
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bioactive polyaniline-b-polyacrylic acid copolymer nanofibrils as an effective antibacterial and anticancer agent in cancer therapy, especially for HT29 treatment
Nazanin Bagheria,
Moslem Mansour Lakouraj *a,
Seyed Reza Nabavib,
Hamed Tashakkorianc and
Mojtaba Mohsenid
*a,
Seyed Reza Nabavib,
Hamed Tashakkorianc and
Mojtaba Mohsenid
aPolymer Chemistry Laboratory, Department of Organic-Polymer Chemistry, Faculty of Chemistry, University of Mazandaran, Babolsar 47416, Iran. E-mail: lakouraj@umz.ac.ir
bDepartments of Applied Chemistry, University of Mazandaran, Babolsar 47416, Iran
cCellular and Molecular Biology Research Center (CMBRC), Health Research Institute, Babol University of Medical Sciences, Babol, Iran
dDepartments of Microbiology, Faculty of Basic Science, University of Mazandaran, Babolsar 47416, Iran
First published on 2nd July 2020
Abstract
In this work, a new highly water-soluble copolymer of polyacrylic acid with polyaniline is introduced. Acrylic acid was polymerized via the Reversible Addition Fragmentation Chain Transfer method (RAFT) in the presence of an initiator and the obtained polyacrylic acid was copolymerized with aniline at room temperature. As the main achievements of this work, the resulting block copolymer with nanosized structure revealed favorable solubility in polar solvents, as well as excellent antibacterial and anticancer activities. Therefore, it is an appropriate candidate for medical applications such as wound healing and cancer therapy, especially in HT29 treatment.
1. Introduction
Nowadays, there are many challenges for scientists to find efficient methods and materials in order to fight and minimize potential health risks in the medical area. Many attempts have been conducted toward preparing anticancer and anti-microbial agents in the medical community. In this regard, polymeric nanomaterials are the most widely used materials. These types of nanoparticles provide a promising landscape for researchers to improve the quality of treatment using modern nanomedical methods. Based on the research, polymeric treatments include polymer drugs,1 modified polymers with protein and pharmaceuticals,2 and polymeric micelles.3 Due to the variability in the methods of modification and macromolecular synthesis, polymeric nanomaterials have found a wide range of applications in treatment programs.For improving the therapeutic performance of anticancer drugs and the effectiveness of medication on cancer cells, the anti-cancer agents should be controllably delivered to the tumors.4 Polymeric materials compared with conventional chemotherapy play an effective role in cancer therapies.5 In fact, polymer nanomaterials not only reduce the toxicity of chemotherapy drugs to natural tissues around tumors, but they can also improve the solubility of anticancer drugs.6 For example, Takahashi et al. introduced new synthetic methacrylate random copolymers as anticancer agents by simulating the action of anticancer drugs. In this research, the anticancer polymers were designed to disrupt the cancer cell membranes. The polymers exhibited cytotoxicity to proliferating prostate cancer, along with killing (eliminating) dormant prostate cancer cells that were resistant to docetaxel.7
Undoubtedly, there is a close relationship between infection and cancer. As a matter of fact, patients suffering from cancer are susceptible to bacterial infections. Therefore, it is important to investigate the medical care applicable to cancer patients in terms of antibiotic effects. Several materials have been introduced for antibacterial activity and are commonly applied as antimicrobial agents that are non-volatile such as silver or copper nanoparticles8 and quaternary ammonium compounds (QACs).9 However, silver and copper, particularly in the form of nanoparticles, have high cytotoxicity and low biocompatibility.10 In the case of QACs, some bacteria resist these materials, as well as cyto-compatibility with these quaternary ammonium compounds.11 Hence, researchers have focused on the modified biologically active polymers with more stability to achieve higher performances in antibacterial properties. In fact, the utilization of antibacterial non-vaporizable polymers leads to the improved efficacy of the antimicrobial activity along with diminished environmental risk.12,13
Among antimicrobial polymers, polycationic materials provide effective antimicrobial activity due to the disruption of negative charges on bacterial cell membranes, which lead to cell death.14 In these antimicrobial polymers, cationic groups such as ammonium, phosphonium and sulfonium ions interact with the bacterial cell membranes; simultaneously, the polymeric residue may penetrate the microbial cell membranes, leading to the distribution of cytoplasmic components and finally resulting in the death of the bacterial cells.15–18 Consequently, cationic polymers have stimulated significant interest for application for medical purposes,19 in the food industry,20 and in water sanitation21 to prevent the growth of bacteria.
Among the various types of antibacterial cationic polymers, polyaniline is the most favorable bioactive polymer owing to its good chemical stability and convenient synthesis.22 For example, researchers have recently investigated the synthesis and the antibacterial activity of the PANI–WO3 nanocomposites.23 The nanocomposites of PANI are more effective against all the tested pathogenic strains. In addition, it was proved that because of the greater surface area and higher interactions of the polymer with the medium, nanostructured polyaniline exhibits excellent dispersion in water, with low toxicity to living systems.24 However, there are many disadvantages regarding the use of these polymeric materials such as deficient solubility in most solvents due to the rigidity of the conjugated benzenoid and quinoid rings, π–π stacking and hydrogen bonding of polyaniline chains,25 which lead to low processability of this compound. In this trend, researchers have improved the solubility of polyaniline using different substituents on the polymer chains to prepare copolymers, or blending polyaniline with other water-soluble polymers.26 For instance, Shao et al. synthesized water-soluble polyaniline films using poly(2-acrylamido-2-methylpropane sulfonic acid) (PAMPS) as a water-soluble dopant. Because of the hydrophilic nature of the PAMPS, the final blend film was completely soluble in water.27 In another work, the synthesis of aqueous dispersions of PANI–cellulose derivatives by polymerization of aniline monomer in cellulose solution was reported. This cellulose derivative revealed better conductivity and solubility, due to mutual hydrogen bonding between the amine groups of polyaniline chains and hydroxyl groups of cellulose. This phenomenon has promoted the dispersion of PANI in the solution and prevents its aggregation in the reaction medium.28
Considering the aforementioned points, and in order to introduce a new water-soluble polymeric bioactive nanomaterial, in this work, the nano-sized hydrophilic PANI copolymer was synthesized by copolymerization of aniline with polyacrylic acid. For this purpose, the aniline-functionalized RAFT agent (CTA) was prepared and used to initiate the controlled radical polymerization of acrylic acid in the presence of AIBN to produce CTA-capped polyacrylic acid (CTA-PAA). The sequential copolymerization of CTA-PAA with aniline was followed through oxidative radical polymerization to afford the corresponding block copolymer (PANI-b-PAA) in the nanometer range. The prepared nanomaterials were characterized by spectral data and physical properties. Thermal analysis was conducted and the morphology, conductivity and biological properties of the obtained copolymer were examined.
The main achievement of this work is that the resulting polymeric nanomaterial exhibited extra solubility in polar solvents such as ethanol, H2O and DMSO, and surprisingly indicated excellent inherent antibacterial and anticancer activity, making this synthetic copolymer a desirable candidate to substitute for traditional chemotherapeutic drugs and also as a strong antimicrobial agent in wound-dressing applications.
