
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew possible candidate structure for phase IV of solid hydrogen†
Guo-Jun Li ab,
Yun-Jun Gub,
Zhi-Guo Lib,
Qi-Feng Chen*b and
Xiang-Rong Chen*a
ab,
Yun-Jun Gub,
Zhi-Guo Lib,
Qi-Feng Chen*b and
Xiang-Rong Chen*a
aCollege of Physics, Sichuan University, Chengdu 610065, China. E-mail: xrchen@scu.edu.cn
bNational Key Laboratory for Shock Wave and Detonation Physics Research, Institute of Fluid Physics, China Academy of Engineering Physics, Mianyang 621900, China. E-mail: chenqf01@gmail.com
First published on 15th July 2020
Abstract
It has been proved in experiments that there are at least five phases of solid hydrogen at high pressure, however, only the structure of phase I has been absolutely determined. We revisited the phase space of solid hydrogen in the pressure range of 200–500 GPa using the particle swarm optimization technique combined with first-principles simulations. A novel orthorhombic structure named Ama2 is proposed as a possible candidate structure for phase IV. The Ama2 structure is a ‘mixed structure’ with two different types of layers and is distinctly different from the previously reported Pc structure. Enthalpies and Gibbs free energies show that Ama2 and Pc are competitive in the pressure region of phase IV. Nevertheless, the Raman and infrared vibron frequencies of Ama2 calculated by using density functional perturbation theory based on first-principles lattice dynamics show a better agreement with the experimental measurements than those of the Pc structure. And the pressure dependence of these low-frequency Raman vibrons of Ama2 obtained from the first-principles molecular dynamics simulation shows a steeper slope, which resolves the long-standing issue of large discrepancies between the calculated Raman frequencies and the experimental ν1 [P. Loubeyre, F. Occelli and P. Dumas, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134101 and C. S. Zha, R. E. Cohen, H. K. Mao and R. J. Hemley, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 4792]. Structural and vibrational analyses show that the hydrogen molecules in the weakly bonded molecular layer of Ama2 form distorted hexagonal patterns, and their vibration can be used to explain the experimental ν1 vibron. It is found that the weakly bonded layer is almost the same as the layers in the C2/c structure. This confirms the experimental conclusion [P. Loubeyre, F. Occelli and P. Dumas, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134101] that the ordering of hydrogen molecules in the weakly bonded molecular layers of the ‘mixed structure’ for phase IV is similar to that in the layers of the C2/c structure.
1 Introduction
Although the hydrogen atom consists of a single proton and electron, it exhibits complex behavior under high pressure, especially in the solid phase. Since Wigner and Huntington predicted that solid hydrogen transforms into a metallic atomic crystal above 25 GPa,1 it has attracted wide attention in theory and experiments due to the importance of developing and testing computer simulation methods, as well as its relevance to astrophysics.2 Later, some important quantum effects were also predicted, such as a metallic liquid ground state at high pressure and low temperature,3,4 high-Tc superconductivity,5–7 and metallic superfluid and superconducting superfluid states.8,9Early static diamond anvil cell (DAC) experiments showed that there were three different low temperature phases of solid hydrogen.2,10,11 Phase I is a molecular solid of quantum rotors, which adopts a hexagonal close packed lattice. It transforms into the broken-symmetry phase II at about 110 GPa. Phase III is characteristic of a large discontinuity in the vibron frequency, and exists above 150 GPa. Due to the development of static compression technologies, more phases of solid hydrogen have been recently discovered at higher temperatures and pressures. Phase IV, discovered at 300 K and above 220 GPa, exhibits a distinctly different spectrum compared with phase III, and has two vibron frequencies in its Raman and infrared (IR) spectra. The structure of phase IV is inferred as an anisotropic, mixed layer structure.12–14 Similarly, a series of new possible phases of solid hydrogen were proposed according to the change in the optical spectrum.15–17
Because of the very weak X-ray scattering by hydrogen atoms and small sample sizes, until now the only structure of solid hydrogen determined clearly by experimental measurements is the hexagonal close packed lattice of phase I.2 Therefore, much effort has been directed toward searching for candidate structures of solid hydrogen by combining DFT with optimization algorithms at high pressures and low temperatures, which has improved our understanding of the experimental observations18–22 and predicted some peculiar properties for hydrogen at pressures beyond those explored experimentally.23–26 The C2/c structure was proposed for phase III above 200 GPa and its Raman and IR vibrons exhibit good agreement with the experimental observations. Compared with phase III, the experimental Raman (infrared) spectrum of phase IV is more complex, and has two distinct vibron frequencies. This indicates a more complex structure for phase IV. The ‘mixed structure’ Pbcn was first proposed to explain the experimental Raman spectrum of phase IV.13 Later, Pickard et al. suggested that the ‘mixed structure’ Pc was more suitable for phase IV, because Pc was more stable than Pbcn based on the phonon dispersion calculations.20 The ‘mixed structure’ of Pc, characterized by two distinctly different layered structures, can qualitatively explain the two Raman and IR vibron peaks observed in phase IV. Nevertheless, the calculated values of the vibron frequencies are far from the experimentally measured values for phase IV. This may arise from significant differences between the Pc structure and the real structure.27,28 Moreover, Azadi et al.29 found that finite temperature and nuclear quantum effects, in addition to a strongly correlated band-gap energy and vibron modes, can reduce the band-gap substantially so that the Pc structure enters a metallic state, which is inconsistent with most experimental evidence. They concluded that the Pc structure was not a good candidate for phase IV. However, the recent single-crystal X-ray diffraction experiments of solid hydrogen indicate that the transitions from phase I to phases III and IV are possible an isostructural phase transition.30 Hence, for the structural model of phase IV, theoretical studies and experimental measurements have not yet achieved consistent conclusions.
