
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring stereoselective excretion and metabolism studies of novel 2-(2-hydroxypropanamido)-5-trifluoromethyl benzoic acid enantiomers
Rong Rong†
a,
Qi-li Zhang†b,
Rui-zhen Zhanga,
Yu-han Dana,
Xin Wanga,
Yun-li Zhao*a and
Zhi-guo Yu *a
*a
aDepartment of Pharmaceutical Analysis, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, 110016, China. E-mail: zhiguo-yu@163.com; yunli76@163.com; Tel: +86-24-43520580
bDepartment of Pharmaceutical Engineering, College of Life Science, Shandong University of Technology, Zibo, 255049, China
First published on 21st July 2020
Abstract
R-/S-2-(2-Hydroxypropanamido)-5-trifluoromethyl benzoic acid (R-/S-HFBA), as a novel COX inhibitor, was firstly reported to have remarkable anti-inflammatory and antiplatelet aggregation activities by our group. In our previous study, stereoselective differences in pharmacokinetics were found between HFBA enantiomers after oral and intravenous administration of each enantiomer to rats. The discrepancies might be associated with the excretion and metabolism of the two enantiomers. In this research, an UHPLC-MS/MS method was established and validated for quantification of R-/S-HFBA in rats urine, feces and bile samples in excretion study. Moreover, an ultra high performance liquid chromatography coupled with Fourier transform ion cyclotron resonance mass spectrometry (UHPLC-FT-ICR-MS) method was employed to understand the metabolism of R-/S-HFBA in rats. Results showed that the total cumulative excretion of R-/S-HFBA in three routes were 65.8% and 58.5% of the dose, respectively. The urinary excretion of R-/S-HFBA was the main route, which accounted for 40.2% and 31.7% respectively; the cumulative biliary excretion of R-/S-HFBA were 11.3% and 7.4%; the cumulative amounts of R-/S-HFBA excreted directly via feces without absorption from the gastrointestinal tract were 14.3% and 19.4%, respectively. R-/S-HFBA existed stereoselective discrepancy in excretion. In addition, 8 metabolites of S-HFBA were detected and tentatively identified including glucuronidation, glycine and N-acetyl conjugation while R-HFBA existed 7 metabolites without glycine conjugation. Formation of metabolites of R-/S-HFBA also exhibited stereoselectivity. In summary, these new findings on excretion and metabolism of R-/S-HFBA provided valuable information for stereoselective pharmacokinetics and were greatly helpful for further investigation, such as safety and mechanism of action.
1. Introduction
Inflammation, one of the most important biological protective processes against injury, infection or irritants, is able to induce cellular damage and release pro-inflammatory mediators.1,2 Prolonged existence of uncontrolled inflammation can cause various fatal diseases such as inflammatory bowel disease, psoriasis, multiple sclerosis.3,4 Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used therapeutic agents due to their anti-inflammatory, analgesic antipyretic effect.5 The pharmacological action of NSAIDs is attributed to the suppression of cyclooxygenase (COX). COX owns two most important isoforms: COX-1 is the naturally expressed enzyme involved in prostanoid biosynthesis and maintenance functions such as gastric cytoprotection. COX-2 is considered as inducibly expressed by inflammatory irritant from injuries or infections.6,7 However, long-term clinical use of NSAIDs, especially aspirin, can cause serious side effects, such as gastrointestinal, cardiovascular, and kidney complications.8,9 Thus, it is extremely necessary to develop more safe and effective anti-inflammatory agents to replace or supplement the current therapies.2-(2-Hydroxypropanamido)-5-trifluoromethyl benzoic acid (HFBA, Fig. 1), with one chiral center and two enantiomers, R-/S-HFBA, was initially synthesized by our group. R-/S-HFBA presented remarkable anti-inflammatory and antiplatelet aggregation activities superior to aspirin, with less gastrointestinal toxicity.10 The action mechanism of HFBA was found to inhibit COX enzyme resulting in a reduction of thromboxane A2 and inflammatory mediators. HFBA could be a effective and safe agent for anti-inflammatory and antiplatelet aggregation therapy with prolonged pharmacological activity. Besides, in our laboratory, the different pharmacokinetic profiles of the two enantiomers in rats were observed after the oral administration of R-HFBA and S-HFBA. R-HFBA exhibited greater t1/2, AUC and lower clearance (CL) than those of S-HFBA.10
 | ||
| Fig. 1 Chemical structures of R-HFBA (A), S-HFBA (B) and IS (C). | ||
Metabolite identification and excretion studies play a crucial role in the discovery and development of new drugs.11 Metabolism is the biochemical process to detoxify and eliminate xenobiotics and endogenous compounds through converting them into more hydrophilic species. Metabolism could have an impact on many aspects of a drug, such as distribution, excretion and drug activation, which usually determines the ability of a new substance to reach clinical use.12–14 Early information on the identification of metabolites formed in vivo is extraordinarily useful for improving the biopharmaceutical and early safe assessment in drug discovery research.15,16 Proper study on the excretion is significant for investigating the major target sites and detailed understanding of the in vivo disposition is also required. These findings can supply valuable information for further research. However, detailed knowledge of metabolism and excretion of R-/S-HFBA has not been reported yet.
Nowadays, high resolution mass spectrometers such as Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS), linear ion trap quadrupole (LTQ),17 or time-of-flight (TOF)18–20 have been becoming extensively used for detection and identification of metabolites of small molecules. Among them, FT-ICR-MS has achieved increasingly attention and become powerful tools for various analyses due to its high mass accuracy and unsurpassed resolution.21,22 In this present study, the excretion study of HFBA enantiomers was performed using UHPLC-MS/MS after a single oral dose. Subsequently, an ultra high performance liquid chromatography coupled with Fourier transform ion cyclotron resonance mass spectrometry (UHPLC-FT-ICR-MS) method was also employed for the metabolite identification of R-/S-HFBA in rats after oral administration.
2. Materials and methods
2.1 Chemicals and reagents
R-HFBA and S-HFBA were synthesized separately in our laboratory (Shenyang Pharmaceutical University, Shenyang, China). The optical purity of each HFBA enantiomer was over 99%.10 Acetaminophen (internal standard, IS, purity > 99%, Fig. 1) was obtained from Sigma (St. Louis, MO, USA). HPLC-grade methanol and acetonitrile were bought form Fisher Scientific (Fair Lawn, NJ, USA). Formic acid (HPLC grade) was obtained from Concord Technology CO. Ltd (Tianjin, China). Other chemicals were all of analytical grade.2.2 Animals
SPF degree male Sprague-Dawley rats (220 ± 20 g) were supplied by Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China). Animals were housed in standard environmental conditions at a temperature of 25 ± 2 °C and relative humidity of 50 ± 10% with free access to standard food and water. They were fasted for 12 h and only have access to water before the experimentation. All experimental studies were performed according to the Guideline for Animal Experimentation of Shenyang Pharmaceutical University and the approval from Animal Ethics Committee of Shenyang Pharmaceutical University (Approval Number: 211002000310215).2.3 Instrument and analytical conditions
The mass spectrometer was operated in positive electrospray mode using multiple reaction monitoring (MRM). The desolvation temperature and source temperature were 350 °C and 150 °C, respectively. The capillary voltage was 3.0 kV. The desolvation gas flow rate and the cone gas flow rate were 700 L h−1 and 150 L h−1, respectively. The ion transitions (m/z) were 278.0 > 187.9 for R-/S-HFBA, 151.9 > 109.9 for IS. The collision energy were 18 eV for R-/S-HFBA and 14 eV for IS, respectively. The cone voltage were 21 V for R-/S-HFBA and 22 V for IS, respectively. The product ion spectra for R-/S-HFBA and IS were shown in our previous report.23
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22, v/v) with a flow rate of 0.3 mL min−1. The auto-sampler and the column temperature was set at 4 °C and 35 °C, respectively. The injection volume was 5 μL.
:22, v/v) with a flow rate of 0.3 mL min−1. The auto-sampler and the column temperature was set at 4 °C and 35 °C, respectively. The injection volume was 5 μL.The mass spectrometric detection was operated on a Solarix 7.0T FT-ICR mass spectrometer equipped with electrospray ionization interface and performed in positive ion mode. The ion source parameters were set as following: capillary voltage, 4500 V; dry gas temperature, 250 °C; dry gas flow, 8 L min−1; nebulizing gas, high-purity nitrogen (N2); nebulizer gas pressure, 4 bar; collision gas, high-purity argon (Ar). Mass spectra recorded was in the range of m/z 50–1000. The collision energy was 10 eV for the MS/MS experiments.
2.4 Drug administration and sample collection
For excretion study, twenty Sprague-Dawley rats were weighed and randomly divided into four groups (5 rats in each group). R-HFBA and S-HFBA was dissolved in 0.5% CMC-Na (croscarmellose sodium) aqueous solution, respectively. Animals in two groups for R-HFBA or S-HFBA were held in individual metabolic cage after administrated with a single dose of 50 mg kg−1 R-HFBA or S-HFBA enantiomer by gastric gavage. Urine and feces were collected at the indicated intervals: 0–2 h, 2–4 h, 4–6 h, 6–8 h, 8–10 h, 10–12 h, 12–24 h, 24–36 h and 36–48 h. Other two groups were used for bile collection. The rats were anesthetized with ether and cannulated in the bile duct. After their recovery from anesthesia, R-HFBA or S-HFBA (50 mg kg−1) was orally administered to the cannulated rats and bile was collected every 2 h up to 12 h. At every interval, the volume of bile and urine samples were measured and feces samples were weighed after natural drying. Blank urine, feces and bile samples were also collected before oral administration. All samples were stored at −80 °C before use.For metabolism study, the rats were randomly divided into four groups with five in each group. R-HFBA and S-HFBA (dissolved in 0.5% CMC-Na) was respectively administered to two groups by gastric gavage with a single dose of 100 mg kg−1. After then the rats were housed in separate metabolic cages to collect the feces and urine samples within 0–24 h after treatment. Blood samples were also collected in heparinized centrifuge tubes at 0.5, 3 and 6 h after administration and then centrifuged at 12000 rpm for 5 min to get plasma. Other two groups were catheterized in common bile ducts under general anesthesia. After recovering from the anesthesia, the rats were orally treated with R-HFBA or S-HFBA at a single dose of 100 mg kg−1 and bile samples were collected 0–12 h after administration.
2.5 Sample preparation
000 rpm for 5 min at 4 °C and the supernatant was carefully transferred to another tube and then evaporated to dryness by nitrogen flow. The residue was redissolved in 200 μL mobile phase and vortex-mixed for 30 s. After centrifuged at 12000 rpm for 3 min at 4 °C, 2 μL of the supernatant was injected into the UHPLC-MS/MS system for analysis.The powdered feces samples (0.25 g, if less than 0.25 g, get all and weighed) were diluted with 2 mL physiological saline and ultrasonic to homogenize. The homogenates were centrifuged at 12000 rpm for 10 min to obtain the supernatants. The supernatants were further processed as the urine and bile samples.
000 rpm for 5 min. The supernatant was collected and stored at 4 °C before injected into the UHPLC-FT-ICR-MS system for analysis. Feces samples were mixed, dried in the dark at ambient temperature and then powdered. 0.2 g of blended feces samples were added into 2.5 mL methanol and ultrasonic extracted for 30 min and subsequently the mixture was centrifuged at 12000 rpm for 10 min. The supernatant was collected and stored at 4 °C before analysis. Urine and bile samples were prepared according to our previous report.222.6 Method validation
The UHPLC-MS/MS method used for quantitative analysis of R-/S-HFBA in biological matrices (urine, bile and feces) was validated by R-HFBA enantiomer following the guidelines of the US Food and Drug Administration (FDA) for a bioanalytical method validation.24 The thorough and complete method validation included specificity, lower limit of quantification (LLOQ), linearity, precision, accuracy, extraction recovery, matrix effect and stability.2.7 Statistical analysis
3. Results
3.1 Method validation
For specificity, under the developed method, no significant endogenous interference in drug-free urine, bile and feces samples was observed at the retention times of HFBA and IS. Typical chromatograms of blank rat bile sample, blank bile sample spiked with HFBA at LLOQ and IS, and rat bile sample collected at 4–6 h after administration of HFBA are shown in Fig. 2.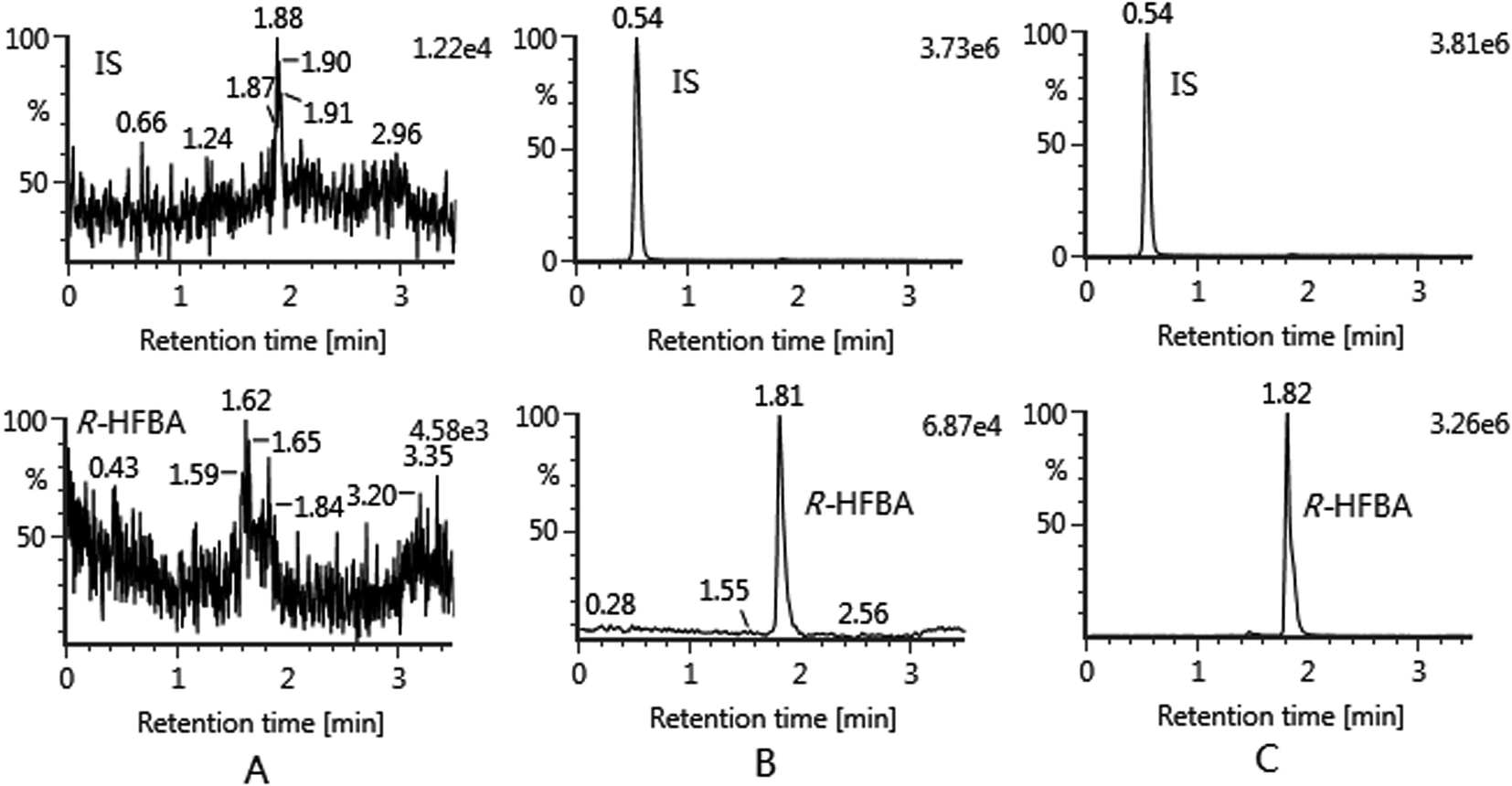 | ||
| Fig. 2 Typical chromatograms of HFBA in rat bile: (A) blank rat bile sample; (B) blank bile sample spiked with R-HFBA at LLOQ and IS; (C) rat bile sample collected at 4–6 h after administration of R-HFBA. | ||
The linearity was evaluated by plotting the peak area ratio of HFBA to the IS versus the nominal concentration using least square linear regression with 1/x2 as weight factor. The calibration curves for HFBA showed good linearity over the range of 0.005–10 μg mL−1 in all matrices. Results are summarized in Table 1. The LLOQ of current assay for HFBA in all matrices was 0.005 μg mL−1 with S/N ratio more than 5 and acceptable accuracy (RE, %; −5.6 to −0.7%) and precision (RSD, %; ≤14.9%).
| Samples | Calibration curve | Regression coefficient (r) | LLOQ (μg mL−1) | RSD (%) |
|---|---|---|---|---|
| Urine | y = 3.26x + 4.49 × 10−3 | 0.9945 | 0.005 | 15.2 |
| Feces | y = 3.29x + 8.94 × 10−3 | 0.9968 | 0.005 | 7.6 |
| Bile | y = 3.36x + 1.55 × 10−3 | 0.9974 | 0.005 | 5.7 |
Precision and accuracy were determined in all matrices by analyzing six replicates of QC (quality control) samples at four different concentrations (LLOQ, low, middle, and high) on three continuous days. The intra-day and inter-day precision (RSD, %) were asked for no more than 15% except for LLOQ, whose precision was ≤20%. The accuracy (RE, %) was required to be within ±15% except for LLOQ (±20%). The results are presented in Table 2 and showed that all values were in the acceptable range.
| Matrix | Concentration (μg mL−1) | Precision RSD (%) | Accuracy RE (%) | |
|---|---|---|---|---|
| Inter-day | Intra-day | |||
| Urine | 0.005 | 14.9 | 10.2 | −5.6 |
| 0.01 | 9.8 | 7.8 | 1.4 | |
| 0.4 | 10.3 | 8.5 | 5.4 | |
| 8 | 5.2 | 9.1 | 4.1 | |
| Feces | 0.005 | 11.4 | 8.3 | −0.7 |
| 0.01 | 4.8 | 7.5 | −5.2 | |
| 0.4 | 10.7 | 5.7 | −6.3 | |
| 8 | 9.6 | 8.9 | 1.2 | |
| Bile | 0.005 | 13.7 | 10.4 | −4.5 |
| 0.01 | 9.2 | 11.8 | 2.8 | |
| 0.4 | 5.4 | 6.2 | 3.9 | |
| 8 | 6.8 | 5.4 | −0.6 | |
Extraction recoveries were determined using low, middle and high concentration levels of QC samples and evaluated by comparing peak area of HFBA obtained from processed QC samples with those from spike-after-extraction samples. The extraction recovery of HFBA in urine, feces and bile ranged 96.6–101.4%, 92.6–97.8% and 92.8–101.2%, respectively. The matrix effect was assessed by comparing peak areas of HFBA dissolved with blank post-extraction matrix supernate at three levels with those dissolved in the initial mobile phase at corresponding concentrations. IS (2 μg mL−1) was also evaluated similarly. Results are listed in Table 3. These results demonstrated that the extraction and purification method was reliable and suitable for HFBA and IS in urine, bile and feces.
| Matrix | Concentration (μg mL−1) | Recovery (%, mean ± SD) | Matrix effect (%, mean ± SD) |
|---|---|---|---|
| Urine | 0.01 | 98.9 ± 5.2 | 96.5 ± 3.6 |
| 4 | 101.4 ± 6.7 | 90.9 ± 5.4 | |
| 8 | 96.6 ± 3.4 | 101.5 ± 2.6 | |
| IS | 103.6 ± 4.8 | 92.6 ± 11.5 | |
| Feces | 0.01 | 97.8 ± 3.2 | 85.0 ± 11.4 |
| 4 | 93.7 ± 6.8 | 92.8 ± 7.9 | |
| 8 | 92.6 ± 10.2 | 101.6 ± 5.4 | |
| IS | 99.1 ± 5.9 | 103.2 ± 3.6 | |
| Bile | 0.01 | 101.2 ± 4.7 | 94.1 ± 3.2 |
| 4 | 92.8 ± 3.8 | 91.7 ± 6.8 | |
| 8 | 95.4 ± 2.3 | 90.4 ± 6.7 | |
| IS | 103.1 ± 2.9 | 92.6 ± 3.9 |
The stability of HFBA in urine, bile and feces were investigated by analyzing three levels of QC samples at 4 °C for 12 h (autosampler stability), at room temperature for 6 h (temperature stability), three freeze–thaw cycles (freeze–thaw stability) and at −80 °C for 30 days (long-term stability). The results shown in Table 4 indicated that HFBA were stable in all matrices under those different conditions.
| Matrix | Concentration (μg mL−1) | Frozen for 30 days | Room temperature for 6 h | Autosampler for 12 h | Three freeze–thaw cycles | ||||
|---|---|---|---|---|---|---|---|---|---|
| RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | ||
| Urine | 0.01 | 6.7 | 7.1 | 8.6 | 6.7 | 8.0 | −3.6 | 10.4 | −7.3 |
| 4 | 2.9 | 3.8 | 3.4 | 2.0 | 3.1 | 1.9 | 4.8 | 4.0 | |
| 8 | 9.4 | 10.8 | 11.5 | 4.9 | 7.9 | 7.2 | 9.3 | 8.1 | |
| Feces | 0.01 | 5.9 | −3.2 | 6.9 | −2.1 | 7.3 | −0.9 | 8.2 | 0.8 |
| 4 | 6.8 | 1.9 | 6.1 | −1.8 | 4.8 | −3.5 | 8.6 | −3.5 | |
| 8 | 5.7 | 3.7 | 5.8 | 7.4 | 8.2 | 4.6 | 4.1 | 7.7 | |
| Bile | 0.01 | 11.2 | 3.5 | 6.3 | −9.8 | 9.3 | −8.0 | 13.5 | −6.8 |
| 4 | 7.4 | 4.5 | 9.9 | 3.9 | 7.0 | 4.7 | 7.8 | 7.6 | |
| 8 | 2.8 | 6.3 | 4.2 | 6.4 | 4.8 | 7.9 | 4.2 | 9.9 | |
3.2 Excretion study
In this present study, the urine, feces and bile excretions of R-/S-HFBA in rats were studied respectively. The excretion ratio of R-/S-HFBA in three routes at different time periods was shown in Fig. 3. The cumulative excretion amount of R-/S-HFBA in three routes were listed in Table 5. In urine, the highest excretion ratio for R-/S-HFBA was found at 0–2 h. The urinary excretion ratios of R-HFBA at 0–2 h, 2–4 h and 6–8 h were significantly higher than those of S-HFBA (p < 0.01, p < 0.05), respectively. The cumulative excretion of R-/S-HFBA in urine during 48 h was 4425 ± 478 μg and 3489 ± 225 μg, which accounted for 40.2% and 31.7% of the total dose. Fecal excretion ratio of R-/S-HFBA was highest at 6–8 h and 10–12 h respectively, and did not find statistic difference at any time period between the enantiomers. The 48 h accumulative fecal excretion of R-/S-HFBA accounted for 25.6% and 26.8% of the total dose respectively. Meanwhile, there was a significant difference (p < 0.05) in biliary excretion ratio between R-HFBA and S-HFBA except for 0–2 h and 2–4 h. The unchanged R-HFBA and S-HFBA, excreted via bile during 12 h after oral administration, accounted for 11.3% and 7.4%. The total cumulative excretion of R-HFBA and S-HFBA were 65.8% and 58.5%, respectively. Of these, 14.3% and 19.4% of the oral dose were excreted directly from feces without absorption respectively. These results indicated that the total cumulative amount of three excretory pathways for R-/S-HFBA was relatively high and R-/S-HFBA existed stereoselective discrepancy in excretion.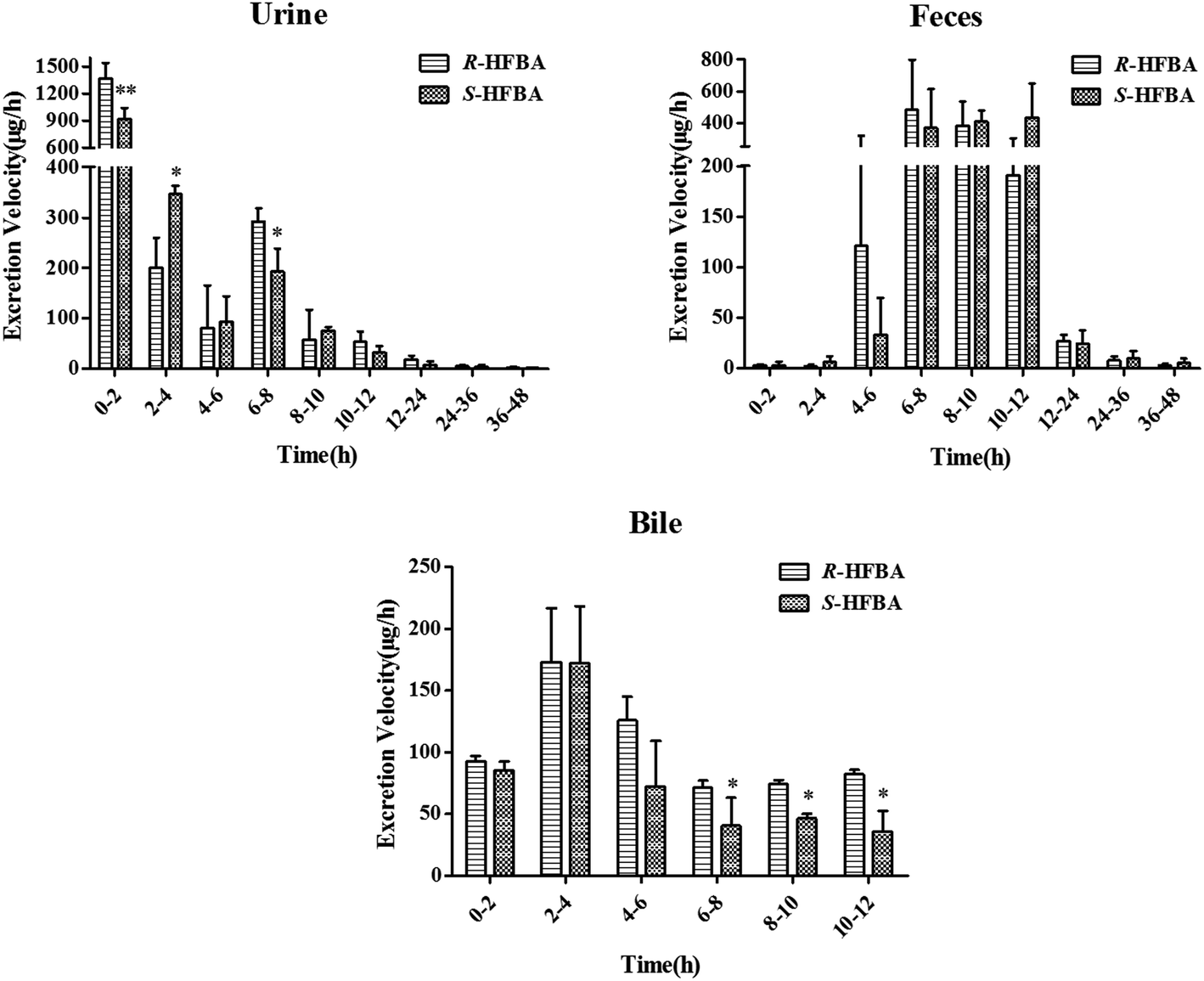 | ||
| Fig. 3 Excretion rate of unchanged R-/S-HFBA in rats urine, feces and bile at different time intervals following intragastric administration of a single dose at 50 mg kg−1. Data are represent as mean ± SD (n = 5). *p < 0.05, **p < 0.01 vs. R-HFBA at the same time intervals in the same samples. | ||
| Time (h) | Urine (μg) | Feces (μg) | Bile (μg) | |||
|---|---|---|---|---|---|---|
| R-HFBA | S-HFBA | R-HFBA | S-HFBA | R-HFBA | S-HFBA | |
| a *p < 0.05, **p < 0.01 vs. R-HFBA at the same times in the same samples; (—) not detected. | ||||||
| 2 | 2747 ± 332 | 1831 ± 253** | 6.300 ± 1.60 | 5.800 ± 7.70 | 185.3 ± 8.20 | 170.8 ± 14.1 |
| 4 | 3149 ± 292 | 2525 ± 285* | 11.00 ± 3.50 | 17.60 ± 18.9 | 531.7 ± 95.4 | 515.4 ± 77.5 |
| 6 | 3309 ± 457 | 2713 ± 185* | 254.1 ± 400 | 89.90 ± 80.3 | 783.1 ± 56.6 | 571.8 ± 95.4* |
| 8 | 3894 ± 504 | 3098 ± 279 | 1216 ± 518 | 791.6 ± 519 | 925.9 ± 68.1 | 652.9 ± 50.7* |
| 10 | 4010 ± 450 | 3249 ± 294* | 1980 ± 512 | 1604 ± 459 | 1075 ± 62.6 | 745.6 ± 57.7* |
| 12 | 4117 ± 435 | 3313 ± 319* | 2362 ± 629 | 2470 ± 203 | 1239 ± 54.6 | 817.0 ± 90.9* |
| 24 | 4333 ± 518 | 3413 ± 251* | 2687 ± 598 | 2764 ± 248 | — | — |
| 36 | 4394 ± 485 | 3469 ± 216* | 2782 ± 635 | 2878 ± 213 | — | — |
| 48 | 4425 ± 478 | 3489 ± 225* | 2817 ± 626 | 2944 ± 252 | — | — |
3.3 Identification of metabolites
The composition and accurate mass for the precursor or fragment product ions were analyzed using the Bruker Compass-Hystar and Data Analysis Software (Bruker, Germany). The maximum mass errors between the theoretical mass of molecular ions and determined values should be below 2 ppm, which demonstrating a high level of confidence in the proposed elemental compositions of the metabolites. Structures of the metabolites were further identified according to the retention time, MS and MS/MS data, the fragmentation pattern of the parent compound relevant and drug biotransformation knowledge.Compared with blank samples, parent compound R-/S-HFBA (M0) and their metabolites (M1–M8) were detected in the plasma, feces, bile, and urine samples. Representative extracted ion chromatograms of R-/S-HFBA and its metabolites in rats urine sample were shown in Fig. 4.
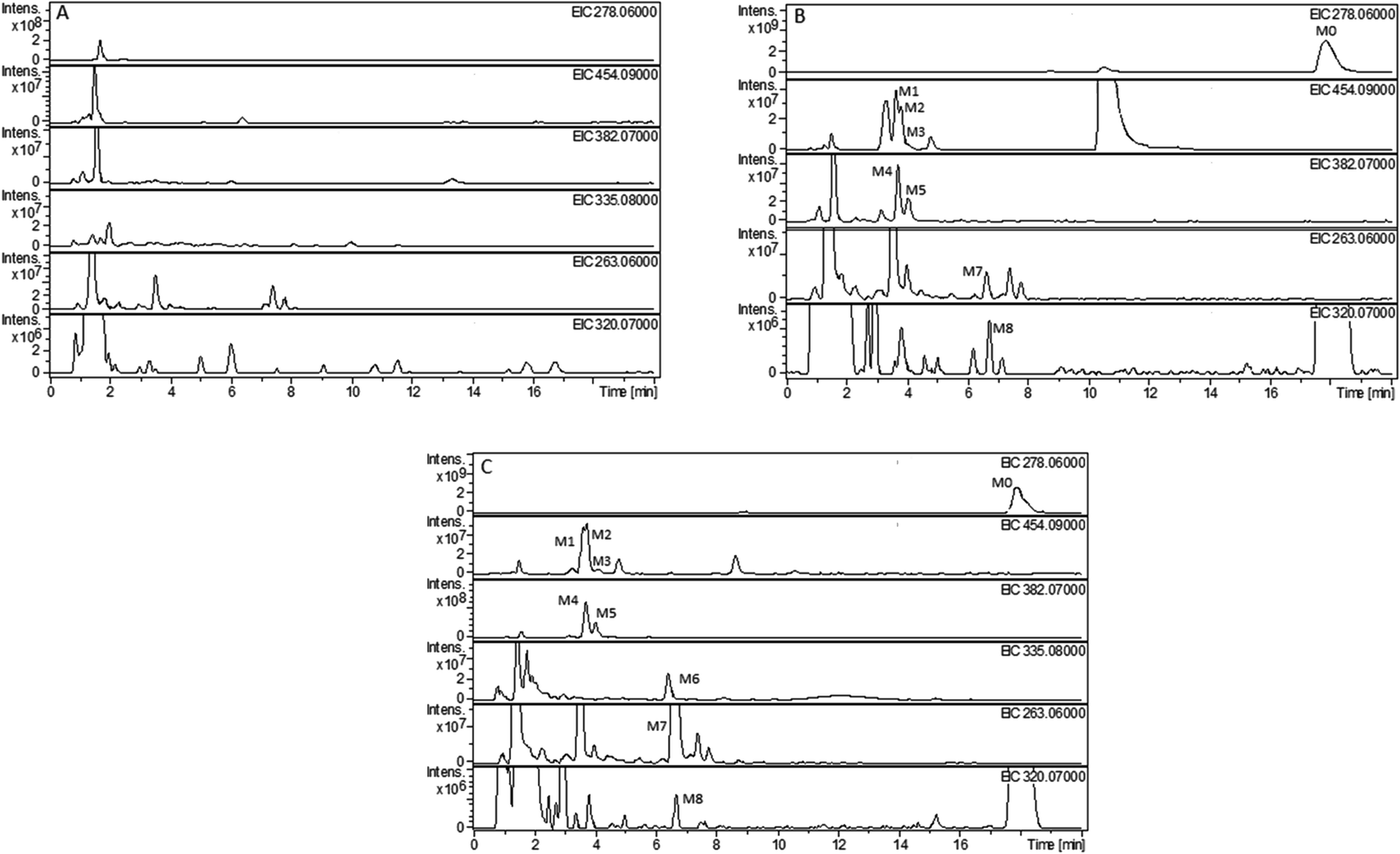 | ||
| Fig. 4 Representative extracted ion chromatograms (EICs) of R-/S-HFBA and its metabolites in rats urine sample after intragastric administration of R-/S-HFBA at a single dose of 100 mg kg−1: (A) blank urine sample from rats; (B) 0–24 h urine sample from rats after intragastric administration of R-HFBA; (C) 0–24 h urine sample from rats after intragastric administration of S-HFBA. | ||
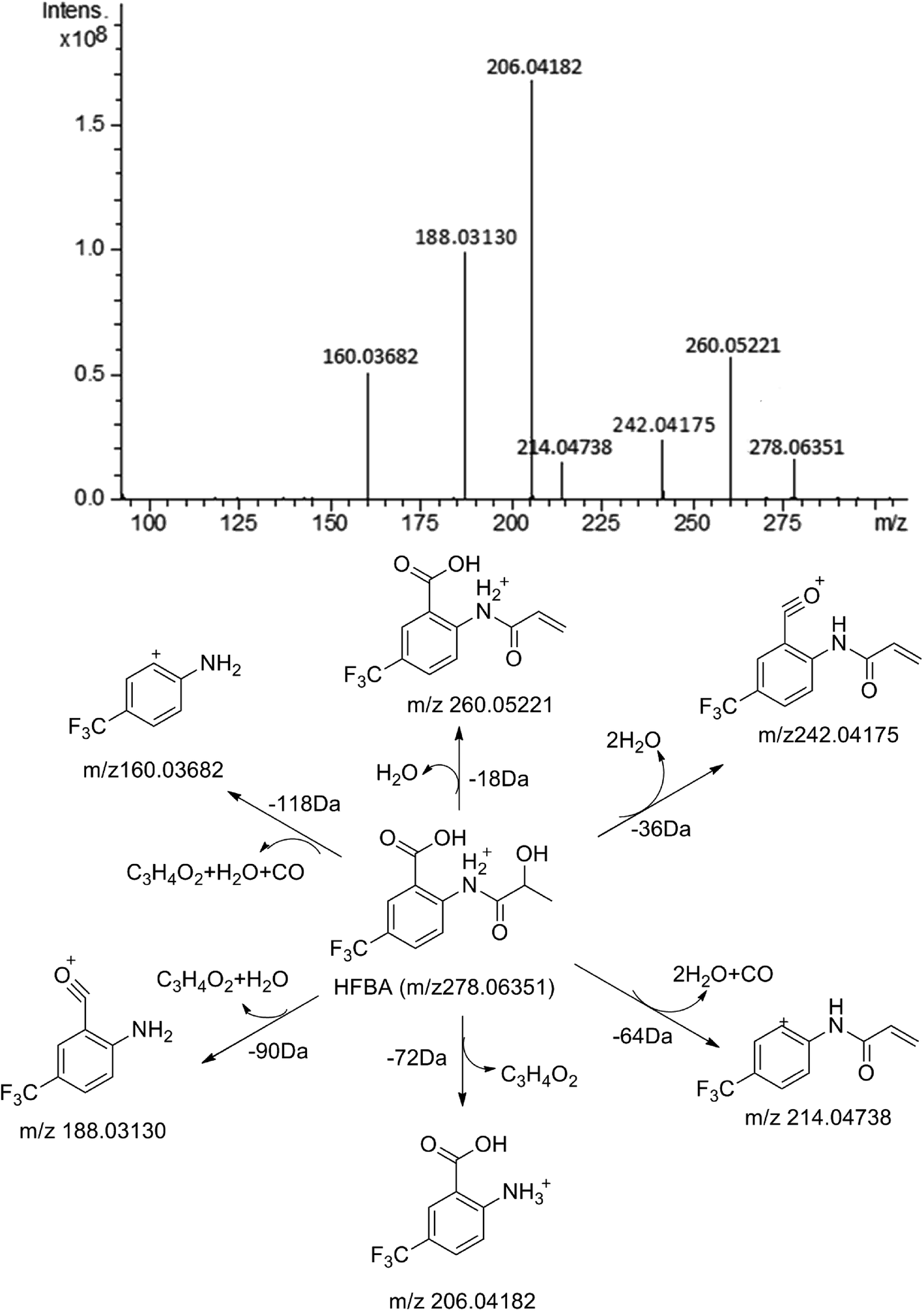 | ||
| Fig. 5 MS/MS spectrum of HFBA and its proposed fragmentation pathways. | ||
MS/MS spectrum of the metabolites were present in Fig. 6–8. The retention times, observed and calculated mass, mass errors, proposed elemental compositions and characteristic fragment ions of these metabolites are shown in Table 6. According to the above results, the possible metabolic pathways of HFBA were shown in Fig. 9.
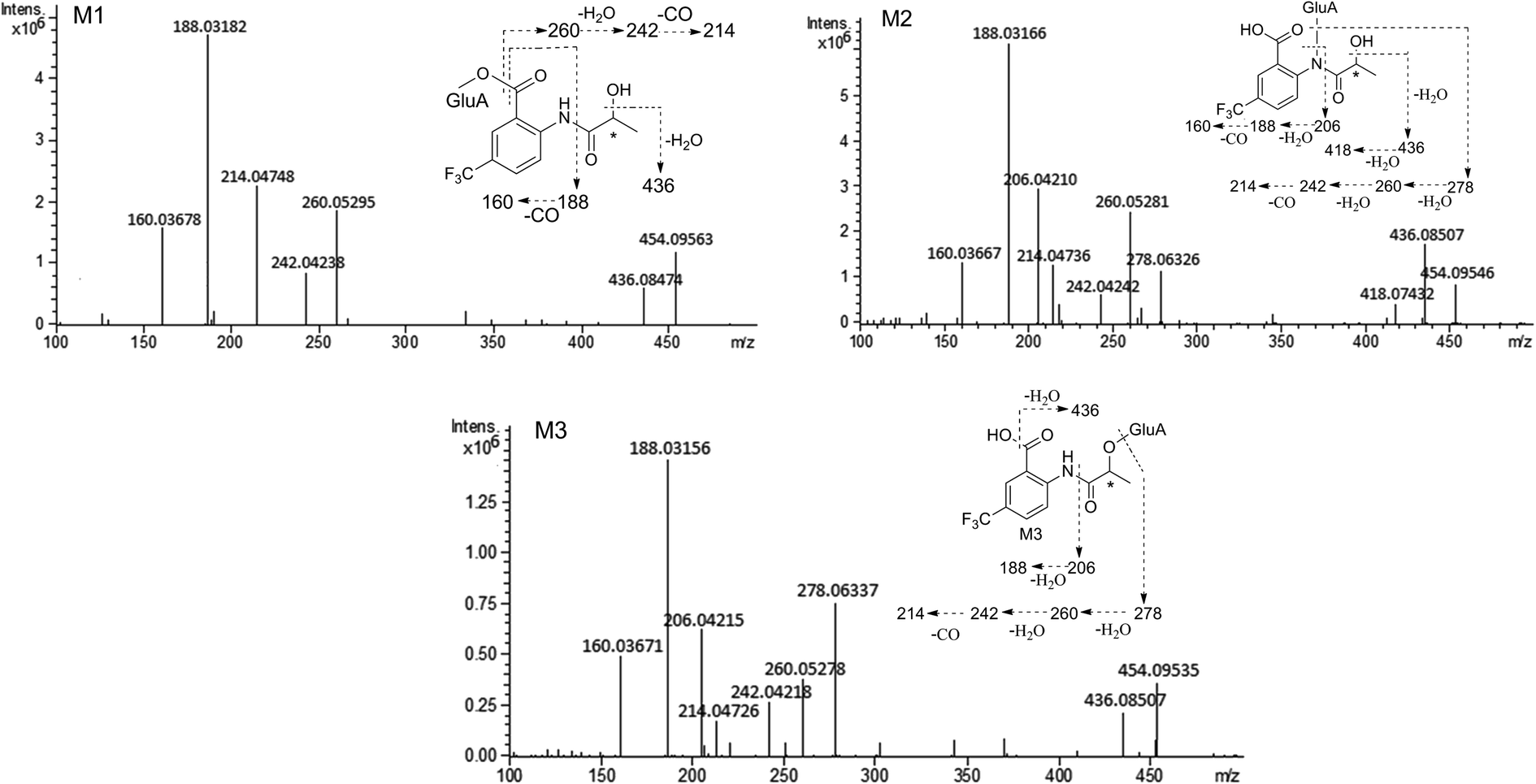 | ||
| Fig. 6 The MS/MS spectrum and major proposed fragment structures of M1, M2 and M3. | ||
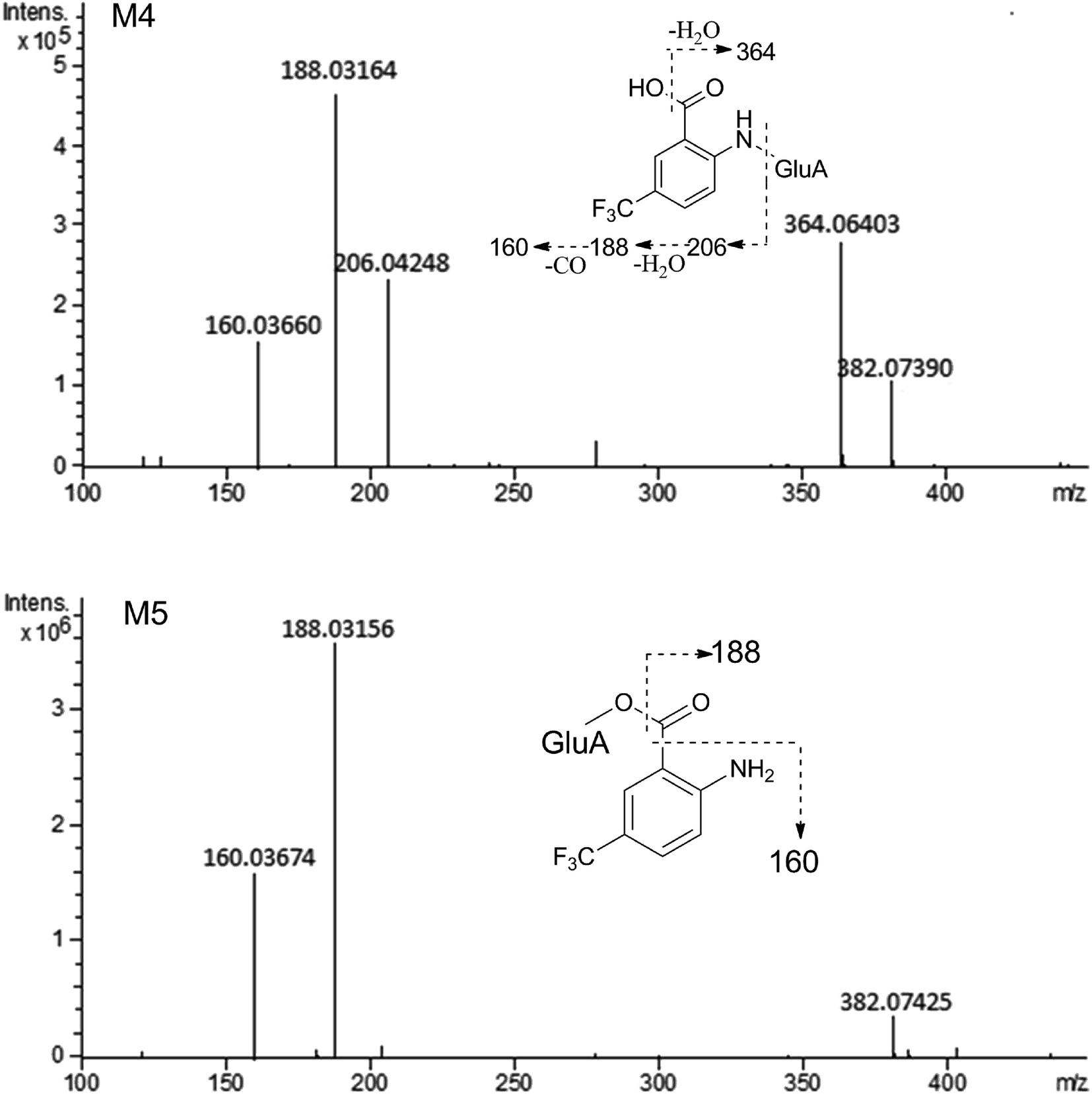 | ||
| Fig. 7 The MS/MS spectrum and major proposed fragment structures of M4 and M5. | ||
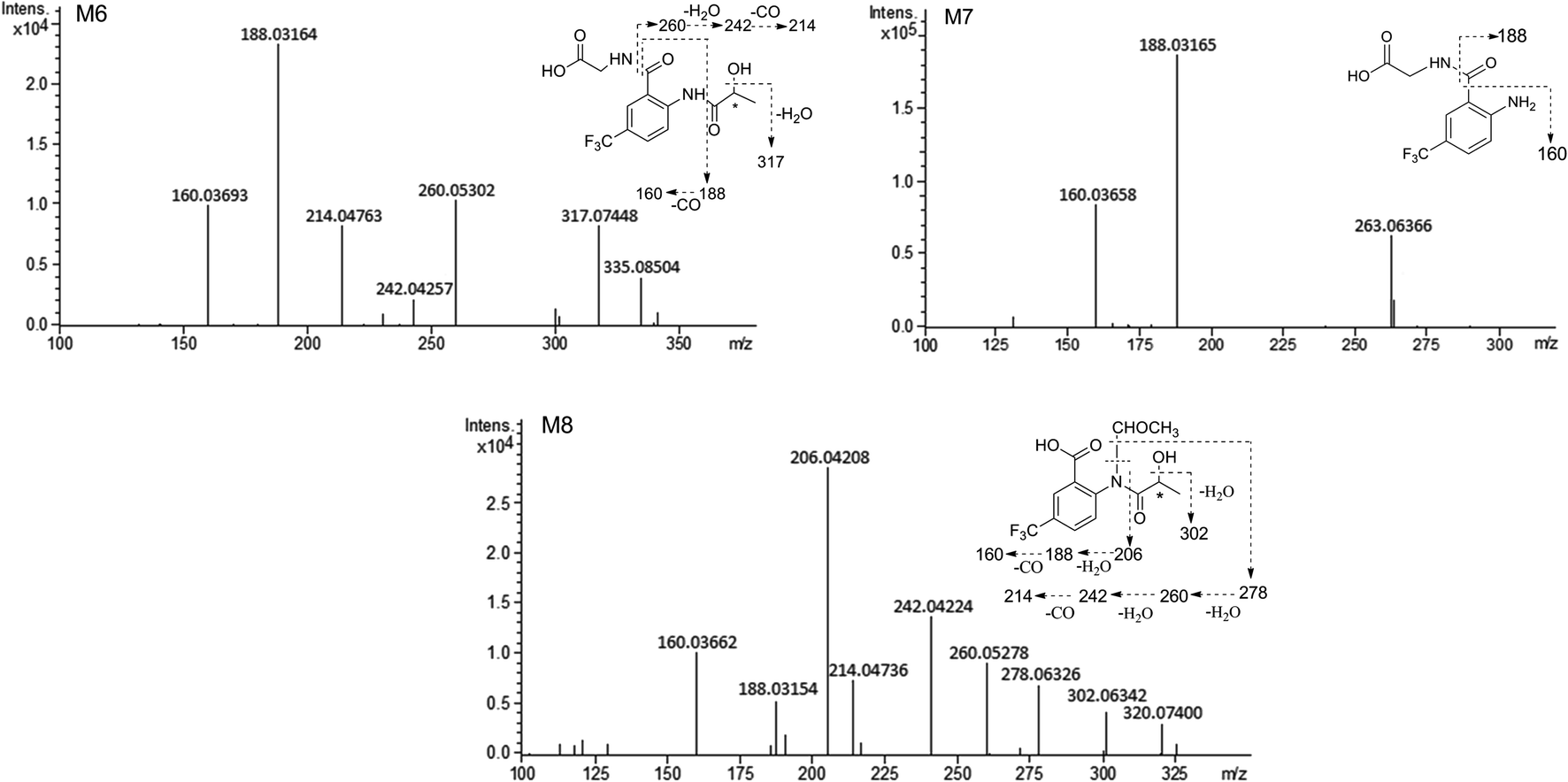 | ||
| Fig. 8 The MS/MS spectrum and major proposed fragment structures of M6, M7 and M8. | ||
| No. | RT (min) | Observed mass (Da) | Calculated mass (Da) | Error (ppm) | Formula | Product ion m/z (Da) | Metabolite description | Source |
|---|---|---|---|---|---|---|---|---|
| a u: urine samples, p: plasma samples, f: feces samples, b: bile samples; R: R-HFBA, S: S-HFBA. | ||||||||
| M0 | 17.87 | 278.06351 | 278.06347 | −0.17 | C11H10F3NO4 | 260.05221, 242.04175, 214.04738, 206.04182, 188.03130, 160.03682 | Parent | p (R, S), u (R, S),f (R, S), b (R, S) |
| M1 | 3.59 | 454.09563 | 454.09556 | −0.15 | C17H18F3NO10 | 436.08474, 260.05295, 242.04238, 214.04748, 188.03182, 160.03678 | Glucuronidation | u (R, S), b (R, S) |
| M2 | 3.76 | 454.09546 | 454.09556 | 0.22 | C17H18F3NO10 | 436.08507, 418.07432, 278.06326, 260.05281, 242.04242, 214.04737, 206.04210, 188.03166, 160.03667 | Glucuronidation | u (R, S), f (R, S), b (R, S) |
| M3 | 4.14 | 454.09535 | 454.09556 | 0.46 | C17H18F3NO10 | 436.08507, 278.06337, 260.05278, 242.04218, 214.04726, 206.04215, 188.03156, 160.03671 | Glucuronidation | u (R, S), f (R, S), b (R, S) |
| M4 | 3.69 | 382.07390 | 382.07443 | 0.14 | C14H14F3NO8 | 364.06403, 206.04248, 188.03164, 160.03660 | Hydrolysis + glucuronidation | p (S), u (R, S), b (R, S) |
| M5 | 4.03 | 382.07425 | 382.07443 | 0.47 | C14H14F3NO8 | 188.03156, 160.03674 | Hydrolysis + glucuronidation | u (R, S), b (R, S) |
| M6 | 6.43 | 335.08504 | 335.08493 | −0.33 | C13H13F3N2O5 | 317.07448, 260.05302, 242.04257, 214.04763, 188.03164, 160.03693 | Glycine conjugation | u (S), b (S) |
| M7 | 6.82 | 263.06366 | 263.06380 | 0.53 | C10H9F3N2O3 | 188.03165, 160.03658 | Hydrolysis + glycine conjugation | u (R, S), b (R, S) |
| M8 | 6.70 | 320.07400 | 320.07403 | 0.09 | C13H12F3NO5 | 302.06342, 278.06326, 260.05278, 242.04224, 214.04736, 206.04208, 188.03154, 160.03662 | N-Acetyl conjugation | u (R, S) |
 | ||
| Fig. 9 Proposed metabolic pathways of R-/S-HFBA in rats. | ||
4. Discussion
In excretion study, renal excretion was the main route of R-/S-HFBA elimination in rats after a single oral administration at 50 mg kg−1. This may be related to the smaller molecular weight and good water solubility of R-/S-HFBA.25 The total cumulative excretion of R-HFBA and S-HFBA in rats were 65.8% and 58.5%, respectively, suggesting that R-/S-HFBA was mainly excreted as the parent compounds and the rest portion could be eliminated by metabolism. This investigation presents some information on R-/S-HFBA excretion, which is important to research enterohepatic circulation, develop opportune dosage forms and devote pre-clinical studies of R-/S-HFBA. Furthermore, the excretion results demonstrated that significant differences in the excretion were observed between the two enantiomers. The cumulative excretions of R-HFBA in urine were higher than those of S-HFBA at the same time periods except for 6–8 h. These phenomena may be relatived with the stereoselective differences in the interaction between R-/S-HFBA and receptor transporter or enzymes during the renal tubular reabsorption and metabolism. There were also significant differences in the cumulative biliary excretion between R-HFBA and S-HFBA except for 0–2 h and 2–4 h, which could be associated with the participation of efflux transporters in hepatic epithelial cells at lower concentrations of R-/S-HFBA within 4–12 h. Besides, the different plasma protein binding may be another factor that lead to differences in excretion between R-/S-HFBA. Recent research in our laboratory had found stereoselective plasma protein binding for R-/S-HFBA.23The metabolism study adopted UHPLC-FT-ICR-MS method in positive ion mode for achieving higher mass resolution of HFBA and possible metabolites in the MS spectra. A total number of 8 metabolites of HFBA were firstly identified in the rat bile, urine, plasma and feces. The metabolic pathways included glucuronidation, glycine conjugation and acetylation. As reported in our previous study, 2-(2-hydroxypropanamido)benzoic acid (HPABA), the precursor compound of HFBA, underwent extensive metabolic reactions in plasma, urine, feces and bile, especially hydroxylation metabolism in the position of phenyl ring.22 In order to decrease significant metabolism to improve pharmacological activity and bioavailability, trifluoromethyl group was introduced into HPABA to synthesize HFBA.10 So according to this study, we found that HFBA was more metabolically stable than HPABA. The variety and the number of HFBA metabolites are significantly reduced compared with that of HPABA metabolites. Hydroxylation metabolite and glucuronic acid derivatives of ring-hydroxylated phase I metabolite could not be detected and there were only one or two metabolites found in rat plasma and feces after oral administration of HFBA. Drugs can be eliminated from the biological system through biotransformation into metabolites. This will affect both its bioavailability and its pharmacokinetic half-life after oral administration. Compounds that are stable in metabolism are likely to show good pharmacokinetic characteristics.26,27 Our previous research had confirmed that HFBA possessed better bioavailability and longer half-life than HPABA, as well as increased activity.10
In this present study, we found that glucuronidation was the main metabolic pathway of HFBA, including glucuronidation of HFBA (M1–M3) and glucuronidation of the hydrolysis product of HFBA (M4–M5). Glucuronidation is a type of phase II metabolic conjugation reaction which is catalyzed by the uridine 5′-diphospho-glucuronosyl transferases (UGTs). The glucuronidated drugs are much more hydrophilic than before and can be excreted via renal or biliary routes, which confers the detoxic effects to this pathway. The UGTs contain 22 enzymes, among which the members of the UGT1A and UGT2B subfamilies are responsible for drug metabolism.28 In order to further understand the metabolic mechanism of HFBA and predict potential drug metabolism interactions, our subsequent research will identify the specific UGT enzymes participated in the glucuronidation of HFBA by the selective inhibitors or single UGT isoenzyme.
Chirality is an intrinsic property to all biological organism. Macromolecules such as enzymes, proteins, polysaccharides and nucleic acids often own characteristic chiral structures.29 The stereospecificity of these macromolecules might result in different biochemical procedures (e.g. absorption, excretion and metabolism) between the enantiomers when interacting with chiral drugs. In our this present study, 8 metabolites of S-HFBA were identified in rats bile, urine, plasma and feces samples while R-HFBA existed 7 metabolites in such samples. There was no glycine conjugation metabolite of R-HFBA found. The amount of those metabolites between the enantiomers was also at a different level. Formation of metabolites exhibited stereoselectivity. Overall, R-HFBA underwent higher excretion and lower metabolism than S-HFBA in rats, which might be a new explanation why R-HFBA had significantly lower CL, longer t1/2 and larger AUC than S-HFBA after oral administration or intravenous to rats in pharmacokinetic studies.10 Different avenues may lead to the stereoselective metabolism of chiral drugs: (1) the metabolic enzymes may react with both enantiomers at different rates to generate different amounts of metabolites; (2) some stereoselective enzymes react with only one enantiomer; (3) sequential reaction with the substrate enantiomers by one enzyme. Our results indicated that R-HFBA was more metabolically stable than S-HFBA. Further study will concentrate on researching such enzymes more intensively to explore the stereoselective metabolism mechanism of R-/S-HFBA.
5. Conclusion
A rapid, sensitive and reliable UHPLC-MS/MS method for determination of R-/S-HFBA in rat urine, feces and bile samples was developed and fully validated. Following this method, the excretion of R-/S-HFBA in rats were completely revealed. Meanwhile, the metabolism of R-/S-HFBA in rats was also investigated for the first time though UHPLC-FT-ICR-MS method. A total of 8 metabolites were discovered and identified. Information obtained in this study provided valuable data for understanding R-/S-HFBA more clearly. The work is of significant value for the further investigation and development of R-/S-HFBA as an innovative drug.Conflicts of interest
All authors declare there is no conflict of interest to disclose.References
- J. M. Dias, T. V. Brito, D. A. Magalhães, P. W. Santos, J. A. Batista, E. G. Dias, H. Fernandes, S. R. Damasceno, R. O. Silva, K. S. Aragao, M. H. Souza, J. V. Medeiros and A. L. Barbosa, Gabapentin, a synthetic analogue of gamma aminobutyric acid, reverses systemic acute infammation and oxidative stress in mice, Inflammation, 2014, 37, 1826–1836 CrossRef CAS PubMed.
- K. Maurent, C. Vanucci-Bacqué, M. Baltas, A. Nègre-Salvayre, N. Augé and F. Bedos-Belval, Synthesis and biological evaluation of diarylheptanoids as potential antioxidant and anti-inflammatory agents, Eur. J. Med. Chem., 2017, 144, 289–299 CrossRef PubMed.
- S. Talwar, K. Nandakumar, P. G. Nayak, P. Bansal, J. Mudgal, V. Mor, C. M. Rao and R. Lobo, Anti-inflammatory activity of Terminalia paniculata bark extract against acute and chronic inflammation in rats, J. Ethnopharmacol., 2011, 134, 323–328 CrossRef PubMed.
- S. Arham, A. B. Syeda, I. B. Muhammad, S. Muhammad, S. Kishwar, B. T. Rasool and I. Javed, Ziziphora clinopodioides ameliorated rheumatoid arthritis and inflammatory paw edema in different models of acute and chronic inflammation, Biomed. Pharmacother., 2018, 97, 1710–1721 CrossRef PubMed.
- P. P. Rao, S. N. Kabir and T. Mohamed, Nonsteroidal Anti-inflammatory Drugs (NSAIDs): progress in small molecule drug development, Pharm, 2010, 3, 1530–1549 CAS.
- K. J. Woo and T. K. Kwon, Sulforaphane suppresses lipopolysaccharide-induced cyclooxygenase-2 (COX-2) expression through the modulation of multiple targets in COX-2 gene promoter, Int. Immunopharmacol., 2007, 7, 1776–1783 CrossRef CAS PubMed.
- J. L. Masferrer, B. S. Zweifel, P. T. Manning, S. D. Hauser, K. M. Leahy, W. G. Smith, P. C. Isakson and K. Seibert, Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3228–3232 CrossRef CAS PubMed.
- I. A. Alsarra, M. O. Ahmed, F. K. Alanazi, K. E. ElTahir, A. M. Alsheikh and S. H. Neau, Influence of cyclodextrin complexation with NSAIDs on NSAID/cold stress induced gastric ulceration in rats, Int. J. Med. Sci., 2010, 7, 232–239 CrossRef CAS PubMed.
- S. M. El-Moghazy, F. F. Barsoum and H. M. Abdel-Rahman, Synthesis and anti-inflammatory activity of some pyrazole derivatives, Med. Chem. Res., 2012, 21, 1722 CrossRef CAS.
- R. Rong, R. Z. Zhang, X. Wang, Y. H. Dan, Y. L. Zhao and Z. G. Yu, Synthesis, pharmacological evaluation and molecular docking of novel R-/S-2-(2-hydroxypropanamido)-5-trifluoromethyl benzoic acid as dual anti-inflammatory anti-platelet aggregation agents, Naunyn Schmiedebergs Arch Pharmacol., 2020, 393, 967–978 CrossRef CAS PubMed.
- N. J. Clarke, D. Rindgen, W. A. Korfmacher and K. A. Cox, Peer Reviewed: systematic LC/MS metabolite identification in drug discovery, Anal. Chem., 2001, 73, 430A–439A CAS.
- J. H. Lin and A. Y. Lu, Role of pharmacokinetics and metabolism in drug discovery and development, Pharmacol. Rev., 1997, 49, 403–449 CAS.
- E. J. Oliveira and D. G. Watson, Liquid chromatography-mass spectrometry in the study of the metabolism of drugs and other xenobiotics, Biomed. Chromatogr., 2000, 14, 351–372 CrossRef CAS PubMed.
- H. W. Darwish, A. A. Kadi, M. W. Attwa and H. S. Almutairi, Investigation of metabolic stability of the novel ALK inhibitor brigatinib by liquid chromatography tandem mass spectrometry, Clin. Chim. Acta, 2018, 480, 180–185 CrossRef CAS PubMed.
- S. C. Yeo, V. M. Sviripa, M. Huang, L. Kril, D. S. Watt, C. M. Liu and H. S. Lin, Analysis of trans-2,6-difluoro-4′-(N,N-dimethylamino)stilbene (DFS) in biological samples by liquid chromatography-tandem mass spectrometry: metabolite identification and pharmacokinetics, Anal. Bioanal. Chem., 2015, 407, 7319–7332 CrossRef CAS PubMed.
- G. Shankar, R. M. Borkar, S. Udutha, S. P. Anagoni and R. Srinivas, Identification and structural characterization of in vivo metabolites of balofloxacin in rat plasma, urine and feces samples using Q-TOF/LC/ESI/MS/MS: in silico toxicity studies, J. Pharm. Biomed. Anal., 2018, 159, 200–211 CrossRef CAS PubMed.
- S. Liu, Y. Che, F. Wang, Z. P. Shang, J. Q. Liu, S. Y. Dai, J. Y. Zhang and W. Cai, Identification of metabolites of 6′-Hydroxy-3,4,5,2′,4′-pentamethoxychalcone in rats by a combination of ultra-high-performance liquid chromatography with linear ion trap-orbitrap mass spectrometry based on multiple data processing techniques, Molecules, 2016, 21, 1266 CrossRef PubMed.
- R. Deng, Y. H. Xu, F. Feng and W. Y. Liu, Identification of poliumoside metabolites in rat feces by high performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 969, 285 CrossRef CAS PubMed.
- X. Zhang, C. J. Liang, J. T. Yin, Y. Q. Sun and L. T. Zhang, Identification of metabolites of liquiritin in rats by UHPLC-Q-TOF-MS/MS: metabolic profiling and pathway comparison in vitro and in vivo, RSC Adv., 2018, 8, 11813–11827 RSC.
- N. Yang, H. Wang, H. Q. Lin, J. L. Liu, B. S. Zhou, X. L. Chen, C. Z. Wang, J. P. Liu and P. Y. Li, Comprehensive metabolomics analysis based on UPLC-Q/TOF-MSE and the anti-COPD effect of different parts of Celastrus orbiculatus Thunb., RSC Adv., 2020, 10, 8396–8420 RSC.
- D. F. Smith, A. Kiss, F. E. Leach, E. W. Robinson, L. Pasa-Tolic and R. M. Heeren, High mass accuracy and high mass resolving power FT-ICR secondary ion mass spectrometry for biological tissue imaging, Anal. Bioanal. Chem., 2013, 405, 6069 CrossRef CAS PubMed.
- J. Guan, Q. L. Zhang, R. Rong, F. Han, H. Y. Zhu, Y. L. Zhao, A. H. Song and Z. G. Yu, Metabolic profile of 2-(2-hydroxypropanamido) benzoic acid in rats by ultrahigh performance liquid Chromatography combined with Fourier transform ion cyclotron resonance mass Spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 993–994, 60–68 CrossRef CAS PubMed.
- R. Rong, X. Wang, Y. H. Dan, R. Z. Zhang, Y. L. Zhao and Z. G. Yu, Determination of protein binding for novel 2-(2-hydroxypropanamido)-5-trifluoromethyl benzoic acid enantiomers to rats, dogs, and humans plasma by UPLC-MS/MS, Chirality, 2020, 32, 524–534 CrossRef CAS PubMed.
- US-FDA Guidance for Industry, Bioanalytical Method Validation. Guidance for Industry: Bioanalytical Method Validation, 2018, E-mail: https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf Search PubMed.
- J. P. Liu, Biopharmaceutics and Pharmacokinetics, People's Medical Publishing House, 4th edn, 2011, p. 157 Search PubMed.
- R. S. Obach, Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecifc binding to microsomes, Drug Metab. Dispos., 1999, 27, 1350–1359 CAS.
- V. Tong and F. S. Abbott, In vitro investigation of the hepatic extraction of RSD1070, a novel antiarrhythmic compound, J. Pharm. Pharm. Sci., 2001, 4, 15–23 CAS.
- A. Rowland, J. O. Miners and P. I. Mackenzie, The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification, Int. J. Biochem. Cell Biol., 2013, 45, 1121–1132 CrossRef CAS PubMed.
- K. Wiberg, Enantiospecific analysis and environmental behavior of chiral persistent organic pollutants (POPs), 2002, www.fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Evaluati Search PubMed.
Footnote |
| † Rong Rong and Qi-li Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |