
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorohydride catalyzed redistribution reaction of hydrosilane and chlorosilane: a potential system for facile preparation of hydrochlorosilanes†
Yi Chenab,
Liqing Aiab,
Yongming Lia and
Caihong Xu *ab
*ab
aBeijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: caihong@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 5th May 2020
Abstract
Various borohydrides were found to catalyze the redistribution reaction of hydrosilane and chlorosilane in different solvents to produce hydrochlorosilanes efficiently and facilely. The redistribution reaction was affected by solvent and catalyst. The substrate scope was investigated in HMPA with LiBH4 as catalyst. A possible mechanism was proposed to explain the redistribution process.
Hydrochlorosilanes, with both Si–H and Si–Cl bonds on the silicon center, have attracted much attention in past decades for their importance in the production of functional polysiloxanes, polysilanes, polysilazanes, and other silicon-containing materials.1 However, the facile and highly selective preparation of this type of compound has remained a great challenge, especially for bulk production. The important commercially available hydrochlorosilanes, MeSiHCl2 and MeSiH2Cl, are obtained as by-products from the Müller–Rochow Direct Process.2
Regarding the preparation of hydrochlorosilanes, much research work has been focused on the redistribution reaction between hydrosilane and chlorosilane, selective chlorination of hydrosilane, and partial reduction of chlorosilane.3 In 1947, Sommer et al. first reported the redistribution reaction between hydrosilane and chlorosilane catalyzed by AlCl3.3a Then the quaternary ammonium salt and tertiary amine were found also can catalyze the redistribution.3b–d However, both of them require relatively high temperature and strict operation conditions, which cause extra cost and limitation of available substrates. Preparation of hydrochlorosilane by partial reduction of chlorosilane has been an attractive subject. NaBH4 was reported to partially reduce dialkyldichlorosilane to dialkylchlorosilane in hexamethyl phosphoric triamide (HMPA), but the mechanism of the selectivity in this reduction system was not well understood.4d Attempts to produce hydrochlorosilane by reduction of chlorosilane with LiAlH4, the commonly used reductants, have not been realized facilely and efficiently because of the intractable over reduction.4
The research on the preparation of hydrochlorosilanes by selective chlorination of hydrosilanes has made great progress in past decades. In 1992, Kunai reported a simple method for selective chlorination of hydrosilane using stoichiometric CuCl2 in the presence of CuI.5a,b The catalytic efficiency was further improved by adding ceramic spheres into the system.5c Chulsky and Dobrovetsky studied the selective chlorination of Si–H bond with HCl gas in the presence of B(C6F5)3 or B(C6F5)3/Et2O catalyst, and proposed the corresponding mechanisms in 2017.6a Recently, Sturm reported that the Si–H can be activated by Lewis base, such as ethers, amines, and chloride ions. The activated Si–H was then selectively chlorinated by the HCl/ether solution.6b Though various catalytic systems for selective chlorination of hydrosilane have been developed, most of them have been limited in laboratory synthesis, due to disadvantages of high cost or strict operation conditions. It is still an important issue to achieve simple and economic preparation of hydrochlorosilanes.
Herein we demonstrate a borohydride catalyzed redistribution reaction system, which leads to a facile and flexible preparation of hydrochlorosilane. During the preparation of Cl2CHSiMeH2 (1) by reduction of Cl2CHSiMeCl2 (2) using borohydrides in THF, hydrochlorosilane Cl2CHSiMeHCl (3) was unexpectedly detected when chlorosilane 2 was accidentally mixed with an ether solution of hydrosilane 1 that contained some LiBH4 residue. We guessed that the formation of 3 may be from the redistribution reaction of 1 and 2, while LiBH4 served as a catalyst. To verify the conjecture and to comprehend the possible solvent effect, we examined the reaction of ClCH2SiCl3 and ClCH2SiH3 in different solvents in the presence of LiBH4 (Table 1). As expected, the redistribution reaction proceeded in various solvents, particularly those with relatively high polarity, e.g., tetrahydrofuran (THF), CH3CN, diethylene glycol dimethyl ether (diglyme), 1,3-dimethyl-2-imidazolidinone (DMI), and HMPA (Table 1, entries 2–6). The relatively high conversion and selectivity encouraged us to further investigate the redistribution reaction, which may be a useful way to synthesize hydrochlorosilane. Although CH3SiHCl2 was unexpectedly formed in a yield of 6% in diglyme, suggesting the C–Cl bond also participated in the redistribution reaction with Si–H bond (Table 1, entry 2), there were few detected CH3SiHCl2 in THF, DMI, or HMPA. It may be attributed to the different solvation effects of LiBH4, which resulted in their different reductive activity toward the chloromethyl group.
|
||
|---|---|---|
| Entry | Solvent | Product (yield)b |
| a Reaction conditions: ClCH2SiH3 (0.01 mol), ClCH2SiCl3 (0.02 mol), LiBH4 (3.0 mol/%), THF (5 mL), room temperature.b Yields were determined by 1H NMR. | ||
| 1 | None | No reaction |
| 2 | Diglyme | CH3SiHCl2: 6%, ClCH2SiHCl2: 63%, ClCH2SiH2Cl: 12% |
| 3 | THF | ClCH2SiHCl2: 63%, ClCH2SiH2Cl: 16% |
| 4 | CH3CN | ClCH2SiHCl2: 71%, ClCH2SiH2Cl: 11% |
| 5 | DMI | ClCH2SiHCl2: 71%, ClCH2SiH2Cl: 14% |
| 6 | HMPA | ClCH2SiHCl2: 72%, ClCH2SiH2Cl: 9% |
| 7 | Bu2O | No reaction |
| 8 | Et2O | No reaction |
| 9 | Toluene | No reaction |
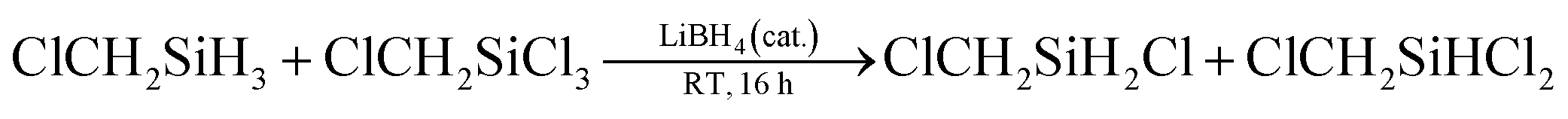
We further optimized the catalyst with the substrate system of ClCH2SiCl3/ClCH2SiH3 as a model (Table 2). It is found that in THF, all examined lithium salt, LiBH4, LiBEt3H, and LiAlH4, can catalyze the redistribution reaction (Table 2, entries 1–5), but NaBH4 and KBH4 which have poor solubility cannot (Table 2, entries 6 and 7). Changed the solvent from THF to diglyme, a better solvent for NaBH4 and KBH4, the redistribution reaction proceeded, though the yield of product is low (see ESI, Fig. 35 and 36†). This result indicated that the solubility of catalyst in solvent is important for the reaction. With the increase of catalyst dosage, the yield of hydrochlorosilane increased (Table 2, entries 1–4). Besides, the metal counterion of BH4− also affects the redistribution reaction. With 4 mol% of LiBH4 or NaBH4 as catalyst, the conversion efficiencies for the redistribution reaction of Cl2CHSiMeCl2/Cl2CHSiMeH2 are 67% and 15%, respectively, though both of them can be dissolved in diglyme completely (see ESI, Fig. 36 and 37†). The possible reason may be (a) the different solvation effects of LiBH4 and NaBH4 in diglyme, which is related to the character of metal ion, and (b) the different solubility of the formed metal chloride, LiCl and NaCl.7
|
|||
|---|---|---|---|
| Entry | Cat. | Cat. (mol%) | Product (yield)b |
| a Reaction conditions: ClCH2SiH3 (0.01 mol), ClCH2SiCl3 (0.02 mol), THF (5 mL), room temperature.b Yields were determined by 1H NMR. | |||
| 1 | LiBH4 | 0.4 | ClCH2SiHCl2: 10%, ClCH2SiH2Cl: 20% |
| 2 | LiBH4 | 0.8 | ClCH2SiHCl2: 32%, ClCH2SiH2Cl: 27% |
| 3 | LiBH4 | 1.5 | ClCH2SiHCl2: 55%, ClCH2SiH2Cl: 18% |
| 4 | LiBH4 | 3.0 | ClCH2SiHCl2: 63%, ClCH2SiH2Cl: 16% |
| 5 | LiBEt3H | 3.0 | ClCH2SiHCl2: 68%, ClCH2SiH2Cl: 12% |
| 6 | NaBH4 | 3.0 | No reaction |
| 7 | KBH4 | 3.0 | No reaction |
| 8 | LiAlH4 | 3.0 | ClCH2SiHCl2: 18%, ClCH2SiH2Cl: 21% |
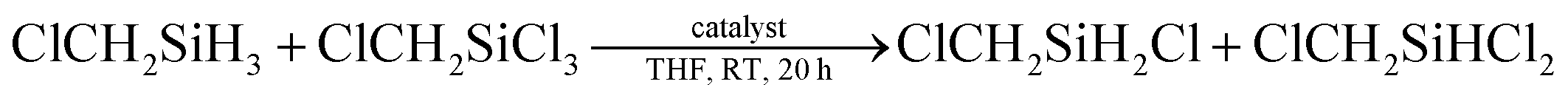
Then under the optimized condition, we investigated the substrate scope of the redistribution reaction in the LiBH4/HMPA system (Table 3). The redistribution reaction of Si–H and Si–Cl proceeded efficiently for various substituted hydrosilane and chlorosilane substrates. The substituents include chloroalkyl, alkyl, and phenyl groups. Especially, for α-chloromethyl silane substrates, e.g., ClCH2SiCl3/ClCH2SiH3, ClCH2SiMeCl2/ClCH2SiMeH2, Cl2CHSiMeCl2/Cl2CHSiMeH2, the redistribution was completed in half an hour (Table 3, entries 1–4). The stronger electron-withdrawing effect of the substituents on silicon center may be the reason. For the reaction system of RSiCl3/RSiH3, namely, the substrate has more available redistribution groups, different predominated product was achieved through adjustment of the proportion of hydrosilane and chlorosilane in reactants (Table 3, entries 1, 2, 6 and 7). The LiBH4 catalyzed redistribution reaction in HMPA has yields ranging from 56% to 85%. Further work is still needed to realize a thorough and selective redistribution, however, the advantages of its very simple and mild reaction condition, easily acquired catalysts, relatively fast reaction rate, and high conversion efficiency suggest a great potential of the redistribution method in achieving more economic and efficient preparation of hydrochlorosilanes.
|
|||||
|---|---|---|---|---|---|
| Entry | R4−xSiHx/R4−xSiClx | Ratio | Cat. (mol%) | t (h) | Product (yield)b |
| a Reaction conditions: [entry 3–5, 8 and 9] hydrosilane (0.01 mol), chlorosilane (0.01 mol); [entry 1 and 6] hydrosilane (0.01 mol), chlorosilane (0.02 mol); [entry 2 and 7] hydrosilane (0.02 mol), chlorosilane (0.01 mol); HMPA (5 mL), room temperature.b Yields determined by 1H NMR.c Isolated yield in a larger scale reaction. Reaction conditions: hydrosilane (0.2 mol), chlorosilane (0.2 mol), HMPA (10 mL), LiBH4 (2 mol%), room temperature. | |||||
| 1 | ClCH2SiH3/ClCH2SiCl3 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
3 | 0.5 | ClCH2SiHCl2: 75%, ClCH2SiH2Cl: 9% |
| 2 | ClCH2SiH3/ClCH2SiCl3 | 2:1 |
3 | 0.5 | ClCH2SiHCl2: 23%, ClCH2SiH2Cl: 56% |
| 3 | ClCH2SiMeH2/ClCH2SiMeCl2 | 1:1 |
2 | 0.5 | ClCH2SiMeHCl: 74% (62%c) |
| 4 | Cl2CHSiMeH2/Cl2CHSiMeCl2 | 1:1 |
4 | 0.5 | Cl2CHSiMeHCl: 85% (72%c) |
| 5 | Et2SiH2/Et2SiCl2 | 1:1 |
4 | 8 | Et2SiHCl:72% |
| 6 | PhSiH3/PhSiCl3 | 1:2 |
3 | 2 | PhSiHCl2: 61%PhSiH2Cl: 10% |
| 7 | PhSiH3/PhSiCl3 | 2:1 |
3 | 2 | PhSiHCl2:12%PhSiH2Cl: 61% |
| 8 | PhMeSiH2/PhMeSiCl2 | 1:1 |
4 | 2 | PhMeSiHCl: 66% |
| 9 | Ph2SiH2/Ph2SiCl2 | 1:1 |
4 | 8 | Ph2SiHCl: 63% |
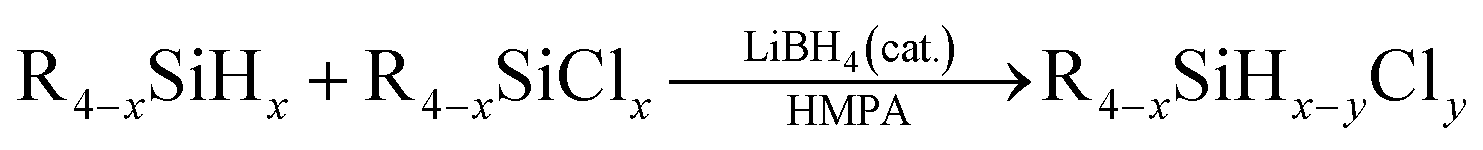
To demonstrate the utility of the redistribution strategy, larger scale syntheses of hydrochlorosilane ClCH2SiMeHCl and ClCH2SiMeHCl were carried out. After finishing the reaction, all the volatiles were first removed by vacuum distillation to give a silane mixture, which was then submitted to a packed column fractional distillation to produce pure hydrochlorosilane product, such as ClCH2SiMeHCl or Cl2CHSiMeHCl. The simple manipulation and good yield indicated its high potential for industrial application.
On the other hand, the redistribution system also provides a possible reference for the partial reduction of chlorosilane to prepare hydrochlorosilane with NaBH4, LiAlH4, and other common reductants. For the NaBH4/HMPA partial reduction system reported by Hiiro,4d the reduction selectivity of chlorosilane can be easily understood from the perspective of the redistribution system herein, i.e., in the presence of deficient amount of borohydrides, hydrochlorosilanes can be produced from the redistribution reaction between the in situ formed hydrosilane products and the chlorosilane substrates. It is also easy to comprehend that partial reduction of chlorosilane can be easily achieved with LiAlH4 as reductant, if the solvents which are effective for the redistribution system are used.
To make clear the mechanism, we further investigated the redistribution reaction. The Si–Cl bond of chlorosilane substrates can be reduced to Si–H bond by borohydrides, with BH3 and chloride salt as the by-products (see ESI, Fig. 51†). Thus, our initial consideration is that BH3, which is formed in situ during the reduction reaction, may play the role of catalyst. However, the experiment proved that BH3 alone cannot catalyze the redistribution reaction (Table 4, entry 1). The Si–Cl reduction process of chlorosilane substrate with borohydrides is easily understood by referring to the well-studied substitution reaction at silicon center using nucleophiles,8 i.e., the negative BH4− attacks to the partial positive silicon center to form a pentacoordinate intermediate of chlorosilane in the first step, then looses BH3 and Cl− to form corresponding silane product. We assumed that the chlorination process of Si–H may also undergo a similar process, i.e., Si–H was first activated through the formation of pentacoordinate intermediate with Cl− that formed in the Si–Cl reduction process. Then the in situ formed Lewis acid BH3 during the Si–Cl reduction process interacted with the activated Si–H bond, which further promoted the Si–H bond dissociation. The early reported research on the ability of B(C6F5)3 and Cl− to activate Si–H bond6b,9 provides a good proof for reasonability of the envisaged chlorination process.
|
|||
|---|---|---|---|
| Entry | Catalyst | t (h) | Product (yield)b |
| a Reaction conditions: ClCH2SiH3 (0.01 mol), ClCH2SiCl3 (0.02 mol), catalyst (3.0 mol/%), THF (5 mL), room temperature.b Yields were determined by 1H NMR. | |||
| 1 | BH3 | 20 | No reaction |
| 2 | LiCl | 5 | ClCH2SiHCl2: 40%, ClCH2SiH2Cl: 23% |
| 3 | BH3, LiCl | 5 | ClCH2SiHCl2: 62%, ClCH2SiH2Cl: 18% |
| 4 | LiBH4 | 5 | ClCH2SiHCl2: 62%, ClCH2SiH2Cl: 18% |
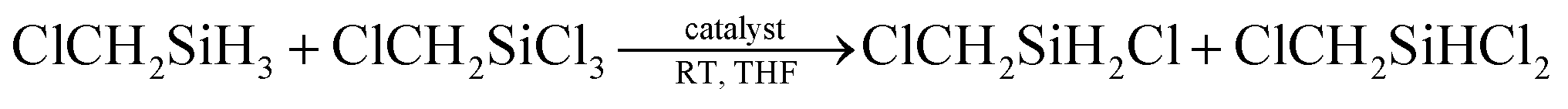
According to the above hypothesis, we proposed a possible Si–H/Si–Cl redistribution mechanism demonstrated as Scheme 1, in which the process of Si–H chlorination and Si–Cl reduction occur simultaneously. To verify it, the redistribution reaction of ClCH2SiCl3/ClCH2SiH3 catalyzed by BH3/LiCl or LiCl alone was performed. As expected, LiCl can catalyze the redistribution of ClCH2SiCl3/ClCH2SiH3, and introduction of BH3 can further accelerate the reaction obviously (Table 4, entry 2 and 3). Besides, almost identical results were achieved when the same amount of LiBH4 or BH3/LiCl was used (Table 4, entry 3 and 4). The results indicate the similar catalytic effect of BH4− and BH3/LiCl, which can be well explained by the mechanism proposed in Scheme 1.
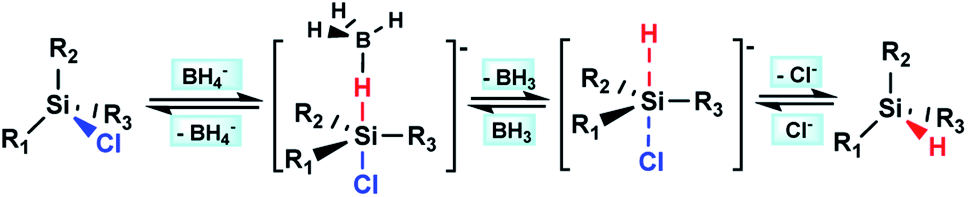 | ||
| Scheme 1 The possible mechanism of borohydrides catalyzed Si–H/Si–Cl redistribution reaction. | ||
To get a better understanding of the possible mechanism in Scheme 1, a DFT calculation at the B3LYP/6-311G (d, p)10 level of theory was performed on the pentacoordinate intermediates [ClCH2SiCl3–H–BH3]− (I) and [ClCH2SiCl3–H]− (II), which was formed in the redistribution of ClCH2SiH3/ClCH2SiCl3 according to the mechanism we proposed. The solvation effect of THF was also considered with SMD model. The calculation result shows that the apical Si–H bond in intermediate II lengthens from l.47 Å to 1.49 Å, while it is elongated from 1.49 Å to 1.65 Å in intermediate I. Calculation results on the other possible pentacoordinate intermediates in the redistribution of ClCH2SiH3/ClCH2SiCl3 were similar. It may reflect the role BH3 or Cl− plays in the activation of Si–H bond in the chlorination process as the mechanism we proposed.
In conclusion, we have demonstrated that borohydrides catalyzed redistribution reaction between hydrosilane and chlorosilane in different solvents efficiently. The new catalytic redistribution system works for a broad scope of substituted hydrosilane and chlorosilane substrates. The very simple and mild reaction condition, the easily acquired catalysts, the relatively fast reaction rate, and high conversion efficiency are all advantages of the redistribution system. Further improvement of the redistribution system is currently under investigation in our laboratory.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. Arkles, in Kirk-Othmer Encyclopedia of Chemical Technology, American Cancer Society, 2000 Search PubMed; (b) R. M. Laine and A. Sellinger, in The Chemistry of Organic Silicon Compounds, John Wiley & Sons, Ltd, 2003, pp. 2245–2316 Search PubMed; (c) W. Xue, M. C. Kung and H. H. Kung, Chem. Commun., 2005, 2164–2166 RSC; (d) M. N. Missaghi, C. M. Downing, M. C. Kung and H. H. Kung, Organometallics, 2008, 27, 6364–6366 CrossRef CAS.
- (a) E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963–965 CrossRef CAS; (b) D. Seyferth, Organometallics, 2001, 20, 4978–4992 CrossRef CAS.
- (a) F. C. Whitmore, E. W. Pietrusza and L. H. Sommer, J. Am. Chem. Soc., 1947, 69, 2108–2110 CrossRef CAS; (b) A. Benouargha, D. Boulahia, B. Boutevin, G. Caporiccio, F. Guida-pietrasanta and A. Ratsimihety, Phosphorus, Sulfur, Silicon Relat. Elem., 1996, 113, 79–87 CrossRef CAS; (c) I. N. Jung, B. R. Yoo, J. S. Han and W.-C. Lim, US Pat., US5965762A, 1999; (d) W. Katsuyu and T. Hidenori, European Patent, EP2308884 (A1), 2011.
- (a) P. A. McCusker and E. L. Reilly, J. Am. Chem. Soc., 1953, 75, 1583–1585 CrossRef CAS; (b) H. E. Opitz, J. S. Peake and W. H. Nebergall, J. Am. Chem. Soc., 1956, 78, 292–294 CrossRef CAS; (c) A. Glüer, J. I. Schweizer, U. S. Karaca, C. Würtele, M. Diefenbach, M. C. Holthausen and S. Schneider, Inorg. Chem., 2018, 57, 13822–13828 CrossRef PubMed; (d) H. Takeshi, S. Hideki and K. Fumihiko, US Pat., US4115426A, 1978.
- (a) A. Kunai, T. Kawakami, E. Toyoda and M. Ishikawa, Organometallics, 1992, 11(7), 2708–2711 CrossRef CAS; (b) A. Kunai and J. Ohshita, J. Organomet. Chem., 2003, 686(1), 3–15 CrossRef CAS; (c) W. Wang, Y. Tan, Z. Xie and Z. Zhang, J. Organomet. Chem., 2014, 769, 29–33 CrossRef CAS.
- (a) K. Chulsky and R. Dobrovetsky, Angew. Chem., Int. Ed., 2017, 56, 4744–4748 CrossRef CAS PubMed; (b) A. G. Sturm, J. I. Schweizer, L. Meyer, T. Santowski, N. Auner and M. C. Holthausen, Chem. - Eur. J., 2018, 24, 17796–17801 CrossRef CAS PubMed.
- (a) H. Hagemann and R. Černý, Dalton Trans., 2010, 39, 6006–6012 RSC; (b) H. C. Brown, Boranes in Organic Chemistry, Cornell University Press, 2019, pp. 217–218 Search PubMed.
- (a) R. J. P. Corriu and C. Guerin, in Advances in Organometallic Chemistry, eds. F. G. A. Stone and R. West, Academic Press, 1982, vol. 20, pp. 265–312 Search PubMed; (b) R. J. P. Corriu and C. Guerin, J. Organomet. Chem., 1980, 198, 231–320 CrossRef CAS.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (b) J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed.
- (a) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (c) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS; (d) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (e) G. W. Spitznagel, T. Clark, P. von Ragué Schleyer and W. J. Hehre, J. Comput. Chem., 1987, 8, 1109–1116 CrossRef CAS; (f) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03536j |
| This journal is © The Royal Society of Chemistry 2020 |