
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTough, stretchable and compressive alginate-based hydrogels achieved by non-covalent interactions†
Zhanxin Jing *,
Xiangyi Dai,
Xueying Xian,
Xiaomei Du,
Mingneng Liao,
Pengzhi Hong and
Yong Li*
*,
Xiangyi Dai,
Xueying Xian,
Xiaomei Du,
Mingneng Liao,
Pengzhi Hong and
Yong Li*
College of Chemistry and Environment, Guangdong Ocean University, Zhanjiang, Guangdong 524088, People's Republic of China. E-mail: jingzhan_xin@126.com; yongli6808@126.com
First published on 22nd June 2020
Abstract
In this study, two alginate-based hydrogels with good mechanical strength, toughness and resilience were synthesized by hydrophobic interaction and coordination bonding. Sodium alginate/poly(acrylamide) semi-interpenetrating network (NaAlg/PAM semi-IPN) hydrogels were first synthesized through the micelle copolymerization of acrylamide and stearyl methacrylate in the presence of sodium alginate, then calcium alginate/poly(acrylamide) double network (CaAlg/PAM DN) hydrogels were prepared by immersing the as-prepared NaAlg/PAM semi-IPN hydrogels in a CaCl2 solution. FT-IR and XPS results revealed NaAlg/PAM semi-IPN hydrogels and CaAlg/PAM DN hydrogels were successfully synthesized through non-covalent interactions. The tensile strength of CaAlg/PAM DN hydrogels could reach 733.6 kPa, and their compressive strengths at 80% strain are significantly higher than those of the corresponding NaAlg/PAM semi-IPN hydrogels, which is attributed to the alginate network crosslinked by Ca2+. The dual physically crosslinked CaAlg/PAM DN hydrogels can achieve fast self-recovery, and good fatigue resistance, which is mainly assigned to energy dissipation through dynamic reversible non-covalent interactions in both networks. The self-healing ability, swelling behavior and morphology of the synthesized alginate-based hydrogels were also evaluated. This study offers a new avenue to design and construct hydrogels with high mechanical strength, high toughness and fast self-recovery properties, which broadens the current research and application of hydrogels.
1. Introduction
Sodium alginate, which could be obtained mainly from marine brown algae belonging to the Phaeophyceae, is a naturally occurring polysaccharides.1,2 It is a polyanionic linear copolymer of 1,4-linked-α-L-guluronic acid and β-D-mannuronic acid residues in varying proportions, and is considered as a unique biocompatible, biodegradable and non-toxic polymer.3 Sodium alginate-based materials have been extensively studied and used in biomedical applications, such as drug delivery,4 tissue engineering5 and wound healing.6 Various material forms based on sodium alginate, such as films, fibers, microspheres and hydrogels, have been designed to meet the needs of the applications.7 Among these material forms, hydrogels are known for their similarity to the natural extracellular matrix and are being increasingly used in the field of tissue engineering.8,9Hydrogels possess the ability to absorb and retain large volumes of water or biological fluids, and don't dissolve in the solvent.10 Due to their soft and rubbery consistency, water uptake capacity, biocompatibility and similar properties to human tissue, hydrogels are extremely suitable as tissue engineering materials, such as contact lenses, engineering scaffolds and biosensors.11,12 However, traditional hydrogels exhibit brittleness and inefficient energy consumption, which originates from their low resistance to crack propagation due to the lack of an efficient energy dissipation mechanism in the gel network.13,14 This leads to poor mechanical performances of hydrogels, and limits their applications.15 Therefore, imparting excellent mechanical properties to the hydrogels is an urgent problem that must be solved. To obtain a hydrogel with excellent mechanical properties, it is necessary to increase the total viscoelastic dissipation along the hydrogel by introducing a dissipation mechanism at the molecular level.13,15 Recently, many techniques for synthesizing hydrogels with excellent mechanical properties have been proposed, including interpenetrating network hydrogels,16 double network hydrogels,17,18 nanocomposite hydrogels19,20 and topological hydrogels.21 Among them, double network hydrogels have demonstrated their excellent mechanical properties. In the double network hydrogels, both networks with contrasting structures are separately cross-linked, and the interpenetration of two networks makes the hydrogels both tough and soft.22 The toughening mechanism for double network hydrogels is mainly based on sacrificial bonds that break from the first network to effectively dissipate energy and protect the second network, thereby sustain stress and store elastic energy. So the mechanical properties of hydrogels are reinforced. But the fracture of the first network leads to the cleavage of irreversible and permanent bond, so it is difficult to repair and recover the hydrogel from damage and fatigue.23 The self-healing and self-recovery properties of hydrogels are also especially important for expanding their applications, such as cartilage, tendon, muscle, and blood vessel.
The introduction of the reversible non-covalent interaction in place of the sacrificial covalent bond in the first network is considered to be an important way to improve the self-healing and fatigue properties of double network hydrogels caused by permanent bond cleavage. Various non-covalent interactions, such as hydrogen bond,24 metal-coordination25,26 and hydrophobic interaction,27,28 have been incorporated into the double network hydrogels, resulting in a range of attractive properties, such as high strength, fatigue resistance, self-healing, self-recovery, shape memory, and remoldability/recyclability/reusability. A fully, physically crosslinking double network hydrogels were synthesized using a hydrogen bond-associated agar gel as the first network and a hydrophobically associated polyacrylamide gel as the second network, and found that the synthesized hydrogels not only exhibit excellent mechanical strength and high toughness, but also have rapid self-recovery, remarkable fatigue resistance, and notable self-healing performances.22 Yuan et al.27 reported a dual physically cross-linked polyacrylamide/xanthan gum double network hydrogels. The results revealed that the prepared hydrogels possess fracture stresses high as 3.64 MPa and compressive stresses at 99% strain of more than 50 MPa. In addition, the introduction of the physically crosslinked structure also gives the hydrogels excellent fatigue resistance and self-healing properties.
In this study, alginate-based hydrogels with semi-interpenetrating structure were first synthesized by hydrophobic interaction in the presence of sodium alginate. Then the synthesized hydrogels were soaked in an aqueous solution of CaCl2, causing that α-L-guluronic acids (G) residues of sodium alginate could couple with Ca2+ to form metal-coordinate bond. This acts as a physical cross-linking point, leading to the formation of alginate network.17,29 So alginate-based hydrogels with double network structure were constructed by hydrophobic interaction and metal-coordination. The mechanical strength, fatigue resistance, self-recovery, self-healing properties and pH-sensitivity of two different structures of alginate-based hydrogels were discussed. As a result, alginate-based hydrogels with double network structure showed excellent mechanical strength, fatigue resistance and self-recovery properties, while alginate-based hydrogels with semi-interpenetrating network structure exhibited excellent self-healing properties. Therefore, the synthesized alginate-based hydrogel with excellent properties has potential application value in many fields.
2. Experimental section
2.1 Materials and reagents
Sodium alginate (SA, viscosity (10 g L−1, 20 °C) >0.02 Pa S) was purchased from Sinopharm Group Reagent Co., Ltd; acrylamide (AM, 99.0%), stearyl methacrylate (SMA, 96%) and ammonium persulfate (APS, >99%) were provided by Shanghai Macklin Biochemical Co., Ltd; sodium dodecyl sulfate (SDS, CP) was obtained from Shantou Guanghua Chemical Factory (Guangdong, China); calcium chloride (CaCl2, AR) was purchased from Guangzhou Chemical Reagent Factory (Guangdong, China); all other reagents used were of analytical grade.2.2 Synthesis of alginate-based hydrogels
A certain amount of acrylamide and stearyl methacrylate were added to 5 mL of distilled water, and stirred at 45 °C for 60 min. Then 0.56 g of sodium dodecyl sulfate was added, and stirred at this temperature. After about 30 min, sodium alginate was added, and stirred until completely dissolved. Next, ammonium persulfate (20 mg) was dissolved in 1 mL of distilled water, and the prepared APS aqueous solution was added to the above mixture, and stirred quickly. Afterwards, the obtained mixture was poured into a mold, and evacuated at room temperature to remove air bubbles. Finally, the mold was placed in the incubator for 6 h at 60 °C, and the obtained sample was marked as NaAlg x%/PAM y%, where x% represents the used amount of sodium alginate with respect to the total amount of acrylamide and stearyl methacrylate; y% represents the molar ratio of stearyl methacrylate in the total amount of acrylamide and stearyl methacrylate. x% was set to 0%, 2.5%, 5.0%, 7.5% and 10.0%, while y% was specified as 1%, 2%, 3% and 4%. The synthesized sample NaAlg x%/PAM y% was immersed in a 0.5 mol L−1 calcium chloride solution for 12 h, and obtained sample was recorded as CaAlg x%/PAM y%.2.3 Characterization
Fourier transform infrared spectroscopy (FT-IR) was performed on a Nicolet Fourier transform infrared spectrometer (USA). The dried sample was ground into powder and mixed with KBr, and then compressed into thin pellet form before being tested. The X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo ESCALAB 250 with Al Kα X-rays. C 1s binding energy value of 284.8 eV was used as standard correcting the observed spectra to eliminate sample charging effects. The surface morphologies of the synthesized samples were observed by a scanning electron microscope (SEM, TESCAN VEGA3 LMH). The sample swollen to equilibrium in phosphate buffer solution (pH = 7.4, I = 0.1) at 37 °C for 24 h was frozen at −20 °C, and then freeze-dried at −50 °C. After the obtained sample was sprayed with gold, it was placed in an SEM to observe the morphology.2.4 Swelling behavior and pH-sensitivity
Swelling behavior and pH-sensitivity of the synthesized hydrogels were evaluated by the gravimetric method. The sample was immersed in aqueous solutions with desired pH (I = 0.1) at 37 °C. At regular period of time, the sample was removed from aqueous solution, blotted with filter paper to remove surface water, and weighted. Swelling ratio (SR, g g−1) at a given moment and equilibrium swelling ratio (ESR, g g−1) can be calculated by the following equations, respectively: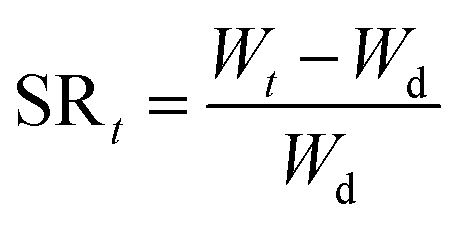
where Wd and Wt are weights of the dry sample and swollen sample at time t; We represents the weight of the sample to achieve the swelling equilibrium. All experiments were repeated at least three times to calculate the mean and variation.