2. Materials
Aniline (Merck Co.) was purified by double distillation under vacuum. All solvents, ammonium persulfate (APS), carbon disulfide (CS2), benzyl chloride, Na3PO4·12H2O, azobisisobutyronitrile (AIBN), and hydrochloric acid (HCl) were purchased from Merck and Sigma Companies and used without further purification. Acrylic acid was supplied by Fluka Company. The cell lines were purchased from the Pasteur Institute of Iran (Department of Cell Bank).3. Experimental
3.1. Synthesis of the chain transfer agent (CTA)
To a solution of aniline (0.016 mol) and hydrated sodium phosphate (0.014 mol) (Na3PO4·12H2O) in 20 mL acetone, CS2 (0.06 mol) was added drop-wise and the mixture was allowed to stir at room temperature for 2 hours. Benzyl chloride (0.02 mol) was then added and the reaction mixture was kept stirring for 3 hours to give a light yellow precipitate. The resultant was filtered and precipitated in cyclohexane. Finally, the product was dried overnight at room temperature in a vacuum desiccator. The chemical structure was confirmed by IR and NMR spectra that showed high purity.Melting point: 68–70 °C.
FTIR (KBr, ν cm−1): 1026, 1327, 1503, 1591, 2926 and 3118 cm−1.
1H-NMR (400 MHz, CDCl3): 4.58, 7.28, 7.30, 7.34, 7.38, 7.41, 7.43, 7.45 and 8.91 ppm.
3.2. Synthesis of CTA-capped polyacrylic acid (CTA-PAA)
The (CTA-PAA) macro-initiator was synthesized based on the modified previously reported research.29 In a typical procedure, acrylic monomer (5 mL) and CTA (0.12 g) were mixed in 25 mL of THF. The mixture solution was degassed and stirred for 30 minutes at room temperature. Then, 0.014 g initiator (AIBN) was added and the mixture was heated to 70 °C and kept in these conditions overnight. After completion of the reaction, the resulting solution was poured into diethyl ether that led to the formation of a sticky material. The solvent was decanted and the product was dried in a vacuum oven at 60 °C for 12 hours. The obtained white powder was characterized by IR and NMR spectroscopies.FTIR (KBr, cm−1): 1248, 1454, 1709, 2932 and 3436 cm−1.
1H-NMR (400 MHz, DMSO-d6): 1.76, 2.50, 3.60, 5.88, 6.07, 6.22, 12.25 ppm.
3.3. Synthesis of polyacrylic acid-b-polyaniline copolymer (PANI-b-PAA)
The synthesis of the PANI-b-PAA copolymer was carried out according to the following procedure: to a mixture of CTA-PAA (1 g) and 30 mL HCl (1 M), ammonium persulfate (0.001 mol) was added and stirred for 30 minutes at room temperature. Then, aniline (0.01 mol) was added and the mixture was stirred for over 4 hours till the color changed to dark green. After that, the resultant was washed with large quantities of ethanol and distilled water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) for 2 hours and then it was centrifuged to separate the insoluble homopolymer of aniline. The final supernatant was concentrated to give the PANI-b-PAA copolymer as a green precipitate. The chemical structure of the copolymer was identified by FTIR and NMR spectroscopy.
:50 v/v) for 2 hours and then it was centrifuged to separate the insoluble homopolymer of aniline. The final supernatant was concentrated to give the PANI-b-PAA copolymer as a green precipitate. The chemical structure of the copolymer was identified by FTIR and NMR spectroscopy.
FTIR (KBr, cm−1): 1135, 1483, 1556, 1648, 1710, 2920 and 3436 cm−1.
1H-NMR (400 MHz, DMSO-d6): 2.50, 4.02, 7.14, 7.16, 7.19, 7.34, 7.45, 10.06 ppm.
4. Characterization
