
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic conversion of ethane to valuable products through non-oxidative dehydrogenation and dehydroaromatization
Hikaru Saito
 *ab and
Yasushi Sekine
b
*ab and
Yasushi Sekine
b
aDepartment of Materials Molecular Science, Institute for Molecular Science, 38 Nishigo-Naka, Myodaiji, Okazaki, Aichi 444-8585, Japan. E-mail: hsaito@ims.ac.jp; hikaru.3110.saito@gmail.com; Tel: +81 564 55 7287
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 169-8555, Japan
First published on 4th June 2020
Abstract
Chemical utilization of ethane to produce valuable chemicals has become especially attractive since the expanded utilization of shale gas in the United States and associated petroleum gas in the Middle East. Catalytic conversion to ethylene and aromatic hydrocarbons through non-oxidative dehydrogenation and dehydroaromatization of ethane (EDH and EDA) are potentially beneficial technologies because of their high selectivity to products. The former represents an attractive alternative to conventional thermal cracking of ethane. The latter can produce valuable aromatic hydrocarbons from a cheap feedstock. Nevertheless, further progress in catalytic science and technology is indispensable to implement these processes beneficially. This review summarizes progress that has been achieved with non-oxidative EDH and EDA in terms of the nature of active sites and reaction mechanisms. Briefly, platinum-, chromium- and gallium-based catalysts have been introduced mainly for EDH, including effects of carbon dioxide co-feeding. Efforts to use EDA have emphasized zinc-modified MFI zeolite catalysts. Finally, some avenues for development of catalytic science and technology for ethane conversion are summarized.
 Hikaru Saito | Hikaru Saito received his PhD at Waseda University in 2020 under the supervision of Prof. Yasushi Sekine. His doctoral thesis focuses on catalytic conversion of ethane to ethylene and aromatic hydrocarbons. Since April 2020, he has worked as a JSPS research fellow at Institute for Molecular Science in the group of Assoc. Prof. Toshiki Sugimoto. His current research focuses on photocatalytic conversion of methane. |
 Yasushi Sekine | Yasushi Sekine received his PhD from The University of Tokyo, 1998. He was appointed as an Assistant Professor of Department of Chemistry of The University of Tokyo in 1998, Waseda University in 2001, and was promoted to Associate Professor in 2007 and to Professor in 2012. His research interests are in the areas of catalysts, hydrogen production, methane/ethane activation, and catalysis in electric fields. |
1. Introduction: chemical utilization of ethane
Chemical utilization of ethane (C2H6) has had great effects on the modern petroleum industry. Inexpensive ethane derived from shale gas and associated petroleum gases is widely available, respectively, in North America and in the Middle East.1 Ethane is converted to ethylene (C2H4) by steam cracking without a catalyst in an ethane cracker. Ethylene is the most fundamental chemicals in petrochemical industry: 149.7 million tons of ethylene were produced in 2017 worldwide2 to synthesize polyethylene, ethylene oxide, styrene, and other ethylene derivatives.Ethylene production from ethane is far superior in terms of cost to production from naphtha, which is a conventional feedstock for ethylene production by the steam cracking. According to the literature,1 costs of ethane were €46.28 and €145.96 per ton in Saudi Arabia and the United States, respectively, while those of naphtha were €708.56 and €669.58 per ton. As shown in Fig. 1, the global demand for ethylene will be expanded to 182.5 million tons in 2023 worldwide2 due to the global increase in population and economic growth. Consequently, the feedstock for ethylene production will be shifted much from naphtha to ethane.
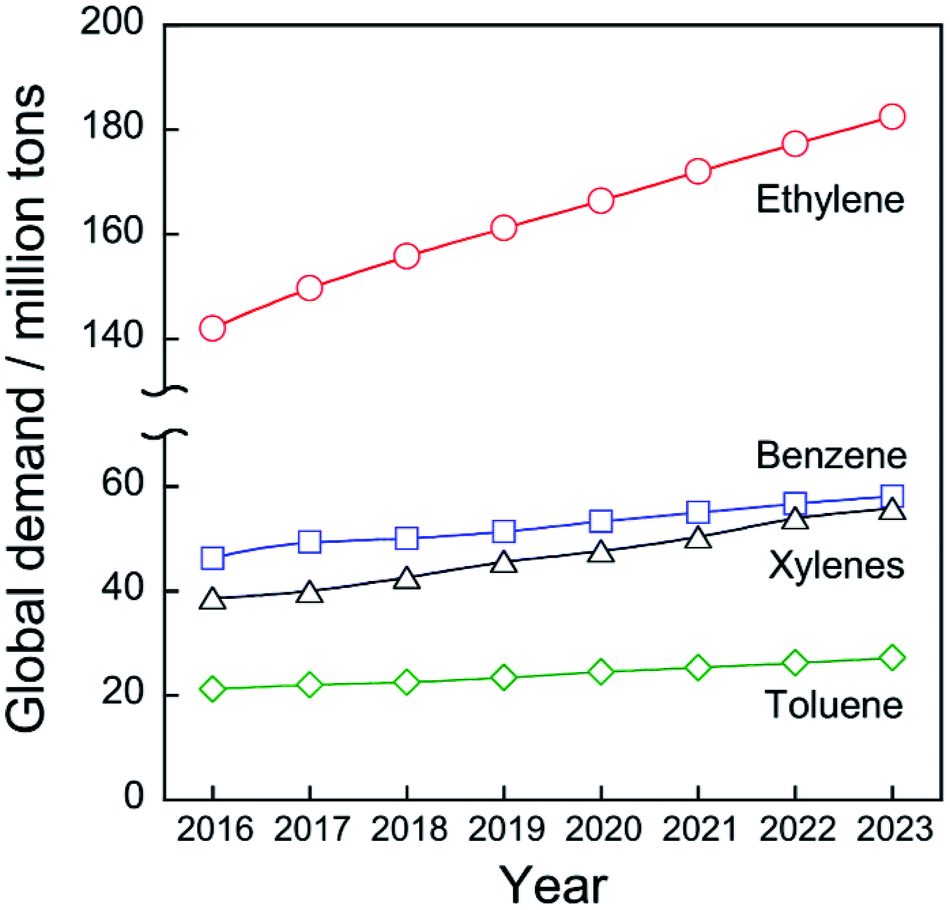 | ||
| Fig. 1 History and projection of global demand for ethylene, benzene, toluene, and xylenes. Data originated from ref. 2. | ||
However, ethane cracking is an energy-intensive process because it necessitates high reaction temperatures (>1073 K). Therefore, ethylene production is favorably conducted at low temperatures to lower the energy consumption and carbon dioxide (CO2) emission. Furthermore, development of ethylene production from ethane instead of naphtha has led to another difficulty. In addition to ethylene, fundamental chemicals including propylene, 1,3-butadiene, and aromatic hydrocarbons are produced by the steam cracking of naphtha. In contrast, these important building blocks are not obtained when using the ethane cracker.1 Therefore, other methods to produce fundamental chemicals aside from ethylene must be used. For instance, propylene must then be produced through dehydrogenation of propane,3 methanol to propylene,4,5 or olefin metathesis of ethylene and 2-butenes.6
Catalytic conversion of ethane to ethylene and aromatic hydrocarbons is anticipated as key technologies to address those needs. Non-oxidative catalytic ethane dehydrogenation (EDH) over oxide or supported-metal catalysts has been investigated widely to achieve environmentally friendly ethylene production. As is the case in propylene production, aromatics including benzene, toluene, and xylenes (BTX), must be produced from feedstocks other than naphtha. BTX are important chemicals used for plastics production and pharmaceuticals synthesis. The global demand for BTX will increase from 111.2 million tons (BTX total) in 2017 to 141.0 million tons in 2023 worldwide (Fig. 1).2 Catalytic ethane dehydroaromatization (EDA) is an attractive alternative to steam cracking of naphtha because valuable aromatics are produced from inexpensive ethane. In addition, EDA can be performed at relatively low temperatures with high BTX selectivity. Therefore, EDA is a potentially promising process for efficient BTX production from natural gas resources. Catalytic EDH and EDA under non-oxidative conditions are summarized in this review because some reviews of oxidative EDH have been published earlier.7,8 Initially, steam cracking of ethane is introduced briefly based on its reaction mechanism. Next, details of non-oxidative EDH and EDA are described. Finally, perspectives for catalytic ethane conversion are discussed.
2. Steam cracking of ethane
2.1. Radical chain reaction mechanism
Steam cracking of ethane is a non-catalytic process for ethane conversion to ethylene through pyrolysis. Before pyrolysis, ethane is mixed with effluent steam to supply reaction heat and to suppress carbonaceous deposits on cracking coils, where the reaction proceeds at ca. 1073 K. Ethane pyrolysis proceeds through a radical chain mechanism first proposed by Rice and Herzfeld.9 This mechanism comprises various elementary reactions such as homolysis, hydrogen abstraction, hydrogen transfer, and recombination.10 The reaction is initiated by homolytic cleavage of the C–C bond in an ethane molecule because enthalpy and the activation energy for the C–C bond dissociation is lower than that of the C–H bond dissociation.11–14 The C–C bond cleavage in an ethane molecule leads to the formation of two methyl radicals as shown below.| C2H6 → 2·CH3 | (1) |
The methyl radical induces the subsequent propagation steps.
| ·CH3 + C2H6 → CH4 + ·C2H5 | (2) |
| ·C2H5 + ·CH3 → C2H4 + CH4 | (3) |
| ·CH3 + C2H6 → ·C3H9 | (4) |
| ·CH3 + C2H4 → ·C3H7 | (5) |
| ·C3H9 + ·CH3 → C3H8 + CH4 | (6) |
| ·C3H7 + ·CH3 → C3H6 + CH4 | (7) |
A hydrogen atom in an ethane molecule is first abstracted by the methyl radical, resulting in the formation of a methane and an ethyl radical (reaction (2)). Subsequent hydrogen transfer in the ethyl radical engenders the formation of ethylene and methane molecules (reaction (3)). The methyl radical also attacks ethane and ethylene (reactions (4) and (5)), resulting in the formation of propane and propylene (reactions (6) and (7)). Heavy hydrocarbons are obtainable through analogous reactions. Furthermore, termination steps proceed through reactions between two radicals.
| ·CH3 + ·CH3 → C2H6 | (8) |
Based on the temperature programmed reaction, pyrolysis of ethane to ethylene initiates at higher than 873 K.15 Under a practical situation, many reactions proceed in addition to the reactions (1)–(8). Sundaram and Froment considered 49 reactions to simulate the product distribution and concentration of radicals.13 The cracking coil is a tubular reactor. Therefore, the temperature gradient along the radial direction is an important factor to be incorporated into calculation of the product distribution and concentration of the radicals. Marin and co-workers reported that the methyl radical concentration is higher near the cracking coil wall than at its center because of high temperatures near the wall, which are the result of endothermicity.14 This phenomenon results in methane formation in great quantities if the simulation is based on a two-dimensional model.
Formation of by-products such as methane, C3, and heavier hydrocarbons is unavoidable in the case of ethylene production through the radical chain mechanism because the reaction initiates from the C–C bond cleavage; it also terminates with methane formation. Particularly, the formation of methane and coke, which are thermodynamically stable, is remarkable at high conversion (temperature) levels.16,17 This remarkable formation decreases the efficiency for ethylene production.
2.2. Coke formation and its suppression
Coke formation on the internal wall of the cracking coils induces severe difficulties such as a pressure drop and a decrease in heat flux to the reactor.18 The latter particularly decreases the ethylene yield because of a kinetic restriction by decreased temperature. To maintain high ethylene yields, the outlet gas temperature is generally operated as constant, which indicates that the cracking coil is heated much more than usual to supply sufficient heat flux, thereby creating a higher temperature of the internal wall than the reaction temperature. Therefore, taking the thermal resistance of the cracking coil and the tolerable pressure drop into consideration, periodic removal of the carbonaceous deposits (decoking) is conducted every 20–60 days depending on operational conditions.17,19 In general, coke formation mechanisms of three types have been proposed: the heterogeneous catalytic mechanism, the heterogeneous radical mechanism, and the homogeneous condensation mechanism.20 The heterogeneous catalytic mechanism is based on coke formation on the surface of the cracking coils. The iron (Fe) and nickel (Ni) included in iron–chromium–nickel (Fe–Cr–Ni) alloys, which are used as cracking coil materials, catalytically promote coke formation on their surface. This phenomenon is observed at the initial stage of the reaction until the cracking coil surface is covered with carbonaceous deposits, indicating that the coke formation rate decreases gradually and reaches an asymptotic value.17,21 The heterogeneous radical mechanism is based on the growth of the carbonaceous deposits through reactions with the radical species. Wauters and Marin proposed that this mechanism comprises five steps: hydrogen abstraction, substitution, addition by radicals, addition to olefins, and cyclization.22 Once the coke forms on the wall, the growth of coke proceeds throughout the reaction. The carbonaceous deposits can also be formed through the homogeneous condensation mechanism as a result of the chain reaction. Therefore, the coke formation via this route is concomitant with the pyrolysis of ethane.From a metallurgic perspective, coating the surface with inert oxides is an effective means of mitigating the coke formation. In principle, aluminum (Al) or manganese (Mn) is included in the Fe–Cr–Ni alloys. After oxidation of the alloy at high temperatures (1323 K for Al; 1023 K for Mn), alumina (Al2O3) or manganese chromate (MnCr2O4), which has high coking suppression capability, is deposited on the surface.20,23 Recently, coating a catalyst for the water gas reaction has been developed to achieve operations without coke formation.
| C(s, graphite) + H2O(g) → CO(g) + H2(g), ΔrHθ (298 K) = 131 kJ mol−1 | (9) |
For instance, BASF developed the CAMOL® process to mitigate coke formation. In this case, tungsten-containing oxides are used as catalysts.24 General Electric also developed gasification catalysts that include perovskite oxides containing cerium (Ce) and alkaline-earth metals.25 However, introduction of the catalysts to cracking coils remains an immature technology. Further research and development are necessary in the domain of catalytic science.
3. Dehydrogenation of ethane under non-oxidative conditions
Catalytic ethane dehydrogenation, denoted as EDH, enables ethylene production at lower temperatures than those of pyrolysis of ethane.| C2H6(g) → C2H4(g) + H2(g), ΔrHθ (298 K) = 137 kJ mol−1 | (10) |
However, the reaction is endothermic. Therefore, high reaction temperatures must achieve higher equilibrium conversion, as portrayed in Fig. 2. Furthermore, low pressure of ethane results in high equilibrium conversion based on Le Chaterlier's principle.
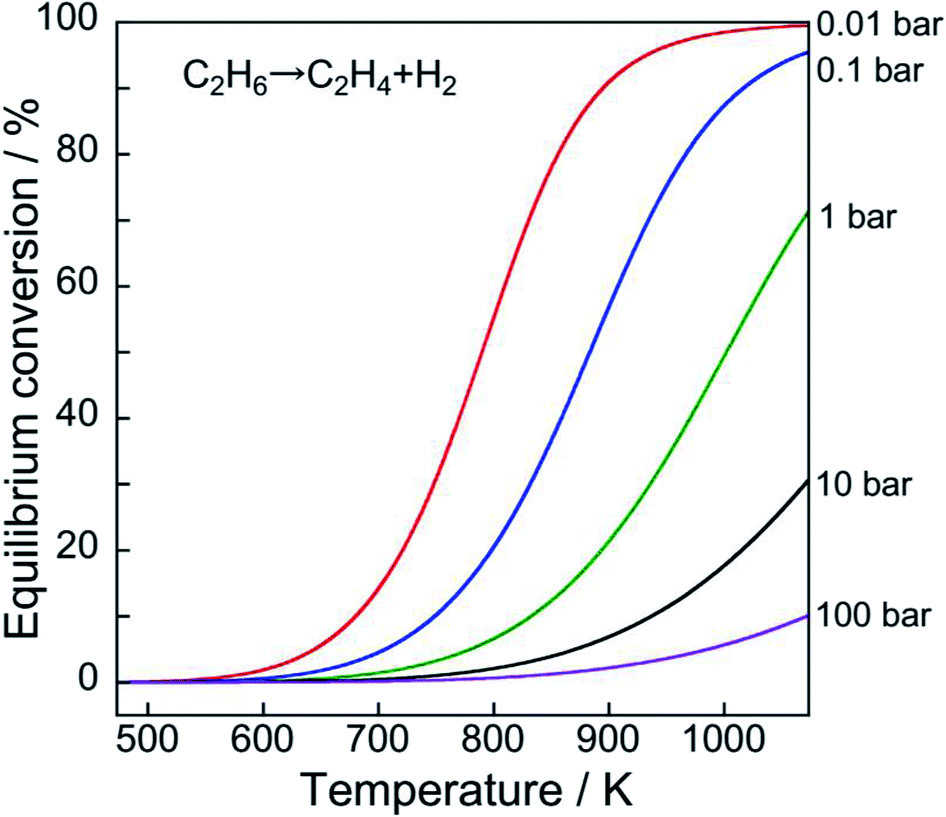 | ||
| Fig. 2 Temperature dependence of equilibrium conversion of EDH at various ethane pressures. | ||
The EDH reaction mechanism is intrinsically unlike pyrolysis of ethane: catalysts should have ability of selective C–H bond dissociation. Side reactions such as hydrogenolysis of ethane and decomposition of ethane and ethylene to coke must be prohibited to achieve high and stable selectivity to ethylene. In this chapter, representative catalysts for EDH are described in terms of their active sites and reaction mechanisms.
3.1. Platinum-based catalysts
For dehydrogenation of alkanes (ethane, propane and butanes), platinum (Pt)-based catalysts have been used widely: one example is known as the Oleflex process.26 In this process, the dehydrogenation reaction are performed by multiple fluidized bed reactors. The spent catalysts are continuously regenerated after the reaction. The regeneration processes are composed of removing carbonaceous deposits and re-dispersion of the Pt particles with chlorine treatment.27 The regenerated catalysts are, then, reused for the reaction. Since dehydrogenation reactions are endothermic, the Pt catalysts are used at high temperatures, which accelerate catalyst deactivations because of coke formation and agglomeration. Therefore, supported-Pt catalysts are modified with promoters such as tin (Sn) for improving their catalytic performance. In this section, we first introduce overview of supported-Pt catalysts. Subsequently, effects of the promoters on the nature of active sites are described in detail.Surface active sites of two types are known to exist for Pt nanoparticles: coordinatively saturated Pt located at terrace sites and coordinatively unsaturated Pt (cus-Pt) such as step and edge sites. Both Pt sites are active for the C–H bond cleavage. However, the latter sites easily induce side reactions such as hydrogenolysis of ethane34,35 and subsequent dehydrogenation of ethylene to coke36,37 as shown in Fig. 3. However, Wu et al. calculated the turnover frequency (TOF) of ethane consumption using Pt/Mg(Al)O with different Pt particle sizes.38 The TOF increased concomitantly with increased Pt particle size, whereas selectivity to ethylene was nearly constant, indicating that EDH proceeds at the terrace sites. This trend is not the case for propane dehydrogenation, where TOF decreases concomitantly with increased Pt particle size.39
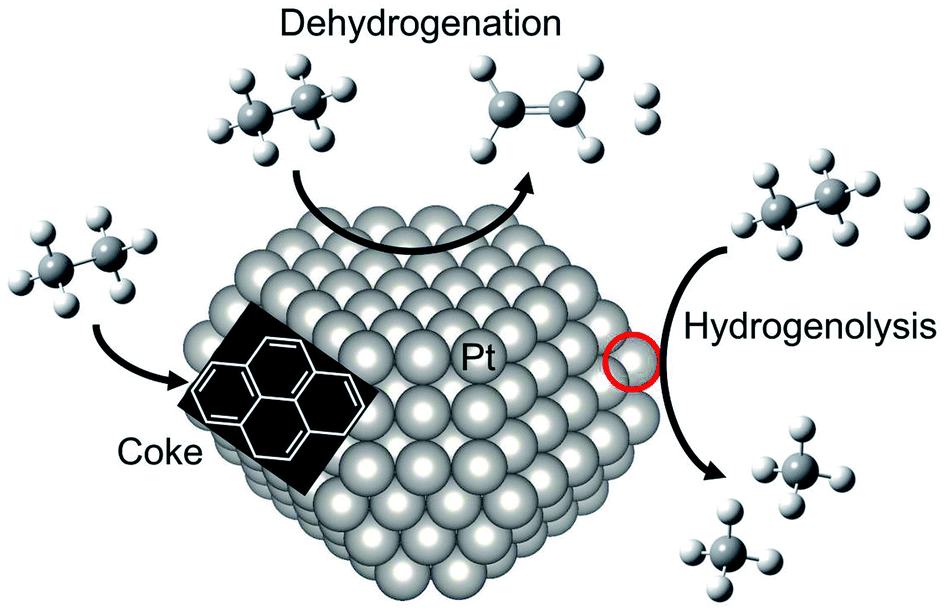 | ||
| Fig. 3 Schematic image of reactivity of ethane on Pt particle. EDH proceeds on the Pt terrace sites, whereas coke formation and hydrogenolysis of ethane proceed on the “cus-Pt” sites circled in red. | ||
The supported-Pt catalysts deactivate in two ways: coke formation and sintering (agglomeration). The carbonaceous deposits formed during the reaction cover the Pt surface and inhibit the access of reactants to active sites. To remove the carbon deposit, the spent catalysts are regenerated under oxidative atmospheres. In addition, the sintering of the Pt nanoparticles decreases the Pt surface area and the number of active sites. In contrast with dehydrogenation of propane and butanes, EDH necessitates the use of high reaction temperatures (>873 K), at which sintering of the Pt nanoparticles is accelerated. Consequently, Pt catalysts are exposed to severe environments: reductive (dehydrogenation) and oxidative (regeneration) atmospheres at high temperatures.
