
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium nanoparticles on a pyridinium supported ionic liquid phase: a recyclable and low-leaching palladium catalyst for aminocarbonylation reactions†
Bernadett Adamcsika,
Enikő Nagya,
Béla Urbána,
Péter Szabób,
Péter Pekkerc and
Rita Skoda-Földes *a
*a
aInstitute of Chemistry, Department of Organic Chemistry, University of Pannonia, Egyetem u. 10, Veszprém, 8200, Hungary. E-mail: skodane@almos.uni-pannon.hu
bInstitute of Chemistry, Department of Analytical Chemistry, University of Pannonia, Egyetem u. 10, Veszprém, 8200, Hungary
cResearch Institute of Biomolecular and Chemical Engineering, NANOLAB, University of Pannonia, Egyetem u. 10, Veszprém, 8200, Hungary
First published on 23rd June 2020
Abstract
A new SILP (Supported Ionic Liquid Phase) palladium catalyst was prepared and characterized by 13C and 29Si CP MAS NMR, DTG, FTIR and TEM. The presence of the grafted pyridinium cations on the surface of the support was found to result in the formation of highly dispersed Pd nanoparticles with their diameter in the range of 1–2 nm. The catalyst was proved to be active not only in the aminocarbonylation of some model compounds but also in the synthesis of active pharmaceutical ingredients. Catalyst recycling and palladium leaching studies were carried out for the first time in aminocarbonylations leading to CX-546(1-(1,4-benzodioxan-6-ylcarbonyl)piperidine), Moclobemide, Nikethamide and a precursor of Finasteride. The latter reaction proves that not only aryl iodides but also an iodoalkene can be converted into the products with the help of the heterogeneous catalyst. The results show that the conditions should be always fine-tuned in the reactions of different substrates to achieve optimal results. Palladium loss was also observed to depend considerably on the nature of the reaction partners.
Introduction
Palladium-catalysed carbonylation reactions serve as powerful tools for the synthesis of carbonyl compounds and carboxylic acid derivatives.1 The reaction of aryl halides with amine nucleophiles and carbon monoxide can be considered an especially important procedure as the amide functionality is a key element in numerous pharmacologically active substances.2 At the same time the relatively high price of palladium-catalysts and metal contamination of crude products3 are the main drawbacks that hinder the application of this methodology in the pharmaceutical industry. In spite of the vast number of heterogeneous catalysts developed in the past decades for C–C coupling reactions,4 their use in carbonylations has come to the front only in the last 10–15 years.5 In the latter case, the catalytic situation is more complex: heterogeneous carbonylations are three-phase reactions, highly influenced by the solubility of CO in the solvent as well as by its adsorption on the surface of the catalyst. In the reactions of amine nucleophiles, both amides and α-ketoamides can be formed via mono- and double carbon monoxide insertion, respectively.1 It should be emphasized that α-ketoamides are also useful products: they can serve as the starting material in Mannich-type reactions and in the synthesis of heterocycles.6 Their functional group is also an important motif in some biologically active compounds.6 At the same time, the selectivity problem may lower the efficiency of the catalytic reaction if the target is either the mono- or the double carbonylated product.A great variety of special supports were used for the preparation of Pd-catalysts for aminocarbonylation and double carbonylation reactions, such as carbon nanotubes,7 graphene oxide nanosheets,8 Y-zeolite,9 polymers with various structure,10–17 a siliceous mesocellular foam,18 metal organic frameworks19–21 or a zeolitic imidazole framework.22,23 Pd/C catalysts were also tested in a number of reactions,24–31 but in some cases a rapid loss of activity was reported when the catalyst was reused.9,31 Another possibility is the application of silica-based catalysts.32–38 Good stability, high porosity and easy modification of surface functional groups are the main features that make silica an ideal support for palladium catalysts. Silica with grafted As-,32,33 Se-,34 and P-ligands35–37 or silica-based supported ionic liquid phases (SILPs) with imidazolium39–44 and phosphonium ions45 were found to exert good stabilising effect to obtain recyclable catalysts for aminocarbonylation reactions.
In our earlier studies it was shown that in case of SILP phases decorated with imidazolium ions, a dissolution–reprecipitation mechanism was also operating in the heterogeneous systems.43 The introduction of dicationic moieties was proved to enhance the stability of the palladium catalysts and lower the loss of palladium.43 Phosphonium-type SILP phases made it possible to carry out the reactions in less polar solvents that led to truly heterogeneous systems with a considerable decrease in Pd leaching.45 At the same time, poor solubility of some of the products in toluene limited the applicability of the latter methodology. These observations necessitate a further quest for the development of more efficient SILP phases that can be used as supports for the catalysts.
In spite of the fact that catalytically active colloidal palladium was shown to be stabilised better by pyridinium- than by imidazolium ionic liquids in alkoxycarbonylations,46 to the best of our knowledge, there is no example for the application of pyridinium SILPs in carbonylation reactions. Moreover, there are only sporadic reports on the synthesis and application of pyridinium SILPs in the literature.47
In the present report it is shown that a considerably more efficient stabilisation of palladium particles can be achieved by a pyridinium SILP compared to its imidazolium and phosphonium counterparts. Also, the pyridinium SILP-Pd phase is proved to be a recyclable and low leaching catalyst not only in simple model reactions but also in the synthesis of substances with pharmacological importance.
Palladium immobilised on different supports was used efficiently by some research groups in the catalytic synthesis of the antidepressant drug, Moclobemide,18,20,22,43 and there is also an example for the application of palladium nanoparticles deposited on ZIF-8 to obtain other compounds of pharmaceutical importance, too.22 At the same time, no exact data on palladium leaching are reported for these synthetic routes. Also, catalyst recirculation experiments were carried out only during the synthesis of Moclobemide in our group.43
Bäckvall18 proved that the aryl iodide in combination with the amine reagent was responsible for the dissolution of palladium in an aminocarbonylation reaction carried out with Pd-nanoparticles supported on a siliceous mesocellular foam. That means that recyclability of the catalyst and metal contamination of the product may greatly depend on the structure of the reagents. In the present work, an optimisation of reaction conditions, investigation of palladium leaching and catalyst recycling were carried out during the synthesis of Moclobemide, Nikethamide, CX-546, and a precursor of the 5α-reductase inhibitor, Finasteride.
Results and discussion
Preparation and characterization of the catalysts
Pyridinium ionic liquids with 3-triethoxypropyl functionality (1, 2) that enables condensation with surface OH groups of silica, could be synthesised only after a nucleophilic substitution of the starting 3-chloropropyltriethoxysilane (Scheme 1). | ||
| Scheme 1 Preparation of ionic liquids (1, 2), supported ionic liquid phases (SILP-1 and SILP-2) and catalysts (CAT-1 and CAT-2). | ||
Modification of the silica support with the pyridinium salts 1, 2 was proved by solid phase NMR measurements. In the 13C CP MAS NMR spectra of SILP-1 and SILP-2 (Fig. S1†) signals of the carbon atoms of the propyl group (around 9 ppm, 25 ppm and 63 ppm) as well as those assigned to the carbon atoms of the aromatic ring (in the region of 129–160 ppm) could be distinguished.
In the spectrum of SILP-2 the singlet of the 4-Me group appeared at 23.0 ppm. The lack of peaks of ethoxy groups proved the successful grafting of the ionic liquid on the surface in both cases. In the 29Si CP MAS NMR spectrum of SILP-1 (Fig. S2†), the peaks situated at −67.6 and −57.0 ppm were assigned to T3 (Si(OSi)3R) and T2 (Si(OSi)2ROH) organosiloxane moieties which supported the formation of covalent bonds between the pyridinium cation and silica through the propylene spacer.48
The FT-IR spectra of 1 and 2 correspond well to those of similar pyridinium salts49 and do not change considerably after the grafting process leading to SILP-1 and SILP-2 (Fig. S5†). The absorbance at 1630 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibrations of the quaternary nitrogen atom in a heterocyclic ring. The strong band around 1470–1480 cm−1 can be attributed to the conjugated CC and CN bonds.
N vibrations of the quaternary nitrogen atom in a heterocyclic ring. The strong band around 1470–1480 cm−1 can be attributed to the conjugated CC and CN bonds.
Thermogravimetric analysis showed that aside from the loss of some residual water, the SILPs were stable up to 250 °C (Fig. S6†).
Palladium was immobilised on the SILPs using Pd2(dba)3⋅CHCl3 as the precursor to obtain CAT-1 and CAT-2 (Scheme 1). The palladium content of the catalysts was determined by ICP-AES (0.70 m/m% for CAT-1 and 0.68 m/m% for CAT-2). The FT-IR spectrum of CAT-1 (Fig. 1) shows the presence of some free dba as a result of decomposition of the precursor complex during immobilisation.
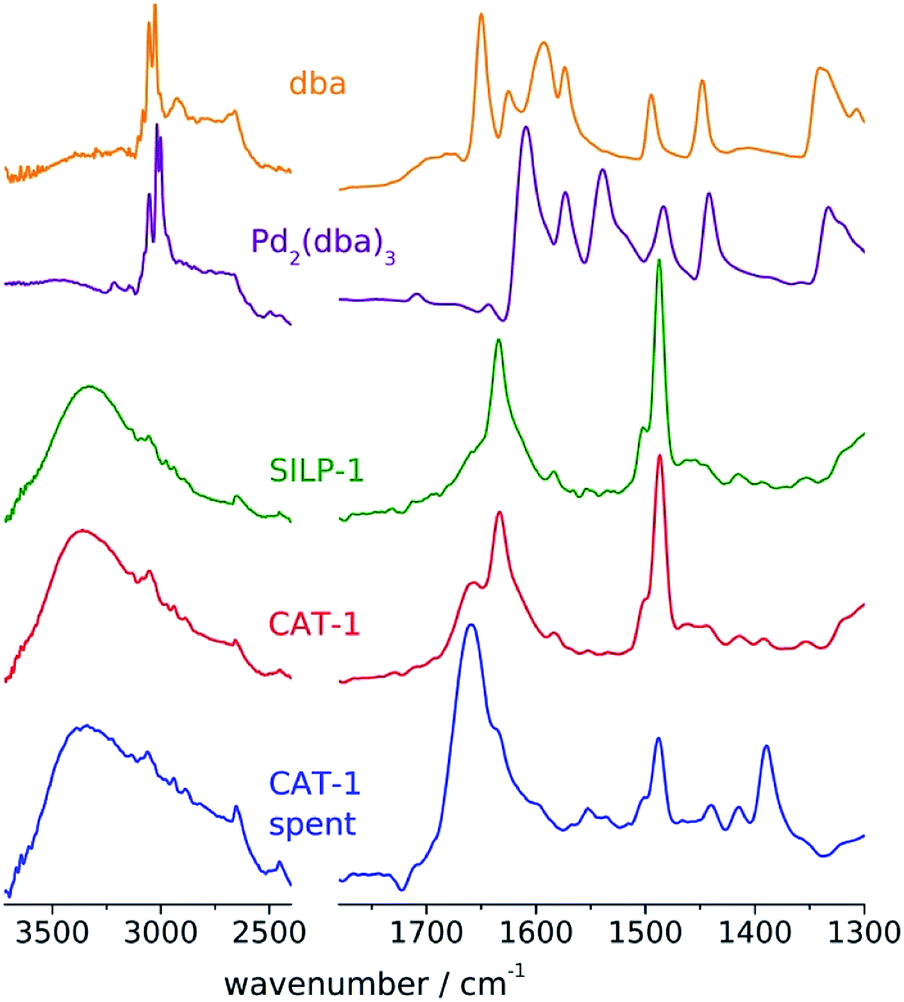 | ||
| Fig. 1 FT-IR spectra of SILP-1 and fresh and spent catalyst CAT-1. | ||
TEM measurements of CAT-1 (Fig. 2) proved the formation of palladium nanoparticles in the diameter range of 1–2 nm.
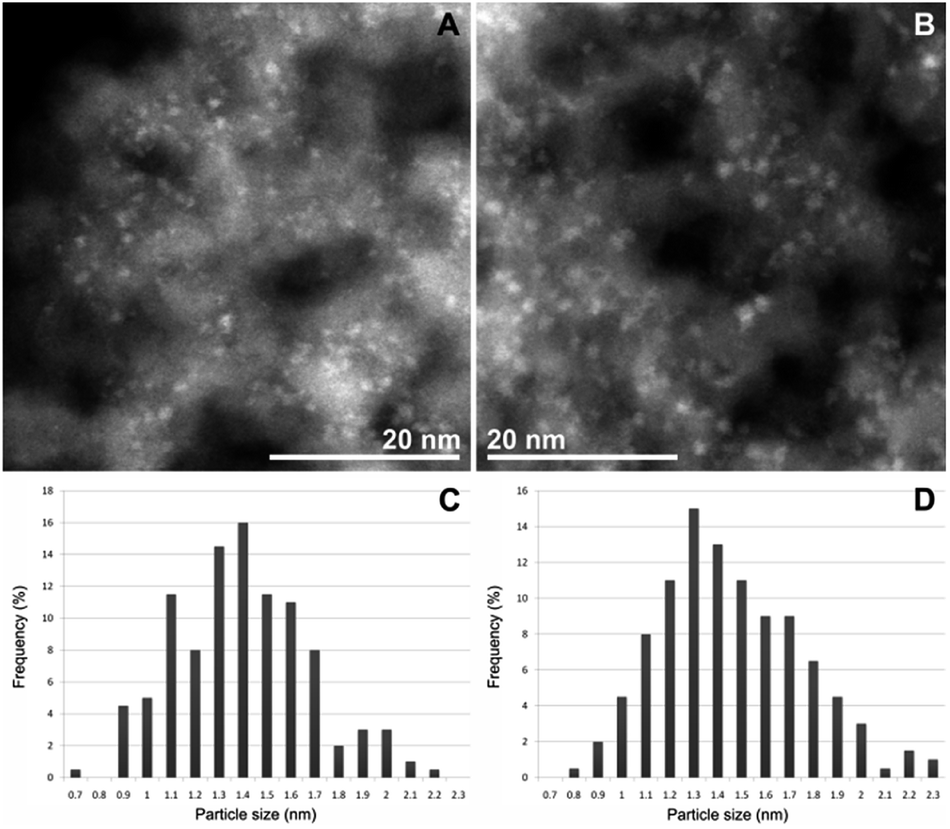 | ||
| Fig. 2 Particle size comparison of the two samples studied with TEM. The silica support appears in gray tone and the bright grains are the Pd nanoparticles on the STEM HAADF images of the CAT-1 fresh (A) and spent (B) samples. The charts show the particle size distribution of the Pd nanoparticles in fresh (C) and spent (D) samples calculated from the measured sizes of 200 nanoparticles in both of the samples. | ||
Palladium content on the surface was found to be too low for XPS analysis.
Catalytic tests
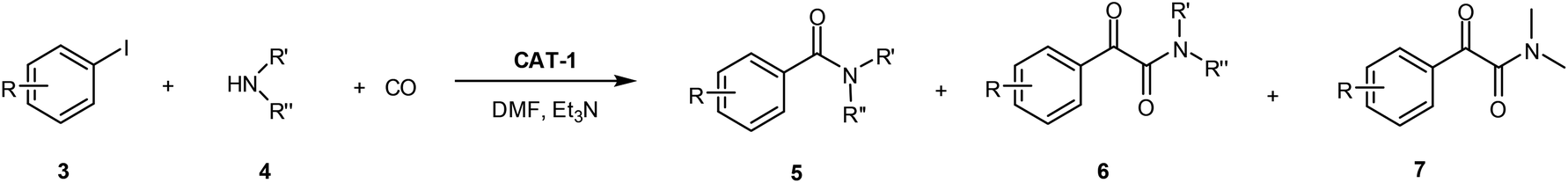
First, activity and recyclability of CAT-1 were tested in the carbonylation reaction of iodobenzene (3a) with morpholine (4a) under different conditions (Table 1). Beside the amide (5a) and α-ketoamide (6a) products, the presence of compound 7 (Scheme 2), formed via decomposition of DMF,41 could be detected in reaction mixtures obtained in this solvent.| Entry | Solvent | Base | Temperature [°C] | CO pressure [bar] | Run 1 | Run 2 | Run 3 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Conversionb [%] | 5a/6a/7c | Conversionb [%] | 5a/6a/7c | Conversionb [%] | 5a/6a/7c | |||||
| a Reaction conditions: 0.2 mmol 3a, 0.5 mmol 4a, 0.25 mmol base, catalyst CAT-1 (Pd/substrate = 1.4 mol%) and 1 mL solvent. Reaction time: 3 h.b Conversion of 3a, determined by GC.c Determined by GC.d After 8 h. | ||||||||||
| 1 | DMF | Et3N | 100 | 1 | 31 | 58/42/0 | 28 | 59/41/0 | 15 | 60/40/0 |
| 2 | DMF | Et3N | 80 | 5 | 42 | 10/88/2 | 38 | 8/87/5 | 39 | 8/87/5 |
| 3 | DMF | Et3N | 100 | 5 | 53 | 13/86/1 | 48 | 12/84/4 | 28 | 13/83/4 |
| 4 | DMF | Et3N | 120 | 5 | 59 | 18/78/4 | 25 | 28/67/5 | 13 | 17/83/0 |
| 5 | DMF | Et3N | 80 | 30 | 36 | 3/95/2 | 43 | 2/95/3 | 26 | 4/92/4 |
| 6 | DMF | Et3N | 100 | 30 | 98 | 4/91/5 | 100 | 5/92/3 | 100 | 4/95/1 |
| 7d | DMF | Et3N | 100 | 30 | 100 | 3/92/5 | 100 | 5/92/3 | 100 | 2/95/3 |
| 8 | DMF | Et3N | 120 | 30 | 100 | 23/74/3 | 96 | 18/78/4 | 96 | 10/85/5 |
| 9 | Acetonitrile | Et3N | 100 | 30 | 46 | 8/92/0 | 45 | 6/94/0 | 34 | 7/93/0 |
| 10 | Acetonitrile | DBU | 100 | 30 | 99 | 9/91/0 | 90 | 7/93/0 | 78 | 6/94/0 |
| 11 | Toluene | Et3N | 100 | 30 | 35 | 62/38/0 | 44 | 65/35/0 | 34 | 64/36/0 |
| 12 | Toluene | DBU | 100 | 30 | 92 | 5/95/0 | 81 | 7/93/0 | 80 | 7/93/0 |
| 13 | 1,4-Dioxane | Et3N | 100 | 30 | 79 | 65/35/0 | 36 | 61/39/0 | 16 | 62/38/0 |
| 14 | 1,4-Dioxane | DBU | 100 | 30 | 98 | 3/97/0 | 94 | 4/96/0 | 97 | 3/97/0 |
| 15 | 1,4-Dioxane | DBU | 120 | 5 | 83 | 84/16/0 | 81 | 83/17/0 | 84 | 85/15/0 |
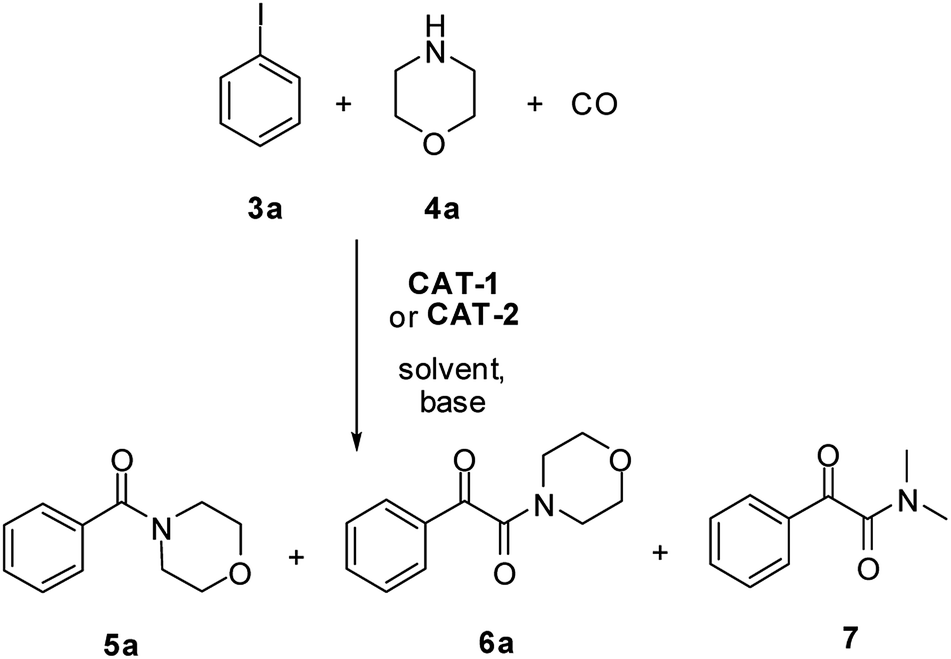 | ||
| Scheme 2 Aminocarbonylation of iodobenzene (3a) with morpholine (4a). Side product 7 is formed in DMF-containing mixtures by the decompositions of DMF. | ||
High pressure together with relatively high temperature was found to be necessary to achieve excellent conversion of iodobenzene (3a) and good recyclability of the catalyst (Table 1, entries 6–8). A rise in the temperature resulted in higher conversions even under 5 bar CO pressure (entries 2–4), but a progressively increasing loss of catalytic activity was observed upon reuse at temperatures higher than 80 °C (entries 3 and 4). As it could be expected based on our previous results42 the application of a higher temperature led to an increase in the ratio of the amide product 5a. At the same time, except for the reaction carried out under atmospheric conditions (entry 1), the α-ketoamide (6a) was the main product in polar solvents, such as DMF (entries 2–8) or acetonitrile (entries 9 and 10). Good activity together with acceptable recyclability of the catalyst could be achieved by replacing the base, Et3N with DBU in solvents such as acetonitrile (entries 9 and 10), toluene (entries 11 and 12) or 1,4-dioxane (entries 13 and 14). In apolar solvents the latter change in the reaction conditions led to a reversal of selectivity. While amide 5a was the main product in the presence of Et3N in toluene (entry 11) and 1,4-dioxane (entry 13), the application of DBU led to α-ketoamide (6a) in excellent yields (entries 12 and 14).
Amide 5a could be produced with good selectivity in the 1,4-dioxane/DBU system at higher temperature and lower pressure (entry 15).
Palladium leaching and catalyst recycling
Recyclability of the two catalysts (CAT-1 and CAT-2) was compared using the best conditions (Table 1, entry 6) for the selective synthesis of α-ketoamide 6a (Fig. 3). They showed very similar performance and could be recycled four times without appreciable loss of activity. The incomplete conversion in the first cycle in both cases shows an initial activation of the catalysts under the reaction conditions. Palladium leaching was found to be 0.20% and 0.24% of the original loading in the first runs with CAT-1 and CAT-2, respectively. After that, loss of palladium remained below the detection limit (0.13% of the original loading) for both catalysts. Accordingly, only a little change could be observed in the Pd content of the heterogeneous catalyst CAT-1 after the 5th run (Pd contents: fresh CAT-1: 0.699 m/m%, spent CAT-1 0.695 m/m%).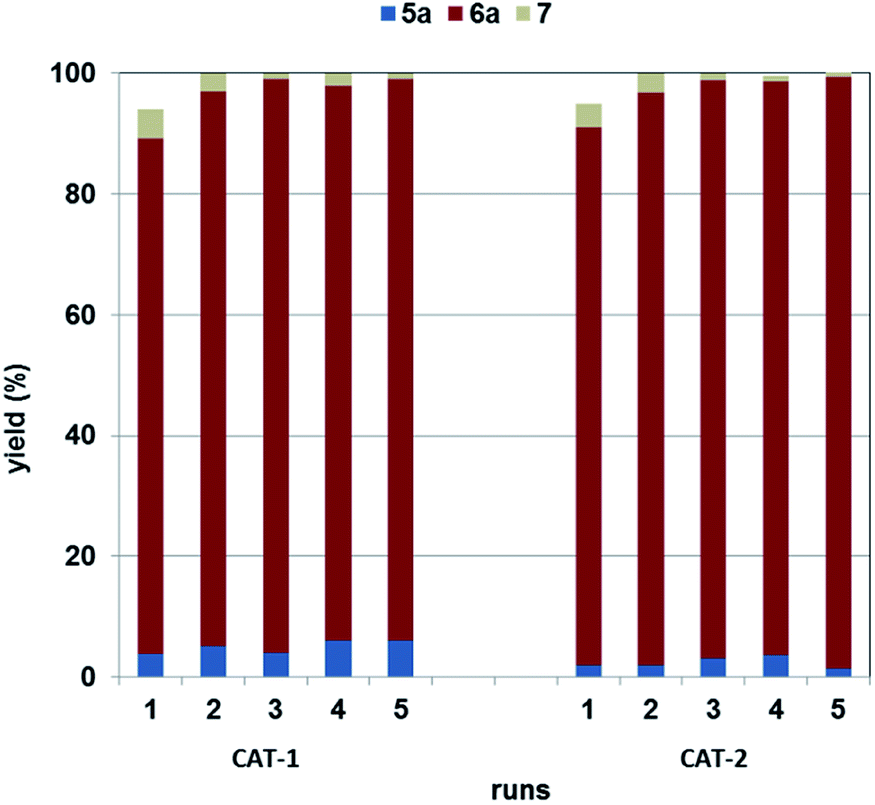 | ||
| Fig. 3 Recirculation experiments with CAT-1 and CAT-2. Reaction conditions: 0.2 mmol 3a, 0.5 mmol 4a, 0.25 mmol Et3N, catalyst (Pd/substrate = 1.4 mol%), 1 mL DMF, 100 °C, 30 bar CO, 3 h; (Yields are determined by GC). | ||
These results are appreciably superior to those obtained with palladium immobilised on SILPs with imidazolium-50 and phosphonium ions.45 Using palladium deposited on an imidazolium-type SILP, metal leaching was found to be 7.6% in the first run and an average of 3.2% loss was measured in the subsequent 5 runs under identical conditions.50 Although palladium-content of the reaction mixture was below the detection limit (i.e. <1%) in the same reaction carried out with a phosphonium SILP-Pd catalyst, it increased above 4.0% in the further runs.45 Also, some loss of activity had been observed with these catalysts under the same conditions.
It should be mentioned that the amount of leached palladium did not change noticeably in a longer reaction using the present system (Table 1, entry 7), so in contrast to the imidazolium SILP-Pd catalyst,50 no reprecipitation of the metal could be observed by heating the reaction mixture for a longer time.
To obtain information about the homogeneous or heterogeneous nature of the catalytic reaction, hot filtration and mercury poisoning tests were carried out in the reactions of CAT-1 and CAT-2 (Table 2). The catalytic mixture was filtered after half an hour. One half of the mixture was heated further under CO pressure, the other was treated similarly but in the presence of mercury.
| Entry | Catalyst | First stepb | Second stepc | |||
|---|---|---|---|---|---|---|
| Conv. of 3ad (%) | Ratio of 5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6ad :6ad |
Hg | Conv. of 3ad (%) | Ratio of 5a:6ad |
||
| a Reaction conditions: 0.2 mmol 3a, 0.5 mmol 4a, 0.25 mmol Et3N, catalyst (Pd/substrate = 1.4 mol%), 1 mL DMF, 30 bar CO, 100 °C.b Reaction time: 0.5 h.c Reaction time: 3 h.d Determined by GC.e Recycled catalyst after a three hour-long first run. | ||||||
| 1 | CAT-1 | 51 | 0:100 |
− | 65 | 2:98 |
| 2 | + | 55 | 1:99 |
|||
| 3 | CAT-1e | 42 | 0:100 |
− | 50 | 1:99 |
| 4 | + | 43 | 0:100 |
|||
| 5 | CAT-2 | 35 | 0:100 |
− | 51 | 1:99 |
| 6 | + | 40 | 0:100 |
|||
| 7 | CAT-2e | 32 | 0:100 |
− | 43 | 1:99 |
| 8 | + | 32 | 0:100 |
|||
Although some iodobenzene was consumed even after the removal of the heterogeneous catalyst (entries 1 and 5), the conversion was much lower than in a typical experiment (compare to Fig. 3). Similar procedures carried out with spent catalysts (entries 3 and 7) resulted in even slower reactions in the absence of the solid phase, which is in accordance with the decrease in the amount of leached palladium after the first run. In reactions carried out with fresh catalysts, carbonylation took place both in the absence (entries 1 and 5) and in the presence of mercury (entries 2 and 6), though with a lower rate in the latter cases. In the experiments with spent catalysts, the reaction could be stopped in the filtrate with the addition of mercury (entries 4 and 8).
No noticeable change in the structure of CAT-1 could be observed by FT-IR and STEM measurements after the catalytic reaction. The FT-IR spectrum (Fig. 1) of the spent catalyst preserved the bands attributed to the pyridinium cation at 1630 and 1480 cm−1 (It should be mentioned that the DMF solvent could not be removed completely and the signal at 1660 cm−1 can be assigned to the amide band of this compound). STEM pictures of spent CAT-1 showed no aggregation of Pd nanoparticles (Fig. S7†). Some redistribution of Pd particles could be observed on the size histogram (Fig. 1), but size range remained essentially the same.
Carbonylation of other substrates
Because of the very similar performance of the two catalysts, CAT-1 was chosen to carry out further experiments and it was used efficiently during the aminocarbonylation of other substrates. Recyclability of the catalyst was investigated in three subsequent cycles in each case (Table 3). The application of cyclic secondary amines as reaction partners led to the ketoamide product with an excellent ketoamide/amide ratio (entries 1–3), at the same time, poor selectivity was obtained with dibutylamine (entry 4). As it was expected from previous results45 selective formation of amide products was observed during carbonylation with aromatic amine nucleophiles (entries 5–7). Other iodobenzene derivatives, such as the 4-methoxy- (entry 8) and 3,4-dimethyl derivative (entry 9) could be converted to the corresponding α-ketoamide with morpholine as the reaction partner with excellent results. In contrast, the amide derivative was the main product in the carbonylation of 4-nitro-iodobenzene (entry 10). Interestingly, a reduction of the nitro group also took place leading to compounds 8 and 9 (Fig. 4) as side products. It should be mentioned that no carbonylation involving the aromatic amino group could be detected, possibly because of the excess of the more reactive morpholine.
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | R′ | R′′ | Run 1 | Run 2 | Run 3 | Yield of main productc (%) | ||||
| Conv.b (%) | Ratio of 5/6/7b | Conv.b (%) | Ratio of 5/6/7b | Conv.b (%) | Ratio of 5/6/7b | 5 | 6 | ||||
| a Reaction conditions: 0.2 mmol iodoarene, 0.5 mmol amine, 0.25 mmol Et3N, CAT-1 (Pd/substrate = 1.4 mol%) and 1 mL DMF, 100 °C, 30 bar CO, 3 h.b Determined by GC.c Yield of product isolated from the combined reaction mixtures of the three runs.d 8 and 9 were formed as side products. | |||||||||||
| 1 | H | (CH2)2–O–(CH2)2 | 98 | 4/91/5 | 100 | 5/92/3 | 100 | 4/95/1 | 82 | ||
| 2 | H | (CH2)5 | 95 | 3/93/4 | 97 | 3/93/4 | 96 | 4/93/4 | 82 | ||
| 3 | H | (CH2)4 | 93 | 7/87/6 | 94 | 6/86/8 | 95 | 6/86/8 | 78 | ||
| 4 | H | Bu | Bu | 78 | 35/65/0 | 89 | 44/56/0 | 83 | 31/69/0 | — | — |
| 5 | H | H | Ph | 91 | 98/2/0 | 100 | 98/2/0 | 100 | 98/2/0 | 95 | |
| 6 | H | H | 4-Me-C6H4 | 100 | 100/0/0 | 95 | 100/0/0 | 86 | 100/0/0 | 78 | |
| 7 | H | H | 4-Bu-C6H4 | 100 | 97/3/0 | 98 | 95/5/0 | 97 | 93/7/0 | 89 | |
| 8 | 4-OCH3 | (CH2)2–O–(CH2)2 | 94 | 3/97/0 | 90 | 4/96/0 | 83 | 5/95/0 | 80 | ||
| 9 | 3,4-(CH3)2 | (CH2)2–O–(CH2)2 | 96 | 4/98/0 | 100 | 8/92/0 | 100 | 7/93/0 | 90 | ||
| 10 | 4-NO2 | (CH2)2–O–(CH2)2 | 100 | 100/0/0 | 100 | 92/0/0d | 100 | 90/0/0d | 84 | ||
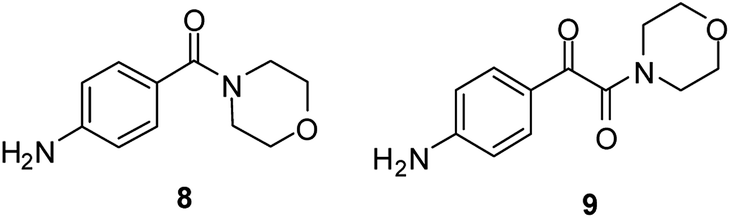 | ||
| Fig. 4 Side products in the aminocarbonylation of 4-nitro-iodobenzene and morpholine. | ||
Synthesis of pharmaceutically active derivatives
As the above results clearly show, the structure of the substrates can greatly affect the outcome of the reaction. Their involvement in palladium leaching was proved by Bäckvall,18 so a detailed investigation of the selectivity of the carbonylation reaction, efficiency of catalyst recycling and amount of leached palladium was carried out in aminocarbonylations leading to four compounds of pharmaceutical importance.CX-546 (1-(1,4-benzodioxan-6-ylcarbonyl)piperidine) (11), developed for the treatment of schizophrenia,51 had been prepared in 95% yield by an aminocarbonylation reaction catalysed by palladium nanoparticles deposited on ZIF-8, but no recycling experiments were carried out.22 In order to test the applicability of catalyst CAT-1 in the same reaction, 6-iodo-1,4-benzodioxane (10) was reacted with piperidine (4b) and CO under different conditions (Scheme 3 and Table 4). The application of high pressure resulted in the selective formation of the corresponding α-ketoamide derivative (12) together with a noticeable loss of catalytic activity of the recycled catalyst in DMF (entry 1). Even a change in the reaction conditions to higher temperature and lower pressure resulted in the formation of the target product (11) with only 33% selectivity (entry 2). Moreover, a constant decrease in the ratio of amide 11 was observed when the catalyst was recycled.
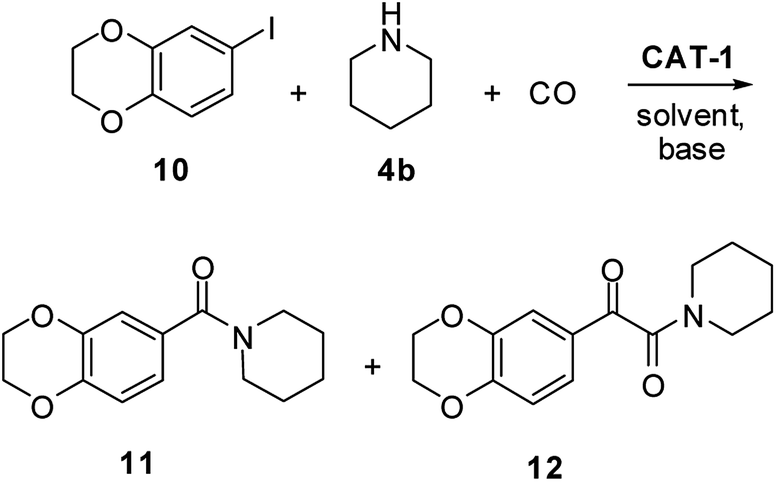 | ||
| Scheme 3 Synthesis of CX-546 (11) by the aminocarbonylation of 6-iodo-1,4-benzodioxane (10) with piperidine (4b). | ||
| Entry | Solvent/baseb | Temp. [°C] | CO pressure [bar] | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conv.c [%] | 11/12 c | Conv.c [%] | 11/12 c | Conv.c [%] | 11/12 c | Conv.c [%] | 11/12 c | Conv.c [%] | 11/12 c | ||||
| a Reaction conditions: 0.2 mmol 10, 0.5 mmol 4b, 0.25 mmol base, catalyst CAT-1 (Pd/substrate = 1.4 mol%) and 1 mL solvent.b A: DMF/Et3N; B:1,4-dioxane/DBU, C: —/piperidine.c Determined by GC. | |||||||||||||
| 1 | A | 100 | 30 | 93 | 0/100 | 99 | 0/100 | 87 | 0/100 | 82 | 0/100 | 78 | 0/100 |
| 2 | A | 120 | 5 | 100 | 33/67 | 95 | 20/80 | 89 | 13/87 | 81 | 12/88 | 76 | 12/88 |
| 3 | B | 100 | 30 | 96 | 6/94 | 95 | 4/96 | 96 | 7/93 | 95 | 7/93 | 94 | 11/89 |
| 4 | B | 120 | 5 | 100 | 82/18 | 100 | 85/15 | 100 | 81/19 | 99 | 87/13 | 99 | 81/19 |
| 5 | C | 100 | 30 | 98 | 18/82 | 100 | 16/84 | 100 | 20/80 | n.d. | n.d. | n.d. | n.d. |
| 6 | C | 120 | 5 | 94 | 92/8 | 83 | 77/23 | 81 | 78/22 | n.d. | n.d. | n.d. | n.d. |
In the next experiments, carbonylation was carried out in the 1,4-dioxane/DBU solvent/base system that also showed good stability in the model reaction of iodobenzene (3a) and morpholine (4a) (see Table 1, entries 13 and 14). As it could be expected based on these results, the α-ketoamide (12) was the main product at 30 bar pressure. Fortunately, CX-546 (11) was obtained with selectivity above 80% at 120 °C and 5 bar CO pressure. Besides, no considerable change in the activity of the catalyst and selectivity of the reaction could be observed in five successive runs.
The palladium content of the combined reaction mixtures was below the detection limit. This means that the total palladium loss was less than 0.25% of the original load after the 5 cycles. Chromatographic separation of the combined reaction mixtures of the five runs led to CX-546 in 78% isolated yield.
As preferred formation of amides from iodoaromatics and secondary amines had been observed before in solvent-free reactions in the presence of a palladium catalyst supported on an imidazolium SILP,42 the application of the same conditions was attempted here. Although CX-546 (11) could be produced with good conversion and excellent selectivity at 120 °C and 5 bar CO pressure (entry 6), a considerable drop of activity and selectivity was noticed when the catalyst was reused.
Previously, the antidepressant drug Moclobemide52 (4-chloro-N-(2-morpholinoethyl)benzamide) (Scheme 4, 15), had been prepared from 1-chloro-4-iodobenzene (13) and 2-morpholinoethanamine (14) in 76–94% yield using different supported catalysts.18,20,22,43 but no information had been reported on catalyst recycling and palladium leaching.
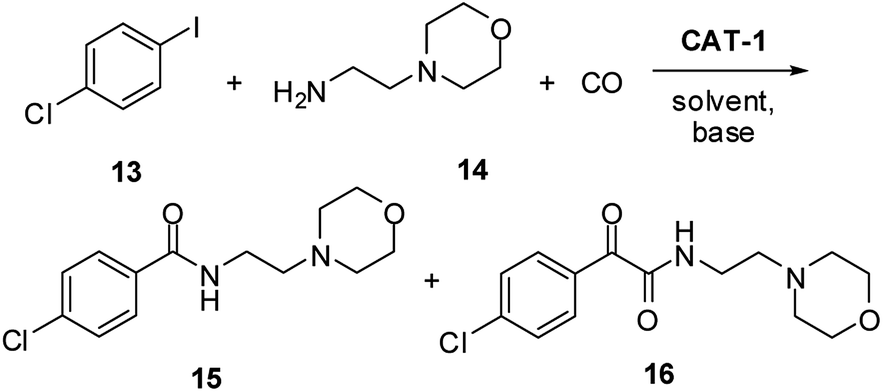 | ||
| Scheme 4 Synthesis of Moclobemide (15) by the aminocarbonylation of 1-chloro-4-iodobenzene (13) and 2-morpholinoethanamine (14). | ||
During the present work, carbonylation of 1-chloro-4-iodobenzene (13, Scheme 4) was carried out first under the standard conditions (30 bar CO, 100 °C) using the DMF/Et3N (Table 5, entry 1) and 1,4-dioxane/DBU (entry 2) systems. Similarly to the reaction of iodobenzene with the non-cyclic dibutylamine (Table 3, entry 4), an increase in the ratio of the amide product (15) was observed compared to the results of the model reaction (Table 1, entries 6 and 14). Still, the double carbonylated product 16 was formed in a considerable amount. Interestingly, only a moderate conversion could be achieved in DMF solvent (entry 1). As it was expected, selectivity could be pushed towards the desired product 15 with the application of lower pressure and higher temperature (entries 3 and 8). An efficient catalyst recycling could be achieved both at 15 bar (entries 3–7) and 5 bar (entries 8–12). Under the latter conditions Moclobemide (15) could be produced with excellent selectivity. According to the ICP-AES measurements, the palladium content of the reaction mixtures was below 0.8% of the original load in each of the five successive runs. Chromatographic separation of the combined reaction mixtures led to Moclobemide (15) in 91% isolated yield.
| Entry | Temp. [°C] | CO press. [bar] | Run | Conv.b [%] | 15/16 |
|---|---|---|---|---|---|
| a Reaction conditions: 0.2 mmol 13, 0.5 mmol 14, 0.25 mmol base (DBU), catalyst CAT-1 (Pd/substrate = 1.4 mol%) and 1 mL solvent (1,4-dioxane).b Determined by GC.c Solvent: DMF, base: Et3N. | |||||
| 1c | 100 | 30 | 1 | 55 | 71/29 |
| 2 | 100 | 30 | 1 | 96 | 51/49 |
| 3 | 120 | 15 | 1 | 100 | 82/18 |
| 4 | 120 | 15 | 2 | 100 | 87/13 |
| 5 | 120 | 15 | 3 | 100 | 84/16 |
| 6 | 120 | 15 | 4 | 100 | 79/21 |
| 7 | 120 | 15 | 5 | 100 | 87/13 |
| 8 | 120 | 5 | 1 | 100 | 97/3 |
| 9 | 120 | 5 | 2 | 100 | 96/4 |
| 10 | 120 | 5 | 3 | 100 | 99/1 |
| 11 | 120 | 5 | 4 | 100 | 96/4 |
| 12 | 120 | 5 | 5 | 100 | 92/8 |
Nikethamide (19, Scheme 5), a respiratory stimulant,53 could also be produced with excellent selectivity using the 1,4-dioxane/DBU solvent/base system (Fig. 5). The catalyst could be recycled in 4 further experiments without a noticeable change in activity or selectivity. Loss of palladium was found to be 1% of the original load per run, according to the ICP-AES measurements.
 | ||
| Scheme 5 Synthesis of Nikethamide (19) by the aminocarbonylation of 3-iodopyridine (17) and diethyl-amine (18). | ||
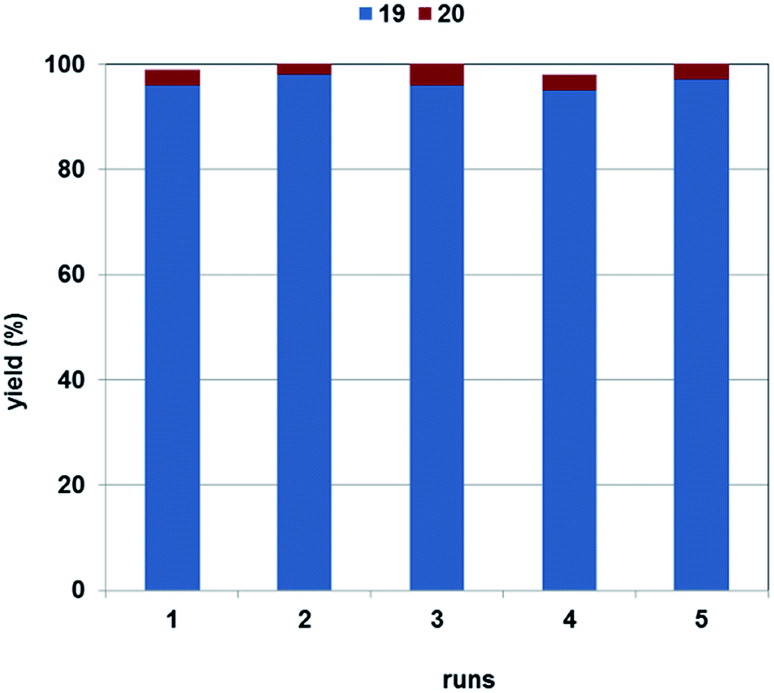 | ||
| Fig. 5 Recirculation of CAT-1 in the carbonylation of 3-iodopyridine and diethyl-amine. Reaction conditions: 0.2 mmol 17, 0.5 mmol 18, 0.25 mmol DBU, catalyst (Pd/substrate = 1.4 mol%), 1 mL 1,4-dioxane, 120 °C, 5 bar CO. (Yields are determined by GC). | ||
Chromatographic separation of the combined reaction mixtures of the five runs led to Nikethamide (19) in 84% isolated yield.
Palladium-catalysed aminocarbonylation under homogeneous conditions was reported to be a key step54 in the synthesis of the 5α-reductase inhibitor Finasteride,55 used in the treatment of benign prostatic hyperplasia. Synthesis of the intermediate 23 (Scheme 6) was attempted under heterogeneous conditions using either the 1,4-dioxane/DBU or the DMF/Et3N solvent/base systems. Similarly to the previous experiments, the possibility of catalyst recycling and the extent of palladium leaching were investigated. Although total conversions of the substrate 21 was observed in five successive runs in both solvents, amide 23 could be isolated only in 46% yield from the reaction mixtures in 1,4-dioxane, due to the formation of a considerable amount of side products. At the same time, amide 23 was formed selectively in DMF and could be isolated in 82% yield (It should be mentioned that no double carbonylation was observed during the conversion of alkenyl iodide 21 in either solvents). Unfortunately, the loss pf palladium was much higher in the polar solvent DMF (2% of the original load per run) than in 1,4-dioxane (0.05% per run). However, these experiments proved that the heterogeneous catalyst can be used not only for the conversion of aryl iodides but also for the aminocarbonylation of an iodoalkene.
 | ||
| Scheme 6 Synthesis of 17-(N-t-butyl-carbamoyl)-4-aza-5α-androst-16-en-3-one (23) by the aminocarbonylation of 17-iodo-4-aza-5α-androst-16-en-3-one (21) and t-butyl-amine (22) (Reaction conditions: 0.2 mmol 21, 0.5 mmol 22, 0.25 mmol base (Et3N or DBU), catalyst CAT-1 (Pd/substrate = 1.4 mol%) and 1 mL solvent (DMF or 1,4-dioxane), 120 °C, 5 bar CO, 3 h). | ||
Conclusions
A better stabilisation of catalytically active palladium nanoparticles can be achieved on SILP phases modified by pyridinium cations compared to imidazolium and phosphonium type SILPs. The heterogeneous catalyst could be recycled with excellent results in the model reaction of iodobenzene and morpholine. It could be used for the selective and high-yielding synthesis of α-ketoamides starting from different iodoaromatics and aliphatic amines under elevated CO pressure. The application of aromatic amines as reaction partners led to the formation of amide products under the same conditions. The possibility of the reuse of the catalyst was proved in all cases. Detailed catalyst-recycling and palladium-leaching tests were carried out for the first time during the synthesis of pharmaceutically active derivatives CX-546 (11), Moclobemide (15), Nikethamide (19) and an intermediate (23) of the synthesis of Finasteride. The catalyst could be recycled in five successive experiments without a change in the catalytic performance and the products could be isolated in good to excellent yields. With the exception of the aminocarbonylation of iodoalkene 21, palladium leaching remained below 1% in all of the reactions.It was found that a tuning of reaction conditions is necessary in most cases to obtain optimal results. Also, in accordance with the investigations of Bäckvall,15 palladium-leaching was found to be greatly dependent on not only the choice of the solvent, but also on the nature of the substrates.
Experimental
General information
17-Iodo-4-aza-5α-androst-16-en-3-one was prepared as described before.51 All other reagents were commercially available and were used without further purification. All solvents were distilled from the appropriate drying agents and were stored under argon atmosphere before use.Reaction mixtures were analysed by gas chromatography (Hewlett Packard 5890) and GC-MS (Hewlett Packard 5971A GC-MSD, HP-1 column). Conversion and selectivity of the reactions were determined by GC using ferrocene as internal standard. The palladium-content of the catalysts and palladium leaching were determined by ICP-AES. Solid phase NMR spectra were recorded on a Bruker Avance 400 spectrometer. 1H and 13C NMR spectra of the products were obtained at 500.15 MHz and 125.78 MHz, respectively, using a Bruker Avance 500 spectrometer or at 400.13 MHz and 100.62 MHz, respectively on a Bruker Avance 400 spectrometer. FT-IR spectra were measured on a BRUKER Vertex 70 type spectrometer with a Bruker Platinum ATR adapter without sample preparation. The spectra were recorded at a resolution of 2 cm−1 with a room temperature DTGS detector (512 scans were co-added). Thermogravimetric analysis was performed on a Netzsch Thermische Analyse TG209 Cell instrument (heating rate 5 °C min−1).
Samples for transmission electron microscopy (TEM) were prepared by depositing a drop of aqueous suspension of the sample on copper TEM grids covered by an amorphous lacey carbon film. TEM analyses were performed using a Talos F200X G2 instrument (Thermo Fisher), operated at 200 kV accelerating voltage, equipped with a field-emission gun and a four-detector Super-X energy-dispersive X-ray spectrometer, and capable of working in both conventional TEM and scanning transmission (STEM) modes. Low-magnification bright-field (BF) images, high-resolution (HRTEM) images and selected-area electron diffraction (SAED) patterns were obtained in TEM mode. STEM high-angle annular dark-field (HAADF) images were collected both for Z-contrast imaging and for mapping elemental compositions by coupling STEM imaging with energy-dispersive X-ray spectrometry (EDS).
Preparation of catalysts
N-((3-triethoxysilyl)propyl)pyridinium iodide (1). 1H NMR (400.13 MHz, DMSO-d6) δ: 9.05–9.11 (m, 1H); 8.62 (t, J = 7.6 Hz, 1H); 8.17 (t, J = 7.6 Hz, 2H); 4.58 (t, J = 7.3 Hz, 1H); 3.73 (q, J = 7.0 Hz, 6H); 1.92–1.99 (m, 2H); 1.13 (t, J = 7.0 Hz, 9H); 0.52–0.56 (m 2H). 13C NMR (100.62 MHz, DMSO-d6) δ: 146.0; 145.2, 128.6; 63.3; 58.4; 25.4; 18.6; 7.1.
4-Methyl-N-((3-triethoxysilyl)propyl)pyridinium iodide (2). 1H NMR (400.13 MHz, DMSO-d6) δ: 8.86 (d, J = 6.6 Hz, 2H); 7.95 (d, J = 6.6 Hz, 2H); 4.45 (t, J = 7.2 Hz, 2H); 3.71 (q, J = 7.0 Hz, 6H); 2.58 (s, 3H); 1.85–1.93 (m, 2H); 1.11 (t, J = 7.0 Hz, 9H); 0.46–0.51 (m, 2H).
General procedures for catalytic reactions
After the evaporation of the solvent of the reaction mixture, the products were purified by column chromatography (silica, eluent: ethyl acetate (Table 3, entry 1), toluene:tert-butyl alcohol 4:1 (entries 2, 5–7), toluene:ethyl acetate = 2:1 (entry 3), toluene:ethyl acetate = 3:4 (entries 8 and 9), toluene:acetone = 5:2 (entry 10), toluene:acetone = 5:2 (11, 12) acetone (15), hexane:ethyl acetate = 2:1 (19), chloroform:methanol = 20:1 (23)).
The 1H-, 13C NMR and MS spectra of the products correspond well to those reported in the literature (Table 3 entries 1–3 and 5–9; 11,40 12, 15, 19).19
1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2-(piperidin-1-yl)ethane-1,2-dione (12) is a new product and its spectra correspond to the proposed structure.
1H NMR (400.13 MHz, CDCl3) δ: 7.45 (dd, J = 0.3 Hz, 2.1 Hz, 1H); 7.43 (dd, J = 2.1 Hz, 8.1 Hz, 1H); 6.91 (dd, J = 0.3 Hz, 8.1 Hz, 1H); 4.23–4.32 (m, 4H); 3.63–3.69 (m, 2H); 3.22–3.28 (m, 2H); 1.62–1.70 (m, 4H); 1.48–1.54 (m, 2H). 13C NMR (100.62 MHz, CDCl3) δ: 190.7; 165.7; 149.6; 143.8; 127.1; 124.0; 118.7; 117.8; 64.8; 64.1; 47.1; 42.1; 26.2; 25.5; 24.4. MS (m/z/rel. int.): 275(M+)/4; 163/100; 135/14; 112/12; 107/22; 84/9; 79/14; 51/18. Yield: 75% (Table 4, entry 1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the GINOP-2.3.2-15-2016-00049 and OTKA K120014 grants. TEM studies were performed at the electron microscopy laboratory of the University of Pannonia, established using grant no. GINOP-2.3.3-15-2016-0009 from the European Structural and Investments Funds and the Hungarian Government.References
- R. Skoda-Földes and L. Kollár, Curr. Org. Chem., 2002, 6, 1097 CrossRef CAS
; C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef
- S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867 CrossRef CAS
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS
- Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed
- B. Urbán, M. Papp and R. Skoda-Földes, Curr. Green Chem., 2019, 6, 78 CrossRef
- C. De Risi, G. P. Pollini and V. Zanirato, Chem. Rev., 2016, 116, 3241 CrossRef CAS PubMed
- B. O. Algin, K. T. Yuanting, N. S. Hosmane and Z. Yinghuai, J. Organomet. Chem., 2013, 747, 184 CrossRef
- Y. Zhang, H. Sun, W. Zhang, Z. Gao, P. Yang and J. Gu, Appl. Catal., A, 2015, 496, 9 CrossRef CAS
- H. Mei, J. Hu, S. Xiao, Y. Lei and G. Li, Appl. Catal., A, 2014, 475, 40 CrossRef CAS
- S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986 RSC
- A. Mansour and M. Portnoy, J. Mol. Catal. A: Chem., 2006, 250, 40 CrossRef CAS
- M. B. Ibrahim, R. Suleiman, M. Fettouhi and B. El Ali, RSC Adv., 2016, 6, 78826 RSC
- T. Suzuka, H. Sueyoshi and H. K. Ogihara, Trans. Mater. Res. Soc. Jpn., 2016, 41, 225 CrossRef CAS
- R. A. Molla, M. A. Iqubal, K. Ghosh, A. S. Roy, Kamaluddin and S. M. Islam, RSC Adv., 2014, 4, 48177 RSC
- B. Chen, F. Li, Z. Huang, T. Lua and G. Yuan, Appl. Catal., A, 2014, 481, 54 CrossRef CAS
- Y. Lei, X. Zhang, Y. Gu, J. Hu, G. Li and K. Shi, Transition Met. Chem., 2016, 41, 1 CrossRef CAS
- Y. Lei, Y. Wan, G. Li, X. Zhou, Y. Gu, J. Feng and R. Wang, Mater. Chem. Front., 2017, 1, 1541 RSC
- F. Tinnis, O. Verho, K. P. Gustafson, C. W. Tai, J. E. Bäckvall and H. Adolfsson, Chem. - Eur. J., 2014, 20, 5885 CrossRef CAS PubMed
- P. Gautam, M. Dhiman, V. Polshettiwar and B. M. Bhanage, Green Chem., 2016, 18, 5890 RSC
- T. T. Dang, Z. Yinghuai, S. C. Ghosh, C. Anqi, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805 RSC
- J. Niu, M. Liu, P. Wang, Y. Long, M. Xie, R. Li and J. Ma, New J. Chem., 2014, 38, 1471 RSC
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406 CrossRef CAS
- T. T. Dang, A. Chen and A. M. Seayad, RSC Adv., 2014, 4, 30019 RSC
- J. Salvadori, E. Balducci, S. Zaza, E. Petricci and M. Taddei, J. Org. Chem., 2010, 75, 1841 CrossRef CAS PubMed
- R. S. Mane and B. M. Bhanage, RSC Adv., 2015, 5, 76122 RSC
- R. S. Mane and B. M. Bhanage, J. Org. Chem., 2016, 81, 1223 CrossRef CAS PubMed
- P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2221 CrossRef CAS
- M. V. Khedkar, S. R. Khan, D. N. Sawant, D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2011, 353, 3415 CrossRef CAS
- A. Satapathy, S. T. G. Gadge, T. Sasaki and B. M. Bhanage, RSC Adv., 2015, 5, 93773 RSC
- K. Natte, H. Neumann and X. F. Wu, Catal. Sci. Technol., 2015, 5, 4474 RSC
- J. Liu, S. Zheng, W. Sun and C. Xia, Chin. J. Chem., 2009, 27, 623 CrossRef CAS
- M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 208, 17 CrossRef CAS
- M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 212, 151 CrossRef CAS
- M. Cai, J. Zhou, H. Zhao and C. Song, React. Funct. Polym., 2002, 50, 191 CrossRef CAS
- M. Cai, H. Zhao and Y. Huang, J. Mol. Catal. A: Chem., 2005, 238, 41 CrossRef CAS
- W. P. Miller, J. N. Long, J. A. de Mello, R. Vilar, H. Audrain, D. Bender, J. Passchier and A. Gee, Angew. Chem., Int. Ed., 2007, 46, 2875 CrossRef PubMed
- W. Hao, J. Sha, S. Sheng and M. Cai, Catal. Commun., 2008, 10, 257 CrossRef CAS
- Q. Hu, L. Wang, C. Wang, Y. Wu, Z. Ding and R. Yuan, RSC Adv., 2017, 7, 37200 RSC
- M. V. Khedkar, A. R. Shindea, T. Sasaki and B. M. Bhanage, J. Mol. Catal. A: Chem., 2014, 385, 91 CrossRef CAS
- B. Urbán, M. Papp, D. Srankó and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2015, 397, 150 CrossRef
- M. Papp, B. Urbán, E. Drotár and R. Skoda-Földes, Green Process. Synth., 2015, 4, 103 CAS
- M. Papp, P. Szabó, D. Srankó and R. Skoda-Földes, RSC Adv., 2016, 6, 45349 RSC
- M. Papp, P. Szabó, D. Srankó, G. Sáfrán, L. Kollár and R. Skoda-Földes, RSC Adv., 2017, 7, 44587 RSC
- B. Dutta, S. Omar, S. Natour and R. Abu-Reziq, Catal. Commun., 2015, 61, 31 CrossRef CAS
- B. Urbán, P. Szabó, D. Srankó, G. Sáfrán, L. Kollár and R. Skoda-Földes, Mol. Catal., 2018, 445, 195 CrossRef
- W. Wojtków, A. M. Trzeciak, R. Choukroun and J. L. Pellegatta, J. Mol. Catal. A: Chem., 2004, 224, 81 CrossRef
- K. P. Boroujeni and M. Jafarinasab, J. Chem. Res., 2012, 36, 429 CrossRef CAS
- L. Han, M. S. Park, S. J. Choi, Y. J. Kim, S. M. Lee and A. W. Park, Catal. Lett., 2012, 142, 259 CrossRef CAS
- D. Cook, Can. J. Chem., 1961, 39, 2009 CrossRef CAS
- M. Papp and R. Skoda-Földes, J. Mol. Catal. A: Chem., 2013, 378, 193 CrossRef CAS
- N. Nagarajan, C. Quast, A. R. Boxall, M. Shahid and C. Rosenmund, Neuropharmacology, 2001, 41, 650 CrossRef CAS PubMed
- U. Bonnet, CNS Drug Rev., 2002, 8, 283 CrossRef CAS PubMed
- M. J. Dulfano and M. S. Segal, J. Am. Med. Assoc., 1963, 185, 69 CrossRef CAS
- Z. Tuba, J. Horváth, J. Széles, M. Lovas Marsai and G. Balogh, Ep. Pat. EP 0705273 B1, 1993
- G. H. Rasmusson, G. F. Reynolds, N. G. Steinberg, E. Walton, G. F. Patel, T. Liang, M. A. Cascieri, A. H. Cheung, J. R. Brooks and C. Berman, J. Med. Chem., 1986, 29, 2298 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: TG measurements of SILP-1 and SILP-2, STEM picture of spent catalyst CAT-1, characterisation of the products and 1H and 13C-NMR spectra of isolated products. See DOI: 10.1039/d0ra03406a |
| This journal is © The Royal Society of Chemistry 2020 |