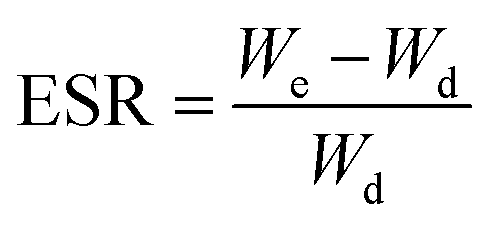
2.5 Rheological measurement
Rheological behaviors were performed on an Anton Paar MCR Rheometer (Austria) using parallel plates of 25 mm diameter. The dynamic strain sweep was first measured in strain range from 0.01% to 500% at frequency 10 rad s−1. Then the dynamic frequency sweep was measured over the frequency range of 0.1–100 rad s−1 at strain 1.0%. To analyze the recovery properties of the sample, two different continuous step strain modes were measured at frequency 10 rad s−1 by the following conditions: (1) the amplitude oscillatory strain was switched from 1% to 100% with 100 s for every strain interval; (2) the amplitude oscillatory strain was stepped from 1% to larger strain (100%, 200%, 300% and 400%) with 100 s for every strain interval. All measurements were performed at room temperature.2.6 Mechanical properties
Tensile test was measured using an electronic universal tester (Shanghai Yihuan Technology Co., Ltd) at room temperature. The film sample (60 mm in length, 6 mm in width and 1 mm in thickness) was tested with 20 mm initial distance between two fixtures at crosshead speed of 100 mm min−1. For the compression test, the cylindrical sample (about 12 mm in diameter × 20 mm in height) was performed at room temperature. The rate of compression testing was set at 20 mm min−1 until the sample fractured. Each sample was tested in triplicate at least to give the mean value and standard deviation.For hysteresis measurement, the sample (60 mm in length, 6 mm in width and 1 mm in thickness) was first stretched to an extension ratio (λ1) at 100 mm min−1, and then unloaded at similar speed. The sample was reloaded, and stretched to an increased extension ratio (λ2) as first loading and unloaded again. The loading–unloading processes were repeated to conduct on the same sample with increasing extension ratio.
For self-recovery test, the sample was initially stretched to a determined strain (600%) at 100 mm min−1, and then unloaded at the similar speed. After each loading–unloading cycle, the sample was relaxed for certain time (0–30 min), and processed to the next cycle. The residual strain ratio was calculated by a ratio of length change of sample after removal of stress with respect to the original length. The recovery ratio was also defined by a ratio of energy dissipation after various waiting time with respect to the initial energy dissipation.
The elastic energy stored (U) when the sample is loaded elastically to a stress δmax can be calculated by area below the stress–strain curve, using the following equation:
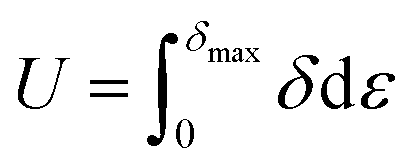
The dissipated energy (ΔU), which is defined as the area of hysteresis loop encompassed by the loading–unloading curve, is calculated by integrating the area between loading–unloading curves, using the following equation:
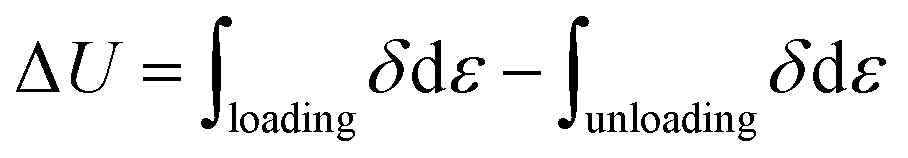
2.7 Self-healing properties
The rectangular sample (60 mm in length, 6 mm in width and 1 mm in thickness) was first cut in half from the middle. Then the two separate interfaces were immediately brought into contact again, and placed in an incubator at 40 °C for a certain period of time. A high humidity was maintained in the incubator to prevent the evaporation of moisture. The tensile properties of the healed sample were evaluated by the above tensile measurement. The healed efficiency was calculated by the ratios of tensile strength and elongation at break of the healed sample to those of origin sample.3. Results and discussion
3.1 Synthesis and characterization of alginate-based hydrogels
Scheme 1 displays the schematic diagram for the synthesis of alginate-based hydrogels. As shown in Scheme 1, SMA is first dissolved in the SDS micelles to form polymerizable micelles, which are hydrophobic micelles.27 Then sodium alginate is added, and stirred to completely dissolve. After that, poly(acrylamide)-based network is constructed by the micelle copolymerization of acrylamide and stearyl methacrylate in the presence of sodium alginate, where the formed polymerizable micelles act as the crosslinking points. Therefore, NaAlg/PAM hydrogels with semi-interpenetrating network structure are synthesized. The synthesized NaAlg/PAM semi-IPN hydrogels are further immersed in an aqueous solution of CaCl2. A ionic bonding cross-linked network is formed through the metal-coordination between carboxyl groups of alginate chain and Ca2+ ions.17,29 Because the PAM network intersects with the alginate network each other, the non-covalent interactions, such as van der Waals force and hydrogen bonding, could be formed between PAM and alginate. Therefore, CaAlg/PAM double network hydrogels are synthesized.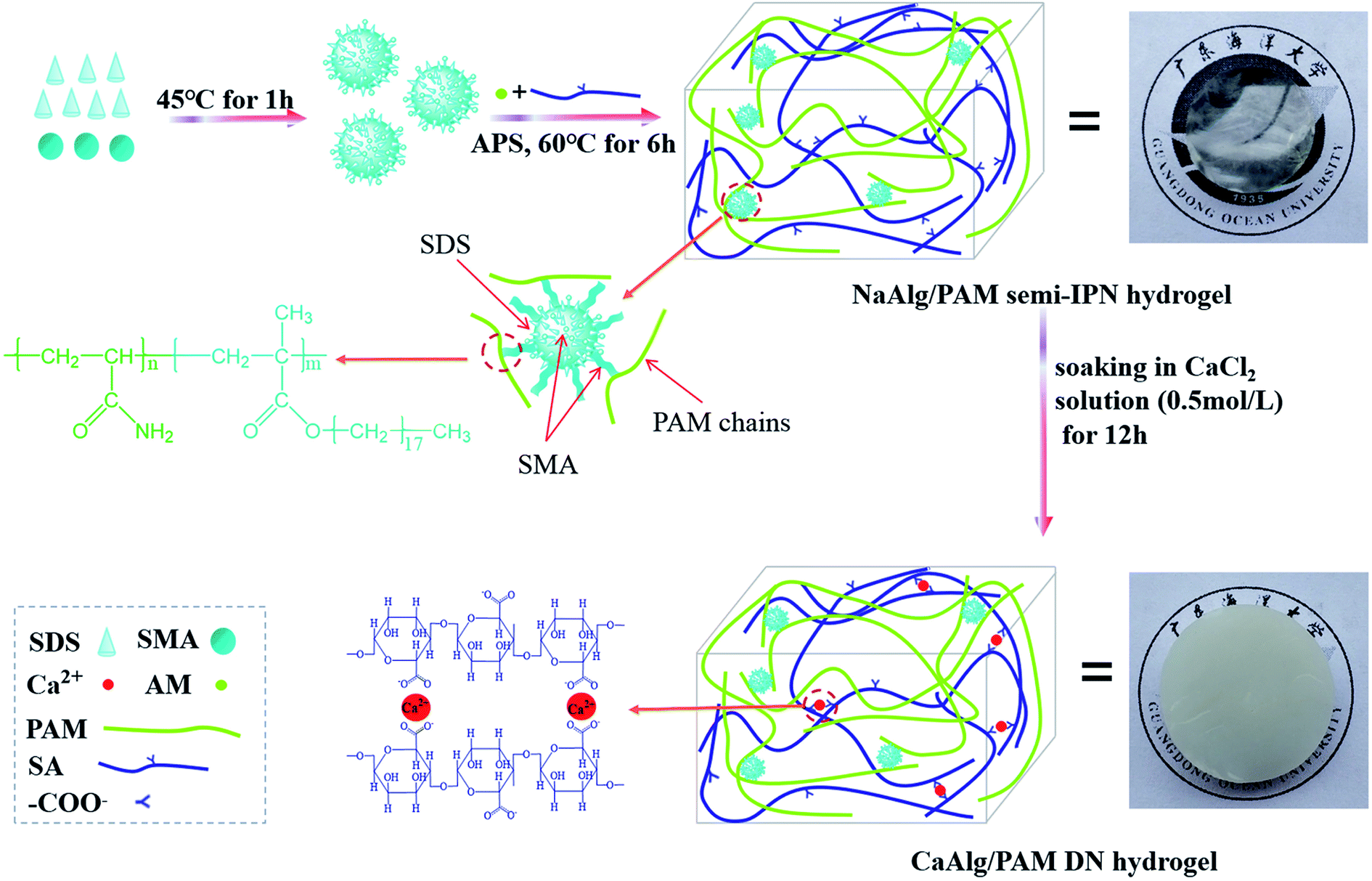 | ||
| Scheme 1 Schematic diagram of NaAlg/PAM semi-interpenetrating network hydrogels and CaAlg/PAM double network hydrogels. | ||
Fig. 1(a) shows FT-IR spectra of sodium alginate, PAM, NaAlg/PAM and CaAlg/PAM hydrogels. In the spectrum of sodium alginate, the broad peak at 3400 cm−1 is due to OH stretching vibration. The obvious absorption peaks at 1612 cm−1 and 1415 cm−1 are attributed to asymmetric and symmetric stretching vibrations of –COO− group of alginate, respectively.30 The peak at 1030 cm−1 is assigned to the C–O stretching vibration of polysaccharide structure.30,31 The spectrum of PAM clearly shows the stretching vibration of N–H at 3357 and 3194 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1661 cm−1, and N–H deformation at 1611 cm−1.27 In the spectra of NaAlg/PAM and CaAlg/PAM hydrogels, the characteristic peaks of both alginate and polyacrylamide could be observed, and no new distinct peaks are observed. Compared with sodium alginate, the wavenumber for asymmetric –COO− stretching of alginate in the hydrogels decreases, which may be attributed to the electrostatic interaction between metal ions and carboxy anions, hydrogen bonding between alginate and polyacrylamide.32,33 Especially for CaAlg/PAM hydrogel, the intensity of the peaks corresponded to O–H, asymmetric –COO− stretching and symmetric C–O stretching in C–O–C structure decrease. The possible reason is that the alginate chains are cross-linked to form a network through the strong electrostatic attraction between alginate chains and Ca2+ ions.32,33 These results indicate that alginate-based hydrogels with different structures can be synthesized through physically cross-linking interactions.
O stretching at 1661 cm−1, and N–H deformation at 1611 cm−1.27 In the spectra of NaAlg/PAM and CaAlg/PAM hydrogels, the characteristic peaks of both alginate and polyacrylamide could be observed, and no new distinct peaks are observed. Compared with sodium alginate, the wavenumber for asymmetric –COO− stretching of alginate in the hydrogels decreases, which may be attributed to the electrostatic interaction between metal ions and carboxy anions, hydrogen bonding between alginate and polyacrylamide.32,33 Especially for CaAlg/PAM hydrogel, the intensity of the peaks corresponded to O–H, asymmetric –COO− stretching and symmetric C–O stretching in C–O–C structure decrease. The possible reason is that the alginate chains are cross-linked to form a network through the strong electrostatic attraction between alginate chains and Ca2+ ions.32,33 These results indicate that alginate-based hydrogels with different structures can be synthesized through physically cross-linking interactions.