The Fourier transform infrared (FTIR) spectra of all samples were recorded at 400 to 4000 cm−1 from KBr pellets on a Bruker infrared Fourier transform spectrometer. The 1H-NMR spectra of all compounds were accomplished using a Bruker 400 MHz Ultra-shield Advance DRX spectrometer in dimethyl sulfoxide (DMSO-d6) and CDCl3 as solvents. The X-ray powder diffraction patterns were recorded on a PHILIPS (PW1730) diffractometer fitted with CuK α radiation (λ = 1.5404 nm) at 40 kV and 30 mA in the 2θ range of 5–60°. Thermal analyses (DSC and TGA) were performed using a TA (Q600) instrument operating at a heating rate of 20 °C min−1, from room temperature up to 350 °C for TGA and 600 °C for DSC under a N2 atmosphere. Melting points of low molecular weight organic compounds were measured using the Electrothermal apparatus. Scanning electron microscopy (SEM) images of samples were taken by KYKY-EM3200. Moreover, the standard four-point probe (Azar Electrode Company, Tabriz, Iran) method was applied to measure the conductivity at room temperature. The electrospinning of the final copolymer was investigated and eventually, biological tests such as antibacterial and cytotoxicity assays (MTT test) were examined for the copolymer.5. Results and discussion
Over the last few decades, living radical polymerization has been utilized as one of the most striking and applicable methods for the synthesis of diverse novel polymers. Among the various techniques for radical polymerization,30,31 the Reversible Addition Fragmentation Chain Transfer (RAFT) is the most attractive one in living or controllable radical polymerizations.32–34 This kind of controlled radical polymerization is associated with thiocarbonylthio compounds as chain transfer agents and, in particular, has been applied for the preparation of various structures such as block, graft, hyperbranched and star copolymer with ideal dispersity and narrow molar mass distribution.35,36 Therefore, the RAFT polymerization method was applied for the synthesis of polyacrylic acid-b-polyaniline to produce an amphiphilic bioactive water-soluble polymeric system (Scheme 1).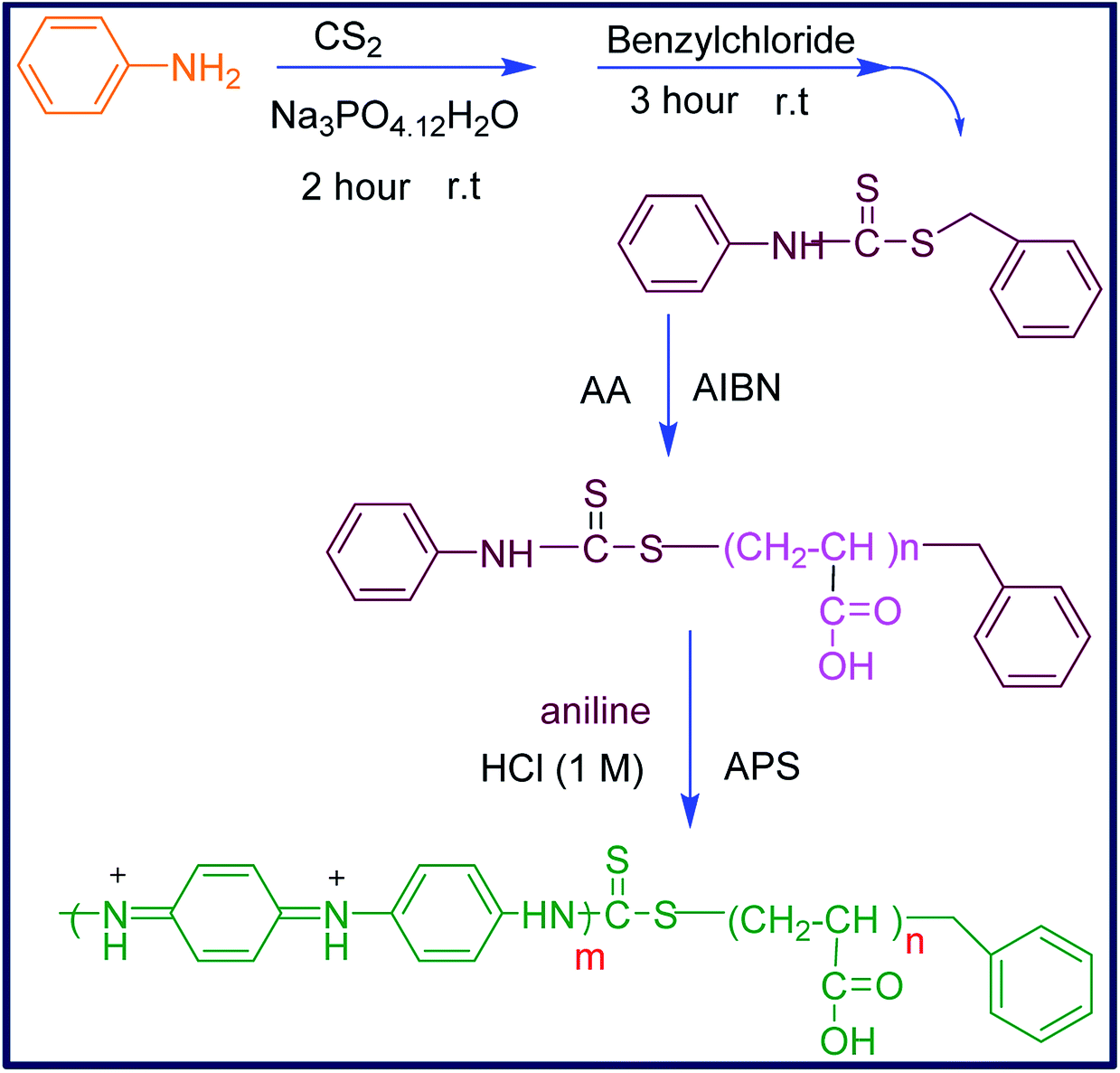 | ||
| Scheme 1 Synthetic pathway for the polyacrylic acid-b-polyaniline copolymer. | ||
5.1. Fourier transform infrared spectroscopy (FTIR)
FTIR spectra of CTA, CTA-PAA and the block copolymer (PANI-b-PAA) are shown in Fig. 1. The spectrum of CTA (Fig. 1a) showed two sharp bands about 1026 and 1326 cm−1, which are attributed to the stretching vibrations of C–S and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds, respectively. Also, peaks at around 1503 and 3118 cm−1 indicated vibrations of CN and N–H.
S bonds, respectively. Also, peaks at around 1503 and 3118 cm−1 indicated vibrations of CN and N–H.
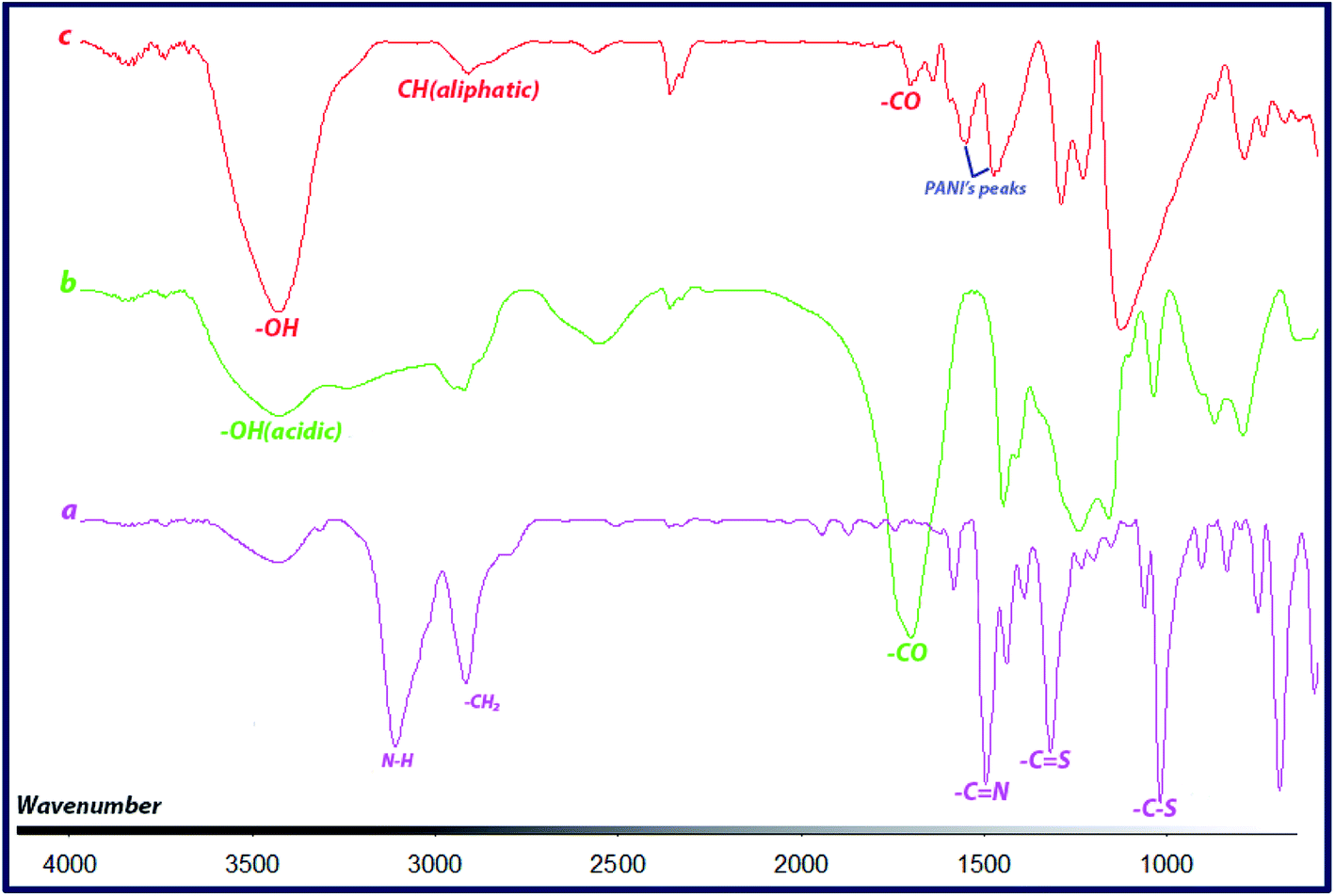 | ||
| Fig. 1 FTIR spectra of CTA (a), CTA-PAA (b) and PANI-b-PAA (c). | ||
As can be seen in Fig. 1b, CTA-PAA displayed peaks at around 1248, 1710 cm−1 and a broad band from 2800 to 3500 cm−1, which clearly demonstrate the characteristic stretching vibrations of C–O, CO (carboxylic acid) and acidic O–H, respectively. In the case of the PANI-b-PAA copolymer, the spectrum (Fig. 1c) exhibited corresponding stretching vibrations of CO, aliphatic C–H and O–H bonds at around 1710, 2920 and 3436 cm−1, respectively. Also, the formation of the PANI segment in the copolymer was confirmed by (or through) the clear peaks at 1556 cm−1, corresponding to the CC stretching of the quinoid ring, and 1483 cm−1 for the CC stretching vibration of the benzenoid ring, as well as 1297 cm−1 related to C–N stretching.