It should also be mentioned that previous structure predictions were generally based on the potential energy surface calculated using the Perdew–Burke–Ernzerhof (PBE) functional.31 Recent theoretical calculations showed that the phase transition and metallization pressure of solid hydrogen strongly depended on the exchange–correlation (XC) functional.32–34 The XC functionals that take into account the van der Waals (vdW) interactions, such as optB88-vdW,35 vdW-DF1,36 and vdW-DF2,37 have an important effect on the phase stability and transition pressure of solid hydrogen, reducing further the discrepancy between the theoretical and experimental phase diagrams compared with the PBE functional.33,34,38,39
Given the above considerations, it is necessary to revisit the candidate structures for phase IV. The particle swarm optimization (PSO) technique has been demonstrated as a successful method to predict stable or metastable structures of various systems under the given external conditions (pressure).24,25,40–42 PSO combined with first-principles total energy calculations adopting the vdW-DF2 functional are used to search extensively and systematically for possible candidate structures at pressures from 200 GPa to 500 GPa. An orthorhombic structure named Ama2 is proposed as a possible candidate for phase IV, whose energy is competitive with that of the Pc structure. Furthermore, the properties of phonon dispersion, bond length, vibration mode, Raman spectra , and infrared spectra for Ama2 were studied in detail. The paper is organized as follows. The calculation method and details are described in Section 2. Section 3 presents the results and discussion. The final conclusions are presented in Section 4.
2 Method
We combined the PSO technique within the evolutionary scheme as implemented in CALYPSO code40,43 with first-principles total energy calculations achieved with the Vienna Ab initio Simulation Package (VASP)44 to search for structures of solid hydrogen. All 1200 simulation cells with 48 atoms per cell were produced at 300 GPa by the CALYPSO code during the structure evolution, which is enough to ensure convergence of the searched structures. This search was repeated at pressures of 400 GPa and 500 GPa. In the structure relaxations and electronic calculations, the projector augmented-wave (PAW) potential45,46 was adopted and the exchange–correlation functional was described by the vdW-DF2 functional. The basis set of plane waves with energy cutoff 1200 eV and the Monkhorst–Pack47 Brillouin zone sampling with a k-point grid with spacing 0.2 Å−1 were found to be sufficient for electronic structure calculation and structure relaxation. The convergence of total energy and force were set to less than 0.001 meV per proton and 0.1 meV Å−1, respectively.Force constants calculated with density functional perturbation theory (DFPT)48,49 were processed using the Phonopy code50 to get the phonon dispersion relation. In the dynamical matrix calculation, the value of the energy cutoff and the size of the k-point grid were the same as in the electronic structure calculation. Then, the vibrational free energy was calculated on a denser Monkhorst–Pack grid with a size of 41 × 41 × 41, which ensured that the vibration free energy converged to 0.01 meV per proton. To ensure that the zero-point (ZP) vibration energy (EZP) variation of all calculated structures converged to 0.1 meV per proton, a supercell with 96 atoms was adopted for the Ama2, C2/c, Pbcn, and Pc structures, while supercells with 144 and 128 atoms were adopted for the Cmca-12 and Cmca-4 structures, respectively (see Table S1 of the ESI† for more details). The total EZP and Gibbs free energy G of the candidate structures were calculated according to the harmonic approximation,
 | (1) |
 | (2) |
The Raman and IR spectra of solid hydrogen were calculated with the CASTEP code using the DFPT method based on first-principles lattice dynamics (LD) calculations.51 The PBE functional and norm-conserving pseudo-potentials, which were generated from the optimization scheme of Lin et al.,52 were adopted. The Raman spectra of the Ama2 structure were calculated using the vdW-DF2 functional within VASP code and post-processed with a python script.53,54