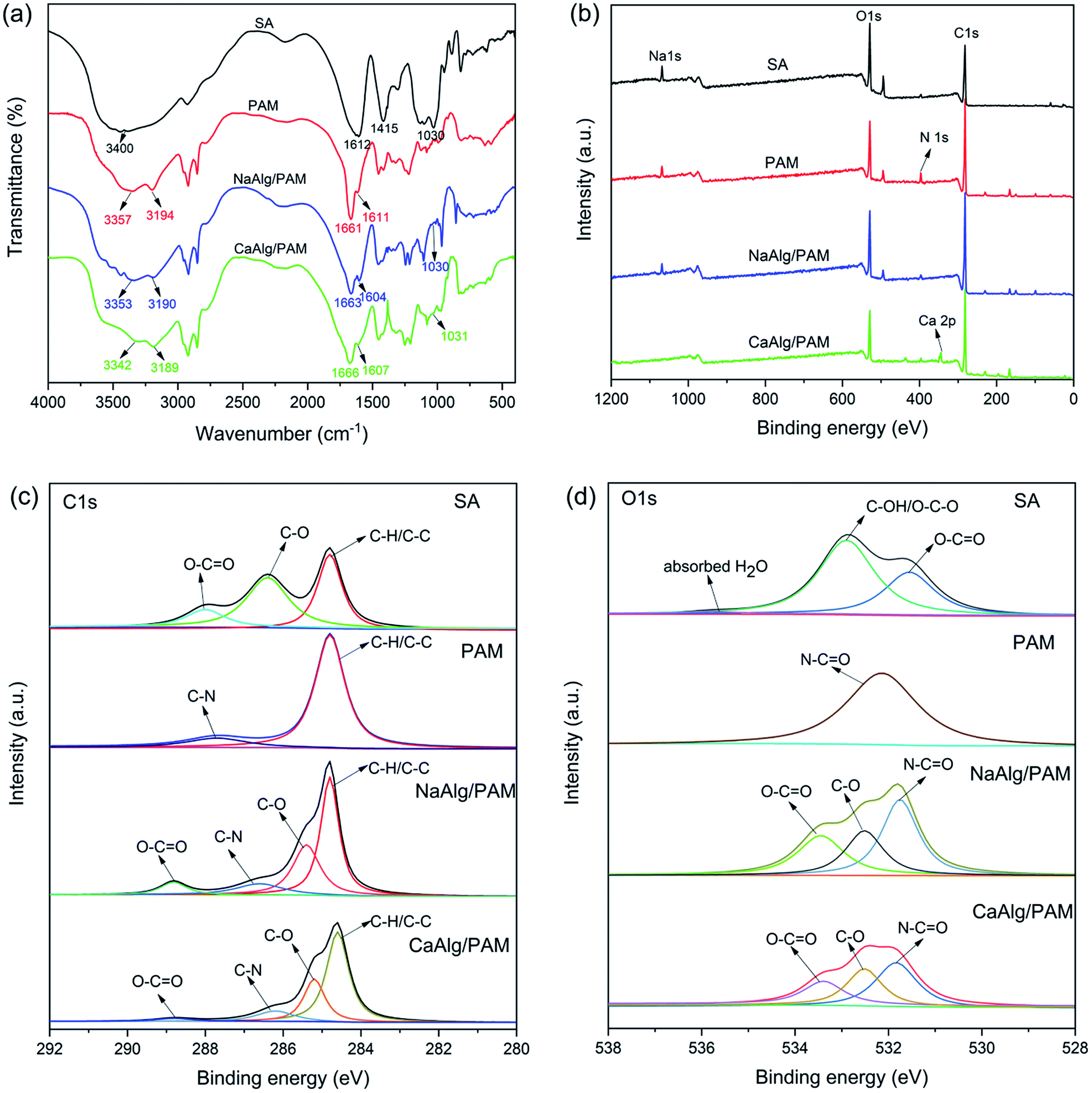 | ||
| Fig. 1 FT-IR (a) and XPS wide-scan (b) spectra of SA, PAM, NaAlg/PAM and CaAlg/PAM hydrogels; C 1s (c) and O 1s (d) level spectra of SA, PAM, NaAlg/PAM and CaAlg/PAM hydrogels. | ||
More detailed structure analysis about NaAlg/PAM semi-IPN hydrogels and CaAlg/PAM double network hydrogels was obtained by XPS. As displayed in Fig. 1(b), C, O, N and Na elements are presented on the surface of the NaAlg/PAM hydrogel. While Na element is not detected on the surface of CaAlg/PAM hydrogel, but there presents Ca element. This suggests that the ion-exchange reaction occurs when NaAlg/PAM hydrogel is soaked in CaCl2 solution. The high-resolution C 1s and O 1s spectra are shown in Fig. 1(c) and (d), respectively. For SA, the C–C/C–H, C–O, and O–CO peaks appeared at 284.8, 286.4 and 288.0 eV, respectively.34 In the O 1s spectrum of SA, the peaks situated at 531.5 and 532.9 eV indicated the presence of O–CO and C–OH/O–C–O,35 which is consistent with the C 1s spectrum of SA. The peak at 535.8 eV is attributed to the water absorbed by SA. The peaks at 284.8 and 287.7 eV in the C 1s spectrum of PAM are assigned to C–H/C–C and C–N, respectively. The O 1s spectrum of PAM shows a peak at 532.1 eV, corresponded to N–CO.36,37 For NaAlg/PAM semi-IPN hydrogel, the C 1s spectrum has four distinctive chemical states: at 284.8 eV related to C–H/C–C, at 285.4 eV for C–O, at 286.4 eV for C–N and 288.8 eV corresponded to O–CO. The corresponding O 1s spectrum exhibits three peaks at 531.8, 532.5 and 533.4 eV, which are attributed to N–CO, C–O and O–CO, respectively. It is observed that the chemical states of C and O in NaAlg/PAM semi-IPN hydrogel contain the chemical states of SA and PAM, and no new chemical states are formed. This reveals that NaAlg/PAM semi-IPN hydrogels are synthesized by the non-covalent interactions. It is clear that the C 1s and O 1s spectra of CaAlg/PAM DN hydrogel are similar to those of NaAlg/PAM semi-IPN hydrogel. However, this is significant that the peak intensity at 288.8 eV corresponded to O–CO reduces, implying the strong coordination between –COO− and Ca2+. This indicates that CaAlg/PAM DN hydrogels are prepared by immersing NaAlg/PAM semi-IPN hydrogels in CaCl2 solution. These results are consistent with the FT-IR result, further confirming that the NaAlg/PAM semi-IPN hydrogel and CaAlg/PAM DN hydrogel based on non-covalent interactions are successfully prepared.
The synthesized NaAlg/PAM semi-IPN hydrogels and CaAlg/PAM DN hydrogels present an excellent performance in ductility and mechanical properties, as displayed in Fig. S1† and 2. It is clear in Fig. 2(a) and (b) that the CaAlg/PAM DN hydrogel sample (60 mm × 10 mm) is easily stretched to 200% of its original length, and the central notched (Φ 4 mm) hydrogel sample also remains stable without crack propagation. It can be observed that the sample can be restored to the original length. A rod sample of 8 mm diameter not only can be able to a certain amount of pressure, but also can resist to slicing with a knife, as displayed in Fig. 2(c) and (d). When the external force is removed, the sample can return to its original state. The sample film can lift a block of 100 g without breaking (as Fig. 2(e)). The synthesized NaAlg/PAM semi-IPN hydrogels can also go through a similar process without damaging, as displayed in Fig. S1.† Therefore, the synthesized hydrogels display stretchability, flexibility, and self-recovery properties. Next, we would further analyze the performance of the synthesized hydrogels through tensile test, compression test, loading–unloading cycle test and rheological test.
 | ||
| Fig. 2 The photographs of CaAlg/PAM DN hydrogel demonstrating the excellent mechanical behaviors: under stretching (a) without and (b) with central notch, compressing (c), slicing with a knife (d) holding a weight of 100 g (e). | ||
3.2 Mechanical properties
Mechanical properties of the synthesized hydrogels were evaluated by tensile test and compression test, and the obtained results are shown in Fig. 3 and Table S2.† The previous literature has reported that the mechanical properties of hydrogels are closely related to their water content, their tensile strength decreases as the water content increases.38 Therefore, the water content of the synthesized hydrogels was measured by gravimetric method, the obtained results are displayed in Table S1.† It is clear that the water content of NaAlg/PAM semi-IPN hydrogels is 39.9–57.5%, while CaAlg/PAM DN hydrogels have a higher water content (67.0–73.4%). This is because the prepared NaAlg/PAM semi-IPN hydrogels don't reach the swelling equilibrium, so it would further swell at the CaCl2 solution. Therefore, the water content of CaAlg/PAM DN hydrogels is higher than that of the corresponding NaAlg/PAM semi-IPN hydrogels. As shown in Fig. 3 and Table S2,† tensile strength of CaAlg/PAM DN hydrogels with higher water content is larger than that of the corresponding NaAlg/PAM semi-IPN hydrogels, while their elongation at break is significantly less than that of the corresponding NaAlg/PAM semi-IPN hydrogels, which is mainly due to the different network structures. Because the interaction between Ca2+ and –COO− in alginate chains increases the cross-linking density of polymer network, so the CaAlg/PAM DN hydrogels cross-linked by Ca2+ exhibit higher mechanical properties.39 As listed Table S2,† with increasing of the SMA concentration from 1% to 4%, the tensile strength of NaAlg/PAM semi-IPN hydrogels increases from 202.7 kPa to 529.7 kPa and the elongation at break also increases from 18.5 mm mm−1 to 33.6 mm mm−1. Fig. 3(a) shows the tensile stress–strain curves of CaAlg/PAM DN hydrogels with various SMA concentrations, and the tensile strength and elongation at break obtained from Fig. 3(a) are shown in Fig. 3(b). CaAlg/PAM DN hydrogels with various SMA concentrations exhibit excellent tensile performances (575.1–733.6 kPa tensile strength and 11.5–17.6 mm mm−1 elongation at break). The tensile strength and elongation at break of CaAlg/PAM DN hydrogels first increase and then decrease as the SMA concentration increases. For CaAlg/PAM DN hydrogel with 2% of SMA, its tensile strength could reach 733.6 kPa, while the elongation at break of the hydrogel with 3% of SMA is largest (17.6 mm mm−1). The increased SMA concentration results in the increasing of cross-linking density of the PAM network. Because the higher the density of cross-linking is, the more rigid network is, and the smaller the elasticity is.40 Therefore, with increasing of the SMA concentration, the tensile strength and elongation at break first increase and then decrease. Compressive stress–strain curves of CaAlg/PAM DN hydrogels with various SMA concentrations are displayed in Fig. 3(c). It can be observed that the compressive strength at strain 80% first increases and then decreases as the SMA concentration increases, and the compressive strength of CaAlg/PAM DN hydrogel with 2% of SMA could reach 1.354 MPa. However, with increasing of the SMA concentration from 1% to 4%, the compressive strength of NaAlg/PAM semi-IPN hydrogels decreases from 0.528 MPa to 0.396 MPa.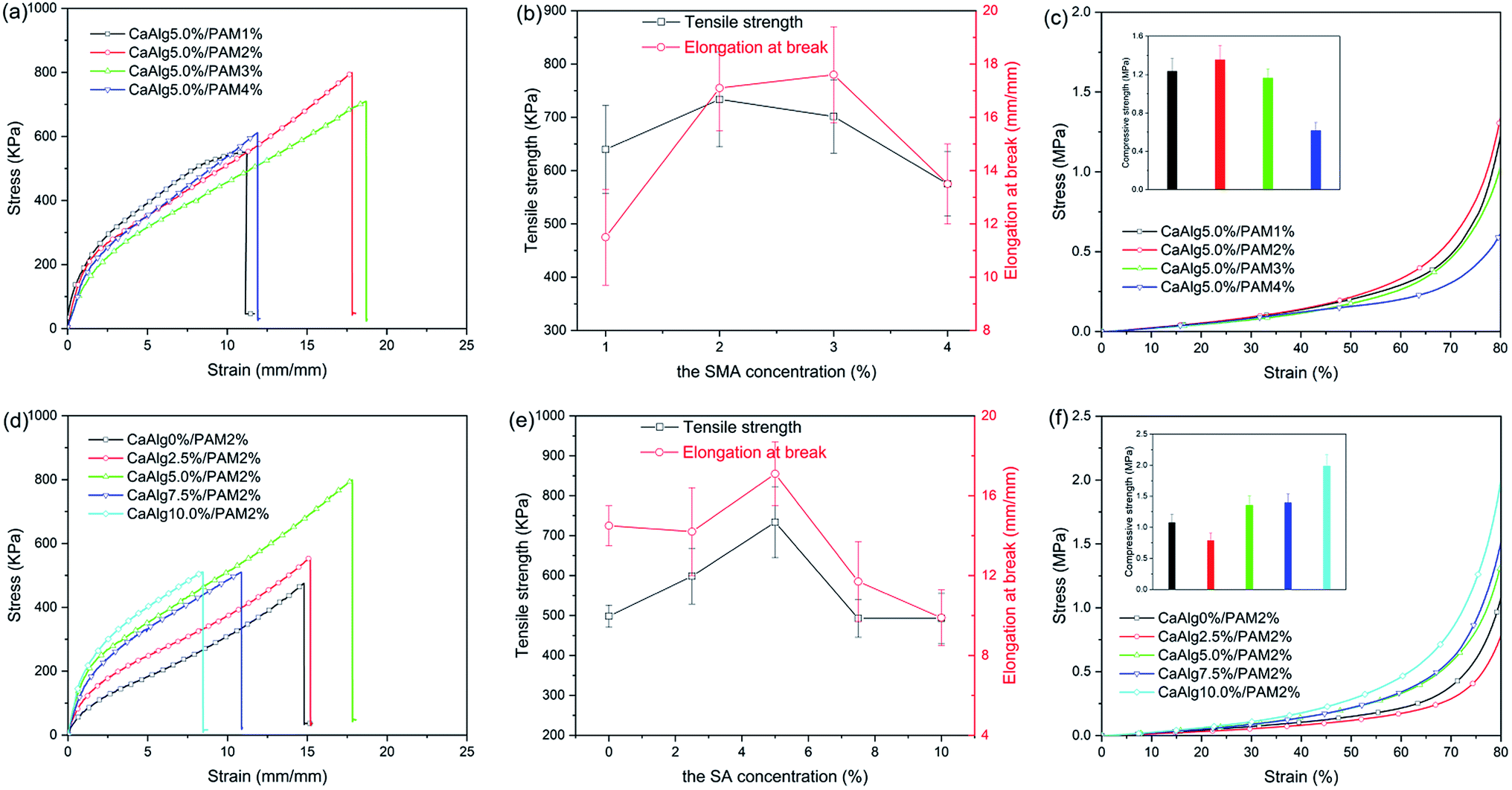 | ||
| Fig. 3 (a and d) Tensile stress–strain curves of CaAlg/PAM DN hydrogels; (b and e) tensile strength and elongation at break of CaAlg/PAM DN hydrogels; (c and f) compressive stress–strain curves of CaAlg/PAM DN hydrogels: (a, b and c) hydrogels with various SMA concentrations; (d, e and f) hydrogels with various SA concentrations. | ||
The influence of the sodium alginate concentration on mechanical properties of prepared hydrogels was also investigated. As listed in Table S2,† with increasing of the SA concentration, the tensile strength of NaAlg/PAM semi-IPN hydrogels first increases and then decreases. Tensile strength of NaAlg/PAM semi-IPN hydrogel with 7.5% of SA could reach 678.2 kPa. And NaAlg/PAM hydrogels exhibit larger elongation at break (24.3–38.2 mm mm−1). Fig. 3(d) shows the tensile stress–strain curves of CaAlg/PAM DN hydrogels with various SA concentrations, and the obtained results from Fig. 3(d) are displayed in Fig. 3(e). The tensile strength of CaAlg/PAM DN hydrogels with different SA concentrations is 492.6–733.6 kPa, their elongation at break is 9.9–17.1 mm mm−1. It is observed that with increasing of the SA concentration, tensile strength and elongation at break first increase and then decrease. The carboxyl anion of alginate can be coupled with Ca2+, so the second network based on alginate is formed. As the concentration of SA increases, the second network becomes much denser. The previous literatures39,41 have reported that because alginate chains could interpenetrate with covalently cross-linked PAM network, the alginate network ionically cross-linked by multivalent cationic could be zipped. For alginate network with lower cross-linking density, the loose alginate network is prone to deformation during stretching, improving the tensile strength and elongation at break. For alginate network with higher cross-linking density, the dense alginate network results in a decrease in the elasticity of the materials, which reduces both tensile strength and elongation at break. As listed in Table S2,† with increasing of the SA concentration, the compressive strength of NaAlg/PAM semi-IPN hydrogels at strain 80% first increases and then decreases. The compressive strength of NaAlg/PAM semi-IPN hydrogel with 5.0% of SA could reach 0.516 MPa. Fig. 3(f) displays the compressive stress–strain curves of CaAlg/PAM DN hydrogels with different SA concentrations. It is clear that the compressive strength of the DN hydrogels first decreases and then increases as the SA concentration increases. For CaAlg/PAM DN hydrogel with 10% of SA, its compressive strength at strain 80% could reach 1.985 MPa. The mechanical properties of alginate-based hydrogels with similar systems are summarized in Table S3.† It is clear that compared with alginate-based hydrogels reported in the literatures, the synthesized alginate-based hydrogels exhibit excellent mechanical properties.
On the basis of the above results, we found that the mechanical properties of the synthesized alginate-based hydrogels are not only related to their composition, but also to their network structure. For NaAlg/PAM hydrogel, the interpenetration of alginate chains and PAM network forms a semi-interpenetrating network structure. The increased SMA concentration enhances the cross-link density of the PAM network, and the increased SA could increase the entanglement of alginate and PAM network. So the cross-linking density of alginate-based hydrogel could be improved as the SMA or SA concentrations increase, resulting in the formation of more robust and flexible network structure. This could lead to the increasing of the strength and stretchability of the hydrogels.42 For CaAlg/PAM hydrogel, alginate chains were cross-linked and formed interpenetrated ionic network due to strong electrostatic attraction between positively charged Ca2+ ions and negatively charged alginate chains. So the CaAlg/PAM hydrogel exhibits double network structure. With increasing of the concentrations of SMA and SA, the cross-linking densities of the first network and the second network increase, respectively. At lower cross-linking density, the increased cross-linking density could obviously improve the strength and stretchability of the materials. At higher cross-linking density, the increased cross-linking point makes the hydrogel network more denser, so that the polymer chain is not easy to move when the sample is deformed, which could cause the sample to break. Therefore, to construct a double network hydrogel with excellent mechanical properties, when one network has a high cross-linking density, the other must be a loose network. When the synthesized DN hydrogel is deformed under stress, the loose network ruptures to efficiently dissipate energy, protect the soft and ductile network from crack propagation, thereby improving the mechanical properties of the hydrogels.43,44
3.3 Hysteresis and self-recovery
The loading–unloading test was used to evaluate the energy dissipation of hydrogels with different structures. Fig. 4(a) displays the loading–unloading curves of PAM, NaAlg/PAM and CaAlg/PAM hydrogels at strain 600%. For PAM and NaAlg/PAM hydrogels, there is no hysteresis loop on the loading–unloading curves, revealing that they show a typical rubber elastic behavior.20 The loading–unloading curve of CaAlg/PAM DN hydrogel shows an obvious hysteresis loop, indicating CaAlg/PAM DN hydrogel dissipates energy effectively. The dissipated energies calculated by Fig. 4(a) are shown in Fig. 4(b). For CaAlg/PAM DN hydrogel, its dissipated energy could reach 540.7 kJ m−3, which is greater than that of PAM (62.1 kJ m−3) and NaAlg/PAM semi-IPN hydrogels (134.1 kJ m−3). These results reveal that CaAlg/PAM DN hydrogel has more effective energy dissipation pathways with respect to PAM and NaAlg/PAM semi-IPN hydrogels, which may be ascribed to the interaction between Ca2+ and –COO− of alginate chains. When the external loading is applied on the CaAlg/PAM hydrogel, the ionic coordination bonds serve as reversible sacrificial bonds and are cracked to effectively dissipate energy.43 Furthermore, the cycle tensile loading–unloading tests of CaAlg/PAM DN hydrogel at different strains during loading–unloading cycles were measured, and the obtained results are shown in Fig. 4(c) and (d). It is evident that the area of loop increases with the increasing strain (as Fig. 4(c)); i.e., the dissipated energy increases from 73.0 kJ m−3 at strain 200% to 655.7 kJ m−3 at strain 1200% (Fig. 4(d)). This further confirms that CaAlg/PAM DN hydrogel has high energy dissipation. However, the reloading curve of CaAlg/PAM DN hydrogel is close, but does not completely overlap with the previous loading curve. The similar phenomenon has been reported in agar/PAMAAc-Fe3+ double network hydrogels.25 The possible reason is that the previous loading has induced a change in the gel network, and a portion of dynamic reversible non-covalent interactions can be restored before the next loading.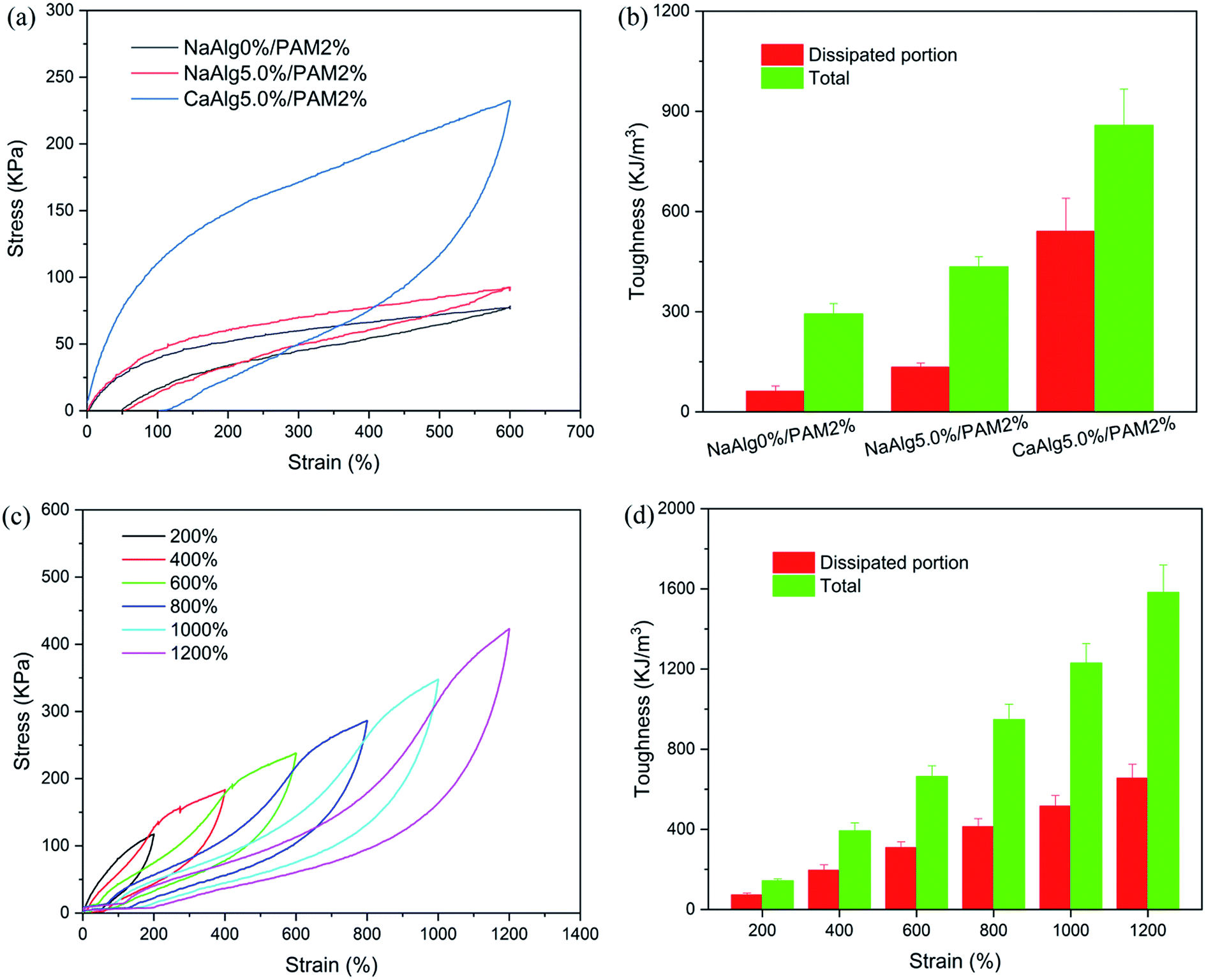 | ||
| Fig. 4 (a) Loading–unloading curves and (b) the corresponding toughness of NaAlg 0%/PAM 2% hydrogel, NaAlg 5.0%/PAM 2% hydrogel and CaAlg 5.0%/PAM 2% hydrogel; (c) cycle loading–unloading curves and (d) the corresponding toughness of CaAlg 5.0%/PAM 2% hydrogel at various strains. | ||
Self-recovery capacity of CaAlg/PAM DN hydrogels was evaluated by the cycle loading–unloading tests, and the obtained results are shown in Fig. 5(a) and (b). As shown in Fig. 5(a), no resting time is applied between two consecutive loadings, the area of hysteresis loop significantly reduces. With increasing of the resting time, the area of hysteresis loop increases. It is observed from Fig. 5(b) that toughness recovery ratio increases as the resting time increases. After 30 min of resting, CaAlg/PAM DN hydrogel can recover ∼60% of toughness. With increasing of the resting time from 0 min to 30 min, the residual strain decreases from 38.6% to 13.7%. Furthermore, the fatigue resistance of CaAlg/PAM DN hydrogels was analyzed by the ten cycles loading–unloading at strain of 600%. As shown in Fig. 5(c), the hysteresis loop and stress of CaAlg/PAM DN hydrogels display an obvious decrease with respect to original cycle, and these figures further decrease as the increase of number of cycle. This indicates that the hydrogel network has been destroyed during loading–unloading process, and it could not be fully recovered quickly. The sample subjected to 10 cycles of loading–unloading was allowed to recover at room temperature for 24 hours, and then the sample was measured again by the ten loading–unloading cycles at strain of 600% (as Fig. 5(d)). It is observed that maximum stress of the recovered sample is slightly larger than its original stress. This reveals that the destroyed network can be formed again, which is mainly attributed to the dynamic reversible non-covalent interaction. And the reformed network could achieve the optimal reorganization with relieving internal stress of the hydrogel.45 These demonstrate that CaAlg/PAM DN hydrogels exhibit the excellent fatigue resistance.