5.2. NMR spectroscopy
1H-NMR spectrum of the synthesized CTA represents the characteristic peaks at corresponding chemical shifts, i.e., 4.58 ppm related to aliphatic CH2, 7.28 to 7.45 ppm for aromatic hydrogens and 8.91 ppm attributed to NH. Furthermore, for the CTA-PAA sample, the chemical shifts in the range of 1.50 to 2.50 ppm are related to the aliphatic CH and CH2 units of the polyacrylic acid chain. Additionally, the peaks observed from 5.90 to 6.29 ppm pertain to the aniline ring, and the characteristic peak at 12.25 ppm is ascribed to the acidic protons of carboxylic acid groups. Finally, for the synthesized PANI-b-PAA copolymer, the chemical shift of 4.01 ppm is related to aliphatic CH, and the triplet peaks located at 7.16, 7.29 and 7.42 ppm are associated with polyaniline. Moreover, the chemical shift at 10.06 ppm refers to acidic protons in the acrylic acid units. These data clearly indicate the successful RAFT copolymerization of aniline and acrylic acid (Fig. 2). Also, the molecular weight of the polymers was calculated based on 1HNMR findings as reported in previous research37,38 using the integration of peaks from Fig. 2B and C, which are equivalent to 13180 and 9950 kDa for polyacrylic acid and polyaniline, respectively.
 | ||
| Fig. 2 1H-NMR spectra of CTA (A), CTA-PAA (B) and PANI-b-PAA (C). | ||
5.3. Conductivity measurements
The electrical conductivity of the copolymer (PANI-b-PAA) was studied by a four-probe technique. In this way, the powdered copolymer (0.3 g) was prepared in a tablet form (diameter: 13 mm, thickness: 1 mm) at a pressure of 14 MPa. The conductivity was measured by the following equation:where σ is the electrical conductivity, V is the voltage measured across inner probes, (I) is the applied current in the outer probes, and (d) is the tablet thickness (cm). The electrical conductivity of the copolymer was 18 S cm−1, whereas the electrical conductivity of the polyaniline homopolymer reported in the literature was lower than in the present work.39,40 It seems that the increase in the electrical conductivity of the copolymer is due to the presence of the polyacrylic acid block in the PANI-b-PAA copolymer, which acts as an internal dopant. It was suggested that carboxylate groups might cooperate in the charge transfer through the internal doping of carboxylic acid. Also, it was assumed that owing to the existence of the polyacrylic acid chain, the solubility of the copolymer was improved, which led to an increment in the amount of aniline units in the polyaniline segments during polymerization.
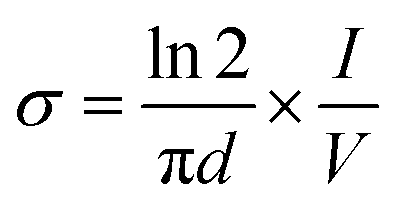
5.4. Scanning electron microscopy (SEM)
The nanostructure of the synthesized PANI-b-PAA copolymer and the size of the nanoparticles were evaluated by SEM imaging (Fig. 3). SEM images demonstrated that the PANI-b-PAA copolymer has a nano-rod shape with a diameter range of 52 to 100 nm. | ||
| Fig. 3 SEM images of PANI-b-PAA. | ||
5.5. Electrospinning of the copolymer
Electrospinning is a method in which materials in solution or melt states are transformed into nano- or micro-sized continuous fibers. To study the presumption of electrospinning of the copolymer, the resulting PANI-b-PAA solution was loaded into two 10 mL glass syringes equipped with a needle. The syringe was then put in a syringe pump and the needle was connected to the positive output of a high voltage power supply. The collector was covered with aluminum foil and placed at a fixed distance of 20 cm from the needle. The flow rate of the solution and applied voltage were fixed at 1 mL per hour and 18–20 kV at room temperature, respectively. Finally, the prepared PANI-b-PAA nanofibers on the foil were dried overnight at room temperature. The morphology of the nano-fibrous scaffolds was examined using scanning electron microscopy. To estimate the diameter, the images were captured, which indicated that the diameters of the fibers were around 80 nm.SEM images show the morphology and distribution of the prepared nanofibers. As is evident, the non-woven fibers were distributed by chance throughout the electrospun structures and formed interconnected pores. As mentioned elsewhere, it is expected that with the increase in the pore numbers and the volume, the cell adhesion and proliferation will be enhanced.41 SEM images proved that the electrospinning process of PANI-b-PAA was successful for the presentation of electrically conducting domains at the surface, which may be effective for transferring electrical signals (Fig. 4).
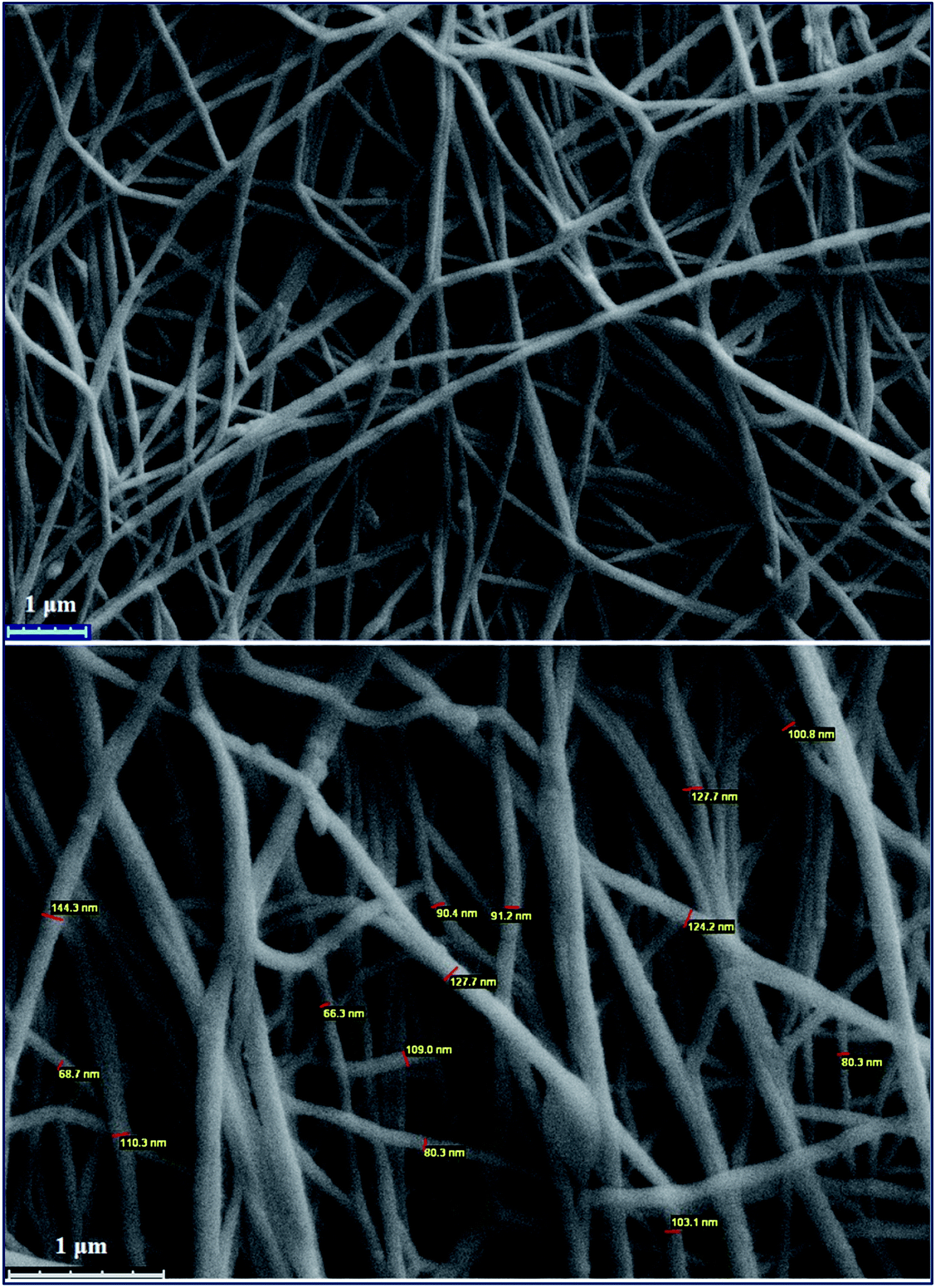 | ||
| Fig. 4 SEM images and the distribution of the prepared nanofibers of electrospun PANI-b-PAA. | ||
5.6. XRD analysis
The X-ray diffraction pattern of CTA-PAA and PANI-b-PAA copolymers are given in Fig. 5. The XRD pattern of PANI-b-PAA (the violet curve) contains some sharp peaks representing the crystal planes. The peak centered at 2θ of about 21.20° may be attributed to the conjugated chain of the emeraldine base form of PANI.42 | ||
| Fig. 5 The XRD patterns of CTA-PAA (red curve) and PANI-b-PAA (violet curve). | ||
As shown in Fig. 5, the red curve, the CTA-RAFT showed a moderate degree of crystallinity (broad peaks in the spectrum).
5.7. Thermal analysis
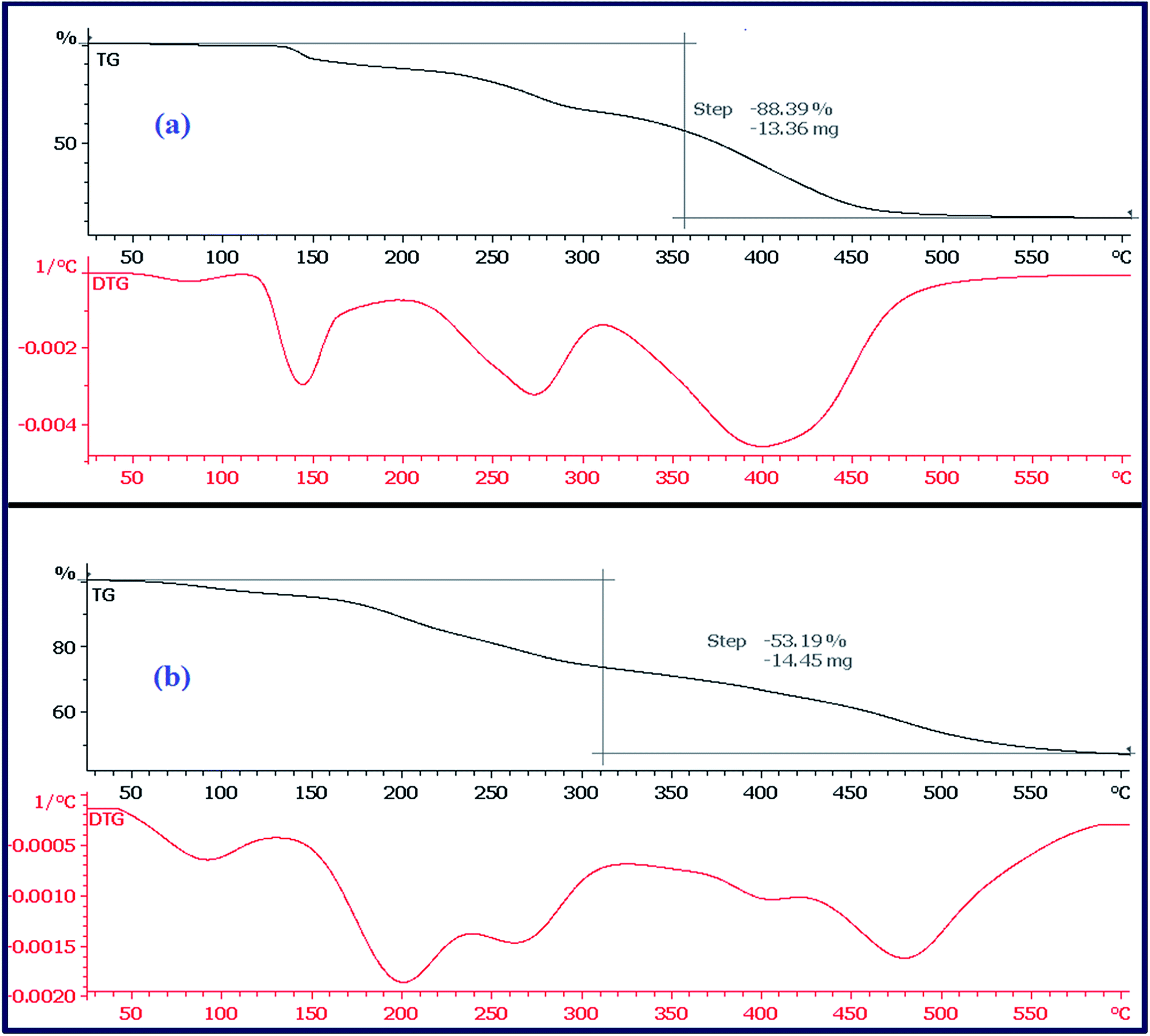 | ||
| Fig. 6 TGA-DTG curves of CTA-PAA (a) and PANI-b-PAA (b). | ||
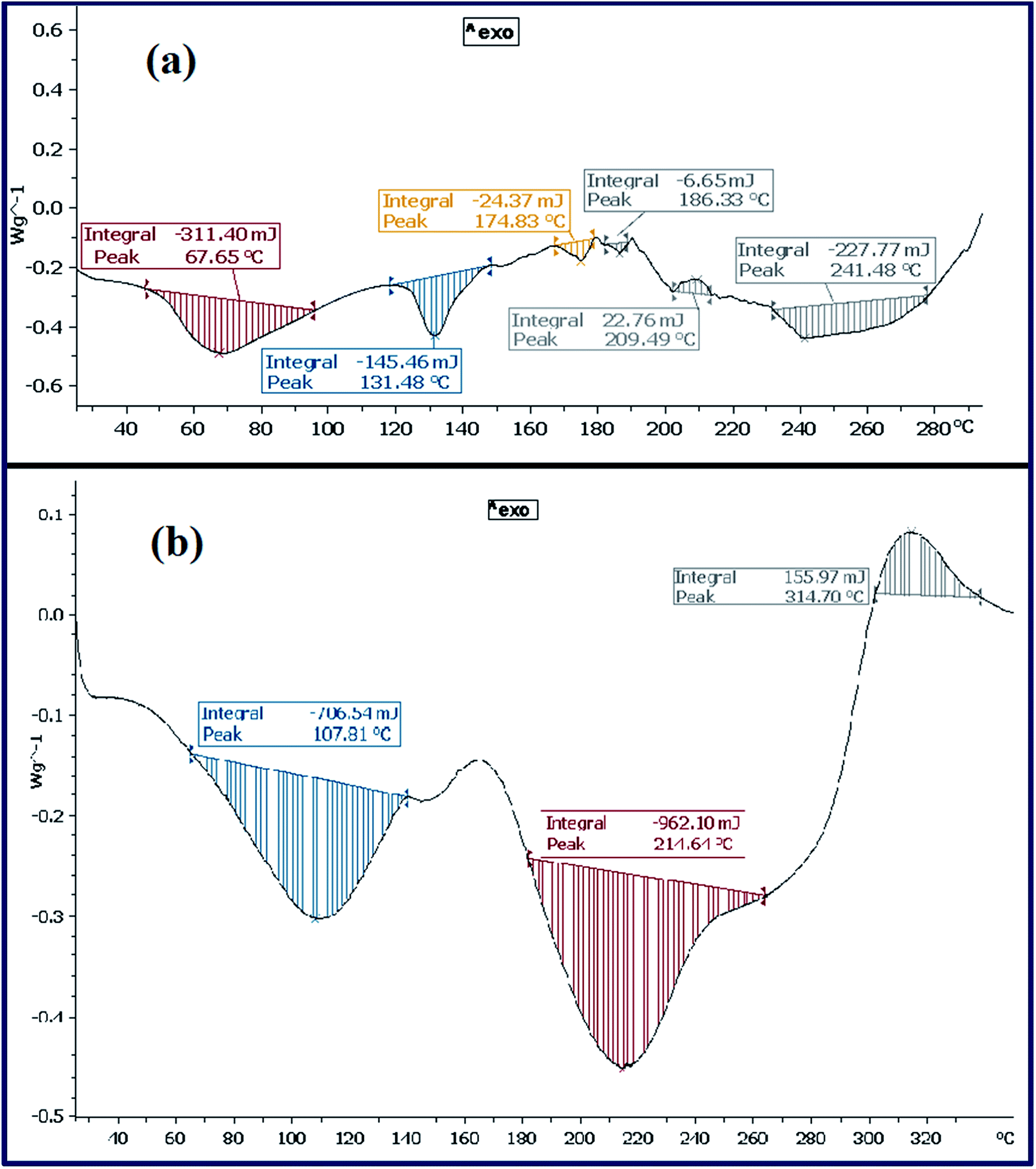 | ||
| Fig. 7 DSC curves of CTA-PAA (a) and PANI-b-PAA (b). | ||
5.8. Determination of antibacterial activity
The antibacterial activity of the synthesized materials was assessed by a Kirby–Bauer disk diffusion method where a disk was placed on the surface of agar dishes.44 The PANI-b-PAA (0.13 g) was prepared in tablet form (diameter: 13 mm, thickness: 1 mm) at a pressure of 14 MPa. Next, 8 prepared tablets were placed on agar dishes infected with both Gram-negative and Gram-positive species of bacteria. The antibacterial activity of the synthesized copolymer was studied against four bacterial species (Escherichia coli PTCC 1330, Pseudomonas aeruginosa PTCC 1074, Staphylococcus aureus ATCC 35923 and Bacillus subtilis PTCC 1023). All the microorganisms were protected (37 °C, 24 h) by inoculation in a Mueller–Hinton broth.The cultures were standardized with a final cell density of around 108 cfu mL−1. The Mueller–Hinton agar (Merck) samples were prepared and inoculated from the standardized cultures of the test organisms, which were spread as regularly as possible throughout the entire media. The tablets were presented on the upper layer of the seeded agar plates and were incubated at 37 °C for 24 h. The antibacterial activity of the copolymer was compared to the famous antibiotics gentamicin (10 mg per disk) and chloramphenicol (30 mg per disk) as positive controls. The antibacterial activity was estimated by measuring the diameter of the inhibition zone (mm) on the surfaces of the plates and the results are reported as the mean ± SD values after two repeated measurements.
The results are summarized in Table 1. As can be seen, the antibacterial activity evaluation of all the copolymeric tablets was performed on four different bacterial strains (Gram-positive: Staphylococcus aureus (S. aureus) and Bacillus subtilis (B. subtilis) and Gram-negative: Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa)) using an agar disc diffusion technique [Fig. 8].
| Test strain | Zone of growth inhibitiona (mm) | ||
|---|---|---|---|
| PANI-b-PAA (filmb) | Gentamicin (10 μg per disc) | Chloramphenicol (30 μg per disc) | |
| a Strong activity > 16 mm, moderate activity 10–16 mm, weak activity < 10 mm.b Diameter of film: 10 mm, Mueller–Hinton agar plate.c No effect. | |||
| E. coli | 28.0 ± 1.4 | 19.6 ± 1.1 | 20.7 ± 1.5 |
| P. aeruginosa | 28.5 ± 0.7 | 15.6 ± 0.5 | NEc |
| S. aureus | 31.5 ± 0.7 | 20.3 ± 1.5 | 21.7 ± 0.6 |
| B. subtilis | 27.5 ± 0.7 | 26.0 ± 1.7 | 22.3 ± 1.2 |
 | ||
| Fig. 8 Photograph of the antibacterial activity assays of the PANI-b-PAA. | ||
The results showed that the antibacterial activities of the PANI-b-PAA block are obviously stronger than standard antibiotics. Furthermore, by comparing these results with previous reports, it was found that the present synthesized copolymer disclosed reasonable antibacterial activity [Table 2].
As shown in Fig. 9, it was suggested that the antimicrobial mechanism of the copolymer may be due to the presence of nano-sized amphiphilic polyaniline fibrils in the copolymer structure. It was assumed that polyaniline segments with positive charges disrupted the bacterial cell membranes, causing leakage of the cytoplasmic contents or complete lysis of the cells and consequently the death of the bacteria as was reported previously in the literature.48
 | ||
| Fig. 9 Leakage of the cytoplasmic contents of the cells by the copolymer. | ||
5.9. Cytotoxicity assay (MTT test)
5.10. Anti-cancer activity
Colorectal cancer is the third most common cancer in the world that causes cancer death every year.50 As reported in previous works, the common drugs available for colorectal cancer (CRC) treatment include 5-fluorouracil (5-FU)/Leucovorin, the first-line treatment, Avastin, Oxaliplatin and Irinotecan.51 Fluoropyrimidines, Oxaliplatin and Irinotecan have been made public as chemotherapy drugs for CRC. Unfortunately, one of the most serious problems of chemotherapy treatment for colorectal cancer involves using high doses of medicines, leading to the cytotoxicity of healthy cells, especially in the case of the mixture of 5-FU and Irinotecan.52 Also, it is noteworthy that these traditional medicines always cause severe hematological reactions because of their undefined distribution in the bone marrow, liver and other living tissues.53 Moreover, most drugs that have been applied in the treatment of colorectal cancer exhibit many side effects of chemotherapy such as hair loss, mouth sores, loss of appetite, vomiting, increased chance of infections and fatigue. Thus, scientists have attempted to develop novel nanomaterial systems with multifunctional characteristics that control the release of drugs.For this purpose, to determine the cytotoxic effect of the synthesized PANI-b-PAA sample, the treatments of two kinds of cancer cell lines, HT29 and PC3 cell lines and fibroblasts were investigated and the results are demonstrated in Fig. 10.
 | ||
| Fig. 10 Proliferation decrement of fibroblasts, colonic cancer cells (HT29), and prostate cancer cells (PC3). | ||
The outcomes attained from Fig. 11 revealed that cell proliferation was decreased in the fibroblast cell lines from concentrations of 250 μg mL−1 to 500 μg mL−1. Moreover, as shown in Fig. 11, by increasing the copolymeric sample concentration, PC3 cell lines did not show any phenomenal decrement but in the case of HT29, an impressive decrease in cell proliferation was observed after 72 h. This means that, fortunately, the synthesized PANI-b-PAA copolymer in a maximum concentration of 125 μg mL−1 shows effective activity for the reduction of human colon cancer cell growth. Therefore, the PANI-b-PAA copolymer is a satisfactory candidate for human colon cancer treatment as an effective, simple preparation and cheap treatment protocol.
