentration of Lucentis for the calculation of dissociation constants for aptamers 1 ( ) and 25 ( ) and 25 ( ) using NFIS. ) using NFIS. | ||
Estimation of the secondary structures of selected aptamers
The analysis and study of the secondary structures of aptamers is required to estimate the common site of interaction between a specific aptamer and Lucentis. For this, the secondary structures of aptamers were predicted while fixing the parameters used by the mfold web server to the same values as were used during SELEX selection (Fig. S8†). The main purpose of this prediction approach was to identify common regions in both aptamers responsible for interactions with the heavy chain of Lucentis. As we did not obtain any specific aptamers targeting the light chain of Lucentis, our focus was on the heavy chain of Lucentis. Due to their RMSD values lower than 1 Å and interactions with passive sites of the heavy chain of Lucentis, only aptamers 1, 9, 14, and 25 fulfilled the conditions used in the docking program and were selected for further analysis. Hence, for secondary structure analysis, we analysed only these four sequences. The best two aptamers (1 and 25) were found to have one big central loop with one common region in the format of a four-base–pair stem with a loop (Fig. 6). This stem (large and small stem loops were present) was absent in aptamer 9 (Fig. S8†), hence, this might be the reason for its low affinity for the heavy chain in comparison with aptamers 1 and 25. Similarly, in aptamer 14 (Fig. S8†), the four base–pair-stem is present with its loop, but the size of the central loop is approximately double those of aptamers 1 and 25. This might present some hindrance towards binding to the heavy chain of Lucentis in comparison to aptamers 1 and 25. Therefore, we can conclude that the four-base–pair stem loop with a central loop (approximately 35 nucleotides) might be responsible for the better binding with the heavy chain of Lucentis shown by aptamers 1 and 25.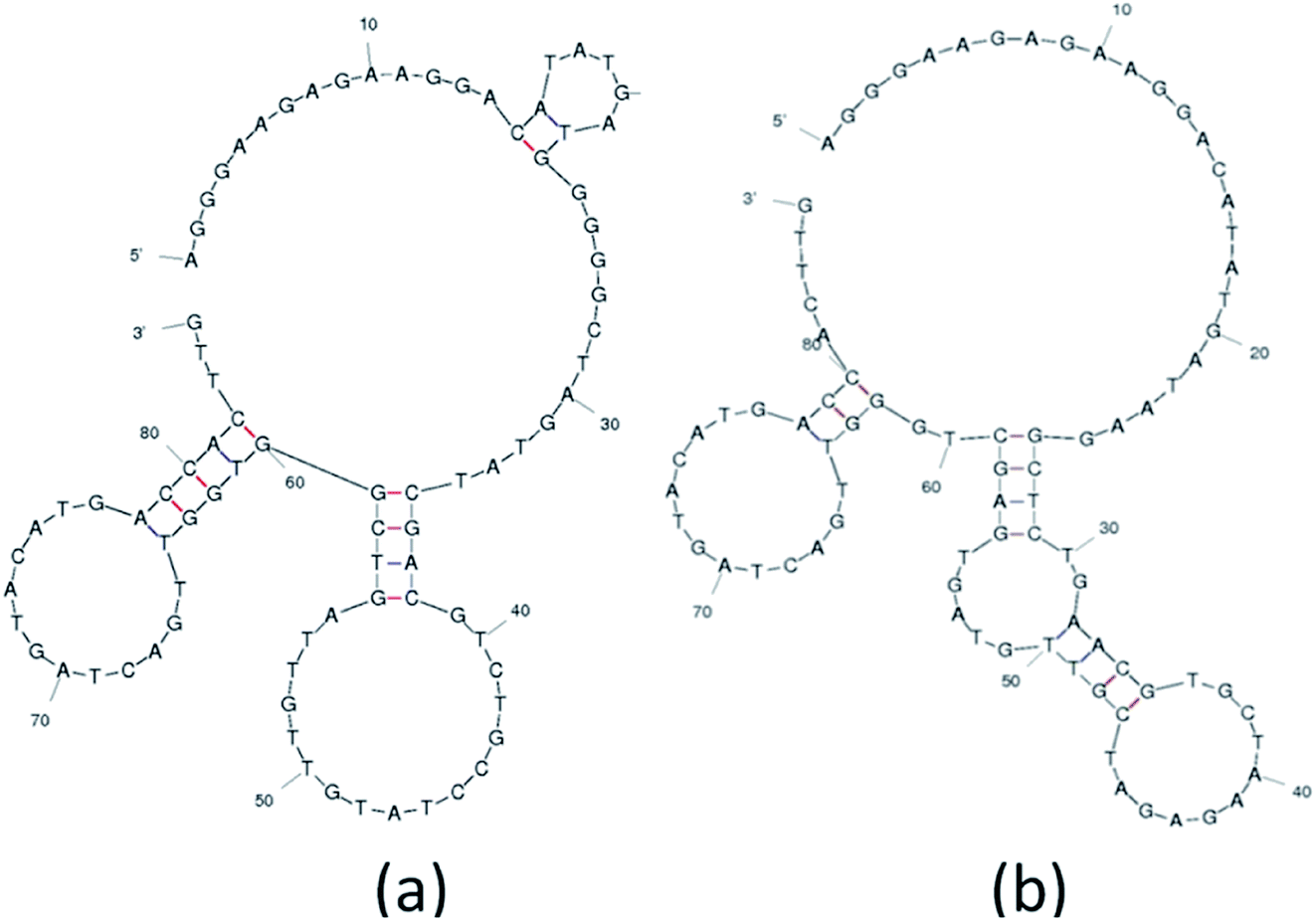 | ||
| Fig. 6 The secondary structures of (a) aptamer 1 and (b) aptamer 25 obtained using the mfold web server. | ||
Aptamer selectivity tests using NFIS
Our selected aptamers, 1 and 25, from SELEX were proven to be specific with high binding affinity (a low dissociation constant in the nM range) for Lucentis using docking simulations, thermofluorimetric and NFIS analysis. Now, to check the selectivity of the aptamers, sensing electrodes modified with aptamer 1 or 25 were dipped in solutions of granulocyte-colony stimulating factor (GCSF), glucose oxidase (GOx), urease, bovine serum albumin (BSA), media broth proteins (formed during the production of Lucentis in a bioreactor), and Lucentis in media broth proteins containing GCSF, GOx, urease and BSA also. The other proteins were used as simulated interferents and media broth was used as a source of interference for real samples. Significant signals from both aptamers were obtained only with Lucentis present in media broth proteins (containing added GCSF, GOx, urease and BSA also), showing minimal signals toward the other interfering proteins (Fig. 7). Although both aptamers can be used for different applications involving Lucentis due to their high specificity for Lucentis, aptamer 1 was found to be slightly more selective than aptamer 25 due to its lower response toward interfering proteins and higher response to Lucentis in media broth proteins (containing GCSF, GOx, urease and BSA). (The immobilization of the aptamers on gold electrodes was characterised using FTIR, as explained in Section S3 and Fig. S9 in the ESI.†)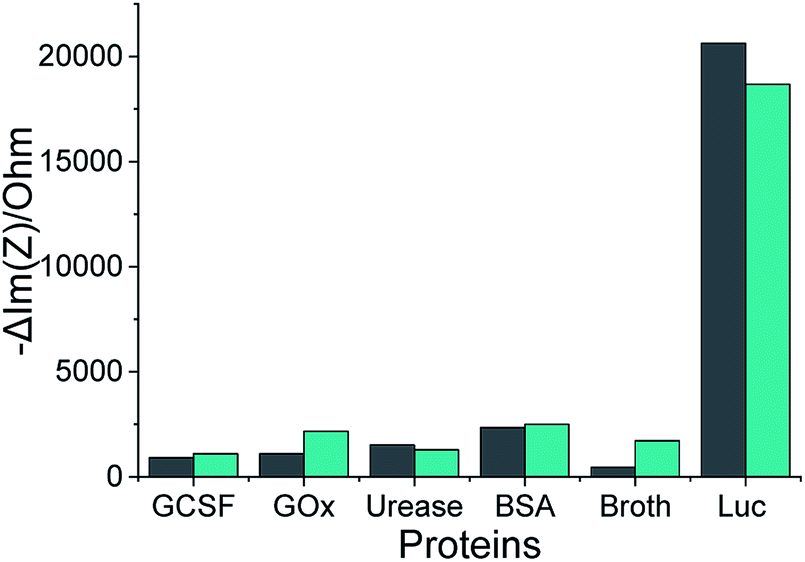 | ||
Fig. 7 The impedimetric responses of aptamers 1 ( ) and 25 ( ) and 25 ( ) following incubation with different proteins, where Luc stands for Lucentis. ) following incubation with different proteins, where Luc stands for Lucentis. | ||
The application of the selected aptamers for the detection of Lucentis
As already discussed, major changes were seen in the capacitive component (−Im(Z)/ohm) due to double-layer capacitance; the increase in the capacitive component (−Im(Z)/ohm) after the immobilization of Lucentis on IDT gold electrodes, as observed in Fig. 5(a and b), might be explained based on changes in the dielectric properties and thickness of the double layer. According to the Stern layer model, the application of charge to an electrode gives rise to the formation of a layer of opposite charge in solution separated by high-permittivity water molecules. Lucentis could displace the solvated ions and water outwards from the electrode surface, thus increasing the thickness (d) of the double layer and decreasing the dielectric permittivity (ε), thereby resulting in a decrease in the double-layer capacitance (Cdl), as shown in eqn (1), and an increase in the capacitive component (−Im(Z)/ohm).25,26 Similarly, interactions between Lucentis and the aptamer on the electrode would further increase the thickness and decrease the dielectric permittivity of the double layer. This phenomenon would enhance with an increase in the concentration of aptamer–Lucentis complex present, resulting in a proportional increase in the capacitive component (−Im(Z)/ohm), as shown in Fig. 5(a and b):26| Cdl = εεoA/d | (1) |
The same phenomenon could also be used to explain the behavioural changes when aptamers 1 and 25 were immobilized on sensing electrodes for the detection of different concentrations of Lucentis. The immobilization of the aptamer and increase in the concentration of Lucentis led to an increase in the thickness (d) of the double layer and a decrease in the dielectric permittivity (ε), resulting in a decrease in the double-layer capacitance (Cdl) and, hence, an increase in the capacitive component (−Im(Z)/ohm) of the impedance (Fig. 8(a and b)). Calibration curves at 4.64 Hz between the change in the capacitive component (−Im(Z)/ohm) and the concentration of Lucentis for both aptamers are shown in Fig. 8(c).
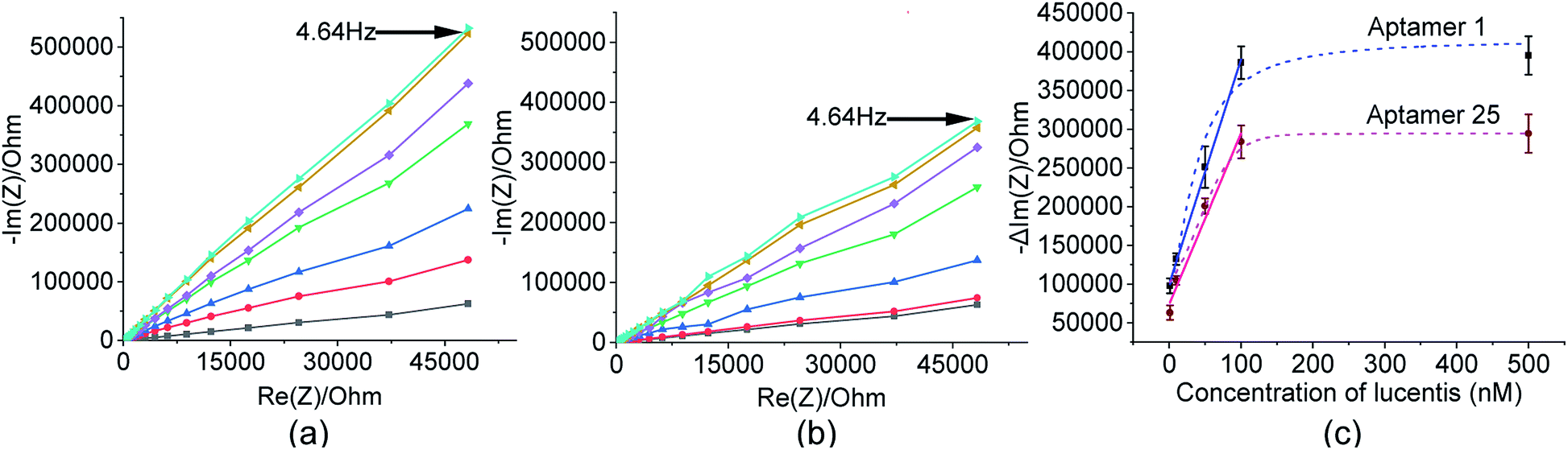 | ||
Fig. 8 Nyquist plots from a gold electrode ( ), an aptamer-immobilized gold electrode/blank ( ), an aptamer-immobilized gold electrode/blank ( ) dipped in phosphate buffer at pH 5.7, and aptamer-immobilized electrodes dipped in 1 nM ( ) dipped in phosphate buffer at pH 5.7, and aptamer-immobilized electrodes dipped in 1 nM ( ), 10 nM ( ), 10 nM ( ), 50 nM ( ), 50 nM ( ), 100 nM ( ), 100 nM ( ), and 500 nM ( ), and 500 nM ( ) Lucentis for (a) aptamer 1 and (b) aptamer 25. (c) Calibration curves between the capacitive component and concentration of Lucentis for aptamers 1 and 25. ) Lucentis for (a) aptamer 1 and (b) aptamer 25. (c) Calibration curves between the capacitive component and concentration of Lucentis for aptamers 1 and 25. | ||