Co-feeding hydrogen (H2) with ethane in the reaction system can inhibit coke formation. Galvita et al. demonstrated that positive effects can be produced for co-feeding H2 on catalytic stability, but selectivity to ethylene decreases because of methane formation through hydrogenolysis.40 Based on density functional theory (DFT) calculations,41–43 high hydrogen coverage on the Pt surface inhibits the formation of coke precursors such as ethylidyne (CH3C) and methylidyne (CH) species. In addition to suppression of the coke precursors, H2 addition to the reaction system promotes ethylene formation under the kinetic conditions.40 Hansen et al. applied DFT calculations to find that H2 addition stabilizes the adsorbed ethylene and ethyl groups, leading not only to suppression of the coke precursors but to the facilitation of ethylene formation.43
Their practical use requires realization of the highly stable and dispersed Pt nanoparticles with coke formation in small amounts. To overcome these shortcomings, Pt catalysts are modified with promoters including boron (B), Al, copper (Cu), zinc (Zn), gallium (Ga), indium (In), and Sn.38,40,44–58 Based on theoretical studies, silicon (Si) and germanium (Ge) are also good candidates for use as promoters.59,60 Irrespective of the elements, the promoter effects are fundamentally classifiable into two types: geometric and electronic. In the following subsection, we describe the respective effects on the catalytic performance. Details of preparation methods, characterizations, and other applications of Pt alloy catalysts are reviewed elsewhere.61,62
The major role of the promoter is alloy formation. Addition of the promoters induces formation of Pt-based alloys or intermetallic compounds, as shown in Fig. 4(a). In addition to Sn,38,40,56–58 other post-transition metals such as Zn, Ga, and In are used as promoters.46–53 Fundamentally, Pt-based alloys are prepared by addition of the promoter and by subsequent reductive treatment. For Mg(Al)O support, some fraction of the Al can be replaced with Ga or In.46,48,49,52 During the pre-reduction step, the dopants move from the partially reduced support to Pt, resulting in Pt-based alloy formation.
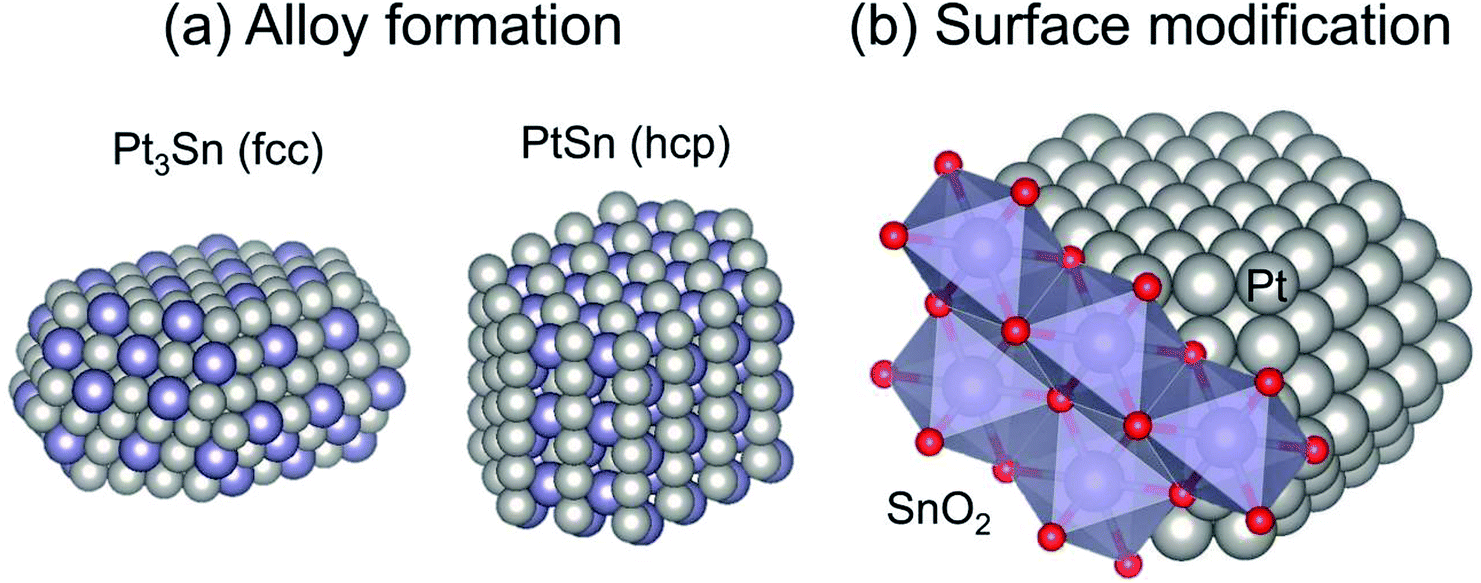 | ||
| Fig. 4 Typical geometric effects of Sn addition to Pt: (a) alloy formation and (b) surface modification. Face centered cubic and hexagonal close packing are denoted respectively as fcc and hcp. | ||
Catalytic performance, particularly selectivity to ethylene and stability, is enhanced by virtue of alloy formation. Alloy formation not only decreases the number of “cus-Pt” through replacement of “cus-Pt” atoms with the promoter: it also varies the surface composition (ensemble) of terrace Pt sites. Celik and co-workers used DFT calculations to elucidate coke formation from ethane on Pt(111) and Pt–Sn(111), representative terrace sites.42 They constructed the alloy models by replacing the surface Pt atoms with Sn atoms, indicating that the model of PtSn has a face centered cubic (fcc) structure. They reported that CH3C (coke precursor) is formed at three-fold Pt sites of Pt(111). For the PtSn(111) surface, where no three-fold Pt exists, CH3C is unstable, leading to suppression of coke formation. They also predicted that a similar effect of alloy formation would be obtained using post-transition metals such as lead (Pb).63
In addition to the ensemble effect, alloy formation can alter the crystalline structure of Pt. Various Pt-based alloys form depending on the preparation conditions, including Pt/promoter ratios and pre-treatment conditions. For instance, Pt3Sn and PtSn alloys can be prepared under different pre-treatment or reduction conditions, as investigated thoroughly by Deng et al.64–67 For the dehydrogenation of ethane and propane, Pt3Sn is suitable; it has the same crystalline structure (fcc) as that of metallic Pt,38,65 whereas PtSn with a hexagonal close packing (hcp) structure is not active for propane.65 The activity of PtSn for EDH has not been evaluated clearly yet. This is also the case in Pt–In catalysts, where Pt3In exhibits higher activity than PtIn2 (a fluorite structure).53 Based on several studies, it seems plausible that the crystalline structure is an important factor for EDH. However, PtZn with a tetragonal gold–copper alloy (AuCu) structure is highly active for EDH.51 Additional studies must be undertaken to elucidate the influence of the crystalline structure on EDH activity.
Surface modification is a geometric effect, as shown in Fig. 4(b). As described in Subsection 3.1.1, side reactions such as coke formation and hydrogenolysis are facile at the “cus-Pt” sites. These active sites can be covered by Sn addition if Pt–Sn alloys are formed or not, leading to suppression of the side reactions.64 Similar effects of Al addition were also reported by Peng et al.37 The surface Sn species, which do not contribute to Pt–Sn alloy, exist on “cus-Pt” as Sn oxides such as SnO2. The location of Sn is evaluated using infrared (IR) spectroscopy with carbon monoxide (CO) as a probe molecule.55,64,67,68 Broad absorption bands for linearly adsorbed CO and weak absorption bands for bridged CO on monometallic Pt are observable, respectively, at around 2080 and 1850 cm−1.55,64 In this case, CO linearly adsorbed onto “cus-Pt” shows the absorption bands at slightly lower wavenumbers than that on Pt terrace because “cus-Pt” interacts strongly with CO, resulting in much back donation from Pt to 2π* (anti-bonding) orbitals of CO. Deng et al. reported that absorption bands attributed to CO2 are observed in the spectrum of Pt modified with SnO2.64 However, no CO2 formation was verified without SnO2. The Pt modified with SnO2 exhibits higher selectivity for dehydrogenation of propane and butane. Therefore, they concluded that SnO2 exists on the Pt surface, where it covers “cus-Pt”. On Pt3Sn and PtSn surfaces, linearly adsorbed CO shows symmetric absorption bands because of the few “cus-Pt” sites. Furthermore, the absorption bands for bridged CO were not apparent, even on Pt3Sn. The existence of multi-coordinated CO was verified on well-defined Pt3Sn(111).69 This difference is ascribed to the surface enrichment of Sn on Pt3Sn and PtSn nanoparticles. Deng et al. applied X-ray photoelectron spectroscopy (XPS) and found that the surface Sn/Pt ratios are higher than those of bulk material,66,67 indicating that surface modification of SnO2 occurs concomitantly with alloy formation. It is worth noticing that segregation of Sn from Pt–Sn alloys70,71 and surface reconstruction of metallic Pt nanoparticles72 occur through CO adsorption. These phenomena alter the wavenumbers and shape of the CO absorption bands. Therefore, the information obtained from the IR spectra would not appropriately reflect the working state of the Pt and Pt–Sn surfaces.
The Pt particle size (dispersion) can be varied by promoter addition. For example, small Pt particles are obtained by Sn addition using silica supports.64,73 Such is not the case for γ-Al2O3 and Mg(Al)O because these Al containing supports stabilize Pt particles,28,29 leading to small Pt particles (<ca. 3 nm) even without Sn.38,74 In addition, Sn plays a crucially important role in re-dispersion of the Pt particles through oxidative and subsequent reduction treatments for catalyst regeneration, as presented in Fig. 5. Weckhuysen and co-workers reported characteristic behavior of the re-dispersive effect of Sn on the Pt particles during regeneration based on CO adsorption, in situ X-ray absorption fine structure (XAFS) spectroscopy, and transmission electron microscopy.74,75 They observed atomically dispersed Sn on γ-Al2O3, which plays a role in nucleation sites for re-dispersion of Pt (Fig. 5(a)). During the oxidative treatment, Sn segregates from the alloy; platinum oxide (PtO) is formed on the support (Fig. 5(b)).76 Subsequent reductive treatment reconstructs the Pt–Sn alloy.75 Therefore, they argued that the agglomeration of Pt is more responsible for deactivation than carbon deposition.74 The phenomena occurring during oxidation are expected to be crucially important for the re-dispersion of Pt–Sn alloy. However, the nucleation mechanism and the role of segregated Sn have not been clearly proved.
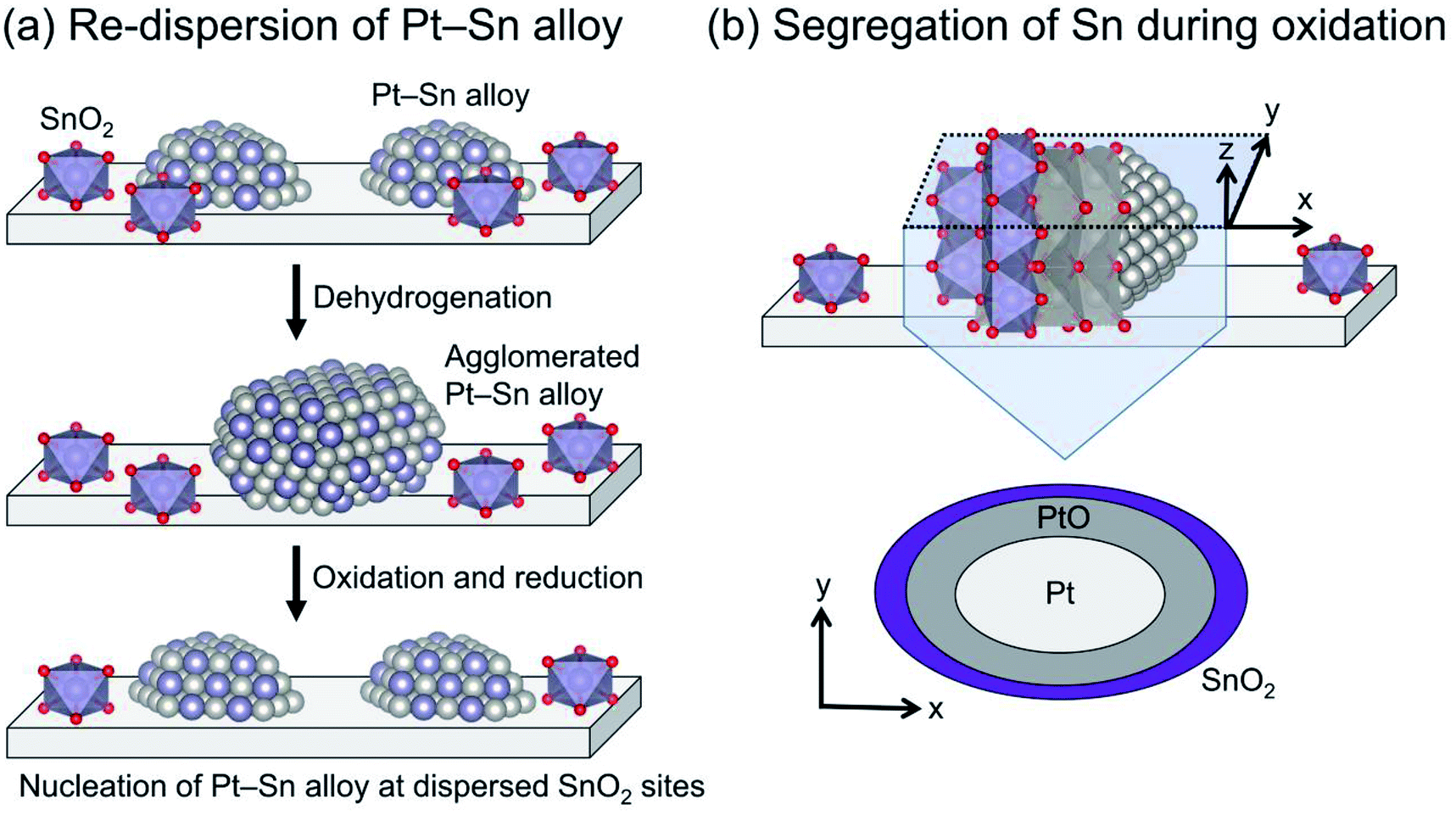 | ||
| Fig. 5 Schematic images of (a) re-dispersion of Pt–Sn alloy through oxidation and subsequent reduction (regeneration) and (b) segregation of Sn from Pt–Sn alloy during oxidation. | ||
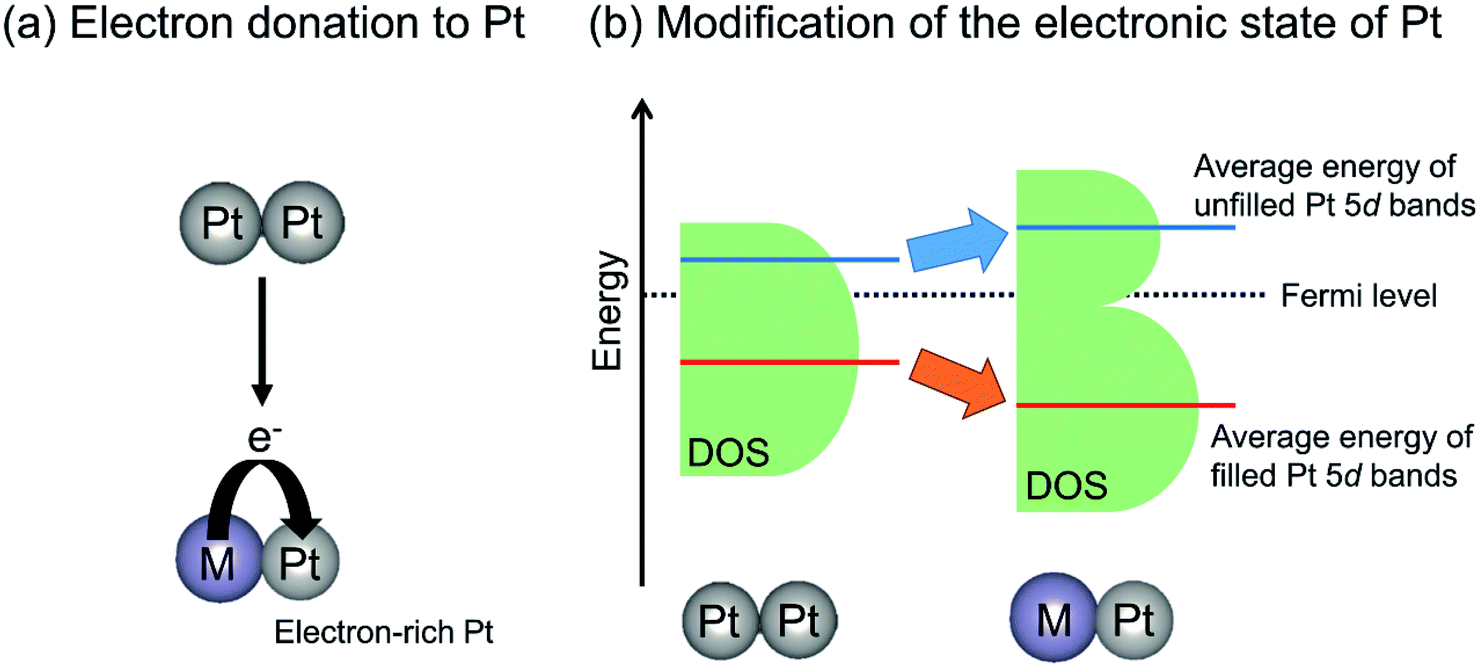 | ||
| Fig. 6 Proposed effects of the promoters on the electronic state of Pt: (a) electron donation from the promoter (M) to Pt and (b) modification of the energy level of Pt 5d bands. Densities of states are denoted as DOS. | ||
The electron donation from Sn to Pt can be supported by CO adsorption using IR spectroscopy. The absorption bands for CO linearly adsorbed onto Pt3Sn or PtSn show at lower wavenumbers than that on monometallic Pt terrace.64,67,68 The shift of the absorption bands is based on formation of electron-rich Pt by alloy formation, resulting in much back-donation from Pt to CO. However, the following two factors must be considered: (i) Pt–Sn alloy has no “cus-Pt” and (ii) the spectra for CO adsorption are measured under the same coverage of CO.81 In the former case, the absorption band shape corresponding to CO adsorbed onto Pt–Sn alloy is sharp compared to that on monometallic Pt. Ideally, the bands should be symmetric to demonstrate that Pt–Sn has environmentally uniform active sites. The latter case is related to dipole–dipole interactions between CO and adjacent CO adsorbed at Pt sites. On the Pt–Sn alloy surface, the number of Pt sites for CO adsorption is diluted with Sn. This dilution induces low coverage of CO and weak dipole–dipole interactions, resulting in low wavenumbers of bands for linearly adsorbed CO.
In contrast with electron donation, Xin et al. argued that the electronic effects of Sn on Pt are not electron donation but the change in the energy level of d-band center of Pt through the hybridization of Pt 5d bands with Sn valence (5s and 5p) bands.77,82 Recently, Miller and co-workers investigated the electronic structure of Pt in PtZn and Pt–Fe (Pt3Fe, PtFe and PtFe3) intermetallic compounds using resonant inelastic X-ray scattering to evaluate the energy level of Pt 5d orbitals.51,83 They reported that the average energy gap separating the filled and unfilled Pt 5d bands is expanded compared with that of monometallic Pt. In other words, Zn or Fe addition stabilized the energy level of the occupied 5d band, whereas the unoccupied 5d band is shifted to higher energy levels, as shown in Fig. 6(b). Additionally, they calculated the projected d density of states localized on Pt atoms.83 The mean energy of filled (below the Fermi level) Pt 5d bands is stabilized by virtue of alloy formation, whereas that of unfilled (above the Fermi level) Pt 5d bands is shifted upward. Therefore, they proposed that the alloy formation effect on the electronic state of Pt is the modification of the energy level of the Pt 5d bands rather than electron donation from the promoters to Pt. This change in the Pt 5d band energy is expected to affect the platinum–ethane interaction and to enhance the catalytic activity.
The change in the electronic state of Pt modified with the promoters influences the adsorption properties of intermediates and products. Hook et al. calculated the binding energy of the intermediates on Pt and Pt–Sn(111) surfaces based on DFT.44 From their results, they reported that ethylene adsorbs weakly on the Pt–Sn surface, indicating that re-adsorption of ethylene, which induces ethylene decomposition to coke, can be inhibited on the surface. Such is also the case in the binding energy of the coke precursors. Generally, ethylene adsorbs on Pt in either π-bonded or di-σ-bonded configuration.84 The latter adsorbs on the Pt surface more strongly, resulting in ethylene decomposition. Sn addition to Pt inhibits ethylene adsorption on Pt, as demonstrated by Liwu et al.85 However, large amounts of carbon deposition were verified in the case of Pt–Sn/γ-Al2O3.74,86 This phenomenon is based on the drain-off mechanism proposed by Lieske et al. and modified by Kumar et al.73,86 As shown in Fig. 7, the coke precursors weakly adsorbed onto Pt–Sn are facile to transfer from the Pt–Sn surface to γ-Al2O3. Then, coke formation proceeds on the support. This coke is burnable at higher temperatures than that deposited on Pt.85 In this way, Sn addition to Pt not only contributes to the suppression of coke formation; it also keeps the Pt–Sn surface clean.