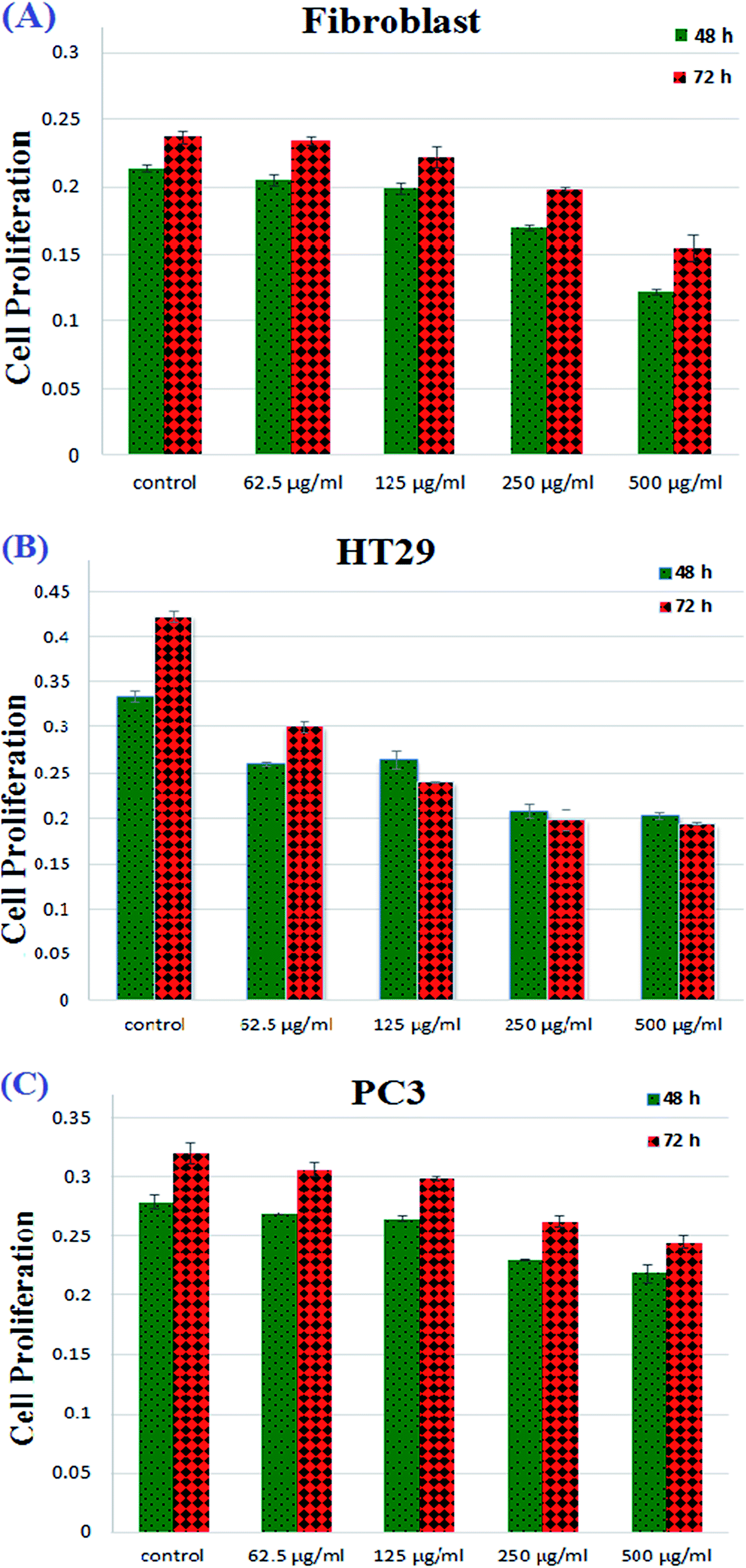 | ||
| Fig. 11 Fibroblast (A), HT29 (B) and PC3 (C) cell proliferation under treatment with different concentrations of PANI-b-PAA samples in concentrations of 62.5, 125, 250 and 500 μg mL−1 in 48 and 72 hours. | ||
6. Conclusion
In the present work, the novel water-miscible conducting diblock copolymer (PANI-b-PAA) was successfully synthesized by sequential RAFT and oxidative radical polymerizations. The obtained products were characterized by different analyses. The nanocrystalline structure of PANI-b-PAA was proved by XRD and SEM imaging. The final sample, PANI-b-PAA, showed reasonable solubility in polar solvents such as H2O, ethanol, a mixture of H2O and ethanol and DMSO. Also, further investigations revealed excellent antibacterial activity for the PANI-b-PAA copolymer on four bacteria, namely, E. coli, P. aeruginosa, B. subtilis, and S. aureus. Moreover, the anti-cancer activity of the synthesized copolymer was investigated. Regarding the MTT assay, the outcomes showed promising results in decreasing the cancer cell proliferation of HT29 at a concentration of 125 μg mL−1. The combination of the antibacterial properties with anticancer features makes the PANI-b-PAA copolymer a desirable polymeric nanomaterial for cancer treatment. In chemotherapy, since the infection may occur in and around cancerous organs, antibacterial activity makes the introduced material more operative for cancer treatment. All the results demonstrate that this charged amphiphilic block copolymer is a proper candidate for biological applications such as the anticancer agent in colorectal cancer. Furthermore, owing to excellent antibacterial properties, it possesses potential activity for wound dressing applications. Therefore, we are hoping that by employing this synthesized copolymer we will be able to reduce the side effects of traditional anti-cancer medicines without using any additional drugs.Data availability
There are no raw/processed data to reproduce these findings.Conflicts of interest
There are no confilict of interest in any form to declare.Acknowledgements
We appreciate the partial financial support of the research Council of the University of Mazandaran.References
- E. F. Connor, I. Lees and D. Maclean, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3146–3157 CrossRef CAS.
- Y. Wang and C. Wu, Biomacromolecules, 2018, 19, 1804–1825 CrossRef CAS PubMed.
- Q. Zhou, L. Zhang, T. Yang and H. Wu, Int. J. Nanomed., 2018, 13, 2921 CrossRef CAS PubMed.
- V. Taghipour-Sabzevar, T. Sharifi and M. M. Moghaddam, Ther. Delivery, 2019, 10, 527–550 CrossRef CAS PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 1–19 CrossRef CAS PubMed.
- M. Narvekar, H. Y. Xue, J. Y. Eoh and H. L. Wong, AAPS PharmSciTech, 2014, 15, 822–833 CrossRef CAS PubMed.
- H. Takahashi, K. Yumoto, K. Yasuhara, E. T. Nadres, Y. Kikuchi, R. S. Taichman and K. Kuroda, Sci. Rep., 2019, 9, 1–11 CrossRef PubMed.