In these measurements, phosphate buffer at pH 5.7 was treated as a blank, and this background signal did not affect the experimental data obtained for different concentrations of Lucentis because it was subtracted from all the experimental data. The LOD was also calculated while considering the background signal, and there was negligible influence from the buffer while detecting the lower limits of measurement, which were found to be 22 nM and 40 nM for aptamers 1 and 25, respectively. The linear fitting models and equations are shown in Table S2,† with linear ranges from 22–100 nM and 40–100 nM for aptamers 1 and 25, respectively. The R2 values demonstrate that the linear fitting models fit the obtained data set. The LOD values were reasonably low in comparison to the standard HPLC method (nearly 200 ng ml−1 or the 4 nM range after 60-fold sample pre-concentration, or about 240 nM without sample pre-concentration).27
For real sample analysis, media broth samples from five different fermenters (five different Escherichia coli clones, unpublished data) were taken, and sensing electrodes were dipped in the media broth. Different concentrations of Lucentis were analysed using NFIS and compared with HPLC. Significant correlation was seen between the data from both methods (Fig. 9). Of both aptamers, better calibration curve sensitivity (Fig. 8(c)) and correlation with HPLC results during real sample analysis (Fig. 9) were shown by aptamer 1 in comparison to aptamer 25. In addition, a lower LOD was also shown by the sensing electrode with aptamer 1.
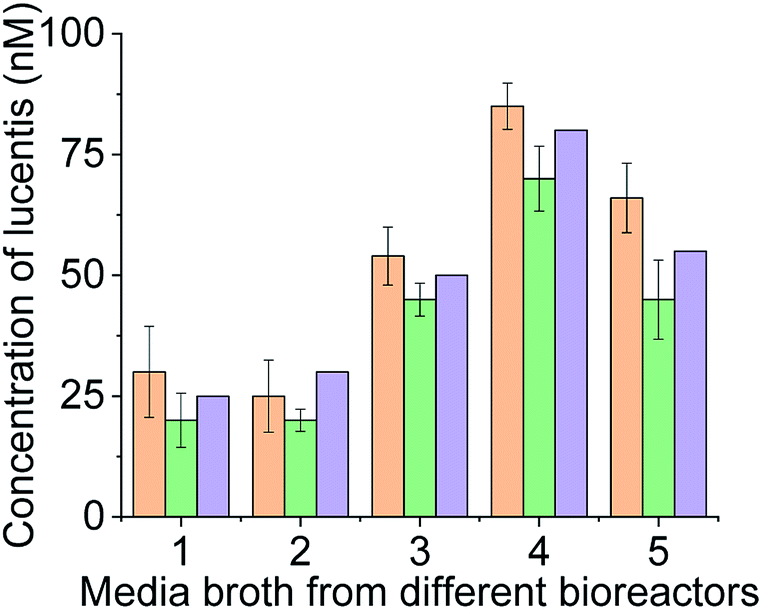 | ||
Fig. 9 Real sample analysis of Lucentis using aptamer 1 ( ), aptamer 25 ( ), aptamer 25 ( ), and HPLC ( ), and HPLC ( ). ). | ||
Hence, from these observations, we can conclude that the two aptamers were highly specific and selective for Lucentis, as tested using various techniques like docking simulations, and thermofluorimetric and NFIS analysis.
Both aptamers 1 and 25 worked efficiently but aptamer 1 performed comparatively better in terms of selectivity, sensitivity, LOD, linear range and for the detection of Lucentis in real samples. Thus, aptamer 1 (Table 1) can be employed further for various applications, like the purification of Lucentis in affinity columns, microfluidic chip-based detection, clone selection, process optimization, and inline monitoring during commercial manufacturing.
Experimental
Chemicals and reagents
(3-Aminopropyl)triethoxysilane (APTES), PCR buffer, MgCl2, dNTP mix and Taq polymerase were purchased from Himedia Lab. Pvt. Ltd. 4B Sepharose beads, glutaraldehyde, aminoethanethiol HCl, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were procured from Sigma-Aldrich, India. Acetone, isopropyl alcohol (IPA), and ethanol were obtained from Merck, India. ssDNA binding dye SYBR green II was obtained from Thermo Fischer Scientific. The glass substrates were obtained from University Wafer, USA. The AZ1512 HS photoresist was obtained from Microchemicals, USA. Lucentis and real samples (culture broth) were obtained from Bioseparations and Bioprocessing Laboratory, Department of Chemical Engineering, IIT Delhi, India.The immobilization of Lucentis on Sepharose beads for affinity chromatography column in the SELEX process
Fig. 1 illustrates the major steps involved in the immobilization of Lucentis on Sepharose beads for aptamer selection during the SELEX process. A fixed Sepharose bead matrix for the immobilization of Lucentis was selected and an affinity column was prepared. At first, 10 g of Sepharose beads was washed successively with 100 ml of water, 100 ml of acetone:water (30:70, 60:40, and 80:20), 300 ml of acetone, and 100 ml of dried acetone. Then, the beads were suspended in 25% APTES solution with nitrogen purging and stirring. The solution was continuously magnetically stirred for 24 h at room temperature. Then, the beads were washed with distilled water thoroughly. Next, 2.5% glutaraldehyde was added dropwise to the beads, followed by continuous stirring for 24 h at room temperature. Then, washing of the beads with 0.1 M sodium acetate buffer at pH 5.7 was performed and, finally, for immobilization, Lucentis (Table S1†) was added to the beads. The solution was stirred at 4 °C for 24 h and then the beads were thoroughly washed with 0.1 M sodium acetate buffer at pH 5.7.
Affinity chromatography column preparation
The glass column was washed properly with ethanol and then equilibrated with 0.1 M sodium acetate buffer at pH 5.7. Modified Sepharose beads were pumped into a 5 cm glass column at a low constant flow rate using a peristaltic pump at 4 °C to maintain the integrity and stability of Lucentis. After the preparation of the column, it was stored at 4 °C. Qualitative and quantitative analysis of the immobilization of Lucentis was carried out using FTIR and Lowry protein estimation methods. In addition, the leaching of Lucentis from column was checked using the Lowry protein estimation method from time to time.The optimization of asymmetric PCR for the regeneration of ssDNA
In SELEX, the product of each cycle (ssDNA) was amplified (dsDNA) for the preparation of a ssDNA library for the next cycle. Hence, the regeneration of ssDNA from dsDNA is one of the crucial steps in the SELEX process. In our protocol, we used asymmetric PCR for the regeneration of ssDNA from dsDNA, and various conditions were optimized to attain the best possible yield of ssDNA for every subsequent cycle of SELEX. Primarily, the number of PCR cycles and annealing temperature required for symmetric PCR to achieve a high yield of dsDNA for asymmetric PCR were optimized. Later, the primer concentration and number of cycles were optimized for asymmetric PCR. A reaction volume of 50 μl was prepared with 5 ng of ssDNA, final forward and reverse primer concentrations of 2 μM, 1× PCR buffer, MgCl2, dNTP mix, and Taq polymerase. The PCR conditions were as follows: initial denaturation at 95 °C for 5 minutes; optimized cycles of denaturation at 95 °C for 30 seconds; annealing at the optimized temperature for 30 seconds; elongation at 72 °C for 30 seconds; and final elongation at 72 °C for 5 min.Preparation of the ssDNA library
The ssDNA N40 library (PO DNA, 5′ TAG GGA AGA GAA GGA CAT ATG AT-N40-TTG ACT AGT ACA TGA CCA CTT GA 3′), and forward (PO DNA, 5′ TAG GGA AGA GAA GGA CAT ATG AT 3′) and reverse primers (PO DNA, 5′ TCA AGT GGT CAT GTA CTA GTC AA 3′) were purchased from TriLink BioTechnologies. The ssDNA library (1 nM) was dissolved in 0.1 M sodium acetate buffer at pH 5.7, heated at 95 °C for 10 minutes, and allowed to cool to room temperature (for every SELEX cycle). Then, the ssDNA library was passed through the affinity chromatography column during the SELEX process.The in vitro selection of aptamers via SELEX
For the selection of aptamers from the ssDNA library, affinity chromatography was used for performing the binding, partitioning, and elution steps of the SELEX process. Fig. 1 shows a schematic diagram illustrating the major steps involved in the SELEX process. The prepared ssDNA library was pumped into the affinity chromatography column for binding with Lucentis and incubated for the time mentioned in Table S1.† After the incubation period, unbound ssDNA was partitioned from the column via passing through 0.1 M sodium acetate washing buffer at pH 5.7. Finally, specifically bound ssDNA was eluted from the column using elution buffers. Then, standard phenol chloroform extraction and ethanol precipitation procedures were performed to get a pellet of eluted ssDNA aptamers. Afterwards, the eluted aptamers were amplified using symmetric PCR and then the generated dsDNA was used to regenerate ssDNA aptamers for the next SELEX cycle using asymmetric PCR. This completed one SELEX cycle, which was repeated until a set of specific aptamers for Lucentis was obtained. The SELEX approach followed for the selection of highly specific aptamers for Lucentis is shown in Table S1.†Sequence analysis after SELEX