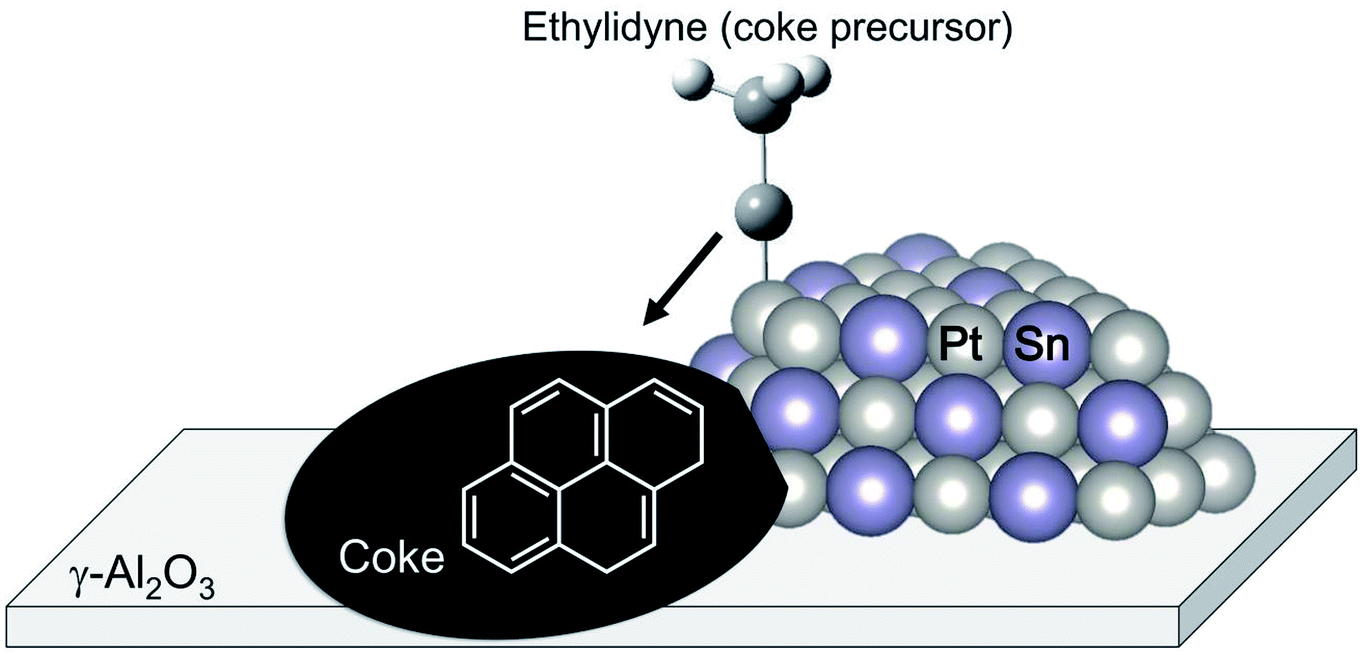 | ||
| Fig. 7 Schematic image of the drain-off mechanism. Coke precursors such as ethylidyne move to the supports on which coke formation proceeds. | ||
3.2. Chromium-based catalysts
Chromium (Cr) oxide catalysts have been used for dehydrogenation of light alkanes, as represented by the Catofin process (Mcdermott-Lummus).26 The dehydrogenation reactions are performed by multiple fixed bed reactors operated in the cyclic mode (reaction and regeneration).87 During the operation, several reactors are used for the reactions while catalyst regeneration is conducted in the others. Among the products, hydrogen is separated by a pressure swing adsorption unit. The unreacted feed is separated from the products and recycled for the reactions. The Catofine process is employed for dehydrogenation of propane and butanes, similarly to the Oleflex process. In terms of cost, the Cr oxide catalysts is superior to Pt-based catalysts. As described in this section, thorough investigations have been conducted of the active sites, effects of support, and influences of co-feeding CO2 in order to use the Cr oxide catalysts for EDH.The Cr species can be various structures. Generally, the Cr species are classifiable into four types: monochromate (isolated mononuclear Cr), polychromate (Cr oxide cluster), amorphous Cr oxide (Cr2O3) and crystalline α-Cr2O3.90–92,94 The Cr species structure is mainly dependent on the amount of Cr loading. At the high loading amount (>5 wt%), the amorphous Cr2O3 and crystalline α-Cr2O3 are easily formed. These Cr species are regarded as inactive for dehydrogenation.90,97 In contrast, monochromate and polychromate species are dominant at low loading amount. The formation of Cr2O3 is distinguishable from the color of the catalysts: actually, the color is varied from yellowish to greenish because of the Cr2O3 formation.95,98,99
Investigations of the nature of monochromate and polychromate have been conducted widely because these Cr species are active sites for EDH.87,100,101 Images of them are presented in Fig. 8. To identify the existence of the Cr species, UV-Vis and Raman spectroscopy are useful techniques. However, the Cr species are reduced under the reaction conditions from Cr6+ or Cr5+ to Cr3+ or Cr2+. Therefore, evaluating the active sites under in situ conditions is important. For fresh Cr oxide catalysts, Cr6+ is dominant. Many studies have specifically examined Cr6+ because the existence of Cr5+ was identified only by electron spin resonance (ESR) and IR spectroscopy.91,98 In addition, the reduced Cr species are mainly Cr3+. Although Cr2+ was identified using XPS and XAFS,96,100 the proportion of Cr2+ in the spent catalysts was rather small.100 Therefore, Cr6+ is considered to be reduced to Cr3+ during the dehydrogenation reaction. The nature of Cr5+ and Cr2+ remains under discussion.
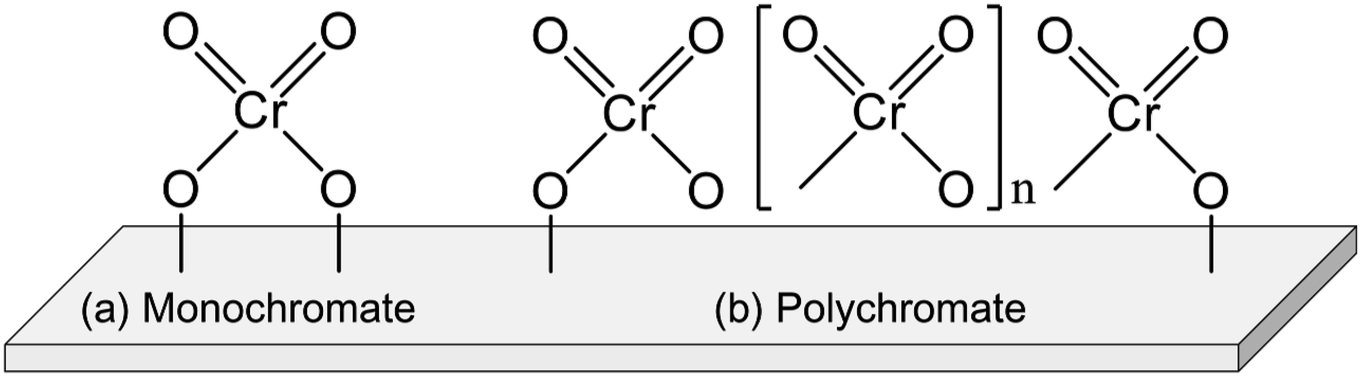 | ||
| Fig. 8 Structure of (a) monochromate and (b) polychromate supported on a metal oxide. | ||
The EDH over Cr oxide catalysts proceeds on the reduced monochromate and polychromate. As presented in Fig. 9(a), Olsbey et al. reported based on results of an isotopic study that end-on and dissociative adsorption of ethane occurs on the active sites.88 However, Shee and Sayari observed adsorbed acetaldehyde using diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy.102 They proposed that the ethoxide species formation is the first step in the dehydrogenation reaction (Fig. 9(b)). Determination of the adsorbed species on the respective Cr species is a challenging subject. Therefore, additional investigations based on, for instance, DFT calculations are expected to elucidate the reaction mechanism.
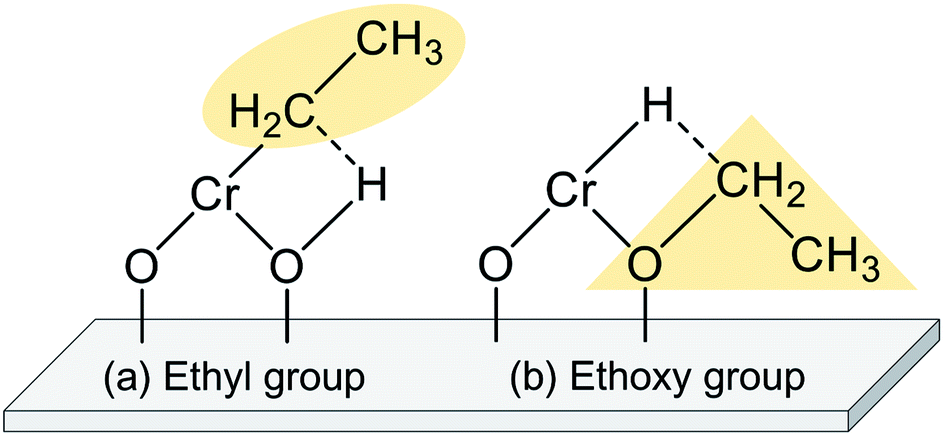 | ||
| Fig. 9 Schematic adsorption of ethane on reduced monochromate supported on a metal oxide: formation of (a) ethyl group and (b) ethoxy group. | ||
Alumina is a typical support for this purpose.88–92,95,102 Recently, Fridman et al. thoroughly examined the Cr species on the alumina support.95 They reported that a part of the monochromate and polychromate was unstable and reduced during dehydrogenation of isobutane. After the reduction, small Cr2O3 clusters were agglomerated; large Cr2O3 clusters were formed. Furthermore, regeneration of the catalyst under oxidative conditions induced formation of aluminum–chromium–chromate species around the periphery of the Cr2O3 clusters, as shown in Fig. 10. Agglomeration of the monochromate and polychromate leads to irreversible deactivation of the catalysts.
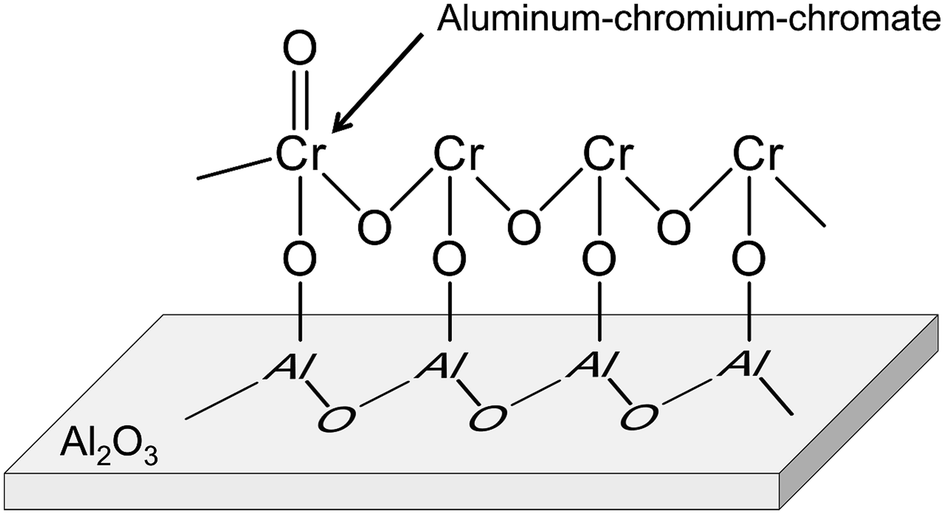 | ||
| Fig. 10 Structure of aluminum–chromium–chromate. | ||
Silica (SiO2) is an alternative support to alumina. Silica support of various kinds, including silicious-zeolites and mesoporous silica, were examined.94–100,103–106 These silica supports have a large surface area, resulting in highly dispersed Cr species on the supports. The amount of silanol (SiOH) groups is expected to play an important role in stabilizing the dispersed Cr species (monochromate or polychromate). Cheng et al. reported that the initial activity of Cr/SBA-15 (mesoporous silica) is proportional to the amount of the SiOH groups contributing to the abundantly dispersed Cr species.105 Botavina et al. proposed detailed structures of monochromate based on XAFS measurements.96 The reduced monochromate was bound with two short Si–O–Cr bonds and a long Si–O–Cr bond. Its electronic state is expected to be Cr2+ rather than Cr3+. These studies indicate that the density of the SiOH groups influences the Cr active site characteristics.
| C2H6(g) + CO2(g) → C2H4(g) + CO(g) + H2O(g), ΔrHθ (298 K) = 178 kJ mol−1 | (11) |
This chemical formula is interpreted as EDH (reaction (10)) in combination with the reverse water gas shift (RWGS) reaction.
| CO2(g) + H2(g) → CO(g) + H2O(g), ΔrHθ (298 K) = 41.2 kJ mol−1 | (12) |
Therefore, the reaction (11) characteristics differ completely from those of oxidative EDH using oxygen as an oxidizing agent, which is an exothermic reaction. Effects of the CO2 introduction to the dehydrogenation system are generally explained based on the following three reasons:108–110 decrease in the partial pressure of ethane (see also Fig. 2); removal of H2 from the reaction system through RWGS, resulting in the promotion of EDH; and improvement of catalytic stability by virtue of removal of carbonaceous deposits through the reverse Boudart reaction.
| C(s, graphite) + CO2(g) → 2CO(g), ΔrHθ (298 K) = 172 kJ mol−1 | (13) |
The promotion of ethane conversion and suppression of coke formation have been reported.99,109 However, the promotive effect on EDH over Cr-based catalysts is attributed not only to the contribution of RWGS but to differences in the nature of active sites and reaction mechanism. Without CO2, the active sites are presumed to be Cr3+ reduced from monochromate or polychromate (Cr6+). At the initial stage of the reaction, where the reduction of Cr6+ proceeds, ethane conversion tends to decrease,100,101 indicating that Cr6+ is more active than Cr3+ for EDH. Indeed, Mimura et al. reported that the apparent activation energy of EDH decreased from 119.3 (without CO2) to 91.3 (with CO2) kJ mol−1 using a Cr/ZSM-5 (zeolite) catalyst.98 Therefore, it is proposed that EDH proceeds through the redox mechanism as shown in Scheme 1. In this scheme, ethane is activated oxidatively at Cr6+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites, resulting in the formation of ethylene and water (H2O). Then, the catalytic cycle is completed through oxidation of the reduced Cr3+ species with CO2. Among the Cr species, Baek et al. proposed based on TPR that the large amount of polychromate results in a high catalytic activity for dehydrogenation of propane in the presence of CO2.111 Therefore, polychromate is expected to be the active site for EDH via the redox mechanism.
O sites, resulting in the formation of ethylene and water (H2O). Then, the catalytic cycle is completed through oxidation of the reduced Cr3+ species with CO2. Among the Cr species, Baek et al. proposed based on TPR that the large amount of polychromate results in a high catalytic activity for dehydrogenation of propane in the presence of CO2.111 Therefore, polychromate is expected to be the active site for EDH via the redox mechanism.
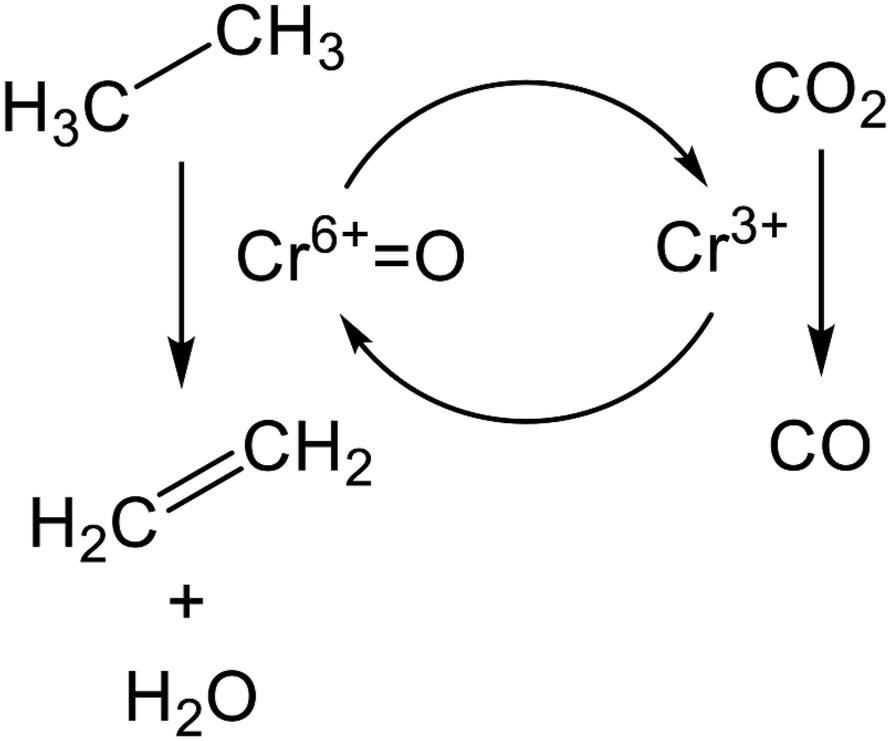 | ||
| Scheme 1 Redox mechanism for EDH in the presence of CO2 over Cr oxide catalysts. | ||
The redox behavior of the Cr species was verified using various techniques such as TPR, XPS, and IR spectroscopy. Based on TPR and XPS, the reduced Cr3+ can be oxidized to Cr6+ under a CO2 atmosphere.99,104,109,110 In addition, Mimura et al. evaluated CrO bonds by IR spectroscopy,98 demonstrating that the intensity of the absorption band attributed to CrO was decreased and increased respectively by ethane and CO2 treatments. However, the reducible Cr6+ species are not re-oxidized completely with CO2, probably because of agglomeration of monochromate and polychromate and because of the low oxidizing ability of CO2.112 Therefore, it is necessary for the catalyst to stabilize the reducible Cr6+ species and to promote oxidation of the reduced Cr3+. The silica supports are expected to be useful to stabilize the Cr active sites because of the presence of SiOH groups as described above. Recently, Al-Awadi et al. reported that modification of Cr/MCM-41 (mesoporous silica) with titania (TiO2) nanoparticles enhances the concentration of Cr6+, leading to high activity for EDH with CO2.113 To promote the oxidation of the reduced Cr species, the use of additives with oxygen storage capacity is one option.114,115 In addition, CO2 activation can be expected to be a key factor because of its low reactivity. Zirconia (ZrO2)-supported Cr catalysts exhibited a high activity for EDH in the presence of CO2 because of the basicity of ZrO2 which would contribute to CO2 activation.110,116 The Zr promoter also increases the amount of reducible Cr6+.117
Some difficulties remain unresolved in the case of EDH in the presence of CO2. For instance, the contribution of CO2 to oxidation of the reduced Cr species and RWGS, is not discussed in detail. One must elucidate the role of respective Cr species in dehydrogenation through the redox mechanism and RWGS. In addition, operando analyses in combination with isotopic CO2 should be performed to prove the redox mechanism.
3.3. Gallium-based catalysts
Several Ga oxide catalysts are known to have catalytic activity for EDH.118 Actually, Ga catalysts are classified into two types: zeolite-supported Ga oxide catalysts and other metal oxide-supported Ga oxide catalysts. Comparison of zeolite supports with other metal oxide supports such as Al2O3 and TiO2 reveals that characteristics of the active sites differ because of the cation-exchange ability of zeolite. In this regard, differences in the characteristics of the active sites, which are derived from properties of the supports, are described.From a practical perspective, Ga/H-ZSM-5 catalysts prepared using various methods such as physical mixing, impregnation, and chemical vapor deposition (CVD) are used to elucidate the characteristics of the Ga species.121–123 The pristine Ga/H-ZSM-5 usually contains highly dispersed Ga oxide species in the micropore system. At this stage, the Ga species are exchanged only insufficiently with the protons. To promote the ion-exchange, the Ga/H-ZSM-5 catalyst must be reduced under a H2 atmosphere. This phenomenon was verified using in situ IR spectroscopy. Intensity of the absorption band at ca. 3610 cm−1 derived from acidic hydroxy groups (Si–OH–Al) decreases under H2 atmospheres.121,124 Simultaneously, a new absorption band attributed to GaO–H appeared at ca. 3660 cm−1, clearly indicating the ion-exchange of the reduced Ga species with the acidic protons. Furthermore, formation of Ga–H bonds, which is unstable at high temperatures such as 573 K, was observed at room temperature. Based on findings obtained using IR spectroscopy, Ga+, GaO+, and [GaH2]+ are candidates for active sites of the reduced Ga/H-ZSM-5.124,125 In addition, Rane et al. evaluated the electronic state of Ga using in situ Ga K-edge XANES spectroscopy.123 At high temperatures (>673 K) under a H2 atmosphere, a new feature was observed in a lower energy region (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 371 eV), indicating the formation of Ga+. Subsequent cooling to 373 K induces the increase of a feature at 10377 eV with disappearance of the spectrum at the low energy feature, indicating the formation of [GaH2]+. However, Getsoian et al. reported that the absorption edge energy was dependent not only on the electronic state of Ga but also on the type of ligand.126 For instance, the absorption energy can be shifted to the lower position through the formation of low-coordinated Ga3+ alkyl or hydride species. However, Schreiber et al. performed quantitative analyses to elucidate the Ga species.127 They conducted H2 pulse tests at 873 K and measured the amount of the consumed H2 and formed H2O. As a result, they concluded that Ga+ was formed after reduction at 873 K.
371 eV), indicating the formation of Ga+. Subsequent cooling to 373 K induces the increase of a feature at 10377 eV with disappearance of the spectrum at the low energy feature, indicating the formation of [GaH2]+. However, Getsoian et al. reported that the absorption edge energy was dependent not only on the electronic state of Ga but also on the type of ligand.126 For instance, the absorption energy can be shifted to the lower position through the formation of low-coordinated Ga3+ alkyl or hydride species. However, Schreiber et al. performed quantitative analyses to elucidate the Ga species.127 They conducted H2 pulse tests at 873 K and measured the amount of the consumed H2 and formed H2O. As a result, they concluded that Ga+ was formed after reduction at 873 K.
The activity of respective Ga species was evaluated experimentally. Rane et al. performed dehydrogenation of propane after reduction or oxidation treatments.123 After reduction treatment to form [GaH2]+, the activity increased gradually with time on stream. In contrast, the activity of the oxidized Ga/H-ZSM-5 containing GaO+ decreased rapidly in 1 h. These activities were close to that of Ga+ species. They concluded that [GaH2]+ and GaO+ are unstable active sites during the dehydrogenation of propane. Furthermore, the intrinsic activities of the Ga species are in the order of GaO+ > Ga+ > [GaH2]+ based on the initial activities. However, Ausavasukhi and Sooknoi conducted EDH in a pulse reactor.122 They demonstrated that the activity was enhanced by the introduction of H2, indicating higher activity of [GaH2]+ for EDH than that of either Ga+ or GaO+. Moreover, the activity of [GaH2]+ decreased with subsequent C2H6 pulses under He, whereas high activity was maintained under H2, indicating the unstable characteristics of the [GaH2]+. Therefore, the Ga+ species would be stable during dehydrogenation reactions, although the activities of the Ga species for dehydrogenation of light alkanes would depend on the reactants. Nascimento and co-workers proposed that the reaction mechanisms were varied because of the size and type (linear or branched) of alkanes based on DFT studies.128,129 In addition, differences in the reactor systems (flow or pulse reactor) must be examined because the adsorbed species, which would influence the activity, exist in a steady state using the flow reactor.
The reaction mechanism of EDH on the Ga species was investigated mainly using DFT calculations. As portrayed in Scheme 2, three reaction mechanisms have been proposed by some research groups:128–133 the alkyl mechanism, the carbenium mechanism, and the concerted mechanism. The differences are based on the mode of ethane activation.
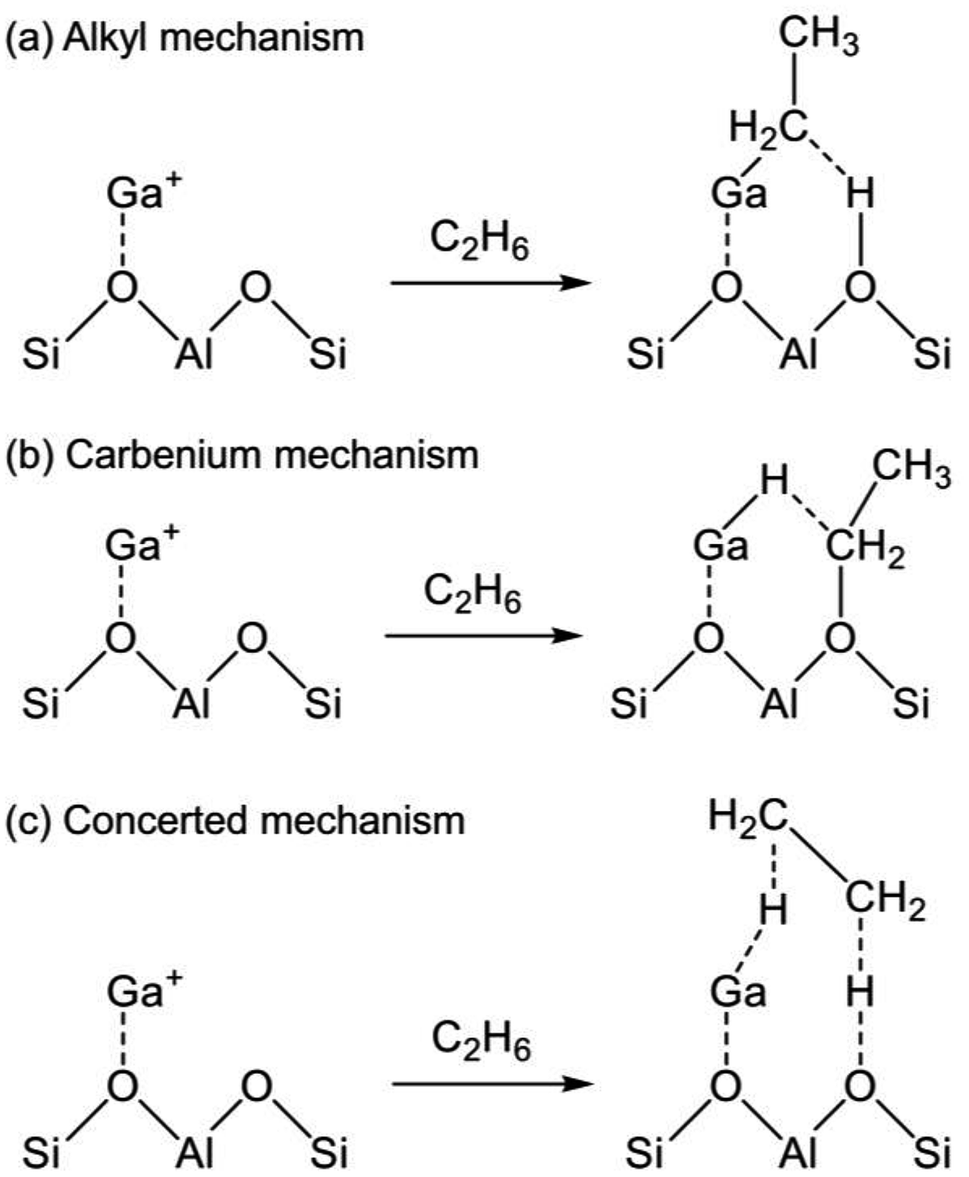 | ||
| Scheme 2 Mechanism of ethane activation via (a) the alkyl mechanism, (b) the carbenium mechanism, and (c) the concerted mechanism. Ga+ is a representative active site of Ga/H-ZSM-5. | ||
The alkyl mechanism begins from formation of Ga–C2H5 by hydrogen abstraction. By contrast, the carbenium mechanism proceeds through formation of an ethoxy group concomitantly with that of Ga–H. In the concerted mechanism, two hydrogen atoms are abstracted simultaneously from an ethane molecule, leading to the formation of ethylene and hydrogen in one step. The activation of ethane at Ga+, [GaH]2+, and [GaH2]+ sites is investigated by DFT calculations using cluster models. The alkyl mechanism is facile at any site.128–132 However, Mansoor et al. recently reported that the carbenium mechanism is preferable at the [GaH]2+ site.133 In addition, Schreiber et al. proposed that the proximity between the Ga and Brønsted acid sites promotes the dehydrogenation of propane.127 Experimentally, Kazansky et al. observed the adsorbed ethane on the Ga+ sites using DRIFT spectroscopy.125,134 They demonstrated the formation of Ga–C2H5 concomitantly with that of Ga–H. After heating, the absorption band of Ga–H at 2057 cm−1 was shifted to 2040 cm−1, which is attributed to H–Ga–H, with the appearance of olefinic C–H. Therefore, EDH at the Ga+ sites can be expected to proceed as the following Scheme 3.
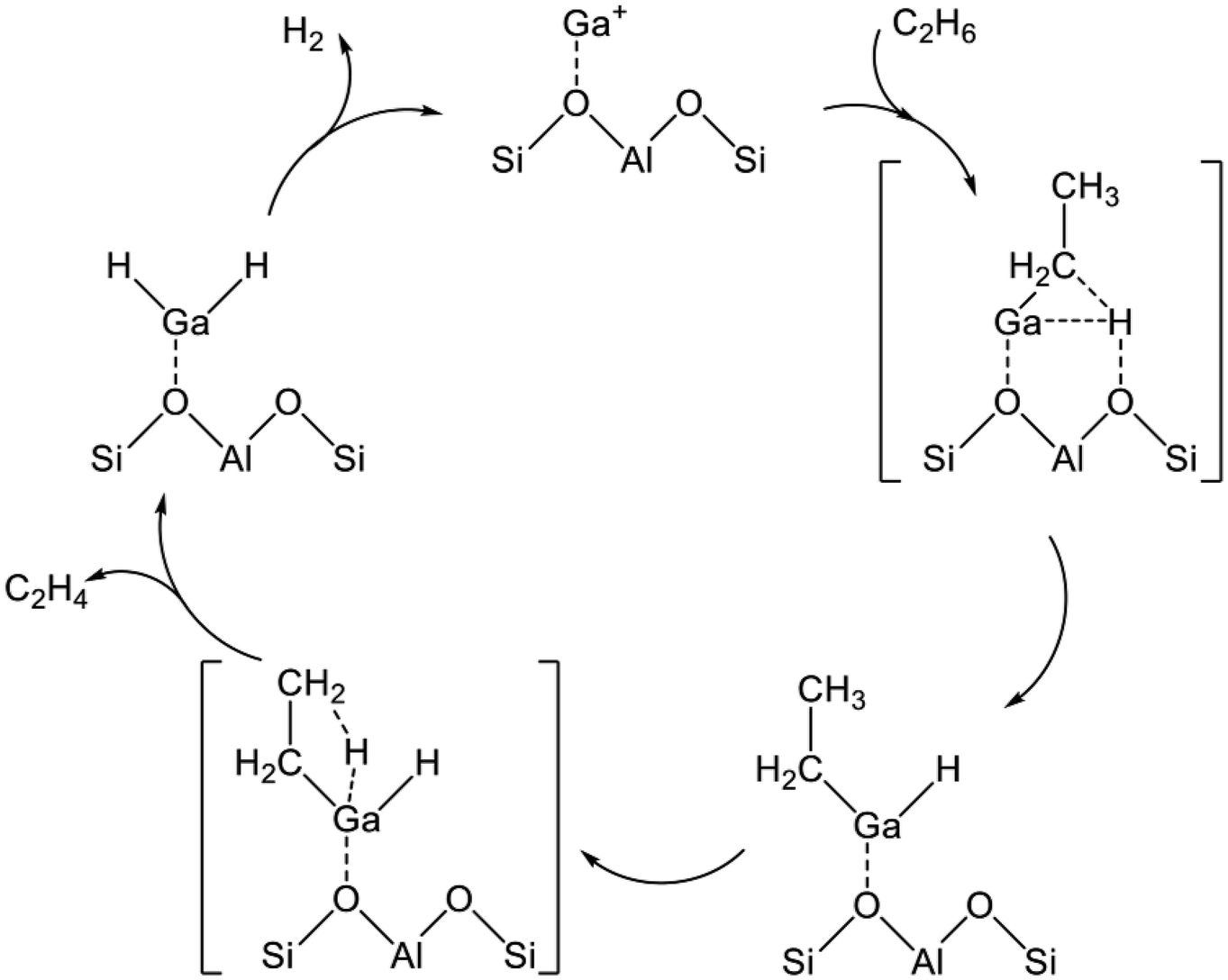 | ||
| Scheme 3 Reaction mechanism of EDH at the Ga+ site. | ||
The dehydrogenation activity of the GaO+ species was evaluated in the presence of steam because the GaO+ species are reduced under the reaction conditions.123 Hensen et al. performed dehydrogenation of propane over Ga/H-ZSM-5 in the presence of steam.135 As a result, stable activity was achieved. At the mononuclear GaO+ site, the high activation barrier for hydrogen recombination was calculated among the elementary steps, resulting in the formation of a H2O molecule and the Ga+ site.131 Therefore, they proposed that multinuclear GaO clusters are the active sites for the dehydrogenation reaction.135–138 The Ga cations included in the clusters are favorable for tetrahedral coordination.135 The proposed EDH at Ga2O2 clusters, for instance, is shown in Scheme 4. The dehydrogenation reaction proceeds at a Ga–O Lewis acid–base pair. Hydrogen recombination is facile at the Ga oxide clusters, although the H2O formation is still favorable. Strong Lewis basicity of the bridged oxygen stabilizes the adsorbed hydrogen, resulting in the inhibition of the hydrogen recombination.138 Therefore, Lewis basicity should be moderate to facilitate hydrogen desorption.
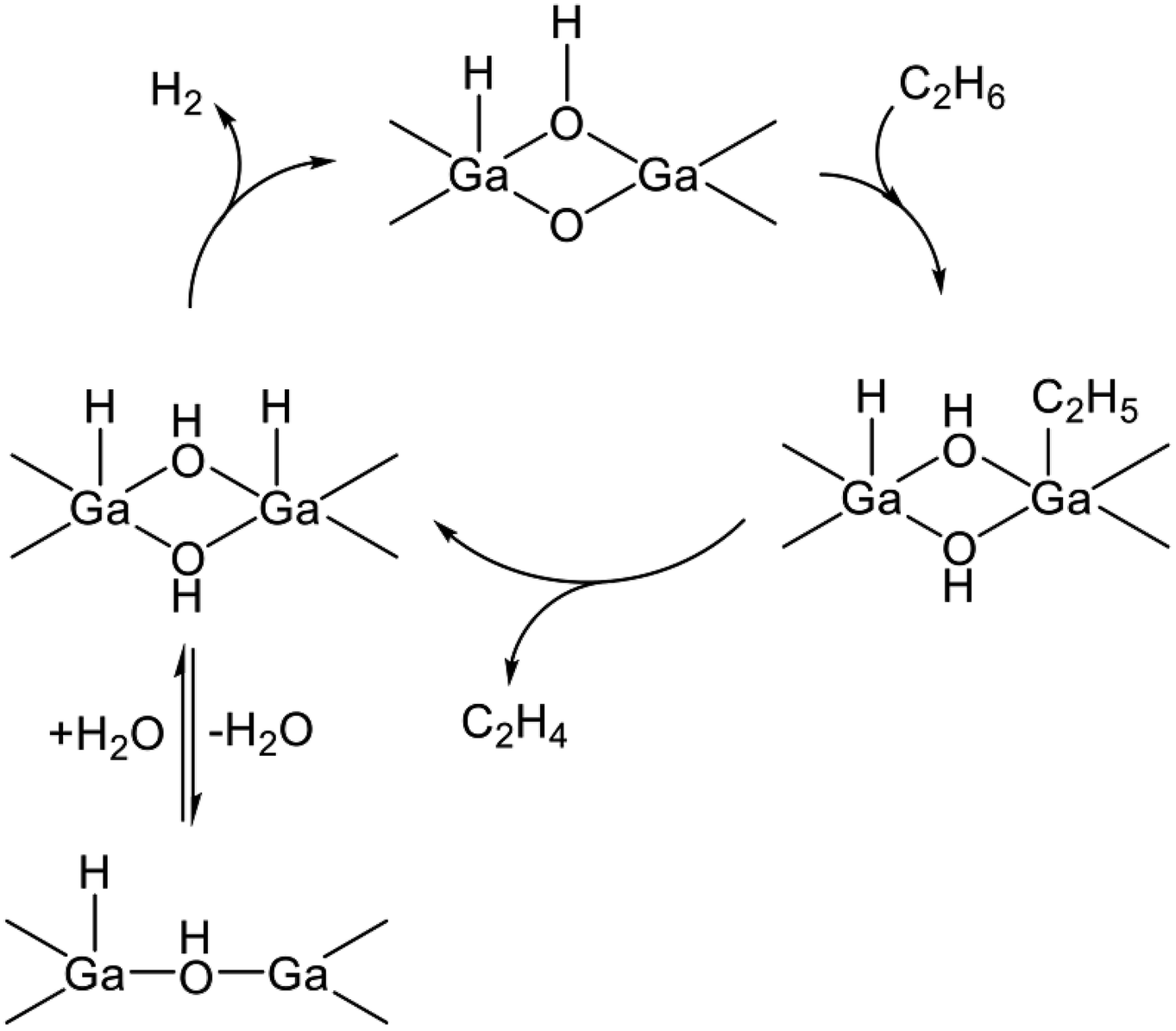 | ||
| Scheme 4 Reaction mechanism of EDH in the presence of steam on a Ga oxide cluster. | ||
The coordination environment of Ga can be evaluated using Ga K-edge XANES. Nishi et al. conducted a quantitative analysis of tetrahedrally and octahedrally coordinated Ga using XANES spectroscopy.143 Hereinafter, these Ga species are denoted respectively as Ga(T) and Ga(O). The analysis is based on deconvolution of the XANES spectra. It is applicable to Ga polymorphs and supported Ga oxide catalysts.143,144 In addition, the coordination environment of Ga2O3 can be evaluated by IR spectroscopy using H2 as a molecular probe. Collins et al. reported that the absorption bands attributed to Ga–H bonds are observed at around 2000 cm−1.145 They further measured the spectra for hydrogen adsorbed onto α-, β-, and γ-Ga2O3.146 The spectra consisted of absorption bands at 2003 and 1980 cm−1. The contribution of the band at 1980 cm−1 was in the order of α-Ga2O3 > γ-Ga2O3 > β-Ga2O3. This order agrees with that of the fraction of Ga(O) in their structures. Therefore, they concluded that the Ga(T)–H and Ga(O)–H bonds are observed respectively at 2003 and 1980 cm−1. The proportion of the surface Ga(T) to Ga(O) is calculable from the absorption band area. However, the band intensity is dependent on the temperatures.147,148 At high temperatures such as 573 K, the intensity increased because of the formation of oxygen defects, resulting in the formation of new Ga–H bonds. Therefore, the coordination environment of surface Ga is expected to be evaluated appropriately at temperatures higher than 573 K because the dehydrogenation reaction is usually conducted at such higher temperatures under reductive conditions.
The reaction mechanism of EDH over Ga2O3 is similar to the alkyl mechanism over Ga/H-ZSM-5 (Scheme 3). Kazansky et al. measured the spectra for ethane adsorbed onto Ga2O3 using DRIFT spectroscopy.149 In this case, the polymorph of Ga2O3 is not described clearly. They observed the formation of Ga–C2H5 in addition to C–H corresponding to CH2 groups derived from ethylene, and Ga–H. The formation of Ga–C2H5 was also verified by 13C cross-polarization magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy.150 In some cases, the formation of Ga–H and alkoxy groups was assumed to occur via the activation of light alkanes.151,152 Nevertheless, no clear evidence of the formation of the alkoxy groups has been reported to date.
The dehydrogenation of ethane over Ga oxide catalysts is performed mainly in the presence of CO2.118 Nakagawa et al. reported that Ga/TiO2 exhibited high activity when compared to other supports including Al2O3, SiO2, ZrO2, and zinc oxide (ZnO).139 In the case of Ga/Al2O3, a negative effect on the catalytic activity was verified in the presence of CO2. The promotive effects of CO2 on EDH are attributed respectively to hydrogen and coke removal through the RWGS and the reverse Boudart reaction (reactions (12) and (13)).153,154 In contrast, selectivity to ethylene tends to decrease with the increase in ethane activation.155 Methane formation is promoted instead of the decrease in ethylene selectivity, indicating the subsequent conversion of ethylene to methane. Therefore, appropriate conditions such as space velocity and partial pressure of CO2 are necessary to achieve high ethylene yield.
In contrast to Cr oxides, Ga2O3 is a non-reducible oxide. Therefore, no report has described the redox mechanism over Ga oxide catalysts in the presence of CO2, although Ga species in H-ZSM-5 might have redox ability. To improve catalytic performance, designing Ga active sites is indispensable. For instance, Ga/H-ZSM-5 catalysts exhibited a higher selectivity to ethylene than β-Ga2O3.156 Also, Ga/H-ZSM-5 exhibits the activity for RWGS, indicating that H2O co-exists during EDH with CO2. Therefore, the Ga oxide clusters, which exhibit high dehydrogenation activity via Scheme 4, would be maintained during the reaction.
3.4. Other catalysts and performance of EDH catalysts
Aside from Pt-, Cr- and Ga-based catalysts, cobalt (Co)- and molybdenum (Mo)-based catalysts exhibit catalytic activities for EDH.157–163 Although Zn catalysts also have ethane activation ability,134 they are applied for aromatization of light alkanes. Both are used mainly in the presence of CO2. Among Co-based catalysts, the electronic state of Co is expected to be important. The existence of Co4+ promotes EDH via the redox mechanism similarly to the Cr catalysts.157 However, Co2+ species facilitate RWGS analogously to the Ga catalysts.158,159 For Mo-based catalysts, molybdenum carbide (Mo2C) is rather more active for ethane activation than molybdenum oxide (MoO3) in the presence of CO2.160 Chen and co-workers evaluated the electronic state of Mo using in situ XANES spectroscopy, revealing that Mo2C is oxidized with CO2.161 They reported further that CO2 is converted to CO and adsorbed oxygen over β-Mo2C.162 Therefore, the oxygen-modified Mo2C (oxycarbide) would be formed and would promote EDH with CO2.Recently, lanthanum manganite (LaMnO3) perovskites were applied for EDH. Yang et al. performed EDH without co-feed over reduced Ni-doped LaMnO3.164 They found that the proportion of Mn4+ increases and that Ni nanoparticles are deposited after reduction treatment with H2. Therefore, they proposed that EDH proceeds on oxygen vacancies around Mn4+ and Ni nanoparticles. In contrast, Sekine and co-workers investigated EDH with H2O over Ba-doped LaMnO3.165,166 They demonstrated clearly that EDH is promoted in the presence of steam because the reaction proceeds via the Mars–van Krevelen mechanism. They proposed that the amount of reducible Mn3+ is a key factor for ethylene formation. Ba doping enhances the redox properties of LaMnO3 and the oxygens coordinated with Ba promote C–H activation of ethane.
Catalytic performance of the catalysts for EDH in the absence and presence of CO2 is summarized respectively in Tables 1 and 2. In addition, ethylene selectivity vs. ethane conversion is shown in Fig. 11. The noble metal catalysts are used at low temperatures without CO2, probably to suppress agglomeration and dry reforming. Yan et al. reported that modification of ceria (CeO2)-supported Ni catalysts with Fe could mitigate dry reforming of ethane by virtue of iron oxide (FeOx) located around periphery of Ni particles (Ni–CeO2 interface).167 Among the noble metals, palladium (Pd) and gold (Au) also exhibit the catalytic activity for EDH.168,169 In order to replace the expensive noble metals, Ni3Ga alloys,170 which exhibited comparable catalytic performance of EDH to noble metal catalysts,171 are good candidates. In recent studies, zeolite-supported metal catalysts appeared as new candidates for EDH without CO2. The characteristic micropores and cation-exchange ability contribute to the formation of active sites such as highly dispersed iron carbides and indium hydrides.172,173 In the presence of CO2, Cr-based catalysts supported on various materials including oxidized diamond174 and titanosilicate175 exhibit high ethane conversion thanks to the redox mechanism (Scheme 1), which indicates that other materials with redox properties can be good catalysts. The metal oxides with redox properties are applicable to a chemical looping process.176 Thorium (Th)- and Ni-based oxides are also reported to exhibit the activities177,178 although comparative investigations are necessary to elucidate advantages of these materials.