3 Results and discussion
3.1 Ama2 structure and relative stability
We found a series of low-energy structures (except for the Pbcn structure) that had been proposed in previous work.18–20 In addition, a new candidate Ama2 structure (space group 40) named as its short Hermann–Mauguin space-group symbol is found for the first time and has 24 atoms in the primitive unit cell. Due to the constraints of the periodic boundary conditions, layer A seems to split into two layers, but in fact, it all belongs to the same layer which is similar to layer C, as shown in Fig. 1. Therefore, Ama2 adopts an ABCDA… structure (see Table S2 of the ESI† for more detailed structural information). Ama2 has two distinct types of layers and is a ‘mixed structure’. One type of layer consists of weakly bonded hydrogen molecules (labeled as layers B and D) forming distorted hexagonal patterns (Fig. 1(c)) that can also be found in the C2/c structure, but are different from the graphene-like weakly bonded molecular layers in Pc.55 The other type of layer contains strongly bonded molecules (labeled as layers A and C) forming highly distorted hexagonal patterns (Fig. 1(d)). Therefore, Ama2 is completely different from Pc and Pbcn, and provides another possible arrangement of hydrogen molecules (H2) in solid hydrogen. | ||
| Fig. 1 The Ama2 structure (supercell with 96 atoms along the c axis) calculated by using the vdW-DF2 functional at 300 GPa. Numbers and red dashed lines in (c) and (d) represent the bond lengths and close contacts between atoms which are smaller than 1.2 (1.31) Å, respectively. Based on the bond lengths of the hydrogen molecules contained in the layers, layers A and C are classified as strongly bonded molecular layers, and layers B and D are classified as weakly bonded molecular layers. (a) The view of the Ama2 structure along the c axis. (b) The view of the Ama2 structure along the a axis. (c) The weakly bonded molecular layers where the molecules lie flat within a plane and form a distorted hexagonal pattern, similar to the layers in C2/c. (d) In the strongly bonded molecular layers, there are two different bond lengths which are 0.680 and 0.684 Å. Hydrogen molecules form a highly distorted hexagonal pattern, which seems to be similar to the layers in Cmca-12. | ||
At the static lattice level, the relative enthalpies of the candidate structures calculated by the vdW-DF2 functional are illustrated in Fig. 2(a). C2/c is the most stable structure and Cmca-4, considered as the metallic molecular phase, always has the highest enthalpy within the pressure range of 200–500 GPa. It is noted that the difference in enthalpy between Pc and Ama2 is smaller than 1 meV per proton above 250 GPa. Besides, the Ama2 phase is more competitive than Pbcn in the range 200–500 GPa. Phase IV of solid hydrogen occurs above 220 GPa, and Pc and Pbcn were both considered as candidate structures in previous literature.13,20 Based on the above comparison of the calculated energies, Ama2 can also be a candidate structure for phase IV. In order to investigate the effects of different XC functionals, we calculated the energies of the candidate structures at the static lattice level using the PBE functional, as shown in Fig. 2(b). The general trends in the energies for different structures coincide with the results of Pickard et al.,18 but are different from the results obtained with vdW-DF2 calculations. Compared with Pc, Ama2, and Pbcn, Cmca-12 is energetically uncompetitive at the vdW-DF2 level, but gets more competitive at the PBE level. The PBE calculations also show that Pc exhibits a slight energetic advantage compared with Ama2 and Pbcn, however, the energy difference between Ama2, Pc, and Pbcn is really small. All in all, the different XC functionals have an effect on the relative stability order of the candidate structures. Nevertheless, the PBE and vdW-DF2 calculations both show that the static lattice energies of Ama2 and Pc differ very little.
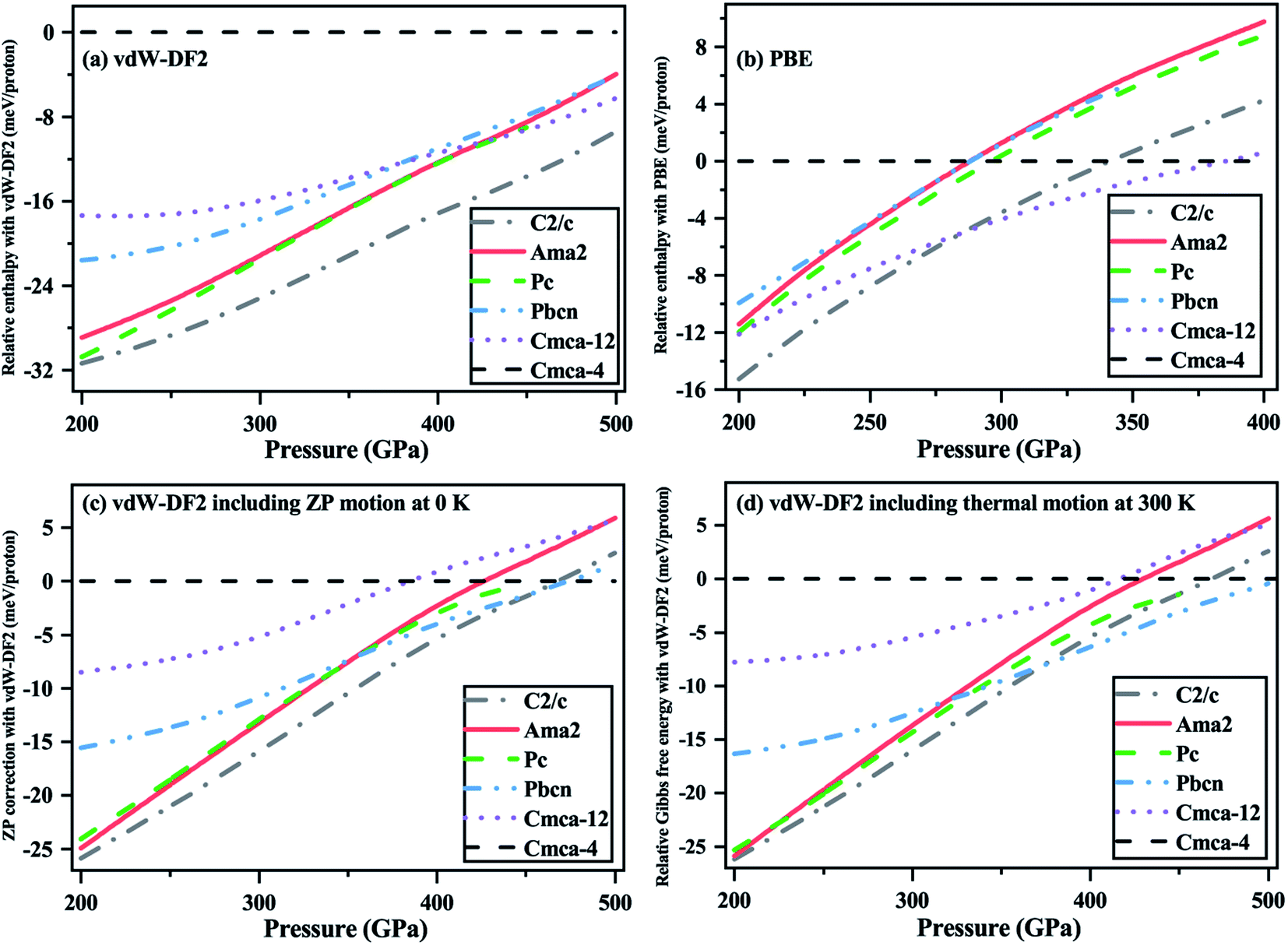 | ||
| Fig. 2 Enthalpies without and with ZP correction, and the Gibbs free energies of the Cmca-12 (purple short dashed line), Pbcn (blue dash-dot line), Pc (green dashed line), Ama2 (red solid line), and C2/c (gray dash-dot line) structures with respect to those of Cmca-4 (black dashed line). (a) and (b) present energies of the static lattice structures calculated using the vdW-DF2 and PBE functionals, respectively. Energies of the static lattice with proton ZP motion correction and Gibbs free energies calculated with the vdW-DF2 functional at 300 K are presented in (c) and (d), respectively. | ||
Due to the small mass of hydrogen nuclei, a strong ZP motion is expected. When the ZP vibrational energy is added to the static lattice energy, as illustrated in Fig. 2(c), C2/c remains stable below 453 GPa, which agrees with the previous predictions by diffusion Monte Carlo (DMC) calculations34 and the recent infrared spectroscopic measurements.56 The Ama2 structure is more competitive energetically compared with Pc and Pbcn below 345 GPa, whereas the Pbcn phase has the lowest energy compared with Ama2 and Pc above 355 GPa. As phase IV was discovered at 225 GPa and 300 K experimentally, we calculated the Gibbs free energies for these structures at 300 K within the harmonic approximation and focused on the difference in energy between the Ama2, Pbcn, and Pc structures. As shown in Fig. 2(d), owing to lattice vibration contributing to the global energy of the candidate structures, the pressure at which C2/c transforms to the Pbcn phase is reduced to 370 GPa. The difference in Gibbs free energy between Pc and Ama2 is smaller than about 1 meV, and Pbcn is energetically uncompetitive compared with Pc and Ama2 in the range of 200–320 GPa. The differences between Pc, Pbcn, and Ama2 become obvious above 350 GPa. In general, under the four different conditions considered above, Ama2 has relatively low energies, and is energetically degenerate with the previously highlighted Pc structure, especially for the pressure region of 200–350 GPa. Therefore, Ama2 is a possible candidate structure for phase IV from an energy point of view.