The elutant from the final SELEX cycle was amplified via PCR, cloned, and DNA sequenced by Chromous Biotech Pvt. Ltd to obtain the sequences of the aptamers specific to Lucentis. Protein Data Bank (PDB) format files of the chains of Lucentis were obtained from the RCSB PDB website. The active and passive sites of the heavy and light chains of Lucentis were discovered and analyzed using I-TASSER. Then, the 25 obtained aptamer sequences were converted into 3D models and docked with each chain of Lucentis (heavy and light) using the HADDOCK web server.28 The RMSD values from every docking pair were checked and compared.The fabrication of IDT gold electrodes, immobilization technique and NFIS measurements
IDT gold electrodes were patterned on a glass substrate using standard photolithography and thermal evaporation techniques. First, gold was deposited on RCA1, RCA2, and a piranha-cleaned glass substrate through a sequence of AZ1512 HS photoresist spin-coating, patterning using UV photolithography, chromium and gold deposition (25 nm of chromium and 85 nm of gold) using a thermal evaporation technique, and, finally, a lift-off process. The width, spacing, and number of IDT electrodes were 0.1 mm, 0.65 mm, and 14, respectively, in the fabricated device (Fig. S10†).At first, the IDT electrode was dipped in ethanol and incubated for 2 h at room temperature. Similarly, the electrode was kept in 1 mM aminoethanethiol HCl solution, with 10 h of incubation at room temperature, followed by washing with Milli-Q water and ethanol. For binding assay experiments, after air drying, the electrode was suspended in a solution of 5 mM EDC, 8 mM NHS, and a known concentration of Lucentis in phosphate buffer at pH 5.7 and incubated for 5 h at room temperature. For the detection of Lucentis, the electrode was dipped in a solution of 5 mM EDC, 8 mM NHS, and 1 μM aptamer in phosphate buffer at pH 7 and incubated for 5 h at room temperature. Then, the washing of gold electrodes was performed five times using phosphate buffer to remove any Lucentis/aptamer that was not immobilized. Schematic representation of aptamer immobilization steps on gold electrode are shown in Fig. 10.
NFIS measurements were performed in a frequency range of 4 Hz-1 MHz with a sinusoidal voltage amplitude of 10 mV and at open circuit potential. NFIS measurements were recorded on a blank sample via dipping the IDT electrode in phosphate buffer at pH 5.7 (Fig. 10). Next, the electrode was kept in the sample and incubated for half an hour at room temperature. Then, the electrode was washed with an abundant amount of phosphate buffer at pH 5.7 and the response was recorded in the presence of phosphate buffer at pH 5.7.
 | ||
| Fig. 10 The immobilization of aptamers on gold sensing electrodes and the impedance spectroscopy detection setup. (a) Bare gold electrode on glass substrate. (b) Formation of thiol layer on gold electrode after aminoethanethiol treatment. (c) Aptamer immobilization on gold electrode using NHS & EDC. (d) Impedance spectroscopy detection setup. | ||
Aptamer–Lucentis binding assays and the evaluation of the dissociation constant (Kd)
After docking simulations, the best four aptamers were tested for their binding affinity towards Lucentis in the laboratory using thermofluorimetric analysis to confirm the docking results.23 In this technique, each of the four aptamers was incubated with Lucentis in the presence of the fluorescent ssDNA binding dye SYBR green II. The protocol is discussed in Section S2 in the ESI.† The best aptamer sequences were used for the estimation of Kd values via thermofluorimetric analysis at different concentrations of Lucentis (13.33 nM, 33.33 nM, 66.66 nM, 333.3 nM, 666.6 nM, 3500 nM, and 6666 nM). The fluorescence intensity of each reaction mixture was measured using real-time PCR, and a saturation curve was plotted for calculating Kd. Reactions were carried out in sextuplicate to calculate the standard deviation of measurements.In NFIS-based binding assays, different concentrations of Lucentis were immobilized on interdigitated (IDT) gold electrodes patterned on a glass substrate (13 nM, 33 nM, 66 nM, 333 nM, 666 nM, and 3500 nM). The immobilization of Lucentis was confirmed using the Lowry protein estimation test. Then, the modified electrode with Lucentis was connected to an electrochemical quartz crystal microbalance and dipped in a solution containing 100 nM aptamer. The NFIS measurement parameters were kept the same as discussed previously.
Estimation of the secondary structures of selected aptamer sequences
After the selection of the two aptamers with the best binding affinities, their secondary structures were investigated to analyze the structural similarities between them using the mfold web server.Selectivity testing of aptamers using NFIS
In this test, various proteins were used for checking the selectivity of the selected aptamers 1 and 25 towards Lucentis. IDT gold electrodes, fabricated in the same way as for studying the binding of aptamers and Lucentis, were used for this test. Instead of Lucentis, 1 μM aptamer was immobilized in phosphate buffer at pH 7, keeping all other steps of immobilization the same. Then, the electrode was dipped in solutions of different proteins, like GCSF, GOx, urease, BSA, media broth containing cellular proteins, and Lucentis in media broth (with cellular proteins, GCSF, GOx, urease and BSA). Then, incubation at 25 °C for half an hour was carried out and impedimetric measurements were recorded, as done in the case of the binding assays, using an electrochemical quartz crystal microbalance.Application of the selected aptamers for the detection of Lucentis
Detection experiments were performed with aptamers 1 and 25 only, due to their satisfactory performance and dissociation constants found via docking simulations, thermofluorimetric and NFIS analysis. To employ the selected aptamers for the detection of Lucentis, the aptamers were immobilized on IDT gold electrodes as discussed before. The immobilization of aptamers on the gold electrodes was confirmed via FTIR.Then, the electrode modified with aptamer was connected to an electrochemical quartz crystal microbalance, as shown in Fig. 10, using all previously discussed parameters, and dipped in a solution containing a fixed concentration of Lucentis (1 nM, 10 nM, 50 nM, 100 nM, and 500 nM). Then, after analysing spiked Lucentis samples, real samples from bioreactors (media broth) were collected and analysed using NFIS. The concentrations obtained with our method were cross-checked using the HPLC standard method.
Conclusions
In this work, we developed highly specific and selective aptamers for Lucentis using a modified and stringent SELEX process (only 10 cycles), and then employed them for the detection of Lucentis. The Kd values for the two best aptamers were found to be in the nanomolar range using thermofluorimetric and NFIS analysis, representing the excellent binding affinities (specificities) of the aptamers for Lucentis. Both aptamers were also found to show selective binding to Lucentis upon testing them in the presence of different simulated interfering proteins, like GCSF, GOx, urease, and BSA, and under real sample conditions using media broth. Though neither of the two aptamers showed prominent responses to interferents, better selectivity was shown by aptamer 1. From secondary structure analysis via the mfold web server, the presence of four-base–pair stem loop structure in the aptamers was found to be responsible for their better affinity for Lucentis. The two chosen aptamers, 1 and 25, were tested for the detection of Lucentis in spiked and real media broth samples; both aptamers succeeded, but aptamer 1 showed slightly better sensitivity, LOD and linear range for Lucentis over aptamer 25. From this proof-of-concept, using these aptamers for the detection of Lucentis on gold electrodes, it can be concluded that the selected aptamer 1 can easily find applications in affinity chromatography for the purification of Lucentis, clone selection, and the development of detection kits/chips and subsequent process analytical techniques.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported partially by the Department of Science & Technology SERB, grant no. EMR/2015/002354, and Department of Biotechnology, Ministry of Science and Technology, grant no. BT/COE/34/SP15097/2015.References
- Q. D. Nguyen, D. M. Brown, D. M. Marcus, D. S. Boyer, S. Patel, L. Feiner, A. Gibson, J. Sy, A. C. Rundle, J. J. Hopkins, R. G. Rubio and J. S. Ehrlich, Ophthalmology, 2012, 119, 789 CrossRef PubMed.
- J. E. Frampton, Drugs Aging, 2013, 30, 331 CrossRef CAS.
- D. M. Brown, P. K. Kaiser, M. Michels, G. Soubrane, J. S. Heier, R. Y. Kim, J. P. Sy and S. Schneider, N. Engl. J. Med., 2006, 355, 1432 CrossRef CAS PubMed.
- A. K. A. S. Neubauer, F. G. Holz, W. Schrader, E. I. Back, T. Kühn and C. Hirneiss, Klin. Monatsbl. Augenheilkd., 2007, 224, 727 CrossRef CAS PubMed.
- R. Sigl, US Pat., 2016/0297877A1, 2016, vol. 1.
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628 CrossRef CAS.
- J. O. Lee, H. M. So, E. K. Jeon, H. Chang, K. Won and Y. H. Kim, Anal. Bioanal. Chem., 2008, 390, 1023 CrossRef CAS PubMed.
- J. Pultar, U. Sauer, P. Domnanich and C. Preininger, Biosens. Bioelectron., 2009, 24, 1456 CrossRef CAS PubMed.
- J. A. Degrasse, PLoS One, 2012, 7, e33410 CrossRef CAS PubMed.
- C. Wang, G. Yang, Z. Luo and H. Ding, Acta Biochim. Biophys. Sin., 2009, 41, 335–340 CrossRef CAS PubMed.
- D. L. Wang, Y. L. Song, Z. Zhu, X. L. Li, Y. Zou, H. T. Yang, J. J. Wang, P. Sen Yao, R. J. Pan, C. J. Yang and D. Z. Kang, Biochem. Biophys. Res. Commun., 2014, 453, 681 CrossRef CAS PubMed.
- S. C. B. Gopinath, Anal. Bioanal. Chem., 2007, 387, 171 CrossRef CAS.
- M. Moreno, E. Rincon, D. Pineiro, G. Fernandez, A. Domingo, A. Jimenez-Ruiz, M. Salinas and V. M. Gonzalez, Biochem. Biophys. Res. Commun., 2003, 308, 214–218 CrossRef CAS PubMed.
- T. S. Misono and P. K. R. Kumar, Anal. Biochem., 2005, 342, 312–317 CrossRef CAS PubMed.
- E. Vianini, M. Palumbo and B. Gatto, Bioorg. Med. Chem, 2001, 9, 2543–2548 CrossRef CAS.
- M. B. Murphy, S. T. Fuller, P. M. Richardson and S. A. Doyle, Nucleic Acids Res., 2003, 31, e110 CrossRef PubMed.
- D. Levesque, J.-D. Beaudoin, S. Roy and J.-P. Perreault, Biochem. J., 2007, 403, 129–138 CrossRef CAS PubMed.
- K. Song, M. Cho, H. Jo, K. Min, S. Ho, T. Kim, M. Su, J. Kang and C. Ban, Anal. Biochem., 2011, 415, 175 CrossRef CAS PubMed.
- Z. Zhuo, Y. Yu, M. Wang, J. Li, Z. Zhang, J. Liu, X. Wu, A. Lu, G. Zhang and B. Zhang, Int. J. Mol. Sci., 2017, 18, 2142 CrossRef PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381 CrossRef CAS PubMed.
- M. Citartan, T. H. Tang, S. C. Tan, C. H. Hoe, R. Saini, J. Tominaga and S. C. B. Gopinath, Songklanakarin J. Sci. Technol., 2012, 34, 125 CAS.
- K. E. Hevener, W. Zhao, D. M. Ball, K. Babaoglu, J. Qi, S. W. White and R. E. Lee, J. Chem. Inf. Model., 2009, 49, 444 CrossRef CAS PubMed.
- J. Hu, J. Kim and C. J. Easley, Anal. Methods, 2015, 7, 7358 RSC.
- G. Wang, J. Liu, K. Chen, Y. Xu, B. Liu, J. Liao, L. Zhu, X. Hu, J. Li, Y. Pu, W. Zhong, T. Fu, H. Liu and W. Tan, Sci. Rep., 2017, 7, 1 CrossRef PubMed.
- F. Malvano, D. Albanese, A. Crescitelli, R. Pilloton and E. Esposito, Biosensors, 2016, 6, 33 CrossRef PubMed.
- P. Ihalainen, F. Pettersson, M. Pesonen, T. Viitala, A. Määttänen, R. Österbacka and J. Peltonen, Nanotechnology, 2014, 25, 094009 CrossRef PubMed.
- A. Martínez-Ortega, A. Herrera, A. Salmerón-García, J. Cabeza, L. Cuadros-Rodríguez and N. Navas, J. Pharm. Anal., 2016, 6, 117–124 CrossRef.
- G. C. P. Van Zundert, J. P. G. L. M. Rodrigues, M. Trellet, C. Schmitz, P. L. Kastritis, E. Karaca, A. S. J. Melquiond, M. Van Dijk, S. J. De Vries and A. M. J. J. Bonvin, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03542d |
| This journal is © The Royal Society of Chemistry 2020 |