| No. | Catalyst | Temperature/K | C2H6 conversion/% | C2H4 selectivity/% | C2H4 yield/% | Ref. |
|---|---|---|---|---|---|---|
| 1 | PtSn/Mg(Al)O | 873 | 2.6 | 100 | 2.6 | 40 |
| 2 | Pt–In/SiO2 | 873 | 15 | 99 | 15 | 53 |
| 3 | PtSn–MgGaAlO | 823 | 28 | 99 | 28 | 54 |
| 4 | Pd–In/SiO2 | 873 | 15 | 100 | 15 | 168 |
| 5 | Au/SiO2-doped TiO2 | 923 | 16 | 95 | 15 | 169 |
| 6 | Ni3Ga/Al2O3 | 873 | 10 | 94 | 9.4 | 171 |
| 7 | Cr/SBA-15/Al2O3/FeCrAl | 1023 | 47 | 87 | 41 | 99 |
| 8 | Cr/MCM-41 | 923 | 19 | 98 | 18 | 100 |
| 9 | Cr–Ce/SBA-15 | 973 | 41 | 83 | 34 | 114 |
| 10 | Cr2O3/Oxidized diamond | 923 | 7.0 | 97 | 6.8 | 174 |
| 11 | Cr/TS-1 | 923 | 52 | 86 | 45 | 175 |
| 12 | Ga2O3/Al2O3 | 923 | 28 | 93 | 26 | 139 |
| 13 | Ga/SiO2-doped TiO2 | 923 | 46 | 85 | 39 | 154 |
| 14 | La0.9Mn0.8Ni0.2O3 | 1023 | 42 | 98 | 41 | 164 |
| 15 | La0.7Ba0.3MnO3−δ | 973 | 4.8 | 88 | 4.2 | 165 |
| 16 | Th0.75Ca0.25O2 | 998 | 48 | 78 | 37 | 177 |
| 17 | Fe/ZSM-5 | 873 | 22 | 72 | 16 | 172 |
| 18 | In–CHA | 933 | 27 | 97 | 26 | 173 |
| No. | Catalyst | Temperature/K | C2H6 conversion/% | C2H4 selectivity/% | C2H4 yield/% | CO2/C2H6 | Ref. |
|---|---|---|---|---|---|---|---|
| 19 | Cr/SBA-15/Al2O3/FeCrAl | 1023 | 67 | 96 | 64 | 2 | 99 |
| 20 | Cr2O3/SO4–SiO2 | 923 | 67 | 82 | 55 | 5 | 103 |
| 21 | Cr/MSU-1 | 973 | 68 | 82 | 56 | 3 | 104 |
| 22 | Cr/SBA-15 | 923 | 41 | 92 | 38 | 5 | 105 |
| 23 | Cr/ZSM-5 | 923 | 66 | 75 | 49 | 5 | 106 |
| 24 | Cr2O3/SiO2 | 923 | 56 | 93 | 52 | 5 | 108 |
| 25 | Cr/H-ZSM-5 | 923 | 68 | 70 | 47 | 9 | 109 |
| 26 | Cr/Ti/MCM-41 | 973 | 52 | 93 | 48 | 5 | 113 |
| 27 | Cr–Ce/SBA-15 | 973 | 55 | 96 | 53 | 3 | 114 |
| 28 | Cr2O3/HZSM-5–ZrO2 | 973 | 65 | 87 | 56 | 5 | 115 |
| 29 | Fe–Cr/ZrO2 | 923 | 54 | 93 | 50 | 3 | 116 |
| 30 | Cr2O3/Oxidized diamond | 923 | 27 | 87 | 24 | 5 | 174 |
| 31 | Cr/TS-1 | 923 | 62 | 81 | 50 | 4 | 175 |
| 32 | Ga2O3 | 923 | 29 | 87 | 25 | 5 | 139 |
| 33 | Ga2O3/ZSM-5 | 923 | 25 | 92 | 23 | 5 | 151 |
| 34 | Ga/SiO2-doped TiO2 | 923 | 47 | 78 | 37 | 5 | 154 |
| 35 | Ga/TiO2 | 973 | 38 | 57 | 22 | 2.5 | 155 |
| 36 | Co–BaCO3 | 923 | 48 | 92 | 44 | 3 | 157 |
| 37 | CoOx/SiO2 | 973 | 46 | 85 | 39 | 2.5 | 159 |
| 38 | Mo2C | 873 | 2.0 | 60 | 1.2 | 1 | 161 |
| 39 | Th0.75Ca0.25O2 | 998 | 46 | 97 | 45 | 0.78 | 177 |
| 40 | CaO–NiO/Al2O3 | 973 | 28 | 56 | 16 | 0.75 | 178 |
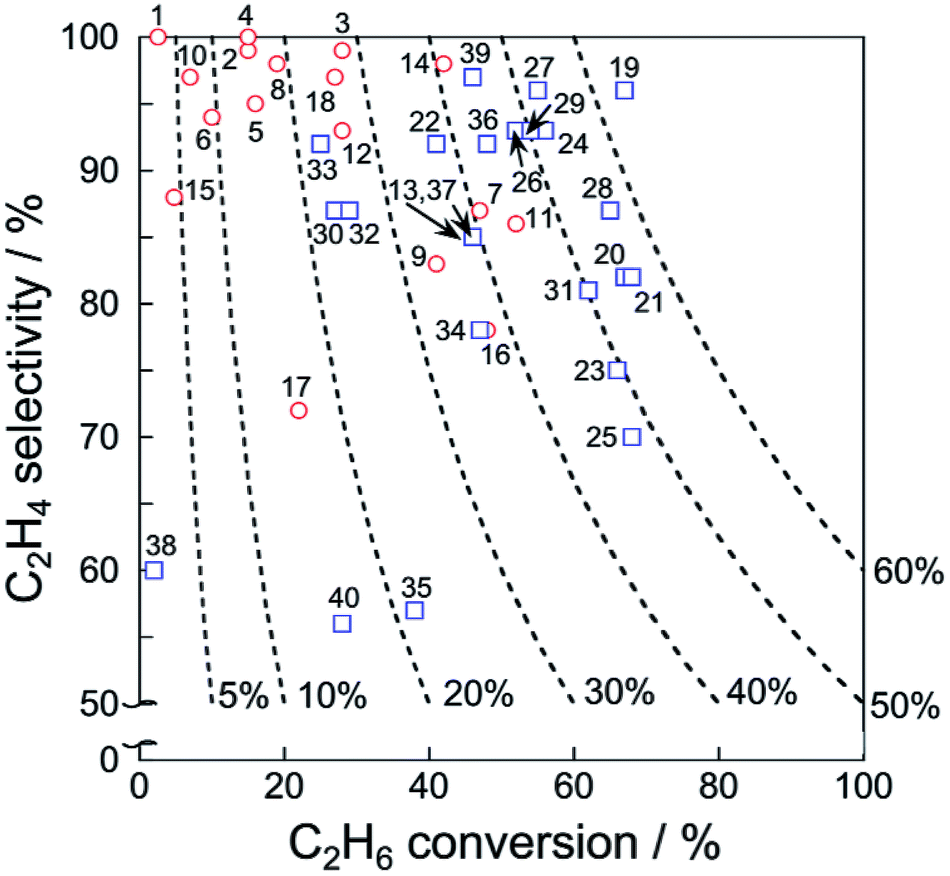 | ||
| Fig. 11 Ethylene selectivity vs. ethane conversion of each catalyst presented in Tables 1 and 2. Dashed lines represent ethylene yield. Also, EDH in the absence and presence of CO2 are shown respectively with red circles and blue squares. | ||
3.5. Non-thermo-catalytic EDH
Recently, electrochemical and photocatalytic EDH was emerged as new approaches for ethylene production. These systems can be operated using renewable energy; ethylene can be produced by renewable electricity and sunlight. Therefore, electrochemical and photocatalytic EDH would play a crucial role in producing petrochemicals in a sustainable society.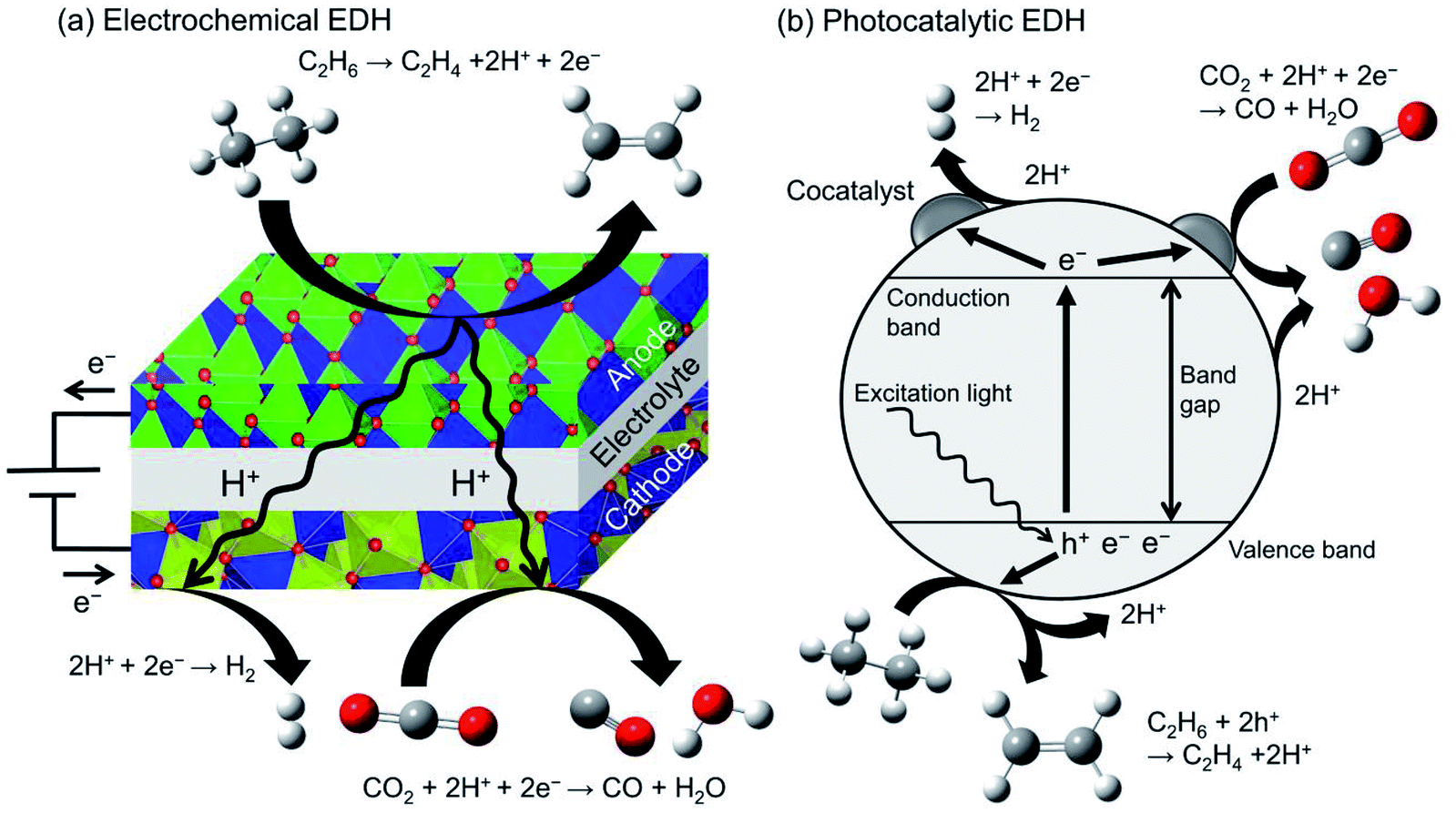 | ||
| Fig. 12 Schematic images of (a) electrochemical EDH by a solid oxide electrolysis cell and (b) photocatalytic EDH over a particulate semiconductor with a cocatalyst. | ||
Ding et al. performed electrochemical EDH at 673–773 K using BaCeO3-based proton conducting materials as the anode.180 They reported that electrochemical EDH proceeds without coke formation at 673 K. Increasing the reaction temperature induced coke formation and low ethylene selectivity. To mitigate coke formation, Zhang et al. used Ni and Cu doped niobium-based complexed oxides (proton conducting material) as the anode.181 Ni–Cu alloys formed through the reduction of the anode contribute to the suppression of coke formation even at 973 K. Ethane conversion and ethylene selectivity are 66.3 and 99.7% respectively at 0.8 V. In combination with CO2 reduction in the cathode, ethane conversion increased up to 75.2%. By contrast, Song et al. performed electrochemical EDH with CO2 reduction using oxygen anion conducting materials.182 They reported that γ-Al2O3 addition to lanthanum-based complexed oxide enhanced ethane conversion and ethylene selectivity without coke formation.
Researches on electrochemical EDH are focused on the performance and electric properties of anode materials. However, elucidation of activity–conductivity relationship is necessary for further improvement of the performance.
Photocatalytic EDH is performed by Zhang et al. using Pd/TiO2 catalysts in combination with CO2 reduction (Fig. 12(b)).184 Pd/TiO2 exhibited ethane conversion of 0.14% at 308 K and 0.2 MPa. The obtained conversion is corresponded to the equilibrium conversion at 498 K. It is explained that the forward and backward reactions require different energy and reaction mechanisms (irreversible).185 They also conducted spin-trapping ESR and demonstrated clearly that the hydroxyl radicals (·OH) are formed on TiO2 surface through oxidation of hydroxy groups with the holes. In addition to the holes, the hydroxyl radicals contribute to ethylene formation.
Producing chemicals from ethane through photocatalytic reactions are still immature research field. Practically using photocatalysts, the photocatalytic reactions must be visible light-driven. In addition to researches on materials science for controlling band gap energy, understanding the reaction mechanism is essential to design photocatalysts exhibiting high ethylene selectivity.
4. Dehydroaromatization of ethane
Ethane dehydroaromatization, denoted as EDA, forms BTX in one step.| 3C2H6(g) → C6H6(g) + 6H2(g), ΔrHθ (298 K) = 337 kJ mol−1 | (14) |
This reaction is an endothermic reaction similar to EDH. Therefore, high reaction temperatures at around 873 K are favorable to achieve high equilibrium conversion. From a practical perspective, the products are readily handled in terms of separation, storage and transportation because the gaseous reactant is converted to liquid products, potentially leading to a feasible process. However, commercial processes have not been developed yet using the natural gas resources (methane and ethane) in contrast with aromatization of C3 and C4 hydrocarbons.120 This lack of development is attributable to high reaction temperatures, resulting in the rapid deactivation of zeolite-supported metal catalysts through coke formation.186–189 Effective zeolite topology and active metals are briefly explained at the beginning of this chapter. Next, details of the nature of Zn/H-ZSM-5, the reaction pathways and deactivation by coke formation are described. Finally, aspects of the catalytic performance of the representative EDA catalysts are summarized.
4.1. Zeolite-supported metal catalysts
The MEL-type zeolite (ZSM-11) is another candidate for the support.193,194 Actually, ZSM-11 has similar micropore channel systems to those of ZSM-5, which consist of two straight channels.190,191 However, the superiority of ZSM-11 compared to ZSM-5 has not been demonstrated clearly. The MWW-type zeolite presented by MCM-22 has not been used in contrast with methane dehydroaromatization.195 Reportedly, MCM-22 has two independent micropore channel systems.190,191 One comprises 12-membered ring cages connected to 10-membered ring windows. The other is a two-dimensional micropore channel system. Difference between methane and ethane dehydroaromatization are attributable to the kinds of active metals. Zn or Ga catalysts are used mainly for EDA in contrast with Mo catalysts for methane dehydroaromatization. The use of Mo might be promising for BTX formation in the case of MCM-22 support because of the different reaction pathways, as described in Section 4.2.
| C2H6(g) + H2(g) → 2CH4(g), ΔrHθ (298 K) = −64.9 kJ mol−1 | (15) |
Also, H-ZSM-5-supported Mo and rhenium (Re) catalysts exhibit high performance for EDA201–203 in addition to dehydroaromatization of methane.204,205 However, these two metals are unsuitable for practical use because of sublimation of MoO3 and rhenium oxide (Re2O7),206,207 which probably exist in the pristine and regenerated catalysts, resulting in irreversible deactivation of the catalysts.
Preparation of Zn/H-ZSM-5 can be done using various methods such as impregnation, ion-exchange, physical mixing, and CVD.209–212 The active Zn species on H-ZSM-5 are classifiable into two types: ZnO species and ion-exchanged Zn species. The first, ZnO, exists on the external and internal surfaces denoted respectively as macrocrystalline ZnO and ZnO clusters. These ZnO species are analyzed conveniently using UV-Vis spectroscopy.209,212 Macrocrystalline ZnO exhibits an absorption band at ca. 370 nm attributed to its band gap energy. By contrast, ZnO clusters exhibit an absorption band at around 270 nm. Chen et al. reported that ZnO clusters were identified using laser-induced luminescence spectroscopy.213 When excited by a 244 nm laser, Zn/H-ZSM-5 containing ZnO clusters showed a purple luminescence band at ca. 440 nm. However, the ZnO species are reduced under reductive conditions at elevated temperatures. Some are exchanged with BAS (solid state ion-exchange), resulting in the formation of ion-exchanged Zn species and H2O.214
| ZnO + 2H+–Oz → Oz–Zn2+–Oz + H2O | (16) |
The existence of the ion-exchanged Zn species is roughly verified by temperature-programmed desorption of NH3 (NH3-TPD). In the TPD profile, two ammonia desorption peaks are observed respectively in low-temperature and high-temperature regions, denoted respectively as l-peak and h-peak.215 The l-peak is attributed to the desorption of ammonia bonded to the ammonia adsorbed onto acid sites. The h-peak is ascribed to desorption of ammonia adsorbed onto the acid sites, which indicates that the peak area of l-peak does not reflect the amount of acid sites. Additionally, the desorbed ammonia derived from BAS is indistinguishable from that from Lewis acid sites.216 Although the h-peak contains ammonia desorbed from BAS and Lewis acid sites, the h-peak intensity decreases after the ion-exchange,209 indicating the exchange of BAS with Zn cations. The decrease in BAS can be verified using 1H MAS NMR spectroscopy or NH3-TPD with IR and mass spectrometry.212,216 Formation of the ion-exchanged Zn species is verified by evaluating the electronic state of Zn because of its difference from that of ZnO.210 XPS studies revealed that the ion-exchanged Zn species show a peak at ca. 1024 eV in the Zn 2p3/2 XP spectrum. In contrast, a peak corresponding to ZnO is observed at ca. 1021 eV.
Structures of the ion-exchanged Zn species are controversial. Berndt et al. proposed the structure of the ion-exchanged Zn.211 They used temperature-programmed surface reaction (TPSR) with CO and observed formation of CO2 and H2, indicating the existence of [Zn(OH)]+, as shown in Scheme 5. During TPSR with CO, the water gas shift reaction proceeds at [Zn(OH)]+ and the nearest BAS, resulting in formation of the isolated Zn2+ (Oz–Zn2+–Oz).
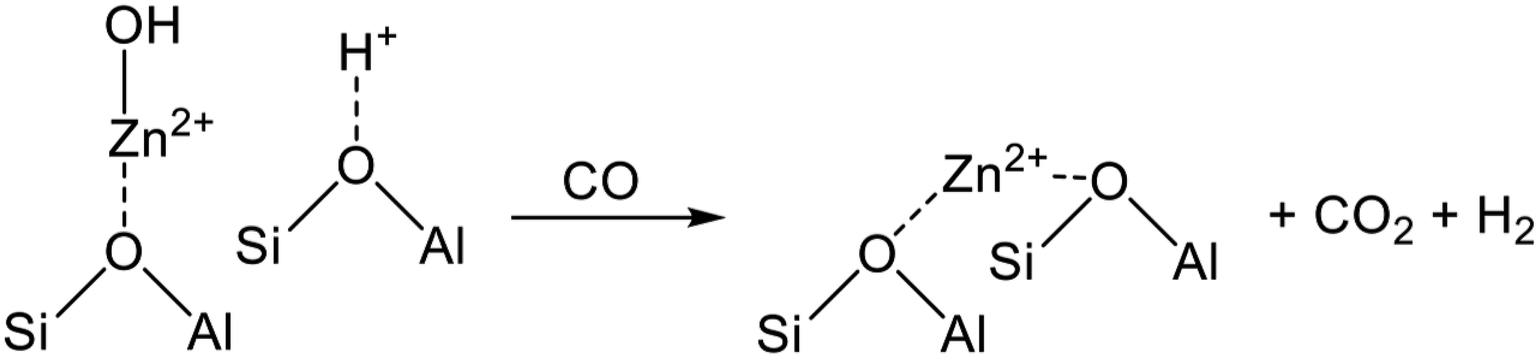 | ||
| Scheme 5 Structural change from [Zn(OH)]+ to isolated Zn2+ after the water gas shift reaction. | ||
In addition, Biscardi et al. analyzed Zn/H-ZSM-5 using in situ Zn K-edge XANES spectroscopy.217 The measurements were conducted in the presence of helium, hydrogen or propylene at up to 773 K. They concluded that [Zn(OH)]+ in pristine Zn/H-ZSM-5 is dehydrated at the nearest BAS, resulting in the isolated Zn2+ interacting with two ion-exchange sites under the propane dehydrogenation atmosphere. In their study, Zn/H-ZSM-5 with a small loading amount of Zn (<1.3 wt%) was used and, therefore, they described that two Zn2+ cations bridged by an oxygen atom (Oz–Zn2+–O–Zn2+–Oz), denoted as binuclear Zn species, could be formed at high Zn loading amount. Almutairi et al. investigated the structures of Zn in H-ZSM-5 prepared by CVD, impregnation, and ion-exchange methods.212 The isolated Zn species are formed by CVD. In contrast, the impregnation or ion-exchange method induces formation of Zn species of various kinds, including the isolated Zn and oxygenated Zn clusters. The specific structure of the oxygenated Zn clusters was not elucidated. According to Penzien et al.,218 the structure of Zn in *BEA-type zeolite depends on the Zn loading amount. Concentration of the binuclear Zn species increases at Zn/Al ≥ 0.15. Therefore, the large amount of Zn would result in formation of the binuclear Zn species in the case of H-ZSM-5. Furthermore, Tamiyakul et al. reported that the ion-exchanged Zn species existing in pristine Zn/H-ZSM-5 was reduced under H2 atmospheres based on XPS.210 They proposed formation of ZnH+ in the presence of H2. Indeed, Zn hydrides were observed at room temperature using DRIFT spectroscopy.219 Formation of ZnH+ would be attributed to the promotive H2 desorption at Zn sites through hydrogen backspillover.220–222 In contrast, Gao et al. also evaluated the electronic state of Zn by XPS after reduction with H2 at various temperatures.223 They described that ZnO clusters are reduced to [Zn(OH)]+ at 573 K, and reported that conversion of [Zn(OH)]+ to Zn2+ occurs at 673 K.
As described above, Zn species existing in H-ZSM-5 are diverse: ZnO, [Zn(OH)]+, Oz–Zn2+–Oz, Oz–Zn2+–O–Zn2+–Oz, and ZnH+. Under EDA conditions, ZnO and [Zn(OH)]+ are converted respectively to metallic Zn and Oz–Zn2+–Oz. The former is vaporized, resulting in the loss of Zn. The latter is anticipated as a major candidate for the active sites. Aleksandrov and Vayssilov investigated EDH at the isolated Zn2+, [Zn(OH)]+ and ZnH+ sites based on DFT.224 The isolated Zn2+ at a paired Al site is more active for ethane activation than ZnH+ at one Al site because of the vicinity of two Al centers. The existence of the binuclear Zn species has not been demonstrated clearly. Pidko and van Santen calculated energy diagrams of EDH at binuclear Zn sites using DFT calculations.225 The binuclear Zn species is not favorable for EDH, which is the initial reaction of EDA, because a stable intermediate is formed on the binuclear sites. However, the binuclear Zn species are presumed to be the active sites for EDA in recent studies.226,227 Therefore, further studies to elucidate the structures of the Zn active sites must be conducted to clarify the nature of the structure–activity relation.
4.2. Reaction pathway of BTX formation
Reaction pathways of BTX formation were investigated using Pt, Zn, Ga, and Re/H-ZSM-5.196,228–230 Because EDA is a complex reaction, the elucidation of each elementary step is challenging. Irrespective of the kind of active metal involved, the proposed reaction pathway is almost identical. As presented in Scheme 6, ethane is first dehydrogenated to ethylene at metal sites on the external or internal surface. Subsequently, ethylene is oligomerized, cyclized, and dehydrogenated to BTX in micropore channel systems. Under low conversion levels (low residence time), ethylene is the main product. However, selectivity to BTX and methane increases with decreased selectivity to ethylene under high conversion levels (high residence time). Also, C3 and C4 hydrocarbons, which are composed mainly of olefins, form during the reaction.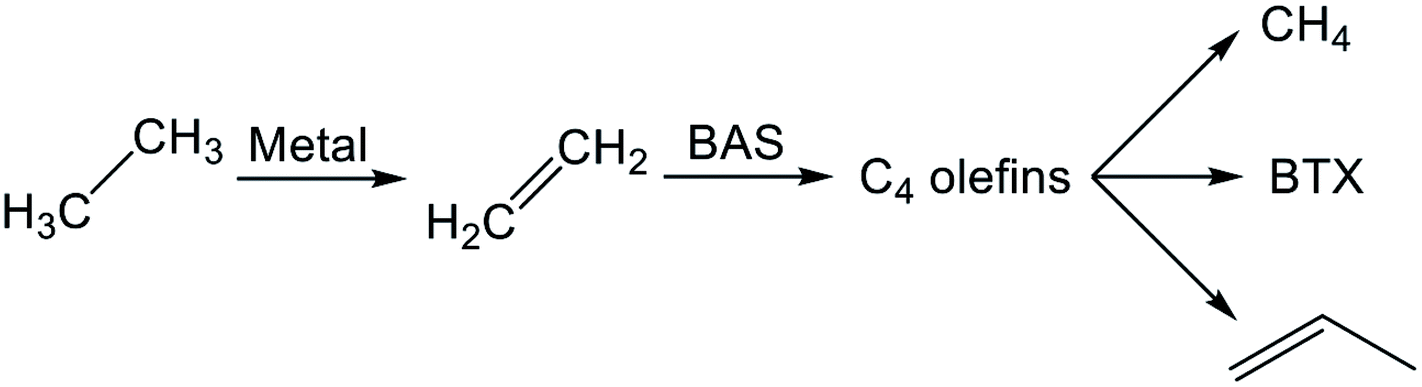 | ||
| Scheme 6 Reaction pathway of EDA. | ||
Hagen et al. demonstrated the formation of linear C4 olefins using a recirculation reactor.231 They proposed that the linear C4 olefins are intermediates for BTX. Reportedly, oligomerization of ethylene proceeds even at room temperature.232,233 In addition, ethylene is converted to propylene through oligomerization and cracking catalyzed by BAS.234,235 Therefore, ethylene would first be oligomerized to C4 olefins and would subsequently be cracked to propylene. Propylene could also be converted to BTX through a similar pathway of ethylene aromatization.236,237 In contrast with Scheme 6, a recent study of aromatization of ethylene proposed that hydrocarbon species also contribute to the reaction, similarly to the methanol conversion to aromatic hydrocarbons.238 It can be demonstrated based on 13C MAS NMR that aliphatic sp3 carbon species attributable to alkylated aromatic hydrocarbons formed through 13C2H4 aromatization are consumed after 12C2H4 aromatization.
Actually, the distribution of BTX depends on the reaction temperature.196 At lower temperatures (<723 K), their proportion is in the order of xylenes > toluene > benzene. In a middle temperature range (773–823 K), the proportion of toluene increases. Furthermore, benzene formation increases at higher temperatures. This tendency is the case in aromatization of ethylene, propylene, and methanol.236,239,240 Iglesia et al. performed dehydroaromatization of propane using 13C-propane (12CH3–13CH2–12CH3).241 They revealed that the number of 13C atoms included in benzene and toluene are statistically distributed, indicating that the propane carbon chain is rearranged randomly during the reaction. This is true probably because interconversion among olefins, which are the intermediates for aromatic hydrocarbons, occurs quite rapidly. Haag et al. reported that an approximate equilibrium distribution was attained for propylene conversion to olefins at 723 and 823 K.240 Depending on the reaction temperatures, light olefins are produced at higher temperatures, resulting in formation of light aromatic hydrocarbons such as benzene and toluene. Based on these results, Keipert et al. proposed that an olefin pool in which mutual olefins are equilibrated is formed during EDA.229 Recently, Liang et al. reported the finding of BTX formed from different olefin intermediates based on transient kinetic studies of ethane and ethylene aromatization.242 They proposed that the distribution of BTX might depend on the partial pressure of the olefin intermediates. In addition, Choudhary et al. reported that the proportion of BTX in aromatization of olefins is dependent on the space velocity.237,243 Dependence of the proportion of BTX on space velocity (conversion) is similar to dependence on temperature. The partial pressure of the intermediates varies with conversion of reactants, which can be controlled by the space velocity and reaction temperature because aromatization reactions are sequential reactions. Inclusively, the distribution of BTX can be controlled in the case in which the interconversion of olefins occurs under kinetic conditions. Under such conditions, other possible reactions such as hydro-dealkylation and trans-alkylation of aromatic hydrocarbons, which are catalyzed by BAS, might also affect the distribution of BTX. Fundamentally, these reactions are not considered for EDA. In addition to elucidation of the reaction pathway, the side reactions described above should be regarded as maximizing selectivity to valuable chemicals such as p-xylene.
The nature of active metals is a crucially important factor in the aromatization of light alkanes. Generally, the first dehydrogenation step (C–H bond activation) is a rate-determining step over H-ZSM-5. The active metals promote dehydrogenation of the alkanes. In contrast, Biscardi and Iglesia proposed the hydrogen desorption step as rate-determining in dehydrogenation of propane over Zn/H-ZSM-5.220 Indeed, Zn/H-ZSM-5 and ZnO exhibit hydrogen spillover and back-spillover effects,221,222 indicating that hydrogen desorption is promoted by virtue of Zn. In addition, the active metals influence BTX formation. Over H-ZSM-5, dienes and cyclodienes are intermediates in aromatization of ethylene, as reported by Batchu et al.244 As presented in Scheme 7, aromatization over H-ZSM-5 fundamentally proceeds through oligomerization, cyclization, and dehydrogenation via hydrogen transfer with formation of alkanes.245 In contrast, modification of H-ZSM-5 with Zn, Ga and silver (Ag) enhances selectivity to BTX in aromatization of ethylene,223,226,238,246–248 indicating the promotive effect of the active metals on formation of aromatic hydrocarbons. Bandiera and Taȃrit proposed that dehydrogenation of cycloalkenes to form BTX competes with ethane activation.249 They demonstrated that EDA was inhibited by addition of cyclohexadiene, regarded as a precursor of BTX, to the ethane feed.250 Therefore, the active metals contribute not only activation of light alkanes but the formation of BTX because aromatization of the intermediates would readily proceed in comparison to hydrogen transfer catalyzed by BAS. A recent study of methanol to aromatic hydrocarbons also revealed that introduction of Zn to H-ZSM-5 enhances dehydrogenation ability, which leads to high yield of aromatic hydrocarbons, and which inhibits hydrogen transfer because of the small amount of BAS induced by ion-exchange with Zn.251
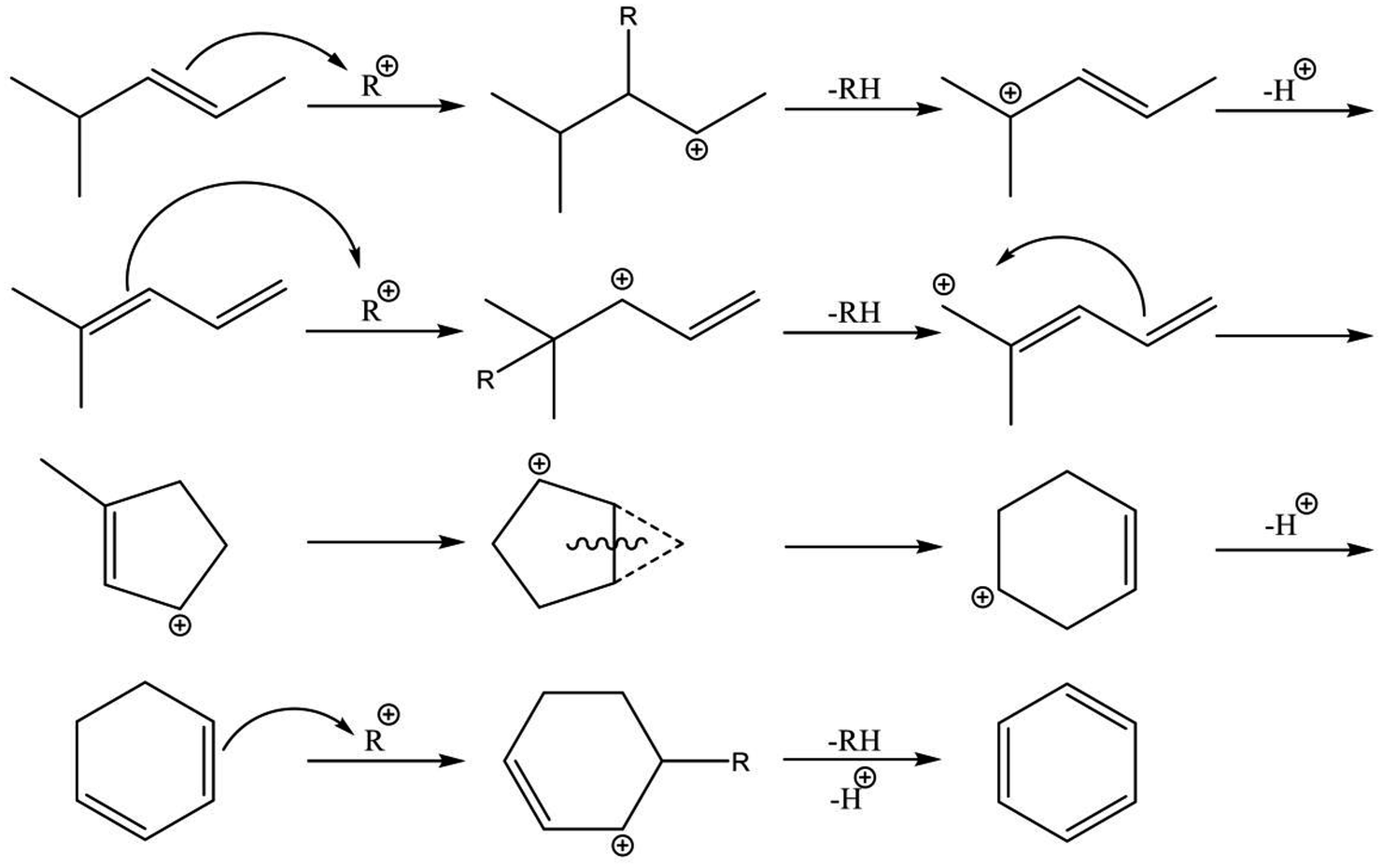 | ||
| Scheme 7 Aromatization of oligomers over H-ZSM-5. | ||
Recent studies of Mo/H-ZSM-5 for dehydroaromatization of methane have elicited new insights into the formation of aromatics.252–255 Generally, Mo/H-ZSM-5 is presumed to be a bifunctional catalyst, as reviewed in the literature.256–258 Briefly, activation of methane proceeds on carburized Mo species; subsequently, BAS promote aromatization of intermediates. Kosinov et al. demonstrated that methane conversion to benzene proceeds over Mo/Silicalite-1 with no Brønsted acidity.252 They proposed that BAS contribute to high dispersion of Mo species, which efficiently convert methane to benzene. Their studies further revealed the existence of radical carbon species in micropores.253 These confined carbon species play a role as intermediates in the dehydroaromatization of methane.254 Therefore, in analogous to methane, EDA over Mo/H-ZSM-5 is expected to proceed through the radical carbon species, as depicted in Scheme 8. Indeed, Uslamin et al. demonstrated clearly in EDA that reduction of Mo and subsequent building up the carbon species in micropores proceed through ethane decomposition.259 Benzene formation is initiated after the two-step induction period. Such was not observed in case of Ga/H-ZSM-5.
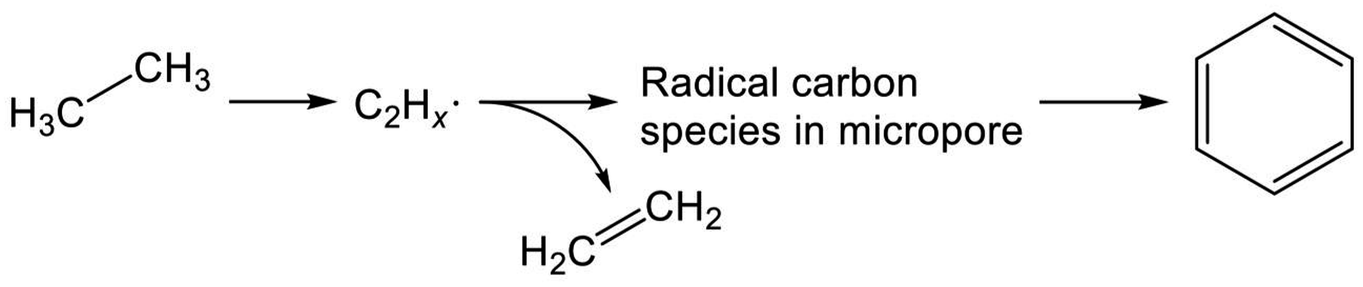 | ||
| Scheme 8 Possible reaction pathway of EDA over Mo/H-ZSM-5. | ||
In addition, Gascon and co-workers explored the nature of the carbon included in Mo (oxy-)carbide.260 They performed 12CO treatment to form Mo (oxy-)carbide and conducted pulse experiments using 13CH4. They demonstrated that 12C included in Mo (oxy-)carbide is consumed to form ethylene and benzene. The nature of Mo species is also investigated elsewhere.261–263 It is noteworthy that the characteristic nature of Mo (oxy-)carbide might not be exhibited in the aromatization of ethylene. After dehydroaromatization of methane and ethylene over Mo/H-ZSM-5, stable selectivity to aromatic hydrocarbons and reactive carbonaceous deposits was achieved in the case of methane.264 They concluded that ethylene is unlikely to be the main intermediate for dehydroaromatization of methane. Therefore, the reaction pathway of BTX formation would be considered carefully in EDA over Mo/H-ZSM-5.
4.3. Catalyst deactivation by coke formation
Deactivation of H-ZSM-5 modified with active metals presents great difficulty. Coke formation over H-ZSM-5 is catalyzed by BAS through over-oligomerization and aromatization. In addition, elements of the active metals and their amounts influence the ethylene to aromatic hydrocarbons step.238 Various techniques including thermogravimetric analysis, and Raman and 13C MAS NMR spectroscopy have been used to analyze coke species. Aspects of coke formation on zeolite catalysts are summarized in the literature.265 The coke species are mainly polycyclic aromatic hydrocarbons under aromatization conditions. They induce blocking of micropore channels, leading to eventual deactivation of the catalysts. Therefore, the catalysts must be periodically regenerated under oxidative conditions.266Addition of a promoter to a metal-modified H-ZSM-5 is one effective means of suppressing coke formation. Robinson et al. performed Fe addition to Mo/ZSM-5 for EDA.266 The Fe promoter improved the catalytic stability because carbon nanotubes were formed on the H-ZSM-5 external surface. Formation of the carbon nanotubes, which have ordered structures, is expected to maintain openings of the micropore channels. Another example is Pt addition to Ga/H-ZSM-5.267 In addition to the stability, Ga–Pt/H-ZSM-5 exhibited higher ethane conversion and selectivity to aromatic hydrocarbons than Ga/H-ZSM-5 and Pt/H-ZSM-5. The high stability of Ga–Pt/H-ZSM-5 would be attributed to consecutive removal of the coke species through hydrogenolysis during the reaction. Indeed, Marecot et al. performed TPSR with H2 using coke-deposited Pt, Re and iridium (Ir) catalysts.268 Methane formation through hydrogenolysis of the deposited coke was verified. The methane formation temperature increased in the order of Ir < Re < Pt.
Modification of BAS is an alternative means of inhibiting coke formation. The BAS are located on the external and internal surfaces. However, the BAS on the external surface are not expected to exhibit shape selectivity to BTX formation because they are not located in the micropore channels. The formation of coke precursors such as polycyclic aromatic hydrocarbons is rather favorable. Epelde et al. investigated the deactivation pathways of H-ZSM-5 during ethylene conversion to propylene.269 They inferred that the coke species composed of condensed aromatics and long aliphatic chains are deposited on the external surface, which indicates that the removal of BAS located at the external surface is effective. Inagaki et al. reported that nitric acid (HNO3) treatment of H-ZSM-5 synthesized without organic structure-directing agent removes Al (BAS) selectively from its external surface.270 In addition, control of the distribution of framework Al would also be an effective means. Generally, the MFI topology has 12 T-sites, which are candidates for Al sites, as shown in Fig. 13. Among them, T1, T4, and T6 do not face the intersections. Although T4 is located at the sinusoidal channels, T1 and T6 are located at the straight channels. The distribution of the Al sites can be controlled by the synthesis conditions.271,272 Liu et al. synthesized H-ZSM-5 with different Al distribution and performed ethane and ethylene aromatization over Pt/H-ZSM-5.273 They concluded that BAS located at the intersections tend to contribute to the formation of aromatic hydrocarbons. H-ZSM-5 with much Al located at the intersections exhibited high and stable conversion and BTX yield. The remaining Al sites located at the straight and sinusoidal channels can be expected to induce coke formation, resulting in the blocking of micropore channels and the inhibition of diffusion of the products.
 | ||
| Fig. 13 Twelve T-sites of MFI topology. | ||
The proximity of a Brønsted acid site to the nearest one is regarded as important for the suppression of coke formation. Zhang et al. prepared Zn/H-ZSM-5 using nano-ZSM-5 with crystalline size of approximately 100 nm.227 Because of the suppression of carbon deposition, Zn/nano-ZSM-5 exhibits higher stability for EDA than Zn/micro-ZSM-5 does. In micropores of nano-ZSM-5, the short diffusion path can be expected to facilitate the escape of aromatic hydrocarbons, leading to mitigation of consecutive conversion of the products to coke. It is noteworthy that nano-ZSM-5 has a larger external surface area than micro-ZSM-5. Assuming that the density of BAS on the external surface is equal to that on the internal surface, the contribution of BAS located on the external surface would be enhanced in the case of nano-ZSM-5. Therefore, selectivity to products would also depend on the crystalline size because BAS on the external surface do not exhibit shape selectivity to BTX formation. Introduction of mesopores, which probably improve mass diffusion, to ZSM-5 enhances the catalytic performance.274 It also contributes to an increase in coke capacity; ZSM-5 with the mesopores exhibited high activity and stability in spite of the large amount of carbon deposition.275 In another study, Ye et al. synthesized pillared ZSM-5 with various lamellar thickness.276 They performed EDA over Mo/pillared-ZSM-5 and demonstrated that ZSM-5 with thick layers (long diffusion path) and moderate Si/Al ratios exhibited high activity, selectivity to aromatic hydrocarbons, and stability. In their study, ZSM-5 with low Si/Al ratios (the large amount of BAS) is unfavorable because of its rapid deactivation. However, ZSM-5 with low Si/Al ratios has high ion-exchange capacity, which is rather convenient to incorporate large amounts of ion-exchanged metal cations such as Zn. In this case, dealumination as a post-synthetic method is a useful means to control the amount of BAS. Saito et al. performed steam treatment of Zn/H-ZSM-5 for EDA.277 They revealed that the framework Al with proton (BAS) is preferentially dealuminated through the steam treatment after ion-exchange with Zn. However, the framework Al with Zn cations was unaffected by the steam treatment. The steam-treated Zn/H-ZSM-5 exhibited high and stable activity for EDA, respectively, by virtue of optimal and large amounts of BAS and Zn.
4.4. Performance of EDA catalysts
Representative performance of EDA catalysts is presented in Table 3. The reaction is conducted at around 873 K using zeolites with low Si/Al ratios. Further studies of the properties of active metals and reaction mechanism are indispensable to enhance their catalytic performance. In addition, the improvement of catalytic stability is crucially important for the practical use of EDA on a large scale. As described above, studies of various kinds have been conducted, examining promoters, zeolite synthesis, and post-synthetic methods to elucidate methods of inhibiting coke formation. From a practical perspective, the reaction system would be pressurized to make the reactor volume small because large amounts of hydrogen are formed through EDA (reaction (14)). Recent study of dehydroaromatization of methane255 has revealed that catalytic stability can be improved at 15 bar through the rapid hydrogenation of coke at high pressure. Additionally, hydrogen removal from the reaction system can enhance BTX yield.278 Therefore, further studies conducted from chemical engineering perspectives must be undertaken to support EDA process development.| Catalyst | Si/Al | Temperature/K | C2H6 conversion/% | Aromatics selectivity/% | Aromatics yield/% | Ref. |
|---|---|---|---|---|---|---|
| Ga/H-ZSM-5 | 15 | 873 | 28.2 | 57.3 | 16.2 | 199 |
| Pt–Ga/H-ZSM-5 | 21 | 823 | 34.4 | 62.0 | 22.1 | 278 |
| Zn–H-ZSM-5 | 11.5 | 773 | 15.4 | 48.0 | 7.4 | 226 |
| Zn–H-ZSM-5 | 15 | 873 | 41.9 | 48.0 | 20.1 | 242 |
| Mo/ZSM-11 | 27 | 973 | 99.7 | 56.7 | 56.5 | 193 |
| Re/ZSM-5 | 30 | 873 | 48.0 | 59.0 | 28.2 | 203 |
5. Conclusion and perspectives
Recent progress in catalysts for EDH and EDA has been described from the perspective of the nature of active sites and reaction mechanisms. In terms of EDH, supported Pt-, Cr- and Ga-based catalysts have been intensively investigated.Generally speaking, Pt-based catalysts exhibit high activity and selectivity at low temperatures (<873 K). Fundamentally, promoters such as Sn are used for Pt catalysts to enhance their performance. Modification of Pt with Sn decreases the number of “cus-Pt”, at which sites hydrogenolysis and coke formation proceed, if Pt–Sn alloys are formed or not. In addition, the promoters affect the electronic state of Pt, although the intrinsic effects are under discussion (electron donation or modification of the Pt 5d bands). The change in the electronic state of Pt enhances TOF concomitantly with fast desorption of ethylene. For Pt-based catalysts, structure–activity and electronic state–activity relations can be expected to be mutually interactive. Practically, the structure of Pt active sites (fcc or hcp) must be examined with the surface composition of Pt/promoter ratios. Determination of the active sites contributes to construction of calculation models. In theoretical studies, coverage of adsorbates such as hydrogen affects the stability of the intermediates. Therefore, calculation conditions must be close to the realistic ones to predict the active site characteristics.
By contrast to Pt catalysts, Cr-based catalysts are cost-effective. Among the Cr species, trivalent monochromate and polychromate are candidates for use as active sites in the absence of CO2. On the other hand, CO2 co-feed enhances activity by virtue of redox properties. For the Cr catalysts, it is challenging to evaluate the activities of respective Cr species because both monochromate and polychromate usually exist. In addition, the reaction mechanisms that take place in the presence of CO2 must be evaluated carefully: for instance, kinetic analyses at low conversion levels and elucidation of CO2 roles using isotopes must be supported. Additional studies are expected to be indispensable for the assessment of Cr catalysts. However, these findings reveal that ethane activation is rather more favorable through the redox mechanism than through the Langmuir–Hinshelwood-like mechanism. Such is the case with other metal oxides with redox properties, such as La–Mn perovskites. These redox materials can be applied to EDH using chemical looping.279
Because of their cost-effectiveness and non-toxicity compared to Pt and Cr, Ga-based catalysts are also candidates for use for EDH. However, additional improvement of the catalytic performance must be done to make the Ga-based catalysts alternative candidates for them. It is noteworthy that zeolite supports in addition to the conventional supports including Al2O3, SiO2 and TiO2 are used for Ga in contrast with Pt and Cr. Zeolite supports contribute to the formation of characteristic Ga active sites, as described in Subsection 3.3.1. The Ga species in zeolite micropores have various structures such as [GaH2]+ and GaO clusters dependent on the reaction atmospheres. In recent studies, zeolite-supported Fe and In catalysts are applied to EDH. The characteristic nature of zeolite can increase the feasible options of active metals.
As for EDA catalysts, Ga, Zn or Mo supported on ZSM-5 has been investigated. Studies of EDA have specifically pursued elucidation of the structure of ion-exchanged Zn in ZSM-5 and methods to inhibit coke formation. Among the proposed structures, the isolated Zn (Oz–Zn2+–Oz) is a probable active site. The specific structures of oxygenated Zn species such as the binuclear Zn (Oz–Zn2+–O–Zn2+–Oz) are not demonstrated clearly. However, it is plausible that the properties of Zn depend on, for example, the preparation method and loading amount. To suppress coke formation, the Al location and the proximity of BAS in ZSM-5 should be controlled. For Mo, the reaction pathway is expected to be different from that of Zn. Therefore, further studies of the reaction pathways of BTX formation are expected to be indispensable for improvement of the catalytic performance.
To implement EDA practically, the activity and selectivity to aromatic hydrocarbons must be enhanced. Considering studies of EDH, CO2 co-feed is one option. Indeed, EDA with CO2 was performed over Ga and Zn/H-ZSM-5.280,281 Furthermore, other active metals with redox properties are expected to exhibit high catalytic performance for EDA with CO2 because the ethane activation step can proceed via the redox mechanism (Scheme 1). In the presence of CO2, Ga/H-ZSM-5 is apparently preferable to Zn/H-ZSM-5, probably because H2O is formed through RWGS. The existence of H2O promotes dehydrogenation in case of Ga/H-ZSM-5.135 The isolated Zn can be converted to [Zn(OH)]+, which would be inactive for aromatization, in the presence of H2O. In addition, a recent study of dehydroaromatization of methane has demonstrated that addition of metallic Zr to Mo/H-ZSM-5 enhances catalytic performance by virtue of efficient removal of H2 through hydrogen absorption with ZrH1.75 formation.282 Removal of H2 from the reaction system can shift the equilibrium of EDH forward, leading to high catalytic activity.
In conclusion, fundamental research efforts must be undertaken to develop catalytic processes for converting ethane to valuable chemicals. Recent progress in catalytic science and technology for ethane conversion has gradually revealed the intrinsic characteristics of the active sites indispensable to ethane activation. These new findings are expected to develop not only methods for efficient production of the valuable chemicals from ethane but for bridging technologies to guide future petrochemical industry development to production of a sustainable society.
Notes
The molecules and crystalline structures were visualized by GaussView 5 and VESTA softwares.283,284Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. S. acknowledges financial support from JSPS KAKENHI (Grant-in-Aid for JSPS Fellows) Grant number 19J10015. Y. S. acknowledges support by JST-CREST (grant no. JPMJCR1423).References
- J. Boulamanti and A. Moya, Renewable Sustainable Energy Rev., 2017, 68, 1205 CrossRef.
- Forecast of Global Supply and Demand Trends for Petrochemical Products (from 2010 to 2023), METI Japan, https://www.meti.go.jp/english/press/2019/<?pdb_no 1017?>1017<?pdb END?>_001.html, 2019, accessed on 20/5/2020 Search PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613 CrossRef CAS PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155 RSC.
- N. Popoff, E. Mazoyer, J. Pelletier, R. M. Gauvin and M. Taoufik, Chem. Soc. Rev., 2013, 42, 9035 RSC.
- R. Grabowski, Catal. Rev.: Sci. Eng., 2006, 48, 199 CrossRef CAS.
- C. A. Gärtner, A. C. van Veen and J. A. Lercher, ChemCatChem, 2013, 5, 3196 CrossRef.
- F. O. Rice and K. F. Herzfeld, J. Am. Chem. Soc., 1934, 56, 284 CrossRef CAS.
- M. L. Poutsma, J. Anal. Appl. Pyrolysis, 2000, 54, 5 CrossRef CAS.
- M. W. M. van Goethem, S. Barendregt, J. Grievink, J. A. Moulijn and P. J. T. Verheijen, Ind. Eng. Chem. Res., 2007, 46, 4045 CrossRef CAS.
- C. Batiot and B. K. Hodnett, Appl. Catal., A, 1996, 137, 179 CrossRef CAS.
- K. M. Sundaram and G. F. Froment, Ind. Eng. Chem. Fundam., 1978, 17, 174 CrossRef CAS.
- K. M. Van Geem, G. J. Heynderichx and G. B. Marin, AIChE J., 2004, 50, 173 CrossRef CAS.
- E. Heracleous and A. A. Lemonidou, Appl. Catal., A, 2004, 269, 123 CrossRef CAS.
- G. F. Froment, B. O. van de Steene, P. S. van Damme, S. Narayanan and A. G. Goossens, Ind. Eng. Chem. Process Des. Dev., 1976, 15, 495 CrossRef CAS.
- K. M. Sundaram, P. S. van Damme and G. F. Froment, AIChE J., 1981, 27, 946 CrossRef CAS.
- S. Mahamulkar, K. Yin, P. K. Agrawal, R. J. Davis, C. W. Jones, A. Malek and H. Shibata, Ind. Eng. Chem. Res., 2016, 55, 9760 CrossRef CAS.
- S. H. Symoens, N. Olahova, A. E. M. Gandarillas, H. Karimi, M. R. Djokic, M. Reyniers, G. B. Marin and K. M. Van Geem, Ind. Eng. Chem. Res., 2018, 57, 16117 CrossRef CAS.
- A. E. M. Gandarillas, K. M. Van Geem, M. Reyniers and G. B. Marin, Ind. Eng. Chem. Res., 2014, 53, 6358 CrossRef.
- S. A. Sarris, N. Olahova, K. Verbeken, M. Reyniers, G. B. Marin and K. M. Van Geem, Ind. Eng. Chem. Res., 2017, 56, 1424 CrossRef CAS.
- S. Wauters and G. B. Marin, Ind. Eng. Chem. Res., 2002, 41, 2379 CrossRef CAS.
- M. Takahashi, Y. Enjo and S. Uramaru, US Pat., US0318593A1, 2011.
- S. S. A. Petrone, R. L. Deuis, F. Kong, and Y. Chen, US Pat., US9421526B2, 2016.
- S. Wang, W. Peng, Q. Fu, Z. Deng, Z. Wu, C. Lin, Y. Gu, X. Zhang, and L. B. Kool, US Pat., US10138431B2, 2018.
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397 CrossRef CAS.
- P. R. Cottrell and M. E. Fettis, US Pat., US5087792, 1992.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670 CrossRef CAS PubMed.
- L. Shi, G.-M. Deng, W.-C. Li, S. Miao, Q.-N. Wang, W.-P. Zhang and A.-H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994 CrossRef CAS PubMed.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- J. Feng, Y. He, Y. Liu, Y. Du and D. Li, Chem. Soc. Rev., 2015, 44, 5291 RSC.
- K. Takehira, Appl. Clay Sci., 2017, 136, 112 CrossRef CAS.
- D. Akporiaye, S. F. Jensen, U. Olsbye, F. Rohr, E. Rytter, M. Rønnekleiv and A. I. Spjelkavik, Ind. Eng. Chem. Res., 2001, 40, 4741 CrossRef CAS.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192 CrossRef CAS PubMed.
- H. Song, R. M. Rioux, J. D. Hoefelmeyer, R. Komor, K. Niesz, M. Grass, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3027 CrossRef CAS PubMed.
- Z. Peng, F. Somodi, S. Helveg, C. Kisielowski, P. Specht and A. T. Bell, J. Catal., 2012, 286, 22 CrossRef CAS.
- G. Peng, D. Gerceker, M. Kumbhalkar, J. A. Dumesic and M. Mavrikakis, Catal. Sci. Technol., 2018, 8, 2159 RSC.
- J. Wu, Z. Peng and A. T. Bell, J. Catal., 2014, 311, 161 CrossRef CAS.
- J. Zhu, M.-L. Yang, Y. Yu, Y.-A. Zhu, Z.-J. Sui, X.-G. Zhou, A. Holmen and D. Chen, ACS Catal., 2015, 5, 6310 CrossRef CAS.
- V. Galvita, G. Siddiqi, P. Sun and A. T. Bell, J. Catal., 2010, 271, 209 CrossRef CAS.
- S. Saerens, M. K. Sabbe, V. V. Galvita, E. A. Redekop, M.-F. Reyniers and G. B. Marin, ACS Catal., 2017, 7, 7495 CrossRef CAS.
- A. Hook, J. D. Massa and F. E. Celik, J. Phys. Chem. C, 2016, 120, 27307 CrossRef CAS.
- M. H. Hansen, J. K. Nørskov and T. Bligaaed, J. Catal., 2019, 374, 161 CrossRef CAS.
- H.-A. Ha, E. T. Baxter, A. C. Cass, S. A. Anderson and A. N. Alexandrova, J. Am. Chem. Soc., 2017, 139, 11568 CrossRef PubMed.
- Z. Han, S. Li, F. Jiang, T. Wang, X. Ma and J. Gong, Nanoscale, 2014, 6, 10000 RSC.
- P. Sun, G. Siddiqi, M. Chi and A. T. Bell, J. Catal., 2010, 274, 192 CrossRef CAS.
- G. Siddiqi, P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200 CrossRef CAS.
- E. A. Redekop, V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, ACS Catal., 2014, 4, 1812 CrossRef CAS.
- M. Filez, E. A. Redekop, H. Poelman, V. V. Galvita, R. K. Ramachandran, J. Dendooven, C. Detavernier and G. B. Marin, Chem. Mater., 2014, 26, 5936 CrossRef CAS.
- Z. Yu, J. A. Sawada, W. An and S. M. Kuznichi, AIChE J., 2015, 61, 4367 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, ACS Catal., 2017, 7, 4173 CrossRef CAS.
- P. Sun, G. Siddiqi, W. C. Vining, M. Chi and A. T. Bell, J. Catal., 2011, 282, 165 CrossRef CAS.
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Catal. Today, 2018, 299, 146 CrossRef CAS.
- S. Fang, K. Zhnag, C. Wang, L. Ma, Q. Zhang, Q. Liu, L. Chen, Q. Zhang and Z. Tian, RSC Adv., 2017, 7, 22836 RSC.
- Q. Zhang, K. Zhang, S. Zhang, Q. Liu, L. Chen, X. Li, C. Wang and L. Ma, J. Catal., 2018, 368, 79 CrossRef CAS.
- A. Virnovskaia, S. Jørgensen, J. Hafizovic, Ø. Prytz, E. Kleimenov, M. Hävecher, H. Bluhm, A. Knop-Gericke, R. Schlögl and U. Olsbye, Surf. Sci., 2007, 601, 30 CrossRef CAS.
- A. Virnovskaia, S. Morandi, E. Rytter, G. Ghiotti and U. Olsbye, J. Phys. Chem. C, 2007, 111, 14732 CrossRef CAS.
- A. Virnovskaia, E. Rytter and U. Olsbye, Ind. Eng. Chem. Res., 2008, 47, 7176 CrossRef.
- E. Jimenez-Izal, H. Zhai, J.-Y. Liu and A. N. Alexandrova, ACS Catal., 2018, 8, 8346 CrossRef CAS.
- E. Jimenez-Izal, J.-Y. Liu and A. N. Alexandrova, J. Catal., 2019, 374, 93 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780 CrossRef CAS PubMed.
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735 CrossRef CAS.
- A. Hook and F. E. Celik, J. Phys. Chem. C, 2017, 121, 17882 CrossRef CAS.
- L. Deng, H. Miura, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, J. Catal., 2018, 365, 277 CrossRef CAS.
- L. Deng, T. Shishido, K. Teramura and T. Tanaka, Catal. Today, 2014, 232, 33 CrossRef CAS.
- L. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, ChemCatChem, 2014, 6, 2680 CrossRef CAS.
- L. Deng, H. Miura, T. Ohkubo, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, Catal. Sci. Technol., 2019, 9, 947 RSC.
- A. Moscu, Y. Schuurman, L. Veyre, C. Thieuleux and F. Meunier, Chem. Commun., 2014, 50, 8590 RSC.
- C. Dupont, D. Loffreda, F. Delbecq and Y. Jugnet, J. Phys. Chem. C, 2007, 111, 8524 CrossRef CAS.
- Q. Wang, D. Tichit, F. Meunier and H. Guesmi, J. Phys. Chem. C, 2020, 124, 9979 CrossRef CAS.
- A. Moscu, C. Theodoridi, L. Cardenas, C. Thieuleux, D. Motta-Meria, G. Agostini, Y. Schuurman and F. Meunier, J. Catal., 2018, 359, 76 CrossRef CAS.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551 CrossRef CAS PubMed.
- M. S. Kumar, D. Chen, A. Holmen and J. C. Walmsley, Catal. Today, 2009, 142, 17 CrossRef.
- H. N. Pham, J. J. H. B. Sattler, B. M. Weckhuysen and A. K. Datye, ACS Catal., 2016, 6, 2257 CrossRef CAS PubMed.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, J. Catal., 2010, 276, 268 CrossRef CAS.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, Phys. Chem. Chem. Phys., 2011, 13, 15833 RSC.
- H. Xin, N. Schweitzer, E. Nikolla and S. Linic, J. Chem. Phys., 2010, 132, 111101 CrossRef PubMed.
- D. Bazin, D. Sayers, J. J. Rehr and C. Mottet, J. Phys. Chem. B, 1997, 101, 5332 CrossRef CAS.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222 CrossRef CAS.
- M. W. Tew, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. C, 2009, 113, 15140 CrossRef CAS.
- D. Tibiletti, A. Goguet, D. Reid, F. C. Meunier and R. Burch, Catal. Today, 2006, 113, 94 CrossRef CAS.
- H. Xin, A. Holewinski, N. Schweitzer, E. Nikolla and S. Linic, Top. Catal., 2012, 55, 376 CrossRef CAS.
- E. C. Wegener, B. C. Bukowski, D. Yang, Z. Wu, A. J. Kropf, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, ChemCatChem, 2020, 12, 1325 CrossRef CAS.
- R. G. Windham and B. E. Koel, J. Phys. Chem., 1990, 94, 1489 CrossRef CAS.
- L. Liwu, Z. Tao, Z. Jingling and X. Zhusheng, Appl. Catal., 1990, 67, 11 CrossRef.
- H. Lieske, A. Sárkány and J. Völter, Appl. Catal., 1987, 30, 69 CrossRef CAS.
- Catofin® Propane/Butane Dehydrogenation, McDermott, https://www.mcdermott.com/What-We-Do/Technology/Lummus/Petrochemicals/Olefins/Propylene-Production/Propane-Butane-Dehydrogenation, 2018, accessed on 18/5/2020.
- U. Olsbye, A. Virnovskaia, Ø. Prytz, S. J. Tinnemans and B. M. Weckhuysen, Catal. Lett., 2005, 103, 143 CrossRef CAS.
- F. Cavani, M. Koutyrev, F. Trifirò, A. Bartolini, D. Ghisletti, R. Iezzi, A. Santucci and G. Del Piero, J. Catal., 1996, 158, 236 CrossRef CAS.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223 CrossRef CAS.
- R. L. Puurunen and B. M. Weckhuysen, J. Catal., 2002, 210, 418 CrossRef CAS.
- S. D. Yim and I.-S. Nam, J. Catal., 2004, 221, 601 CrossRef CAS.
- Y. Wang, Y. Ohishi, T. Shishido, Q. Zhang, W. Yang, Q. Guo, H. Wan and K. Takehira, J. Catal., 2003, 220, 347 CrossRef CAS.
- K. Takehira, Y. Ohishi, T. Shishido, T. Kawabata, K. Takaki, Q. Zhang and Y. Wang, J. Catal., 2004, 224, 404 CrossRef CAS.
- V. Z. Fridman, R. Xing and M. Severance, Appl. Catal., A, 2016, 523, 39 CrossRef CAS.
- M. Botavina, C. Barzan, A. Piovano, L. Braglia, G. Agostini, G. Martra and E. Groppo, Catal. Sci. Technol., 2017, 7, 1690 RSC.
- D. He, Y. Zhang, S. Yang, Y. Mei and Y. Luo, ChemCatChem, 2018, 10, 5434 CrossRef CAS.
- N. Mimura, M. Okamoto, H. Yamashita, S. T. Oyama and K. Murata, J. Phys. Chem. B, 2006, 110, 21764 CrossRef CAS PubMed.
- X. Shi, S. Ji, K. Wang and C. Li, Energy Fuels, 2008, 22, 3631 CrossRef CAS.
- T. V. M. Rao, E. M. Zahidi and A. Sayari, J. Mol. Catal. A: Chem., 2009, 301, 159 CrossRef CAS.
- H. H. Shin and S. McIntosh, ACS Catal., 2015, 5, 95 CrossRef CAS.
- D. Shee and A. Sayari, Appl. Catal., A, 2010, 389, 155 CrossRef CAS.
- S. Wang, K. Murata, T. Hayakawa, S. Hamakawa and K. Suzuki, Catal. Lett., 1999, 63, 59 CrossRef CAS.
- L. Liu, H. Li and Y. Zhang, Catal. Today, 2006, 115, 235 CrossRef CAS.
- Y. Cheng, L. Zhou, J. Xu, C. Miao, W. Hua, Y. Yue and Z. Gao, Microporous Mesoporous Mater., 2016, 234, 370 CrossRef CAS.
- Y. Cheng, C. Miao, W. Hua, Y. Yue and Z. Gao, Appl. Catal., A, 2017, 532, 111 CrossRef CAS.
- S. Wang and Z. H. Zhu, Energy Fuels, 2004, 18, 1126 CrossRef CAS.
- S. Wang, K. Murata, T. Hayakawa, S. Hamakawa and K. Suzuki, Appl. Catal., A, 2000, 196, 1 CrossRef CAS.
- N. Mimura, I. Takahara, M. Inaba, M. Okamoto and K. Murata, Catal. Commun., 2002, 3, 257 CrossRef CAS.
- T. A. Bugrova, V. V. Dutov, V. A. Svetlichnyi, V. C. Corberán and G. V. Mamontov, Catal. Today, 2019, 333, 71 CrossRef CAS.
- J. Baek, H. J. Yun, D. Yun, Y. Choi and J. Yi, ACS Catal., 2012, 2, 1893 CrossRef CAS.
- T. Shishido, K. Shimamura, K. Teramura and T. Tanaka, Catal. Today, 2012, 185, 151 CrossRef CAS.
- A. S. Al-Awadi, A. M. El-Toni, S. M. Al-Zahrani, A. E. Abasaeed, M. Alhoshan, A. Khan, J. P. Labis and A. Al-Fatesh, Appl. Catal., A, 2019, 584, 117114 CrossRef.
- X. Shi, S. Ji and K. Wang, Catal. Lett., 2008, 125, 331 CrossRef CAS.
- F. Rahmani, M. Haghighi and B. Mohammadkhanai, Microporous Mesoporous Mater., 2017, 242, 34 CrossRef CAS.
- S. Deng, H. Li, S. Li and Y. Zhang, J. Mol. Catal. A: Chem., 2007, 268, 169 CrossRef CAS.
- X. Li, S. Liu, H. Chen, S. Luo, F. Jing and W. Chu, ACS Omega, 2019, 4, 22562 CrossRef CAS PubMed.
- K. Nakagawa, M. Okamura, N. Ikenaga, T. Suzuki and T. Kobayashi, Chem. Commun., 1998, 1025 RSC.
- A. Bhan and W. N. Delgass, Catal. Rev.: Sci. Eng., 2008, 50, 19 CrossRef CAS.
- G. Giannetto, R. Monque and R. Galiasso, Catal. Rev.: Sci. Eng., 1994, 36, 271 CrossRef CAS.
- K. M. Dooley, T. F. Guidry and G. L. Price, J. Catal., 1995, 157, 66 CrossRef CAS.
- A. Ausavasukhi and T. Sooknoi, Catal. Commun., 2014, 45, 63 CrossRef CAS.
- N. Rane, A. R. Overweg, V. B. Kasansky, R. A. van Santen and E. J. M. Hensen, J. Catal., 2006, 239, 478 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, R. A. van Santen and E. J. M. Hensen, J. Catal., 2004, 227, 263 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, R. A. van Santen and E. J. M. Hensen, J. Catal., 2005, 233, 351 CrossRef CAS.
- A. B. Getsoian, U. Das, J. Camocho-Bunquin, G. Zhang, J. R. Gallagher, B. Hu, S. Cheah, J. A. Schaidle, D. A. Ruddy, J. E. Hensley, T. R. Krause, L. A. Curtiss, J. T. Miller and A. S. Hock, Catal. Sci. Technol., 2016, 6, 6339 RSC.
- M. W. Schreiber, C. P. Plaisance, M. Baumgärtl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849 CrossRef CAS PubMed.
- M. S. Pereira and M. A. C. Nascimento, J. Phys. Chem. B, 2006, 110, 3231 CrossRef CAS PubMed.
- M. S. Pereira, A. M. da Silva and M. A. C. Nascimento, J. Phys. Chem. C, 2011, 115, 10104 CrossRef CAS.
- E. A. Pidko, V. B. Kazansky, E. J. M. Hensen and R. A. van Santen, J. Catal., 2006, 240, 73 CrossRef CAS.
- E. A. Pidko, E. J. M. Hensen and R. A. van Santen, J. Phys. Chem. C, 2007, 111, 13068 CrossRef CAS.
- Y. V. Joshi and K. T. Thomson, J. Catal., 2007, 246, 249 CrossRef CAS.
- E. Mansoor, M. Head-Gordon and A. T. Bell, ACS Catal., 2018, 8, 6146 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, N. Rane, R. A. van Santen and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2005, 7, 3088 RSC.
- E. J. M. Hensen, E. A. Pidko, N. Rane and R. A. van Santen, Angew. Chem., Int. Ed., 2007, 46, 7273 CrossRef CAS PubMed.
- E. A. Pidko, E. J. M. Hensen, G. M. Zhidomirov and R. A. van Santen, J. Catal., 2008, 255, 139 CrossRef CAS.
- E. A. Pidko, R. A. van Santen and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2009, 11, 2893 RSC.
- E. A. Pidko and R. A. van Santen, J. Phys. Chem. C, 2009, 113, 4246 CrossRef CAS.
- K. Nakagawa, C. Kajita, Y. Ide, M. Okamura, S. Kato, H. Kasuya, N. Ikenaga, T. Kobayashi and T. Suzuki, Catal. Lett., 2000, 64, 215 CrossRef CAS.
- M. Zinkevich and F. Aldinger, J. Am. Ceram. Soc., 2004, 87, 683 CrossRef CAS.
- B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2005, 232, 143 CrossRef CAS.
- H. Seki, H. Saito, K. Toko, Y. Hosono, T. Higo, J. G. Seo, S. Maeda, K. Hashimoto, S. Ogo and Y. Sekine, Appl. Catal., A, 2019, 581, 23 CrossRef CAS.
- K. Nishi, K. Shimizu, M. Takamatsu, H. Yoshida, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, J. Phys. Chem. B, 1998, 102, 10190 CrossRef CAS.
- K. Shimizu, M. Takamatsu, K. Nishi, H. Yoshida, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, J. Phys. Chem. B, 1999, 103, 1542 CrossRef CAS.
- S. E. Collins, M. A. Baltanás, J. L. G. Fierro and A. L. Bonivardi, J. Catal., 2002, 211, 252 CrossRef CAS.
- S. E. Collins, M. A. Baltanás and A. L. Bonivardi, Langmuir, 2005, 21, 962 CrossRef CAS PubMed.
- W. Jochum, S. Penner, K. Föttinger, R. Kramer, G. Rupprechter and B. Klötzer, J. Catal., 2008, 256, 268 CrossRef CAS.
- Y. Pan, D. Mei, C. Liu and Q. Ge, J. Phys. Chem. C, 2011, 115, 10140 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, A. A. Pronin, R. Schlögl and F. C. Jentoft, J. Phys. Chem. B, 2006, 110, 7975 CrossRef CAS PubMed.
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev and A. G. Stepanov, Chem. Phys. Lett., 2010, 496, 148 CrossRef CAS.
- Y. Cheng, T. Lei, C. Miao, W. Hua, Y. Yue and Z. Gao, Microporous Mesoporous Mater., 2018, 268, 235 CrossRef CAS.
- H. Xiao, J. Zhang, P. Wang, X. Wang, F. Pang, Z. Zhang and Y. Tan, Catal. Sci. Technol., 2016, 6, 5183 RSC.
- K. Nakagawa, C. Kajita, K. Okumura, N. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Catal., 2001, 203, 87 CrossRef CAS.
- T. Lei, Y. Cheng, C. Miao, W. Hua, Y. Yue and Z. Gao, Fuel Process. Technol., 2018, 177, 246 CrossRef CAS.
- R. Koirala, R. Buechel, F. Krumeich, S. E. Pratsinis and A. Baiker, ACS Catal., 2015, 5, 690 CrossRef CAS.
- Z. Shen, J. Liu, H. Xu, Y. Yue, W. Hua and W. Shen, Appl. Catal., A, 2009, 356, 148 CrossRef CAS.
- X. Zhang, Q. Ye, B. Xu and D. He, Catal. Lett., 2007, 117, 140 CrossRef CAS.
- R. Koirala, R. Buechel, S. E. Pratsinis and A. Baiker, Appl. Catal., A, 2016, 527, 96 CrossRef CAS.
- R. Koirala, O. V. Safonova, S. E. Pratsinis and A. Baiker, Appl. Catal., A, 2018, 552, 77 CrossRef CAS.
- F. Solymosi and R. Németh, Catal. Lett., 1999, 62, 197 CrossRef CAS.
- M. D. Porosoff, M. N. Z. Myint, S. Kattel, Z. Xie, E. Gomez, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2015, 54, 15501 CrossRef CAS PubMed.
- S. Yao, B. Yan, Z. Jiang, Z. Liu, Q. Wu, J. H. Lee and J. G. Chen, ACS Catal., 2018, 8, 5374 CrossRef CAS.
- Z. Ji, H. Lv, X. Pan and X. Bao, J. Catal., 2018, 361, 94 CrossRef CAS.
- X. Yang, T. Wei, B. Chi, J. Pu and J. Li, J. Catal., 2019, 377, 629 CrossRef CAS.
- H. Saito, H. Seki, Y. Hosono, T. Higo, J. G. Seo, S. Maeda, K. Hashimoto, S. Ogo and Y. Sekine, J. Phys. Chem. C, 2019, 123, 26272 CrossRef CAS.
- K. Toko, K. Ito, H. Saito, Y. Hosono, K. Murakami, S. Misaki, T. Higo, S. Ogo, H. Tsuneki, S. Maeda, K. Hashimoto, H. Nakai and Y. Sekine, J. Phys. Chem. C, 2020, 124, 10462 CrossRef CAS.
- B. Yan, S. Yao and J. G. Chen, ChemCatChem, 2020, 12, 494 CrossRef CAS.
- Z. Wu, E. C. Wegener, H.-T. Tseng, J. R. Gallagher, J. W. Harris, R. E. Diaz, Y. Ren, F. H. Ribeiro and J. T. Miller, Catal. Sci. Technol., 2016, 6, 6965 RSC.
- Q. Xie, T. Lei, C. Miao, W. Hua, Y. Yue and Z. Gao, Catal. Lett., 2020, 150, 2013 CrossRef CAS.
- Y. He, Y. Song, D. A. Cullen and S. Laursen, J. Am. Chem. Soc., 2018, 140, 14010 CrossRef CAS PubMed.
- Y. He, Y. Song and S. Laursen, ACS Catal., 2019, 9, 10464 CrossRef CAS.
- L.-C. Wang, Y. Zhang, J. Xu, W. Diao, S. Karakalos, B. Liu, X. Song, W. Wu, T. He and D. Ding, Appl. Catal., B, 2019, 256, 117816 CrossRef.
- Z. Maeno, S. Yasumura, X. Wu, M. Huang, C. Liu, T. Toyano and K. Shimizu, J. Am. Chem. Soc., 2020, 142, 4820 CrossRef CAS PubMed.
- K. Nakagawa, C. Kajita, N. Ikenaga, T. Suzuki, T. Kobayashi, M. Nishitani-Gamo and T. Ando, J. Phys. Chem. B, 2003, 107, 4048 CrossRef CAS.
- X. Zhao and X. Wang, Catal. Commun., 2006, 7, 633 CrossRef CAS.
- M. H. Jeong, J. Sun, G. Y. Han, D. H. Lee and J. W. Bae, Appl. Catal., B, 2020, 270, 118887 CrossRef CAS.
- T. Baidya, N. van Vegten and A. Baiker, Top. Catal., 2011, 54, 881 CrossRef CAS.
- T. V. Sagar, M. Surendar, D. Padmakar, G. Parameswaram, N. Lingaiah, K. S. R. Rao, I. A. K. Reddy, C. Sumana and P. S. S. Prasad, Catal. Lett., 2017, 147, 82 CrossRef CAS.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333 CrossRef CAS.
- D. Ding, Y. Zhang, W. Wu, D. Chen, M. Liu and T. He, Energy Environ. Sci., 2018, 11, 1710 RSC.
- X. Zhang, L. Ye, H. Li, F. Chen and K. Xie, ACS Catal., 2020, 10, 3505 CrossRef CAS.
- Y. Song, L. Lin, W. Feng, X. Zhang, Q. Dong, X. Li, H. Lv, Q. Liu, F. Yang, Z. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 16043 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919 CrossRef CAS PubMed.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280 CrossRef CAS.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592 RSC.
- Y. Ono, Catal. Rev.: Sci. Eng., 1992, 34, 179 CrossRef CAS.
- M. A. Bañares, Catal. Today, 1999, 51, 319 CrossRef.
- A. Hagen and F. Roessner, Catal. Rev.: Sci. Eng., 2000, 42, 403 CrossRef CAS.
- Y. Xiang, H. Wang, J. Cheng and J. Matsubu, Catal. Sci. Technol., 2018, 8, 1500 RSC.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 6th edn, 2007 Search PubMed.
- Database of Zeolite Structure, Structure Commission of the International Zeolite Association, http://www.iza-structure.org/databases/, 2017, accessed on 14/4/2020 Search PubMed.
- M. Stöcker, Microporous Mesoporous Mater., 2005, 82, 257 CrossRef.
- O. A. Anunziata, G. A. Eimer and L. B. Pierella, Appl. Catal., A, 1999, 182, 267 CrossRef CAS.
- O. A. Anunziata, G. A. Eimer and L. B. Pierella, Catal. Lett., 2001, 75, 93 CrossRef CAS.
- Y. Shu, D. Ma, L. Xu, Y. Xu and X. Bao, Catal. Lett., 2000, 70, 67 CrossRef CAS.
- K.-H. Steinberg, U. Mroczek and F. Roessner, Appl. Catal., 1990, 66, 37 CrossRef CAS.
- A. Hagen, F. Roessner, I. Weingart and B. Spliethoff, Zeolites, 1995, 15, 270 CrossRef CAS.
- V. I. Yakerson, T. V. Vasina, L. I. Lafer, V. P. Sytnyk, G. L. Dykh, A. V. Mokhov, O. V. Bragin and Kh. M. Minachev, Catal. Lett., 1989, 3, 339 CrossRef CAS.
- P. Schulz and M. Baerns, Appl. Catal., 1991, 78, 15 CrossRef CAS.
- F. Roessner, A. Hagen, U. Mroczek, H. G. Karge and K.-H. Steinberg, Stud. Surf. Sci. Catal., 1993, 75, 1707 CrossRef CAS.
- F. Solymosi and A. Szőke, Appl. Catal., A, 1998, 166, 225 CrossRef CAS.
- A. Krogh, A. Hagen, T. W. Hansen, C. H. Christensen and I. Schmidt, Catal. Commun., 2003, 4, 627 CrossRef CAS.
- F. Solymosi and P. Tolmacsov, Catal. Lett., 2004, 93, 7 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35 CrossRef CAS.
- L. Wang, R. Ohnishi and M. Ichikawa, Catal. Lett., 1999, 62, 29 CrossRef CAS.
- R. W. Borry III, Y. H. Kim, A. Huffsmith, J. A. Reimer and E. Iglesia, J. Phys. Chem. B, 1999, 103, 5787 CrossRef.
- C. Bolivar, H. Charcosset, R. Frety, M. Primet, L. Tournayan, C. Betizeau, G. Leclercq and R. Maurel, J. Catal., 1975, 39, 249 CrossRef CAS.
- G. Chen, Y. Zhao, L. Shang, G. I. N. Waterhouse, X. Kang, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2016, 3, 1500424 CrossRef PubMed.
- X. Niu, J. Gao, Q. Miao, M. Dong, G. Wang, W. Fan, Z. Qin and J. Wang, Microporous Mesoporous Mater., 2014, 197, 252 CrossRef CAS.
- S. Tamiyakul, T. Sooknoi, L. L. Lobban and S. Jongpatiwut, Appl. Catal., A, 2016, 525, 190 CrossRef CAS.
- H. Berndt, G. Lietz and J. Völter, Appl. Catal., A, 1996, 146, 365 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, P. C. M. M. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71 CrossRef CAS.
- J. Chen, Z. Feng, P. Ying and C. Li, J. Phys. Chem. B, 2004, 108, 12669 CrossRef CAS.
- A. Hagen, E. Schneider, A. Kleinert and F. Roessner, J. Catal., 2004, 222, 227 CrossRef CAS.
- F. Lónyi and J. Valyon, Microporous Mesoporous Mater., 2001, 47, 239 CrossRef.
- N. Katada, Mol. Catal., 2018, 458, 116 CrossRef CAS.
- J. A. Biscardi, G. D. Meitzner and E. Iglesia, J. Catal., 1998, 179, 192 CrossRef CAS.
- J. Penzien, A. Abraham, J. A. van Bokhoven, A. Jentys, T. E. Müller, C. Sievers and J. A. Lercher, J. Phys. Chem. B, 2004, 108, 4116 CrossRef CAS.
- V. B. Kazansky and A. I. Serykh, Phys. Chem. Chem. Phys., 2004, 6, 3760 RSC.
- J. A. Biscardi and E. Iglesia, J. Catal., 1999, 182, 117 CrossRef CAS.
- L. A. Dufresne and R. Le Van Mao, Catal. Lett., 1994, 25, 371 CrossRef CAS.
- S. Triwahyono, A. A. Jalil, R. R. Mukti, M. Musthofa, N. A. M. Razali and M. A. A. Aziz, Appl. Catal., A, 2011, 407, 91 CrossRef CAS.
- J. Gao, C. Wei, M. Dong, G. Wang, Z. Li, Z. Qin, J. Wang and W. Fan, ChemCatChem, 2019, 11, 3892 CrossRef CAS.
- H. A. Aleksandrov and G. N. Vayssilov, Catal. Today, 2010, 152, 78 CrossRef CAS.
- E. A. Pidko and R. A. van Santen, J. Phys. Chem. C, 2007, 111, 2643 CrossRef CAS.
- A. Mehdad and R. F. Lobo, Catal. Sci. Technol., 2017, 7, 3562 RSC.
- Y. Zhang, S. Wu, X. Xu and H. Jiang, Catal. Sci. Technol., 2020, 10, 835 RSC.
- A. Hagen, F. Roessner and W. Reschetilowski, Chem. Eng. Technol., 1995, 18, 414 CrossRef CAS.
- O. P. Keipert, D. Wolf, P. Schulz and M. Baerns, Appl. Catal., A, 1995, 131, 347 CrossRef CAS.
- L. Ma and X. Zou, Appl. Catal., B, 2019, 243, 703 CrossRef CAS.
- A. Hagen, O. P. Keipert and F. Roessner, Stud. Surf. Sci. Catal., 1996, 101, 781 CrossRef CAS.
- A. G. Stepanov, M. L. Luzgin, V. N. Romannikov, V. N. Sidelnikov and E. A. Paukshtis, J. Catal., 1998, 178, 466 CrossRef CAS.
- J. N. Kondo and K. Domen, J. Mol. Catal. A: Chem., 2003, 199, 27 CrossRef CAS.
- T. Koyama, Y. Hayashi, H. Horie, S. Kawauchi, A. Matsumoto, Y. Iwase, Y. Sakamoto, A. Miyaji, K. Motokura and T. Baba, Phys. Chem. Chem. Phys., 2010, 12, 2541 RSC.
- Y. Iwase, Y. Sakamoto, A. Shiga, A. Miyaji, K. Motokura, T. Koyama and T. Baba, J. Phys. Chem. C, 2012, 116, 5182 CrossRef CAS.
- M. Shibata, H. Kitagawa, Y. Sendoda and Y. Ono, Stud. Surf. Sci. Catal., 1986, 28, 717 CrossRef CAS.
- V. R. Choudhary, D. Panjala and S. Banerjee, Appl. Catal., A, 2002, 231, 243 CrossRef CAS.
- E. A. Uslamin, H. Saito, N. Kosinov, E. Pidko, Y. Sekine and E. J. M. Hensen, Catal. Sci. Technol., 2020, 10, 2774 RSC.
- V. R. Choudhary, S. Banerjee and D. Panjala, Microporous Mesoporous Mater., 2002, 51, 203 CrossRef CAS.
- W. O. Haag, R. M. Lago and P. G. Rodewald, J. Mol. Catal., 1982, 17, 161 CrossRef CAS.
- E. Iglesia, J. E. Baumgartner and G. L. Price, J. Catal., 1992, 134, 549 CrossRef CAS.
- T. Liang, H. Toghiani and Y. Xiang, Ind. Eng. Chem. Res., 2018, 57, 15301 CAS.
- V. R. Choudhary, P. Devadas, S. Banerjee and A. K. Kinage, Microporous Mesoporous Mater., 2001, 47, 253 CrossRef CAS.
- R. Batchu, V. V. Galvita, K. Alexopoulos, K. Van der Borght, H. Poelman, M.-F. Reyniers and G. B. Marin, Appl. Catal., A, 2017, 538, 207 CrossRef CAS.
- M. Guisnet, N. S. Gnep, D. Aittaleb and Y. J. Doyemet, Appl. Catal., A, 1992, 87, 255 CrossRef CAS.
- P. Qiu, J. H. Lunsford and M. P. Rosynek, Catal. Lett., 1998, 52, 37 CrossRef CAS.
- H. Coqueblin, A. Richard, D. Uzio, L. Pinard, Y. Pouilloux and F. Epron, Catal. Today, 2017, 289, 62 CrossRef CAS.
- M.-F. Hsieh, Y. Zhou, H. Thirumalai, L. C. Grabow and J. D. Rimer, ChemCatChem, 2017, 9, 1675 CrossRef CAS.
- J. Bandiera and Y. B. Taȃrit, Appl. Catal., A, 1997, 152, 43 CrossRef CAS.
- J. Bandiera and Y. B. Taarit, Appl. Catal., A, 1997, 161, L43 CrossRef CAS.
- I. Pinilla-Herrero, E. Borfecchia, J. Holzinger, U. V. Mentzel, F. Joensen, K. A. Lomachenko, S. Bordiga, C. Lamberti, G. Berlier, U. Olsbye, S. Svelle, J. Skibsted and P. Beato, J. Catal., 2018, 362, 146 CrossRef CAS.
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijplema, B. Mezari and E. J. M. Hensen, ACS Catal., 2017, 7, 520 CrossRef CAS.
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016 CrossRef CAS PubMed.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459 CrossRef CAS PubMed.
- N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068 CrossRef CAS PubMed.
- Z. R. Ismagilov, E. V. Matus and L. T. Tsikoza, Energy Environ. Sci., 2008, 1, 526 RSC.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792 RSC.
- I. Vollmer, I. Yarulina, F. Kapteijn and J. Gascon, ChemCatChem, 2019, 11, 39 CrossRef CAS.
- E. A. Uslamin, H. Saito, Y. Sekine, E. J. M. Hensen and N. Kosinov, Catal. Today DOI:10.1016/j.cattod.2020.04.021 , in press.
- I. Vollmer, B. van der Linden, S. Ould-Chikh, A. Aguilar-Tapia, I. Yarulina, E. Abou-Hamad, Y. G. Sheider, A. I. O. Suarez, J.-L. Hazemann, F. Kapteijn and J. Gascon, Chem. Sci., 2018, 9, 4801 RSC.
- I. Vollmer, N. Kosinov, Á. Szécsényi, G. Li, I. Yarulina, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, A. Aguilar-Tapia, J.-L. Hazemann, E. Pidko, E. Hensen, F. Kapteijn and J. Gascon, J. Catal., 2019, 370, 321 CrossRef CAS.
- I. Vollmer, S. Ould-Chikh, A. Aguilar-Tapia, G. Li, E. Pidko, J.-L. Hazemann, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2019, 141, 18814 CrossRef CAS PubMed.
- G. Li, I. Vollmer, C. Liu, J. Gascon and E. A. Pidko, ACS Catal., 2019, 9, 8731 CrossRef CAS.
- I. Vollmer, E. Abou-Hamad, J. Gascon and F. Kapteijn, ChemCatChem, 2020, 12, 544 CrossRef CAS.
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69 CrossRef CAS.
- B. Robinson, X. Bai, A. Samanta, V. Abdelsayed, D. Shekhawat and J. Hu, Energy Fuels, 2018, 32, 7810 CrossRef CAS.
- A. Samanta, X. Bai, B. Robinson, H. Chen and J. Hu, Ind. Eng. Chem. Res., 2017, 56, 11006 CrossRef CAS.
- P. Marecot, S. Peyrovi, D. Bahloul and J. Barbier, Appl. Catal., 1990, 66, 181 CrossRef CAS.
- E. Epelde, M. Ibañez, A. T. Aguayo, A. G. Gayubo, J. Bilbao and P. Castaño, Microporous Mesoporous Mater., 2014, 195, 284 CrossRef CAS.
- S. Inagaki, S. Shinoda, Y. Kaneko, K. Takeuchi, R. Komatsu, Y. Tsuboi, H. Yamazaki, J. N. Kondo and Y. Kubota, ACS Catal., 2013, 3, 74 CrossRef CAS.
- J. Dĕdeček, Z. Sobalík and B. Wichterlová, Catal. Rev.: Sci. Eng., 2012, 54, 135 CrossRef.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303 CrossRef CAS.
- H. Liu, H. Wang, A.-H. Xing and J.-H. Cheng, J. Phys. Chem. C, 2019, 123, 15637 CrossRef CAS.
- Y. Wang, A. Caiola, B. Robinson, Q. Li and J. Hu, Energy Fuels, 2020, 34, 3100 CrossRef CAS.
- F. Goodarzi, R. P. Thumbayil, K. Enemark-Rasmussen, J. Mielby, T. T. M. Nguyen, P. Beato, F. Joensen and S. Kegnæs, ChemCatChem, 2020, 12, 1519 CrossRef CAS.
- J. Ye, L. Bai, B. Liu, H. Tian, J. Hu, F. Polo-Garzon, R. T. Mayes, Z. Wu and Y. Fang, Ind. Eng. Chem. Res., 2019, 58, 7094 CrossRef CAS.
- H. Saito, S. Inagaki, K. Kojima, Q. Han, T. Yabe, S. Ogo, Y. Kubota and Y. Sekine, Appl. Catal., A, 2018, 549, 76 CrossRef CAS.
- O. V. Chetina, T. V. Vasina and V. V. Lunin, Appl. Catal., A, 1995, 131, 7 CrossRef CAS.
- X. Zhu, Q. Imtiaz, F. Donat, C. R. Müller and F. Li, Energy Environ. Sci., 2020, 13, 772 RSC.
- E. Gomez, X. Nie, J. H. Lee, Z. Xie and J. G. Chen, J. Am. Chem. Soc., 2019, 141, 17771 CrossRef CAS PubMed.
- A. Mehdad, N. S. Gould, B. Xu and R. F. Lobo, Catal. Sci. Technol., 2018, 8, 358 RSC.
- A. Kumar, K. Song, L. Liu, Y. Han and A. Bhan, Angew. Chem., Int. Ed., 2018, 57, 15577 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView (Version 5), Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |