
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic C–H aerobic and oxidant-induced oxidation of alkylbenzenes (including toluene derivatives) over VO2+ immobilized on core–shell Fe3O4@SiO2 at room temperature in water†
Pegah Mohammadpour and
Elham Safaei*
and
Elham Safaei*
Department of Chemistry, College of Sciences, Shiraz University, Shiraz, 7194684795, Iran. E-mail: e.safae@shirazu.ac.ir
First published on 22nd June 2020
Abstract
Direct C–H bond oxidation of organic materials, and producing the necessary oxygenated compounds under mild conditions, has attracted increasing interest. The selective oxidation of various alkylbenzenes was carried out by means of a new catalyst containing VO2+ species supported on silica-coated Fe3O4 nanoparticles using t-butyl hydroperoxide as an oxidant at room temperature in H2O or solvent-free media. The chemical and structural characterization of the catalyst using several methods such as FTIR spectroscopy, XRD, FETEM, FESEM, SAED, EDX and XPS showed that VO2+ is covalently bonded to the silica surface. High selectivity and excellent conversion of various toluene derivatives, with less reactive aliphatic (sp3) C–H bonds, to related benzoic acids were quite noticeable. The aerobic oxygenation reaction of these alkylbenzenes was studied under the same conditions. All the results accompanied by sustainability of the inexpensive and simple magnetically separable heterogeneous catalyst proved the important criteria for commercial applications.
Introduction
One of the most attractive and valuable fields in chemistry is the direct functionalization of C–H bonds.1 Generally, this type of bond is very strong and relatively unreactive, and can be found in organic materials and, in special cases, in natural hydrocarbons and petroleum contaminants. Hence, the activation of C–H bonds in these kinds of materials and consequent transformation of these worthless materials into useful and valuable compounds is a significant economical and green step in synthetic chemistry.2 Actually, if C–H bonds are activated selectively, the multistep syntheses of complex molecules can be transformed into facile one-step syntheses. In addition, waste reduction can be supposed from this straightforward and ecofriendly synthetic tool, which makes it a long-standing goal to find new organic scaffolds.3 It is worth mentioning that, because of high C–H bond dissociation energy, especially in terminal methyl groups, functionalization of C–H bonds is usually carried out in harsh conditions such as high temperatures, acidic or basic environment, and using strong oxidants or reductants.4 Therefore, designing a potential catalyst to achieve the goals of good C–H bond activation efficiency, low cost and low energy consumption is a challenging goal for scientific and industrial communities. Among various hydrocarbons, the benzene family and some of its structural relatives including toluene, ethylbenzene, and xylenes are members of the BTEX group of pollutants which are simply released into the environment by the petroleum and chemical industries and combustion processes.5 The selective aerobic oxidation of these compounds to their corresponding alcohol, aldehyde or acid would be a very desirable but challenging research project.6 Metal oxides often catalyze these types of reactions at high temperatures in which regulating the reactivity of oxygen is difficult.7 For example, bimetallic Pd/Au catalysts were developed to promote the air oxidation of toluene to provide suitable performance at a temperature of 160 °C.8 Solvent-free aerobic oxidation of toluene was investigated using anion modified mesoporous mixed oxides at a high temperature of 120 °C and 6 h with low toluene to benzaldehyde conversion value of 15.5%.9 Recently, selective C–H oxidation of toluene to benzaldehyde using manganese tungstate (MnWO4) nanobars in the presence of H2O2 as oxidant at 80 °C has been reported.10 It has been shown that the most effective transition metals such as palladium,11 platinum,12 rhenium,13 iridium,14 ruthenium15 and gold16 facilitate these efficient transformations through C–H activation pathways. However, the relatively high price, low natural abundance and partly strong toxicity have limited their application. Generally, in chemical reactions, heterogeneous catalysts have advantages over homogeneous catalysts due to their recyclability, easy separation from the reaction mixture and cost effectiveness which make them environmentally benign catalysts. One of the most important classes of heterogeneous catalysts is easily recoverable and reusable magnetic nanoparticles. The most popular magnetic nanoparticles are silica-coated iron oxide nanoparticles because of their high abundance, good physical and thermal stability against degradation, having dielectric property and hydrophobicity, safety and cheapness. Additionally, they are biocompatible and allow easy surface modification due to the presence of abundant silanol groups on their surface.17 Supported vanadium oxide catalysts constitute a very important class of heterogeneous catalysts that exhibit a notable ability to catalyze various reactions such as oxidation of sulfides,18 oxidative dehydrogenation,19 reduction of toxic NOx20 and epoxidation of olefins.21 On the other hand, some types of C–H functionalization have been reported using these catalysts as uneconomical reactions which often proceed in difficult situations with relatively low conversion and selectivity with high energy consumption.22 According to mentioned considerations, this research is concerned with a newly synthesized vanadyl species (VO2+) supported on silica-coated magnetic nanoparticles and its catalytic property evaluation toward C–H activation of alkylbenzenes (especially toluene derivatives).Results and discussion
Catalyst overview
Recently, the attention of research in green chemistry has focused on sustainable and recyclable catalysts which possess high activity and selectivity, cost effectiveness, and are easily available and structurally simple. Concerning the concept of green catalysts, silica-coated Fe3O4 magnetic nanoparticles (SiO2@Fe3O4) were synthesized by co-precipitation method and coated with silica. The presence of –OH groups on the silica surface and the oxophilic character of vanadium facilitated the immobilization of VO2+ species and produced VO2+ supported on silica-coated Fe3O4 (VO2+@SiO2@Fe3O4). It has been reported that vanadium species can immobilize on silica surfaces in different forms.23 The monomeric form of VO42+ species has a terminal V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and three V–O legs connected to the silica surface (Scheme 1A). Upon increasing the vanadium loading on silica surface, the vanadium species aggregate to oligomer species connected by V–O–V bridges (Scheme 1B) but the isolated monomeric species are supposed to be the most active species during the catalytic process owing to the higher accessibility of the vanadium site compared to other polymeric types.