3.2 Phonon spectra
Phonon dispersion relations can be used to identify the stability of a candidate structure of solid hydrogen. Here, we depict the phonon dispersion relations of the C2/c, Ama2, Pc, and Pbcn phases at 300 GPa according to the harmonic approximation in Fig. 3. The Ama2 phase has no imaginary frequencies along the high symmetry point in the Brillouin zone, which suggests that the Ama2 structure is dynamically stable. But the Pc and Pbcn structures have negative frequencies in the vicinity of some high symmetry points, and the Pbcn phase has more unstable modes in reciprocal space than the Pc phase as suggested by Pickard et al.20 Based on a large number of phonon dispersion calculations, the Ama2 phase is stable over the investigated pressure range of 250–500 GPa, whereas Pc is unstable. Therefore, in terms of the phonon dispersion properties, Ama2 seems to be more competitive than the Pc structure for phase IV. To further confirm these structures, the Raman and IR properties of the Pc and Ama2 structures should be examined against the experimentally measured data, which will be discussed in detail in the next section. | ||
| Fig. 3 Phonon dispersions of (a) C2/c, (b) Ama2, (c) Pc, and (d) Pbcn calculated by using the vdW-DF2 functional at 300 GPa. | ||
3.3 Raman and IR spectra
Raman and infrared spectroscopy are crucial tools for identifying the candidate structures of phase IV. In Fig. 4 we show the Raman spectra of Ama2 at 300 GPa calculated by LD and the molecular projection method based on first-principles molecular dynamics (FPMD) simulations,57–59 together with the experimental Raman spectrum at 303 GPa. The FPMD calculation details can be found in the ESI.† At the LD level, the Raman spectra of Ama2 at 300 GPa calculated by vdW-DF2 and PBE are used to illustrate the dependence of the peak positions on the XC functional. Both functionals predict that Ama2 has two strong Raman vibron modes, which are consistent with the two intense Raman vibrons observed in experiments for phase IV.12,13,28 Based on the vibrational analysis, the lower and higher frequencies of Raman vibrons correspond to the hydrogen molecular vibrations of weakly bonded molecular layers and strongly bonded molecular layers, respectively (see Fig. S1 in the ESI†). The Raman vibron frequencies estimated by PBE are obviously smaller than those predicted by vdW-DF2. The lower and higher Raman vibron frequencies calculated by PBE and vdW-DF2 at 300 GPa are 3182/3939 and 4461/4817 cm−1, respectively. This is closely related to the bond lengths (BLs) calculated by PBE and vdW-DF2. The BLs of Ama2 calculated by the PBE functional are larger than those calculated by the vdW-DF2 functional. For instance, at 300 GPa, the BLs in the weakly (strongly) bonded molecular layers of Ama2 are 0.689/0.699 (0.68/0.684) Å according to vdW-DF2 and 0.766/0.78 (0.73/0.735) Å according to PBE (see the ESI† for more details). These results confirm the empirical formula relating the Raman frequency and bond length r, i.e. νr3 = constant.13 Although the results calculated by the PBE functional seem to be more reasonable than those calculated by the vdW-DF2 functional, the results still disagree with the experimental observation to some extent. This is due to the fact that the LD method, which is used to calculate the Raman spectrum at 0 K, doesn’t include the anharmonic effect (finite-temperature effect) on the proton. The FPMD method can effectively include the finite-temperature effect. Therefore, the Raman peaks calculated by the FPMD method coincide with the experimental measurement very well. And the experimentally broadened linewidth of the ν1 vibron12,13,28 is also captured by the FPMD method (see Fig. S2 in the ESI† for more details). | ||
