
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-less amperometric sensor manufactured using a Nafion–LaNiO3 nanocomposite for hydrogen peroxide†
Elahe Ahmadia,
Mohammad Bagher Gholivand *b and
Changiz Karamic
*b and
Changiz Karamic
aDepartment of Chemistry, Kermanshah Branch, Islamic Azad University, Kermanshah, Iran
bAnalytical Chemistry Department, Chemistry Faculty, Razi University, Kermanshah, Iran. E-mail: mbgholivand2013@gmail.com; Fax: +98 833 4274559; Tel: +98 833 4274557
cNano Drug Delivery Research Center, Kermanshah University of Medical Sciences, Kermanshah, Iran
First published on 19th June 2020
Abstract
In the present study, an enzyme-less amperometric sensor based on Nafion (NF) and a LaNiO3 (LNO) nanocomposite was constructed for H2O2 detection. LNO from the perovskite group was mixed with NF as an effective solubilizing and stabilizing agent that was used as a novel modifier for modification of the glassy carbon electrode (GCE). The designed sensor showed a desirable electrocatalytic response toward H2O2 reduction. The calibration curve revealed two linear portions in the concentration ranges of 0.2–50 μM and 50–3240 μM, and the detection limit was 0.035 μM. The accuracy of the interference-free sensor was checked by recovery analysis in serum samples.
1. Introduction
Hydrogen peroxide (H2O2) is known as a chemical threat to the environment. Furthermore, H2O2 has a great effect on the development of many diseases as well as the reactions of enzyme generation.1,2 Therefore, detection and analysis of H2O2 matter a lot in different fields, including environmental,3 pharmaceutical,4 food industry,5,6 and clinical diagnostics.7 Reliable analysis of H2O2 has been extensively done using chromatographic,8 spectrophotometric,9 and electrochemical10–12 techniques. Electroanalytical methods as low-cost techniques that are simple, selective, and sensitive with short analysis time have found a wider range of uses. The electrochemical monitoring of H2O2 can be done through enzymatic and nonenzymatic approaches. Although enzymatic electrochemical sensors have outstanding performance in terms of selectivity and sensitivity, their production is often complex and costly, they have a limited lifetime, and the response of these designed sensors depends on environmental conditions such as pH and temperature.13 So, to eliminate these disadvantages, enzyme-less biosensors can be used as a detection option. Several substances such as metal and metal oxide/hydroxide,14,15 metal complex,16 nanoparticles (NPs),17 polymeric films,18 and Prussian blue (PB)19 have been employed to construct non-enzymatic H2O2 sensors.Perovskite-type oxides as mixed metal oxides with unique properties such as good thermal stability, porosity, ion exchangeability, catalytic potential, high surface-volume ratio, and redox properties20 can be a candidate as an electrode modifier. These compounds have been utilized as catalysts for fuel cells,21 batteries,22 and also sensing materials.23 LaNiO3 (LNO) is one of these mixed metal oxides which its activity has been theoretically considered for both oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) among the perovskite-type oxides.24,25 Also, the catalytic activity of LNO toward H2O2 reduction reaction (HPRR), with a potential application in the cathode side of fuel cells, has been studied by Amirfakhri et al.26 LNO nanoparticles (NPs) have a phase with high purity, petite size (typically about 30–40 nm), and very large and active surface area (about 22 m2 g−1).27 These properties encouraged us to use this mixed oxides as an electrode modifier for H2O2 detection.
In the present research, the porous LNO was synthesized in the presence of zinc nitrate by sol–gel procedure, and then a nanocomposite consisting of NF and synthesized porous LNO was utilized for modification of the glassy carbon electrode (GCE) and construction of a new sensor for H2O2 analysis. The electrocatalytic activity of LNO enhanced the amperometric signal of the analyte and created a sensor with higher sensitivity and selectivity. The electrode has remarkable stability, so that during this work, only one electrode was used. The applicability of the sensor was checked by monitoring of H2O2 in serum samples.
2. Experiential
2.1. Reagents and chemicals
The whole chemical and reagents consumed in this study were acquired as an analytical grade and applied without additional purification. Hydrogen peroxide (H2O2), ethylene glycol (C2H4(OH)2), lanthanum nitrate salt (La(NO3)2·6H2O), zinc nitrate salt (Zn (NO3)2·6H2O), nickel nitrate salt (Ni (NO3)2·6H2O), citric acid (C6H8O7) and ammonium chloride (NH4Cl) were gained from Merck Co. (Darmstadt, Germany). Nafion was purchased from Fluka. The total aqueous solutions were prepared using double-distilled water. Sodium hydroxide (NaOH, 0.1 M) solution was utilized as a supporting electrolyte. The whole of the experiments was performed at ambient temperature (25 ± 1 °C).2.2. Equipment
A typical system comprising of the three-electrode cell was utilized, that was consist of bare GCE (Azar electrode, 2 mm diameter) or modified GCEs as working, a platinum wire as a counter and a silver/silver chloride (Ag/AgCl) in saturated potassium chloride as reference electrodes. Electrochemical impedance spectroscopy (EIS), amperometry, and cyclic voltammetry (CV) techniques were done via a potentiostat and galvanostat (Autolab, PGSTAT 302N, Netherlands) with FRA 2 impedance analysis module and the NOVA 1.11 software. The pH measurements were carried out by a Metrohm pH meter (model 780). The synthesized mixed metal oxides and electrode modifiers were characterized by an X-ray diffractometer (XRD, Phillips PW 1730), transmission electron microscopy (TEM, PHILIPS CM200 FEG) and field emission scanning electron microscopy (FE-SEM) (MIRA Ш model, TESCAN Co., Czech). Also, for appraising of the LNO surface area, the BET procedure was applied using of BEL (BELSORP MINI II, Japan) apparatus.2.3. Preparation and characterization of LaNiO3
The reported sol–gel procedure28 was utilized for LNO synthesis. In the beginning, the ethylene glycol along with citric acid was dissolved in distilled water, while the solution was stirring vigorously. Following that, the nickel nitrate salt was appended to this solution with molar ratios of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 citric acid, ethylene glycol, and nickel nitrate, respectively. After dissolving the nickel nitrate salt, the stoichiometric quantity of lanthanum nitrate salt was added to this solution. Finally, the solution was leisurely stirred for 12 h at 60 °C to convert the solution to a high viscosity gel. After the formation of a gel, zinc nitrate salt was used in the next step to create the LNO with prose structure.
:1:1 citric acid, ethylene glycol, and nickel nitrate, respectively. After dissolving the nickel nitrate salt, the stoichiometric quantity of lanthanum nitrate salt was added to this solution. Finally, the solution was leisurely stirred for 12 h at 60 °C to convert the solution to a high viscosity gel. After the formation of a gel, zinc nitrate salt was used in the next step to create the LNO with prose structure.
Thus Zn(NO3)2 was introduced into the precursor after the gel formation step. During calcination, Zn(NO3)2 was changed into ZnO. For dissolving and etching of ZnO from LNO perovskite structure, an ammonium chloride solution (2 M) under vigorous agitation was used. After etching of ZnO for 2 h, and washing with deionized water it was finally re-calcined at 800 °C for 2 h for stabilization of the catalyst structure.
The identification of prepared LNO NPs was made by XRD and TEM methods. The X-ray diffraction method was applied to recognize the phases in the LNO compound. Fig. 1A demonstrates the configuration of a rhombohedra structure which was confirmed with comparatively sharp peaks at the 2θ = 33°, 47°, 59°.28 Nevertheless, in the XRD patterns, minor amounts of nickel oxide and zinc oxide compounds were observed. The results were consistent with the results presented in previous studies.28,29
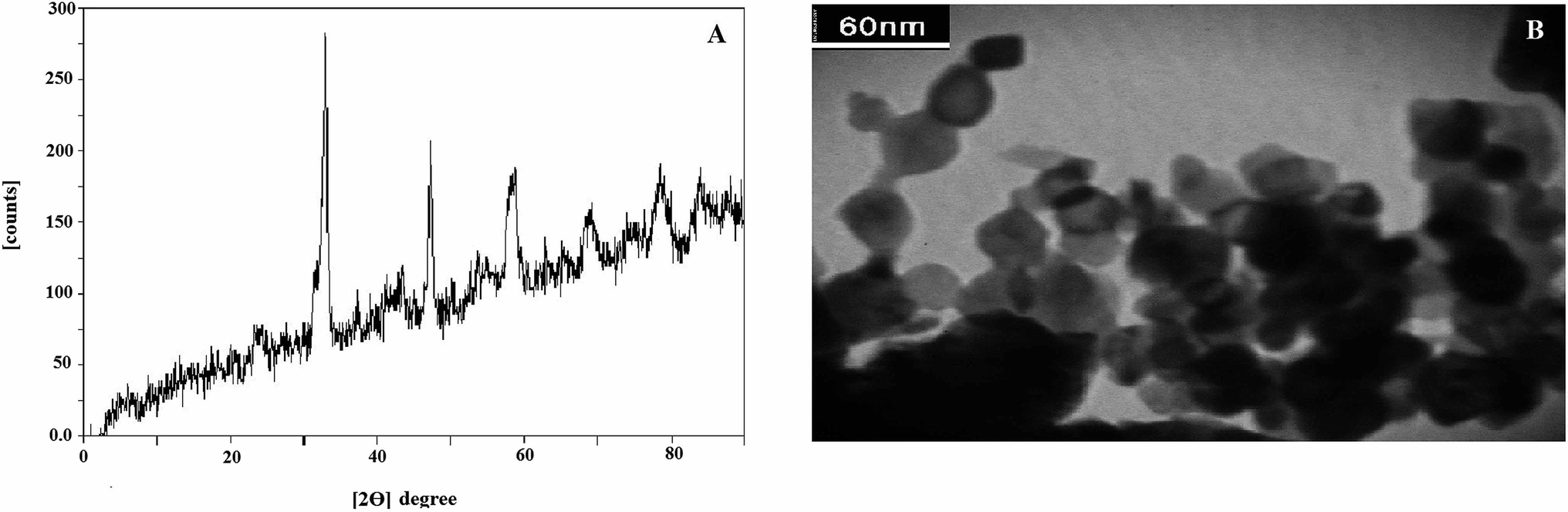 | ||
| Fig. 1 The XRD pattern (A) and tem form (B) of synthesized LNO. | ||
The TEM method was used for the evaluation of prepared LNO NPs. The acquired TEM image in Fig. 1B indicates an identical structure for synthesized LNO NPs that particles have an almost spherical shape. It also appears that the spherical particles are agglomerated and have a mean particle size of between the 20–40 nm.
Also, the surface area of this compound was estimated by the BET method and found to be almost 9 m2 g−1. According to previous research on the LNO structure, there is a claim that adding Zn with an efficient ionic radius of 0.75 A in the etching step leads to the creation of artificial holes in the final structure of LNO compound, which ultimately increases the active surface of the nanoparticle.
2.4. Construction of NF-LNO/GCE
In the beginning, the bare GCE was cleaned with alumina slurry (0.05 μm) as long as a mirror-like surface was achieved. Afterward, the cleaned electrode was washed using ethanol and double-distilled water in an ultrasonic bath, consecutively. The NF-LNO homogenous solution was prepared with an appending of 4 mg LNO NPs to 1 mL NF solution (0.3%) and was ultrasonicated for 2 h. Subsequently, 5 μL of the prepared nanocomposite was cast on the surface of the cleaned electrode and was dried at room temperature. The fabricated electrode was considered as NF-LNO/GCE. Before each experiment, electrolyte solutions were purged with high purity nitrogen for 5 min to remove dissolved oxygen, and then a nitrogen atmosphere was kept over the solution during measurements.To optimize the NF and LNO values as ingredients of the sensor one variable at a time was utilized. On the other hand, the amount of one of these compounds was kept constant, and the other changed to give the best signal. The NF and LNO amounts varied in the range of 0 to 5% and 2 to 6 mg mL−1 respectively, the results showed that the best response was achieved with optimal amounts of 0.3% NF and 4 mg mL−1 LNO.
3. Result and discussion
3.1. Characterization of nanocomposite
The morphology of the modified electrodes surface was evaluated by SEM and their images were depicted in Fig. 2. The bare GC substrate is featureless after polishing and sonication processes (Fig. 2A). While SEM image of NF-LNO/GCE, Fig. 2B shows a homogeneous distribution of spherical nanocomposite at the electrode surface.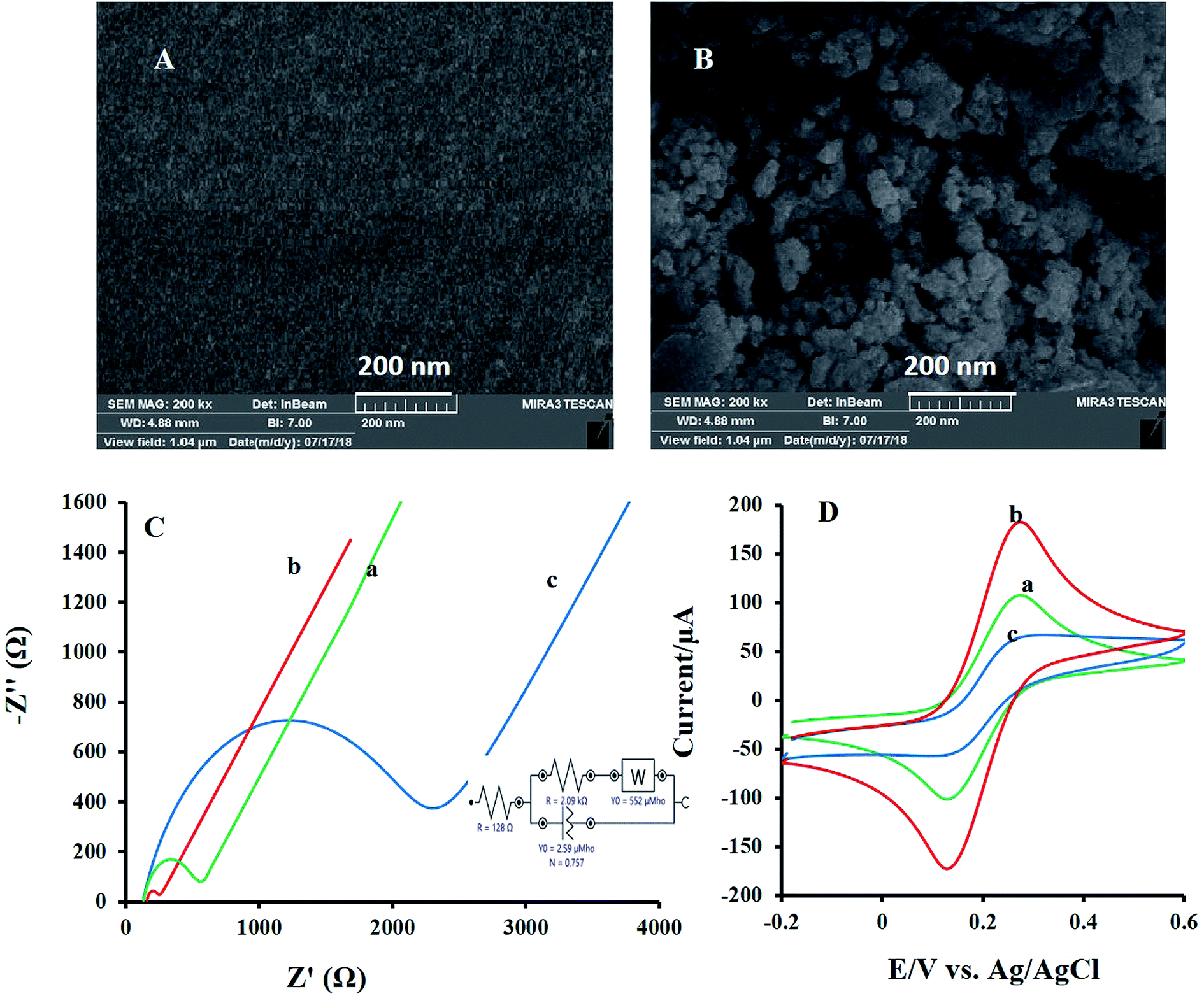 | ||
| Fig. 2 The SEM forms of the bare GCE (A) and NF-LNO/GCE (B). (C) The nyquist plots of (a) GCE, (b) LNO/GCE and (c) NF-LNO/GCE in the solution of KCl (1 M) and K3[Fe(CN)6] (5 mM) with EIS conditions: initial potential, 0.15 V and frequency range, 100 kHz to 0.1 Hz. Inset: an equipollent circuit utilized for modeling of impedance data in the attendance couple of redox. (D) The CVs of the (a) GCE, (b) LNO/GCE, and (c) NF-LNO/GCE in KCl (1 M) and K3[Fe(CN)6] (5 mM); scan rates: 50 mV s−1. | ||
Furthermore, for describing the surface features during the modification of the bare electrode, electrochemical impedance spectroscopy (EIS) was applied. In EIS measurements, mainly [Fe(CN)6]3−/4− was used as a probe. In these measurements, the semicircle diameter of impedance is equal to the electron transfer resistance (Rct), which controls the electron transfer kinetics of the redox probe on the electrode surface. Fig. 2C introduces the Nyquist plots of the bare GCE (curve a), LNO/GCE (curve b), and NF-LNO/GCE (curve c) electrodes in a solution containing [Fe(CN)6]3−/4− (5 mM) and KCl (0.1 M). Nyquist plot of the unmodified electrode (curve a) gives a semicircle domain with Rct about 454 Ω. The shape of the Nyquist plots of LNO/GCE (curve b) and NF-LNO/GCE (curve c) are similar as that of an unmodified GCE, but with different diameters. The Rct, about 80.2 Ω and 2 kΩ, were achieved for LNO/GCE and NF-LNO/GCE, respectively. The enhancement in Rct value at the NF-LNO/GCE (curve b) is due to the presence of NF in the modifier and repulsive force between probe and NF with negative charge surface.30 Also, the utilized circuit for fitting the EIS data is presented in the inset of Fig. 2C. In addition to the EIS technique, the cyclic voltammograms of both electrodes were also acquired in the ([Fe(CN)6]3−/4−) solution (Fig. 2D). However, at the surface of unmodified and modified electrodes in the presence of ferrocyanide-ferricyanide redox couple, a pair of redox peaks were discerned. Based on the observed results, the peak currents of the redox couple at the surface of the modified electrode (b) were significantly decreased relative to that of the bare electrode. These results confirmed the EIS founding.
3.2. The behavior of H2O2 at the prepared sensor in the different supporting electrolytes
In the initial step of experiments, the electrochemical behavior of H2O2 was investigated at the surface of the constructed sensor in several supporting electrolyte. So, the effect of 0.1 M of each electrolyte, such as phosphate, ammonium, and NaOH, was tested, and based on the cyclic voltammetric outcomes of H2O2, the best results were observed in sodium hydroxide solution (0.1 M). As a result, 0.1 M of NaOH was selected as the finest supporting electrolyte for electroanalysis experiments.3.3. Electrocatalytic reduction of H2O2 at NF-LNO/GCE
For the study, the electrocatalytic role of mixed metal oxides to the detection of H2O2, the electrochemical signal of H2O2 at the bare GCE, NF/GCE, LNO/GCE, and NF-LNO/GCE was evaluated by CV. Fig. 3 shows the CV responses of 0.1 M NaOH solution containing 10 μM H2O2 at the modified and unmodified electrodes. Before injecting of H2O2, the solution was purged with pure nitrogen for oxygen removal. At the bare GC electrode (curve b) as well as NF/GCE (curve c) in the presence of H2O2, an ill cathodic peak was observed, which refuse the application of GCE, NF/GCE as a suitable electrode for hydrogen peroxide detection. Using the LNO/GCE (curve d) or NF-LNO/GCE (curve e) in NaOH solution in the presence of H2O2, the cathodic peak was remarkably increased at the tested electrodes which show the electrocatalytic activity of LNO for analyte reduction, while the NF-LNO/GCE doesn't show any peak in the absence of H2O2 (curve a). On the other hand, the electrocatalytic reduction signal of H2O2 was moderately altered to a better shape at the surface of NF-LNO/GCE (curve e) with a small shift in the peak potential toward positive values. In these experiments, LNO did not have excellent stability at the surface of the electrode alone, so for more stability, this catalyst nanoparticle was mixed with the NF polymer.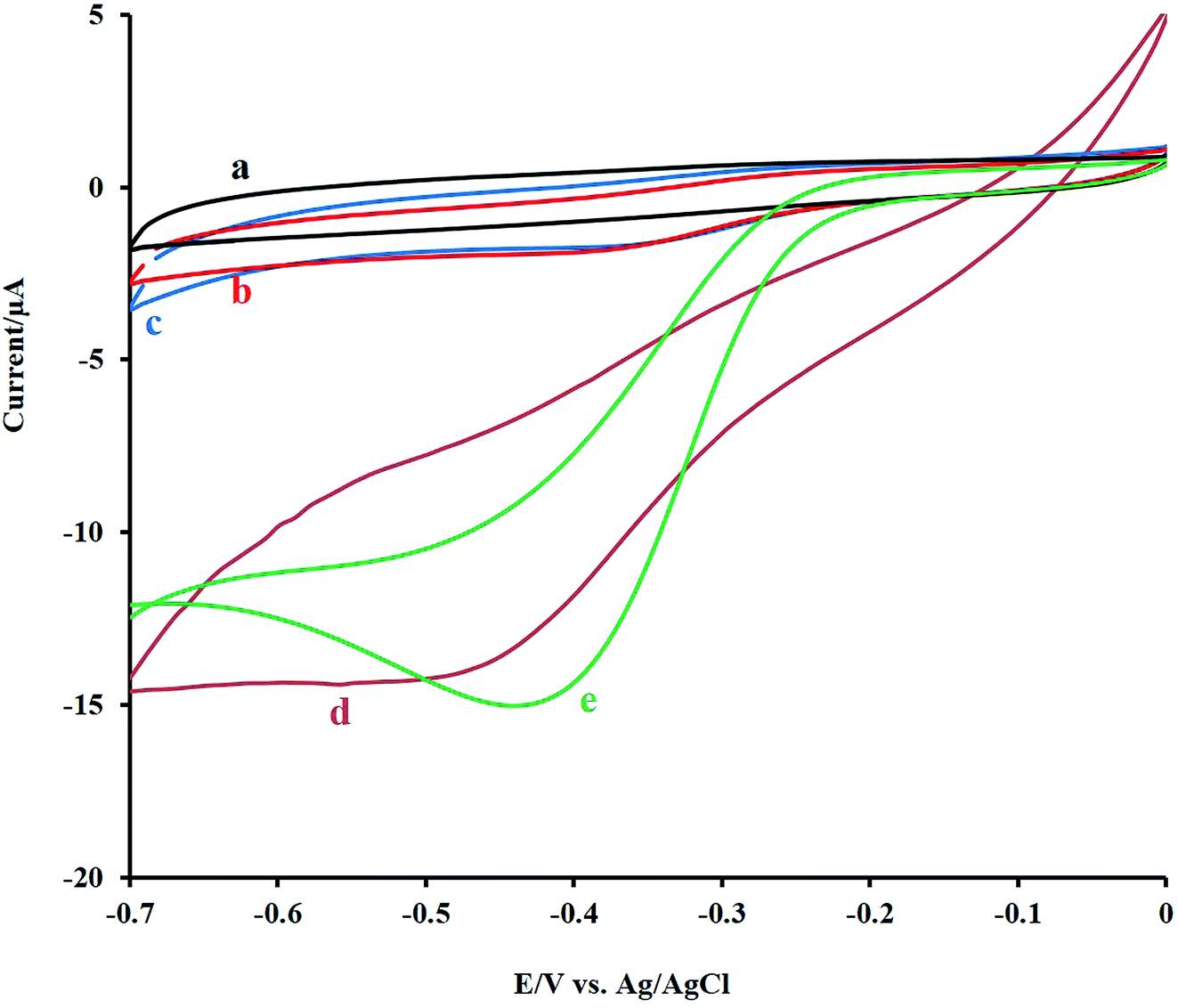 | ||
| Fig. 3 The CV responses of the bare GCE (b), NF/GCE (c), LNO/GCE (d) and NF-LNO/GCE (e) electrodes in 0.1 M NaOH solution containing 10 μM of H2O2 and (a) the CV responses of the NF-LNO/GCE in the absence of H2O2. All of CVs were recorded at a scan rate of 50 mV s−1. | ||
Fig. 4A demonstrates the CVs of 10 μM H2O2 at various sweep rates from 5 to 110 mV s−1. As can be seen, with increasing the sweep rates, the peak currents were increased. The peak intensity was Plotted versus the square root of the scan rate and resulted in a linear line (Fig. 4B), which reveals that the electrodic reaction is a diffusion-controlled process. Also, there was a linear relationship between the logI and logν with a slope of 0.21, which confirms the diffusion control of the electrode mechanism (Fig. 4C).
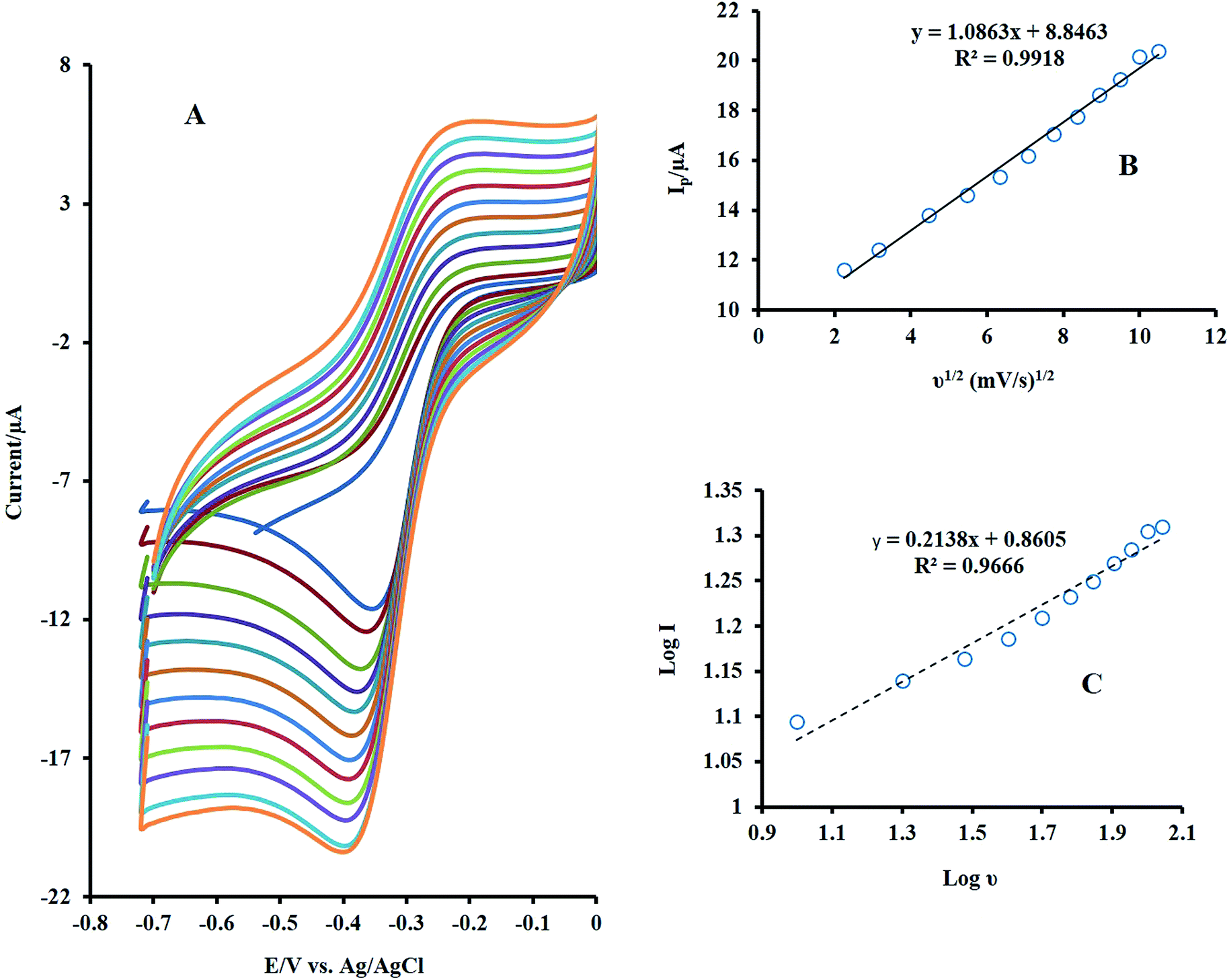 | ||
| Fig. 4 (A) Effect of scan rate on the CVs of NF-LNO/GCE in 0.1 M NaOH solution containing 10 μM H2O2 at various sweep rates from 5 to 110 mV s−1 (B) plot of Ip vs. ν1/2, (C) plot of log Ip vs. logν. | ||
3.4. Amperometric monitoring of H2O2
The dependence of the sensor response to H2O2 concentration was checked by the CV method, and its outcome is presented in Fig. 5. As shown, by increasing the target concentration, the peak current was enhanced linearly in the range of 4 to 30 μM.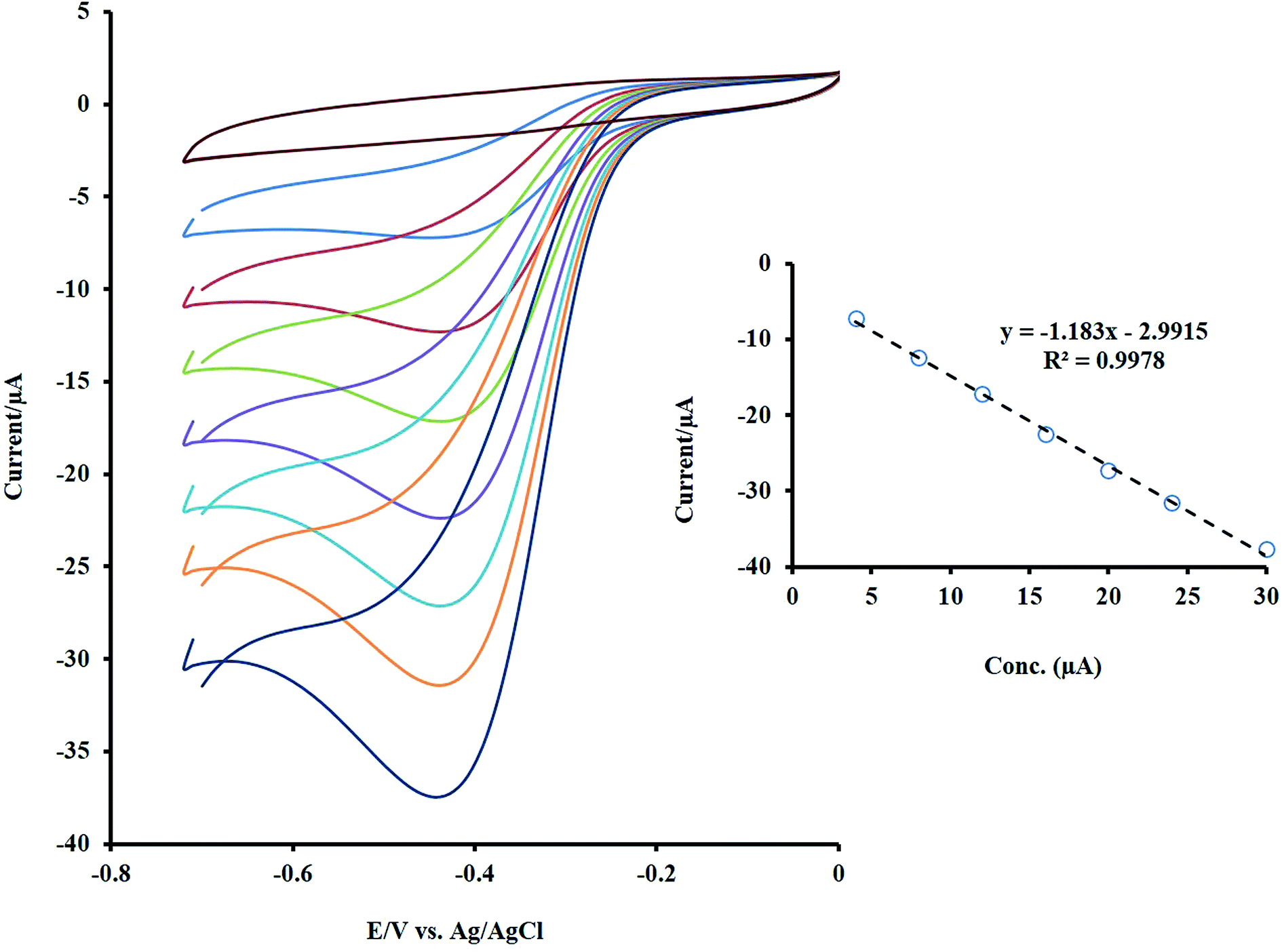 | ||
| Fig. 5 The CVs of NF-LNO/GCE in 0.1 M NaOH solution containing different concentrations of the H2O2 (between 4 to 30 μM). Inset: the linear trend of changes in the Ip with concentration. | ||
However, to assess the higher sensitivity and lower detection limit for the designed sensor, the amperometric method under stirring as a sensitive technique was utilized for H2O2 detection. At first, the applied constant potential for amperometry was selected. The response of the modified electrode was recorded at operating potential ranging from −0.3 to −0.7 V in the presence of a constant amount of H2O2 (Fig. 1S†). The best results, highest signal, was obtained at −0.5 V. The amperometric response of rotating NF-LNO/GCE was obtained by a consecutive appending of H2O2 in the wide range of concentration into the 10 mL stirring solution (1200 rpm) of NaOH 0.1 M at an applied potential of −0.5 V (Fig. 6A and B). As shown during the successive addition of analyte, a well-defined signal was achieved, demonstrating the stable and efficient catalytic ability of the LNO presented in the composite film at the GCE. The calibration graphs of H2O2 at the NF-LNO/GCE are linear in two concentration ranges of 0.2–50 μM and 50–3240 μM with correlation coefficients greater than 0.996 (insets of Fig. 6) and the detection limit of 3.5 nM was acquired based on the signal-to-noise ratio of 3. The linear ranges of the proposed sensor and its detection limit were compared with the analytical parameters of other recently reported sensors, and the results are summarized in Table 1.17,31–45 According to the collected data in Table 1, the constructed sensor exhibits a comparable and even better linear range and detection limit.
 | ||
| Fig. 6 Amperometric measurements of the H2O2 at the rotating designed sensor (1200 rpm) in 0.1 M NaOH solution for successive addition of various amounts of H2O2 between 0.2–50 μM (A) and 50–3240 μM (B) at −0.5 V. Insets of (A and B): calibration curve based on the two various ranges of concentration evaluation. | ||
| Electrode | Linear range (μM) | LOD (μM) | Conditions | Real sample | References |
|---|---|---|---|---|---|
| a Screen printed electrode.b Inkjet-printed Ag electrode under the optimized 20 printed layers.c Reduced graphene oxide.d Carbon paste electrode.e Fe3O4@nitrogen-doped mesoporous carbon microcapsules core/shell composites.f Ferrocene/thionin bimediator/paraffin wax impregnated graphite electrode.g Graphene quantum dots-chitosan/methylene blue.h Reduced graphene oxide nanoribbons.i Prussian blue.j Activated pencil graphite electrodes.k Graphitic carbon nitride/ZnO nanosheets/fluorine-doped tin oxide electrode. | |||||
| Ag–Co/MWCNT/GCE | 50–10000 |
0.74 | PBS (pH = 7.5) | — | 17 |
| CuO/rGOc/Cu2O/Cu foil electrode | 0.5–8300 | 0.10 | PBS (pH = 7.4) | — | 31 |
| Graphene-chitosan/SPEa | 20–60000 |
5 | PBS (pH = 7) | — | 32 |
| g-C3N4/ZnO/FTO electrodek | 50–14150 |
1.7 | PBS (pH = 7.4) | Drinking water, tape water, serum | 33 |
| PBi/MoS2-rGO/GCE | 0.3–1150 | 0.14 | PBS (pH = 6.03) | Tap water | 34 |
| River water | |||||
| MnO2/rGONRsh/GCE | 0.25–2245 | 0.071 | PBS (pH = 7.4) | Fetal bovine serum | 35 |
| Fc-TH bimediator/PIGEf | 0.569–785 | 0.19 | PBS (pH = 7) | Fruit juice | 36 |
| IPAgE-20Lb | 100–6800 | 5 | PBS (pH = 7.4) | — | 37 |
| CuO/APGEj | 5–1600 | 0.21 | NaOH 0.1M | Milk | 38 |
| GO–MgO–Al2O3–nafion/GCE | 0.01–0.05 | 0.005 | PBS (pH = 7) | Hair dye solution | 39 |
| Fe3O4@NMCMse/GCE | 50–33000 |
5.9 | PBS (pH = 7) | Milk | 40 |
| rGO/CuFe2O4/CPEd | Amperometry 2–200 | 0.52 | PBS (pH = 5) | Milk, green tea | 41 |
| DPV 2–1000 | 0.064 | Hair dye cream | |||
| Mouthwash solution | |||||
| CuFe2O4/nickel foam electrode | 500–25000 |
22 | NaOH 0.1M | — | 42 |
| Nafion/Pt NPs/rGO/GCE | 5–3000 | 0.4 | PBS (pH = 7) | Fetal bovine serum | 43 |
| GQDs-CS/MBg/GCE | 1–11780 |
0.7 | PBS (pH = 7.4) | Honey, pineapple juice, tap water, spring water | 44 |
| polyaniline/Cu/GCE | 1.0–500 | 0.33 | PBS (pH = 6) | Tap water | 45 |
| NF-LNO/GCE | 0.2–50 | 0.035 | NaOH 0.1M | Serum | This work |
| 50–3240 | |||||
3.5. Evaluation of sensor stability and reproducibility
The repeatability of the proposed electrode was tested by 100 times recording the cyclic voltammograms (CVs) of 10 μM of H2O2 in NaOH 0.1 M at a scan rate of 50 mV s−1, and the results showed that the last voltammogram had a 5% decrease compared to the first one. This reveals the good repeatability and stability of the designed electrode.The stability of the designed sensor was also evaluated by amperometry methods. For 2 times, 10 μM of H2O2 was added to the NaOH solution (0.1 M), and the amperometric signal was recorded for a long time (about 500 s), the experiment was followed by adding the second injection (10 μM of H2O2). As shown in Fig. 7, no significant change was observed during this time. The initial activity of the mentioned sensor, which was exposed to air at ambient temperature, was preserved for more than one month. After one month, the initial response of this sensor was reduced by 3%. These findings reveal the excellent stability of the electrode for a long time. For evaluation of the sensor reproducibility, five individual electrodes were fabricated, and each was used for monitoring of H2O2 with known concentration using CV. The results depict a standard deviation of less than 2% for peak currents. Therefore, the obtained results indicated that the constructed senor (NF-LNO/GCE) has acceptable reproducibility.
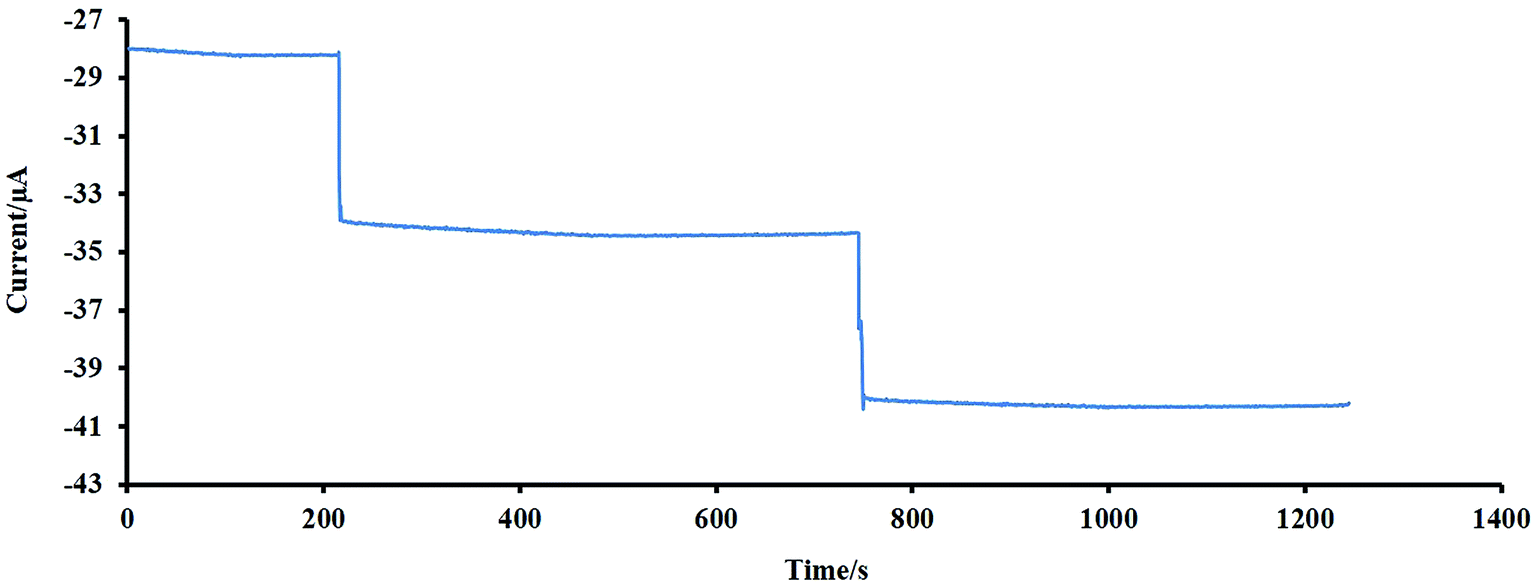 | ||
| Fig. 7 An investigation of the stability of the designed sensor using an amperometry method. | ||
3.6. Evaluation of real sample
To test the applicability of the sensor, determination of H2O2 in serum samples was carried out. After the usual steps of preparation as previous works,46 the serum samples were diluted 10 times, with 0.1 M NaOH as supporting electrolyte. The standard addition method was employed for evaluating the recoveries of the spiked H2O2 with different concentrations (Table 2). Based on these data, the recoveries of the analyte in real spiked samples are admissible. So the fabricated sensor could be effectively applied for the analysis of H2O2 in real samples accompanied by various matrices.| Sample | Added (μM) | Founded (μM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 1 | 10 | 9.7 | 97 | 3.92 |
| 2 | 60 | 58 | 96.6 | 2.57 |
| 3 | 600 | 625 | 104.1 | 3.45 |
3.7. Investigation of the effect of some important interferences
H2O2 as a remarkable material in medical, clinical and biological samples has great importance; thus the effect of some cations, anions and available compounds in physiological samples with electroactivity properties on the current response of the NF-LNO/GCE to successive addition of 10 μM H2O2 and different amounts of interfering species under optimal experimental conditions were evaluated, and the results are presented in Fig. 8. As shown, the interfering compounds including sucrose (Suc, 1 mM), glucose (Glu, 1 mM), arabinose (Ara, 1 mM), alanine (Ala, 1 mM), urea (Ure, 1 mM), citric acid (C.A., 1 mM), acetaminophen (Ac, 500 μM), histidine (Hys, 500 μM), arginine (Arg, 100 μM), L-cysteine (Cys, 100 μM) and fructose (Fru, 1 M) have no significant interference effect on H2O2 signal. According to Fig. 8, the current–time responses of these compounds are significantly lower than that of H2O2, which illustrates that there is no apparent signal related to the mentioned interfering materials in the detection of H2O2 and also indicating the high selectivity of this sensor. | ||
| Fig. 8 An investigation of the effect of some interfering substances (demonstrated by arrows) at the rotating designed sensor in the presence of H2O2 (10 μM). | ||
4. Conclusion
In this research, a new and sensitive sensor for H2O2 analysis was developed based on a composite consisting of NF as an effective solubilizing and stabilizing agent and a mixed metal oxide (LNO) with good electrocatalytic activity on the GCE.This composite can catalyze the H2O2 reduction in 0.1 M NaOH and accelerates the kinetics of the catalytic reaction. The designed sensor displayed high sensitivity, selectivity, fast amperometric response, and excellent stability. The performances of the offered method were compared with those of other studies that previously have been published, and the outcomes were presented in Table 1. As it is seen, some of the analytical parameters of this sensor are comparable or better than those of other reported H2O2 sensors. Also, the offered sensor can be used as an amperometric sensor for H2O2 monitoring in synthetic and real samples with different matrices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out by Elahe Ahmadi (PhD student) as a part of a PhD thesis at Razi University. It was supported by the Razi University Research Council. This work is guided by the supervisor (Mohammad Bagher Gholivand) in Gholivand Lab.References
- P. Poprac, K. Jomova, M. Simunkova, V. Kollar, C. J. Rhodes and M. Valko, Trends Pharmacol. Sci., 2017, 38, 592–607 CrossRef CAS PubMed.
- C. Li, R. Pan, P. Li, Q. Guan, J. Ao, K. Wang, L. Xu, X. Liang, X. Jin, C. Zhang and X. Zhu, Anal. Chem., 2017, 89, 5966–5975 CrossRef CAS PubMed.
- K. Takeda, H. Nojima, K. Kuwahara, R. C. Chidya, A. O. Adesina and H. Sakugawa, Anal. Sci., 2018, 34, 459–464 CrossRef CAS PubMed.
- T. F. Brewer, F. J. Garcia, C. S. Onak, K. S. Carroll and C. J. Chang, Annu. Rev. Biochem., 2015, 84, 765–790 CrossRef CAS PubMed.
- T. C. Chou, K. Y. Wu, F. X. Hsu and C. K. Lee, J. Food Drug Anal., 2018, 26, 662–669 CrossRef CAS PubMed.
- D. Li, M. Wang, N. Cheng, X. Xue, L. Wu and W. Cao, Food Chem., 2017, 237, 225–231 CrossRef CAS PubMed.
- C. C. Winterbourn, Antioxid. Redox Signaling, 2018, 29(6), 541–551 CrossRef CAS PubMed.
- P. Gimeno, C. Bousquet, N. Lassu, A. F. Maggio, C. Civade, C. Brenier and L. Lempereur, J. Pharm. Biomed. Anal., 2015, 107, 386–393 CrossRef CAS PubMed.
- H. Cai, X. Liu, J. Zou, J. Xiao, B. Yuan, F. Li and Q. Cheng, Chemosphere, 2018, 193, 833–839 CrossRef CAS PubMed.
- M. Guler, V. Turkoglu, A. Bulut and M. Zahmakiran, Electrochim. Acta, 2018, 263, 118–126 CrossRef CAS.
- S. A. Kitte, M. N. Zafar, Y. T. Zholudov, X. Ma, A. Nsabimana, W. Zhang and G. Xu, Anal. Chem., 2018, 90, 8680–8685 CrossRef CAS PubMed.
- C. H. Díaz Nieto, A. M. Granero, J. C. Lopez, G. D. Pierini, G. J. Levin, H. Fernández and M. A. Zon, Sens. Actuators, B, 2018, 263, 377–386 CrossRef.
- J. Yuan, S. Xu, H. Y. Zeng, X. Cao, A. Dan Pan, G. F. Xiao and P. X. Ding, Bioelectrochemistry, 2018, 123, 94–102 CrossRef CAS PubMed.
- W. Hooch Antink, Y. Choi, K. dong Seong and Y. Piao, Sens. Actuators, B, 2018, 255, 1995–2001 CrossRef CAS.
- Y. Wang, W. Cao, Q. Zhuang and Y. Ni, Anal. Lett., 2018, 51, 2441–2456 CrossRef CAS.
- Y. H. Tang, N. C. Lo and P. Y. Chen, Electrochem. Commun., 2018, 87, 44–48 CrossRef CAS.
- H. C. Kazici, F. Salman, A. Caglar, H. Kivrak and N. Aktas, Fullerenes, Nanotub. Carbon Nanostruct., 2018, 26, 145–151 CrossRef CAS.
- R. Zhang, C. Jiang, X. Fan, R. Yang, Y. Sun and C. Zhang, Microchim. Acta, 2018, 185(1), 1–9 CrossRef PubMed.
- S. Y. Lu, Y. Chen, X. Fang and X. Feng, Electroanalysis, 2018, 30, 583–592 CrossRef CAS.
- S. Chopra, S. Sharma, T. C. Goel and R. G. Mendiratta, Mater. Chem. Phys., 2005, 91, 161–165 CrossRef CAS.
- V. Neburchilov, H. Wang, J. J. Martin and W. Qu, J. Power Sources, 2010, 195, 1271–1291 CrossRef CAS.
- K.-N. Jung, J.-I. Lee, W. Bin Im, S. Yoon, K.-H. Shin and J.-W. Lee, Chem. Commun., 2012, 48, 9406 RSC.
- M. Ghasdi and H. Alamdari, Sens. Actuators, B, 2010, 148, 478–485 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- S. J. Amirfakhri, J. L. Meunier and D. Berk, J. Power Sources, 2014, 272, 248–258 CrossRef CAS.
- J. Chen, J. Wu, Y. Liu, X. Hu and D. Geng, Phys. Status Solidi, 2018, 1800380 CrossRef.
- A. Jahangiri, M. Saidi, F. Salimi and A. Mohammadi, Res. Chem. Intermed., 2018, 44, 1755–1773 CrossRef CAS.
- R. Pereñíguez, V. M. González-DelaCruz, J. P. Holgado and A. Caballero, Appl. Catal., B, 2010, 93, 346–353 CrossRef.
- N. Amini, M. B. Gholivand and M. Shamsipur, J. Electroanal. Chem., 2014, 714–715, 70–75 CrossRef CAS.
- C. C. Zhao, X. Wu, P. Li, C. C. Zhao and X. Qian, Microchim. Acta, 2017, 184, 2341–2348 CrossRef CAS.
- Q. Zhu, B. Liang, Y. Cai, Q. Cao, T. Tu, B. Huang, L. Fang and X. Ye, Talanta, 2018, 190, 70–77 CrossRef CAS PubMed.
- H. Tian, H. Fan, J. Ma, L. Ma and G. Dong, Electrochim. Acta, 2017, 247, 787–794 CrossRef CAS.
- Z. Cheng, Q. Shen, H. Yu, D. Han, F. Zhong and Y. Yang, Microchim. Acta, 2017, 184, 4587–4595 CrossRef CAS.
- Z. L. Wu, C. K. Li, J. G. Yu and X. Q. Chen, Sens. Actuators, B, 2017, 239, 544–552 CrossRef CAS.
- M. Devendiran, K. Krishna Kumar and S. Sriman Narayanan, J. Electroanal. Chem., 2017, 802, 78–88 CrossRef CAS.
- L. Shi, M. Layani, X. Cai, H. Zhao, S. Magdassi and M. Lan, Sens. Actuators, B, 2018, 256, 938–945 CrossRef CAS.
- M. A. Kamyabi and N. Hajari, J. Braz. Chem. Soc., 2017, 28, 808–818 CAS.
- S. A. Bin Asif, S. B. Khan and A. M. Asiri, J. Taiwan Inst. Chem. Eng., 2017, 74, 255–262 CrossRef.
- Z. Qin, Y. Zhao, L. Lin, P. Zou, L. Zhang, H. Chen, Y. Wang, G. Wang and Y. Zhang, Microchim. Acta, 2017, 184, 4513–4520 CrossRef CAS.
- A. Benvidi, M. T. Nafar, S. Jahanbani, M. D. Tezerjani, M. Rezaeinasab and S. Dalirnasab, Mater. Sci. Eng., C, 2017, 75, 1435–1447 CrossRef CAS PubMed.
- H. Xia, J. Li, L. Ma, Q. Liu and J. Wang, J. Alloys Compd., 2018, 739, 764–770 CrossRef CAS.
- C. Zhang, H. Jiang, R. Ma, Y. Zhang and Q. Chen, Ionics, 2017, 23, 1309–1317 CrossRef CAS.
- F. Mollarasouli, K. Asadpour-Zeynali, S. Campuzano, P. Yáñez-Sedeño and J. M. Pingarrón, Electrochim. Acta, 2017, 246, 303–314 CrossRef CAS.
- J. Liang, M. Wei, Q. Wang, Z. Zhao, A. Liu, Z. Yu and Y. Tian, Anal. Lett., 2018, 51(4), 512–522 CrossRef CAS.
- M. B. M. B. Gholivand, E. Ahmadi and M. Haseli, Anal. Biochem., 2017, 527, 4–12 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03587d |
| This journal is © The Royal Society of Chemistry 2020 |