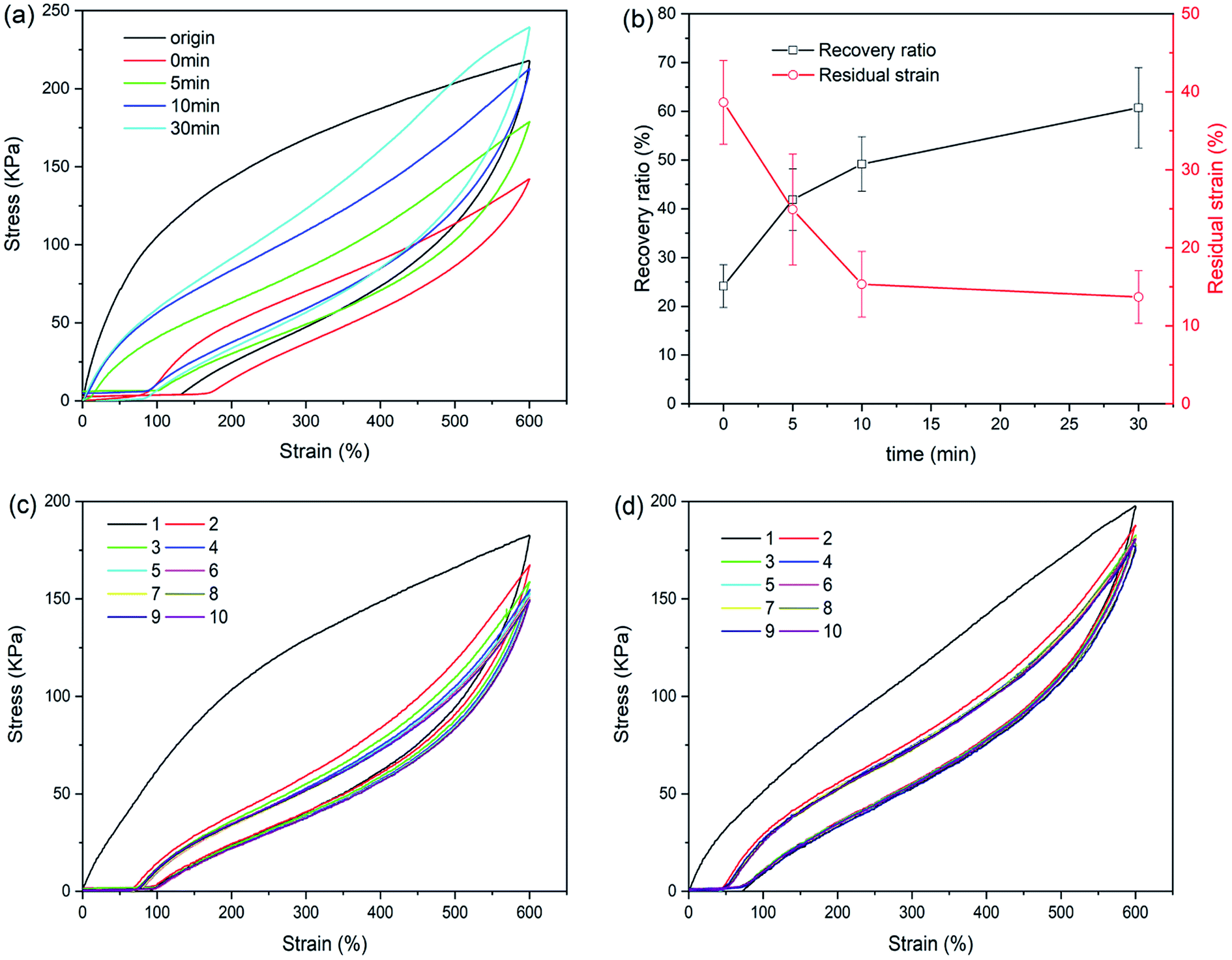 | ||
| Fig. 5 (a) Cyclic loading–unloading curves and (b) toughness recovery ratio and residual strain of CaAlg 5.0%/PAM 2% DN hydrogel at different resting time; the ten successive tensile loading–unloading curves of the (c) as-prepared samples and (d) recovered sample after 24 h resting time. | ||
3.4 Rheological behavior
Fig. 6(a) shows the results of strain sweep for NaAlg 5.0%/PAM 2% semi-IPN hydrogel and CaAlg 5.0%/PAM 2% DN hydrogel. It is clear that the modulus values of NaAlg 5.0%/PAM 2% hydrogel and CaAlg 5.0%/PAM 2% hydrogel have no dependence on the strain when the strain is less than 5%. With further increase of strain, the G′ value decreases. This indicates that the linear viscoelastic domain for CaAlg 5.0%/PAM 2% hydrogel is larger than that of NaAlg 5.0%/PAM 2% hydrogel. It can be observed that the G′ value of CaAlg 5.0%/PAM 2% hydrogel in the linear viscoelastic domain is higher than that of NaAlg 5.0%/PAM 2% hydrogel. This may be closely related to cross-linking density of hydrogel network. The G′′ values of hydrogels display a obvious peak prior to the final decrease, revealing an increasing use of deformation energy to deform subdomains of network structure before the inner structure finally breaks.46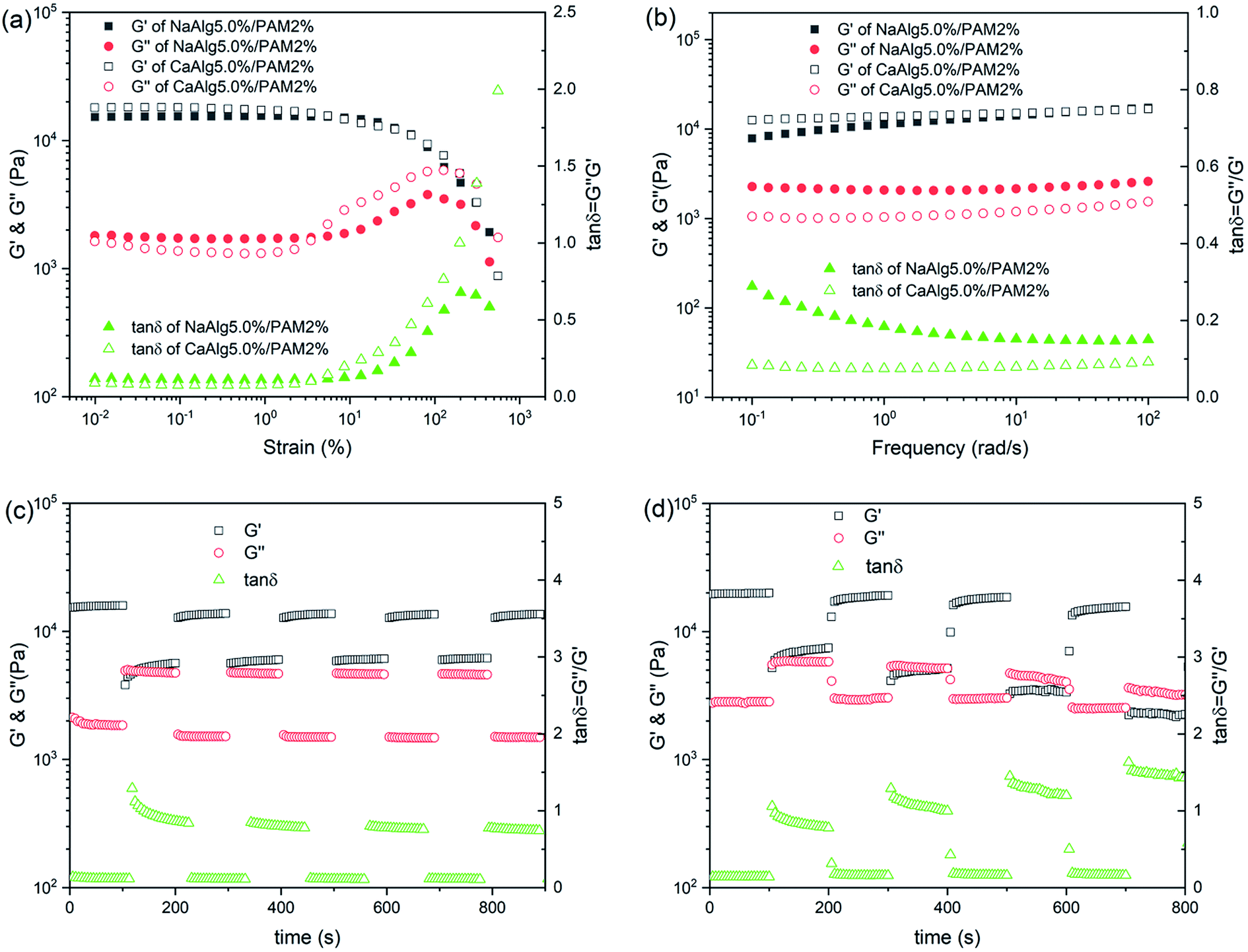 | ||
| Fig. 6 Dynamic rheological behaviors of NaAlg 5.0%/PAM 2% and CaAlg 5.0%/PAM 2% hydrogels: (a) strain sweep by frequency of 10 rad s−1 at 25 °C; (b) oscillatory frequency sweeps by 1.0% strain at 25 °C; (c) cyclic continuous step strain measurements in which the strain was switched from 1% strain for 100 s to 100% strain for 100 s; (d) cyclic continuous step strain measurements in which the strain was switched from 1% strain for 100 s to various larger strains (100%, 200%, 300% and 400%) for 100 s. | ||
Fig. 6(b) displays the frequency dependence of G′ and G′′ for NaAlg 5.0%/PAM 2% semi-IPN hydrogel and CaAlg 5.0%/PAM 2% DN hydrogel. It is observed that the G′ over the measured frequency range is always larger than the G′′, and both of them have poor frequency dependence. This reveals that the NaAlg/PAM semi-IPN hydrogel and CaAlg/PAM double network hydrogel are prepared by physical interactions. Similar result has been reported by Yang et al.47 It is also clear that the G′ value of CaAlg 5.0%/PAM 2% hydrogel is higher than that of NaAlg 5.0%/PAM 2% semi-IPN hydrogel, which is mainly due to the second network formed by Ca2+ crosslinking of alginate. The loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = G′′/G′) represents the lost energy to storage energy during deformation. Compared to NaAlg 5.0%/PAM 2% semi-IPN hydrogel, the tanδ value of CaAlg 5.0%/PAM 2% double network hydrogel is much lower (about 0.1), revealing that the formation of network structure leads to the increasing of elastic property more obvious than that of viscous property.48 And the tanδ value of CaAlg 5.0%/PAM 2% hydrogel is nearly stable and almost independent of frequency.
δ = G′′/G′) represents the lost energy to storage energy during deformation. Compared to NaAlg 5.0%/PAM 2% semi-IPN hydrogel, the tanδ value of CaAlg 5.0%/PAM 2% double network hydrogel is much lower (about 0.1), revealing that the formation of network structure leads to the increasing of elastic property more obvious than that of viscous property.48 And the tanδ value of CaAlg 5.0%/PAM 2% hydrogel is nearly stable and almost independent of frequency.
The changes in moduli of NaAlg 5.0%/PAM 2% semi-IPN hydrogel and CaAlg-5.0%/PAM-2% DN hydrogel with the shear strain jump between 1% and 100% are shown in Fig. S2(a)† and 6(c). As displayed in Fig. 6(c), the initial G′ and G′′ values of CaAlg 5.0%/PAM 2% hydrogel at 1% strain are 15.7 and 1.9 kPa, respectively. The G′ value decreases to 5.3 kPa and the G′′ value increases to 4.8 kPa, and the both almost completely return to their original values upon the strain returning to 1% over all the cycles. NaAlg 5.0%/PAM 2% semi-IPN hydrogel also shows a similar phenomenon. This indicates that the synthesized samples show stable hydrogel properties under low shear strain value. During the abrupt increase in the strain up to larger strain (as Fig. S2(b)† and 6(d)), the G′ value is lower than G′′, revealing that the viscous regime dominates over elastic. Upon reversal of the strain, G′ and G′′ could completely return to their original values, demonstrating the reformation of the gel network. Similar result has been reported by Sadiys Anjum et al.49 As shown in Fig. 6(d), the cycle is repeated with larger strain for up to 400%, the elastic state suddenly retracts without any significant loss in modulus. These results reveal that the synthesized hydrogel exhibits good self-recovery ability, which is consistent with the result of cycle loading–unloading tensile test.
3.5 Self-healing properties
Self-healing properties of materials are important for improving the recycling and extending application of materials. The self-healing properties of the materials benefit from dynamic and reversible bonds, such as hydrogen bonding, metal-coordination, and hydrophobic interaction.50,51 In this study, NaAlg/PAM semi-IPN hydrogels and CaAlg/PAM DN hydrogels were constructed through hydrophobic interaction and metal-coordination. So the self-healing properties of the synthesized hydrogels were evaluated. Fig. 7 shows the self-healing properties of NaAlg 5.0%/PAM 2% semi-IPN hydrogel. As displayed in Fig. 7(a), NaAlg 5.0%/PAM 2% semi-IPN hydrogel was first cut into two pieces and one piece was dyed with methylene blue, and then the cut surfaces were kept in contact at 40 °C for 24 h. The healed sample can be also withstood bending and stretching deformation, as Fig. 7(b). These results reveal that there occurs the migration of the polymer chain and the reconstruction of dynamic reversible hydrophobic interaction at the interface of the hydrogel, which could give the materials self-healing properties. The identical result was acquired by an optical microscopy, as presented in Fig. 7(c). It is clear that the crack of the sample becomes smaller as the self-healing time prolongs, and almost disappears at 48 h. Tensile stress–strain curves of samples subjected to different healing time are shown in Fig. 7(d), and the healing efficiencies obtained from Fig. 7(d) are presented in Fig. 7(e). It is observed that the healing efficiency increases as the healing time prolongs. For the sample healed for 48 h, its stress and elongation at break are 231.9 kPa and 4.06 mm mm−1, respectively, showing a higher self-healing ability (79.5% HE of stress, 16.7% HE of strain). However, CaAlg/PAM DN hydrogels exhibited very low self-healing efficiency, so the stress–strain curves of the samples could not be obtained. Because of the higher binding strength of Ca2+ and –COO− of alginate, thereby the reversibility of dynamic non-covalent association is greatly reduced. This reduces the ability to achieve self-healing through weak non-covalent bond interactions. Therefore, we should consider how to improve the self-healing efficiency of CaAlg/PAM DN hydrogels in future research.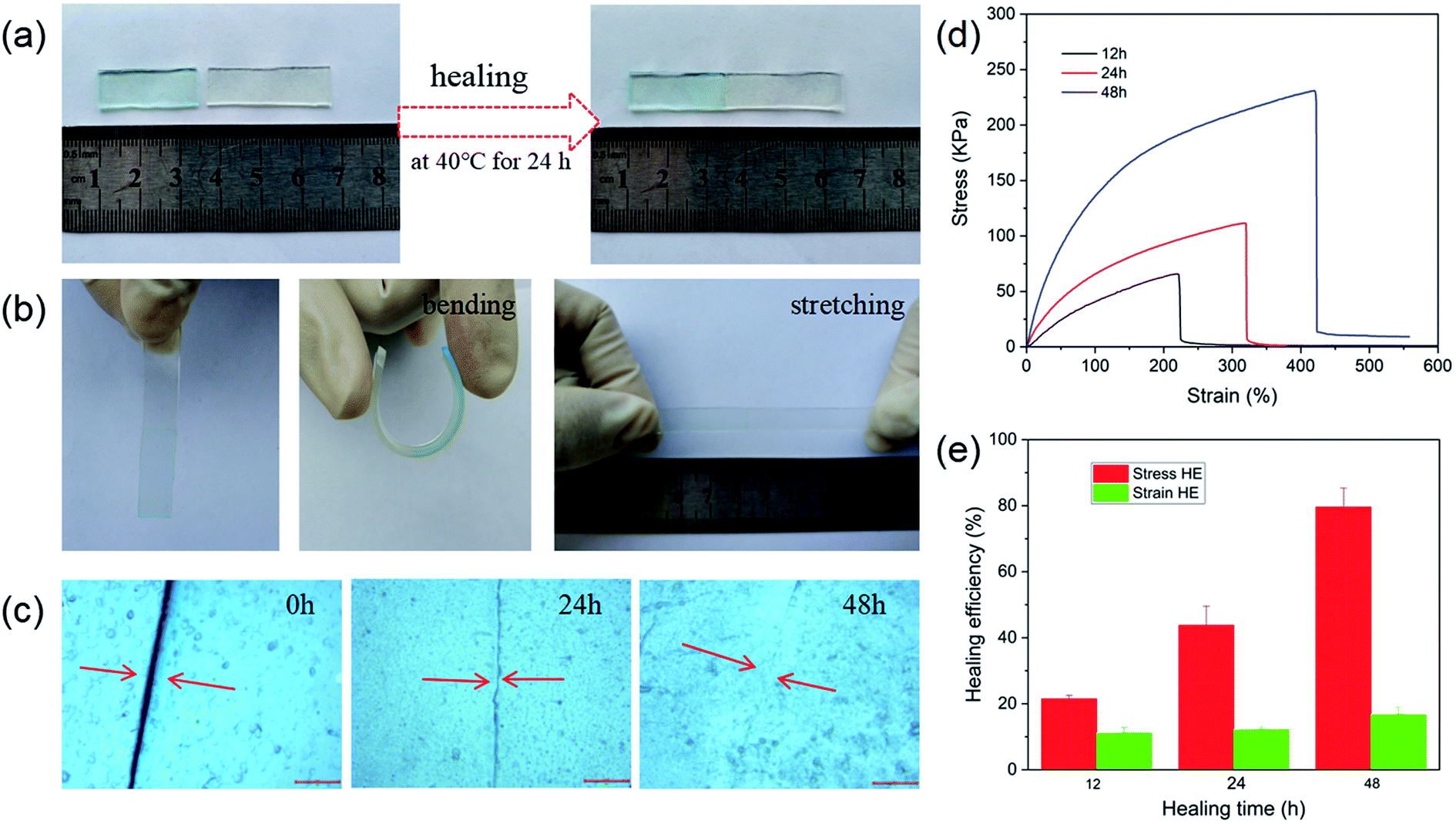 | ||
| Fig. 7 Self-healing properties of NaAlg 5.0%/PAM 2% hydrogel: (a) digital photographs of the process to prepare healed sample; (b) digital photographs of healed sample that can withstand different deformations; (c) optical microscopy images of the sample after being healed for various time; (d) typical stress–strain curves of healed hydrogels; (e) healing efficiency of healed hydrogels. | ||
3.6 Swelling behavior and surface morphology
Fig. 8 shows swelling behavior of NaAlg 5.0%/PAM 2% semi-IPN hydrogel and CaAlg 5.0%/PAM 2% DN hydrogel in pH = 7.4 buffer solution. It is clear in Fig. 8(a) that the swelling ratio of NaAlg/PAM semi-IPN hydrogel is larger than that of CaAlg/PAM DN hydrogel. This further confirms that CaAlg/PAM DN hydrogels have a higher cross-linking density, which is attributed to the metal-coordination between carboxyl anion of alginate and Ca2+. It is observed in Fig. 8(b) that the volume of the sample increases significantly as the pH value of the solution increases, which may be attributed to ionization of hydrophilic COOH groups of hydrogels.52 The influence of the composition on equilibrium swelling ratio of the synthesized hydrogels at various pH buffer solutions (1.5, 7.4 and 12.0) are shown in Fig. 8(c), (d) and S3.† As shown in Fig. 8(c) and S3(a)†, with increasing of the SMA concentration, the equilibrium swelling ratio at similar pH buffer solution decreases. The possible reason is that the increased SMA leads to an increase in the cross-linking density of the PAM network, which limits the swelling of the hydrogel. For CaAlg/PAM DN hydrogels with various SA concentrations (as Fig. 8(d)), its equilibrium swelling ratio in pH 1.5 and 7.4 buffer solutions slight increases as the SA concentration increases from 0% to 5.0%. The possible reason is that the introduction of SA may initially interfere with the PAM network, resulting in the decreasing of physical interactions in the hydrogel. With further increasing of the SA concentration, the equilibrium swelling ratio decreases, which may be assigned to filling up of the void spaces of the network chains by alginate. The similar result has been reported by Samanta, et al.53 In the pH 12.0 buffer solution, the swelling ratio of CaAlg/PAM hydrogels has been increasing as the SA concentration decreases. The physical interactions between the polymer chains in higher pH solution are significantly weakened, so the swelling ratio of the sample is determined by its crosslink density. With increasing of the SA concentration, the alginate network cross-linked by Ca2+ is more denser, resulting in a low swelling ratio. However, NaAlg/PAM hydrogels with various SA concentrations exhibit a different phenomenon from CaAlg/PAM hydrogels, as displayed in Fig. S3(b).† This may be related to their different material structures.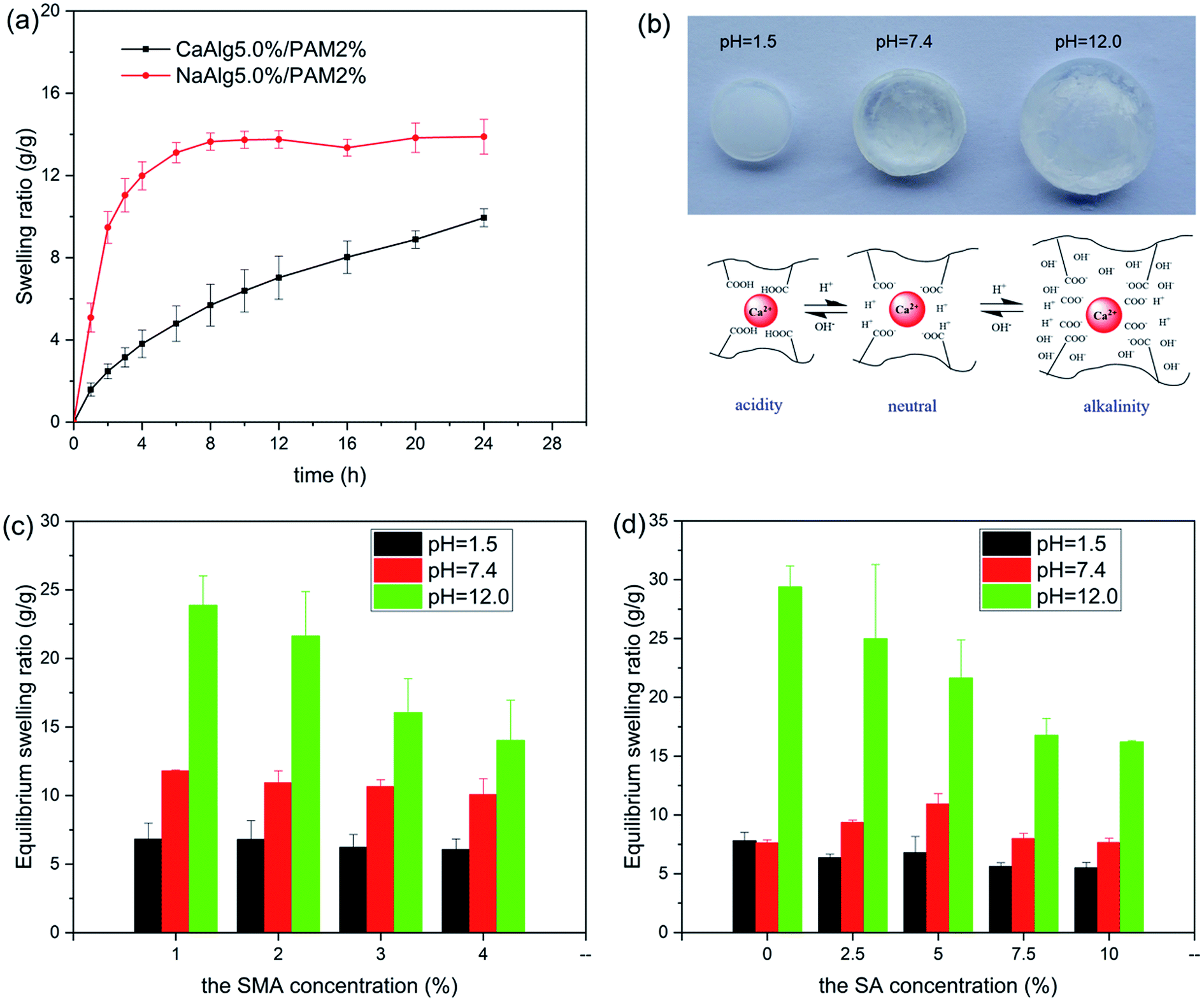 | ||
| Fig. 8 (a) Swelling behaviors of NaAlg/PAM and CaAlg/PAM hydrogels in pH = 7.4 buffer solution; (b) the mechanism of pH-sensitivity swelling behavior of CaAlg/PAM hydrogels; (c and d) equilibrium swelling ratios of CaAlg/PAM hydrogels with different compositions in different buffer solutions: (c) hydrogels with various SMA concentrations, (d) hydrogels with various SA concentrations. | ||
SEM images of hydrogels that reached equilibrium swelling in pH 7.4 buffer solution are displayed in Fig. 9. As displayed in Fig. 9(a) and (b), NaAlg 5.0%/PAM 4% and NaAlg 10.0%/PAM 2% semi-IPN hydrogels show a markedly porous structure and intersected pore channels. However, the corresponding CaAlg/PAM DN hydrogels show a relatively dense surface, as shown in Fig. 9(e) and (f). This may be mainly attributed to the formation of Ca2+ ion cross-linked alginate network. It is clear that the SEM image of CaAlg 5.0%/PAM 4% DN hydrogel presents a honeycomb-like surface, and the pore size clearly reduces with respect to NaAlg 5.0%/PAM 4.0% semi-IPN hydrogel. Compared with NaAlg 10.0%/PAM 2% semi-IPN hydrogel, the pore density of CaAlg 10.0%/PAM 2% DN hydrogel is significantly reduced. This is because CaAlg/PAM DN hydrogels have a higher cross-linking density with respect to the corresponding NaAlg/PAM semi-IPN hydrogels. The results are consistent with the results of swelling test and mechanical performances. Comparing CaAlg 5.0%/PAM 2% (Fig. 9(c)) and CaAlg 5.0%/PAM 4% (Fig. 9(e)) DN hydrogels, it can be found that the sample with higher SMA concentration presents larger pore density. The increased SMA concentration increases the cross-linking density of PAM network, limiting the swelling of the hydrogel. This could result in the reduced pore size and increased pore density. With increasing of the SA concentration, the pore density of the hydrogel decreases, as displayed in Fig. 9(c), (d) and (f). The possible reason is that the increased SA concentration leads to a denser alginate network cross-linked with Ca2+ ions, so that the hydrogel is less swellable, resulting in a decrease in pore density. These results reveal that the surface morphology of the prepared alginate-based hydrogel can be adjusted by the network structure and composition of the hydrogel.
 | ||
| Fig. 9 SEM images of alginate-based hydrogels: (a) NaAlg 5.0%/PAM 4%; (b) NaAlg 10.0%/PAM 2%; (c) CaAlg 5.0%/PAM 2%; (d) CaAlg 0%/PAM 2%; (e) CaAlg 5.0%/PAM 4%; (f) CaAlg 10.0%/PAM 2%. | ||
4. Conclusion
We have successfully proposed a novel strategy to synthesize fully physically cross-linked alginate-based hydrogels with high mechanical strength, toughness and self-recovery capability, which originates from the dynamically reversible non-covalent interactions. These dynamically reversible non-covalent interactions act as sacrificial bonds to dissipate energy during the deformation process. Mechanical properties of alginate-based hydrogels could be regulated through their network structure and composition. CaAlg/PAM DN hydrogels exhibit high toughness with respect to PAM single hydrogel and NaAlg/PAM semi-IPN hydrogel, which may be attributed to the different network structures and the unzipping of coordination interactions between carboxyl anion of alginate and Ca2+. The synthesized alginate-based hydrogels exhibit good mechanical properties, and the CaAlg/PAM DN hydrogels also show fast self-recovery and good fatigue resistance properties. Swelling experiment further demonstrates the formation of coordination interactions between carboxyl anion of alginate and Ca2+. This strategy enriches the exploration of alginate-based hydrogels based on non-covalent interactions, and would expand their applications.Conflicts of interest
There is no conflict of interest.Acknowledgements
This work was supported financially by the Natural Science Foundation of Guangdong Province (2018A030307020), College Youth Innovation Talents Project of Guangdong Province (2017KQNCX089), Science and Technology Plan Project of Zhanjiang City (2019A01006), Project of enhancing school with innovation of Guangdong Ocean University (Q18304), Program for Scientific Research Start-up Funds of Guangdong Ocean University (R19010), National College Student Innovation and Entrepreneurship Training Program (201810566034, CXXL2018034).References
- C. Gao, M. Liu, J. Chen and X. Zhang, Polym. Degrad. Stab., 2009, 94, 1405–1410 CrossRef CAS.
- W. Ding, J. Zhou, Y. Zeng, Y. Wang and B. Shi, Carbohydr. Polym., 2017, 157, 1650–1656 CrossRef CAS PubMed.
- M. A. Abd EI-Ghaffar, M. S. Hashem, M. K. EI-Awady and A. M. Rabie, Carbohydr. Polym., 2012, 89, 667–675 CrossRef PubMed.
- J. Supramaniam, R. Adnan, N. H. M. Kaus and R. Bushra, Int. J. Biol. Macromol., 2018, 118, 640–648 CrossRef CAS PubMed.
- B. Balakrishnan, N. Joshi, A. Jaykrishnan and R. Banerjee, Acta Biomater., 2014, 10, 3650–3663 CrossRef CAS PubMed.
- M. Summ, D. Russo, I. Penna, N. Margaroli, I. S. Bayer, T. Bandiera, A. Athanassiou and R. Bertorelli, Eur. J. Pharm. Biopharm., 2018, 122, 17–24 CrossRef PubMed.
- Y. Ahn, H. Kim and S. Y. Kwak, Eur. Polym. J., 2019, 116, 480–487 CrossRef CAS.
- L. Van den Broeck, S. Piluso, A. H. Soultan, M. D. Volder and J. Patterson, Mater. Sci. Eng., C, 2019, 98, 1133–1144 CrossRef CAS PubMed.
- L. Münster, Z. Capáková, M. Fišera, I. Kuřitka and J. Vícha, Carbohydr. Polym., 2019, 218, 333–342 CrossRef PubMed.
- X. Hu, Y. Wang and M. Xu, Int. J. Biol. Macromol., 2019, 135, 501–511 CrossRef CAS PubMed.
- M. Mandru, M. Bercea, L. M. Gradinaru, C. Ciobanu, M. Drobota, S. Vlad and R. Albulescu, Eur. Polym. J., 2019, 118, 137–145 CrossRef CAS.
- J. Filipecki, M. Sitarz, A. Kocela, K. Kotynia, P. Jelen, K. Filipecka and M. Gaweda, Spectrochim. Acta, Part A, 2014, 131, 686–690 CrossRef CAS PubMed.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449–5455 CrossRef CAS.
- H. Jiang, L. Duan, X. Ren and G. Gao, Eur. Polym. J., 2019, 112, 660–669 CrossRef CAS.
- K. Yasuda, J. P. Gong, Y. Katsuyama, A. Nakayama, Y. Tanabe, E. Konodo, M. Ueno and Y. Osada, Biomaterials, 2005, 26, 4468–4475 CrossRef CAS PubMed.
- G. Du, L. Nie, G. Gao, Y. Sun, R. Hou, H. Zhang, T. Chen and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 3003–3008 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- Q. Chen, D. Wei, H. Chen, L. Zhu, C. Jiao, G. Liu, L. Huang, J. Yang and L. Wang, Macromolecules, 2015, 48, 8003–8010 CrossRef CAS.
- C. W. Chang, A. van Spreeuwel, C. Zhang and S. Varghese, Soft Matter, 2010, 6, 5157–5164 RSC.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- Y. Okumuar and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zhang, Adv. Funct. Mater., 2015, 25, 1598–1607 CrossRef CAS.
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- H. Chen, Y. Liu, B. Ren, Y. Zhang, J. Ma, L. Xu, Q. Chen and J. Zheng, Adv. Funct. Mater., 2017, 27, 1703086 CrossRef.
- Q. Chen, X. Yan, L. Zhu, H. Chen, B. Jiang, D. Wei, L. Huang, J. Yang, B. Liu and J. Zheng, Chem. Mater., 2016, 28, 5710–5720 CrossRef CAS.
- A. Andersen, M. Krogsgaard and M. Birkedal, Biomacromolecules, 2018, 19, 1402–1409 CrossRef CAS PubMed.
- N. Yuan, L. Xu, H. Wang, Y. Fu, Z. Zhang, L. Liu, C. Wang, J. Zhao and J. Rong, ACS Appl. Mater. Interfaces, 2016, 8, 34034–34044 CrossRef CAS PubMed.
- B. Zhang, Z. Gao, G. Gao, W. Zhao, J. Li and X. Ren, Macromol. Mater. Eng., 2018, 303, 1800072 CrossRef.
- J. Li, W. R. K. Illeperuma, Z. Suo and J. J. Vlassak, ACS Macro Lett., 2014, 3, 520–523 CrossRef CAS.
- M. Ionita, M. A. Pandele and H. Lovu, Carbohydr. Polym., 2013, 94, 339–344 CrossRef CAS PubMed.
- J. Wang, C. Liu, Y. Shuai, X. Cui and L. Nie, Colloids Surf., B, 2014, 113, 223–229 CrossRef CAS PubMed.
- B. Huang, R. Hu, Z. Xue, J. Zhao, Q. Li, T. Xia, W. Zhang and C. Lu, Carbohydr. Polym., 2020, 231, 115736 CrossRef PubMed.
- X. Su and B. Chen, Carbohydr. Polym., 2018, 197, 497–507 CrossRef CAS PubMed.
- Y. Fei, Y. Li, S. Han and J. Ma, J. Colloid Interface Sci., 2016, 484, 196–204 CrossRef CAS PubMed.
- S. K. Tam, J. Dusseault, S. Polizu, M. Ménard and J. P. Hallé, Biomaterials, 2005, 26, 6950–6961 CrossRef CAS PubMed.
- R. Bahulekar, T. Tokiwa, J. Kano, T. Matsumura, I. Kojima and M. Kodama, Carbohydr. Polym., 1998, 37, 71–78 CrossRef CAS.
- S. Rudenja, N. Zhao and S. Liu, Eur. Polym. J., 2010, 46, 2078–2084 CrossRef CAS.
- A. Dixt, D. S. Bag and S. J. S. Kalra, Polymer, 2017, 119, 263–273 CrossRef.
- C. H. Yang, M. X. Wang, H. Haider, J. H. Yang, J. Y. Sun, Y. M. Chen, J. Zhou and Z. Suo, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422 CrossRef CAS PubMed.
- J. Chen, R. An, L. Han, X. Wang, Y. Zhang, L. Shi and R. Ran, Mater. Sci. Eng., C, 2019, 99, 460–467 CrossRef CAS PubMed.
- W. Zhang, J. Hu, J. Tang, Z. Wang, J. Wang and T. Lu, ACS Macro Lett., 2019, 8, 17–23 CrossRef CAS.
- D. S. Jones, G. P. Andrews, D. L. Caldwell, C. Lorimer, S. P. Gorman and C. P. McCoy, Eur. J. Pharm. Biopharm., 2012, 82, 563–571 CrossRef CAS PubMed.
- Y. Hu, Z. Du, X. Deng, T. Wang, Z. Yang and W. Zhou, Macromolecules, 2016, 49, 5660–5668 CrossRef CAS.
- X. Li, Q. Yang, Y. Zhao, S. Long and J. Zheng, Soft Matter, 2017, 13, 911–920 RSC.
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed.
- Y. Tao, R. Zhang, W. Xu, Z. Bai, Y. Zhou, S. Zhao, Y. Xu and D. Yu, Food Hydrocolloids, 2016, 52, 923–933 CrossRef CAS.
- X. J. Yang, P. Zhang, P. Li, Z. Li, W. Xia, H. Zhang, Z. Di, M. Wang, H. Zhang and Q. J. Niu, J. Mol. Liq., 2019, 280, 128–134 CrossRef CAS.
- K. Hou, Y. Li, Y. Liu, R. Zhang, B. S. Hsiao and M. Zhu, Polymer, 2017, 123, 55–64 CrossRef CAS.
- S. Anjum, P. Gurave, M. V. Badiger, A. Torris, N. Tiwari and B. Gupta, Polymer, 2017, 126, 196–205 CrossRef CAS.
- Z. Jing, Q. Zhang, Y. Q. Liang, Z. Zhang, P. Hong and Y. Li, Polym. Int., 2019, 68, 1710–1721 CrossRef CAS.
- Z. Jing, A. Xu, Y. Q. Liang, Z. Zhang, C. Yu, P. Hong and Y. Li, Polymers, 2019, 11, 952 CrossRef CAS PubMed.
- P. Mukhopadhyay, K. Sarkar, S. Bhattacharya, A. Bhattacharyya, R. Mishra and P. P. Kundu, Carbohydr. Polym., 2014, 112, 627–637 CrossRef CAS PubMed.
- H. S. Samanta and S. K. Ray, Carbohydr. Polym., 2014, 99, 666–678 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03733h |
| This journal is © The Royal Society of Chemistry 2020 |