
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA proof-of-concept, two-tiered approach for ricin detection using ambient mass spectrometry†
Devin J. Swiner,
George R. Durisek III,
Hannah Osae and
Abraham K. Badu-Tawiah *
*
Department of Chemistry and Biochemistry, The Ohio State University, 100 W 18th Ave, Columbus, OH 43210, USA. E-mail: badu-tawiah.1@osu.edu
First published on 30th April 2020
Abstract
Ricin is a naturally occurring, highly potent toxin native to castor bean plants that has recently been used as a biological weapon in cases of bioterrorism and suicide attempts. Difficulties with direct detection arise from large heterogeneities in ricin glycosylation, which leads to markedly different bioactivity, and the fact that carefully developed and laborious sample preparation steps are required to maintain the activity of the protein during analysis. Herein, we present an alternative, two-tiered approach to identify the presence of ricin by detecting ricinoleic acid and ricinine, which are co-extracted with the protein. This direct mass spectrometric-based technique takes as little as 2 minutes, and we determined its sensitivity to be in the parts-per-trillion range. Our method is applicable to paper substrates from suspected contaminated envelopes and biofluids from at-risk patients. The fact that prior sample preparations are not needed in this procedure means that analysis can be performed in the field for emergency cases.
Introduction
In 2018, the New York Times reported multiple cases of suspicious envelopes confirmed to contain ricin, that were addressed to the White House and other government agencies.1 Similar cases have been described in recent years by major news outlets2 and it appears to be an increasing trend where ricin has been used as a weapon for acts of bioterrorism3 and in cases of (attempted) suicide.2a,4 Ricin is a naturally occurring, highly potent toxin found in the castor bean plant Ricinus communis. It is easy to cultivate from the plant's seeds and, due to its easy dissemination and moderate morbidity rates, the Center for Disease Control and Prevention (CDC) has listed it as a category B agent.5 Fatal exposure to the poison can arise from injection, inhalation, and/or ingestion in quantities as low as 5 μg kg−1.5,6 Because these fatalities can occur in both humans and animals, there is a need to detect its presence in various matrices, including biological fluids,7 seeds,8 and fertilizers.4b,9The toxicity of ricin is limited to the existence of both the active forms of its A and B chains. Therefore, ideal detection methods must determine the activities and the presence of both chains in a short amount of time to enable the development and implementation of effective and proper emergency response plans.10 Popular techniques for determining the bioactivity of ricin are based on the release of free adenine.11 While these offer a means to correlate adenine release to catalytic activity, they often require tedious sample purification steps to maintain the bioactivity of the protein and a minimization of the background noise that can interfer with the fluorescence detection strategy typically employed. Similarly, immunological12 and cytotoxicity9a,13 assays have been used to detect the protein from a variety of matrices and, while warranting sensitivity levels on the order of 100 pg mL−1,5 still require extensive sample preparation and long analysis times (ranging from several hours11b,14 to days9a,15). The key feature of ricin toxicity, which involves rapid distribution to most organs in the body,16 makes therapeutic treatment particularly challenging if diagnoses are delayed. An analytical procedure that transcends prolonged sample treatments to afford results in real-time is a potential solution to this problem. However, no such approach has been developed for rapid determination of the presence of ricin.
To improve turnaround time, the detection of small organic compounds as biomarkers for ricin poisoning is becoming increasingly popular. In particular, the analysis of ricinine has attracted much attention7a,7c,7d,17 since the only natural source for this small alkaloid is from the castor bean.18 Hence, exposure to castor beans, unpurified ricin, or any other castor bean-based products (e.g., castor oil) should result in its presence. While ricinine naturally exists in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio with ricin,8b ricinolein, the triglyceride form of ricinoleic acid, is the main (90%) component of castor oil. Therefore, the simultaneous detection of ricinine and ricinoleic acid can serve as strong indicators for the presence of ricin in a sample (Scheme 1).
:5 ratio with ricin,8b ricinolein, the triglyceride form of ricinoleic acid, is the main (90%) component of castor oil. Therefore, the simultaneous detection of ricinine and ricinoleic acid can serve as strong indicators for the presence of ricin in a sample (Scheme 1).
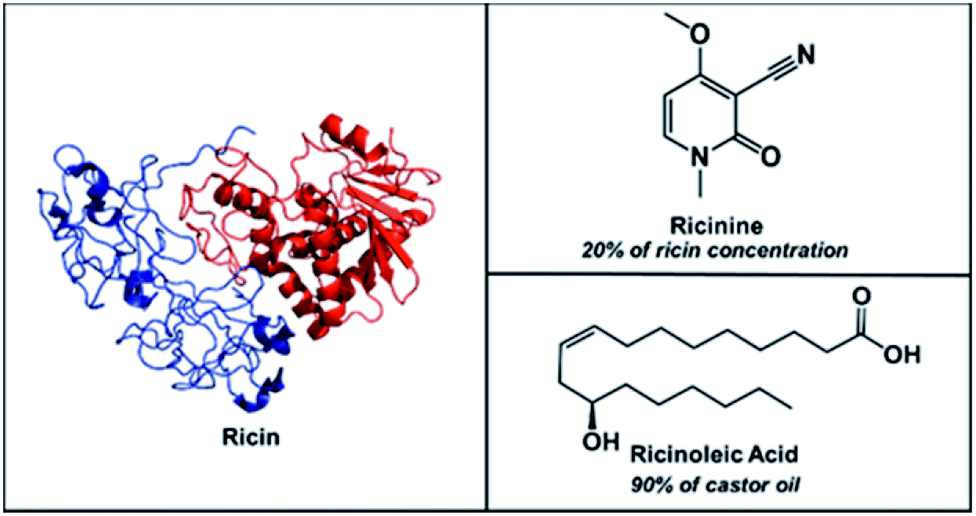 | ||
| Scheme 1 Chemical structures of ricin (MW 65750 Da) accompanied by its small molecule biomarkers, ricinine (MW 164 Da) and ricinoleic acid (MW 298 Da). | ||
Mass spectrometry (MS) is one of the most common analytical methods for detecting ricinine in complex mixtures,7a,7d,17 however, all reported methods utilize liquid chromatographic (LC) separations prior to tandem MS (MS/MS) analysis. Although sensitive, sample preparation steps are required before subjecting the extracted sample to LC-MS/MS. Even though the biosynthesis of both ricinine and ricinoleic acid in the castor bean plant have been well characterized,19 the two compounds are rarely20 used together to infer the presence of ricin. In the present study, we aimed to (1) develop a proof-of-concept, two-tiered approach based on the multiplex detection of ricinine and ricinoleic acid in a single run, (2) detect the two biomarkers directly from the surface of suspected envelopes without prior extraction procedures and (3) establish the analytical merits for ricinine in different untreated biological fluids.
Experimental
Materials and methods
Cellulose material preparation
Thread (35 mm in length) was cut from the spool and placed in a plastic desiccator with 0.5 mL of silanization reagent (trichloro(3,3,3-trifluoropropyl)silane). Vacuum was pulled for 5 minutes, sealed, and then allowed to gas-phase react for 55 minutes, totalling a 60 minute treatment time. For paper strip analysis, Whatman grade 1 filter paper was manually cut to desired dimensions, 35 mm × 1 mm.Mass spectrometry
Mass spectra were acquired on a Thermo Fisher Scientific Finnigan LTQ linear ion trap mass spectrometer (San Jose, CA, U.S.A.). The tip of the thread was positioned parallel to the MS inlet via a copper alligator clip, which was connected to an external high-voltage supply (0–6 kV). The thread spray ionization method generates ions without gas assistance so a close interface distance (0.5–5 mm) between the tip and the MS inlet was used to optimize signal intensity. MS parameters used were as follows: 200 °C capillary temperature, 3 microscans, and 60% S-lens voltage. Thermo Fisher Scientific Xcalibur 2.2 SP1 software was applied for MS data collecting and processing. Tandem MS with collision-induced dissociation (CID) was utilized for analyte identification. 1.5 Th (mass/charge units) for isolation window and 25 (manufacturer's unit) of normalized collision energy was chosen for the CID tests.Results and discussion
Qualitative studies using the two-tiered approach
Our two-tiered detection strategy is as illustrated in Fig. 1, where ricinoleic acid is used in an initial screening test followed by the detection of ricinine to confirm the existence of a ricin-contaminated sample. Fig. 2 demonstrates the actual analytical procedure in which a novel strip-paper spray ionization was used for contaminated envelope samples. For direct analysis of biofluids (urine and serum), we used hydrophobic thread spray ionization.21 Ricinoleic acid (MW 298.46 g mol−1) was detected in the negative-ion mode at m/z 297 while the ion signal m/z 165 from ricinine (MW 164.16 g mol−1) was recorded using positive-ion mode. Both ions were subjected to collisional activation in MS/MS experiments and the diagnostic fragments were used as confirmation for the selected biomarkers. Although the detection of ricinoleic acid does not indicate eminent danger, it does suggest the use of castor bean seed-related products, which gives a good reason to test for the presence of ricinine in the same sample. We can conduct this two-tiered approach in under 2 minutes, indicating that the proposed methodology can be automated and used to analyze numerous samples in a short amount of time. Given the low molecular weights and structures of the two biomarkers, and the fact that ionization occurs without sample pre-treatment, we expect it will be straightforward to employ with portable mass spectrometers for in situ detection in the field.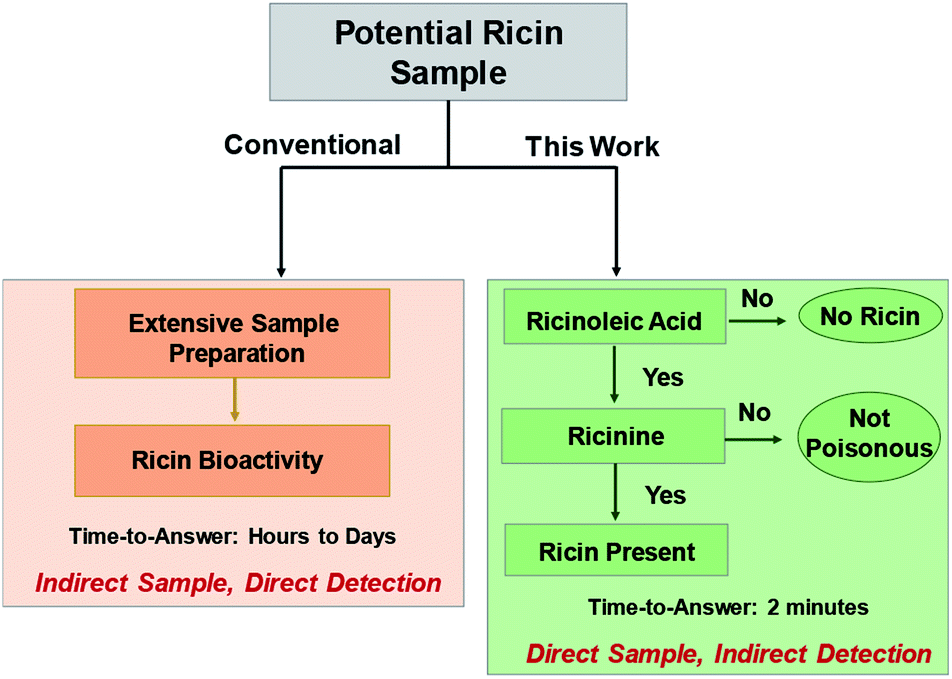 | ||
| Fig. 1 Two-tiered approach to detect the presence of ricin using selected small molecule biomarkers, ricinine and ricinoleic acid, in different matrices. | ||
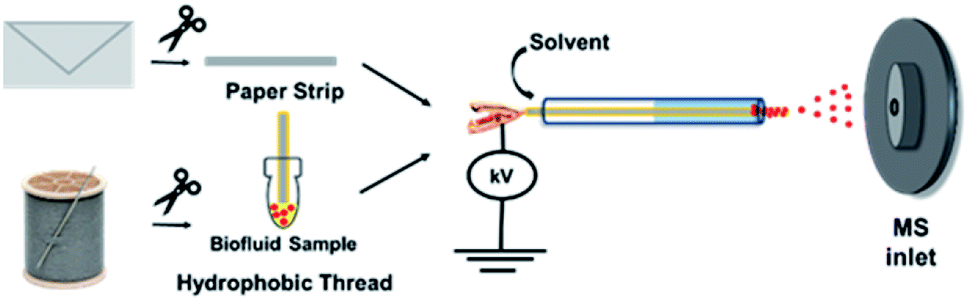 | ||
| Fig. 2 Schematic illustrating the experimental procedures for the ambient MS-based detection of ricin from paper substrates and biofluid samples using strip-paper spray and thread spray ionization methods. | ||
The two samples (contaminated envelopes and biofluids) were selected based on two anticipated scenarios: (i) envelope analysis would be needed for cases where ricin is sent via mail and (ii) biological fluid analysis in cases where ingestion/inhalation/injection had already occurred. For the former scenario, a strip of filter paper was manually cut and placed inside a glass capillary. To initiate MS analysis, the paper strip was wetted with acidified ethyl acetate spray solvent (pH 5.5). Gas-phase ions were generated upon the application of 5 kV (direct current; DC) to the wet paper, which were subsequently sampled by the mass spectrometer. For analysis of biofluids, a single piece of hydrophobic thread was used to absorb 10 μL samples by simply dipping the thread in the biofluids. After, the biofluid-containing thread was placed in a glass capillary and fitted in front of the mass spectrometer inlet. Application of 4 kV and acidified ethyl acetate enabled extraction of the analytes and the subsequent release of charged droplets from the wet thread allowing for analyte characterization by MS. The pH 5.5 solvent (achieved with acetic acid) was used as a compromise to promote the protonation [M + H]+ of ricinine molecules to facilitate detection in the positive-ion mode while also not severely hindering the deprotonation of ricinoleic acid when detected in the negative-ion mode. Other optimization experiments/parameters (e.g., spray solvent, organic acid type, paper spray configuration) are discussed in detail in the ESI Fig. 1–3.†
Collectively, the results show that both ricinoleic acid and ricinine standards can be detected more effectively (10×) from paper strips than when traditional paper spray substrates (paper cut into triangles) were utilized. The increased ion yield from this modified paper spray configuration is due to the existence of a delayed extraction phenomenon where the containment of the paper strip inside the glass capillary limits solvent evaporation and offers both an extended analyte extraction time (60 s) and online pre-concentration effect before DC voltage is applied to ionize the extracted analyte. Such extraction mechanism cannot be implemented with a traditional paper spray experiment because of the high surface area of the planar paper triangle, which facilitates rapid solvent evaporation. Consequently, the conventional experiment requires the spray solvent and voltage to be applied simultaneously, which results in low extraction efficiency and limited sensitivity. Positive identification of both ricinine and ricinoleic acid present on untreated paper strips at 0.25 μg mL−1 levels could be achieved when compared with uncontaminated paper strips when using the optimized 60 s delayed extraction procedure (ESI Fig. 4†). Although hydrophobic thread spray generated lower background signal compared to strip-paper spray, the two ambient ionization methods afforded comparable sensitivity for both analytes when using neat samples (ESI Fig. 5†).
Quantification studies using ricinine via cellulose materials
In order to reduce false negative results, sensitive quantification capabilities are vital for confirmatory tests. We chose to investigate the presence of ricinine in untreated human serum and raw urine. On average, extracts from castor bean seeds are found to contain 1% ricin (based on terrorist's handbook procedure8b) suggesting that an estimated 0.2% of ricinine can be co-extracted. Considering that only a fraction of this amount can be inhaled or ingested in a terrorist attack, it is essential that analytical methods for ricinine detection exert at least parts-per-billion sensitivity levels. Representative product ion spectra for ricinoleic acid and ricinine in 10 μL urine at 250 ng mL−1 concentrations, recorded using hydrophobic thread spray MS, are shown in Fig. 3A and B, respectively. Since the confirmatory test of our method relies on ricinine detection, we further characterized the analyte's signal in terms of limit of detection (LOD) measurements based on the characteristic MS/MS transition m/z 165 → 138. Calibration curves were subsequently constructed for a concentration range of 10–250 ng mL−1 in raw urine and serum, Fig. 3C and D, respectively, after comparison with signal (m/z 168 → 141) from the ricinine-(methyl-d3) internal standard. Based on linear regression analysis, we determined the LODs for ricinine in urine and serum to be 23 and 27 fg mL−1 (parts-per-trillion), respectively. Relative standard deviations less than 10% (shown by small error bars) and high linearity (>0.99) were obtained for each biofluid indicating high accuracy, exceptional precision and excellent reproducibility. These high-quality analytical merits were obtained without the need for sample preparation or offline pre-concentration steps, something essential to chromatographic-based methods. It will also not be surprising to encounter high dosages of ricinine in biofluids, especially in the cases of suicide attempts with crude extracts, making this method's sensitivity suitable for analysis. To further evaluate the robustness of our approach in a more complex mixture, we detected ricinine and ricinoleic acid, in the presence of each other, after spiking equal concentrations (0.25 μg mL−1 each) in raw urine. The results (ESI Fig. 6†) showed that both analytes can be positively confirmed from the same complex sample in under 2 minutes with minimal matrix interferences.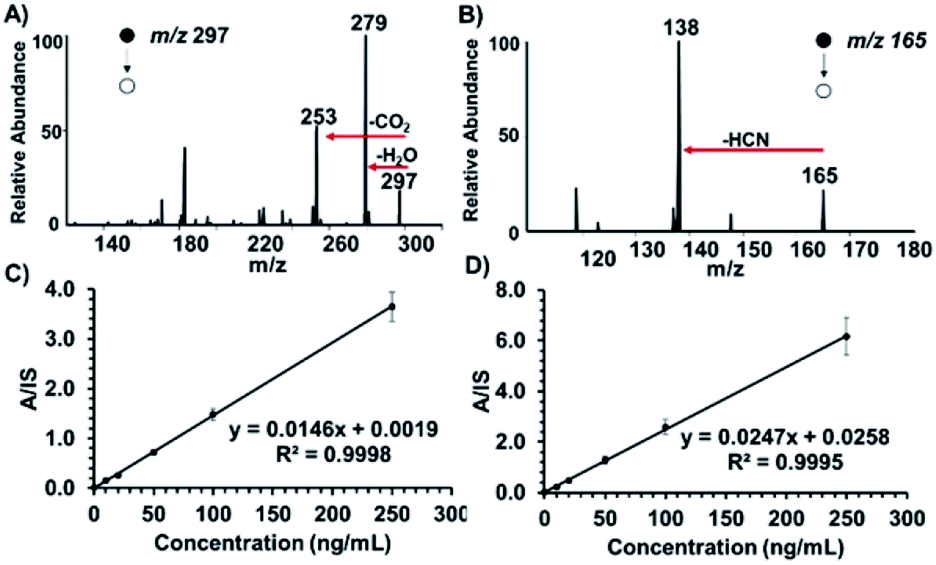 | ||
| Fig. 3 Representative tandem MS spectra of a urine sample containing (A) ricinoleic acid (negative-ion mode) and (B) ricinine (positive-ion mode). Analyte concentrations were 250 ng mL−1 and were analyzed with hydrophobic thread spray using acidified ethyl acetate (pH 5.5) as the spray solvent. Calibration curves (10–250 ng mL−1) for ricinine, with an internal standard concentration of 100 ng mL−1, in (C) raw urine and (D) serum samples. Each concentration was analyzed in replicates of 5, and spray voltage of 4 kV was used in all cases. | ||
Potential implications of the timescale for the approach presented in the current report are greatest when considering large-scale screening in an emergency. In this case, it is vital to confirm the presence of the toxin in at-risk people in as short of amount of time as possible. Given that ricin is slowly excreted from the body (concentration levels of ∼880 ng mL−1 can be reached between 24–48 h),6,9a and the fact that ricinine concentrations in urine and serum after 6 h of exposure is in the 10–35 ng mL−1 range,7a,7d we believe that our two-tiered methodology will be suitable for analysis for an extended period of time post-exposure. With our approach, raw urine samples could be successfully analyzed at the point-of-care at a rate of approximately 30 samples per hour, which would allow a large number of patients to be diagnosed and treated (e.g., breathing support, intravenous fluids, stomach pumping with activated charcoal, etc.22,23) within the window of intervention (∼4–8 hours22,24) when symptoms are beginning to occur. Urine analysis also presents a non-invasive sampling opportunity, which can facilitate screening of large groups of people within a community quickly. Our approach has also shown tremendous sensitivity for more complex serum samples (Fig. 3D), which is important for the analysis of samples collected several hours after ricin exposure when the concentrations begin to decrease. The comparable sensitivity in urine versus serum samples is not surprising, and is attributed to the fact that the organic ethyl acetate spray solvent extracts only the organic components from the biofluid leaving behind aqueous-based contaminants (e.g., inorganic salts, proteins, etc.) that typically exert ion suppression in electrospray. In addition, the absence of proteins and red blood cells in serum suggests that the amount of detectable free analytes are comparable in both urine and serum. Possible binding of analytes with proteins and/or diffusion into haematids might present a challenge when using untreated whole blood samples. Even for complex samples like blood, we have observed part-per-trillion sensitivities for organic compounds such as cocaine and diazepam.21b The main advantage of the current two-tiered approach is the fact that it presents a highly efficient alternative to direct ricin detection by transcending difficulties related to heterogeneity of ricin glycosylation, that often leads to differential toxicity and bioactivity,25 as well as the challenges associated with sample preparation.
Conclusions
In conclusion, we have presented a method for simple, fast, and accurate testing for ricin poisoning, which uses substrate-based ambient ionization mass spectrometry to analyze ricinine and ricinoleic acid in a two-tiered sequential approach from a single untreated sample. Compared with other methods including liquid chromatography mass spectrometry, the strip paper and thread spray ionization methods enable the analysis of a sample without sample processing, which reduces the time needed for analysis, as well as the variation induced in sample preparation steps. The use of acetified ethyl acetate as a spray solvent enabled the detection of both biomarkers in positive and negative ion modes as well as the real-time extraction of analytes from paper substrates and from complex biological fluids. The reduced matrix effects derived from the use of this organic solvent and the online pre-concentration effects associated with the spray mechanism allowed for selective transfer of the analyte molecules and for detection limits of 23 and 27 part-per-trillion in raw urine and serum, respectively, for ricinine analysis. The capacity to simply and quickly detect ricinine and ricinoleic acid biomarkers on paper and in biofluids widens the applicability of ricin detection in various settings, both in the laboratory and in the field. The use of this two-tiered approach also increases confidence in indirect ricin detection and would aid in expediting the process for emergency cases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (Award Number R01-AI-143809).Notes and references
- (a) A. D. Philipps, F. B. I. Arrests Utah Man in Ricin Scare at Pentagon, N. Y. Times, 2018 Search PubMed; (b) B. H. Cooper and M. D. Shear, Ricin Suspected in Mail Sent to Trump and Pentagon Officials, N. Y. Times, 2018 Search PubMed.
- (a) L. Rathke, Judge detains ricin suspect with prior suicide attempts, Wash. Post: National, 2017 Search PubMed; (b) B. J. Goldstein, Letters Threatening Mayor Tested Positive for Ricin, N. Y. Times, 2013 Search PubMed; (c) C. D. Johnston and C. Hulse, RICIN ON CAPITOL HILL: THE OVERVIEW; Finding of Deadly Poison in Office Disrupts the Senate, N. Y. Times, 2004 Search PubMed.
- (a) A. S. Olsnes, Toxicon, 2004, 44, 361–370 CrossRef PubMed; (b) B. E. Janik, M. Ceremuga, J. Saluk-Bijak and M. Bijak, Int. J. Mol. Sci., 2019, 20 Search PubMed; (c) C. P. D. Anderson, J. Pharm. Pract., 2012, 25, 121–129 CrossRef PubMed; (d) D. H. J. Jansen, F. J. Breeveld, C. Stijnis and M. P. Grobusch, Clin. Microbiol. Infect., 2014, 20, 488–496 CrossRef PubMed.
- (a) A. V. Coopman, M. De Leeuw, J. Cordonnier and W. Jacobs, Forensic Sci. Int., 2009, 189, e13–e20 CrossRef PubMed; (b) B. S. Worbs, K. Köhler, D. Pauly, M.-A. Avondet, M. Schaer, M. B. Dorner and B. G. Dorner, Toxins, 2011, 3, 1332–1372 CrossRef PubMed; (c) C. E. I. Hamelin, R. C. Johnson, J. Osterloh and J. Thomas, J. Anal. Toxicol., 2012, 36, 660–662 CrossRef PubMed; (d) D. J. Kopferschmitt, F. Flesch, A. Lugnier, P. Sauder, A. Jaeger and J. M. Mantz, Hum. Toxicol., 1983, 2, 239–242 CrossRef PubMed; (e) E. T. S. Ferguson, W. Indian Med. J., 2011, 60, 596 Search PubMed.
- F. Musshoff and B. Madea, Drug Test. Anal., 2009, 1, 184–191 CrossRef CAS PubMed.
- J. Audi, M. Belson, M. Patel, J. Schier and J. Osterloh, JAMA, J. Am. Med. Assoc., 2005, 294, 2342–2351 CrossRef CAS PubMed.
- (a) A. R. C. Johnson, S. W. Lemire, A. R. Woolfitt, M. Ospina, K. P. Preston, C. T. Olson and J. R. Barr, J. Anal. Toxicol., 2005, 29, 149–155 CrossRef PubMed; (b) B. S. W. Smith, N. M. Graber, R. C. Johnson, J. R. Barr, R. S. Hoffman and L. S. Nelson, Ann. Plast. Surg., 2009, 62, 12 CrossRef PubMed; (c) C. C. T. Pittman, J. M. Guido, E. I. Hamelin, T. A. Blake and R. C. Johnson, J. Anal. Toxicol., 2013, 37, 237–240 CrossRef CAS PubMed; (d) D. B. T. Røen, A. M. Opstad, A. Haavind and J. Tønsager, J. Anal. Toxicol., 2013, 37, 313–317 CrossRef PubMed.
- (a) A. C. S. Brinkworth, E. J. Pigott and D. J. Bourne, Anal. Chem., 2009, 81, 1529–1535 CrossRef PubMed; (b) B. S. M. Darby, M. L. Miller and R. O. Allen, J. Forensic Sci., 2001, 46, 15097J Search PubMed.
- (a) A. D. Pauly, S. Worbs, S. Kirchner, O. Shatohina, M. B. Dorner and B. G. Dorner, PloS One, 2012, 7, e35360 CrossRef PubMed; (b) B. S. Roels, V. Coopman, P. Vanhaelen and J. Cordonnier, J. Vet. Diagn. Invest., 2010, 22, 466–468 CrossRef PubMed; (c) C. S. Krieger-Huber, Kleintierpraxis, 1980, 25, 281–286 Search PubMed.
- (a) A. W. P. Bozza, W. H. Tolleson, L. A. Rivera Rosado and B. Zhang, Biotechnol. Adv., 2015, 33, 117–123 CrossRef PubMed; (b) B. J. G. Schier, M. M. Patel, M. G. Belson, A. Patel, M. Schwartz, N. Fitzpatrick, D. Drociuk, S. Deitchman, R. Meyer, T. Litovitz, W. A. Watson, C. H. Rubin and M. Kiefer, Am. J. Publ. Health, 2007, 97, S152–S157 CrossRef PubMed; (c) C. J. Koplan, Publ. Health Rep., 2001, 116, 9–16 CrossRef; (d) D. A. S. Khan, A. M. Levitt and M. J. Sage, Morb. Mortal. Wkly. Rep., 2000, 49, RR-4 Search PubMed; (e) E. D. M. Schieltz, S. C. McGrath, L. G. McWilliams, J. Rees, M. D. Bowen, J. J. Kools, L. A. Dauphin, E. Gomez-Saladin, B. N. Newton, H. L. Stang, M. J. Vick, J. Thomas, J. L. Pirkle and J. R. Barr, Forensic Sci. Int., 2011, 209, 70–79 CrossRef PubMed.
- (a) A. V. L. H. Bevilacqua, J. M. Nilles, J. S. Rice, T. R. Connell, A. M. Schenning, L. M. Reilly and H. D. Durst, Anal. Chem., 2010, 82, 798–800 CrossRef PubMed; (b) B. F. Becher, E. Duriez, H. Volland, J. C. Tabet and E. Ezan, Anal. Chem., 2007, 79, 659–665 CrossRef PubMed; (c) C. H. B. Hines, E. E. Brueggemann and M. L. Hale, Anal. Biochem., 2004, 330, 119–122 CrossRef PubMed; (d) D. S. R. Kalb, D. M. Schieltz, F. Becher, C. Astot, S.-Å. Fredriksson and J. R. Barr, Toxins, 2015, 7, 4881–4894 CrossRef PubMed.
- (a) A. G. D. Griffiths, H. Newman and D. J. Gee, J. Forensic Sci. Soc., 1986, 26, 349–358 CrossRef PubMed; (b) B. S. T. Gaylord, T. L. Dinh, E. R. Goldman, G. P. Anderson, K. C. Ngan and D. R. Walt, Anal. Chem., 2015, 87, 6570–6577 CrossRef PubMed; (c) C. R.-H. Shyu, H.-F. Shyu, H.-W. Liu and S.-S. Tang, Toxicon, 2002, 40, 255–258 CrossRef PubMed; (d) D. A. Godal, S. Olsnes and A. Pihl, J. Toxicol. Environ. Health, 1981, 8, 409–417 CrossRef PubMed; (e) E. S. R. Kalb and J. R. Barr, Anal. Chem., 2009, 81, 2037–2042 CrossRef PubMed.
- (a) A. P. Sehgal, M. Khan, O. Kumar and R. Vijayaraghavan, Food Chem. Toxicol., 2010, 48, 3171–3176 CrossRef PubMed; (b) B. M. Halter, J. L. Almeida, A. Tona, K. D. Cole, A. L. Plant and J. T. Elliott, Assay Drug Dev. Technol., 2009, 7, 356–365 CrossRef PubMed; (c) C. M. O'Hare, A. N. Brown, K. Hussain, A. Gebhardt, G. Watson, L. M. Roberts, E. S. Vitetta, P. E. Thorpe and J. M. Lord, FEBS Lett., 1990, 273, 200–204 CrossRef; (d) D. F. W. M. B. Preijers, W. J. M. Tax, T. D. Witte, A. Janssen, H. d. Heijden, H. Vidal, J. M. C. Wessels and P. J. A. Capel, Br. J. Haematol., 1988, 70, 289–294 CrossRef PubMed.
- (a) A. N. Koja, T. Shibata and K. Mochida, Toxicon, 1980, 18, 611–618 CrossRef PubMed; (b) B. M. A. Poli, V. R. Rivera, J. F. Hewetson and G. A. Merrill, Toxicon, 1994, 32, 1371–1377 CrossRef PubMed; (c) C. E. Duriez, F. Fenaille, J.-C. Tabet, P. Lamourette, D. Hilaire, F. Becher and E. Ezan, J. Proteome Res., 2008, 7, 4154–4163 CrossRef PubMed.
- (a) A. P. G. Wahome, Y. Bai, L. M. Neal, J. D. Robertus and N. J. Mantis, Toxicon, 2010, 56, 313–323 CrossRef PubMed; (b) B. R. Rasooly and X. He, J. Food Prot., 2012, 75, 951–954 CrossRef PubMed.
- (a) A. C. S. Ramsden, M. T. Drayson and E. B. Bell, Toxicology, 1989, 55, 161–171 CrossRef PubMed; (b) B. R. F. Brown and D. E. White, Int. J. Exp. Pathol., 1997, 78, 267–276 CrossRef PubMed.
- (a) A. N. Verougstraete, D. Helsloot, C. Deprez, O. Heylen, I. Casier and K. Croes, J. Anal. Toxicol., 2019, 43, e1–e5 CrossRef PubMed; (b) B. S. L. Isenberg, M. D. Carter, M. A. Miller, A. I. Noras, M. A. Mojica, S. T. Carlsen, C. P. Bulathsinghala, J. D. Thomas and R. C. Johnson, J. Anal. Toxicol., 2018, 42, 630–636 Search PubMed.
- G. R. Waller and L. M. Henderson, J. Biol. Chem., 1961, 236, 1186–1191 CAS.
- (a) A. T. Robinson and E. Fowell, Nature, 1959, 183, 833–834 CrossRef PubMed; (b) B. P. Broun and C. Somerville, Plant Physiol., 1997, 113, 933–942 CrossRef PubMed.
- G. H. Choi, L. Kim, D. Y. Lee, C. long Jin, S.-J. Lim, B. J. Park, N.-J. Cho and J.-H. Kim, J. Appl. Biol. Chem., 2016, 59, 165–169 CrossRef CAS.
- (a) A. S. Jackson, D. J. Swiner, P. C. Capone and A. K. Badu-Tawiah, Anal. Chim. Acta, 2018, 1023, 81–88 CrossRef PubMed; (b) B. D. J. Swiner, S. Jackson, G. R. Durisek, B. K. Walsh, Y. Kouatli and A. K. Badu-Tawiah, Anal. Chim. Acta, 2019, 1082, 98–105 CrossRef PubMed.
- O. F. Lopez Nunez, A. F. Pizon and K. Tamama, J. Emerg. Med., 2017, 53, e67–e71 CrossRef PubMed.
- Y. Gal, O. Mazor, R. Falach, A. Sapoznikov, C. Kronman and T. Sabo, Toxins, 2017, 9, 311 CrossRef PubMed.
- (a) A. S. Das and V. K. Kataria, Med. J. Armed Forces India, 2010, 66, 255–260 CrossRef; (b) B. L. Polito, M. Bortolotti, M. G. Battelli, G. Calafato and A. Bolognesi, Toxins, 2019, 11, 324 CrossRef PubMed.
- P. Sehgal, O. Kumar, M. Kameswararao, J. Ravindran, M. Khan, S. Sharma, R. Vijayaraghavan and G. B. K. S. Prasad, Toxicology, 2011, 282, 56–67 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03317k |
| This journal is © The Royal Society of Chemistry 2020 |