O and three V–O legs connected to the silica surface (Scheme 1A). Upon increasing the vanadium loading on silica surface, the vanadium species aggregate to oligomer species connected by V–O–V bridges (Scheme 1B) but the isolated monomeric species are supposed to be the most active species during the catalytic process owing to the higher accessibility of the vanadium site compared to other polymeric types.
 | ||
| Scheme 1 Molecular structures of supported VO42+ species. (A) Monomer struture and (B) oligomer structure. | ||
Catalyst characterization
The FT-IR spectra of (A) SiO2@Fe3O4 and (B) VO2+@SiO2@Fe3O4 magnetic nanoparticles are shown in Fig. S1.† The bands around 466, 570–616 and 800–1300 cm−1 were assigned to the stretching vibrations of symmetric Si–O–Si, Fe–O and symmetric and asymmetric Si–O–Si bonds, respectively. Broad absorption bands of OH groups appeared at 3260–3572 cm−1 and the band centered around 1622 cm−1 is assigned to the bending vibrations of water. The stabilization of vanadyl species on magnetic nanoparticles was confirmed by the existence of a weak shoulder of VO stretching at 962 cm−1 as reported in the literature.24 The crystalline structure of VO2+@SiO2@Fe3O4 was explored by using powder XRD (Fig. S2†). All the diffraction peaks corresponded to (220), (311), (400), (422), (511) and (411) pattern, which indicated the cubic spinel crystalline structure of Fe3O4 according to standard data (JCPDS 19-0629).25 This could confirm the stability of the magnetic nanoparticles after coating with silica and immobilization of VO2+ species. To characterize the morphology of the catalyst, field-emission transmission electron microscopy (FETEM) and field-emission scanning electron microscopy (FESEM) were applied at different scales (Fig. 1). The uniform morphology of the catalyst is visible in both FETEM and FESEM images. Possibly van der Waals forces between particles lead to an aggregation process. Fig. 1F shows the selected area electron diffraction (SAED) pattern which confirms the cubic structure of magnetic nanoparticles. The elemental composition of VO2+@SiO2@Fe3O4 was characterized by energy-dispersive X-ray spectroscopy (EDX) (Fig. S3†) which determined the components of the catalyst. Also, the amount of vanadium incorporated on the silica-coated Fe3O4 nanoparticles was estimated to be 10.3 mmol g−1 by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis which confirmed the EDX results. Of note, due to the noticeable oxophilicity properties of vanadium especially in higher oxidation states,26 the notable percentage of vanadium loading is expected. On the other hand, wasting of VOCl3 has been prevented remarkably by the great affinity of vanadyl ions for binding on silica surfaces.
 | ||
| Fig. 1 FETEM images of VO2+@SiO2@Fe3O4: (A) 200 nm, (B) 50 nm, (C) 20 nm, (D) 5 nm, (E) after recycling. (F) SAED image and FESEM images of the catalyst: (G) 1 μm, (H) 500 nm and (I) 200 nm. | ||
Magnetization curve and the hysteresis loops of VO2+@SiO2@Fe3O4 show the superparamagnetic behaviour of the catalyst. As shown in Fig. S4,† the magnetic saturation value of magnetic nanoparticles is 18 emu g−1. It can be seen that the nanomaterials could be efficiently separated from solution by placing an external magnet near the vessel. For further characterization of the catalyst and investigation of vanadium valence state, X-ray photoelectron spectroscopy (XPS) was used (Fig. 2). A V 2p split was created by spin orbit coupling.27 The binding energy levels of 517.7 and 524.8 eV are attributed to V 2p3/2 and V 2p1/2, respectively, which match well with the literature value for V5+.28 In the XPS survey of catalyst, peaks related to Fe 2p, O 1s, Si 2s and Si 2p verify the presence of these elements in the catalyst.
 | ||
| Fig. 2 The XPS spectrum of VO2+@SiO2@Fe3O4. | ||
Catalyst activity
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [mg, mol%] | Solvent | O2/Ar | TBHP [equiv.] | T [h] | Conv. [%] | Sel. [%] to B |
| 1 | — | Solvent free | O2 | 2 | 9 | 5 | 97 |
| 2 | 30, 39 | Solvent free | O2 | 2 | 9 | 64 | 51 |
| 3 | 40, 52 | Solvent free | O2 | 2 | 9 | 81 | 50 |
| 4 | 60, 78 | Solvent free | O2 | 2 | 9 | 79 | 55 |
| 5 | 40, 52 | Solvent free | O2 | 4 | 9 | 85 | 42 |
| 6 | 40, 52 | Solvent free | Ar | 2 | 9 | 81 | 50 |
| 7 | 40, 52 | CH3CN | Ar | 2 | 9 | 90 | 60 |
| 8 | 40, 52 | H2O | Ar | 2 | 9 | 99 | 99 |
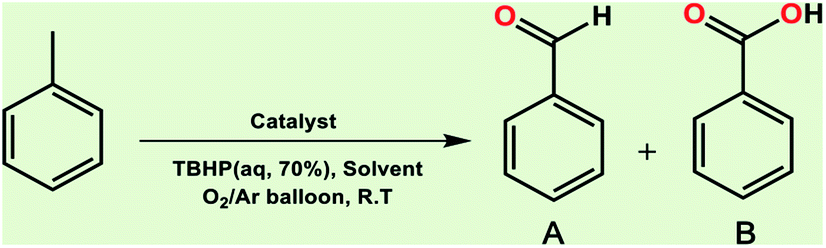
Optimization of conditions for other alkylbenzenes was performed in different situations. Ethylbenzene was chosen as a typical reactant for this reaction (Table 2). In the absence of catalyst, no progress was seen in ethylbenzene oxidation (Table 2, entry 1). Unlike toluene derivatives, a higher level of conversion and selectivity for solvent-free oxidation reactions was seen (Table 2, entries 2–6) and the green solvent systems, especially water, did not help to improve the reaction process (Table 2, entries 8–10). In addition, improvement in the conversion to oxygenated products was observed in the presence of oxygen balloon and not argon atmosphere (Table 2, entry 7), which suggests a different mechanism from that of toluene family. The efficiency of the catalyst in conversion of toluene was examined in the presence of various toluene derivatives including different electron-donating and electron-withdrawing substituents under optimized conditions (Table 3, entries 1–13).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [mg, mol%] | Solvent | O2/Ar | TBHP [equiv.] | T [h] | Conv. [%] | Sel. [%] to A |
| 1 | — | Solvent free | O2 | 2 | 8 | 4 | 97 |
| 2 | 30, 39 | Solvent free | O2 | 2 | 8 | 53 | 93 |
| 3 | 40, 52 | Solvent free | O2 | 2 | 8 | >99 | 99 |
| 4 | 50, 65 | Solvent free | O2 | 2 | 8 | 92 | 87 |
| 5 | 40, 52 | Solvent free | O2 | 1 | 8 | 75 | 100 |
| 6 | 40, 52 | Solvent free | O2 | 3 | 8 | 79 | 96 |
| 7 | 40, 52 | Solvent free | Ar | 2 | 8 | 73 | 93 |
| 8 | 40, 52 | CH3CN | O2 | 2 | 8 | 71 | 50 |
| 9 | 40, 52 | CH3CN/H2O | O2 | 2 | 8 | 74 | 90 |
| 10 | 40, 52 | H2O | O2 | 2 | 8 | 69 | 98 |

| Entry | Substrate | Major product (A) | Time (h) | Yield (%) | Selectivity to A (%) |
|---|---|---|---|---|---|
| a Reaction parameter: catalyst (40 mg, 52 mol%), substrate (1 mmol), H2O (0.5 mL), TBHP (2 equiv. 70% in H2O), argon balloon, T = RT, yields were determined by isolated yield after purification by chromatographic column.b Reaction parameter: catalyst (40 mg, 52 mol%), substrate (1 mmol), TBHP (2 equiv. 70% in H2O), oxygen balloon, T = RT, yields were determined by GC.c Reaction parameter: TBHP (4 equiv. 70% in H2O).d Reaction parameter: catalyst (40 mg, 52 mol%), substrate (1 mmol), H2O (0.5 mL), oxygen balloon, T = RT, yields were determined by GCe.e The GC analysis was performed in the presence of anisole as internal standard and the averages of 3 measurements are reported. GC method: 250 °C inlet, 280 °C detector, follow 1 mL min−1, oven temperature program: 50 °C for 2 min, 10 °C min−1 ramp to 250, and hold at 250 °C for 5 min. | |||||
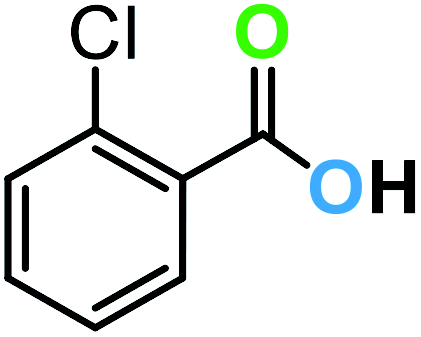
| 1a |  |
 |
8 | 99 | 92 |
| 2a |  |
 |
12 | 90 | 90 |
| 3a |  |
 |
12 | 87 | 89 |
| 4a |  |
 |
10 | >99 | 97 |
| 5a | 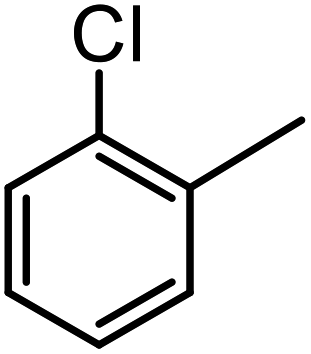 |
 |
14 | 72 | 88 |
| 6a |  |
 |
14 | 76 | 89 |
| 7a |  |
 |
14 | 82 | 97 |
| 8a |  |
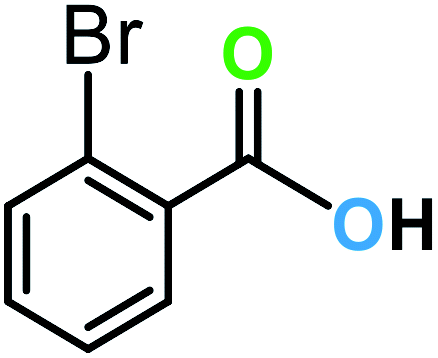 |
14 | 90 | 90 |
| 9a | 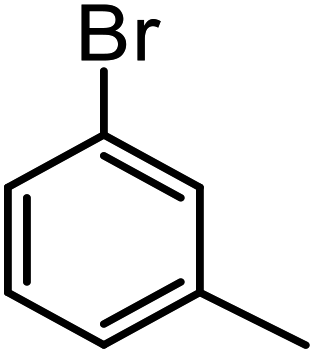 |
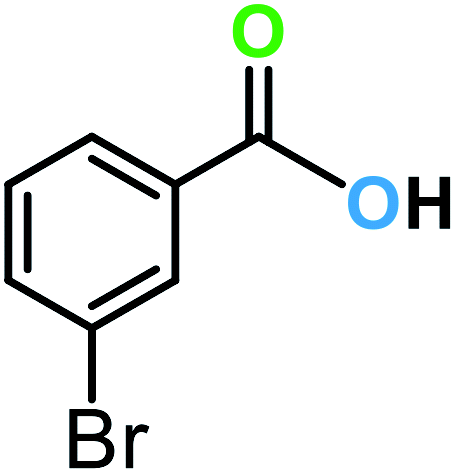 |
15 | 81 | 60 |
| 10a |  |
 |
14 | 99 | 64 |
| 11a |  |
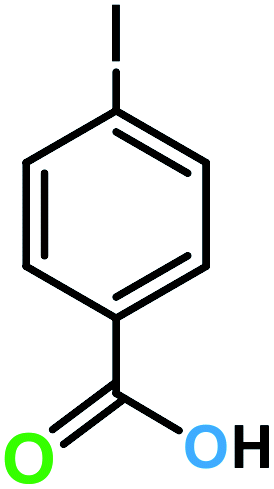 |
14 | >99 | 100 |
| 12a |  |
 |
14 | 91 | 89 |
| 13a |  |
 |
17 | 60 | 80 |
| 14b |  |
 |
8 | >99 | 96 |
| 15b,c |  |
 |
7 | 81 | 100 |
| 16b | 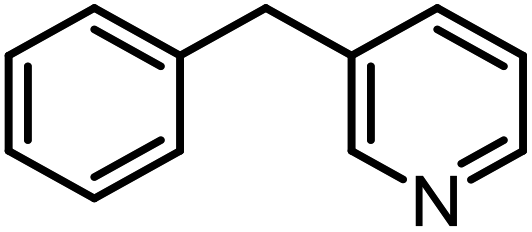 |
 |
7 | >99 | >99 |
| 17b,c |  |
 |
10 | 72 | 100 |
| 18d |  |
 |
10 | 99 | 99 |
| 19d |  |
 |
24 | 99 | 97 |
The best result was seen with toluene to benzoic acid conversion. The results in Table 3 confirmed that the inductive and resonance effects of ortho and para substitution can activate the C–H bond more than in meta derivatives (Table 3, entries 2–10). In comparison with electron-withdrawing groups, the electron-donating substitutions continued the reactions more effectively (Table 3). According to the observations, in the presence of TBHP, the following mechanism is proposed for C–H activation of toluene (Scheme 2). After addition of TBHP, t-BuOOH–vanadyl adduct 1 was formed and the reaction pathway passed through the four-centered transition state of 2 in the presence of toluene. In continuing, t-BuOH was released and the oxygen species of 3 formed at the same time. Detection of t-BuOH by GC technique is a good indication to confirm this claim. Fig. 3 shows the time monitoring of ethylbenzene consumption and t-BuOH production during the reaction process. The presence of H2O accelerated the separation of product toward the formation of benzoic acid and improved the selectivity.
 | ||
| Scheme 2 Proposed mechanism for toluene conversion to benzoic acid using TBHP. | ||
 | ||
| Fig. 3 Time monitoring of ethylbenzene consumption and t-BuOH production. | ||
In another experiment, in the presence of oxygen atmosphere, ethylbenzene was oxidized to acetophenone with high selectivity and full conversion (Table 3, entry 14). As the results show (Table 3, entries 15–17), by increasing the steric effect of substituents on the phenyl ring of ethylbenzene, the conversion percentages were decreased while the selectivity improved remarkably. It seems that molecular oxygen assisted the reaction progress. As the cleavage of C–H bonds in the middle of an alkyl chain is easier than that of terminal ones, aerobic oxidation of ethylbenzene was performed in mild conditions compared to toluene. The proposed mechanism for oxidation of ethylbenzene and other substrates (entries 14–17) is illustrated in Scheme 3. The hydrogen bonding shown in adduct 2 is formed by the approach of alkylbenzene to the catalyst and TBHP which consequently produces the superoxo-vanadium species 3. After O–O bond cleavage, removal of t-BuOH (detected by GC monitoring) and H abstraction step, the oxygenated compound was produced. In the absence of O2, the observed conversion of ethylbenzene oxidation reaction was 73% (Table 2, entry 7) which suggested that the mechanism was completed aerobically by protonated form of catalyst 5. As in previous steps, the reaction progressed to the final step through superoxo-vanadium intermediate 7 assisted by O2 with an accompanying loss of H2O and conversion of remaining alkylbenzenes. For further mechanism investigation, the oxidation reactions of toluene and ethylbenzene were carried out in the presence of TEMPO (1 mmol) and identical results strongly indicated that the reaction did not proceed via a radical chain pathway.
 | ||
| Scheme 3 Proposed mechanism for other alkylbenzene oxidation reactions using TBHP. | ||
The pathway of toluene aerobic oxidation is suggested as shown in Scheme 4. It is expected that the spin-state of O2 in the ground state is a triplet while many organic substrates are singlet and their reactions are spin-forbidden. But transition metal complexes could react with triplet O2 to produce metal–oxygen intermediates. It was proposed that after hydrogen interaction between toluene and VO2+, the molecular oxygen in reaction media facilitated oxygen transfer to toluene. Afterwards, the OO bond in the formed species was dissociated by water and the catalyst was recovered. The aerobic oxidation of other substrates in Table 3 was not quite so successful and the obtained conversion at around 24 h did not exceed the 15% conversion yield.
 | ||
| Scheme 4 Proposed mechanism for aerobic oxidation of toluene to benzyl alcohol. | ||
A comparison of time dependence of conversion in both oxygen- and oxidant-induced reactions is clearly shown in Fig. 4.38
 | ||
| Fig. 4 Time monitoring of toluene conversion. | ||
Results of hot filtration test and reusability of the catalyst
In order to investigate whether VO2+ was released from the solid support and to confirm that the reaction pathway proceeded heterogeneously, a hot filtration test for ethylbenzene oxidation reaction under optimized reaction conditions was done. The reaction was monitored by GC in half of the reaction time and the obtained resulting conversion was 42% (Fig. 5A). | ||
| Fig. 5 (A) Monitoring and hot filtration test. (B) Recycling of catalyst in ethylbenzene oxidation reaction. | ||
According to green chemistry principles, the simple recovery and durability of a catalyst are some of the most important features that also should be considered from an economic viewpoint in industries. To confirm the reusability of the catalyst, it was collected, washed and used several times. After 6 reaction cycles, a 9% reduction of conversion was seen (Fig. 5B). FETEM observation confirmed the maintenance of the morphology of the recovered catalyst (Fig. 1E). Furthermore, ICP analysis determined that the leached amount of VO2+ after completion of cycle 7 was negligible (<1 mol%). Therefore, the stability and reusability of the catalyst were appropriate for its use in important reactions.
Particularly, reports of C–H bond oxidation reactions (especially for toluene and its derivatives) using heterogeneous catalysts at room temperature are rare and a comparison of this work with recent reports indicates distinctly the efficacy of this catalyst (Table 4).
| Entry | Catalyst | Reaction conditions | Conv. (%) | Major product |
|---|---|---|---|---|
| 1 (ref. 29) | MnWO4 nanobars (0.01 g, 5 wt%) | Toluene (0.2 mL), CH3CN (8 mL), H2O2 (3 equiv.), T = 80 °C, time = 24 h | 60 | Benzaldehyde |
| 2 (ref. 30) | Mn0.3Zr0.7O2 | Toluene (1000 ppm), 20% O2 balanced by N2, total flow rate = 50 mL min−1, weight hourly space velocity = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1, T = 235 °C, in a fixed-bed quartz tubular reactor 000 mL gcat−1 h−1, T = 235 °C, in a fixed-bed quartz tubular reactor |
90 | Maleic anhydride |
| 3 (ref. 31) | Cu functionalized nano-crystalline ZSM-5 (1 g, 0.4 wt%) | Toluene (25 mL), deionized water (25 mL), H2O2 (25 mL), T = 180 °C, time = 4 h, in a PARR autoclave reactor, N2 gas pressure = 5.0 bar | 96 | Benzoic acid |
| 4 (ref. 32) | Pd–Ag@CeO2 (100 mg) | Toluene (500 ppm), 20% O2 balanced with N2, T = 88 °C, total gas flow rate = 33.3 mL min−1, visible light intensity = 160 mW cm−2 | 50 | Not identified |
| 5 (ref. 33) | Polystyrene grafted vanadium Schiff base complex (30 mg, 6.58 wt%) | Toluene (5 mmol), CH3CN (10 mL), H2O2 (15 mmol), T = 65 °C, time = 6 h | 79 | Benzaldehyde |
| 6 (ref. 34) | Pt28 subnanocatalyst (10 mg, 0.216 wt%) | Toluene (2 mL), O2 (1 MPa), T = 160 °C, time = 15 h, in an autoclave reactor | 1000 μmol product | Benzoic acid |
| 7 (ref. 35) | MnTPPCl (1.0 × 10−3 mmol) | Toluene (5 mmol), cyclohexene (3 mmol), CH3CN (10 mL), O2 (1.2 MPa), T = 160 °C, time = 4 h | 13 | Benzoic acid |
| 8 (ref. 36) | Fe3O4@SiO2-APTES-MnLGDC (40 mg, 2 mol%) | Toluene (1 mmol), solvent free, TBHP (4 equiv., 70%), T = 60 °C, time = 20 h | 71 | Benzoic acid |
| This work | VO2+@SiO2@Fe3O4 (40 mg, 52 mol%) | Toluene (1 mmol), H2O (0.5 mL), O2 balloon, T = RT, time = 10 h | 99 | Benzyl alcohol |
| Toluene (1 mmol), H2O (0.5 mL), Ar, TBHP (2 equiv., 70%), T = RT, time = 8 h | 99 | Benzoic acid |
Conclusions
In conclusion, to convert aromatic hydrocarbons especially toluene and xylenes which are typical petroleum contaminants to useful compounds for other industries, we have introduced a highly efficient and simple earth-abundant vanadyl-based magnetic nano-catalyst. We applied a green solvent or no solvent and relatively green reagents with the minimum organic material usage under very mild reaction conditions of room temperature to produce important oxygenated compounds through C–H activation reaction. Aerobic and totally green oxidation reactions of toluene and ethylbenzene demonstrated that immobilization of VO2+ on silica-coated Fe3O4 nanoparticles made it an excellent catalyst which can be used in important industrial reactions with minimal energy consumption and pollution. Furthermore, the simplicity of the catalyst structure and good capability of the oxo vanadium(IV) cation in the oxygen transfer process as well as recoverability and reusability of the nanomagnet make the catalyst efficient and sustainable. All these points show that we are standing at the closest point to the dream of green chemistry for the alkylbenzene C–H activation process.Experimental section
Materials and methods
All reagents and solvents were purchased from Merck and Sigma Aldrich sources and used without further purification. FTIR spectra from KBr pellets of the compounds were recorded using a Shimadzu FT-IR-8300 spectrophotometer. The morphological topographies of particles were evaluated using FETEM (JEOL, JEM-2100F, 200 kV). FESEM analysis and EDX were performed with a Tescan Mira (II) at an acceleration voltage of 15 kV. The X-ray powder patterns were obtained using a Philips PW1730 (step size: 0.05, time per step: 1 s). The magnetic properties of the catalyst were measured using VSM with homemade vibrating sample magnetometer apparatus (Meghnatis Daghigh Kavir Company, Iran) at RT from −10000 to +10000 Oe. The electronic states of the powders were investigated using XPS (Multilab 2000, Thermo Scientific, Al Kα radiation). Furthermore, the reactions were followed using thin layer chromatography (TLC) and gas chromatography (GC, Agilent gas chromatograph model 7890A) with an HP-1 methyl siloxane column (30 m in length × 320 μm × 0.25 μm) using a flame ionization detector.
Synthesis of magnetic nanoparticles (SiO2@Fe3O4)
Magnetic nanoparticles (Fe3O4) were prepared using a co-precipitation method according to a literature procedure.39 5.2 g of FeCl3·6H2O and 2.8 g of FeSO4 were dissolved in 25 mL of deionized water, followed by adding 0.85 mL of HCl (37%) under Ar gas. The resulting solution was added dropwise into 25 mL of 1.5 mol L−1 NaOH solution with mechanical stirring under argon atmosphere at 80 °C. The produced Fe3O4 nanoparticles were magnetically separated and rinsed three times with water and ethanol. According to the literature,40 Fe3O4 nanoparticles (0.5 g) were dispersed in a mixture of ethanol (50 mL), water (5 mL), and tetraethoxysilane (0.20 mL). Afterward, 5 mL NaOH (10 wt%) was added dropwise and the reaction was allowed to proceed under mechanical stirring. After stirring at room temperature for 30 min, the SiO2@Fe3O4 particles were separated using a magnetic field and washed with ethanol and water three times and dried under vacuum at 80 °C for 10 h.Synthesis of catalyst
The silica-coated Fe3O4 nanoparticles were dried at 80 °C overnight. Incorporation of VO2+ species was carried out in an argon-filled glove box. To a stirring suspension of SiO2@Fe3O4 (1 g) in dry dichloromethane (40 mL), 0.2 mL of VOCl3 was added dropwise and the reaction mixture continued stirring for 3 days at room temperature. The resulting material was separated from the reaction mixture using an external magnet and washed three times with dichloromethane, acetone and ethanol. The final light-brown to green material was dried at 80 °C for 12 h.General procedure for oxidant-mediated and aerobic oxidation of alkylbenzenes
In a 5 mL round-bottom flask, a mixture of 40 mg catalyst and 1 mmol toluene derivative was stirred in the presence of water (0.5 mL) and 2 mmol TBHP as oxidant was added. The reaction was carried out under argon atmosphere and at room temperature. Other alkylbenzene oxidation processes were carried out under oxygen atmosphere and solvent-free reaction conditions.In order to perform aerobic oxidation reactions, the catalyst (40 mg, 52 mol%) and substrate (1 mmol) were stirred in the presence of 0.5 mL water (saturated with oxygen for 15 minutes), under oxygen balloon atmosphere at room temperature.
The conversion and selectivity were monitored by GC or TLC (Table 3, entries 5–13) and, after completion of the reaction, the mixture was extracted with ethyl acetate and then the catalyst was recovered using an external magnet. The products were collected, purified and identified by 1H NMR spectroscopy.
Hot filtration test and reusability of the catalyst
A hot filtration test for ethylbenzene oxidation reaction under optimized reaction conditions was done. For this purpose, after 4 h, the catalyst was separated easily using an external magnet and the reaction was allowed to go on for a further 5 h. The reaction process was monitored by GC and the obtained resulting conversion was only 42% (Fig. 5A). To confirm the reusability of the catalyst, after the completion of the first ethylbenzene oxidation reaction cycle, the catalyst was removed from the reaction mixture magnetically, washed several times with ethyl acetate and ethanol, dried at 70 °C and used in a new reaction cycle. This was repeated for 6 reaction cycles. FETEM and ICP analysis determined the VO2+ leaching after completion of cycle 7.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the Department of Chemistry, Shiraz University for support. E. Safaei would like to thank Iran National Science Foundation (INSF) for its support (grant no. 98013021).References
- (a) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2010, 111, 1293–1314 CrossRef PubMed; (b) S. Y. Zhang, F. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC; (c) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (d) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (e) C. M. Che, V. K. Lo, C. Y. Zhou and J. S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC; (f) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC; (g) L. McMurray, F. ÓHara and M. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- (a) J. Hess, D. Bednarz, J. Bae and J. Pierce, Am. J. Public Health, 2011, 101, 1568–1579 CrossRef PubMed; (b) B. S. Schwartz, C. L. Parker, J. Hess and H. Frumkin, Am. J. Public Health, 2011, 101, 1560–1567 CrossRef PubMed; (c) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- (a) H. M. Davies, J. Du Bois and J. Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC; (b) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548–6551 CrossRef CAS PubMed.
- (a) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245–1262 CrossRef CAS; (b) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (c) D. Wei, X. Zhu, J. L. Niu and M. P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef CAS; (d) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed; (e) L. Ackermann, J. Org. Chem., 2014, 79, 8948–8954 CrossRef CAS PubMed.
- (a) J. A. Cunningham, H. Rahme, G. D. Hopkins, C. Lebron and M. Reinhard, Environ. Sci. Technol., 2001, 35, 1663–1670 CrossRef CAS PubMed; (b) J. A. MacDonald, Environ. Sci. Technol., 2000, 34, 346A–353A CrossRef CAS PubMed.
- (a) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230–234 CrossRef CAS PubMed; (b) K. T. Smith, S. Berritt, M. González-Moreiras, S. Ahn, M. R. Smith, M.-H. Baik and D. J. Mindiola, Science, 2016, 351, 1424–1427 CrossRef CAS PubMed; (c) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS; (d) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed; (e) Y. Liu, T. N. Gao, X. Chen, K. Li, Y. Ma, H. Xiong and Z. A. Qiao, Inorg. Chem., 2018, 57, 12953–12960 CrossRef CAS PubMed.
- (a) J. Lv, Y. Shen, L. Peng, X. Guo and W. Ding, Chem. Commun., 2010, 46, 5909–5911 RSC; (b) E. Janiszewska, A. Held, K. Nowińska and S. Kowalak, RSC Adv., 2019, 9, 4671–4681 RSC; (c) F. M. Bautista, J. M. Campelo, D. Luna, J. Luque and J. M. Marinas, Appl. Catal., A, 2007, 325, 336–344 CrossRef CAS.
- (a) C. Evangelisti, E. Schiavi, L. A. Aronica, A. M. Caporusso, G. Vitulli, L. Bertinetti, G. Martra, A. Balerna and S. Mobilio, J. Catal., 2012, 286, 224–236 CrossRef CAS; (b) L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- X. Wang, G. Wu, F. Wang, H. Liu and T. Jin, Catal. Commun., 2017, 98, 107–111 CrossRef CAS.
- D. D. Mal, S. Khilari and D. Pradhan, Green Chem., 2018, 20, 2279–2289 RSC.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- J. A. Labinger, Chem. Rev., 2016, 117, 8483–8496 CrossRef PubMed.
- Y. Horino, Angew. Chem., Int. Ed. Engl., 2007, 46, 2144–2146 CrossRef CAS PubMed.
- A. M. Voutchkova and R. H. Crabtree, J. Mol. Catal. A: Chem., 2009, 312, 1–6 CrossRef CAS.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (b) N. S. Upadhyay, V. H. Thorat, R. Sato, P. Annamalai, S. C. Chuang and C. H. Cheng, Green Chem., 2017, 19, 3219–3224 RSC.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J. D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC.
- (a) F. Gregori, I. Nobili, F. Bigi, R. Maggi, G. Predier and G. Sartori, J. Mol. Catal. A: Chem., 2008, 286, 124–127 CrossRef CAS; (b) I. Khedher, A. Ghorbel, J. M. Fraile and J. A. Mayoral, J. Mol. Catal. A: Chem., 2006, 255, 92–96 CrossRef CAS.
- (a) K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvi and M. Taoufik, Organometallics, 2013, 32, 6452–6460 CrossRef CAS; (b) B. S. Liu, G. Rui, R. Z. Chang and C. T. Au, Appl. Catal., A, 2008, 335, 88–94 CrossRef CAS; (c) X. Rozanska, E. V. Kondrateriko and J. Sauer, J. Catal., 2008, 256, 84–94 CrossRef CAS; (d) B. Solsona, T. Blasco, J. M. L. Nieto, M. L. Pena, F. Rey and A. Vidal-Moya, J. Catal., 2001, 203, 443–452 CrossRef CAS.
- (a) A. Boyano, M. C. Iritia, I. Malpartida, M. A. Larrubia, L. J. Alemany, R. Moliner and M. J. Lazaro, Catal. Today, 2008, 137, 222–227 CrossRef CAS; (b) I. E. Wachs, G. Deo, B. M. Weckhuysen, A. Andreini, M. A. Vuurman, M. deBoer and M. D. Amiridis, J. Catal., 1996, 161, 211–221 CrossRef CAS.
- (a) G. L. Linden and M. F. Farona, Inorg. Chem., 1977, 16, 3170–3173 CrossRef CAS; (b) A. Held, J. Kowalska-Kuś, K. Nowińska and K. Góra-Marek, Catal. Lett., 2018, 148, 2058–2068 CrossRef CAS.
- (a) D. A. Bulushev, F. Rainone and L. Kiwi-Minsker, Catal. Today, 2004, 96, 195–203 CrossRef CAS; (b) C. Freitag, S. Besselmann, E. Löffler, W. Grünert, F. Rosowski and M. Muhler, Catal. Today, 2004, 91, 143–147 CrossRef; (c) K. Nomiya, K. Hashino, Y. Nemoto and M. Watanabe, J. Mol. Catal. A: Chem., 2001, 176, 79–86 CrossRef CAS; (d) M. Salavati-Niasari, M. R. Elzami, M. R. Mansournia and S. Hydarzadeh, J. Mol. Catal. A: Chem., 2004, 221, 169–175 CrossRef CAS; (e) G. B. Shul'pin, G. S. Mishra, L. S. Shul'pina, T. V. Strelkova and A. J. Pombeiro, Catal. Commun., 2007, 8, 1516–1520 CrossRef.
- (a) I. Muylaert and P. Van Der Voort, Phys. Chem. Chem. Phys., 2009, 11, 2826–2832 RSC; (b) N. Das, H. Eckert, H. Hu, I. E. Wachs, J. F. Walzer and F. J. Feher, J. Phys. Chem., 1993, 97, 8240–8243 CrossRef CAS.
- (a) L. D. Frederickson and D. M. Hausen, Anal. Chem., 1963, 35, 818–827 CrossRef CAS; (b) G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469–3478 CrossRef CAS.
- (a) M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS PubMed; (b) S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2010, 195, 5768–5774 CrossRef CAS.
- M. C. Biesinger, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- D. D. Mal, S. Khilari and D. Pradhan, Green Chem., 2018, 20, 2279–2289 RSC.
- X. Yang, X. Yu, M. Jing, W. Song, J. Liu and M. Ge, ACS Appl. Mater. Interfaces, 2018, 11, 730–739 CrossRef PubMed.
- N. Viswanadham, S. K. Saxena and H. Ala'a, Mater. Today, 2017, 3, 37–48 CrossRef.
- J. Gao, Z. Si, Y. Xu, L. Liu, Y. Zhang, X. Wu, R. Ran and D. Weng, J. Phys. Chem. C, 2018, 123, 1761–1769 CrossRef.
- P. Paul, A. Ghosh, S. Chatterjee, A. Bera, S. M. Alam and S. M. Islam, Inorg. Chim. Acta, 2019, 492, 198–212 CrossRef CAS.
- M. Huda, K. Minamisawa, T. Tsukamoto, M. Tanabe and K. Yamamoto, Angew. Chem., Int. Ed., 2019, 58, 1002–1006 CrossRef CAS PubMed.
- H. Y. Chen, M. Lv, X. T. Zhou, J. X. Wang, Q. Han and H. B. Ji, Catal. Commun., 2018, 109, 76–79 CrossRef CAS.
- T. Karimpour, E. Safaei and B. Karimi, RSC Adv., 2019, 9, 14343–14351 RSC.
- X. Zheng, L. He, Y. Duan, X. Jiang, G. Xiang, W. Zhao and S. Zhang, J. Chromatogr. A, 2014, 1358, 39–45 CrossRef CAS PubMed.
- (a) K. J. Liu, Z. H. Duan, X. L. Zeng, M. Sun, Z. Tang, S. Jiang, Z. Cao and W. M. He, ACS Sustainable Chem. Eng., 2019, 7, 10293–10298 CrossRef CAS; (b) K. J. Liu, S. Jiang, L. H. Lu, L. L. Tang, S. S. Tang, H. S. Tang, Z. Tang, W. M. He and X. Xu, Green Chem., 2018, 20, 3038–3043 RSC; (c) B. Chen, S. Li, S. Liu, M. Dong, B. Han, H. Liu and L. Zheng, J. Mater. Chem. A, 2019, 7, 27212–27216 RSC; (d) G. Shi, S. Xu, Y. Bao, J. Xu and Y. Liang, Catal. Commun., 2019, 123, 73–78 CrossRef CAS; (e) C. He, L. Yu, N. Lu, W. Wang, W. Chen, S. Lu, Y. Yang, D. Ma and S. Huang, Nano Res., 2020, 13, 646–656 CrossRef CAS.
- K. J. Liu, T. Y. Zeng, J. L. Zeng, S. F. Gong, J. Y. He, Y. W. Lin, J. X. Tan, Z. Cao and W. M. He, Chin. Chem. Lett., 2019, 30, 2304–2308 CrossRef CAS.
- J. Javidi, M. Esmaeilpour and M. Rajabnia Khansari, RSC Adv., 2015, 5, 73268–73278 RSC.
- R. Chlosta, G. Tzolova-Müller, R. Schlögl and C. Hess, Catal. Sci. Technol., 2011, 1, 1175–1181 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03483e |
| This journal is © The Royal Society of Chemistry 2020 |