| Fig. 4 Raman peaks of Ama2 compared with the experimental measurements. Raman peaks were calculated at 0 K and 300 GPa by the LD method using vdW-DF2 (pink line) and PBE (blue line). Raman peaks (red line) were calculated at 220 K and 300 GPa using the molecular projection method FPMD. Experimental data at 303 GPa and room temperature (black line) are taken from Zha et al.28 When the detected Raman frequency is higher, the sensitivity of the experimental detection device is lower, so the calculated vibronic amplitudes are not compared directly to the experimental measurements.13,57 The dashed line guides the eyes. | ||
Moreover, we used the LD method to calculate the pressure dependence of the Raman and IR vibron frequencies of Ama2 and the other candidate structures, which are compared with experimental measurements. The Raman and IR spectra of the Ama2 structure calculated at different pressures are depicted in Fig. S4,† and the derived pressure dependence of the vibron frequencies is presented in Fig. 5 (see Fig. S5† for the pressure dependence of low-frequency Raman frequencies). It is noted that the calculated IR intensity ratio between the two vibrons of Ama2 (see Fig. S4†) is inverse to the experimental one.27 This may be because the IR spectra calculated by LD don’t include the temperature effect. Below about 220 GPa, both Raman and IR measurements show only one vibronic frequency labeled as ν1 for phase III. The experimental vibronic frequency ν2 occurs above 225 GPa in the Raman and IR spectra and is identified as a signal of the transition from phase III to phase IV. Both the Pc and Ama2 structures have two intense vibrons in the Raman and IR spectra, whereas Cmca-12 and C2/c have only one intense vibron and can be excluded from the candidate structures of phase IV. For the lower-frequency vibron ν1, the calculated Raman and IR frequencies of Pc are lower than the experimental frequencies, especially for IR. This is probably because the coupling between the hydrogen molecules in the graphene-like weakly bonded layers of the Pc structure was overestimated, as suggested by Loubeyre et al.27 The low-frequency Raman and IR ν1 vibrons of Ama2 are close to those of C2/c and Cmca-12 (all of them consist of layers with H2 molecules forming distorted hexagonal patterns), and are better reproductions of the experimental observations than those of Pc. But the slope of the Raman ν1 vibron of Ama2 is still too flat. This is because LD calculations do not include the finite-temperature effect. The FPMD calculations are more consistent with the experimental measurements and provide a steeper slope for the Raman ν1 vibron of Ama2. The high-frequency Raman ν2 vibron of Ama2 calculated with FPMD perfectly reproduces the experimental observations, whereas the results of the LD calculations are smaller than the experimental results. The huge difference between the two methods confirms the experimental conclusion13 that the finite-temperature effect is a crucial factor for phase IV. Moreover, the frequencies of the Ama2 ν1 vibron calculated by FPMD show a better agreement with the experimental data compared with those of the Pc structure. This arises from the slight difference in molecule arrangement between Ama2 and Pc at a finite-temperature. To some extent, the Raman and IR properties of Ama2 narrow the gap between theoretical calculations and experiments.
 | ||
| Fig. 5 Pressure dependence of (a) Raman and (b) IR vibron frequencies of solid hydrogen. LD calculations were performed at 0 K for Ama2 (red solid line), Pc (green dashed line), C2/c (gray dash-dot line), and Cmca-12 (purple short dashed line). FPMD calculations were performed for Ama2 (red stars) at 220 K. Raman vibron frequencies of Pc (green stars) are taken from data calculated at 220 K by Magdău and Ackland.57 Experimental data are taken from Howie et al.13 (gray open squares), Loubeyre et al.27 (pink open triangles), and Zha et al.28 (blue open circles). | ||
4 Conclusion
In this work, we revisited the phase space of solid hydrogen in the pressure range of 200–500 GPa by combining the particle swarm optimization technique and first-principles energy calculations. The Ama2 structure was proposed as a possible candidate structure for phase IV of solid hydrogen. The new structure shares features of the C2/c, Cmca-12, Pc, and Pbcn structures. Ama2 adopts a ‘mixed structure’ and has two types of layers. This feature is consistent with the previously highlighted Pc and Pbcn structures. One type of layer with weakly bonded molecules forms distorted hexagonal patterns, which is almost the same as the layers in the C2/c structure. The other type of layer contains strongly bonded molecules which form highly distorted hexagonal patterns and non-hexagonal patterns. Moreover, the frequencies of the Raman and IR ν1 vibron of Ama2, arising from the vibrations of the weakly bonded molecular layers, agree better with the experimental ν1 frequencies than those of Pc. The features of the Ama2 structure confirm the experimental conclusion of Loubeyre et al.27 that the ordering of hydrogen molecules in the weakly bonded molecular layers of ‘mixed structures’ of phase IV is similar to that in the layers in the C2/c structure. The pressure dependent Raman ν1 vibron of Ama2 obtained from FPMD has a steeper slope and is more consistent with the experimental ν1. Phase IV is believed to exhibit strong quantum effects for the protons and a single classical structure may not fully describe phase IV.28 The Ama2 structure is a good addition to the structure model for phase IV in view of the DFT calculations. Moreover, it should be noted that the C2/c structure is more energetically stable than Pc and Ama2 within the pressure and temperature ranges of phase IV according to the DFT simulation. However, more advanced DMC calculations including anharmonic corrections stabilize the Pc structure.55 Therefore, further work will focus on investigating the stability of Ama2 and Pc by using the more advanced method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Hua Y. Geng for valuable discussion. This work is supported by the Foundation of National Key Laboratory of Shock Wave and Detonation Physics (Grant No. 6142A0301020217, and JCKYS2018212001), the National Natural Science Foundation of China (Grant No. 11872057, 11802280, and 11674292), the Science Challenge Project (Grant No. TZ2016001), the Science and Technology Development Foundation of China Academy of Engineering Physics (Grant No. 2013A0101001, and 2015B0102001), the NSAF (Grant No. U1830101), the Youth Program of National Natural Science Foundation of China (11804284) and the Foundation and Frontier Research Project of Chongqing (cstc2017jcyjAX0308). We are particularly grateful for the computational resources of the TianHe-2 at the LvLiang Cloud Computing Center of China.References
- E. Wigner and H. B. Huntington, J. Chem. Phys., 1935, 3, 764–770 CrossRef CAS.
- J. M. McMahon, M. A. Morales, C. Pierleoni and D. M. Ceperley, Rev. Mod. Phys., 2012, 84, 1607–1653 CrossRef CAS.
- S. A. Bonev, B. Militzer and G. Galli, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 014101 CrossRef.
- S. Deemyad and I. F. Silvera, Phys. Rev. Lett., 2008, 100, 155701 CrossRef PubMed.
- N. W. Ashcroft, Phys. Rev. Lett., 1968, 21, 1748–1749 CrossRef CAS.
- P. Cudazzo, G. Profeta, A. Sanna, A. Floris, A. Continenza, S. Massidda and E. K. Gross, Phys. Rev. Lett., 2008, 100, 257001 CrossRef CAS PubMed.
- J. M. McMahon and D. M. Ceperley, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 144515 CrossRef.
- S. A. Bonev, E. Schwegler, T. Ogitsu and G. Galli, Nature, 2004, 431, 669–672 CrossRef CAS PubMed.
- E. Babaev, A. Sudbø and N. W. Ashcroft, Phys. Rev. Lett., 2005, 95, 105301 CrossRef CAS PubMed.
- I. F. Silvera, Rev. Mod. Phys., 1980, 52, 393–452 CrossRef CAS.
- H.-k. Mao and R. J. Hemley, Rev. Mod. Phys., 1994, 66, 671–692 CrossRef CAS.
- M. I. Eremets and I. A. Troyan, Nat. Mater., 2011, 10, 927–931 CrossRef CAS PubMed.
- R. T. Howie, C. L. Guillaume, T. Scheler, A. F. Goncharov and E. Gregoryanz, Phys. Rev. Lett., 2012, 108, 125501 CrossRef PubMed.
- C. S. Zha, Z. Liu and R. J. Hemley, Phys. Rev. Lett., 2012, 108, 146402 CrossRef PubMed.
- R. T. Howie, T. Scheler, C. L. Guillaume and E. Gregoryanz, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 214104 CrossRef.
- P. Dalladay-Simpson, R. T. Howie and E. Gregoryanz, Nature, 2016, 529, 63–67 CrossRef CAS PubMed.
- R. T. Howie, P. Dalladay-Simpson and E. Gregoryanz, Nat. Mater., 2015, 14, 495–499 CrossRef CAS PubMed.
- C. J. Pickard and R. J. Needs, Nat. Phys., 2007, 3, 473–476 Search PubMed.
- C. J. Pickard and R. J. Needs, Phys. Status Solidi B, 2009, 246, 536–540 CrossRef CAS.
- C. J. Pickard, M. Martinez-Canales and R. J. Needs, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 214114 CrossRef.
- B. Monserrat, R. J. Needs, E. Gregoryanz and C. J. Pickard, Phys. Rev. B, 2016, 94, 134101 CrossRef.
- B. Monserrat, N. D. Drummond, P. Dalladay-Simpson, R. T. Howie, P. López Ríos, E. Gregoryanz, C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2018, 120, 255701 CrossRef CAS PubMed.
- J. M. McMahon and D. M. Ceperley, Phys. Rev. Lett., 2011, 106, 165302 CrossRef PubMed.
- H. Y. Geng, H. X. Song, J. F. Li and Q. Wu, J. Appl. Phys., 2012, 111, 063510 CrossRef.
- H. Liu, H. Wang and Y. Ma, J. Phys. Chem. C, 2012, 116, 9221–9226 CrossRef CAS.
- H. Y. Geng, Q. Wu and Y. Sun, J. Phys. Chem. Lett., 2017, 8, 223–228 CrossRef CAS PubMed.
- P. Loubeyre, F. Occelli and P. Dumas, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134101 CrossRef.
- C. S. Zha, R. E. Cohen, H. K. Mao and R. J. Hemley, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4792–4797 CrossRef CAS PubMed.
- S. Azadi, R. Singh and T. D. Kühne, J. Comput. Chem., 2018, 39, 262–268 CrossRef CAS PubMed.
- C. Ji, B. Li, W. Liu, J. S. Smith, A. Majumdar, W. Luo, R. Ahuja, J. Shu, J. Wang, S. Sinogeikin, Y. Meng, V. B. Prakapenka, E. Greenberg, R. Xu, X. Huang, W. Yang, G. Shen, W. L. Mao and H.-K. Mao, Nature, 2019, 573(7775), 558–562 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Azadi and W. M. C. Foulkes, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 014115 CrossRef.
- S. Azadi and G. J. Ackland, Phys. Chem. Chem. Phys., 2017, 19, 21829–21839 RSC.
- J. McMinis, R. C. Clay, D. Lee and M. A. Morales, Phys. Rev. Lett., 2015, 114, 105305 CrossRef PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101(R) CrossRef.
- M. A. Morales, J. M. McMahon, C. Pierleoni and D. M. Ceperley, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184107 CrossRef.
- R. C. Clay, J. McMinis, J. M. McMahon, C. Pierleoni, D. M. Ceperley and M. A. Morales, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 184106 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Chen, H. Y. Geng, X. Yan, Y. Sun, Q. Wu and X. Chen, Inorg. Chem., 2017, 56, 3867–3874 CrossRef CAS PubMed.
- X. Zhong, L. Yang, X. Qu, Y. Wang, J. Yang and Y. Ma, Inorg. Chem., 2018, 57, 3254–3260 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. Giannozzi, S. de Gironcoli, P. Pavone and S. Baroni, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 7231–7242 CrossRef CAS PubMed.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. S. Lin, A. Qteish, M. C. Payne and V. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 4174–4180 CrossRef PubMed.
- A. Fonari and S. Stauffer, https://github.com/raman-sc/VASP/, 2013.
- D. Porezag and M. R. Pederson, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7830–7836 CrossRef CAS PubMed.
- N. D. Drummond, B. Monserrat, J. H. Lloyd-Williams, P. Lòpez Rìos, C. J. Pickard and R. J. Needs, Nat. Commun., 2015, 6, 7794 CrossRef CAS PubMed.
- P. Loubeyre, F. Occelli and P. Dumas, Nature, 2020, 577, 631–635 CrossRef CAS PubMed.
- I. B. Magdău and G. J. Ackland, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 174110 CrossRef.
- I. B. Magdău and G. J. Ackland, J. Phys.: Conf. Ser., 2014, 500, 032012 CrossRef.
- G. J. Ackland and I. B. Magdău, High Pressure Res., 2014, 34, 198–204 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The structural information for the Ama2 phase, vibrational analysis, the FPMD details, and the pressure dependence of the bond lengths are given. See DOI: 10.1039/d0ra03295f |
| This journal is © The Royal Society of Chemistry 2020 |