- S. Fernando, T. Gunasekara and J. Holton, Sri Lankan Journal of Infectious Diseases, 2018, 8, 2–11 CrossRef.
- S. Buffet-Bataillon, P. Tattevin, M. Bonnaure-Mallet and A. Jolivet-Gougeon, Int. J. Antimicrob. Agents, 2012, 39, 381–389 CrossRef CAS PubMed.
- R. Vazquez-Muñoz, B. Borrego, K. Juárez-Moreno, M. García-García, J. D. M. Morales, N. Bogdanchikova and A. Huerta-Saquero, Toxicol. Lett., 2017, 276, 11–20 CrossRef PubMed.
- H. Nagamune, T. Maeda, K. Ohkura, K. Yamamoto, M. Nakajima and H. Kourai, Toxicol. In Vitro, 2000, 14, 139–147 CrossRef CAS PubMed.
- E. Hume, J. Baveja, B. Muir, T. Schubert, N. Kumar, S. Kjelleberg, H. Griesser, H. Thissen, R. Read and L. Poole-Warren, Biomaterials, 2004, 25, 5023–5030 CrossRef CAS PubMed.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef PubMed.
- E.-R. Kenawy, S. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- G. Lu, D. Wu and R. Fu, React. Funct. Polym., 2007, 67, 355–366 CrossRef CAS.
- H.-I. Chang, M.-S. Yang and M. Liang, React. Funct. Polym., 2010, 70, 944–950 CrossRef CAS.
- R. A. Anderson, K. Feathergill, X. Diao, M. Cooper, R. Kirkpatrick, P. Spear, D. P. Waller, C. Chany, G. F. Doncel and B. Herold, J. Androl., 2000, 21, 862–875 CAS.
- J. Guo, J. Qin, Y. Ren, B. Wang, H. Cui, Y. Ding, H. Mao and F. Yan, Polym. Chem., 2018, 9, 4611–4616 RSC.
- M. Râpă, P. Stoica, E. Tănase, E. Grosu and G. Vlad, J. Optoelectron. Adv. Mater., 2013, 15, 807–816 Search PubMed.
- T. Huang, Y. Qian, J. Wei and C. Zhou, Polymers, 2019, 11, 560 CrossRef CAS PubMed.
- E.-R. Kenawy, F. I. Abdel-Hay, A. A. El-Magd and Y. Mahmoud, React. Funct. Polym., 2006, 66, 419–429 CrossRef CAS.
- W. Jia, E. Reitz, H. Sun, H. Zhang and Y. Lei, Mater. Lett., 2009, 63, 519–522 CrossRef CAS.
- B. Manjunatha, A. N. Shetty, S. Kaveri, S. S. Mety, K. Anjaneya, R. Reddy and S. Kalyane, BioNanoScience, 2019, 1–8 Search PubMed.
- E. I. Yslas, L. E. Ibarra, M. A. Molina, C. Rivarola, C. A. Barbero, M. L. Bertuzzi and V. A. Rivarola, J. Nanopart. Res., 2015, 17, 389 CrossRef.
- J. Tarver, J. E. Yoo, T. J. Dennes, J. Schwartz and Y.-L. Loo, Chem. Mater., 2009, 21, 280–286 CrossRef CAS.
- H. Zeghioud, S. Lamouri, Z. Safidine and M. Belbachir, J. Serb. Chem. Soc., 2015, 80, 917–931 CrossRef CAS.
- L. Shao, J. Qiu, M. Liu, H. Feng, L. Lei, G. Zhang, Y. Zhao, C. Gao and L. Qin, Synth. Met., 2011, 161, 806–811 CrossRef CAS.
- H. Hussin, S. N. Gan, S. Mohamad and S. W. Phang, Polym. Polym. Compos., 2017, 25, 515–520 CAS.
- J. Loiseau, N. Doerr, J. Suau, J. Egraz, M. Llauro, C. Ladavière and J. Claverie, Macromolecules, 2003, 36, 3066–3077 CrossRef CAS.
- E. Rizzardo and D. H. Solomon, Aust. J. Chem., 2012, 65, 945–969 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- X. Tian, J. Ding, B. Zhang, F. Qiu, X. Zhuang and Y. Chen, Polymers, 2018, 10, 318 CrossRef PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Chem.–Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- M. C. Iovu, C. R. Craley, M. Jeffries-El, A. B. Krankowski, R. Zhang, T. Kowalewski and R. D. McCullough, Macromolecules, 2007, 40, 4733–4735 CrossRef CAS.
- C. Yang, J. K. Lee, A. J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416–5423 RSC.
- J. U. Izunobi and C. L. Higginbotham, J. Chem. Educ., 2011, 88, 1098–1104 CrossRef CAS.
- K. Paulsen and D. Frasco, Analysis of artists’ pigments by far-infrared microspectroscopy, 2016, pp. 28–31 Search PubMed.
- A. Olad and A. Rashidzadeh, Iranian Journal of Chemical Engineering, 2008, 5, 45–54 Search PubMed.
- B. Taşdelen, Mater. Today: Proc., 2018, 5, 15983–15989 Search PubMed.
- W. J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef CAS PubMed.
- B. Belaabed, J. L. Wojkiewicz, S. Lamouri, N. El Kamchi and T. Lasri, J. Alloys Compd., 2012, 527, 137–144 CrossRef CAS.
- V. d. A. M. Gonzaga, B. A. Chrisostomo, A. L. Poli and C. C. Schmitt, Mater. Res., 2018, 21, 1–7 Search PubMed.
- E. N. Zare, M. M. Lakouraj and M. Mohseni, Synth. Met., 2014, 187, 9–16 CrossRef.
- A. Shalini, R. Nishanthi, P. Palani and V. Jaisankar, Mater. Today: Proc., 2016, 3, 1633–1642 Search PubMed.
- X. Liang, M. Sun, L. Li, R. Qiao, K. Chen, Q. Xiao and F. Xu, Dalton Trans., 2012, 41, 2804–2811 RSC.
- M. Ghaffari-Moghaddam and H. Eslahi, Arabian J. Chem., 2014, 7, 846–855 CrossRef CAS.
- K. P. Jotiram, R. Prasad, V. S. Jakka, R. Aparna and A. Phani, Nano Biomed. Eng., 2012, 4, 144–149 CAS.
- M. Golpour, H. A. Niaki, H. R. Khorasani, A. Hajian, R. Mehrasa and A. Mostafazadeh, Int. J. Mol. Cell. Med., 2014, 3, 74 Search PubMed.
- F. A. Haggar and R. P. Boushey, Clin. Colon Rectal Surg., 2009, 22, 191–197 CrossRef PubMed.
- R. Matuo, F. G. Sousa, A. E. Escargueil, I. Grivicich, D. Garcia-Santos, J. A. B. Chies, J. Saffi, A. K. Larsen and J. A. P. Henriques, J. Appl. Toxicol., 2009, 29, 308–316 CrossRef CAS PubMed.
- L. Fang, Y. Jiang, Y. Yang, Y. Zheng, J. Zheng, H. Jiang, S. Zhang, L. Lin, J. Zheng and S. Zhang, Oncotarget, 2016, 7, 81880 CrossRef PubMed.
- R. Ortiz, L. Cabeza, J. L. Arias, C. Melguizo, P. J. Álvarez, C. Vélez, B. Clares, A. Áranega and J. Prados, AAPS J., 2015, 17, 918–929 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |